

Abstract
Sepsis remains an important health problem worldwide due to inefficient treatments often resulting in multi-organ failure. Neutrophil recruitment is critical during sepsis. While neutrophils are required to combat invading bacteria, excessive neutrophil recruitment contributes to tissue damage due to their arsenal of molecular weapons that do not distinguish between host and pathogen. Thus, neutrophil recruitment needs to be fine-tuned to ensure bacterial killing, while avoiding neutrophil-inflicted tissue damage. We recently showed that the actin-binding protein HS1 promotes neutrophil extravasation; and hypothesized that HS1 is also a critical regulator of sepsis progression. We evaluated the role of HS1 in a model of lethal sepsis induced by cecal-ligation and puncture. We found that septic HS1-deficient mice had a better survival rate compared to WT mice due to absence of lung damage. Lungs of septic HS1-deficient mice showed less inflammation, fibrosis, and vascular congestion. Importantly, systemic CLP-induced neutrophil recruitment was attenuated in the lungs, the peritoneum and the cremaster in the absence of HS1. Lungs of HS1-deficient mice produced significantly more interleukin-10. Compared to WT neutrophils, those HS1-deficient neutrophils that reached the lungs had increased surface levels of Gr-1, ICAM-1, and L-selectin. Interestingly, HS1-deficient neutrophils had similar F-actin content and phagocytic activity, but they failed to polymerize actin and deform in response to CXCL-1 likely explaining the reduced systemic neutrophil recruitment in HS1-deficient mice. Our data show that HS1 deficiency protects against sepsis by attenuating neutrophil recruitment to amounts sufficient to combat bacterial infection, but insufficient to induce tissue damage.
Keywords: Inflammation, HCLS, Cortactin, Diapedesis, Neutrophil extravasation, Actin dynamics
1. Introduction
Sepsis is a highly mortal systemic overwhelming host immune response to infection (Liu et al., 2014), with bacteremia being a defining feature, among others (Mansur et al., 2015). Sepsis is considered a medical emergency requiring immediate treatment to improve the chances of survival (Kumar et al., 2006). To prevent this challenging condition, the innate immune system must provide immediate protection against invading pathogens. To this end, neutrophils are rapidly activated and recruited upon pathogen sensing (Kolaczkowska and Kubes, 2013; Nauseef and Borregaard, 2014). Neutrophil recruitment requires multiple subsequent adhesive interactions with the vascular endothelium including tethering, rolling, firm adhesion and diapedesis (Nourshargh and Alon, 2014; Schnoor et al., 2015; Vestweber, 2015). However, these canonical mechanisms vary in the lung, an organ that often fails during sepsis due to severe tissue damage (Hickey and Westhorpe, 2013; Rossaint and Zarbock, 2013, 2015). Once recruited, neutrophils release an arsenal of anti-microbial substances that kill invading bacteria (Rosales, 2018). However, these secreted products including reactive oxygen species (ROS) and proteases can also inflict serious tissue damage (Kruger et al., 2015; Wang, 2018). Thus, strict regulation of neutrophil recruitment is required to ensure effective clearance of bacteria while avoiding tissue damage. During sepsis, excessive uncontrolled neutrophil recruitment takes place; and evidence exists that attenuating this event can have protective effects during sepsis (Goncalves-de-Albuquerque et al., 2018; Hasan et al., 2011; Lerman et al., 2014; Spite et al., 2009). Of note, the timing of the neutrophil response seems critical for sepsis outcome. Depleting neutrophils at early stages of sepsis increase mortality, whereas depleting neutrophils at later stages showed improved disease outcome (Hoesel et al., 2005). A better understanding of the molecular mechanisms driving this excessive neutrophil recruitment during sepsis is therefore of utmost importance to develop novel treatment strategies.
Neutrophil extravasation is an adhesive and migratory event that depends on actin remodeling. Actin dynamics are known to be critical for neutrophil recruitment during sepsis (Schnoor et al., 2017); and are regulated by different actin-binding proteins (ABP). Neutrophils strongly express the ABP hematopoietic cell-specific lyn substrate (HCLS1 or HS1) (Castro-Ochoa et al., 2019; Cavnar et al., 2012). We recently showed that HS1 deficiency reduced neutrophil extravasation in the inflamed cremaster and peritoneum in vivo by regulating the activation of small GTPases and integrins (Latasiewicz et al., 2017). Given that excessive neutrophil recruitment contributes to severe tissue damage during sepsis, we hypothesized that the absence of HS1 protects against experimental murine polymicrobial sepsis by reducing, but not ablating, neutrophil recruitment.
2. Materials and methods
2.1. Mice and CLP-induced sepsis
All animal experiments have been approved by the institutional animal care and use committee and the bioethics committee of Cinvestav. 8–12 weeks old male littermate WT and HS1-KO mice (Taniuchi et al., 1995) on C57BL/6 background (at least 20 backcrosses) were used for the induction of sepsis by cecal ligation and puncture (CLP) (Toscano et al., 2011). Briefly, mice were anesthetized by i.p. injection of 125 mg/kg ketamine hydrochloride (Anesket, PISA, Mex) and 12.5 mg/kg xylazine (Procin, PISA, Mex); the abdominal area was shaved and dis-infected, a 1.5 cm incision was made in the linea alba, the cecum was carefully exposed, the distal portion of the cecum was ligated using 4–0 sterile silk (between 30% and 40% of the cecum), punctured once with a 21 G needle and 0.5 mm of feces were exposed. Subsequently, the cecum was returned to the peritoneal cavity and the wound closed with silk and disinfected again. Sham control mice were subjected to the same surgical procedure without ligation and puncture of the cecum. 24 h after surgery, mice were anesthetized again, and blood was collected from the heart for further analysis. Mice were sacrificed by anesthesia overdose. Before extraction of organs, mice were perfused by injection of 20 mL PBS through the heart right ventricle to remove all blood. For survival curves, operated mice were observed for five days.
2.2. Histology
Twenty-four h after CLP, mice were sacrificed by anesthesia overdose. For H&E staining, after tissue perfusion, the right lung was ligated from the trachea, cut and lysed for protein and RNA analyzes. The left lung was perfused via the trachea using 1 mL of 10% formalin and incubated for 5 min. Subsequently, the lung was excised from the thoracic cavity and submerged in formalin for 24 h. Paraffin cross-sections of the lung were stained for hematoxylin and eosin using standard protocols (Citalan-Madrid et al., 2017). A semi-quantitative analysis of lung histology was performed in a blinded fashion by a pathologist using a score varying from 0 (no lesion) to 4 (major and extended lesions) for each of the following criteria: fibrosis, edema, alveolar wall thickening, alveolar hemorrhage, vascular congestion, and presence of mucus (Eveillard et al., 2010; Klopfleisch, 2013).
2.3. Intravital microscopy (IVM)
WT and HS1-KO mice were anesthetized as described above. Cremaster muscles of WT or HS1-KO mice were prepared for IVM 24 h after sham or CLP operation. Postcapillary venules with a diameter of 20–40 μm were recorded using an intravital upright microscope (Axioscope A1, Carl Zeiss Germany) with a 40x saline immersion objective (Schnoor et al., 2011). Rolling and arrest were quantified by transillumination IVM; and transmigrated cells were quantified by differential interference contrast (DIC) microscopy. Cells that transmigrated were determined within an area of 100 μm of vessel length over 75 μm on each side of the vessel (Latasiewicz et al., 2017). Videos and images were analyzed using ImageJ (NIH, Bethesda, MD) and ZEN 2 Blue Edition (Carl Zeiss, Germany) softwares. Rolling flux fraction was calculated as percent of total leukocyte flux. Blood flow center line velocity was determined from recorded videos after tail vein injection of 50 μL of sonicated 2 μm Fluospheres (ThermoFisher Scienftific).
2.4. Lung permeability
Fluorescent microbeads (2 μm diameter) were injected into the tail vein, and 2 h later mice were perfused with 20 mL PBS via the right heart ventricle. Then lungs were instilled via the trachea with approximately 1 mL of a 1:1 dilution of Tissue-Tek and PBS, subsequently, the trachea was ligated with silk to avoid leakage before tissue extraction and embedding in Tissue-Tek. 8 μm cross-sections were cut and fixed in 100% ethanol at −20 °C for 20 min. Tissues were analyzed by fluorecence microscopy and mean fluorescence intensities of fluorescent beads that leaked into the lungs were quantified using ImageJ software.
2.5. qRT-PCR
Lung RNA was obtained using TRIzol (Invitrogen, Thermo Fisher Inc.). RNA was quantified using a NanoDrop® ND-1000 spectrophotometer (Invitrogen, Thermo Fisher Inc.). cDNA synthesis was performed using Oligo-dT primers and SuperScript II reverse transcriptase (Invitrogen, Thermo Fisher Inc.) according to the manufacturer’s instructions. For real-time PCR, 100 ng of cDNA of each sample was analyzed using SYBR Green PCR Master Mix in a StepOne™ Real-Time PCR System (Applied Biosystems). The conditions used were: activation 95 °C for 10 min, annealing at 62 °C for 30 s, signal acquisition and extension at 72 °C for 15 s Actb (β-actin) and Gapdh were used as house-keeping genes. Data were analyzed using the ΔΔCt-method (Schnoor et al., 2008).
2.6. Determination of serum cytokine levels
This was done using a Milliplex 25plex-Immunology Multiplex Assay kit (Millipore) according to the manufacturer’s instructions.
2.7. Antibodies
Antibodies against murine antigens (BioLegend, Mexico-City): APC-anti-L-selectin, Cy7-anti-CD45, ICAM-1 (clone YN1.1, uncoupled) with secondary AF488-goat-anti-mouse, PE-anti-CD117 (clone ACK2), APC-anti-Ly6G (clone 1A8), F4/80-PerCP/Cy5.5, CD3-PE, CD19-BV421, CD45-FITC, APC-anti-CD106 (clone 429). Alexa647-anti-HS1 (clone D5A9 XP) was purchased from Cell Signaling Technology.
2.8. Flow cytometry
After sacrification, lung tissues were perfused with PBS by intracardial injection. The left lung was cut into small pieces in 500 μL of cold HBSS containing Ca2+ and Mg2+, 2.5 mg/mL of collagenase A (Sigma), 1 mg/mL of DNase I and 0.5% FBS. Tissues were then incubated in a 24-well plate at 37 °C for 30 min with orbital agitation at 125 rpm. Cell suspensions were further disrupted using a 19 G syringe, passed through a 40 μm cell strainer (Corning) and washed with 5 mL of PBS. Cell suspensions were centrifuged at 1500 rpm for 5 min at 4 °C and resuspended in 1 mL of PBS + 0.5% FBS for flow cytometry analysis.
Peripheral blood (PB) was obtained by cardiac puncture. Cells from lung, blood and peritoneal lavage fluid were counted and an equal number of cells was blocked with TruStain FcX (Biolegend). Leukocyte populations were identified using specific, labeled antibodies: CD45-FITC for total leukocytes, F4/80-PerCP-Cy5.5 for macrophages, CD3-PE for T cells, CD19-BV421 for B cells, and neutrophils were detected using either anti-Gr-1-PE or anti-Ly6G APC-Cy7 antibodies. Stainings were performed on ice for 30 min and then washed with PBS containing 3% FBS. 7-AAD was employed to determine dead cells. Data were acquired using a FACS canto II and analyzed using FlowJo Trelstar V10 software.
2.9. Peritoneal lavage
Twenty four h after CLP, mice were euthanised by anesthesia overdose and the abdominal surface sprayed with 70% ethanol. A total of 10 mL (5 mL, 2 times) of cold sterile PBS was injected into the peritoneum, the peritoneal lavage fluid recovered, and placed on ice until further usage for bacterial counting and flow cytometry to determine neutrophil numbers.
2.10. Bacterial counts
Colony-forming units (CFU) were determined by inoculating peritoneal lavage fluid and blood on tryptase soy agar (MCD Laboratory, Mexico), and incubated at 37°C overnight. CFU were counted after 24 h.
2.11. HoxB8 progenitors
In brief, murine hematopoietic progenitors were isolated from bone marrow with the EasySep Mouse HSPC Enrichment Kit (StemCell Technologies) and transduced with the tamoxifen-inducible expression vector HoxB8 as described previously (Wilson et al., 2020). Cells were cultured in basal media: Opti-MEM-reduced serum media with Gluta-MAX Supplement, 10% FBS, 1% non-essential amino acids, 1% penicillin-streptomycin, and 30 μM 2-mercaptoethanol. For selection of transduced cells, progenitors were cultured in the presence of 100 nM 4-hydroxytamoxifen (4-OHT), 50 ng/mL recombinant murine stem cell factor (mSCF), and 1 μg/mL puromycin (all from BioLegend). The surviving progenitor cells were used as WT progenitor cell lines and maintained in basal media supplemented with mSCF and 4-OHT (Wilson et al., 2020). Progenitors were differentiated into neutrophils by washing the cells three times in 10 mL of PBS and resuspending them in basal media containing 20 ng/mL mSCF and 20 ng/mL recombinant murine granulocyte colony-stimulating factor (mG-CSF, BioLegend) for two days followed by washing the cells three times with 10 mL of PBS and resuspending them in basal media containing 20 ng/mL mG-CSF for another three days. Successfully differentiated neutrophils exhibited multi-lobed nuclei, expression of Ly6G, and loss of CD117 expression.
To generate a HS1 knockout (KO) cell line, HoxB8 progenitors were transduced with a lentiviral vector that expressed Cas9 and single-guide RNA (sgRNA) targeting the HS1 gene. We used the pLentiCRISPR v2 vector (gift from Feng Zhang – Addgene plasmid #52961) modified to confer blasticidin resistance, and the target sgRNA sequence TGTATCCCAGTCATCACCCT. Empty vector expression without targeting sgRNA was used as wild-type control. HS1 knockouts were selected using 5 μg/mL blasticidin. The KO were analyzed by flow cytometry using AlexaFluor647-anti-HS1 (clone D5A9XP, Cell Signaling Technology) and APC-anti-CD106 (clone 429 (MVCAM.A), BioLegend) as the isotype control and confirmed complete HS1-KO.
2.12. Phagocytosis assays
Twenty-four h after CLP, 400 μL of GFP-labeled zymosan (200 μg) (Invitrogen, Thermo Fisher Inc.) were injected into the peritoneal cavity using a syringe with 27 G needle. The abdomen was gently shaken to distribute the zymosan particles and incubated for 15 min. Afterwards, mice were anesthetized for 10 min and peritoneal lavage performed as described above. The recovered peritoneal fluid was analyzed by flow cytometry and the number of neutrophils that phagocytosed zymosan were quantified.
In the case of HoxB8 neutrophils, cells were washed three times in PBS and 2 × 106 neutrophils were resuspended in 100 μL of HBSS++ containing 0.3 mg/mL pHrodo Green S. aureus and 10% FBS. The samples were incubated for 0 or 90 min at 37 °C. To quench phagocytosis, 1 mL of ice-cold PEB (PBS, 1 mM EDTA, 1% BSA) was added to the samples and placed on ice. After washing once and resuspending in 200 μL of PBS, the cells were stained using the Zombie Violet Fixable Viability Kit (BioLegend) and APC anti-Ly6G for 15 min on ice. After a final wash, cells were analyzed by flow cytometry for phagocytosis in the FITC channel in the Ly6Ghigh populations.
2.13. Actin polymerization assay
Peritoneal lavage and peripheral blood were collected as mentioned above. 1 × 106 cells were incubated in 200 μL RPMI medium containing 1% FBS and 300 ng/mL recombinant mouse CXCL1 (BioLegend) for 0.5–5 min and subsequently fixed with 4% PFA for 20 min at RT. Cell suspensions were blocked for 20 min with anti-mouse TruStain FcX, and stained with anti-CD45-PB, anti-Ly6G-PE/Cy7 antibodies and Alexa fluor 488-phalloidin for 30 min at RT in PBS containing 0.5% Triton X-100 + 3% FBS. Stained neutrophils were identified by gating CD45+ Ly6G+ cells using a FACS canto II and the MFI of filamentous actin in these cells were quantified using FlowJo V10 software.
In the case of HoxB8 neutrophils, cells were washed three times in PBS, resuspended in 200 μL of PBS, and stained for 15 min with Zombie Violet. Afterward, the cells were washed once in PBS, and 2 × 106 neutrophils were resuspended in 100 μL of HBSS++ containing 0.1% Pluronic solution and 50 ng of CXCL1. After 10 s, 30 s, 60 s, and 120 s, the reactions were quenched by adding 100 μL ice-cold 8% Formaldehyde, 4% Glutaraldehyde solution (Huebinger et al., 2018), and placing on ice for 10 min. As control (0 s time point), the neutrophils were resuspended in the buffer solution without CXCL1 and quenched immediately. Samples were then washed with PBS and resuspended in 200 μL of permeability buffer (0.1% Tween 20 in PBS) at room temperature for 10 min. The samples were washed once more and resuspended in 200 μL blocking buffer (5% BSA in PBS) at room temperature for 30 min, followed by staining with Phalloidin 488 for 25 min and APC anti-Ly6G for 15 min. The samples were washed and analyzed by flow cytometry. The MFI of the FITC channel was compared across the timepoints and cell lines in the Ly6Ghigh populations.
2.14. Quantification of ROS production
Cells from peritoneal lavage fluid and from digested lung tissue were incubated in 200 μL of RPMI containing 10 μM DHR for 20 min at 37 °C. Cells were then immediately fixed with 4% PFA at RT for 20 min and immunostained with anti-CD45-PB and anti-Ly6G-APC/Cy7 antibodies. Fluorescence generated by DHR oxidation was quantified by flow cytometry using the FITC channel.
2.15. Microfluidic assay
To construct the microfluidic constriction platforms, the devices were designed in AutoCAD and sent out for photolithography to Front Range Photomask (Lake Havasu City, Arizona). The silicon master was then manufactured at the Microfabrication Core Facility at Harvard University. After receiving the master, the silicon wafer was silanized to passivate the surface by dispensing 20 μL of polydimethylsiloxane (PDMS) (Sylard, Dow Corning) onto foil and placing it next to the silicon wafer within a vacuum desiccator for 30 min. Finally, the silicon wafer was placed on a hotplate at 150 °C for 10 min to evaporate the excess silane. The PDMS stamps were cut out with the molded microfluidic design imprinted on them, and holes were punched using a blunt 18 G needle for inlet and outlet tubing. The surface of the PDMS was cleaned with Scotch tape. Glass coverslips were soaked in concentrated sulfuric acid overnight, rinsed with copious amounts of Millipore water, and then rinsed with 70% ethanol and dried. The PDMS stamp (design side up) and the glass coverslip were subject to the corona discharge generated by a Tesla coil (Model BD-20, Electro-Technic Products, INC) for two minutes. The PDMS stamp and the glass coverslip were then sandwiched together (design facing the glass) to form the whole microfluidic device.
For the constriction experiments, microfluidic devices with constrictions of 5 μm across and 8 μm high were used. Transit of neutrophils was followed through the constrictions and their dwell times were recorded using fluorescence microscopy with a frame rate of 0.053 s for 20 min. The flow rate set on the syringe pump (Braintree Scientific) was 250 nl/min for two syringes for a total of 500 nL/min throughout the device with an average fluid velocity of 1.5 mm/s, similar to the reported values for fluid velocity in the pulmonary capillaries (Ekpenyong et al., 2015). Data were exported as TIFF files to ImageJ, where the particle tracks were built with the Plugin Trackmate (Schindelin et al., 2012; Tinevez et al., 2017). The Trackmate files were exported to Mathematica, where final data filtering was applied; namely, removing non-signal cell species, identifying cells that lysed in the constrictions, and collecting dwell times into cumulative distributions. Finally, these dwell times were plotted for statistical analysis as Kaplan-Meier Curves and their hazard ratios reported. Data sets were done in triplicate and repeated independently three times.
For cell preparation on the day of the microfluidic experiment, neutrophils were washed three times in optimal buffer (PBS, 1 mM EDTA, 2% FBS). The EasySep Mouse Neutrophil Enrichment Kit (StemCell Technologies) was used to purify differentiated neutrophils from undifferentiated progenitors. Neutrophils were always kept on ice until used in the microfluidic device. 1 × 106 neutrophils were washed two times with PBS, resuspended in 200 μL PBS, and stained with CSFE for 15 min, washed two times with PBS, and counted. Stained neutrophils were resuspended in HBSS++, 0.1% Pluronic solution to a final concentration of 1 × 106 cells/mL. Neutrophils were stimulated with 1 ng/mL CXCL1 in the microfluidics device via two inlets—one for the neutrophils and the other for the stimulus. The final concentration of CXCL1 on-chip was 0.5 ng/mL. Samples were imaged with a TILL Photonics iMIC TIRF microscope (FEI Company) with an Olympus 10x UPlanFI objective, NA 0.30 and an Andor iXon3 EMCCD camera.
2.16. Statistics
For multi-group comparisons, one-way ANOVA with Bonferroni’s post-hoc test was applied. For comparison of two different groups, Student t-test with Welch’s correction was applied. The non-parametric Mann-Whitney U-test with Bonferroni’s correction has been used to analyze histological scores. p-values < 0.05 were considered statistically significant.
3. Results
3.1. HS1 deficiency improves survival of mice after induction of lethal sepsis
To analyze the effect of HS1 deficiency on sepsis development, mice were subjected to sham or CLP surgeries and monitored for five days. As expected, all sham-operated mice survived the experimental period (Fig. 1A). CLP-operated WT animals showed a mortality rate of 40% after 24 h, which increased to 92% after 48 h, and 100% after 72 h. HS1-KO mice showed improved survival, with mortality rates of 22% after 24 h, 60% after 48 h and 72 h, and 67% after 96 h. Three septic HS1-KO mice recovered completely. We also measured body weights during this experiment. Sham-operated animals barely lost any weight during the entire experimental period of five days (Fig. 1B). Due to lethality, WT CLP mice could only be weighed after 24 h and only one surviving mouse after 48 h. During this period the mice did not lose much weight. However, HS1-KO CLP mice lost 16–18% of weight after 2–3 days, respectively, but afterwards started to gain weight (Fig. 1B), indicating that after three days of sepsis, HS1-KO mice started to recover from the disease.
Fig. 1.
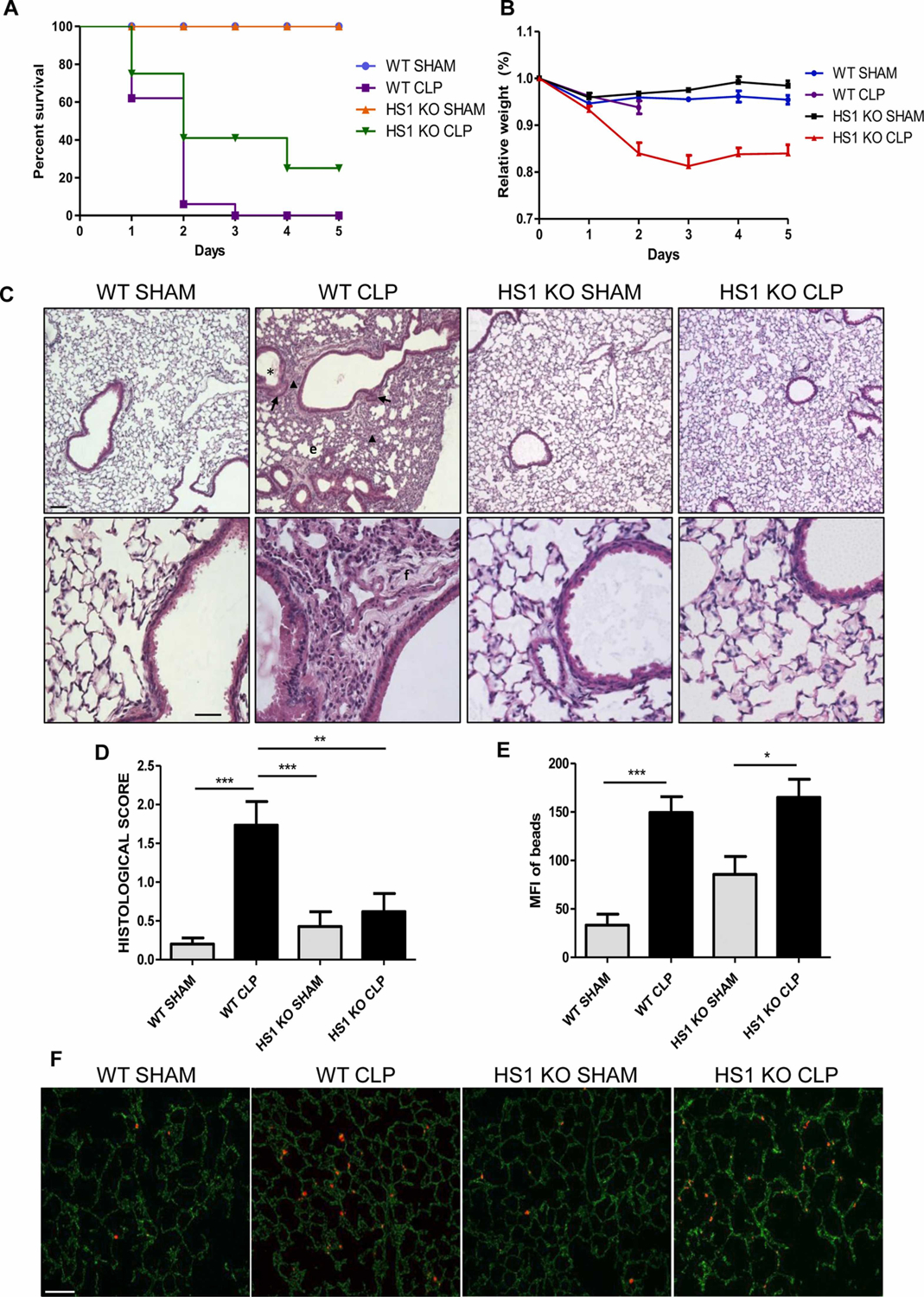
HS1 deficiency protects against CLP-induced sepsis and lung tissue damage. (A) 8–12 week-old C57BL/6 male mice were subjected to sham or CLP surgeries and monitored for 5 days (WT Sham (n = 10), WT CLP (n = 12), HS1-KO Sham (n = 9), and HS1-KO CLP (n = 10) mice). Data are represented as percentage of survival for each group. (B) Body weight of these animals was measured daily. Data are represented as percentage of body weight for each group relative to the body weight before surgeries. (C) Representative hematoxylin and eosin stainings of lung cross-sections from WT and HS1-KO mice, sham, and CLP (n = 5). Upper panels show images taken at 10x. Bar = 60 μm. Lower panels show magnifications at 40x. Bar = 20 μm. Images of WT and HS1-KO lungs show normal lung morphology. WT CLP mice showed clear signs of lung inflammation such as mucus in bronchi lumen (*), congested capillaries (arrow), alveoli wall thickening and collapse (arrowhead), and fibrosis (f). By contrast, HS1-KO CLP mice showed lung morphology similar to WT sham lungs. (D) Lung histopathological scores were quantified by a pathologist in a blinded fashion. A score of zero corresponds to normal histology, while a score of 4 corresponds to the strongest damage as described in methods. Scores are depicted as means +/− SEM; n = 5; **p < 0.01, ***p < 0.001. (E) Quantification of vascular permeability to 2 μm fluorescent beads in the lungs as depicted in the representative fluorescence images (F) revealed a similar increase in permeability after CLP in both WT and HS1-KO mice. Bar = 100 μm. Data are depicted as means +/− SEM; n = 5. *p < 0.05, ***p < 0.001.
3.2. Absence of HS1 protects lungs from CLP-induced tissue damage
Although renal failure is the major cause of morbidity in patients with sepsis (Hotchkiss et al., 2016), severe lung damage is one of the most common manifestations of sepsis (Englert et al., 2019; Kim and Hong, 2016). Thus, we decided to first analyze lung histology. To this end, lungs were perfused to remove blood; and hematoxylin/eosin-stained cross-sections were evaluated by a pathologist in a blinded fashion. Normal lung histology could be observed in both WT and HS1-KO mice after sham surgeries with no apparent signs of inflammation (Fig. 1C). As expected, lungs from WT CLP mice showed severe signs of inflammation and tissue damage including alveoli wall thickening and collapse, presence of mucus in bronchi lumen, immune cell infiltration, capillary congestion, and fibrosis 24 h after CLP (Fig. 1C). By contrast, septic HS1-KO mice showed very few abnormalities, and the overall histology was comparable to sham controls suggesting that HS1-deficiency protects lungs during sepsis (Fig. 1C). Histological scores of the lungs confirmed this impression with average scores of 0.2+/−0.04 and 0.42+/−0.18 for the WT and HS1-KO sham-operated groups, respectively (Fig. 1D). By contrast, WT CLP mice had a significantly increased score of 1.73+/−0.31; whereas the score of HS1-KO CLP mice only increased to 0.61+/−0.22, which was not significantly different when compared to sham controls, but significantly improved when compared to WT CLP mice (Fig. 1D).
We also performed in vivo permeability assays using fluorescent beads. Vascular permeability in the lung significantly increased after CLP (Fig. 1E, representative images shown in Fig. 1F). Of note, this increase was similar in HS1-KO CLP mice when compared to WT CLP mice. Given that HS1 is not expressed in vascular endothelial cells, this result is not surprising and likely depends on unaltered release of permeability-inducing substances such as histamine.
3.3. HS1-KO mice show increased pro- and anti-inflammatory cytokine and chemokine expression in lung tissue after CLP
The inflammatory response is in large part regulated by cytokine production (Chaudhry et al., 2013; Nedeva et al., 2019). High levels of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6 are associated with severe sepsis and these cytokines have been used as biomarkers for the diagnosis and prognosis of the clinical outcome in septic patients (Bozza et al., 2007; Gouel-Cheron et al., 2012; Mera et al., 2011; Wu et al., 2009). On the other hand, the anti-inflammatory cytokine IL-10 is also increased in plasma of patients with sepsis and thought to counteract the exacerbated systemic inflammation to limit organ damage (Gogos et al., 2000). However, persistent overproduction of this cytokine may also cause immunosuppression and thus complicate sepsis (Chaudhry et al., 2013; Gogos et al., 2000; Wu et al., 2009). MCP-1 and KC are major chemoattractants for monocytes and neutrophils, important immune cells that help to combat infection, but high concentrations of these chemoattractants have been associated with septic organ damage (Bozza et al., 2007; Mera et al., 2011). Thus, we determined the levels of these cytokines and chemokines by real-time RT-PCR using lung cDNA.
In lungs derived from WT CLP mice, we detected significantly increased mRNA levels of the proinflammatory cytokines TNF-α, IL1β, IL6, and the chemokines KC and MCP1 24 h post-surgery (Fig. 2A). Importantly, the anti-inflammatory cytokine IL10, which is critical to counteract excessive inflammatory responses, was not induced in WT CLP mice. Lungs from sham-operated HS1-KO mice did not show altered cytokine levels when compared to WT control mice. Lungs from HS1-KO mice showed even higher cytokine and chemokine levels than the WT CLP mice. Of special importance here is the very high IL10 production in septic HS1-KO lungs (Fig. 2A) that likely contributes to attenuating the immune response and lung damage in HS1-KO mice.
Fig. 2.
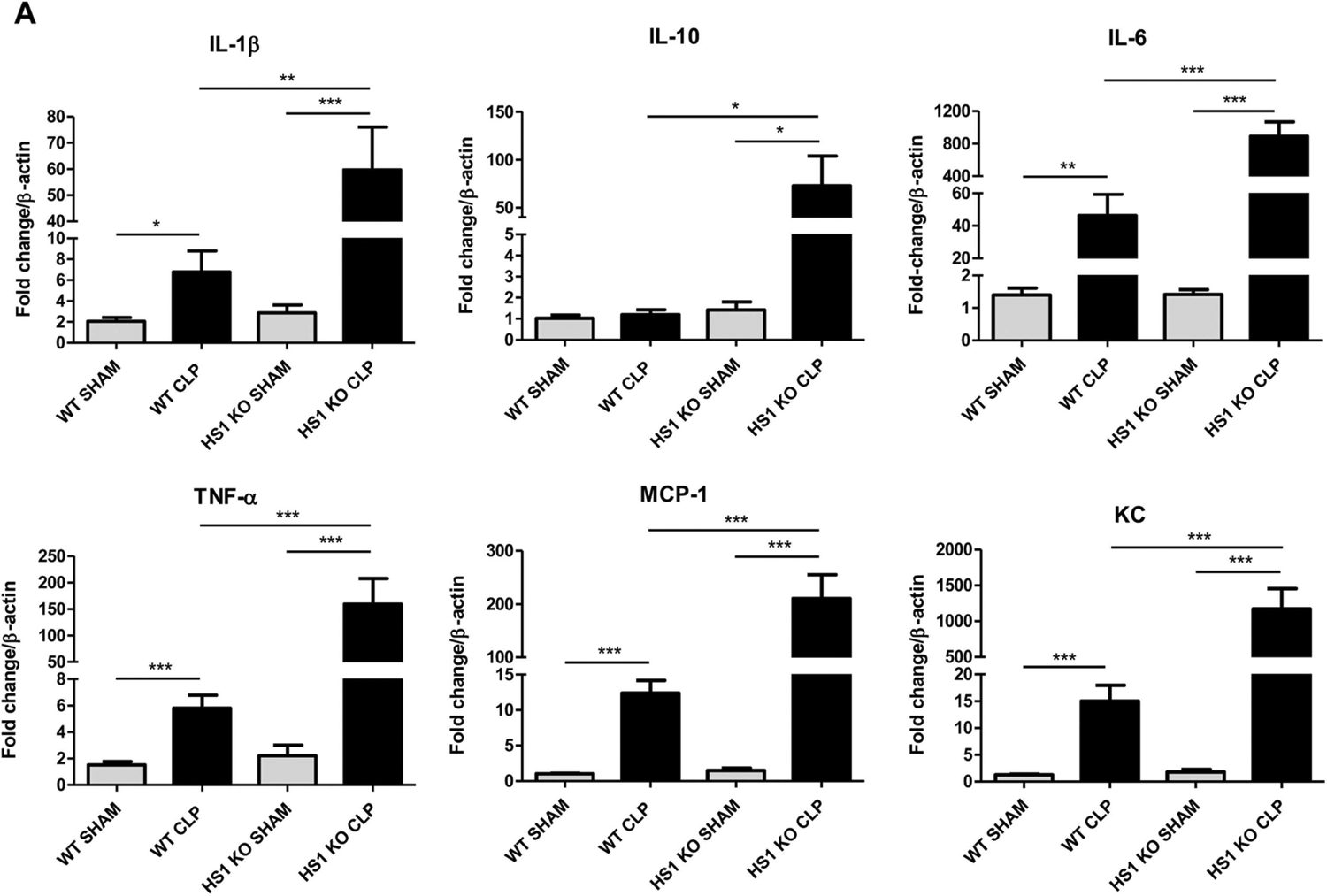
HS1 deficiency modulates lung inflammation. (A) Relative expression of the indicated cytokine and chemokine mRNA levels in lung tissues. Data were calculated using the ΔΔCt method and displayed as fold increase normalized to the expression of β-actin as house-keeping gene. Data are represented as mean+/−SEM; n = 5; *p < 0.05, **p < 0.01, ***p < 0.001.
3.4. Septic HS1-KO mice show increased pro- and anti-inflammatory plasma cytokine levels
To analyze whether HS1 deficiency would also alter the systemic inflammatory response during sepsis, we measured plasma protein levels of various cytokines. As expected, in WT mice, CLP sepsis induced a significant increase in plasma concentrations of the proinflammatory cytokines IL-1β, IL-6 and TNF-α, and the anti-inflammatory cytokine IL-10 when compared to sham-operated animals (Fig. 3). The levels of these cytokines in sham-operated HS1-KO mice were similar to WT controls (Fig. 3). Septic HS1-KO mice had also higher plasma levels of IL-1β, IL-6 and TNF-α; but importantly the IL-10 levels were much higher compared to WT CLP mice (Fig. 3), suggesting that absence of HS1 also alters the systemic inflammatory response during sepsis.
Fig. 3.
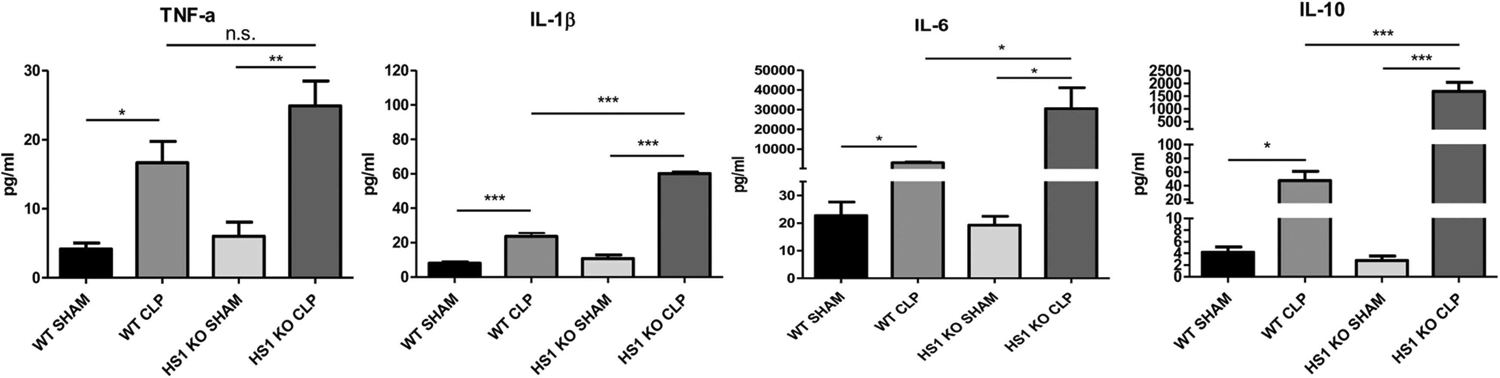
Septic HS1-KO mice show increased pro- and anti-inflammatory plasma cytokine levels. Plasma protein concentrations of the indicated cytokines and chemokines were measured in each experimental group. Data are represented as mean+/−SEM; n = 5; *p < 0.05, **p < 0.01, ***p < 0.001.
3.5. Less neutrophils infiltrate the lungs in HS1-KO mice
Excessive recruitment and hyperactivation of neutrophils leads to organ tissue damage during sepsis thus worsening the outcome of affected patients (Laschke et al., 2007; Lerman et al., 2014; Murdoch et al., 2011; Sarangi et al., 2012; Weaver et al., 2015). Therefore, we speculated that the observed protection of HS1-KO mice during sepsis was a consequence of reduced neutrophil recruitment. To test this idea, we analyzed the number of neutrophils showing high Gr-1 signal in perfused and digested lung tissues by flow cytometry. We found that sham-operated WT and HS1-KO mice had similar low frequencies of lung neutrophils (Fig. 4A). CLP sepsis significantly increased the frequency of lung neutrophils in WT mice, but this increase did not occur in HS1-KO mice (Fig. 4A). Different neutrophil subtypes have been defined according to their surface expression levels of receptors such as Gr-1, ICAM-1 and L-selectin (Woodfin et al., 2016; Zhang et al., 2015). To determine whether lung neutrophils in septic WT and HS1-KO mice show different surface receptor profiles, we sorted and analyzed neutrophils from lung cell suspensions. Interestingly, HS1-KO neutrophils isolated from septic lungs showed significantly higher levels of Gr-1, CD45 and ICAM-1 (Fig. 4B); while levels of L-selectin were similar. This result is of relevance because higher levels of Gr-1 are related to increased neutrophil maturity and higher levels of ICAM-1 to enhanced neutrophil effector functions (Deniset et al., 2017). Thus, it seems that those HS-1 KO neutrophils that reach the lung are capable and sufficiently mature to combat infection.
Fig. 4.
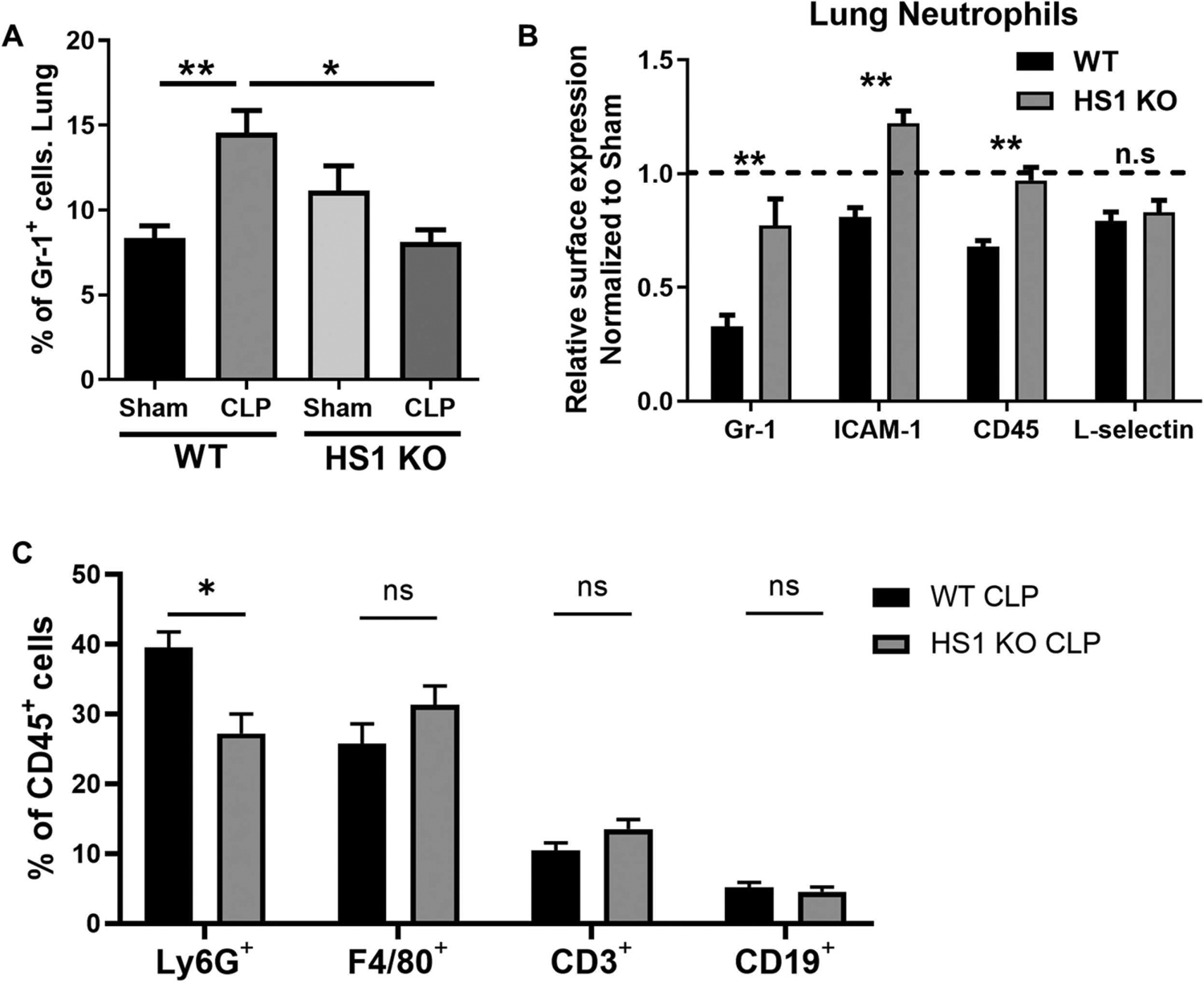
Neutrophil recruitment into the lung is reduced in the absence of HS1. (A) Frequency of Gr-1high neutrophils in lungs from WT and HS1-KO mice 24 h after sham or CLP surgeries. (B) Relative expression (normalized to sham, set as 1) of Gr-1, ICAM-1, CD45 and L-selectin 24 h after sham or CLP surgeries in lung neutrophils. (C) Frequency of different leukocyte populations (Ly6G+-neutrophils, F4/80+-macrophages, CD3+-T cells, CD19+-B cells) analyzed from CD45+ cells in lungs of septic WT and HS1 KO mice. Data represent mean+/−SEM; n = 3–6; *p < 0.05, **p < 0.01.
As HS1 is also expressed in other leukocytes, we analyzed different leukocyte populations in the lungs of septic WT and HS1 KO mice. Using Ly6G as neutrophil marker, we confirmed that within the population of CD45+-leukocytes the frequency of neutrophils was also reduced (Fig. 4C) similar to what we observed with the neutrophil frequency of total lung cells (Fig. 4A). Surprisingly, the frequency of F4/80+-macrophages, CD3+-T cells, and CD19+-B cells was not significantly different in WT and HS1 KO mice (Fig. 4C), suggesting that the observed differences including reduced production of cytokines cannot be attributed to a general reduction of immune cells in the lungs in HS1 KO mice.
3.6. Less neutrophils infiltrate the peritoneum of septic HS1-KO mice
During CLP sepsis, neutrophils are first recruited to the peritoneum by chemotactic cues to control the infection. Thus, we analyzed neutrophil recruitment into the peritoneum after CLP sepsis, a model that starts with peritonitis. Basal cell numbers in the peritoneum were not significantly different in sham WT and HS1-KO mice (Fig. 5A). As expected, significantly more neutrophils were found in the peritoneum of septic WT mice after 24 h, but neutrophil recruitment into the peritoneum of septic HS1-KO mice was significantly lower (Fig. 5A). We then determined bacterial CFU in the peritoneum. Surprisingly, despite reduced neutrophil numbers in septic HS1-KO mice, the bacterial count was not significantly different from that of septic WT mice (Fig. 5B), suggesting that those HS1-KO neutrophils that reach the peritoneum are functional and capable of fighting the infection. This idea was confirmed in an in vivo phagocytosis assay, in which HS1-KO neutrophils under both sham and septic conditions phagocytosed zymosan particles as effectively as WT neutrophils (Fig. 5C and D). Both, the frequency (Fig. 5C) of neutrophils that phagocytosed, and the amount of phagocytosed particles per neutrophil (Fig. 5D) was not significantly different in WT and HS1-KO neutrophils under both basal and septic conditions. These assays also confirmed that septic neutrophils have reduced phagocytic activity; and showed that this reduction is similar in WT and HS1-KO neutrophils (Fig. 5C and D). As the phagocytosis efficiency in different cells in the absence of HS1 is still a matter of debate (Castro-Ochoa et al., 2019), we wanted to confirm this result independently. Using HoxB8 neutrophils, we found that both WT and HS1-KO cells phagocytosed S. aureus efficiently without significant difference in an actin-dependent manner as pretreatment with cytochalasin D abolished the phagocytic activity as expected (Fig. 5E). Moreover, we analyzed ROS production using DHR assays and did not find significant differences between septic WT and HS1 KO neutrophils (Fig. 5F), further highlighting that the absence of HS1 does not affect neutrophil effector functions. Thus, HS1 KO neutrophils are capable of fighting pathogens.
Fig. 5.
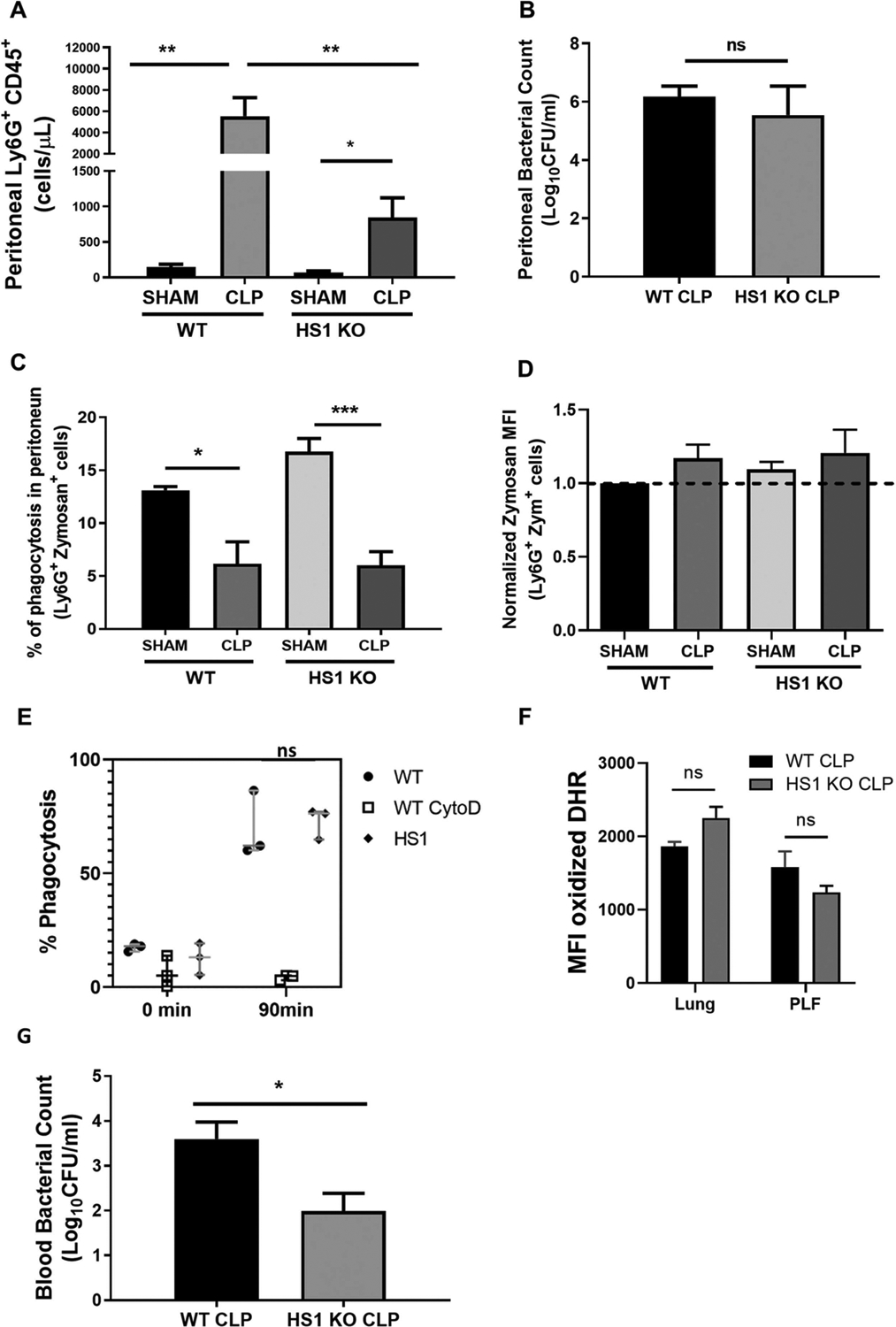
Fewer neutrophils infiltrate the peritoneum in CLP-induced sepsis in the absence of HS1. (A) Frequency of Ly6G+CD45+ cells in the peritoneum 24 h after CLP. (B) Peritoneal bacterial colony-forming units (CFU) were determined 24 h post-CLP by inoculating peritoneal lavage fluid on tryptic soy agar and counting CFU after 24 h. (C) Frequency of Ly6G+CD45+ cells that phagocytosed zymosan particles after 25 min incubation in the peritoneum 24 h post-CLP. (D) Zymosan mean fluorescence intensities (MFI) in Ly6G+ neutrophils (n = 7 in A-D). (E) Frequency of HoxB8 neutrophils that phagocytosed pHrodo Green S. aureus after 90 min incubation. Cytochalasin D treatment as control prevented phagocytosis (n = 3 independent experiments in triplicate). (F) Production of reactive oxygen species as determined by DHR assays is not significantly affected in the absence of HS1 in lung neutrophils and neutrophils from PLF. (G) Bacterial CFU in the peripheral blood (PB) of septic WT and HS1-KO mice (n = 5–8). Data are displayed as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001; ns, non-significant; PLF, Peritoneal Lavage Fluid; zym, zymosan.
As bacteremia is a serious complication of sepsis, we next analyzed bacterial counts in the blood. Importantly, the bacterial count in the peripheral blood of septic HS1-KO mice was significantly lower compared to WT mice (Fig. 5G), suggesting that HS1 deficiency reduces systemic spreading of the infection.
3.7. HS1-KO neutrophils show impaired CXCL1-induced actin polymerization and stiffness
HS1 is an ABP known to regulate actin dynamics. Given that phagocytosis and recruitment are processes highly dependent on actin remodeling, we investigated chemokine-induced F-actin dynamics in neutrophils. The amount of actin filaments was similar in WT and HS1-KO neutrophils, and WT neutrophils responded quickly to CXCL1 with the formation of new filaments as expected (Fig. 6A). Of note, HS1-KO neutrophils failed to produce new actin filaments in response to CXCL1. We confirmed this result independently using HoxB8 neutrophils. WT neutrophils responded quickly to CXCL1 with robust actin polymerization and new filament generation, which was abolished completely in the presence of cytochalasin D as expected (Fig. 6B). By contrast, HS1-KO HoxB8 neutrophils did not produce any actin filaments in response to CXCL1.
Fig. 6.
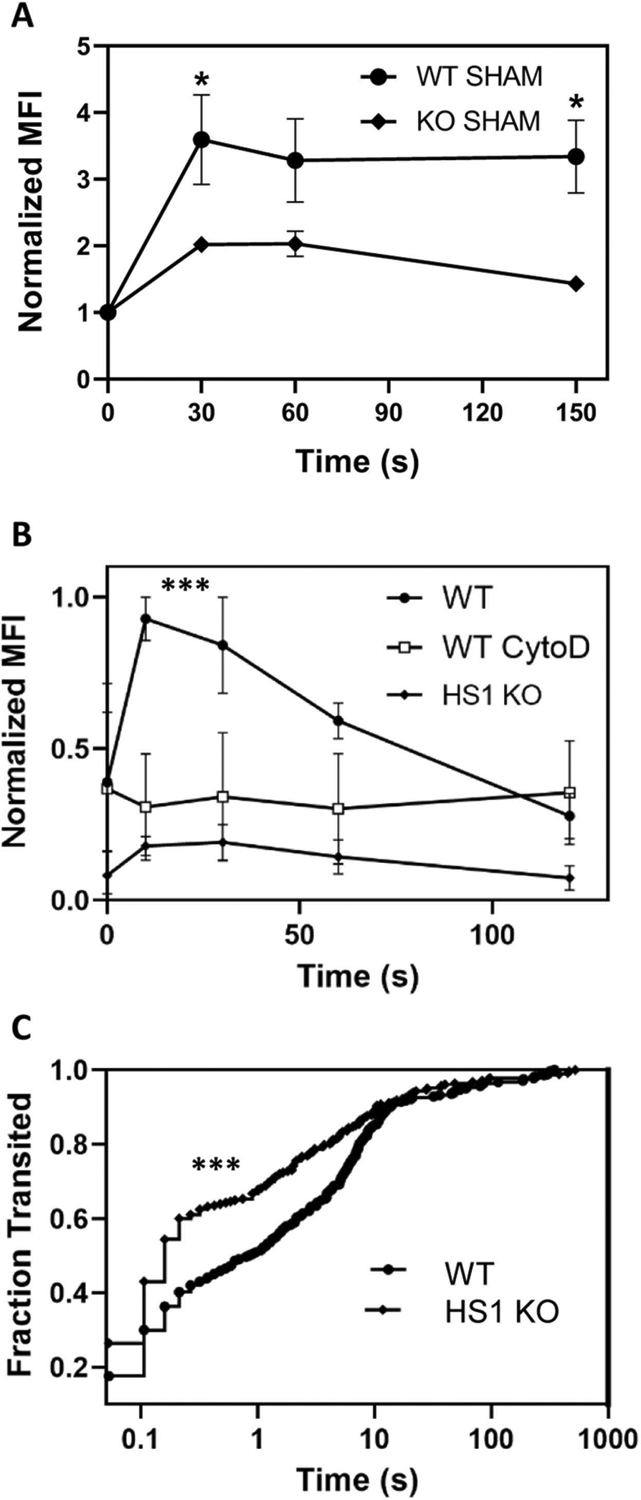
HS1-KO neutrophils show defects in actin polymerization and cell stiffness. (A) Normalized MFI of F-actin as determined by AF488-phalloidin staining in blood neutrophils from WT and HS1-KO mice after treatment with CXCL1 for the indicated times; n = 6. (B) Normalized MFI of F-actin in HoxB8 neutrophils after treatment with CXCL1 for the indicated times; n = 4. Data are displayed as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. (C) Kaplan-Meier curves of neutrophil fractions that transited the 5 μm microfluidics device at the indicated times. The hazard ratio for WT/HS1-KO is 0.7769 [95% CI 0.6515–0.9266; p-value < 0.0001].
Chemokine-induced actin polymerization is known to alter cell stiffness or deformability. To analyze cell deformability, we stimulated HoxB8 neutrophils with CXCL1 and analyzed the impact on the dwell times within 5 μm constrictions using microfluidics devices. Dwell times are a method to evaluate cell deformability (stiffness), where highly deformable cells transit the constrictions more rapidly. The resulting Kaplan-Meier curves showing cumulative distributions of the fraction of cells passing through the constriction over time demonstrated that WT neutrophils stimulated with CXCL1 had a lower fraction of cells transiting between 0.1 s and 10 s as compared to CXCL1-treated HS1-KO neutrophils (Fig. 6C). As CXCL1 is the most important chemoattractant for mouse neutrophils, the observed unresponsiveness of HS1-KO neutrophils to CXCL1 may explain the altered recruitment pattern.
3.8. HS1 deficiency reduces neutrophil recruitment in the cremaster during CLP sepsis
Finally, to analyze neutrophil recruitment in a distant tissue as a systemic response to sepsis, we performed intravital microscopy of the cremaster muscle in CLP and sham-operated WT and HS1-KO mice without any other local inflammatory stimulus (Supplementary videos 1–4) (Bagher and Segal, 2011; Gavins and Chatterjee, 2004). While the rolling flux fraction was not different between the groups (Fig. 7A), rolling velocity was reduced in WT CLP mice, but not in HS1-KO CLP mice when compared to the respective sham controls (Fig. 7B). The number of firmly adherent cells was strongly increased in WT CLP mice, and this increase was much less pronounced in HS1-KO CLP mice (Fig. 7C). The number of transmigrated cells was significantly increased in WT CLP mice compared to sham controls (Fig. 7D and E). Importantly, transmigration did not significantly increase in HS1-KO CLP mice compared to HS1-KO sham controls indicating that during systemic inflammation HS1 deficiency prevents neutrophil recruitment in distant tissues, where no neutrophil presence is needed. Hemodynamic parameters did not show significant differences between WT and HS1-KO mice (Fig. 7F). These results highlight the similarity between innate immune responses after local and systemic inflammatory stimuli and the relevance of HS1 for these responses.
Fig. 7.
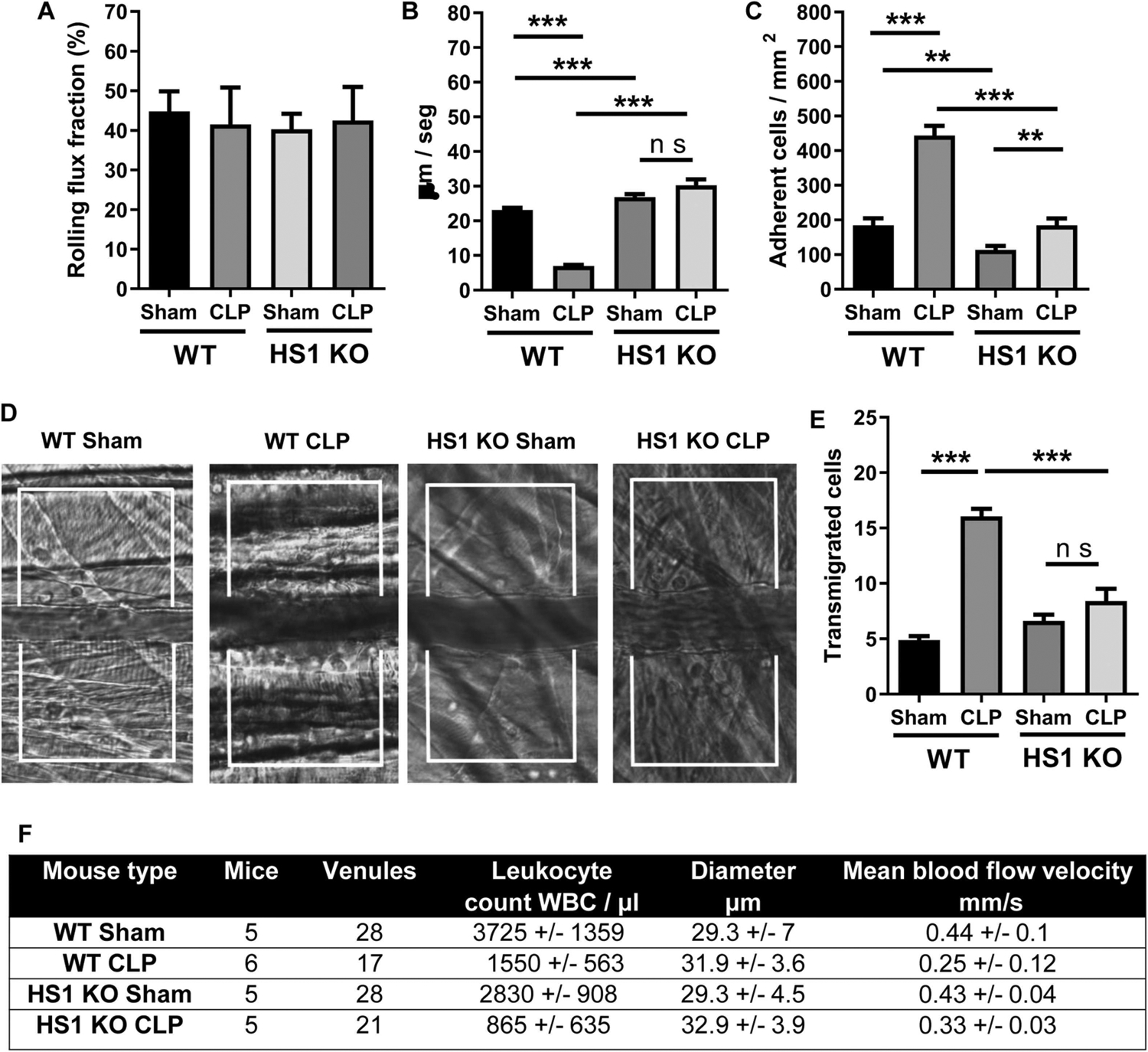
Septic HS1-KO mice show decreased systemic neutrophil extravasation in the cremaster. (A-C) IVM of the cremaster muscle 24 h after sham or CLP operation of male 8–12-week old WT and HS1-KO mice with quantification of the following parameters: Rolling flux fraction (A), rolling velocity (B), firmly adherent cells (C). (D) Representative DIC images of the supplementary videos. (E) Quantification of transmigrated cells from DIC images as shown in (D). Note that mice did not receive an extra intrascrotal stimulus so that recruitment only occurs in response to the systemic stimulus of the CLP surgery. Data are displayed as mean +/− SEM; n = 6. **p < 0.01, ***p < 0.001. (F) Hemodynamic parameters of sham- and CLP-operated WT and HS1-KO mice. Data are shown as mean+/−SEM.
4. Discussion
Here, we analyzed whether the ABP HS1 is involved in CLP sepsis development and sepsis-induced neutrophil recruitment. We found that absence of HS1 was associated with increased survival that was accompanied by reduced local and systemic neutrophil recruitment and lung tissue damage. HS1-KO neutrophils phagocytosed as efficiently as WT neutrophils but did not respond to CXCL1 with actin filament formation. Our results provide evidence that HS1 deficiency does not prevent neutrophil recruitment, but attenuates it to amounts sufficient to fight infections, but insufficient to cause lethal tissue damage.
During CLP sepsis, bacteria can be found in different organs as soon as 2 h post-CLP, where they accumulate over time (Hyde et al., 1990). However, some HS1-KO mice not only survived the 5-day experimental period, but recovered completely suggesting that, despite their known dysfunction in neutrophil recruitment, bacterial infection did not kill these mice. We showed that HS1-deficient neutrophils in the peritoneum phagocytose as efficiently as WT neutrophils; and that systemic translocation of bacteria is reduced in HS1-KO mice. The exact mechanisms by which bacterial translocation is inhibited in the absence of HS1 requires further investigation. A possibility is that non-recruited neutrophils that remain in blood vessels enhance systemic bacterial clearance. Support for this idea comes from a recent study describing that lung capillaries provide a vascular protective niche where neutrophils get stuck, interact with the endothelium and clear sequestered bacteria (Yipp et al., 2017). This could be a potential explanation for pathogen clearance and increased survival after sepsis in the absence of HS1, without having the deleterious side effects of neutrophil-released molecules that contribute to tissue damage after extravasation into affected tissues.
Activation of innate immunity and recruitment of neutrophils is a critical host response during sepsis. The number of recruited neutrophils in the lung, cremaster and peritoneum was significantly reduced in HS1-KO mice after CLP, suggesting altered HS1-dependent local and systemic neutrophil recruitment. Reduced neutrophil influx into the lungs may explain the absence of severe tissue damage in septic HS1-KO mice. There is evidence supporting this idea. For example, isolated neutrophils from septic humans and mice showed upregulation of α3β1 integrin (CD49c/CD29); and administration of an α3β1 blocking peptide or conditional downregulation of the α3-integrin subunit in mice resulted in reduced neutrophil infiltration into the lung and reduced mortality after CLP (Lerman et al., 2014). Blocking CXCR4 and CXCR7 signaling prevented neutrophil migration to the lungs, the peritoneal cavity, and liver (Ngamsri et al., 2020). CXCL1 mobilized neutrophils to control lung bacterial burdens and bacterial dissemination leading to better survival (Paudel et al., 2019). Moreover, resolvin D2 has been shown to reduce neutrophil recruitment and to increase resolution of inflammation and survival in CLP sepsis (Spite et al., 2009). Thus, it will be interesting to study the role of HS1 in chemokine receptor signaling and inflammation-resolving actions of neutrophils. Of special importance in this context is how HS1 translates CXCL1 signals to actin polymerization given the unresponsiveness of septic HS1-KO neutrophils to this chemokine.
Within the septic lung, HS1-KO neutrophils showed much higher levels of CD45, Gr-1 and ICAM-1 than WT neutrophils. Emerging evidence suggests the presence of different neutrophil subsets (Rosales, 2018), with Gr-1 being a marker for maturity (Deniset et al., 2017), and ICAM-1 for increased effector functions in the lung (Woodfin et al., 2016). Thus, it seems reasonable to assume that increased Gr-1 and ICAM-1 levels in HS1-KO neutrophils of septic lungs would render them more efficient in bacterial clearance to compensate for the lower numbers. Support for this idea comes from a recent study showing that inhibition of src family kinases by dasatinib also reduced neutrophil recruitment and protected against sepsis (Goncalves-de-Albuquerque et al., 2018). HS1 is a prominent target of src family kinases, and different tyrosine phosphorylation patterns determine HS1 functions (Butler et al., 2008). Thus, it will be critical to analyze post-translational modifications of HS1 during sepsis and determine if the improved survival observed after dasatinib treatment relates to HS1 inhibition.
We confirmed by IVM of sham and CLP WT and HS1-KO mice that systemic leukocyte recruitment in tissues distant to the original infection is also reduced in septic HS1-KO mice. This is in agreement with our previous study demonstrating that leukocyte infiltration into CXCL1-stimulated cremaster muscles was reduced in HS1-KO mice due to lack of proper activation of the GTPases Rac1 and Rap1 which are fundamental for integrin activation (Latasiewicz et al., 2017). Thus, a systemic inflammatory stimulus (CLP) is sufficient to induce leukocyte recruitment in distant tissues without further local stimulus; and HS1 also prevents this distant organ response, presumably by similar mechanisms, thus highlighting the importance of HS1 for controlling leukocyte recruitment in different inflammatory settings and different organs.
Analyzing plasma levels of different cytokines, we consistently found higher cytokine levels in HS1-KO CLP mice than in WT CLP mice. Whether this is a compensatory mechanism to increase neutrophil presence is yet unknown. Interestingly, production of the anti-inflammatory cytokine IL-10 was much higher in HS1-KO CLP mice compared to WT CLP mice. Previous studies have demonstrated that IL-10 is critical for sepsis survival. For example, administration of IL-10 one hour after CLP and every three hours thereafter resulted in increased survival despite an increase in TNF-α (Rongione et al., 2000). Moreover, IL-10-KO mice had increased mortality in CLP sepsis and showed higher serum levels of TNF-α and IL-6. Of note, administration of recombinant IL-10 improved long-term survival of WT animals and increased survival of IL-10-KO mice (Latifi et al., 2002). Re-expression of IL-10 in IL-10-KO mice also increased sepsis survival by protecting lymphoid cells in the thymus from apoptosis (Tschoeke et al., 2008). On the other hand, IL-10 is associated with inflammation resolution; thus, the massively increased local and systemic IL10 levels in septic HS1-KO mice likely contribute to balancing out the excessive inflammation and neutrophil recruitment; and may induce a pro-resolving phenotype in those neutrophils that reach the inflamed lungs thereby preventing tissue damage. Thus, it is tempting to speculate that there is a causal relationship between increased IL-10 levels, reduced neutrophil recruitment, and increased survival of septic HS1-KO mice. The exact source of IL-10 is currently unknown, but it is likely that different types of tissue-resident and recruited immune cells contribute to IL-10 production. Given that endothelial and epithelial cells and fibroblasts do not express HS1, it is unlikely that these cell types are affected in HS1-KO mice, which is in line with our data of unaltered vascular permeability. The fact that septic HS1-KO mice show increased permeability similar to septic WT mice in spite of significantly reduced neutrophil recruitment is not surprising because it is accepted that neutrophil recruitment and vascular permeability are mechanistically independent responses to inflammation, which we have experimentally proven in cortactin-deficient mice (Schnoor et al., 2011). It will be interesting to analyze the release of permeability-inducing factors such as histamine that might not be affected in the absence of HS1.
In summary, our data show that HS1-KO mice are protected from lethal CLP sepsis due to reduced neutrophil infiltration, tissue damage and actin polymerization; and increased IL-10 levels. HS1 deficiency reduces systemic neutrophil infiltration into different tissues without impairing anti-bacterial host defense during sepsis. It will be interesting to investigate in future studies whether HS1 could serve as useful target to modulate neutrophil recruitment in septic patients.
Supplementary Material
Acknowledgments
This work was supported by the Mexican Council for Science and Technology (CONACYT, 207268 to MS). MS acknowledges funding from the Agencia Mexicana de Cooperación Internacional para el Desarrollo (AMEXCID) of the Secretaría de Relaciones Exteriores (SRE) (AMEX-CID_2020-4); and from CONACYT (CB-2016-284292; and PRONAII Leukemia Grant 302978 of the National Strategic Health Program).
We thank Dr. Klemens Rottner (Technical University of Braunsch-weig, Germany) and Dr. Daisuke Kitamura (Tokyo University of Science, Japan) for providing HS1-KO mice; and Angélica Silva Olivares for excellent technical assistance. AGP, IMGF, NLL and SCP received pre-doctoral stipends from CONACYT. EV received a postdoctoral fellowship from CONACYT.
CTL and BMN acknowledge funding from National Institutes of Health (awards R35GM124911 to CTL, and T32HL134625 to BMN).
This manuscript is dedicated to the memory of our dear colleague Dr. Mineko Shibayama who recently passed away. She helped with the histopathological analysis in this study.
Footnotes
CRediT authorship contribution statement
Idaira M. Guerrero-Fonseca: Investigation, Validation, Formal analysis, Visualization Writing – original draft. Alexander García-Ponce: Investigation, Validation, Formal analysis, Visualization, Writing – original draft. Eduardo Vadillo: Investigation, Validation, Formal analysis, Visualization, Writing – original draft. Nathaniel L. Lartey: Investigation, Validation, Formal analysis, Visualization. Hilda Vargas-Robles: Investigation, Validation, Visualization. Sandra Chánez-Paredes: Investigation, Validation, Visualization. Ramón Castellanos-Martínez: Investigation, Validation, Visualization. Abigail Betanzos: Validation, Formal analysis, Resources. Porfirio Nava: Validation, Formal analysis, Resources. Brittany M. Neumann: Investigation, Validation, Visualization. Kinga Penkala-Auguste: Investigation, Validation, Visualization. Craig T. Lefort: Validation, Resources, Supervision. Michael Schnoor: Validation, Conceptualization, Writing – original draft, Writing – review & editing, Supervision, Project administration, Funding acquisition.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.ejcb.2022.151214.
References
- Bagher P, Segal SS, 2011. The mouse cremaster muscle preparation for intravital imaging of the microcirculation. J. Vis. Exp. JoVE [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza FA, Salluh JI, Japiassu AM, Soares M, Assis EF, Gomes RN, Bozza MT, Castro-Faria-Neto HC, Bozza PT, 2007. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit. Care 11, R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler B, Kastendieck DH, Cooper JA, 2008. Differently phosphorylated forms of the cortactin homolog HS1 mediate distinct functions in natural killer cells. Nat. Immunol 9, 887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Ochoa KF, Guerrero-Fonseca IM, Schnoor M, 2019. Hematopoietic cell-specific lyn substrate (HCLS1 or HS1): A versatile actin-binding protein in leukocytes. J. Leukoc. Biol 105, 881–890. [DOI] [PubMed] [Google Scholar]
- Cavnar PJ, Mogen K, Berthier E, Beebe DJ, Huttenlocher A, 2012. The actin regulatory protein HS1 interacts with Arp2/3 and mediates efficient neutrophil chemotaxis. J. Biol. Chem 287, 25466–25477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry H, Zhou J, Zhong Y, Ali MM, McGuire F, Nagarkatti PS, Nagarkatti M, 2013. Role of cytokines as a double-edged sword in sepsis. In Vivo 27, 669–684. [PMC free article] [PubMed] [Google Scholar]
- Citalan-Madrid AF, Vargas-Robles H, Garcia-Ponce A, Shibayama M, Betanzos A, Nava P, Salinas-Lara C, Rottner K, Mennigen R, Schnoor M, 2017. Cortactin deficiency causes increased RhoA/ROCK1-dependent actomyosin contractility, intestinal epithelial barrier dysfunction, and disproportionately severe DSS-induced colitis. Mucosal Immunol. [DOI] [PubMed] [Google Scholar]
- Deniset JF, Surewaard BG, Lee WY, Kubes P, 2017. Splenic Ly6G(high) mature and Ly6G(int) immature neutrophils contribute to eradication of S. pneumoniae. J. Exp. Med 214, 1333–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekpenyong AE; Toepfner N; Chilvers ER; Guck J, Mechanotransduction in neutrophil activation and deactivation. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2015, 1853 (11, Part B), 3105–3116.4. [DOI] [PubMed] [Google Scholar]
- Englert JA, Bobba C, Baron RM, 2019. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eveillard M, Soltner C, Kempf M, Saint-Andre JP, Lemarie C, Randrianarivelo C, Seifert H, Wolff M, Joly-Guillou ML, 2010. The virulence variability of different Acinetobacter baumannii strains in experimental pneumonia. J. Infect 60, 154–161. [DOI] [PubMed] [Google Scholar]
- Gavins FN, Chatterjee BE, 2004. Intravital microscopy for the study of mouse microcirculation in anti-inflammatory drug research: focus on the mesentery and cremaster preparations. J. Pharmacol. Toxicol. Methods 49, 1–14. [DOI] [PubMed] [Google Scholar]
- Gogos CA, Drosou E, Bassaris HP, Skoutelis A, 2000. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J. Infect. Dis 181, 176–180. [DOI] [PubMed] [Google Scholar]
- Goncalves-de-Albuquerque CF, Rohwedder I, Silva AR, Ferreira AS, Kurz ARM, Cougoule C, Klapproth S, Eggersmann T, Silva JD, de Oliveira GP, Capelozzi VL, Schlesinger GG, Costa ER, Estrela Marins RCE, Mocsai A, Maridonneau-Parini I, Walzog B, Macedo Rocco PR, Sperandio M, de Castro-Faria-Neto HC, 2018. The yin and yang of tyrosine kinase inhibition during experimental polymicrobial sepsis. Front. Immunol 9, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouel-Cheron A, Allaouchiche B, Guignant C, Davin F, Floccard B, Monneret G, AzuRea G, 2012. Early interleukin-6 and slope of monocyte human leukocyte antigen-DR: a powerful association to predict the development of sepsis after major trauma. PLoS One 7, e33095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan Z, Palani K, Rahman M, Thorlacius H, 2011. Targeting CD44 expressed on neutrophils inhibits lung damage in abdominal sepsis. Shock 35, 567–572. [DOI] [PubMed] [Google Scholar]
- Hickey MJ, Westhorpe CL, 2013. Imaging inflammatory leukocyte recruitment in kidney, lung and liver–challenges to the multi-step paradigm. Immunol. Cell Biol 91, 281–289. [DOI] [PubMed] [Google Scholar]
- Hoesel LM, Neff TA, Neff SB, Younger JG, Olle EW, Gao H, Pianko MJ, Bernacki KD, Sarma JV, Ward PA, 2005. Harmful and protective roles of neutrophils in sepsis. Shock 24, 40–47. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL, 2016. Sepsis and septic shock. Nat. Rev. Dis. Prim 2, 16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebinger J, Spindler J, Holl KJ, Koos B, 2018. Quantification of protein mobility and associated reshuffling of cytoplasm during chemical fixation. Sci. Rep 8, 17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde SR, Stith RD, McCallum RE, 1990. Mortality and bacteriology of sepsis following cecal ligation and puncture in aged mice. Infect. Immun 58, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Hong SB, 2016. Sepsis and acute respiratory distress syndrome: recent update. Tuberc. Respir. Dis 79, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfleisch R, 2013. Multiparametric and semiquantitative scoring systems for the evaluation of mouse model histopathology–a systematic review. BMC Vet. Res 9, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P, 2013. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol 13, 159–175. [DOI] [PubMed] [Google Scholar]
- Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, Benarafa C, Roos D, Skokowa J, Hartl D, 2015. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog. 11, e1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D, Kumar A, Cheang M, 2006. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med 34, 1589–1596. [DOI] [PubMed] [Google Scholar]
- Laschke MW, Menger MD, Wang Y, Lindell G, Jeppsson B, Thorlacius H, 2007. Sepsis-associated cholestasis is critically dependent on P-selectin-dependent leukocyte recruitment in mice. Am. J. Physiol. Gastrointest. Liver Physiol 292, G1396–G1402. [DOI] [PubMed] [Google Scholar]
- Latasiewicz J, Artz A, Jing D, Blanco MP, Currie SM, Avila MV, Schnoor M, Vestweber D, 2017. HS1 deficiency impairs neutrophil recruitment in vivo and activation of the small GTPases Rac1 and Rap1. J. Leukoc. Biol 101, 1133–1142. [DOI] [PubMed] [Google Scholar]
- Latifi SQ, O’Riordan MA, Levine AD, 2002. Interleukin-10 controls the onset of irreversible septic shock. Infect. Immun 70, 4441–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman YV, Lim K, Hyun YM, Falkner KL, Yang H, Pietropaoli AP, Sonnenberg A, Sarangi PP, Kim M, 2014. Sepsis lethality via exacerbated tissue infiltration and TLR-induced cytokine production by neutrophils is integrin alpha3beta1-dependent. Blood 124, 3515–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu V, Escobar GJ, Greene JD, Soule J, Whippy A, Angus DC, Iwashyna TJ, 2014. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA 312, 90–92. [DOI] [PubMed] [Google Scholar]
- Mansur A, Klee Y, Popov AF, Erlenwein J, Ghadimi M, Beissbarth T, Bauer M, Hinz J, 2015. Primary bacteraemia is associated with a higher mortality risk compared with pulmonary and intra-abdominal infections in patients with sepsis: a prospective observational cohort study. BMJ Open 5, e006616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mera S, Tatulescu D, Cismaru C, Bondor C, Slavcovici A, Zanc V, Carstina D, Oltean M, 2011. Multiplex cytokine profiling in patients with sepsis. APMIS 119, 155–163. [DOI] [PubMed] [Google Scholar]
- Murdoch EL, Karavitis J, Deburghgraeve C, Ramirez L, Kovacs EJ, 2011. Prolonged chemokine expression and excessive neutrophil infiltration in the lungs of burn-injured mice exposed to ethanol and pulmonary infection. Shock 35, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef WM, Borregaard N, 2014. Neutrophils at work. Nat. Immunol 15, 602–611. [DOI] [PubMed] [Google Scholar]
- Nedeva C, Menassa J, Puthalakath H, 2019. Sepsis: inflammation is a necessary evil. Front. Cell Dev. Biol 7, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngamsri KC, Jans C, Putri RA, Schindler K, Gamper-Tsigaras J, Eggstein C, Kohler D, Konrad FM, 2020. Inhibition of CXCR4 and CXCR7 is protective in acute peritoneal inflammation. Front. Immunol 11, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourshargh S, Alon R, 2014. Leukocyte migration into inflamed tissues. Immunity 41, 694–707. [DOI] [PubMed] [Google Scholar]
- Paudel S, Baral P, Ghimire L, Bergeron S, Jin L, DeCorte JA, Le JT, Cai S, Jeyaseelan S, 2019. CXCL1 regulates neutrophil homeostasis in pneumonia-derived sepsis caused by Streptococcus pneumoniae serotype 3. Blood 133, 1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongione AJ, Kusske AM, Kwan K, Ashley SW, Reber HA, McFadden DW, 2000. Interleukin-10 protects against lethality of intra-abdominal infection and sepsis. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 4, 70–76. [DOI] [PubMed] [Google Scholar]
- Rosales C, 2018. Neutrophil: a cell with many roles in inflammation or several cell types? Front. Physiol 9, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossaint J, Zarbock A, 2013. Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J. Innate Immun 5, 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossaint J, Zarbock A, 2015. Pathogenesis of multiple organ failure in sepsis. Crit. Rev. Immunol 35, 277–291. [DOI] [PubMed] [Google Scholar]
- Sarangi PP, Hyun YM, Lerman YV, Pietropaoli AP, Kim M, 2012. Role of beta1 integrin in tissue homing of neutrophils during sepsis. Shock 38, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoor M, Cullen P, Lorkowski J, Stolle K, Robenek H, Troyer D, Rauterberg J, Lorkowski S, 2008. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J. Immunol 180, 5707–5719. [DOI] [PubMed] [Google Scholar]
- Schnoor M, Lai FP, Zarbock A, Klaver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D, 2011. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J. Exp. Med 208, 1721–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoor M, Alcaide P, Voisin MB, van Buul JD, 2015. Crossing the vascular wall: common and unique mechanisms exploited by different leukocyte subsets during extravasation. Mediat. Inflamm 2015, 946509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoor M, Garcia Ponce A, Vadillo E, Pelayo R, Rossaint J, Zarbock A, 2017. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell. Mol. Life Sci. CMLS 74, 1985–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, 2012. Fiji: an open-source platform for biological-image analysis. Nature Methods 9 (7), 676–682, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN, 2009. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi I, Kitamura D, Maekawa Y, Fukuda T, Kishi H, Watanabe T, 1995. Antigen-receptor induced clonal expansion and deletion of lymphocytes are impaired in mice lacking HS1 protein, a substrate of the antigen-receptor-coupled tyrosine kinases. EMBO J. 14, 3664–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez J-Y, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW, 2017. TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Toscano MG, Ganea D, Gamero AM, 2011. Cecal ligation puncture procedure. J. Vis. Exp. JoVE [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschoeke SK, Oberholzer C, LaFace D, Hutchins B, Moldawer LL, Oberholzer A, 2008. Endogenous IL-10 regulates sepsis-induced thymic apoptosis and improves survival in septic IL-10 null mice. Scand. J. Immunol 68, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestweber D, 2015. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol 15, 692–704. [DOI] [PubMed] [Google Scholar]
- Wang J, 2018. Neutrophils in tissue injury and repair. Cell Tissue Res. 371, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver LC, Bao F, Dekaban GA, Hryciw T, Shultz SR, Cain DP, Brown A, 2015. CD11d integrin blockade reduces the systemic inflammatory response syndrome after traumatic brain injury in rats. Exp. Neurol 271, 409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ZS, Witt H, Hazlett L, Harman M, Neumann BM, Whitman A, Patel M, Ross RS, Franck C, Reichner JS, Lefort CT, 2020. Context-dependent role of vinculin in neutrophil adhesion, motility and trafficking. Sci. Rep 10, 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfin A, Beyrau M, Voisin MB, Ma B, Whiteford JR, Hordijk PL, Hogg N, Nourshargh S, 2016. ICAM-1-expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemia. Blood 127, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HP, Chen CK, Chung K, Tseng JC, Hua CC, Liu YC, Chuang DY, Yang CH, 2009. Serial cytokine levels in patients with severe sepsis. Inflamm. Res 58, 385–393. [DOI] [PubMed] [Google Scholar]
- Yipp BG, Kim JH, Lima R, Zbytnuik LD, Petri B, Swanlund N, Ho M, Szeto VG, Tak T, Koenderman L, Pickkers P, Tool ATJ, Kuijpers TW, van den Berg TK, Looney MR, Krummel MF, Kubes P, 2017. The lung is a host defense niche for immediate neutrophil-mediated vascular protection. Sci. Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, Merad M, Frenette PS, 2015. Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.