

Abstract
As of August 2022, clusters of acute severe hepatitis of unknown aetiology in children have been reported from 35 countries, including the USA1,2. Previous studies have found human adenoviruses (HAdVs) in the blood from patients in Europe and the USA3–7, although it is unclear whether this virus is causative. Here we used PCR testing, viral enrichment-based sequencing and agnostic metagenomic sequencing to analyse samples from 16 HAdV-positive cases from 1 October 2021 to 22 May 2022, in parallel with 113 controls. In blood from 14 cases, adeno-associated virus type 2 (AAV2) sequences were detected in 93% (13 of 14), compared to 4 (3.5%) of 113 controls (P < 0.001) and to 0 of 30 patients with hepatitis of defined aetiology (P < 0.001). In controls, HAdV type 41 was detected in blood from 9 (39.1%) of the 23 patients with acute gastroenteritis (without hepatitis), including 8 of 9 patients with positive stool HAdV testing, but co-infection with AAV2 was observed in only 3 (13.0%) of these 23 patients versus 93% of cases (P < 0.001). Co-infections by Epstein–Barr virus, human herpesvirus 6 and/or enterovirus A71 were also detected in 12 (85.7%) of 14 cases, with higher herpesvirus detection in cases versus controls (P < 0.001). Our findings suggest that the severity of the disease is related to co-infections involving AAV2 and one or more helper viruses.
Subject terms: Phylogenetics, Epidemiology, Adenovirus, Next-generation sequencing, Viral hepatitis
A retrospective analysis using PCR testing, viral enrichment-based sequencing and agnostic metagenomic sequencing finds an association between adeno-associated virus type 2 and paediatric hepatitis of unknown cause.
Main
Viral hepatitis is an inflammation of the liver most commonly caused by one of the main hepatitis viruses (A–E). Since October 2021, clusters of acute non-A–E severe hepatitis of unknown aetiology in children have been reported in 35 countries, including the USA1,2. As of 17 August 2022, 358 people under investigation have been reported in the USA, of whom 22 (6%) required a liver transplant and 13 (4%) died8.
HAdVs are non-enveloped, double-stranded DNA viruses that cause a variety of infections in both adults and children, including respiratory tract infection, conjunctivitis and gastroenteritis (mainly from HAdV type 40 (HAdV-40) and HAdV-41)9. Although a potentially life-threatening disease in immunocompromised patients7,9, hepatitis from adenovirus infection has been thought to be rare in immunocompetent children without underlying comorbidities10. HAdVs, particularly HAdV-41, have been found in blood from clusters of acute severe hepatitis cases from Scotland, the UK and the USA3–7, but whether the virus is causative remains unclear. Adeno-associated viruses (AAVs) are small, single-stranded DNA parvoviruses that are considered non-pathogenic in humans and for this reason have been widely used as vectors for gene therapy11. Importantly, AAVs require a helper virus, such as a herpesvirus or adenovirus, for productive infection of liver tissue12. AAVs have previously been reported in children with acute severe hepatitis in a study of nine patients from the UK13, often in association with adenovirus or human herpesvirus 6 (HHV-6) infection14.
In this study, 27 samples (21 whole blood, 2 plasma, 1 liver tissue, 1 nasopharyngeal swab and 2 stool sample(s)) from 16 children with acute severe hepatitis of unknown aetiology were analysed (Fig. 1). All children met the clinical case definition established by the Centers for Disease Control and Prevention, including lack of a confirmed aetiology, liver enzyme levels (aspartate aminotransferase (AST) or alanine aminotransferase (ALT)) >500 U l−1, age < 10 years and onset on or after 1 October 2021 (refs. 2,15). Cases were enrolled from six states (Alabama, California, Florida, Illinois, North Carolina, and South Dakota) from 1 October 2021 to 22 May 2022. All 16 cases had positive testing for HAdV from blood, and thus HAdV infection was over-represented compared to the overall affected population, of which HAdV is detected in 45–90% (refs. 3–7). The median age of affected children was 3 years; 56% were female and 44% male (Table 1). Mean elevations in ALT and AST were 2,293 ± 1,486 U l−1 (normal range: 4–36 U l−1) and 2,652 ± 1,851 U l−1 (normal range: 8–33 U l−1), respectively (Supplementary Table 1). A liver biopsy was carried out in eight patients; viral PCR testing of the liver biopsies for HAdVs, herpesviruses, enteroviruses and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was incomplete, but yielded positive results for HAdV in three patients with no other viruses detected (Fig. 2b and Supplementary Table 1). Two children underwent liver transplantation, but none died from complications of liver failure.
Fig. 1. Epidemiology of cases and controls.
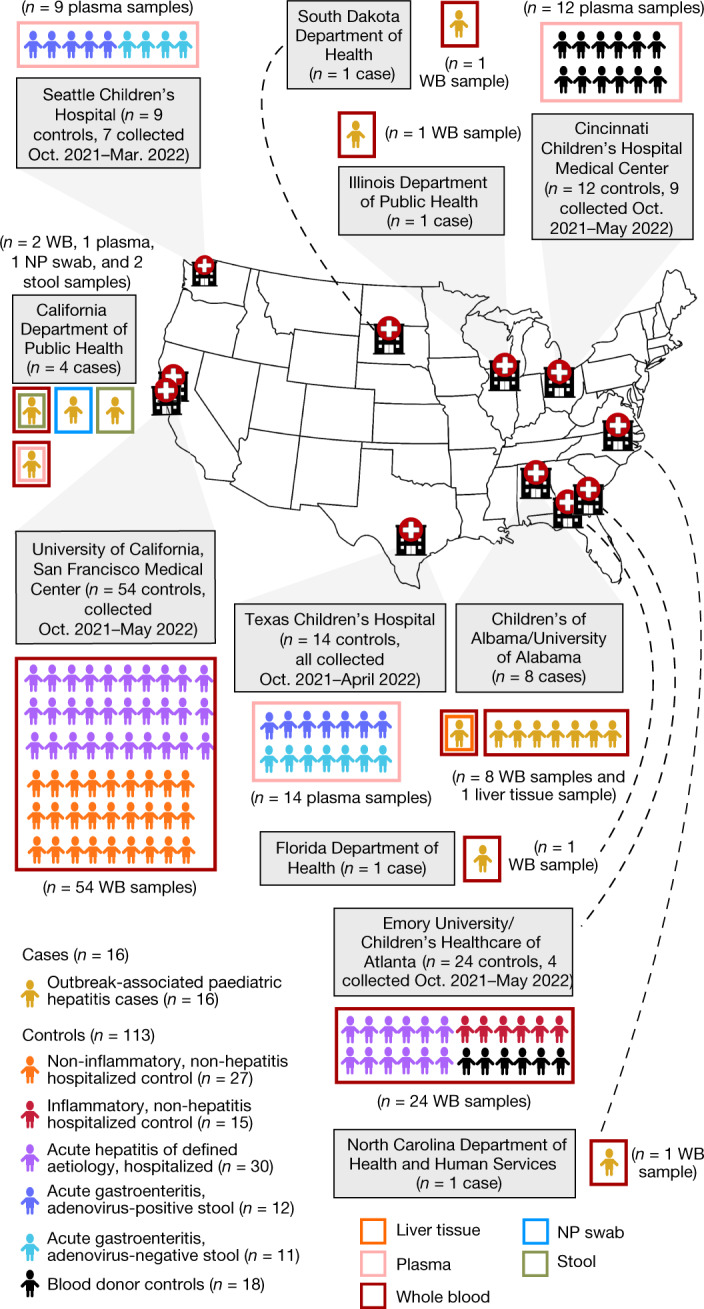
Geographic distribution of the 16 acute severe hepatitis cases and 113 controls in the study, showing the hospital or public health laboratory sites providing samples and associated clinical data from cases and/or controls. WB, whole blood; NP, nasopharyngeal.
Table 1.
Demographic and clinical characteristics of cases and controls
| Characteristic | Cases (n = 16) | Controls, all (n = 113) | Controls, acute hepatitis of defined aetiology (n = 30) | Controls, acute gastroenteritis (n = 23) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Percentage | No. | Percentage | P value | No. | Percentage | P value | No. | Percentage | P value | ||
| Sex | Female | 9 | 56.3% | 56 | 49.6% | 0.79 | 15 | 50.0% | 0.76 | 10 | 43.5% | 0.52 |
| Male | 7 | 43.8% | 57 | 50.4% | 15 | 50.0% | 13 | 56.5% | ||||
| Age (years) mean ± s.d. | 3.8 ± 2.0 | 7.4 ± 6.2 | 0.0239 | 10.7 ± 5.8 | <0.0001* | 1.6 ± 1.6 | <0.0001* | |||||
| Immunocompromised | Yes | 0 | 0.0% | 15 | 13.3% | 0.21 | 6 | 20.0% | 0.08 | 0 | 0.0% | 1 |
| No | 16 | 100.0% | 98 | 86.7% | 24 | 80.0% | 23 | 100.0% | ||||
| ICU stay | Yes | 2 | 12.5% | 30 | 26.5% | 0.50 | 15 | 50.0% | 0.08 | 1 | 3.3% | 0.27 |
| No | 10 | 62.5% | 71 | 62.8% | 15 | 50.0% | 22 | 73.3% | ||||
| Unknown | 4 | 25.0% | 12 | 10.6% | 0 | 0 | 0.0% | |||||
| ALT (U l−1) mean ± s.d. | 2,293 ± 1,486 | N/A | 291 ± 288 | <0.0001* | N//A | |||||||
| AST (U l−1) mean ± s.d. | 2,652 ± 1,851 | N/A | 455 ± 833 | <0.0001* | N/A | |||||||
| Fever | Yes | 6 | 37.5% | 40 | 35.4% | 0.78 | 16 | 53.3% | 0.10 | 13 | 43.3% | 0.51 |
| No | 9 | 56.3% | 46 | 40.7% | 7 | 23.3% | 10 | 33.3% | ||||
| Unknown | 1 | 6.3% | 27 | 23.9% | 7 | 23.3% | 0 | 0.0% | ||||
| Diarrhoea | Yes | 8 | 50.0% | 36 | 31.9% | 0.58 | 10 | 33.3% | 0.74 | 20 | 87.0% | 0.03 |
| No | 7 | 43.8% | 45 | 39.8% | 12 | 40.0% | 3 | 13.0% | ||||
| Unknown | 1 | 6.3% | 32 | 28.3% | 8 | 26.7% | 0 | 0.0% | ||||
| Nausea and/or vomiting | Yes | 11 | 68.8% | 38 | 33.6% | 0.05 | 14 | 46.7% | 0.50 | 17 | 73.9% | 1 |
| No | 0 | 0.0% | 15 | 13.3% | 2 | 6.7% | 0 | 0.0% | ||||
| Unknown | 5 | 31.3% | 60 | 53.1% | 14 | 46.7% | 6 | 26.1% | ||||
| Malaise and/or fatigue | Yes | 10 | 62.5% | 15 | 13.3% | 0.016 | 9 | 30.0% | 0.49 | 0 | 0.0% | N/A |
| No | 5 | 31.3% | 35 | 31.0% | 8 | 26.7% | 0 | 0.0% | ||||
| Unknown | 1 | 6.3% | 63 | 55.8% | 13 | 43.3% | 23 | 100.0% | ||||
| Abdominal pain | Yes | 6 | 37.5% | 22 | 19.5% | 0.24 | 7 | 23.3% | 0.71 | 2 | 8.7% | 0.18 |
| No | 10 | 62.5% | 17 | 15.0% | 7 | 23.3% | 0 | 0.0% | ||||
| Unknown | 0 | 0.0% | 74 | 65.5% | 16 | 53.3% | 21 | 91.3% | ||||
| Anorexia | Yes | 10 | 62.5% | 4 | 3.5% | 0.08 | 3 | 10.0% | 0.12 | 0 | 0.0% | N/A |
| No | 6 | 37.5% | 10 | 8.8% | 8 | 26.7% | 0 | 0.0% | ||||
| Unknown | 0 | 99 | 87.6% | 19 | 63.3% | 23 | 100.0% | |||||
| Jaundice | Yes | 13 | 81.3% | 0 | 0.0% | <0.0001* | 0 | 0.0% | <0.0001* | 0 | 0.0% | N/A |
| No | 2 | 12.5% | 25 | 22.1% | 10 | 33.3% | 0 | 0.0% | ||||
| Unknown | 1 | 6.3% | 88 | 77.9% | 20 | 66.7% | 23 | 100.0% | ||||
| Cough | Yes | 5 | 31.3% | 13 | 11.5% | 0.29 | 10 | 33.3% | 0.74 | 0 | 0.0% | N/A |
| No | 8 | 50.0% | 45 | 39.8% | 12 | 40.0% | 0 | 0.0% | ||||
| Unknown | 3 | 18.8% | 55 | 48.7% | 8 | 26.7% | 23 | 100.0% | ||||
Uncorrected P values were calculated using two-tailed Fisher’s exact test for categorical variables and two-tailed unpaired t-test for continuous variables. A Bonferroni-corrected significance level of P < 0.001 was considered significant (n = 50 comparisons). Significant comparisons are denoted with an asterisk. ICU, intensive care unit; ALT, alanine transaminase; AST, aspartate transaminase.
Fig. 2. Sequencing and molecular-based testing for viruses carried out on cases and controls.

a, Analyses carried out on the different sample groups. The specific assays carried out for each group are shown in green whereas assays that were not carried out are shaded in black. WGS, whole-genome sequencing. mNGS, metagenomic next-generation sequencing. b, Graphical chart showing results of sequencing or PCR-based assays for virus detection. Each circle represents a sequenced sample with at least three non-overlapping reads29 aligning to a viral reference sequence. Circles are colour-coded on the basis of sample type and scaled according to normalized viral read counts. Viral PCR positive and negative results are denoted by plus and minus symbols, respectively. For the HAdV-F PCR, the HAdV subtype identified by hexon sequencing is shown to the left of the viral PCR result. PBV, picobirnavirus; bx, biopsy; ND, not determined. c, Associations between viruses detected in blood and cases of acute severe hepatitis of unknown aetiology in children. Red and blue shading indicate positive and negative detection, respectively. Uncorrected P values were calculated using two-tailed Fisher’s exact test. A Bonferroni-corrected significance level of P < 0.002 was considered significant (n = 24 comparisons). Exact P values are provided in Supplementary Table 2. Ca, cases; Co, controls; AL, Alabama; CA, California; FL, Florida; GA, Georgia; IL, Illinois; NC, North Carolina; OH, Ohio; SD, South Dakota; TX, Texas; WA, Washington; NS, not significant; ***P < 0.001; ****P < 0.0001.
Controls (n = 113) consisted of 78 whole blood, 1 serum and 34 plasma samples. Many controls were enrolled from California (n = 54) and Georgia (n = 24) to be geographically similar to the cases, with the remaining controls enrolled from Ohio (n = 12), Texas (n = 14) and Washington (n = 9). Of the 113 controls, 69 (61.0%) were collected over the same time frame as that for the cases (that is, collected between 1 October 2021 and 22 May 22). The 113 controls consisted of 42 patients (37%) without hepatitis, 30 (26.5%) patients with acute hepatitis (ALT > 100 U l−1) of defined aetiology, 23 (21%) patients with acute gastroenteritis (12 with positive HAdV stool testing) and 18 (16%) blood donors. Differences in age and sex between cases and controls were non-significant overall, although cases were significantly younger and older, respectively, than controls with hepatitis of defined aetiology and those with acute gastroenteritis (Table 1). Among reported symptoms, only jaundice was significantly more associated with cases than controls (Table 1). Mean elevations in ALT and AST in the 30 hepatitis controls were 291 ± 288 U l−1 and 455 ± 833 U l−1, respectively, and significantly lower than in cases (P < 0.0001; Table 1 and Supplementary Table 1).
Virus detection in hepatitis cases
Metagenomic sequencing for agnostic detection of all viruses16, tiling multiplex PCR amplicon sequencing for AAV2 and HAdV-41 (ref. 17), metagenomic sequencing with probe capture viral enrichment18 and virus-specific polymerase chain reaction (PCR) testing for HAdV with genotyping by hexon gene sequencing were carried out to identify AAV2 and HAdV viruses in clinical samples from cases (Fig. 2a). We detected AAV2 from available whole-blood or plasma samples in 13 (93%) of 14 cases and confirmed detection of HAdV in all 14 cases (Fig. 2b). Of the 14 cases, 10 (71%) were typed as HAdV-41, 1 (7%) was typed as HAdV-40, and 1 (7%) was typed as HAdV-2; 1 (7%), sample NC_14, was untypable. AAV2 and HAdV-41 enrichment were carried out using either tiled multiplex PCR amplicon sequencing targeting the genomes of HAdV-41 and AAV (n = 9 samples, with a mean 6.6 ± 1.5 (s.d.) million raw reads generated per sample) or probe capture viral enrichment targeting 3,153 species (n = 4, with a mean 128 ± 34 (s.d.) million raw reads generated per sample). Mean normalized viral counts expressed as reads per kilobase per million mapped reads for AAV2 ( = 4,063) were approximately 12.7 times greater than for HAdV-41 ( = 320; Supplementary Table 1). AAV2 was detected in both liver tissue and whole blood from one case and in one of two plasma samples, but neither AAV2 nor HAdV-41 were detected in nasopharyngeal swab and stool samples. No reads from AAVs other than AAV2 were detected, despite using methods that would enrich for other AAV subtypes. Among the four cases analysed using metagenomic sequencing with probe capture viral enrichment (13_FL, 14_NC, 15_IL and 16_SD), Epstein–Barr virus (EBV) was also detected in the blood sample from 13_FL.
Metagenomic sequencing was less sensitive than viral enrichment and targeted sequencing for detection of AAV2 (8 of 14 cases, 57%) and HAdV-41 (0 of 13 cases, 0%; Fig. 2b and Supplementary Table 1). However, reads from additional viruses were identified including EBV (n = 2), cytomegalovirus (CMV; n = 1), HAdV-2 (adenovirus type C; n = 1) and enterovirus A71 (EV-A71; n = 1) from whole blood, HAdV-1 (adenovirus type C; n = 1) from nasopharyngeal swab, and picobirnavirus (n = 1) from stool. EV-A71 was detected as a co-infection in an HAdV-41–AAV2 case, and HAdV-2 was detected in the single AAV2-negative case.
Among the 113 controls, AAV2 was detected in 4 (3.5%), including 2 (16.7%) of 12 children with acute HAdV-positive gastroenteritis, 1 (9.1%) of 11 hospitalized children with HAdV-negative gastroenteritis, and 1 donor control who was also positive for HAdV (Fig. 2b and Supplementary Table 1). The AAV2-positive child with HAdV-negative gastroenteritis was HAdV-negative from stool but both HAdV41-positive and CMV-positive from blood and had been discharged from the hospital with liver failure (although additional clinical details were not available). Of note, neither AAV2 nor HAdV-41 was detected among the 30 paediatric controls with acute hepatitis of defined aetiology and 42 hospitalized children without hepatitis.
We next carried out virus-specific PCR testing for EBV, CMV, HHV-6 and SARS-CoV-2 for all available cases and controls (Fig. 2b and Supplementary Table 1). EBV and HHV-6 were detected from blood in 11 (79%) of 14 and 7 (50%) of 14 cases, respectively, versus 1 (0.88%) of 113 controls for each virus. No tested cases were positive for CMV compared to 2 (1.8%) of 113 controls. SARS-CoV-2 was not detected in blood from cases or controls (Fig. 2b), or in liver biopsy tissue (Supplementary Table 1).
Virus associations with hepatitis cases
We used Fisher’s exact test on cases (n = 14) and controls to investigate associations between detected viruses in blood and cases of acute severe hepatitis. Controls were stratified into four groups for comparison against cases (Fig. 2c and Supplementary Table 2). Three viruses, AAV2, EBV and HHV-6, were significantly associated with cases when compared to each control group, except for HHV-6 detection between cases and donors (P = 0.010, Bonferroni-corrected significance level of P < 0.002). As all 14 cases were known to be HAdV-positive a priori, statistical analysis was not carried out for HAdV-41.
Phylogenetic and substitution analyses
We carried out multiple sequence alignment and phylogenetic analysis of 13 recovered AAV2 genomes from 12 cases with >25% breadth of coverage (Fig. 3 and Extended Data Fig. 1). Multiple sequence alignment was carried out in parallel with all complete 119 AAV2 reference genomes deposited in GenBank as of 18 August 2022. A whole-genome nucleotide phylogenetic tree revealed that the 13 genomes were located within a distinct subgroup of a large human-infecting AAV2 clade (Fig. 3a). Other previously sequenced AAV2 genomes from France and the USA from patients without hepatitis were also found within this subgroup. Amino acid phylogenetic trees of the VP1 protein and the assembly-activating protein (AAP) showed slightly different topologies (Supplementary Fig. 1), but the 13 genomes were still positioned within a distinct subgroup.
Fig. 3. Phylogenetic and substitution analysis of AAV2 genomes.

a, Phylogenetic tree of 119 AAV2 genomes available from GenBank as of 18 August 2022. The 12 recovered genomes from this study with >25% coverage are denoted in red. The location of the AAV2 reference genome (NC_001401.2) is marked with a black asterisk. The phylogenetic tree was constructed by multiple sequence alignment of the AAV genomes or amino acid sequences using the MAFFT algorithm30, followed by maximum-likelihood-based tree construction using IQ-TREE31. b, Multiple sequence alignment of 13 AAV2 genomes recovered from cases. Nucleotide mismatches are represented by black-coloured vertical lines, whereas areas of missing coverage are represented by dark grey rectangles. Amino acid variants with respect to the reference genome (NC_001401.2) are denoted in blue and red arrows; red arrows indicate shared substitutions that were reported in another study from the UK and Scotland14. Substitution sites that were identified in 100% of cases with sufficient coverage in both studies are highlighted in bold red text. ITR, inverted terminal repeat.
Extended Data Fig. 1. Amino acid phylogenetic analysis of AAV2 genomes from cases, related to Fig. 3.

(A) Phylogenetic tree corresponding to the Assembly Activating Protein (AAP) protein. (B) Phylogenetic tree corresponding to the VP1 protein. Phylogenetic trees was constructed by multiple sequence alignment of the AAV genomes or amino acid sequences using the MAFFT algorithm30, followed by maximum likelihood based tree construction using IQ-TREE31.
To explore the underlying basis behind the observed phylogenetic clustering, we carried out multiple sequence alignment of the 13 genomes from cases. We then searched for shared coding substitutions relative to the AAV2 reference genome that were found in ≥50% of genomes with coverage at that substitution site. This analysis yielded 35 substitutions that were unevenly distributed across the viral genome, with 15 of 35 (42.9%) in the AAP, 14 of 35 (40%) in the VP1 protein, and 6 of 35 (17.1%) in the Rep78 protein (Fig. 3b and Supplementary Table 3). Clusters of substitutions were found to be located within hypervariable regions of the capsid VP1 and AAP proteins13. Of note, 25 (71.4%) of the 35 substitutions were shared with those identified in an independent study of severe paediatric hepatitis from the UK14.
Discussion
Here we report virus findings from PCR, metagenomic and targeted sequencing of samples from 16 paediatric cases of acute severe hepatitis of unknown aetiology that were first identified in the USA in October 2021. After combining these three diagnostic modalities, AAV2 was detected in 93% (13 of 14) of cases for whom wholeblood or plasma samples were available (P < 0.001). Conversely, AAV2 was detected in only 4 (3.5%) of 113 controls and HAdV-41 was detected in 9 (8%) of 113. Notably, AAV2 was not detected in whole blood from 30 paediatric controls with hepatitis of defined aetiology. HAdV sequences were detected in all 14 cases, of which 11 (79%) were genotyped as HAdV-41. The 100% detection rate of HAdV is expected given that all 14 cases were known to be HAdV positive by clinical testing a priori. Other co-infecting viruses, including EBV and HHV-6, were detected by PCR in many cases but rarely in controls (P < 0.001); EV-A71 was also detected by metagenomic sequencing in one case but not in any control. Taken together, these findings show a significant association between co-infection by AAV2 and one or more hepatotropic viral pathogens and the clinical manifestations of severe acute hepatitis, although a direct causal link has yet to be confirmed.
Initial epidemiological investigation and molecular testing have found adenovirus in 45–90% of severe acute hepatitis cases3–7. In the UK, 116 of 179 (64.8%) reported cases as of May 2022 have tested positive for HAdV (ref. 1). This is in comparison to 8 of 9 (88.9%) positive cases in the USA corresponding to a cluster of cases who presented at a children’s hospital in Alabama between October 2021 and March 2022 (ref. 5), for which there is a substantial overlap with cases from our study. An expanded national investigation in the USA detected HAdV in slightly less than half (44.6%) of cases3. Consistent with previous reports5,14, HAdV genotyping of our cases by sequencing confirmed that most of the detected serotypes were HAdV-41 (11 of 14, 79%).
Our results suggest that co-infection with AAV2 may cause more severe liver disease than infection by an adenovirus and/or herpesvirus alone. This condition may be analogous to the fulminant liver failure that can occur when hepatitis D virus (HDV, ‘delta’) infection is superimposed on a chronic hepatitis B virus (HBV) infection19. Notably, AAV2 was seen in only 2 (16.7%) of 12 paediatric controls with HAdV-associated acute gastroenteritis in the absence of liver inflammation, suggesting that co-infection with AAV2 may predispose patients to more severe disease. Here we also found that in cases of AAV2–HAdV co-infection, viral loads for AAV2 were higher than for HAdV, with an approximately 12.7× increase in mean normalized read counts by targeted sequencing. This observation may be partially explained by previously published data showing that AAVs can suppress the replication of other hepatotropic viruses, including adenoviruses and herpesviruses12. Similarly, patients co-infected with HDV usually exhibit low HBV viral loads owing to suppression of HBV replication by HDV-induced interferons20.
Among the AAVs, AAV2 is the most well-characterized AAV and has been shown to replicate to high titres in the liver and spleen11. Seroprevalence data from infants, children and adults demonstrate that 30–80% of the general population is seropositive and that natural infection with AAV can occur at all ages21. However, there is a peak of AAV2 infection between the ages of 1 to 5 years old22, consistent with the observed distribution of ages of cases in the current study.
Phylogenetic analysis reveals that the recovered AAV2 genomes from all cases map to a distinct subclade. The positioning is driven by groups of substitutions located within hypervariable regions of the capsid VP1 and AAP proteins13. Notably, two of these substitutions in the VP1 protein, R585S and R588T, are arginine-to-serine and arginine-to-threonine substitutions that probably impact receptor binding, as these residues are necessary for the interaction of AAV2 with its heparan sulfate proteoglycan receptor23. These are not only shared among the US genomes in the current study but also overlap substantially with the substitutions found in an independent study of acute severe hepatitis cases in children from the UK14. Notably, several of these shared capsid substitutions (V151A, Q457M, S492A, E499D, F533Y, R585S and R588T) are also found in a sublineage of AAV2 (AAVv66) that exhibits increased replication, virion stability, central nervous system transduction and evasion of neutralizing antibodies relative to wild-type AAV2 (ref. 24). However, as other contemporary AAV2 genomes are not readily available, it is unclear whether the relatedness in AAV2 genomes across geographically dispersed regions and to AAVv66 merely represents detection of a predominant global circulating strain.
In the current study, hepatotropic viruses other than HAdV, including EBV, HHV-6 and EV-A71, were detected, albeit in a smaller proportion of cases. Infection by EBV or HHV-6 alone has been implicated in cases of liver failure requiring transplantation25,26. However, although we found substantial differences in the relative proportions of EBV and HHV-6 in cases compared to controls, we do not believe it likely that these viruses are the primary cause of acute severe hepatitis in these children. First, the viral loads for these herpesviruses were very low, with median PCR cycle threshold (Ct) values of 38.1 and 38 for EBV and HHV-6, respectively, and thus the borderline positive PCR results may represent detection of integrated proviral DNA rather than bona fide low-level herpesvirus viraemia. Second, herpesvirus reactivation can be seen in various cytokine-associated inflammatory conditions such as coronavirus disease 2019 (COVID-19)27, and whether such reactivation contributes to disease pathogenesis is unclear. Third, herpesviruses were not detected in available liver biopsy tissue from cases, and the only viruses detected were HAdV (two of six, 33%) and AAV2 (one of one, 100%). Nevertheless, it is striking that among the 16 cases, triple or quadruple infections with AAV2, adenovirus, EBV and/or HHV-6 were detected in whole blood from at least 12 cases (75%). We postulate that the COVID-19 pandemic and more than 2 years of school and childcare closures and decreased social interactions with other children may have generated a vulnerable population of young children who failed to develop broad immunity to common viral pathogens owing to lack of exposure. Notably, among the nine cases with available information, five of nine (55.6%) children had never attended school or a childcare centre, and the remaining four were probably hindered from interacting with other children by social isolation measures enacted at the onset of the pandemic. Decreased immunity from lack of exposure to common viral pathogens may have predisposed cases to infection by multiple viruses, thus increasing the likelihood of more severe disease manifestations such as hepatitis.
Limitations of the study include: lack of availability and/or incomplete testing of liver biopsies, particularly for the presence or absence of AAV2, making it difficult to ascertain the pathogenetic mechanisms underlying the viral infection; low titres associated with detected viral pathogens; and the retrospective study design and the inclusion of only cases that had previously tested positive for HAdV.
In summary, here we identify a distinct strain of AAV2 and co-infection with at least one helper virus in blood from US paediatric cases of acute severe hepatitis of unknown aetiology. These results are consistent with findings from independent and contemporary studies of acute severe hepatitis in children from Scotland and the UK14,28. AAV2 infection may contribute to the pathogenesis and/or severity of the hepatitis, or alternatively, may be a non-pathogenic marker of liver inflammation. Further studies including serologic surveillance, viral culture and animal models are needed to investigate the potential role that AAV2 infection may play in this disease.
Methods
Ethics statement
Remnant clinical samples from cases with acute severe hepatitis were collected and analysed under ‘no subject contact’ protocols with waiver of informed consent approved by the institutional review boards (IRBs) of the University of Alabama, Birmingham, the California Department of Public Health, the New York State Department of Health and the Centers for Disease Control and Prevention (CDC). Whole-blood samples from paediatric controls (age < 18) from Children’s Healthcare of Atlanta were prospectively collected and analysed under a protocol approved by the Emory IRB (STUDY00000723); parents or guardians of these children provided oral consent for study enrolment and collection and analysis of their samples. Remnant whole-blood samples from paediatric controls (age < 18) at University of California, San Francisco (UCSF) were collected, biobanked and analysed under a ‘no subject contact’ protocol with waiver of informed consent approved by the UCSF IRB (protocol no. 11-05519). A subset of the control samples was provided by the CDC from children enrolled in the National Vaccine Surveillance Network (NVSN) study. Approval for the NVSN study was obtained from the institutional review board at each participating site and from the CDC (protocol no. 6164). Parents or guardians of eligible children provided written informed consent for participant enrolment. Blood specimens were also collected as remnant samples from clinical procedures.
Participant recruitment and sample collection
This was a retrospective observational case–control study using all available samples from cases and controls. A severe acute hepatitis case enrolled in this study was a person under investigation by local, state or federal public health agencies, defined as a person <10 years of age with elevated (>500 U l−1) AST or ALT, an unknown aetiology for the hepatitis, and onset on or after 1 October 2021 (ref. 8). All cases (n = 16) were hospitalized with acute elevation in liver enzymes, AST or ALT, and one or more of the following symptoms on presentation: nausea, vomiting, jaundice, generalized weakness and abdominal pain. Cases were selected for inclusion into the current study if blood samples had previously tested positive for adenovirus by clinical testing, resulting in a selection bias that made case–control comparisons related to adenovirus detection not meaningful. Blood samples from cases meeting the criteria for a person under investigation but testing negative for adenovirus were not available.
Controls from UCSF in California (n = 54) included children hospitalized with hepatitis of defined aetiology (>100 U l−1) or another inflammatory or non-inflammatory condition. These controls were selected on the basis of availability of convenience samples from a study of biomarkers of acute inflammatory disease, including severe COVID-19, in hospitalized patients. Controls were selected to be geographically similar (located within the same state) to cases from California. Remnant whole-blood samples from controls from UCSF (n = 54) were retrospectively biobanked and aliquoted with addition of 2× DNA/RNA Shield (Zymo Research) in a 1:1 ratio by volume and stored at −80 °C until use.
Controls from Children’s Healthcare of Atlanta (n = 24) in Georgia included children hospitalized with hepatitis of defined aetiology (>100 U l−1) or another inflammatory or non-inflammatory condition (n = 18) or blood donors (n = 6). These controls were selected from available biobanked samples from consented participants enrolled in a prospective study of COVID-19 and multi-system inflammatory syndrome in children. Controls were selected to be geographically similar (located within a neighbouring state with similar demographic characteristics) to the cases from Alabama and Florida32. Collected samples were stored at −80 °C until use.
Serum or plasma samples from children enrolled in the NVSN study were also included as controls. Three groups of children were selected: children admitted for acute gastroenteritis who tested positive for adenovirus in the stool (n = 12), children admitted for acute gastroenteritis who tested negative for adenovirus in the stool (n = 11) and blood donors (n = 12). These children were enroled from three sites (Seattle, Houston and Cincinnati) from June 2021 to May 2022 and enrolment criteria have previously been described33. Collected samples were stored at −80 °C until use.
Nucleic acid extraction
Whole-blood and plasma samples collected from cases from Alabama were extracted at the Wadsworth Center laboratory using the NucliSENSÒ easyMAG ‘specific B’ protocol (bioMerieux) according to the manufacturer’s instructions. Samples from cases from Florida, Illinois, North Carolina and South Dakota were extracted using the Zymo Direct-zol DNA/RNA Miniprep Kit (Zymo Research) following the manufacturer’s instructions. Briefly, 200 µl of sample was extracted and total nucleic acid was eluted in 60 µl and stored at −80 °C until use.
Samples from cases from California were extracted at the California Department of Public Health. Whole-blood samples (200 µl) were extracted using the Qiagen Blood Mini Kit (Qiagen) according to the manufacturer’s instructions. Total nucleic acid was eluted in 100 µl and stored at −80 °C until use. Respiratory samples, serum, plasma and clarified stool suspensions were extracted using the NucliSENS easyMAG instrument (bioMerieux). Briefly, 300 µl of nasopharyngeal swab, serum or plasma sample or 140 µl of clarified stool suspension was lysed with 1 ml of lysis buffer and incubated for 10 min at room temperature, followed by addition of 100 µl magnetic silica and transfer to the instrument to begin the automated extraction. Total nucleic acid was eluted in 110 µl for nasopharyngeal swab, serum or plasma or 60 µl for stool and stored at −80 °C before use.
For control samples from California, Georgia or the NVSN study, total nucleic acid was extracted at UCSF from the original sample using two different protocols for whole blood, plasma or serum. Whole-blood samples (400 µl) that had been pretreated with DNA/RNA Shield (Zymo Research) were extracted using the Quick-RNA Whole Blood Kit (Zymo Research) according to the manufacturer’s instructions. Total nucleic acid was eluted in 15 µl and stored at −80 °C until use. Plasma or serum samples (200 µl) were extracted using the Mag-Bind Viral DNA/RNA 96 Kit (Omega Bio-Tek) on a KingFisher Flex instrument (Thermo-Fisher) according to the manufacturer’s instructions. Total nucleic acid was eluted in 100 µl and stored at −80 °C until use.
Viral PCR testing
Samples from California cases were screened for adenovirus using a pan-adenovirus PCR targeting all HAdVs34 and/or a group F adenovirus (HAdV-40 or HAdV-41) real-time PCR35. A cycle threshold cutoff of ≤40 was used to call a positive result by PCR. PCR and Sanger sequencing of the HAdV hexon gene targeting hypervariable regions 1–6 (ref. 36) were carried out on all adenovirus-positive samples with purified PCR products sequenced in-house or sent to an outside laboratory (Sequetech, Mountain View, CA) for sequencing. Sanger sequences were assembled and edited in Sequencher 5.2.4 (Gene Codes) and analysed using the nucleotide BLAST aligner37. Samples from cases outside California and all control samples were screened for adenovirus using a pan-adenovirus PCR assay34 and sequenced using a nested PCR assay targeting HAdV hexon hypervariable regions 1–6, followed by Sanger sequencing38. PCR testing for detection of CMV, EBV and HHV-6 was carried out as previously described39.
Viral enrichment-based targeted sequencing
For cases from California and Alabama and all controls, viral enrichment followed by targeted sequencing of viral genomes was carried out using custom spiked primers designed to target HAdV-41 or AAV1–AAV8 genomes. Spiked primers were designed using the metagenomic sequencing with spiked primer enrichment (MSSPE) algorithm17 as follows. For HAdV-41, 22 representative HAdV-41 genomes were aligned using MAFFT, and for AAV, 11 genomes representing AAV1–AAV8 were aligned using MAFFT. Next, automated primer design was carried out by running the MSSPE algorithm with the following parameters: kmer size = 25, segment window = 500, e-value = 0.1, dS = 2, dG = −9,000. The algorithm generated 219 HAdV and 150 AAV spiked primers (Supplementary Table 4). PCR amplification from extracted DNA was carried out as previously described40, with the following modifications: the annealing temperature for PCR was 60 °C for HAdV and 53 °C for AAV. The HAdV primers were amplified in two different reactions using two different sets of primers (Supplementary Table 4, Set1 and Set2), whereas the AAV2 primers were amplified in a single reaction. NGS libraries were prepared using the NEBNext Ultra II DNA Kit (New England Biolabs) and Revelo DNA-Seq Mech Kit on the MagicPrep NGS system (Tecan Genomics). Final libraries were quantified using the Qubit Flex instrument (Invitrogen) with the dsDNA HS Assay Kit (Invitrogen). Libraries were pooled and sequenced on an Illumina NextSeq using 300-base-pair (bp) single-end sequencing. Negative template controls were included in every run to monitor for contamination. No contamination from HAdV-41 or AAV2 reads was detected in the negative template control libraries.
For cases from Florida, Illinois, North Carolina and South Dakota, probe capture target enrichment of viral genomes was carried out on metagenomic libraries using the Twist Comprehensive Viral Research Panel (Twist Biosciences), which covers reference genomes of 3,153 viruses and 15,488 different strains18. Individual DNA libraries or cDNA libraries from RNA were hybridized to Comprehensive Viral Research Panel probes according to the manufacturer’s instructions. Libraries were then barcoded and sequenced on an Illumina MiSeq (250-bp paired-end sequencing) or an Illumina NextSeq (150-bp paired-end sequencing). For plasma sample 14_NC, nucleic acid carrier, either bacteriophage lambda DNA (New England Biolabs) or HeLa total RNA (Thermo-Fisher), was added during the library preparation step according to the manufacturer’s instructions.
Viral metagenomic sequencing and analysis
Metagenomic DNA and RNA libraries were prepared using the Revelo DNA-Seq Mech Kit on the MagicPrep NGS system (Tecan Genomics), NEBNext Ultra II DNA Library Prep Kit (New England Biolabs) and NEBNext Ultra II RNA Library Prep Kit (New England Biolabs), according to the manufacturer’s instructions. Libraries were pooled and sequenced on a NextSeq 550 Sequencing System using 150-bp single-end sequencing. Potential contamination was monitored in each run by processing water and negative controls in parallel with samples.
Sequencing data from all cases and controls were analysed for viral nucleic acids using SURPI+ (v1.0.7-build.4)41, an automated bioinformatics pipeline for pathogen detection and discovery from metagenomic data that has been modified to incorporate enhanced filtering and classification algorithms29. A threshold of ≥3 non-overlapping reads was used for calling a positive virus detection29.
Phylogenetic analysis
Multiple sequence alignments were carried out using MAFFT algorithm (v7.388)30 as implemented in Geneious (version 10.0.9)42. Nucleotide and amino acid phylogenetic trees were inferred using a maximum-likelihood method with ultrafast bootstrap approximation as implemented in IQ-TREE (version 1.6.1)31 using 1,000 bootstrap replicates. Trees were visualized using FigTree (version 1.4.4).
Viral genome assembly and analysis
Binary base call (BCL) files generated by Illumina sequencers were simultaneously demultiplexed and converted to FASTQ files using bcl2fastq (version 2.20.0.422). Custom scripts were used to assemble AdV and AAV2 genomes as follows. Briefly, raw FASTQ reads were filtered using BBDuk (version 38.87)43 for removal of adaptors, primer sequences and low-quality reads, and then HAdV-41 or AAV reads were identified by Bowtie2 (ref. 44) alignment (parameters: -D 20 -R 3 -L11 -N 1) to a reference database consisting of 1,395 HAdV-41 or 3,600 AAV partial sequences or genomes, respectively. These aligned reads were then mapped to the HAdV-41 reference genome (accession DQ315364.2) or consecutively to AAV genomes 1–8. For all AAV genomes, the assembly with the highest breadth of coverage corresponded to the AAV2 reference genome (accession number NC_001401.2). Consensus assemblies were generated using iVar45 (parameters: -t 0.5 -m 1). AAV consensus genomes were further analysed for shared alterations in the nucleotide and translated nucleotide (amino acid) sequences relative to the AAV2 reference by carrying out multiple sequence alignment using the MAFFT algorithm30, followed by visualization of the alignment using Geneious software (version 10.0.9)42.
Statistical analysis
Statistical analyses were carried out using the Python scipy package (version 1.5.2)46 and rstatix package (version 0.7.0) in R (version 4.0.3)47. Uncorrected P values were calculated using two-tailed Fisher’s exact test for categorical variables and two-tailed unpaired t-test for continuous variables, with the significance level determined after a Bonferroni correction for multiple comparisons.
Data visualization
Plots were generated using matplotlib (version 3.3.2), seaborn (version 0.11.0) and plotly (version 5.6.0) packages in Python software (version 3.7.12), Jupyter notebook (version 6.1.4), RStudio (version 1.4) and Adobe Illustrator (version 26.4.1) software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-023-05949-1.
Supplementary information
Detailed demographic, clinical, laboratory and genomic characteristics for individual cases and controls.
Statistical associations of detected viruses in cases compared to controls.
Frequency of single nucleotide variants in AAV2 genomes from cases.
Primer sequences for detection and virus whole-genome sequencing of HAdV-41 and AAV2.
Acknowledgements
We thank the public health officers who provided samples and patient metadata in support of this study: J. Kauerauf and L. Saathoff-Huber, Illinois Department of Public Health; S. Bunch, D. Dessi, A. Gent, C. Panzera, S. Vaccaro and J. Vergara Aguirre, Florida Department of Health; L. DiBiase, University of North Carolina Hospitals; L. Deyneka and R. Pelc, North Carolina Division of Public Health; and the North Carolina State Laboratory of Public Health. We thank H. Guevara, T. Padilla, E. Saguar, C. Wright, B. Molinar, L. Moua and A. Chen from the Respiratory and Gastroenteric Diseases Section of the Viral and Rickettsial Disease Laboratory, California Department of Public Health. We thank staff members at the UCSF Clinical Laboratories and the UCSF Clinical Microbiology Laboratories for their help in identifying and retrieving patient whole-blood samples for biobanking. We thank the Wadsworth Center HAdVanced Genomics Technology Core for carrying out the sequencing reactions, and S. Ogbamikael for carrying out the hexon gene-sequencing assays. We thank the Genomics group at the National Biodefense Analysis and Countermeasures Center for their efforts in creating the genomic libraries at short notice from the samples they were provided. This work was financially supported in part by US CDC contracts 75D30121C12641 and 75D30121C10991 (C.Y.C.), BARDA contract 75A50122C00022 (C.Y.C.), the Chan-Zuckerberg Biohub (C.Y.C.) and National Institutes of Health/National Institute of Child Health and Human Development grant R61HD105618 (C.Y.C. and C.A.R.). This work was also financially supported in part under agreement number HSHQDC-15-C-00064 awarded to Battelle National Biodefense Institute (BNBI) by the Department of Homeland Security (DHS), Science and Technology Directorate, for the management and operation of the National Biodefense Analysis and Countermeasures Center (NBACC), a Federally Funded Research and Development Center. This manuscript has been authored in part by Battelle National Biodefense Institute, LLC under contract with DHS. The publisher, by accepting the article for publication, acknowledges that the US Government retains a non-exclusive, paid up, irrevocable, world-wide licence to publish or reproduce the published form of this manuscript, or allow others to do so, for US Government purposes. The funders had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript; and decision to submit the manuscript for publication. The findings and conclusions in this article are those of the authors and do not necessarily represent the views or opinions of the Alabama Department of Public Health, the California Department of Public Health, the California Health and Human Services Agency, the Florida Department of Health, the Illinois Department of Public Health, the New York State Department of Health/Wadsworth Center, the North Carolina Department of Public Health or the South Dakota Department of Health, nor do they reflect the official policy or position of the Federal Bureau of Investigation (FBI) or CDC. The views and conclusions contained in this article should not be interpreted as necessarily representing the official policies, either expressed or implied, of DHS or the US Government. DHS does not endorse any products or commercial services mentioned in this presentation. In no event shall DHS, BNBI or NBACC have any responsibility or liability for any use, misuse, inability to use or reliance upon the information contained herein. In addition, no warranty of fitness for a particular purpose, merchantability, accuracy or adequacy is provided regarding the contents of this document. This is publication #22.28 of the FBI Laboratory Division. Names of commercial manufacturers are provided for identification purposes alone, and inclusion does not imply endorsement of the manufacturer, or its products or services by the FBI.
Extended data figures and tables
Author contributions
C.Y.C., J.V., R.T.R. and K.S.G. conceived and designed the study. C.Y.C., V.S. and A.S.G. coordinated the study. V.S., A.S.G., A.F., H.J.H., P.E.O. and J.T. carried out metagenomic sequencing and targeted viral enrichment experiments. V.S. and D. Stryke ran the SURPI computational pipeline for detection of viruses from metagenomic data. V.S. and C.Y.C. analysed metagenomic and targeted viral enrichment data, assembled AAV2 genomes and carried out phylogenetic and substitution analyses. A.L.B., N.H.B., R.L.B., K.Y.G., J.S.G., S.P.L., K.P., D.R. and R.T.R. carried out viral probe capture enrichment experiments and assembled AAV2 genomes. R.J., M.T.L., Z.L. and H.D.H. investigated acute severe hepatitis cases and collected samples and clinical metadata. U.P., M.S., K.W., M.S.O., J.E.T., J.M.B., D. Sugerman, C.P., X.L., P.C., J.V., H.L.K. and the CDC Pediatric Hepatitis of Unknown Etiology Working Group investigated acute severe hepatitis cases for the CDC and collected samples and clinical metadata. L.A.I., H.S., W.B. and L.H.G.S. investigated acute severe hepatitis cases in Alabama and collected samples and clinical metadata. C.C. and C.A.R. provided samples and clinical metadata from paediatric controls from Georgia. V.S., A.S.G., J.N. and C.Y.C. provided samples and clinical metadata from paediatric controls from California. A.H., C.-Y.P., K.R., J.B.B., C.M. and D.A.W. investigated acute severe hepatitis cases and carried out viral PCR testing and HAdV genotyping at the California Department of Public Health. D.M.L. and K.S.G. carried out viral PCR testing and HAdV genotyping at the Wadsworth Center/New York State Department of Public Health. C.Y.C. and V.S. wrote the initial draft of the manuscript and prepared the figures. C.Y.C., V.S., A.S.G., U.P., J.E.T., J.M.B., C.P., X.L., J.V., H.L.K. and R.T.R. edited the manuscript. All authors read the manuscript and agreed to its contents.
Peer review
Peer review information
Nature thanks Frank Tacke and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
Raw metagenomic sequencing data with human reads removed have been deposited in the NCBI Sequence Read Archive (Bioproject accession number PRJNA918667, under umbrella BioProject accession number PRJNA171119). Detailed protocols for spiked primer enrichment and metagenomic library preparation, consensus genome files, alignment files, phylogenetic tree files and supplementary tables have been uploaded to a Zenodo data repository (10.5281/zenodo.7089581).
Code availability
Custom scripts for HAdV-41 and AAV genome assembly are available in a Zenodo data repository (10.5281/zenodo.7089581).
Competing interests
C.Y.C. is a founder of Delve Bio and on the scientific advisory board for Delve Bio, Mammoth Biosciences, BiomeSense and Poppy Health. He is also a co-inventor on US patent 11380421, “Pathogen detection using next generation sequencing”, under which algorithms for taxonomic classification, filtering and pathogen detection are used by SURPI+ software. C.Y.C. receives research support unrelated to this manuscript from Abbott Laboratories, Inc. The institution of C.A.R. has received financial support to conduct clinical research unrelated to this manuscript from BioFire Inc., GSK, Janssen, MedImmune, Merck, Moderna, Novavax, PaxVax, Pfizer, Regeneron and Sanofi-Pasteur. She is co-inventor of patented respiratory syncytial virus vaccine technology, which has been licensed to Meissa Vaccines, Inc. K.S.G. receives research support unrelated to this manuscript from Thermo-Fisher and has a royalty-generating collaborative agreement with Zeptometrix. The other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Venice Servellita, Alicia Sotomayor Gonzalez, Daryl M. Lamson
A list of authors and their affiliations appears at the end of the paper
Contributor Information
Charles Y. Chiu, Email: charles.chiu@ucsf.edu
Pediatric Hepatitis of Unknown Etiology Working Group:
Olivia Almendares, Neha Balachandran, Jordan E. Cates, Brandi Freeman, Anita Kambhampati, Rachel Marine, Everardo Vega, Mary E. Wikswo, Erin R. McKeever, David Kelly, Julie A. Boom, Leila C. Sahni, Janet A. Englund, Eileen J. Klein, Mary A. Staat, Maeve Marsh, Erin Ricketts, Joshua L. Clayton, Natalia Cano, Miriam Oseguera, and Robert Blattner
Extended data
is available for this paper at 10.1038/s41586-023-05949-1.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-023-05949-1.
References
- 1.Khader S, Foster I, Dagens A, Norton A, Sigfrid L. Severe acute hepatitis of unknown aetiology in children-what is known? BMC Med. 2022;20:280. doi: 10.1186/s12916-022-02471-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Severe acute hepatitis of unknown aetiology in children - multi-country. WHO Disease Outbreak Newshttps://www.who.int/emergencies/disease-outbreak-news/item/2022-DON400 (2022).
- 3.Cates J, et al. Interim analysis of acute hepatitis of unknown etiology in children aged <10 years - United States, October 2021-June 2022. MMWR Morb. Mortal. Wkly Rep. 2022;71:852–858. doi: 10.15585/mmwr.mm7126e1. [DOI] [PubMed] [Google Scholar]
- 4.Elsheikh R, et al. Acute hepatitis of unknown origin in children: Behind the statistics. Hepatology. 2022 doi: 10.1002/hep.32682. [DOI] [PubMed] [Google Scholar]
- 5.Gutierrez Sanchez LH, et al. A case series of children with acute hepatitis and human adenovirus infection. N. Engl. J. Med. 2022 doi: 10.1056/NEJMoa2206294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelgeri C, et al. Clinical spectrum of children with acute hepatitis of unknown cause. N. Engl. J. Med. 2022 doi: 10.1056/NEJMoa2206704. [DOI] [PubMed] [Google Scholar]
- 7.Marsh K, et al. Investigation into cases of hepatitis of unknown aetiology among young children, Scotland, 1 January 2022 to 12 April 2022. Euro Surveill. 2022;27:2200318. doi: 10.2807/1560-7917.ES.2022.27.15.2200318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Technical Report: Acute Hepatitis of Unknown Cause (CDC, 2022); https://www.cdc.gov/ncird/investigation/hepatitis-unknown-cause/technical-report.html.
- 9.Kager J, et al. Fulminant adenoviral-induced hepatitis in immunosuppressed patients. Viruses. 2022;14:1459. doi: 10.3390/v14071459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hierholzer JC. Adenoviruses in the immunocompromised host. Clin. Microbiol. Rev. 1992;5:262–274. doi: 10.1128/CMR.5.3.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meier AF, Fraefel C, Seyffert M. The interplay between adeno-associated virus and its helper viruses. Viruses. 2020;12:662. doi: 10.3390/v12060662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao G, et al. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl Acad. Sci. USA. 2003;100:6081–6086. doi: 10.1073/pnas.0937739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho, A. et al. Adeno-associated virus 2 infection in children with non-A-E hepatitis. Nature10.1038/s41586-023-05948-2 (2023). [DOI] [PubMed]
- 15.Recommendations for adenovirus testing and reporting of children with acute hepatitis of unknown etiology. CDC Health Alert Networkhttps://emergency.cdc.gov/han/2022/han00462.asp (2022).
- 16.Chiu CY, Miller SA. Clinical metagenomics. Nat. Rev. Genet. 2019;20:341–355. doi: 10.1038/s41576-019-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng X, et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 2020;5:443–454. doi: 10.1038/s41564-019-0637-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KW, et al. Respiratory viral co-infections among SARS-CoV-2 cases confirmed by virome capture sequencing. Sci. Rep. 2021;11:3934. doi: 10.1038/s41598-021-83642-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Negro F. Hepatitis D virus coinfection and superinfection. Cold Spring Harb. Perspect. Med. 2014;4:a021550. doi: 10.1101/cshperspect.a021550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urban S, Neumann-Haefelin C, Lampertico P. Hepatitis D virus in 2021: virology, immunology and new treatment approaches for a difficult-to-treat disease. Gut. 2021;70:1782–1794. doi: 10.1136/gutjnl-2020-323888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.La Bella T, et al. Adeno-associated virus in the liver: natural history and consequences in tumour development. Gut. 2020;69:737–747. doi: 10.1136/gutjnl-2019-318281. [DOI] [PubMed] [Google Scholar]
- 22.Calcedo R, et al. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin. Vaccine Immunol. 2011;18:1586–1588. doi: 10.1128/CVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen CL, et al. Molecular characterization of adeno-associated viruses infecting children. J. Virol. 2005;79:14781–14792. doi: 10.1128/JVI.79.23.14781-14792.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu HL, et al. Structural characterization of a novel human adeno-associated virus capsid with neurotropic properties. Nat. Commun. 2020;11:3279. doi: 10.1038/s41467-020-17047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chevret L, et al. Human herpesvirus-6 infection: a prospective study evaluating HHV-6 DNA levels in liver from children with acute liver failure. J. Med. Virol. 2008;80:1051–1057. doi: 10.1002/jmv.21143. [DOI] [PubMed] [Google Scholar]
- 26.Mellinger JL, et al. Epstein-Barr virus (EBV) related acute liver failure: a case series from the US Acute Liver Failure Study Group. Dig. Dis. Sci. 2014;59:1630–1637. doi: 10.1007/s10620-014-3029-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brooks B, et al. Epstein-Barr virus and human herpesvirus-6 reactivation in acute COVID-19 patients. Viruses. 2022;14:1872. doi: 10.3390/v14091872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morfopoulou, S. et al. Genomic investigations of unexplained acute hepatitis in children. Nature10.1038/s41586-023-06003-w (2023). [DOI] [PMC free article] [PubMed]
- 29.Miller S, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019;29:831–842. doi: 10.1101/gr.238170.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.QuickFacts: Alabama. US Census Bureauhttps://www.census.gov/quickfacts/AL (2021).
- 33.Esona MD, et al. Rotavirus genotype trends and gastrointestinal pathogen detection in the United States, 2014-2016: results from the New Vaccine Surveillance Network. J. Infect. Dis. 2021;224:1539–1549. doi: 10.1093/infdis/jiab177. [DOI] [PubMed] [Google Scholar]
- 34.Heim A, Ebnet C, Harste G, Pring-Akerblom P. Rapid and quantitative detection of human adenovirus DNA by real-time PCR. J. Med. Virol. 2003;70:228–239. doi: 10.1002/jmv.10382. [DOI] [PubMed] [Google Scholar]
- 35.Lyman WH, et al. Prospective study of etiologic agents of acute gastroenteritis outbreaks in child care centers. J. Pediatr. 2009;154:253–257. doi: 10.1016/j.jpeds.2008.07.057. [DOI] [PubMed] [Google Scholar]
- 36.Lu X, Erdman DD. Molecular typing of human adenoviruses by PCR and sequencing of a partial region of the hexon gene. Arch. Virol. 2006;151:1587–1602. doi: 10.1007/s00705-005-0722-7. [DOI] [PubMed] [Google Scholar]
- 37.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 38.Okada M, Ogawa T, Kubonoya H, Yoshizumi H, Shinozaki K. Detection and sequence-based typing of human adenoviruses using sensitive universal primer sets for the hexon gene. Arch. Virol. 2007;152:1–9. doi: 10.1007/s00705-006-0842-8. [DOI] [PubMed] [Google Scholar]
- 39.Dupuis M, et al. Molecular detection of viral causes of encephalitis and meningitis in New York State. J. Med. Virol. 2011;83:2172–2181. doi: 10.1002/jmv.22169. [DOI] [PubMed] [Google Scholar]
- 40.Quick J, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017;12:1261–1276. doi: 10.1038/nprot.2017.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naccache SN, et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014;24:1180–1192. doi: 10.1101/gr.171934.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kearse M, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bushnell, B. BBMap: a fast, accurate, splice-aware aligner. https://www.osti.gov/servlets/purl/1241166 (2014).
- 44.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grubaugh ND, et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Virtanen P, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.R Core Team. R: A Language and Environmental for Statistical Computinghttp://www.R-project.org (R Foundation for Statistical Computing, 2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed demographic, clinical, laboratory and genomic characteristics for individual cases and controls.
Statistical associations of detected viruses in cases compared to controls.
Frequency of single nucleotide variants in AAV2 genomes from cases.
Primer sequences for detection and virus whole-genome sequencing of HAdV-41 and AAV2.
Data Availability Statement
Raw metagenomic sequencing data with human reads removed have been deposited in the NCBI Sequence Read Archive (Bioproject accession number PRJNA918667, under umbrella BioProject accession number PRJNA171119). Detailed protocols for spiked primer enrichment and metagenomic library preparation, consensus genome files, alignment files, phylogenetic tree files and supplementary tables have been uploaded to a Zenodo data repository (10.5281/zenodo.7089581).
Custom scripts for HAdV-41 and AAV genome assembly are available in a Zenodo data repository (10.5281/zenodo.7089581).