

Summary
Proper defense against microbial infection depends on the controlled activation of the immune system. This is particularly important for the RIG-I-like receptors (RLRs), which recognize viral dsRNA and initiate antiviral innate immune responses with the potential of triggering systemic inflammation and immunopathology. Here we show that stress granules (SGs), molecular condensates that form in response to various stresses including viral dsRNA, play key roles in controlled activation of RLR signaling. Without the SG nucleators G3BP1/2 and UBAP2L, dsRNA triggers excessive inflammation and immune-mediated apoptosis. In addition to exogenous dsRNA, host-derived dsRNA generated in response to ADAR1 deficiency is also controlled by SG biology. Intriguingly, SGs can function beyond immune control by suppressing viral replication independent of the RLR pathway. These observations thus highlight the multi-functional nature of SGs as cellular “shock absorbers” that converge on protecting cell homeostasis–by dampening both toxic immune response and viral replication.
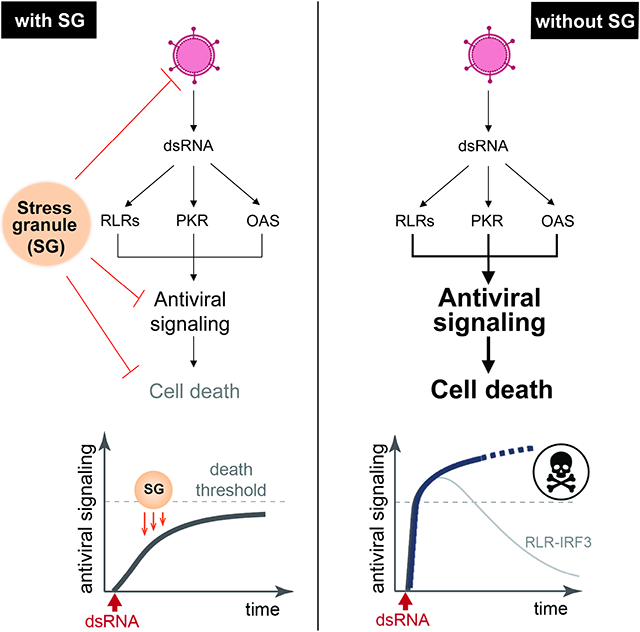
Graphical Abstract
E-toc blurb:
Paget et al. report that stress granules, condensates that form upon cellular stresses, are involved in antiviral innate immunity. Stress granules prevent excessive innate immune activation to protect cells from immune-mediated cell death upon viral infection and in auto-immunopathology disease. This suggests a function for SGs to maintain cellular homeostasis.
Introduction
Detection of foreign nucleic acids is central to innate immune defense in all kingdoms of life.1 Double-stranded RNA (dsRNA) is one such foreign nucleic acid that triggers a wide range of innate immune responses. It has long been thought that dsRNAs are produced only during viral infection as a result of RNA-dependent RNA polymerization of the viral RNA genome or convergent bi-directional transcription of the viral DNA genome.2,3 However, recent studies suggest that dsRNA can also be produced from many dysregulated cellular processes, activating similar innate immune responses as in infected cells.4,5 Accordingly, the innate immune and inflammatory response to dsRNAs underlie diverse pathologies from autoimmunity to neurodegeneration and to metabolic disorders.6-8
One family of innate immune receptors that shape the cellular response to dsRNA are RIG-I-like receptors (RLRs).9 RIG-I and MDA5 in the RLR family function as the first line of defense against a broad range of viruses. Upon dsRNA binding, RLRs multimerize and activate the signaling adaptor molecule MAVS by inducing MAVS multimerization.10-13 Activated MAVS then triggers a cascade of biochemical events culminating in the activation of IRF3 and NF-κB, and subsequent induction of a large group of antiviral genes, including type I interferons (IFNs).
In addition to transcriptional remodeling by the RLR pathway, foreign dsRNA also triggers other cellular changes, including assembly of molecular condensates known as stress granules (SGs).14,15 SG assembly is a highly conserved cellular phenomenon in eukaryotes, and is induced not only by dsRNA, but also by other cellular stress conditions, including heat shock and oxidative stress. These diverse stimuli activate several kinases, for example the dsRNA-dependent kinase PKR, which phosphorylate the translational initiation factor eIF2α and suppress global protein synthesis to help cells recover from stress.16 SGs are formed when stalled ribosome-mRNA complexes accumulate and aggregate together with other cytoplasmic proteins, including the key nucleators G3BP1/2 and UBAP2L. Recent studies showed that G3BP1/2 utilize multivalent interactions with RNA to form a network of protein-RNA interactions in cells, which then drives SG nucleation.17-20 UBAP2L also plays an important role in SG formation, albeit through a poorly understood mechanism.18,21,22
Physiological functions of SG formation are yet unclear. While SGs were initially thought to be the sites of translational suppression, recent studies argued against this notion.23,24 In the context of innate immunity, SGs were proposed to function as the signaling scaffold for RLRs.25,26 This is based on the observations that, RLRs are concentrated at SGs together with viral RNAs, and that knocking down G3BPs diminished induction of type I IFNs. This is in line with the previous reports that SGs are frequently targeted or altered by many viruses.27-29 However, other reports have raised questions whether SGs are in fact the sites of RLR activation. While a subset of viral RNAs are enriched within SGs, dsRNAs are excluded.26,30 Additionally, SG-disrupting pharmacological agents (e.g. cycloheximide) do not impair RLR signaling, while other stressors, such as arsenite or heat-shock, trigger SGs and RLR colocalization without activating RLRs.30,31 Furthermore, SGs were reported to suppress other innate immune pathways, such as NLRP3 inflammasome and MAPK signaling.32,33
Here we report evidence supporting that SGs have at least two distinct functions in antiviral innate immunity. First, SGs prevent excessive activation of RLR signaling and immune-mediated cell death. Second, SGs have cell-intrinsic activity to restrict viral replication independent of RLRs. These findings highlight the multi-functional nature of SGs.
Results
SG-deficient ΔG3BPs cells display hyperactivation of RLR signaling
To understand the role of SGs in RLR signaling, we first examined cellular response to in vitro transcribed 162 bp dsRNA harboring 5’-triphosphate groups (5’ppp), a known ligand for RIG-I.31 Use of a viral dsRNA mimic ensures a potent RLR stimulation without confounding factors such as viral antagonisms. Immunofluorescence (IF) analysis showed that dsRNA transfection of U2OS cells induced formation of cytosolic granules enriched for SG markers G3BP1 or TIAR14,15 (Figures 1A and S1A-B). These granules displayed characteristics of SGs, such as the dependence on the two key nucleators G3BP1 and G3BP2 (G3BPs)17-20 and the sensitivity to cycloheximide treatment (Figure S1C). They were also enriched for RLRs, MAVS and downstream signaling molecules, including TRAF proteins and TBK1 (Figures 1A and S1B), all of which are known features of SGs.26,34,35 Given that MAVS is anchored to the outermembrane of mitochondria, we further examined cellular localization of two other mitochondrial proteins, COXIV and NIX, and found that both proteins colocalized with SGs (Figure S1B), which is also consistent with other reports of mitochondrial association with SGs.35,36 Of note, these dsRNA-triggered SGs appeared different in size and composition from cytosolic granules triggered by polyIC (Figure S1C)—a synthetic dsRNA mimetic formed by a mixture of two polymers (poly-I and poly-C) of heterogeneous length with meshed network structures.37 Further, unlike a previous report,38 polyIC-stimulated granules in our study were also dependent on G3BPs and were incompletely suppressed by PKR/RNase L double knock-out (Figures S1D and S1E), likely reflecting reported variabilities among commercial polyIC reagents.39-41
Figure 1. RLR signaling is hyperactive in SG-deficient ΔG3BPs cells.

A. Immunofluorescence (IF) analysis of RIG-I, MAVS and dsRNA (red) with G3BP1 (green) in U2OS cells. See Figure S1A for antibody validation. Cells were transfected with 162 bp dsRNA containing 5’ppp (500 ng/ml) for 6 hrs prior to imaging. Raw images for RIG-I and MAVS without contrast adjustment (raw) were also shown. For dsRNA imaging, 162 bp dsRNA 3’-labeled with Cy5 was introduced into cells by lipofectamine transfection or electroporation. Unless mentioned otherwise, unlabeled dsRNA and lipofectamine transfection was used throughout the manuscript. Cell nuclei were stained with Hoechst 3342. Bottom right: SG colocalization was measured by Pearson colocalization coefficient (PCC) between G3BP1 foci and indicated molecules from 10 fields of view.
B. Heatmap of z-scores displaying differentially expressed genes in WT vs ΔG3BPs U2OS cells. Cells were transfected with 162 bp dsRNA with 5’ppp (500 ng/ml) for 6 hrs. Genes showing log2-fold change (lfc2) >2 (with p_adj<0.05) upon dsRNA stimulation in a MAVS-dependent manner (based on the analysis in Figure S2B) were shown. All genes were shown in Figure S2A.
C. Levels of IFNβ, IL-6, and RANTES mRNAs. U2OS cells were transfected with dsRNA as in (B) and were analyzed 6 or 24 hr post-dsRNA. Data were normalized to WT 6 hr post-dsRNA.
D. Levels of secreted IFNβ, IL-6, and RANTES as measured by ELISA. U2OS cells were transfected with dsRNA as in (B) and were analyzed at 6 hr post-dsRNA.
E. Level of IFNβ mRNAs in response to the increasing concentrations of dsRNA (50-2000 ng/ml) at 6 hr post-dsRNA. Data were normalized to WT 50 ng/ml.
F. Activation state of IRF3, as measured by its phosphorylation level in U2OS cells.
G. Activation state of IRF3, as measured by its nuclear translocation. U2OS cells were stained with anti-IRF3 antibody at indicated timepoints and the level of nuclear IRF3 signal was quantitated (a.u. indicates arbitrary unit). Each data point represents a nucleus (n=61-179). DAPI staining was used for defining nuclear boundary.
H. Activation state of MAVS, as measured by cell-free IRF3 dimerization assay. Mitochondrial fraction (P5) containing MAVS was isolated from U2OS cells 6 hrs post-dsRNA, and mixed with a common pool of cytosolic extract (S18) from unstimulated WT U2OS cells and in vitro translated 35S-IRF3. Dimerization of 35S-IRF3 was analyzed by native gel assay. * indicates mini-MAVS.
I. Cell-free IRF3 dimerization assay, comparing the activity of the mitochondrial fraction isolated from WT, ΔG3BPs and ΔG3BPs/ΔMAVS U2OS cells.
Data are presented in means ± SD. p values were calculated using two-tailed unpaired Student’s t test (ns, p>0.05). RNA-seq results contain 2 biological repeats and were confirmed by two independent experiments. All other data are representative of at least three independent experiments.). Raw data for the heatmap can be found in the supplemental file (data 1).
We next examined cellular distribution of dsRNA, of which 3’-end was labeled with Cy5. Cy5-dsRNA activated RLRs similarly to non-labeled dsRNA (Figure S1F). SG localization of dsRNA was minimal regardless of whether dsRNA was delivered to the cytoplasm by cationic lipid transfection or by electroporation (Figure 1A, bottom). The lack of dsRNA colocalization is not in line with the notion that SGs are the sites of RLR activation.
We next compared RLR signaling in the wild-type (WT) and SG-deficient G3BP knock-out (ΔG3BPs) background by examining their transcriptome at two time points (6 and 24 hr post-dsRNA). ΔG3BPs cells displayed enhanced antiviral signaling than WT cells at 6 hr post-dsRNA (Figures 1B and S2A). This was confirmed by independent analysis of mRNA levels of select few cytokines (IFNβ, IL-6 and RANTES, Figure 1C) and their secreted protein levels (Figure 1D). Enhanced signaling was observed in ΔG3BPs at all doses of dsRNA tested (Figure 1E). G3BPs complementation in ΔG3BPs cells restored SG formation and suppressed RLR signaling (Figure S2C), further supporting the role of G3BPs in suppressing RLR signaling.
In contrast to the immediate response to dsRNA, RLR signaling at 24 hr post-dsRNA showed more complex, gene-specific patterns in ΔG3BPs cells. While most dsRNA-induced genes increased in expression from 6 to 24 hr post-dsRNA, a subset of genes, including IFNβ, markedly decreased from 6 hr to 24 hr (Figure S2D). Similarly, RT-qPCR measurement showed that the spike in the IFNβ mRNA level in ΔG3BPs cells at 6 hr was followed by a sharp decline at 24 hr to the level comparable to the WT level (Figure 1C). In contrast, RANTES and IL-6 mRNAs remained higher in ΔG3BPs cells at both 6 and 24 hr (Figure 1C).
Given the complex behavior of gene induction, we next examined RLR signaling by measuring the activation state of the upstream signaling molecules IRF3 and MAVS. IRF3 is the transcription factor responsible for IFNβ induction and its activation requires IRF3 phosphorylation, dimerization and nuclear translocation. Analysis of the levels of p-IRF3 and nuclear IRF3 showed that IRF3 was more active in ΔG3BPs than in WT cells at 6 hr post-dsRNA (Figures 1F & 1G). However, at 24 hr post-dsRNA, a sharp drop was seen in ΔG3BPs cells both with p-IRF3 and nuclear IRF3, mirroring the pattern of IFNβ mRNA induction. As will be discussed in Figure 4, this decline of the IRF3 activity was due to negative feedback regulation and cell death that was enhanced in ΔG3BPs cells.
Figure 4. SGs dampen dsRNA-triggered apoptosis and the consequent negative feedback regulation of IRF3.

A-C. Cell death in WT vs. ΔG3BPs U2OS cells at 24 hr post-dsRNA as examined by (A) bright-field microscopy, (B) caspase-3/7 activity and (C) Sytox uptake.
D. Cell death in response to staurosporin (STS) and etoposide. U2OS cells were treated with STS (1 μM) or etoposide (20 μM) for 24 hrs before Sytox analysis.
E. Cell death in WT, ΔUBAP2L and ΔPKR U2OS cells at 24 hr post-dsRNA.
F. Comparison of cell death triggered by dsRNA, etoposide and a combination of caspase-8 inhibitor (Z-IETD-FMK, Casp-8i) and TNFα. Etoposide was used as a known trigger for apoptosis, while Casp-8i+TNFα was for necroptosis.
G. Analysis of PARP and caspase-3 (Casp-3) cleavage using samples from (F).
H. Apoptotic caspase cleavage in U2OS cells at 6 or 24 hr post-dsRNA.
I. Effect of pan-caspase inhibitor (Q-VD-OPh) on dsRNA-triggered cell death, as measured by Sytox uptake at 24 hr post-dsRNA. U2OS cells were treated with Q-VD-OPh (10 μM) 1 hr pre-dsRNA.
J. Effect of Q-VD-OPh (10 μM) on PARP cleavage in U2OSΔG3BPs cells at 24 hr post-dsRNA.
K. Effect of Q-VD-OPh on IRF3 phosphorylation and caspase-3 cleavage.
L. Effect of Q-VD-OPh on IFNβ mRNA induction in U2OSΔG3BPs cells. Data were normalized to 6 hr post-dsRNA in the absence of Q-VD-Oph.
M. Effect of Q-VD-OPh on IFNβ mRNA induction in A549 cells. Data were normalized to 6 hr post-dsRNA in WT A549 in the absence of Q-VD-Oph.
Data are presented in means ± SD. p values were calculated using two-tailed unpaired Student’s t test (ns, p>0.05). All data are representative of three independent experiments.
We next examined the activation state of MAVS using a previously established cell-free assay.10,13 The mitochondrial fraction (P5) containing MAVS was extracted from dsRNA-stimulated WT or ΔG3BPs cells, and the signaling potential of MAVS was measured by incubating P5 with cytosolic fraction (S18) from unstimulated WT cells, which provided a common pool of downstream signaling molecules in the resting state (Figure 1H, left). In vitro-translated 35S-labeled IRF3 was added to the mixture in order to measure MAVS’ ability to activate IRF3, as visualized by the monomer-to-dimer transition of 35S-IRF3 in the native gel. Only P5 from dsRNA-stimulated cells could activate 35S-IRF3 (Figure 1H) and this required MAVS (Figure 1I). Most importantly, P5 from ΔG3BPs cells was more potent than WT P5 (Figure 1H, right). Altogether, these results show that the RLR signaling pathway is more potently activated by dsRNA in ΔG3BPs than in WT cells, as measured by RLR-induced cytokine levels, global transcriptome, and activation states of IRF3 and MAVS.
We next asked whether hyper-activation of RLRs in ΔG3BPs cells at an early timepoint is generalizable to other cell types and whether this is independent of the method of dsRNA delivery or the type of dsRNA. As with U2OS cells, A549, HeLa and human bronchial epithelial cells (HBEC) also formed SGs upon dsRNA introduction (Figure S3A). In all cases, SG formation required G3BPs (Figure S3A). These cells also displayed hyper-activation of RLRs in the ΔG3BPs than in WT background (Figure S3B-D). Comparison of the transcriptome at the basal level did not reveal any obvious and consistent pattern of basal inflammation in ΔG3BP cells (Figure S3E). Comparing different methods of dsRNA delivery, we found that ΔG3BPs cells consistently showed higher levels of RLR signaling either by electroporation (Figure S3F) or lipofectamine transfection of dsRNA (Figure 1). dsRNAs of different duplex lengths or with different sequences also triggered more potent RLR signaling in ΔG3BPs cells (Figure S3G). However, dsRNA-independent signaling activities of gain-of-function (GOF) MDA5, GOF RIG-I or STING were unaffected by ΔG3BPs (Figures S3H and S3I). Given that the dsRNA-independent activation of GOF MDA5, GOF RIG-I or STING occurs without SG formation, these observations suggest that the effect of G3BPs may be specific to SG-forming conditions.
SGs suppress RLR signaling in response to dsRNA
To test whether the observed effect of G3BPs on RLR signaling is indeed mediated by SGs rather than other potential functions of G3BPs, we examined two other genetic models for SG deficiency, ΔUBAP2L and ΔPKR. Like G3BPs, UBAP2L is an essential nucleator for SGs.17,21 PKR, on the other hand, is not directly involved in SG assembly, but is an upstream kinase that blocks translation in response to dsRNA, a pre-requisite for SG nucleation.14,15 Previous studies suggested that certain granules distinct from SGs formed in ΔPKR cells upon polyIC stimulation.38 We also found that ΔPKR cells formed G3BP1 foci, but they differed from SGs in that they did not show enrichment of MAVS or TIAR (Figure 2A) and that they were significantly smaller in size (Figure 2B, top). ΔUBAP2L cells also displayed G3BP1 foci upon dsRNA stimulation, but these foci lacked MAVS (Figure 2A) and were smaller in size and less frequent (Figure 2B). Thus, both ΔPKR and ΔUBAP2L cells can form G3BP1 foci but they are distinct from conventional SGs in WT cells, as measured by the composition, size and frequency. Importantly, both ΔUBAP2L and ΔPKR cells showed hyperactivation of RLR signaling at 6 hr post-dsRNA (Figures 2C-2E), similar to ΔG3BPs cells. Thus, analysis of three distinct SG-deficient backgrounds (ΔG3BPs, ΔUBAP2L and ΔPKR) commonly suggest that SGs suppress RLR signaling, at least at early time points.
Figure 2. SG deficiency leads to hyperactivation of RLR signaling.

A. Immunofluorescence analysis of RIG-I, MAVS, TIAR (red) and G3BP1 (green) in ΔUBAP2L and ΔPKR U2OS cells at 6 hrs post-dsRNA.
B. G3BP1 foci size and frequency in ΔUBAP2L and ΔPKR U2OS cells. Foci size was quantitated for at least 200 randomly selected granules from Z-stack images (0.15 μm step size). Foci frequency was measured from 5 fields of view.
C. Antiviral signaling in U2OS cells (WT vs ΔUBAP2L) in response to dsRNA transfection (500 ng/ml). Data were normalized to WT 6 hr post-dsRNA.
D. Same as (C), comparing WT and ΔPKR U2OS cells.
E. IRF3 phosphorylation in U2OS cells (WT vs ΔPKR) upon dsRNA stimulation.
F. Level of protein synthesis as measured by puromycin incorporation (SUnSET assay72). U2OS cells were transfected with dsRNA (500 ng/ml) for 6 hrs and pulsed with puromycin (1 μg/ml) for 15 mins prior to anti-puromycin WB.
G. Colocalization of RIG-I, MAVS and TIAR (red) with G3BP1 (green) in U2OSΔPKR cells upon treatment with TG (1 μM) without dsRNA. G3BP1 foci size was quantitated for at least 600 randomly selected granules from Z-stack images (0.15 μm step size).
H. Antiviral signaling in U2OS cells (WT vs ΔPKR) in response to dsRNA, in the presence and absence of TG. Cells were treated with TG (1 μM) at 1 hr post-dsRNA and harvested 6 hr post-dsRNA. Data were normalized to WT in the absence of TG.
I. Antiviral signaling in U2OS cells (WT vs ΔPKR) in response to dsRNA, with or without nutrient starvation (N.S.). Cells were incubated with a starvation medium for 2 hrs prior to dsRNA transfection and were harvested 6 hr post-dsRNA. Data were normalized to WT in the absence of nutrient starvation. Left: SGs in ΔPKR cells upon nutrient starvation for 8 hrs as visualized by G3BP1 foci.
Data are presented in means ± SD. p values were calculated using two-tailed unpaired Student’s t test (ns, p>0.05). Data are representative of three independent experiments.
At 24 hr post-dsRNA, however, ΔPKR cells diverged from ΔUBAP2L and ΔG3BPs cells in gene induction patterns. Both ΔUBAP2L and ΔG3BP cells showed sustained activation of IL-6 and RANTES, displaying higher levels than WT cells at both 6 and 24 hr. ΔPKR cells, however, showed dramatic reduction in both genes at 24 hr, dropping below the WT levels (Figures 2C & 2D). We suspect that this divergence between PKR and G3BPs/UBAP2L reflects the fact that ΔPKR affects both translation and SG formation, while ΔG3BPs and ΔUBAP2L selectively affect SG formation without altering translation (Figure 2F). Note that translation alone has a positive effect on innate immune signaling.42-44
We next asked whether the RLR-suppressive function is specific to dsRNA-triggered SGs or whether SGs triggered by other stmuli, such as ER stress or nutrient starvation, can also repress RLR signaling. Thapsigargin (TG) is an ER stressor and triggers SGs by activating PERK, instead of PKR. Accordingly, addition of TG to ΔPKR cells induced SGs, as judged by colocalization of RIG-I, MAVS and TIAR with G3BPs foci (Figure 2G). Since ΔPKR did not form SGs in response to dsRNA, ΔPKR cells offered a unique opportunity to test the effect of TG-triggered SGs on RLR signaling. TG treatment in ΔPKR cell reduced RLR signaling at 6 hr post-dsRNA (Figure 2H). Similar suppression was not observed in WT cells where dsRNA alone was sufficient to induce SGs (Figure 2H). Nutrient starvation also suppressed dsRNA-dependent antiviral signaling only in ΔPKR cells, not in WT cells (Figure 2I). Thus, SGs suppress RLR signaling, regardless of whether SGs are induced by PKR or PERK.
SGs also suppress PKR and OAS pathways
We next asked whether SGs can also affect other innate immune pathways, such as those mediated by PKR and OAS enzymes, which recognize dsRNA and cooperate with RLRs for antiviral immunity (Figure 3A). While both PKR and OASes globally restrict protein synthesis, they do so through different mechanisms; PKR restricts translation through eIF2α phosphorylation, whereas OASes activate RNase L—a highly potent and non-specific ribonuclease that cleaves rRNAs, tRNAs and mRNAs.45
Figure 3. SGs suppress PKR and OAS pathways.

A. Schematic of dsRNA-dependent innate immune pathways, involving the dsRNA sensors RLRs, PKR and OASes.
B. IF analysis of PKR, OAS3, RNase L (red) and G3BP1 (green) in U2OS cells. See Figure S1A for antibody validation.
C. SG colocalization was measured by Pearson colocalization coefficient (PCC) between G3BP1 foci and indicated molecules from 10 fields of view.
D. PKR activity in WT vs. ΔG3BPs U2OS cells as measured by PKR phosphorylation and ATF4 expression at indicated time points.
E. RNase L activity in WT vs. ΔG3BPs U2OS cells as measured by rRNA degradation. Total RNA was isolated 24 hr post-dsRNA and was analyzed by TapeStation.
As previously reported46-49, PKR, OAS3 and RNase L were enriched within SGs (Figures 3B-3C). PKR was more active in ΔG3BPs cells than in WT cells, as measured by the levels of p-PKR and ATF416 upon dsRNA transfection (Figure 3D). Given that PKR is an upstream inducer of SGs, this result suggests that SG is a negative feedback regulator of PKR. Similarly, OASes were also more active in ΔG3BPs cells, as measured by the integrity of 28S/18S rRNAs (Figure 3E). Since the levels of PKR and OASes are induced by type I IFNs and thus the RLR pathway, it is possible that enhanced RLR signaling in ΔG3BPs contributed to the hyperactivation of PKR and RNase L. In keeping with this notion, knocking out MAVS partly rescued RNA integrity of ΔG3BPs cells (Figure 3E). Altogether, these results suggest that SGs inhibit all three dsRNA-dependent innate immune pathways involving RLRs, PKR and OASes (Figure 3A).
Lack of SG leads to apoptosis and the consequent suppression of IRF3 at later time points
In addition to hyperactivation of the innate immune signaling pathways, we also found that ΔG3BPs cells underwent pronounced cell death upon dsRNA stimulation, as measured by the loss of cell-to-surface attachment, Sytox uptake, caspase-3/7 activity and LIVE/DEAD dye staining (Figures 4A-4C and S4A). While a high dose of dsRNA can trigger cell death even in WT cells, ΔG3BPs cells underwent significantly more pronounced cell death at all doses of dsRNA tested (Figure 4B). Cell death progressed gradually over 24 hrs (Figure 4C). The hypersensitivity of ΔG3BPs cell viability was dependent on the cell death trigger, as other stimuli, such as etoposide and staurosporine, did not trigger greater cell death in ΔG3BPs than WT cells (Figure 4D). Similar increase in dsRNA-triggered cell death in the ΔG3BPs background was observed with A549 (Figures S4B-D), HeLa (Figures S4E-G) and HBEC cells (Figure S4H). Moreover, ΔUBAP2L and ΔPKR cells also underwent increased cell death upon dsRNA stimulation (Figure 4E), further supporting the notion that the lack of SGs is responsible for the hypersensitivity to dsRNA.
DsRNA-induced cell death in ΔG3BPs was accompanied by morphologic features similar to apoptotic blebs (Figure 4F, left and center) but distinct from necroptotic cells (Figure 4F, right). We also observed cleavage of PARP and caspase-3, consistent with apoptotic, not necroptotic cell death (Figure 4G). Other apoptotic caspases, such as caspases-8 and -9, were also activated (Figure 4H), but pyroptotic caspase-1 was not (Figure S4I). A pan-caspase inhibitor (Q-VD-Oph) completely blocked dsRNA-dependent cell death and cleavage of PARP in ΔG3BPs (Figures 4I & 4J), but a pyroptosis inhibitor (disulfiram) or necroptosis inhibitor (MLKL inhibitor) did not (Figure S4J). Thus, dsRNA-mediated cell death in ΔG3BPs is caspase-dependent apoptosis.
We next asked whether apoptosis or caspase activation was the reason for the marked decline in the IRF3 activity in ΔG3BPs cells at later time points (Figures 1C and 1E). This hypothesis was based on previous reports that apoptotic caspases can suppress IRF3 signaling by cleaving MAVS, IRF3 and/or RIP1.50-52 While we did not observe clear signs of caspase-dependent cleavage of RIG-I, MAVS and IRF3 (Figure S4K), Q-VD-Oph significantly improved the level of p-IRF3 at 24 hr without significantly altering it at 6 hr (Figure 4K). This was in line with the observation that caspase activity was high at 24 hr but was minimal at 6 hr (Figure 4H). Similar effect of Q-VD-Oph was observed on the level of IFNβ mRNA in U2OSΔG3BPs (Figure 4L) and A549ΔG3BPs cells (Figure 4M). In keeping with minimal activation of caspases in WT U2OS cells, Q-VD-Oph did not affect RLR signaling in WT cells (Figures 4K and 4M). Collectively, these data suggest that the lack of SGs results in hyperactivation of caspases in response to dsRNA, which causes a marked decline in the IRF3 activity at later time points. In other words, SGs prevent dsRNA-induced caspase activation and cell death, minimizing caspase-dependent suppression of the RLR-MAVS-IRF signaling axis and allowing a more sustained immune activation.
SGs minimize cell death by preventing overstimulation of innate immune pathways by dsRNA
We next investigated why SG deficiency leads to increased cell death in response to dsRNA stimulation. We first examined the potential role of the RLR-MAVS pathway, as it has previously been shown to contribute to dsRNA-dependent cell death.53,54 Knocking out MAVS largely rescued cell viability in U2OSΔG3BPs (Figure 5A) and A549ΔG3BPs (Figure S5A) upon dsRNA stimulation. Knocking out MAVS also significantly reduced cleavage of caspase-3, -8 and -9, albeit not to completion (Figure 5B). However, the IRF3-IFNα/β signaling axis downstream of MAVS was not important as knocking out IRF3 did not restore cell viability in ΔG3BPs cells despite the complete abrogation of IFNβ induction (Figures 5A & 5C). Inhibitors of TBK1 and JAK also did not alleviate dsRNA-triggered cell death in ΔG3BPs (Figure S5B), further suggesting that the cell death in ΔG3BPs is mediated by a MAVS-dependent, but IRF3- and IFN-independent mechanism.
Figure 5. SGs prevent dsRNA-triggered cell death by suppressing RLR, PKR and OAS pathways.

A. Cell death in U2OS cells as measured by Sytox uptake (left) and bright field microscopy (right) at 24 hr post-dsRNA.
B. Apoptotic caspase cleavage in Δ U2OS cells at 14 or 24 hr post-dsRNA.
C. Levels of IFNβ and TNFα mRNAs in U2OS cells at 6 hr post-dsRNA. Data were normalized to WT at 6 hr post-dsRNA.
D. Heat map of z-scores for differentially expressed genes in apoptosis pathway (KEGG pathway hsa04210) in U2OS cells at 6 hr post-dsRNA stimulation.
E. Level of secreted TNFα in U2OS cells 6 hr post-dsRNA.
F. Effect of anti-TNFα antibody on dsRNA-triggered cell death in U2OSΔG3BPs. Cells were pre-treated with anti-TNFα antibody (0.01, 0.1 and 1 μg/ml) 30 min prior to transfection with dsRNA. Cell death was measured by Sytox uptake at 24 hr post-dsRNA.
G. Cell death in ΔG3BPs and ΔG3BPsΔPKR at 24 hr post-dsRNA.
H. Cell death in ΔG3BPs and ΔG3BPsΔRNase L at 24 hr post-dsRNA.
I. Schematic for dsRNA-induced cell death in ΔG3BPs cells. The lack of SGs make ΔG3BPs cells hypersensitive to dsRNA, resulting in more potent activation of RLR, PKR and OASes. The TNFα signaling branch (but not the IRF3-IFN branch) downstream of RLR-MAVS makes the primary contribution to cell death in U2OS cells. PKR and OASes-RNase L also contribute, likely by suppressing global protein synthesis.
Data are presented in means ± SD. p values were calculated using two-tailed unpaired Student’s t test (ns, p>0.05). All data are representative of three independent experiments.
To identify the potential apoptosis-triggering factors downstream of MAVS, we looked for apoptosis-related genes among those up-regulated by dsRNA through the RLR pathway (Figure 5D). Several pro-apoptotic genes, including TNFα, FAS and TNFRSF10B (DR5), were hyperinduced in ΔG3BPs than in WT cells, and their induction was dependent on MAVS but not IRF3 (Figures 5C and 5D). We also detected markedly elevated secretion of TNFα in ΔG3BPs cells compared to WT cells (Figure 5E). Blocking TNFα signaling using anti-TNFα significantly relieved dsRNA-induced cell death in ΔG3BPs cells (Figure 5F). The RLR-MAVS-TNFα signaling axis was dependent on NF-kB since NF-kB inhibitors, bengamide B and ACHP, reduced expression of TNFα and cell death (Figures S5C and S5D). However, treatment with TNFα alone did not induce cell death (Figure S5E), suggesting that TNFα cooperates with other factors to induce apoptosis in ΔG3BPs cells.
We next examined the potential role of PKR and OAS-RNase L pathways in cell death, as they lie downstream of MAVS and are also hyperactivated in ΔG3BPs cells. Knocking out PKR or RNase L partially relieved dsRNA-triggered cell death in U2OSΔG3BPs cells, albeit not to the same extent as MAVS knock-out (Figures 5G & 5H). In A549ΔG3BPs cells, knocking out RNase L significantly rescued cell viability, but knocking out PKR did not (Figures S5F & S5G), suggesting that the contribution of each dsRNA-sensing pathway can vary depending on cell type. Another note-worthy observation was that, even in the same cell type, the effect of PKR knock-out was highly context-dependent; in the wild-type U2OS background, knocking out PKR increased dsRNA-triggered cell death (likely due to the inhibition of SG formation) (Figure 4E), while in the G3BPs-deficient background, knocking out PKR decreased dsRNA-triggered cell death (likely due to the relief of translational inhibition) (Figure 5G). This highlights the context-dependent impact of PKR on the life-death decision.
Altogether, our data indicate that SGs minimize dsRNA-induced cell death by suppressing innate immune responses mediated by the RLR-MAVS-TNFα, PKR-eIF2α and OAS-RNase L pathways. The IRF3-mediated type I IFN response does not play a role, likely due to the strong negative feedback regulation through apoptotic caspases (Figure 5I).
SGs suppress RLR signaling during viral infection, while restricting viral replication independent of RLRs.
We next investigated how SGs impact innate immune responses and cell viability during viral infection. We used four viruses: Sendai virus (SeV), influenza A virus (IAV), vesicular stomatitis virus (VSV) and encephalitis myocarditis virus (EMCV). For IAV and VSV, the NS1-deletion variant of IAV (IAVΔNS1) and the M51R variant of VSV (VSVM51R) were used as they trigger more potent immune responses.55,56 SeV, IAVΔNS1 and VSVM51R predominantly activate RIG-I, whereas EMCV activates MDA5.57
SeV infection robustly induced SG formation and RLR signaling in WT U2OS cells (Figures 6A-C), but ΔG3BPs cells mounted a more potent RLR response (Figures 6B and 6C) and increased cell death (Figures 6D and 6E). EMCV, IAVΔNS1 and VSVM51R also showed similar results (Figures 6F and 6G; see also Figures S6A-G). In all cases, RLR signaling and cell death were predominantly dependent on MAVS (Figures 6B, 6F and 6G).
Figure 6. SGs suppress RLR signaling during viral infection, while restricting viral replication independent of RLRs.

A. IF analysis of RIG-I, MAVS (red) and G3BP1 (green) in U2OS cells. Cells were infected with SeV (100 HA/ml) for 20 hrs.
B. Levels of secreted IFNβ, IL-6, RANTES and TNFα as measured by ELISA. U2OS cells were infected with SeV (100 HA/ml) and were analyzed 6 hr post-infection (hpi).
C. Antiviral signaling in U2OS cells upon SeV infection (MOI=1.0). Data were normalized to WT at 6 hpi.
D. Cell death in U2OS cells at 24 hr post-SeV infection (MOI=0, 0.1, and 1.0).
E. Effect of anti-TNF and Q-VD-Oph on cell death in U2OSΔG3BPs cells upon SeV infection. Cells were infected with SeV (MOI=0.1, and 1.0), treated with inhibitors 1 hpi and analyzed at 24 hpi.
F. Antiviral signaling upon infection with IAVΔNS1, EMCV and VSVM51R. A549 cells were infected with IAVΔNS1 (MOI=0.1) and EMCV (MOI=0.1), whereas U2OS cells were infected with VSVM51R (MOI=1). Cells were harvested at 24 hpi for IAVΔNS1 and 6 hpi for EMCV and VSVM51R. Data were normalized to WT in the presence of virus for each graph. See also Figure S6 for more comprehensive analysis with different MOIs and time of analysis.
G. Cell death upon infection with IAVΔNS1, EMCV and VSVM51R at 24 hpi.
H. IF images of SeV proteins (green). U2OS cells were infected with SeV (MOI=1) and stained with anti-SeV serum at 18 hpi.
I. Relative cell-to-cell spreading of SeV (MOI=1). Number of cells above the background fluorescence per field of view were analyzed. Each data point represents a field of view (n=20). Data were normalized against the WT average value.
J. Relative level of SeV protein staining in infected cells (MOI=1). Corrected total cell fluorescence (CTCF) at 18 hpi. Each data point represents infected cell (n=200). Data were normalized against the WT average value.
K. Schematic summarizing the dual function of SGs in (i) suppressing RLR signaling and (ii) restricting viral replication independent of the RLR pathway. Both functions converge on maintaining cell homeostasis.
Data are presented in means ± SD. All data are representative of at least three independent experiments. p values were calculated using two-tailed unpaired Student’s t test (ns, p>0.05).
To examine how SGs impact viral replication and propagation, we measured cell-to-cell spreading and viral protein level per infected cell using immunofluorescence with antibodies against SeV, IAVΔNS1 and VSVM51R proteins (Figure 6H, antibodies against EMCV proteins were not available). With all three viruses, cell-to-cell spreading was more restricted in ΔG3BPs than in WT cells, and this effect of G3BPs required MAVS (Figures 6I and S7A-S7C). This result is consistent with the notion that hyper-active RLR signaling in ΔG3BPs cells restricts viral replication and spreading. Unexpectedly, the level of viral proteins per infected cell was higher in ΔG3BPs than in WT cells for all three viruses (Figures 6J and S7A-S7C), and this effect of G3BPs was independent of MAVS. In keeping with this, overall viral mRNAs were higher in ΔG3BPs cells than in WT cells (Figures S7E-H). These data altogether suggest that SGs have at least two independent functions during infection: (1) suppressing RLR signaling and consequent cell death, and (2) restricting viral replication independent of RLR signaling (Figure 6K).
SGs protect cells from self-derived dsRNA accumulated under the ADAR1 deficiency
Our findings above showed that SGs have immune-suppressive and cell-protective roles against exogenous dsRNA regardless of the specific origins of dsRNA. We next asked whether these functions of SGs can be extended to cellular responses to self-derived dsRNA, which can erroneously accumulate under pathologic conditions. One such condition is the deficiency of RNA-editing enzyme ADAR1, which converts adenosine within dsRNA to inosine and disrupts duplex RNA structure.58 ADAR1 deficiency leads to accumulation of endogenous dsRNAs, aberrant activation of MDA5, PKR and OASes and ultimately pathogenesis of autoinflammatory diseases.59-65 The immunological effect of the ADAR1 deficiency is more evident upon IFNβ treatment (priming), which upregulates the levels of all three types of dsRNA sensors and further sensitizes cells to self-derived dsRNAs.63-65 Consistent with these reports, priming with IFNβ was necessary in WT cells for robust SG formation and RLR-MAVS signaling in ADAR1-knocked-down cells (Figures 7A & 7B). These foci are SGs as evidenced by TIAR colocalization. Note that IFNβ priming without ADAR1 knock-down did not activate RLR signaling, in line with the ligand requirement for RLR signaling. In ΔG3BPs cells, ADAR1 knock-down resulted in a more potent RLR response than in WT cells, both in the presence and absence of IFNβ pre-treatment (Figure 7B). ADAR1 knock-down also led to significantly increased cell death in ΔG3BPs than in WT cells (Figure 7C). As with exogenous dsRNA stimulation, this cell death was predominantly dependent on MAVS and partly on PKR and RNase L (Figure 7C), and could be relieved by anti-TNFα and pan-caspase inhibitor (Figure 7D). ΔUBAP2L cells were also hypersensitive to ADAR1 knock-down than WT cells (Figure 7E), further supporting the notion that SG deficiency increases sensitivity to the ADAR1 deficiency (Figure 7E). These results together suggest that SGs suppress excessive immune activation and cell death, whether they are triggered by exogenous or endogenous dsRNA.
Figure 7. SGs suppress immune response to self-derived dsRNAs under the ADAR1 deficiency.

A. IF analysis of G3BP1 (green) and TIAR (red) in U2OS cells in the presence or absence of ADAR1 knock-down and IFNβ priming. Cells were transfected with siRNA for 24 hrs and then treated with IFNβ (10 ng/ml) for additional 24 hrs prior to imaging.
B. Antiviral signaling in U2OS cells upon ADAR1 knock-down. Data were normalized to WT cells in the presence of IFNβ priming and ADAR1 knock-down.
C. Cell death upon ADAR1 knock-down, as measured by brightfield images (left) and Sytox uptake (right).
D. Effect of anti-TNF and pan-caspase inhibitor (Q-VD-Oph) on cell death upon ADAR1 knock-down.
E. Antiviral signaling and cell death upon ADAR1 knock-down in U2OS cells. All samples were treated with IFNβ (10 ng/ml). Data were normalized to WT cells in the presence of ADAR1 knock-down.
F. Schematic summarizing the roles of SGs in protecting cells from dsRNA. SGs suppress a broad range of dsRNA-triggered innate immune pathways (RLR, PKR and OASes), regardless of the origin of dsRNA. In particular, SGs slow down the ramp-up speed of RLR signaling and help maintain its magnitude below the “death” threshold. In the absence of SGs, RLRs are hyperactivated, leading to an excessive innate immune response and consequent cell death. The IRF3-IFN axis downstream of RLR-MAVS does not contribute to cell death and often displays a dynamic temporal behavior characterized by a sharp peak followed by a strong decline due to caspase-dependent feedback regulation.
Data are presented in means ± SD. p values were calculated using two-tailed unpaired Student’s t test (ns, p>0.05). All data are representative of three independent experiments.
Discussion
Phase separation has recently emerged as a widespread phenomenon that occurs in many biological processes, from transcription to signal transduction.66,67 Functions of phase separation, however, remain unclear in most cases. SGs are one such phase separated entities with poorly characterized functions. We here use three independent SG-deficient genetic—ΔG3BPs, ΔUBAP2L and ΔPKR—to show that SGs exert a negative impact on dsRNA-dependent innate immune pathways. RLRs and downstream signaling molecules were highly enriched within SGs, but dsRNA was not, arguing against the idea that SGs are sites of RLR activation or that SGs selectively recruit activated molecules. Rather, these innate immune molecules appear to be recruited to SGs independent of their activation state, which may exert sequestration effect and retard their activation. It is also possible that transient transit through SGs may alter their signaling activities, perhaps by post-translational modifications or by promoting association with inhibitory molecules. We found that absence of SGs led to overactivation of the RLR pathway, excessive production of TNFα and other pro-apoptotic genes, and consequent cell death. The SG deficiency also resulted in hyperactivation of PKR and OASes, further contributing to dsRNA-dependent cell death. We thus propose that SGs function as a “buffer” or “shock absorber” to enable controlled activation of innate immune pathways upon dsRNA stimulation, by increasing the temporal window for mounting an appropriate immune response while maintaining its magnitude below the “death” threshold (Figure 7F). Given our findings that SGs control cellular response not only to viral dsRNA, but also to self-derived dsRNAs that can be generated from many dysregulated cellular processes, SGs may serve as a key immune modulator in a broad range of pathophysiological conditions.
How can we reconcile our findings with previous reports suggesting that SGs amplify RLR signaling? One potential explanation may be in the complexity with which cells regulate IRF3-dependent IFN induction, one branch of the RLR pathways that is often used as a measure of the overall RLR signaling activity. Unlike other signaling axes downstream of RLR-MAVS, the IRF3-IFNs axis in SG-deficient cells responded to dsRNA with an initial spike followed by a marked decline. This decline, which was seen only in SG-deficient cells, was a result of strong negative feedback regulation by apoptotic caspases that were activated in SG-deficient cells. We speculate that such feedback regulatory mechanism for the IRF3-IFNs axis may account for seemingly conflicting data in the literature. Additionally, specific methods of SG inhibition may have further contributed to the confusion. Unlike depleting SG nucleators G3BPs and UBAP2L, depletion of PKR resulted in a more complex, time-dependent signaling behavior. This likely reflects the fact that PKR regulates both translation and SG formation, while G3BPs and UBAP2L only affect SGs without altering translational control. Additionally, PKR is not only an upstream inducer of SGs, but is also subject to SGs-mediated feedback regulation. These complex relationships between PKR and SGs caution using ΔPKR as the sole genetic model for the SG-deficiency.
Our results show a dual role of SGs in antiviral immunity—suppressing RLR-mediated excessive inflammation and restricting viral replication independent of RLRs. Previous studies showed that many viral RNAs and proteins are localized within SGs, which may exert sequestration and inhibitory functions independent of RLRs.68,69 Additionally, all viruses rely on the host ribosome to synthesize viral proteins, which may be affected by having a large pool of the 40S subunit enriched within SGs.70,71 Our observations thus highlight the multi-functional nature of SGs that cannot be simply categorized into anti- or pro-viral activities. This is in line with the observation that different viruses cope with SGs differently; some inhibit SGs, while others alter or take advantage of SGs.27-29 The diverse functions of SGs may instead be understood as a cellular mechanism for maintaining cell homeostasis—a common consequence of dampening toxic immune response and viral replication.
Limitations of the Study:
It is currently unclear precisely how SGs regulate both antiviral innate immune response and viral replication. We also do not know the fraction of RLR signaling molecules localized within SGs. What we reported here are the common effect of the three different genetic perturbations (ΔG3BPs, ΔUBAP2L and ΔPKR) and the three chemical perturbations (dsRNA, TG and nutrient starvation), which all converge on the notion that SGs suppress antiviral signaling. However, it is possible that other biological processes besides SGs are commonly affected by all of these genetic and chemical perturbations and could have contributed to the observed effect on antiviral signaling and viral replication.
STAR*Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to and will be fulfilled by the corresponding author Sun Hur (Sun.Hur@crystal.harvard.edu).
Materials availability
All unique materials will be available upon publication and request.
Data and Code Availability
Data Availability: RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Imaging data have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-G3BP1 | Cell Signaling | Cat #17798 |
| Mouse anti-G3BP1 | Santa Cruz Biotechnology | Cat #sc-365338 |
| Rabbit anti-RIG-I | In-house | Takashi Fujita lab (Oh et al., 2016) |
| Rabbit anti-MAVS | Fortis Life Sciences | Cat #A300-782A |
| Mouse anti-TIAR | Santa Cruz Biotechnology | Cat #sc-398372 |
| Rabbit anti-phospho-IRF3 | Cell Signaling | Cat #4947 |
| Rabbit anti-IRF3 (WB) | Cell Signaling | Cat #4302 |
| Rabbit anti-IRF3 (IF) | In-house | Takashi Fujita lab (Oh et al., 2016) |
| Rabbit anti-beta-actin | Cell Signaling | Cat #4302 |
| Mouse anti-vinculin | Cell Signaling | Cat#4650S |
| Rabbit anti-UBAP2L | Fortis Life Sciences | Cat #A300-533A |
| Rabbit anti-PKR | Cell Signaling | Cat #12297 |
| Rabbit anti-G3BP2 | Fortis Life Sciences | Cat #A302-040A |
| Rabbit anti-NIX | Cell Signaling | Cat #12396 |
| Rabbit anti-COXIV | Cell Signaling | Cat #4850 |
| Rabbit anti-VCP | Abcam | Cat #ab111740 |
| Rabbit anti-NDP52 | Abcam | Cat #ab68588 |
| Rabbit anti-MDA5 | Cell Signaling | Cat #5321 |
| Mouse anti-TRAF2 | Santa Cruz Biotechnology | Cat #sc-136999 |
| Mouse anti-TRAF6 | Santa Cruz Biotechnology | Cat #sc-8409 |
| Rabbit anti-TBK1 | Cell Signaling | Cat #3054 |
| Mouse anti-puromycin | EMD Milipore | Cat #MABE343 |
| Rabbit anti-PKR | Cell Signaling | Cat #12297 |
| Mouse anti-OAS3 | Santa Cruz Biotechnology | Cat #sc-398225 |
| Rabbit anti-RNase L (WB) | Cell Signaling | Cat #2728 |
| Mouse anti-RNase L (IF) | Santa Cruz Biotechnology | Cat #sc-74405 |
| Rabbit anti-phospho-PKR | Abcam | Cat #ab32036 |
| Rabbit anti-ATF4 | Cell Signaling | Cat #11815 |
| Rabbit anti-Caspase-8 | Cell Signaling | Cat #4790 |
| Rabbit anti-Caspase-8 | Sigma Aldrich | Cat #NB100-56116 |
| Rabbit anti-Caspase-9 | Cell Signaling | Cat #3493 |
| Rabbit anti-Caspase-3 | Cell Signaling | Cat #9662 |
| Rabbit anti-PARP | Cell Signaling | Cat #9532 |
| Rabbit anti-Sendai Virus pAb | MBL | Cat #PD029 |
| Rabbit anti-Influenza Virus A NP pAb | Thermo Fisher | Cat #PA5-32242 |
| Mouse anti-VSV-G | Kerafast | Cat #EB0010 |
| Mouse anti-ADAR1 | Santa Cruz Biotechnology | Cat # sc-73408 |
| Rabbit anti-Caspase-1 | Cell Signaling | Cat #2225 |
| Anti-Rabbit IgG-HRP | Cell Signaling | Cat #7074P2 |
| Anti-Mouse IgG-HRP | GE Healthcare | Cat #NA931V |
| Alexa Fluor 647® Affinipure Donkey anti-Rabbit IgG | Jackson ImmunoResearch Laboratories | Cat #711-605-152 |
| Alexa Fluor® 488 Affinipure Donkey anti-mouse IgG | Jackson ImmunoResearch Laboratories | Cat #715-545-150 |
| Alexa Fluor® 488 Affinipure Donkey anti-rabbit IgG | Jackson ImmunoResearch Laboratories | Cat #711-545-152 |
| Alexa Fluor® 647 Affinipure Donkey anti-mouse IgG | Jackson ImmunoResearch Laboratories | Cat #715-605-151 |
| Bacterial and virus strains | ||
| Sendai Virus Cantell Strain | ATCC | Cat #VR-907 |
| Sendai Virus Cantell Strain | Charles River Laboratories | Cat #VR-907 |
| Encephalomyocarditis virus (Cell culture adapted) | ATCC | Cat #VR-1762™ |
| IAVΔNS1 | Laboratory of Benjamin TenOever | Benjamin TenOever lab (Blanco-Melo et al., 2020) |
| VSV M51R | Laboratory of Benjamin TenOever | Benjamin TenOever lab |
| Chemicals, peptides, and recombinant proteins | ||
| Lonza NucleofectorTM 2b | Lonza Bioscience | Cat #: AAB-1001 |
| Nucleofector Kit L | Lonza Bioscience | Cat #VCA-1005 |
| Mammalian Protease Arrest | G-Biosciences | Cat #768-108 |
| TnT T7 Coupled Reticulocyte Lysate System | Promega | Cat #L4610 |
| Thapsigargin | Selleck Chem | Cat #S7895 |
| Puromycin | Selleck Chem | Cat #S7417 |
| CellEventTM Caspase 3-7 Green detection reagent | Invitrogen | Cat #C10423 |
| Hoechst 33342 | Thermo Fisher | Cat #H3570 |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermo Fisher | Cat #D1306 |
| Fluoromount G | SouthernBiotech | Cat#0100-01 |
| Etoposide | Selleck Chem | Cat #S1225 |
| Staurosporine | Selleck Chem | Cat #S1421 |
| Z-IETD-FMK | Selleck Chem | Cat #S7314 |
| Human TNF-α | Cell Signaling | Cat #8902 |
| Q-VD-Oph | Selleck Chem | Cat #S7311 |
| Human TNF-α Neutralizing Rabbit mAb | Cell Signaling | Cat #7321 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Invitrogen | Cat #13778150 |
| Recombinant IFN-β | Peprotech | Cat #300-02BC |
| Doxycycline | Fisher Scientific | Cat #AC446060050 |
| Cycloheximide | Selleck Chem | Cat#S7418 |
| Calf intestinal phosphatase (CIP) | New England Biolabs | Cat #M0525S |
| 2’-3’-cGAMP | Invivogen | Cat #tlrl-nacga23 |
| Tetraethylthiuram disulfide (Disulfiram) | Sigma-Aldrich | Cat #86720 |
| Necrosulfonamide | Sigma-Aldrich | Cat #480073 |
| Ruxolitinib | Selleck Chem | Cat #S1378 |
| BX-795 | Selleck Chem | Cat #S1274 |
| ACHP | Fisher Scientific | Cat#45-471-0 |
| Bengamide B | Fisher Scientific | Cat#52-731-00U |
| Pierce™ Sodium meta-periodate | Thermo Fisher | Cat #20504 |
| Zeba desalting columns | Thermo Fisher | Cat #89882 |
| Alt-R® S.p. Cas9 Nuclease | IDT | Cat #1081058 |
| Neon™ Transfection System | Invitrogen | Cat # MPK5000 |
| Neon™ Transfection System 10 μL Kit | Invitrogen | Cat #MPK1025 |
| Lipofectamine 3000 | Invitrogen | Cat #L3000001 |
| Digitonin | Calbiochem | Cat #CAS 11024-24-1 |
| Amersham ECL Prime Western Blotting Detection Reagent | Cytiva Life Sciences | Cat#RPN2236 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher | Cat #34094 |
| 4–15% Mini-PROTEAN® TGX™ Precast Protein Gels, 15-well | Bio-Rad | Cat #4561086 |
| TRI reagent (Trizol) | Zymo Research | Cat #R2050-1-200 |
| RNA Clean & Concentrator | Zymo Research | Cat #R1017 |
| High-Capacity cDNA reverse transcription kit | Applied Biosystems | Cat #4368813 |
| RNase Inhibitor, Human Placenta | New England Biolabs | Cat#M0307L |
| SYBR Green Master Mix | Applied Biosystems | Cat #4309155 |
| DMEM, high glucose, pyruvate | Cellgro | Cat #10-013-CV |
| DMEM, no glucose, no glutamine, no phenol red | Gibco | Cat #A1443001 |
| Airway epithelial cell basal medium | ATCC | Cat #PCS-300-030 |
| Bronchial epithelial cell growth kit | ATCC | Cat #PCS-300-040™ |
| Direct-zol RNA Miniprep kit | Zymo Research | Cat #R2052 |
| OPTI-MEM | Gibco | Cat #31985070 |
| Critical commercial assays | ||
| LumiKine™ Xpress hIFN-β 2.0 | Invivogen | Cat code luex-hifnbv2 |
| Human CCL5/RANTES Quantikine ELISA Kit | R&D systems | Cat #DRN00B |
| Human IL-6 Quantikine ELISA Kit | R&D systems | Cat #D6050 |
| Human TNF-alpha Quantikine ELISA Kit | R&D systems | Cat #DTA00D |
| Live/Dead ™ cell imaging kit | Thermo Fisher | Cat #R37601 |
| Sytox Green Nucleic Acid stain | Thermo Fisher | Cat #S7020 |
| Deposited Data | ||
| Raw data from RNA-seq | This study | GSE173953 |
| Raw imaging data | This study | 10.17632/ndwddpcyzm.1 |
| Experimental models: Cell lines | ||
| U2OS (WT, ΔG3BPs, ΔPKR, ΔUBAP2L) | Laboratory of Dr. Paul J. Anderson | Paul Anderson lab (Aulas et al., 2017; Kedersha et al., 2016; Sanders et al., 2020) |
| U2OS (ΔMAVS, ΔRIG-I, ΔG3BPs ΔRNase L, ΔPKR, ΔRNase L, ΔG3BPs ΔIRF3 and ΔG3BPs ΔPKR) | This study | N/A |
| A549 WT | Laboratory of Dr. Susan Weiss | Susan Weiss lab (Li et al., 2017) |
| A549 (ΔG3BPs, ΔG3BP ΔMAVS and ΔG3BPs ΔRNase L) | This study | N/A |
| HBEC3-KT | ATCC | CRL-4051 |
| HBEC3ΔG3BPs | This study | N/A |
| HeLa WT | Laboratory of Dr. Gracjan Michlewski | Gracjan Michlewski lab (Choudhury et al., 2014) |
| HeLaΔG3BPs | This study | N/A |
| HEK293T | ATCC | CRL-11268 |
| Oligonucleotides | ||
| 112 or 162 bp dsRNA | In-house | (Cadena et al., 2019) |
| Cy5-Monohydrazide | Cytiva | Cat #PA15121 |
| Poly I:C | Invivogen | Cat #tlrl-pic |
| ON-TARGETplus Human ADAR1 (103) siRNA Set | Horizon Discovery | Cat #LQ-008630-00-0020 |
| ON-TARGETplus Non-targeting Pool | Horizon Discovery | Cat #001810-10-20 |
| Primer sequences | See table S1 | N/A |
| Recombinant DNA | ||
| plentiCRISPRv2 | Addgene | Cat #98290 |
| psPAX2 | In-house | James DeCaprio lab (Cheng et al., 2017) |
| pMD2.G VSV-G | In-house | James DeCaprio lab (Cheng et al., 2017) |
| pInducer20 | In-house | Hidde Ploegh Lab (Ashour et al., 2015) |
| pInducer20-hG3BP1-IRES-hG3BP2 | This study | N/A |
| pFLAG-CMV4 empty vector | In-house | Ahmad et al., 2018 |
| pFLAG-CMV4-MDA5 (R337G) | In-house | Ahmad et al., 2018 |
| pFLAG-CMV4-MDA5 (G495R) | In-house | Ahmad et al., 2018 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
U2OS cells
Cells were maintained in DMEM (High glucose, L-glutamine, Pyruvate) with 10% fetal bovine serum
A549 cells
Cells were maintained in DMEM (High glucose, L-glutamine, Pyruvate) with 10% fetal bovine serum
HEK293T cells
Cells were maintained in DMEM (High glucose, L-glutamine, Pyruvate) with 10% fetal bovine serum
HeLa cells
Cells were maintained in DMEM (High glucose, L-glutamine, Pyruvate) with 10% fetal bovine serum
HBEC cells:
HBEC3-KT cells were maintained in Airway Epithelial Cell Basal Medium supplemented with the contents of a Bronchial Epithelial Cell Growth kit.
METHOD DETAILS
Material Preparation
Cell lines
U2OS, A549, HeLa, HBEC3-KT and HEK293T cells were used for all experiments in this paper. The parental wild-type U2OS, G3BP1 and G3BP2 double knock-out cell lines were kindly provided by Dr. Paul J. Anderson and described elsewhere (Kedersha et al., 2016). The U2OS PKR knock-out cell line was generated by Dr. Shawn M. Lyons. Briefly, DNA encoding a guide RNA which targets the 5th exon of PKR were cloned into pCas-Guide (Origene) according to manufacturer’s instructions. U2OS cells were co-transfected with pCas-Guide-PKR and pDonor-D09 using Lipofectamine 2000. The following day, cells were selected with 1.5 mg/ml of puromycin for 48 hrs to select for transfectants. Single cell clones were isolated by limiting dilution and confirmed by western blotting and genomic sequencing. The U2OS UBAP2L knock-out cell line was kindly provided by Dr. Paul J. Anderson (Sanders et al., 2020). HEK293T cells were purchased from ATCC. The parental A549 cells were kindly provided by Dr. Susan Weiss, University of Pennsylvania (Li et al., 2017). The parental Hela cells were kindly provided by Dr. Gracjan Michlewski, University of Edinburgh.
For U2OS cells, for the generation of the PKR (in ΔG3BPs) knock-out cell line, the ribonucleoprotein complex with gRNA and Alt-R® S.p. Cas9 Nuclease V3 was delivered using Lipofectamine 3000. After 48 hrs, media was refreshed, and single clones were isolated by limiting dilution and confirmed by western blotting and genomic sequencing. For the generation of the MAVS (both in WT and ΔG3BPs backgrounds), RNase L (in WT, ΔPKR and ΔG3BPs), RIG-I and IRF3 (in ΔG3BPs) knockout cell lines, gRNA was cloned in the pLentiCRISPRv2 vector using the restriction enzyme BsmBI. 293T cells were transfected with the following plasmids at a 3:1:0.7 ratio: (i) pLentiCRISPRv2 with the gRNA, (ii) pMD2.G VSV-G and (iii) psPAX2. U2OS (WT and ΔG3BPs) were infected with 0.45 μM filtered supernatants harvested at 48 hrs post-transfection for 48 hrs, then selected with 1 μg/ml neomycin.
For HelaΔG3BPs and A549ΔG3BPs, cells were first transduced using a gRNA targeting G3BP1 as described above. After transduction, the ribonucleoprotein complex with gRNA targeting G3BP2 and Alt-R® S.p. Cas9 Nuclease V3 (IDT) was delivered using Lipofectamine 3000, as described above as well. For generation of RNase L, MAVS and PKR knockout in A549 ΔG3BPs the same gRNA and same approach was used as described for the U2OS cells. A list of all gRNAs can be found in Supplementary Table 2. Single cell clones were isolated by limiting dilution and confirmed by western blotting and genomic sequencing. All cells were maintained at 5.0% CO2 in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and 1% L-glutamine. Cell lines were routinely tested for mycoplasma contamination.
For HBEC3-KT, for the generation of the G3BP1 and G3BP2 knock out cell line, the ribonucleoprotein complex with gRNA and Alt-R® S.p. Cas9 Nuclease V3 was delivered using Neon™ Transfection system. Briefly, 5 μl of 100 μM gRNA of G3BP1 and G3BP2 was mixed with 20 μg Alt-R® S.p. Cas9 Nuclease V3 and left at RT for 15 min. Early passage cells were trypsinized and washed twice. 500,000 cells were resuspended in Resuspension buffer R and mixed with the RNP complex and electroporated at 1,400 V, 20 milliseconds, and two pulses. After 3 days, all cells were harvested, and the cells were re-electroporated with RNP complex. Knockout was confirmed by WB, for all experiments a polyclonal population was used.
Plasmids
The plentiCRISPRv2 puro was a gift from Brett Stringer (Addgene plasmid #98290). The plasmids psPAX2 and pMD2.G VSV-G were a kind gift from dr. James DeCaprio, MD, Dana-Farber Cancer institute. To generate the pInducer20-hG3BP1-IRES-G3BP2, hG3BP1 was cloned into pInducer20 (kindly provided by Dr. Hidde Ploegh, Boston Children’s Hospital) using NotI and AscI restriction sites followed by insertion of a SalI digested IRES-G3BP2 fragment generated by overlap PCR. The plasmids pFLAG-CMV4 (empty vector), pFLAG-CMV4-MDA5 (G495R) and pFLAG-CMV4-MDA5 (R337G) containing the gain-of-function MDA5 G495R and R337G were generated by our lab as described previously (Ahmad et al., 2018). The plasmid pFLAG-CMV4-RIG-I (C268F) was generated by inserting RIG-I into the HindIII and KpnI digested backbone. PCR mutagenesis was used to generate the point mutation C268F.
Viruses
Sendai virus (Cantell strain) was purchased from Charles River. Encephalomyocarditis virus (murine) was purchased from ATCC (VR-129B). VSV M51R and IAVdelNS1 were kindly provided by dr. Tristan Jordan and dr. Benjamin TenOever (NYU Langone Health). IAVdelNS1 was grown as previously described (Blanco-Melo et al., 2020). Briefly, MDCK cells expressing IAV-NS1 (MDCK-NS1 cells) were infected with IAVdelNS1 in EMEM containing 0.35% bovine serum albumin (BSA, MP Biomedicals), 4 mM L-glutamine, 10 mM HEPES, 0.15% NaHCO3, 1 μg/mL TPCK-trypsin (Sigma-Aldrich). Infectious titers were determined by TCID50/mL on MDCK-NS1 cells. VSV M51R was grown in in Vero cells in DMEM supplemented with 2% FBS. Viral supernatant was titered by plaque assay on naïve Vero cells. For both IAVdelNS1 and VSVM51R stocks, viral supernatants were spun down to remove cellular debris. For SeV, EMCV, IAVΔNS1 and VSVM51R infection, A549 or U2OS cells were counted on the day of infection and subsequently infected with the listed MOI with 1 hr of absorption.
dsRNA
dsRNAs used in this study were prepared by in vitro T7 transcription as described previously (Peisley et al., 2012). The templates for RNA synthesis were generated by PCR amplification. The sequences of the dsRNA are shown in Table S3. The two complementary strands were co-transcribed, and the duplex was separated from unannealed ssRNAs by 8.5% acrylamide gel electrophoresis in TBE buffer. RNA was gel-extracted using an Elutrap electroelution kit, ethanol precipitated, and stored in 20 mM Hepes, pH 7.0. Qualities of RNAs were analyzed by TBE polyacrylamide gel electrophoresis. For 3’-Cy5 labeling of RNA, the 3’end of RNA was oxidized with 0.1 M sodium meta-periodate (Pierce) overnight in 0.1 M NaOAc pH 5.4. The reaction was quenched with 250 mM KCl, buffer exchanged using Zeba desalting columns into 0.1 M NaOAc pH 5.4 and further incubated with Cy5-hydrazide for 6 hrs at RT. The polyI:C high molecular weight was purchased from Invivogen. For the generation of 162 bp_A (CIP), 162 bp dsRNA was treated with CIP for 1 hour at 37 degrees.
RNA-seq
Cells were seeded in 6-wells plate and stimulated with 1 μg 162 bp dsRNA with 5’ ppp as described above. At indicated timepoints total RNAs were extracted from indicated cells using TRIzol reagent and RNA Clean & Concentrator. Quality control and mRNA-seq library construction were performed by Novogene Co. Libraries were sequenced on the Illumina NovaSeq 6000 instrument with a paired-end read length of 2 x 150 bp, which resulted in ~20 M reads per sample. The raw sequence files were pre-processed using Trimmomatic v. 0.36 to trim Illumina adaptor sequences and low-quality bases. Trimmed reads were mapped to the human genome (UCSC hg38) using STAR aligner v. 2.5.4a. HTseq-count (v. 0.9.1) was used to count gene reads. Gene-count normalization and differential analysis were performed with DESeq25. Heatmaps were generated using Pheatmap. Scatter plots were generated using ggplot2.
RT-qPCR
Cells were transfected at 80% confluency with 500 ng/ml 162 bp dsRNA with 5’ppp unless stated otherwise. Lipofectamine 2000 was used for transfection with 2 μl lipofectamine reagent per μg of dsRNA diluted in 50 ul Opti-MEM per 500 ng of dsRNA. For mock transfection, cells were transfected with only lipofectamine reagent diluted in Opti-MEM. At indicated timepoints, total RNAs were extracted using TRIzol reagent and cDNA was synthesized using High-Capacity cDNA reverse transcription kit according to the manufacturer’s instruction. Real-time PCR was performed using a set of gene-specific primers or random primers, a SYBR Green Master Mix, and the StepOne™ Real-Time PCR Systems (Applied Biosystems). The full list of gene-specific primers can be found in table S1. For electroporation of dsRNA, 162 bp dsRNA with 5’ppp was electroporated into cells using a NucleofectorTM 2b (Lonza). 2 μg/ml of dsRNA was electroporated into cells using the cell line NucleofectorTM Kit L by following the manufacturer’s instructions using the U2OS program. At 8 hrs post-electroporation RNA was harvested for RT-qPCR. To determine the effect of TG on signaling, cells were treated with TG (1 μM) at 1 hr post-dsRNA transfection. In the G3BP1/2 complementation experiment, cells were seeded and expression of G3BP1-IRES-G3BP2 was induced with doxycycline for 24 hours and subsequently stimulated with dsRNA. To determine the effect of Q-VD-Oph on signaling, cells were pre-treated for 1 hr with either DMSO or 10 μM Q-VD-Oph prior to dsRNA transfection. For GOF MDA5 stimulation, A549 WT and ΔG3BPs were transfected with 1 μg/ml pFLAG-CMV4, pFLAG-CMV4-MDA5 (G495R) or pFLAG-CMV4-MDA5 (R337G). At 6 hrs post-transfection the media was replaced with DMEM with 10% FBS. At 24 hpt, total RNA was extracted. For cGAMP stimulation, cells were permeabilized with digitonin buffer (50 mM HEPES pH 7.0, 100 mM KCl, 3 mM MgCl2, 0.1 mM DTT, 85 mM sucrose, 0.2% BSA, 1 mM ATP, 10 μg/ml digitonin with 10 or 20 μg/ml cGAMP for 30 min. After 30 min, complete Dulbecco’s modified Eagle medium with 10% fetal bovine serum was added and total RNA was extracted at 6 hrs post-transfection. For viral RNA measurement, virus-specific primers were used as listed in Table S1. In main figures, qPCR data was normalized to WT cells.
ELISA
Cells were seeded in 6-well plates at 70% confluency. Cells were transfected with 162 bp dsRNA containing 5’ppp (500 ng/ml). At 6 hours post-transfection supernatant was harvested and used for ELISA. For IFN-β ELISA, the LumiKine™ Xpress hIFN-β 2.0 kit was used according to the manufacturer’s instructions. For RANTES, IL-6, TNF-α ELISA, the Human CCL5/RANTES Quantikine ELISA Kit, Human IL-6 Quantikine ELISA Kit, Human TNF-alpha Quantikine ELISA Kit were used respectively according to the manufacturer’s instructions. To determine the cytokine release upon SeV infection, cells were infected with 100 HA/ml. SeV Cantell strain for 6 hours. Fresh supernatant was used for the ELISAs. The results were obtained using a Biotek M1 Synergy microplate reader using the manufacturer’s instructions.
Immunoblotting
Cells were seeded at 80% confluency in 6- or 12-well plates and transfected with 500 ng/ml 162 bp dsRNA with 5’ppp with lipofectamine 2000 as described above. At indicated timepoints, cells were lysed with 1% SDS lysis buffer (10 mM Tris pH 7.5, 150 mM NaCl, 10 mM DTT, 1% SDS), then boiled for 10 min. For the SUnSET assay, cells were pulsed with puromycin (1 μg/mL) at 6 hrs post-dsRNA for 15 min prior to harvesting. Proteins were resolved on 4–15% gradient gels, transferred to PVDF membranes, and blotted using standard procedures. Membranes were visualized using Amersham ECL reagent or SuperSignalTM West Femto Maximum Sensitivity Substrate.
Immunofluorescence microscopy and image analysis
U2OS, Hela, A549 or HBEC cells were seeded on coverslips to reach 60-80% confluency the next day. U2OS, Hela and A549 cells were stimulated with 500 ng/ml 162 bp dsRNA for 6 hrs as mentioned previously unless otherwise stated. HBEC cells were stimulated with 200 ng/ml 162 bp dsRNA for 4 hrs. At indicated timepoints, cells were fixed with 4% paraformaldehyde at RT for 10 min, and permeabilized with 0.2% Triton-X at RT for 10 min. Followed by blocking for 30 min at RT with 1% BSA in PBST, and staining using primary antibody for 1 hr at RT followed by 2 washes and then secondary antibody incubation for 1 hr. Hoechst 33342 was used to stain the nuclei. Coverslips were mounted using Fluoromount-G and imaged with a Zeiss Axio Imager M1 at 40X magnification. For imaging of HBEC cells, a Nikon TI2 motorized inverted microscope was used and images were taken with a 100x oil-immersion lens at the Nikon Imaging Center at Harvard Medical School. For immunofluorescence with Cy5-dsRNA, cells were transfected with 500 ng/mL of 162 bp dsRNA with 3’ Cy5 and 5’ ppp. For electroporation of dsRNA, 1 μg/mL of 162 bp dsRNA with 3’ Cy5 and 5’ppp was electroporated into cells using a NucleofectorTM 2b following the manufacturer’s instructions using the U2OS program. At 8 hrs post-electroporation the cells were fixed as mentioned before. For TG induction, cells were treated with 1 μM TG for 1 hrs prior to fixation. For nutrient starvation, cells were washed 3 times with DMEM minus glucose, FBS, glutamine and pyruvate (starvation media) and then left for 8 hrs prior to fixation. For cycloheximide treatment, cells were stimulated with TG or transfected with dsRNA and subsequently treated with cycloheximide 10 μg/ml for 6 hrs. For SeV SG induction, cells were infected with 100 HA/mL SeV for 20 hrs prior to fixation. For SeV, VSV M51R and IAVdelNS1 protein level determination, cells were seeded for 90% confluency on the day of infection. Prior to infection cells were counted and infected. Cells were fixed at timepoints before cell death occurred. Protein level quantification was done using the same microscope settings using several replicates and multiple FOV. For ADAR1 knock-down, cells were transfected with pooled siRNA for ADAR1 or non-targeting control siRNA (50 nM). At 24 hrs post-trasfection media was changed to media containing 10 ng/mL recombinant human interferon. At 48 hrs post-transfection, cells were fixed and processed for imaging as described above. For G3BP1-G3BP2 complementation, expression was induced with doxycycline for 24 hrs prior to 162 bp dsRNA transfection. For Q-VD-Oph treatment, cells were pre-treated with DMSO or Q-VD-Oph (10 μM) for 1 hr prior to dsRNA stimulation. All images were processed and analyzed with ImageJ. To highlight SGs, contrast adjustment was based on the mock transfected cells. To determine the Pearson Correlation Coefficient, the ImageJ plugin JaCOP was used on 10 separate FOV with at least 40 granules per FOV.
For nuclear IRF3 localization, U2OS WT and ΔG3BPs cells were stimulated with 162 bp dsRNA containing a 5’ppp (100 ng) for 6 or 16 hrs. Cells were prepared for immunofluorescence and stained with IRF3 (provided by Takashi Fujita) or DAPI. Images were taken randomly across the slide and the presence of IRF3 in the nucleus of each cell was quantified using ImageJ. The pixel intensity of nuclear IRF3 signal in each cell (a.u) was used to plot the data points.
For stress granule size analysis, U2OS WT, U2OSΔPKR and U2OSΔUBAP2L cells were stimulated with 162 bp dsRNA with 5’ppp for 6 hrs or 1 μM TG as mentioned previously. Cells were fixed and stained for G3BP1. Z-stack images (0.15 μM step size) were obtained using a Nikon TI2 motorized inverted microscope. All images were taken with a 60x oil-immersion lens. Stress granule size was determined by using the 3D Object Counter plugin in ImageJ. At least 200 SG from multiple fields of view (FOV) were picked randomly and analyzed. The percentage of cells that contained SG was determined by dividing the number of cells containing SG by the total amount of cells as measured by Hoechst 33342 staining for at least 5 FOV.
Cell death analysis and caspase cleavage assay
Cell detachment upon stimulation of cells was assessed at the indicated timepoints using the Nikon Eclipse TS2R at 20X or 40X magnification. For brightfield images, cells were seeded at 90% confluency in 12-well plates and transfected with 500 ng per well of 162 bp dsRNA with 5’ppp. At indicated timepoints, brightfield images were acquired from 3 different wells for each sample in duplicate. For the brightfield images of figure 4E, cells were stimulated with different cell death stimuli. U2OSΔG3BPs cells were transfected with 162 bp dsRNA (500 ng/ml) or treated with etoposide (20 μM) or Z-IETD-FMK (50 μM) plus TNFα (20 ng/ml) for 24 hrs. At 24 hrs post-treatment, the brightfield images were obtained using the Nikon Eclipse TS2R at 40X magnification.
For quantification of cell death, U2OS, Hela and A549 cells were seeded in 12-well plates and transfected with 500 ng per well with 162 bp dsRNA with 5’ppp unless otherwise indicated. At the indicated timepoints, cells were incubated with Sytox Green Nucleic Acid stain (2 μM final) and Hoechst 33342 (3,000-fold dilution) for 30 min. Sytox Green signal was measured with the Nikon Eclipse TS2R at 20X or 40X magnification using a 470 ex filter set. The percentage of dead cells was calculated as the number of Sytox positive cells divided by the total number of Hoechst positive cells using ImageJ. To determine the effect of different reagents on dsRNA-induced cell death, cells were pre-treated with DMSO, disulfiram (10 or 25 μM), Q-VD-Oph (10 μM), Human TNF-α Neutralizing Rabbit mAb (10, 100, 1000 ng/ml), Ruxolitinib (0.5 or 5 μM), BX-795 (0.5 or 5 μM), Necrosulfonamide (MLKL inhibitor) (1 μM), Bengamide B (1 or 5 μM) or ACHP (1 or 5 μM) for 30 min - 1 hr prior to dsRNA transfection. For SeV, VSV M51R, IAVdelNS1 or EMCV infection, cells were counted on the day of infection and infected with the indicated MOI.
Live/Dead TM cell imaging kit was used to quantify cell death in U2OS WT and U2OSΔG3BP1/2 at the indicated timepoints. The manufacturer’s instructions were followed for this analysis. To quantify caspase 3/7 cleavage, cells were stimulated with dsRNA as previously mentioned and caspase 3/7 activity was analyzed using the CellEventTM Caspase 3-7 Green detection reagent. Similar to Sytox, the cells were stained with the Green detection reagent (1:30,000). The caspase 3/7 activity in figure 4B was measured using a Biotek M1 Synergy microplate reader using the manufacturer’s instructions. All experiments were performed at n=3.
IRF3 dimerization assay
This assay was adapted from the method described previously (Ahmad et al., 2018). Briefly, U2OS cells (mock or 112 bp dsRNA-transfected) were homogenized in hypotonic buffer (10 mM Tris pH 7.5, 10 mM KCl, 0.5 mM EGTA, 1.5 mM MgCl2, 1 mM sodium orthovanadate, 1X mammalian Protease Arrest) and centrifuged at 1,000 g for 5 min to pellet the nuclei. The supernatant (S1), containing the cytosolic and the mitochondrial fractions, was further centrifuged at 5,000 g for 15 min to pellet the crude mitochondrial fraction (P5). The P5 fraction was further washed once with isotonic buffer (hypotonic + 0.25 M D-Mannitol). The cytosolic fraction for the experiment was extracted from wild type untransfected U2OS cells using the same procedure as above except that the final spin was done at 18,000 g for 15 min and the supernatant S18 containing the cytosolic fraction was recovered. Subsequently, 35 μg of each P5 pellet was resuspended in 25 μl S18 (3 mg/ml) and used for IRF3 dimerization assay and Western blot analysis. 35S-IRF3 was prepared by in vitro translation using TnT T7 Coupled Reticulocyte Lysate System according to manufacturer’s instructions. The IRF3 dimerization was carried out by adding 16 μl (P5+S18) mix to 2 μl 35S-IRF3 in (20 mM HEPES pH 7.4, 4 mM MgCl2 and 2 mM ATP) in a total reaction volume of 20 μl. The reaction was incubated at 30°C for 1 h followed by centrifugation at 18,000 g for 5 min and the supernatant was subjected to native PAGE analysis. IRF3 dimerization was visualized by autoradiography and phosphorimaging on Amersham Typhoon 5 Biomolecular Imager. The image was quantified using ImageQuant.
RNase L activity analysis
Total RNA was isolated from cells and loaded on an RNA pico chip using an Agilent Bioanalyzer.
ADAR1 siRNA knock-down
Cells were seeded at 60% confluency and transfected with either pooled ADAR1 siRNA or non-targeting control siRNA (50 nM). After 24 hours, media was changed and recombinant human IFN-β was at 1 ng/mL or 10 ng/mL for 24 hours. After 48 hours, RNA was harvested and RT-qPCR and WB were performed to confirm ADAR1 knock-down.
QUANTIFICATION AND STATISTICAL ANALYSIS
Average values and standard deviations were calculated using Microsoft Excel and SPSS (IBM). The values for n represent biological replicates for cellular experiments or individual samples for biochemical assays. For each figure, individual replicate values were plotted together with the average values. The number of replicates is also indicated in the figure legends. Unless otherwise mentioned, all assays were performed in at least 3 independent experiments. p values were calculated using the two-tailed unpaired Student’s t test and are shown in the graphs.
Supplementary Material
SuppData1: Raw data for the heatmap in Figure 1B
SuppData2: Raw data for the heatmap in Figure S2B (Related to Figure 1)
SuppData3: Raw data for the heatmap in Figure S2D (Related to Figure 1)
Highlights.
SGs prevent excessive activation of dsRNA-induced innate immune signaling.
dsRNA triggers MAVS-dependent immune-mediated apoptosis in SG-deficient cells.
SGs regulate viral replication in RLR-dependent and -independent manners.
SGs protect cells from self-derived dsRNA-mediated immunopathology.
Acknowledgement:
We acknowledge Drs. Paul Anderson, Igor Brodsky and the rest of the Hur lab members for helpful discussions. We thank Dr. Takashi Fujita for sharing anti-IRF3 antibody. Cell images for z-stack analysis were collected at the Nikon imaging center at Harvard Medical School. This work was supported by NSF fellowship (CC), Roche post-doctoral fellowship (HW), and NIH grants to SML (R00GM124458), PI (R01GM126150, R01GM146997), BT (R01AI145882, R21AI171083, R01AI170487), and SH (R01AI154653, R01AI111784, DP1AI152074). SH is a Howard Hughes Medical Institute investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
B.T. is a cofounder of Archean Biologics. S.H. is a consultant for Odyssey therapeutics and CJ Cheil Jedang.
References
- 1.tenOever BR (2016). The Evolution of Antiviral Defense Systems. Cell Host Microbe 19, 142–149. 10.1016/j.chom.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Weber F, Wagner V, Rasmussen SB, Hartmann R, and Paludan SR (2006). Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80, 5059–5064. 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Son K-N, Liang Z, and Lipton HL (2015). Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J. Virol 89, 9383–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stok JE, Vega Quiroz ME, and van der Veen AG (2020). Self RNA Sensing by RIG-I-like Receptors in Viral Infection and Sterile Inflammation. J Immunol 205, 883–891. 10.4049/jimmunol.2000488. [DOI] [PubMed] [Google Scholar]
- 5.Chen YG, and Hur S (2022). Cellular origins of dsRNA, their recognition and consequences. Nat Rev Mol Cell Biol 23, 286–301. 10.1038/s41580-021-00430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uggenti C, Lepelley A, and Crow YJ (2019). Self-Awareness: Nucleic Acid-Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol 37, 247–267. 10.1146/annurev-immunol-042718-041257. [DOI] [PubMed] [Google Scholar]
- 7.Saldi TK, Gonzales PK, LaRocca TJ, and Link CD (2019). Neurodegeneration, Heterochromatin, and Double-Stranded RNA. J Exp Neurosci 13, 1179069519830697. 10.1177/1179069519830697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sud N, Rutledge AC, Pan K, and Su Q (2016). Activation of the dsRNA-Activated Protein Kinase PKR in Mitochondrial Dysfunction and Inflammatory Stress in Metabolic Syndrome. Curr Pharm Des 22, 2697–2703. 10.2174/1381612822666160202141845. [DOI] [PubMed] [Google Scholar]
- 9.Ablasser A, and Hur S (2020). Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat Immunol 21, 17–29. 10.1038/s41590-019-0556-1. [DOI] [PubMed] [Google Scholar]
- 10.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, and Chen ZJ (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peisley A, Wu B, Yao H, Walz T, and Hur S (2013). RIG-I Forms Signaling-Competent Filaments in an ATP-Dependent, Ubiquitin-Independent Manner. Mol Cell 51, 573–583. [DOI] [PubMed] [Google Scholar]
- 12.Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, and Hur S (2011). Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci U S A 108, 21010–21015. 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng W, Sun L, Jiang X, Hou F, Adhikari A, Xu M, and Chen ZJ (2010). Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofmann S, Kedersha N, Anderson P, and Ivanov P (2021). Molecular mechanisms of stress granule assembly and disassembly. Biochim Biophys Acta Mol Cell Res 1868, 118876. 10.1016/j.bbamcr.2020.118876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Treeck B, and Parker R (2019). Principles of Stress Granules Revealed by Imaging Approaches. Cold Spring Harb Perspect Biol 11. 10.1101/cshperspect.a033068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanders DW, Kedersha N, Lee DSW, Strom AR, Drake V, Riback JA, Bracha D, Eeftens JM, Iwanicki A, Wang A, et al. (2020). Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 181, 306–324 e328. 10.1016/j.cell.2020.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang P, Mathieu C, Kolaitis RM, Zhang P, Messing J, Yurtsever U, Yang Z, Wu J, Li Y, Pan Q, et al. (2020). G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 181, 325–345 e328. 10.1016/j.cell.2020.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillen-Boixet J, Kopach A, Holehouse AS, Wittmann S, Jahnel M, Schlussler R, Kim K, Trussina I, Wang J, Mateju D, et al. (2020). RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell 181, 346–361 e317. 10.1016/j.cell.2020.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, and Anderson P (2016). G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Biol 212, 845–860. 10.1083/jcb.201508028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cirillo L, Cieren A, Barbieri S, Khong A, Schwager F, Parker R, and Gotta M (2020). UBAP2L Forms Distinct Cores that Act in Nucleating Stress Granules Upstream of G3BP1. Curr Biol 30, 698–707 e696. 10.1016/j.cub.2019.12.020. [DOI] [PubMed] [Google Scholar]
- 22.Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, et al. (2018). Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 172, 590–604 e513. 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bley N, Lederer M, Pfalz B, Reinke C, Fuchs T, Glass M, Moller B, and Huttelmaier S (2015). Stress granules are dispensable for mRNA stabilization during cellular stress. Nucleic Acids Res 43, e26. 10.1093/nar/gku1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mateju D, Eichenberger B, Voigt F, Eglinger J, Roth G, and Chao JA (2020). Single-Molecule Imaging Reveals Translation of mRNAs Localized to Stress Granules. Cell 183, 1801–1812 e1813. 10.1016/j.cell.2020.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, et al. (2013). Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7, e43031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh SW, Onomoto K, Wakimoto M, Onoguchi K, Ishidate F, Fujiwara T, Yoneyama M, Kato H, and Fujita T (2016). Leader-Containing Uncapped Viral Transcript Activates RIG-I in Antiviral Stress Granules. PLoS Pathog 12, e1005444. 10.1371/journal.ppat.1005444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCormick C, and Khaperskyy DA (2017). Translation inhibition and stress granules in the antiviral immune response. Nat Rev Immunol 17, 647–660. 10.1038/nri.2017.63. [DOI] [PubMed] [Google Scholar]
- 28.White JP, and Lloyd RE (2012). Regulation of stress granules in virus systems. Trends Microbiol 20, 175–183. 10.1016/j.tim.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng Q, Langereis MA, and van Kuppeveld FJ (2014). Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev 25, 577–585. 10.1016/j.cytogfr.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langereis MA, Feng Q, and van Kuppeveld FJ (2013). MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J. Virol 87, 6314–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cadena C, Ahmad S, Xavier A, Willemsen J, Park S, Park JW, Oh SW, Fujita T, Hou F, Binder M, and Hur S (2019). Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 177, 1187–1200 e1116. 10.1016/j.cell.2019.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, and Takekawa M (2008). Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol 10, 1324–1332. 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- 33.Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, Guy CS, Briard B, Place DE, Bhattacharya A, et al. (2019). DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594. 10.1038/s41586-019-1551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao B, Gong X, Fang S, Weng W, Wang H, Chu H, Sun Y, Meng C, Tan L, Song C, et al. (2021). Inhibition of anti-viral stress granule formation by coronavirus endoribonuclease nsp15 ensures efficient virus replication. PLoS Pathog 17, e1008690. 10.1371/journal.ppat.1008690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aoyama-Ishiwatari S, Okazaki T, Iemura SI, Natsume T, Okada Y, and Gotoh Y (2021). NUDT21 Links Mitochondrial IPS-1 to RLR-Containing Stress Granules and Activates Host Antiviral Defense. J Immunol 206, 154–163. 10.4049/jimmunol.2000306. [DOI] [PubMed] [Google Scholar]
- 36.Amen T, and Kaganovich D (2021). Stress granules inhibit fatty acid oxidation by modulating mitochondrial permeability. Cell Rep 35, 109237. 10.1016/j.celrep.2021.109237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, and Reis e Sousa C (2009). Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol 83, 10761–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burke JM, Lester ET, Tauber D, and Parker R (2020). RNase L promotes the formation of unique ribonucleoprotein granules distinct from stress granules. J Biol Chem 295, 1426–1438. 10.1074/jbc.RA119.011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mian MF, Ahmed AN, Rad M, Babaian A, Bowdish D, and Ashkar AA (2013). Length of dsRNA (poly I:C) drives distinct innate immune responses, depending on the cell type. J Leukoc Biol 94, 1025–1036. 10.1189/jlb.0312125. [DOI] [PubMed] [Google Scholar]
- 40.Mueller FS, Richetto J, Hayes LN, Zambon A, Pollak DD, Sawa A, Meyer U, and Weber-Stadlbauer U (2019). Influence of poly(I:C) variability on thermoregulation, immune responses and pregnancy outcomes in mouse models of maternal immune activation. Brain Behav Immun 80, 406–418. 10.1016/j.bbi.2019.04.019. [DOI] [PubMed] [Google Scholar]
- 41.Kowash HM, Potter HG, Edye ME, Prinssen EP, Bandinelli S, Neill JC, Hager R, and Glazier JD (2019). Poly(I:C) source, molecular weight and endotoxin contamination affect dam and prenatal outcomes, implications for models of maternal immune activation. Brain Behav Immun 82, 160–166. 10.1016/j.bbi.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, and Ron D (2004). Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 24, 10161–10168. 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McAllister CS, Taghavi N, and Samuel CE (2012). Protein kinase PKR amplification of interferon beta induction occurs through initiation factor eIF-2alpha-mediated translational control. J Biol Chem 287, 36384–36392. 10.1074/jbc.M112.390039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dalet A, Arguello RJ, Combes A, Spinelli L, Jaeger S, Fallet M, Vu Manh TP, Mendes A, Perego J, Reverendo M, et al. (2017). Protein synthesis inhibition and GADD34 control IFN-beta heterogeneous expression in response to dsRNA. EMBO J 36, 761–782. 10.15252/embj.201695000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kristiansen H, Gad HH, Eskildsen-Larsen S, Despres P, and Hartmann R (2011). The Oligoadenylate Synthetase Family: An Ancient Protein Family with Multiple Antiviral Activities. J. Interferon & Cytokine Research 31. [DOI] [PubMed] [Google Scholar]
- 46.Banerjee S, Gusho E, Gaughan C, Dong B, Gu X, Holvey-Bates E, Talukdar M, Li Y, Weiss SR, Sicheri F, et al. (2019). OAS-RNase L innate immune pathway mediates the cytotoxicity of a DNA-demethylating drug. Proc Natl Acad Sci U S A 116, 5071–5076. 10.1073/pnas.1815071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, Donovan J, Korennykh A, Silverman RH, and Weiss SR (2017). Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 6. 10.7554/eLife.25687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, Ivan AP, Buchumenski I, Berger AC, Goldstein JT, et al. (2018). Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat Commun 9, 5450. 10.1038/s41467-018-07824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. 10.1038/s41586-018-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, Zhang R, Huang X, Feng JM, and Jiang Z (2019). Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol Cell 74, 19–31 e17. 10.1016/j.molcel.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 51.Rajput A, Kovalenko A, Bogdanov K, Yang SH, Kang TB, Kim JC, Du J, and Wallach D (2011). RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 34, 340–351. 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 52.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, et al. (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577. 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S, and Hartmann G (2009). Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon independent apoptosis in human melanoma cells. J Clin. Invest 119, 2399–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lei Y, Moore CB, Liesman RM, O'Connor BP, Bergstralh DT, Chen ZJ, Pickles RJ, and Ting JP (2009). MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One 4, e5466. 10.1371/journal.pone.0005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, and Muster T (1998). Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252, 324–330. 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 56.Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, and Lyles DS (2003). Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol 77, 4646–4657. 10.1128/jvi.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loo MY, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, and Garcia-Sastre A (2008). Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol 82, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walkley CR, and Li JB (2017). Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol 18, 205. 10.1186/s13059-017-1347-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, and Stetson DB (2015). Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 43, 933–944. 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rice GI, Kasher PR, Forte G.M.a., Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, and al, e. (2010). Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genetics 44, 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, and Walkley CR (2015). RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014). The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9, 1482–1494. 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, Zhang CZ, and Hur S (2018). Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation Cell 172, 797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Q, Miyakoda W, Yang W, Khillan J, Stachura DL, Weiss MJ, and Nishikura K (2004). Stree-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase. J. Biol. Chem 279, 4952–4961. [DOI] [PubMed] [Google Scholar]
- 65.Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, and Rice CM (2018). Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172, 811–824 e814. 10.1016/j.cell.2017.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alberti S, and Hyman AA (2021). Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat Rev Mol Cell Biol 22, 196–213. 10.1038/s41580-020-00326-6. [DOI] [PubMed] [Google Scholar]
- 67.Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jayabalan AK, Adivarahan S, Koppula A, Abraham R, Batish M, Zenklusen D, Griffin DE, and Leung AKL (2021). Stress granule formation, disassembly, and composition are regulated by alphavirus ADP-ribosylhydrolase activity. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2021719118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emara MM, and Brinton MA (2007). Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci U S A 104, 9041–9046. 10.1073/pnas.0703348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kimball SR, Horetsky RL, Ron D, Jefferson LS, and Harding HP (2003). Mammalian stress granules represent sites of accumulation of stalled translation initiation complexes. Am J Physiol Cell Physiol 284, C273–284. 10.1152/ajpcell.00314.2002. [DOI] [PubMed] [Google Scholar]
- 71.Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, and Anderson P (2002). Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13, 195–210. 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmidt EK, Clavarino G, Ceppi M, and Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6, 275–277. 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SuppData1: Raw data for the heatmap in Figure 1B
SuppData2: Raw data for the heatmap in Figure S2B (Related to Figure 1)
SuppData3: Raw data for the heatmap in Figure S2D (Related to Figure 1)
Data Availability Statement
Data Availability: RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Imaging data have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-G3BP1 | Cell Signaling | Cat #17798 |
| Mouse anti-G3BP1 | Santa Cruz Biotechnology | Cat #sc-365338 |
| Rabbit anti-RIG-I | In-house | Takashi Fujita lab (Oh et al., 2016) |
| Rabbit anti-MAVS | Fortis Life Sciences | Cat #A300-782A |
| Mouse anti-TIAR | Santa Cruz Biotechnology | Cat #sc-398372 |
| Rabbit anti-phospho-IRF3 | Cell Signaling | Cat #4947 |
| Rabbit anti-IRF3 (WB) | Cell Signaling | Cat #4302 |
| Rabbit anti-IRF3 (IF) | In-house | Takashi Fujita lab (Oh et al., 2016) |
| Rabbit anti-beta-actin | Cell Signaling | Cat #4302 |
| Mouse anti-vinculin | Cell Signaling | Cat#4650S |
| Rabbit anti-UBAP2L | Fortis Life Sciences | Cat #A300-533A |
| Rabbit anti-PKR | Cell Signaling | Cat #12297 |
| Rabbit anti-G3BP2 | Fortis Life Sciences | Cat #A302-040A |
| Rabbit anti-NIX | Cell Signaling | Cat #12396 |
| Rabbit anti-COXIV | Cell Signaling | Cat #4850 |
| Rabbit anti-VCP | Abcam | Cat #ab111740 |
| Rabbit anti-NDP52 | Abcam | Cat #ab68588 |
| Rabbit anti-MDA5 | Cell Signaling | Cat #5321 |
| Mouse anti-TRAF2 | Santa Cruz Biotechnology | Cat #sc-136999 |
| Mouse anti-TRAF6 | Santa Cruz Biotechnology | Cat #sc-8409 |
| Rabbit anti-TBK1 | Cell Signaling | Cat #3054 |
| Mouse anti-puromycin | EMD Milipore | Cat #MABE343 |
| Rabbit anti-PKR | Cell Signaling | Cat #12297 |
| Mouse anti-OAS3 | Santa Cruz Biotechnology | Cat #sc-398225 |
| Rabbit anti-RNase L (WB) | Cell Signaling | Cat #2728 |
| Mouse anti-RNase L (IF) | Santa Cruz Biotechnology | Cat #sc-74405 |
| Rabbit anti-phospho-PKR | Abcam | Cat #ab32036 |
| Rabbit anti-ATF4 | Cell Signaling | Cat #11815 |
| Rabbit anti-Caspase-8 | Cell Signaling | Cat #4790 |
| Rabbit anti-Caspase-8 | Sigma Aldrich | Cat #NB100-56116 |
| Rabbit anti-Caspase-9 | Cell Signaling | Cat #3493 |
| Rabbit anti-Caspase-3 | Cell Signaling | Cat #9662 |
| Rabbit anti-PARP | Cell Signaling | Cat #9532 |
| Rabbit anti-Sendai Virus pAb | MBL | Cat #PD029 |
| Rabbit anti-Influenza Virus A NP pAb | Thermo Fisher | Cat #PA5-32242 |
| Mouse anti-VSV-G | Kerafast | Cat #EB0010 |
| Mouse anti-ADAR1 | Santa Cruz Biotechnology | Cat # sc-73408 |
| Rabbit anti-Caspase-1 | Cell Signaling | Cat #2225 |
| Anti-Rabbit IgG-HRP | Cell Signaling | Cat #7074P2 |
| Anti-Mouse IgG-HRP | GE Healthcare | Cat #NA931V |
| Alexa Fluor 647® Affinipure Donkey anti-Rabbit IgG | Jackson ImmunoResearch Laboratories | Cat #711-605-152 |
| Alexa Fluor® 488 Affinipure Donkey anti-mouse IgG | Jackson ImmunoResearch Laboratories | Cat #715-545-150 |
| Alexa Fluor® 488 Affinipure Donkey anti-rabbit IgG | Jackson ImmunoResearch Laboratories | Cat #711-545-152 |
| Alexa Fluor® 647 Affinipure Donkey anti-mouse IgG | Jackson ImmunoResearch Laboratories | Cat #715-605-151 |
| Bacterial and virus strains | ||
| Sendai Virus Cantell Strain | ATCC | Cat #VR-907 |
| Sendai Virus Cantell Strain | Charles River Laboratories | Cat #VR-907 |
| Encephalomyocarditis virus (Cell culture adapted) | ATCC | Cat #VR-1762™ |
| IAVΔNS1 | Laboratory of Benjamin TenOever | Benjamin TenOever lab (Blanco-Melo et al., 2020) |
| VSV M51R | Laboratory of Benjamin TenOever | Benjamin TenOever lab |
| Chemicals, peptides, and recombinant proteins | ||
| Lonza NucleofectorTM 2b | Lonza Bioscience | Cat #: AAB-1001 |
| Nucleofector Kit L | Lonza Bioscience | Cat #VCA-1005 |
| Mammalian Protease Arrest | G-Biosciences | Cat #768-108 |
| TnT T7 Coupled Reticulocyte Lysate System | Promega | Cat #L4610 |
| Thapsigargin | Selleck Chem | Cat #S7895 |
| Puromycin | Selleck Chem | Cat #S7417 |
| CellEventTM Caspase 3-7 Green detection reagent | Invitrogen | Cat #C10423 |
| Hoechst 33342 | Thermo Fisher | Cat #H3570 |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermo Fisher | Cat #D1306 |
| Fluoromount G | SouthernBiotech | Cat#0100-01 |
| Etoposide | Selleck Chem | Cat #S1225 |
| Staurosporine | Selleck Chem | Cat #S1421 |
| Z-IETD-FMK | Selleck Chem | Cat #S7314 |
| Human TNF-α | Cell Signaling | Cat #8902 |
| Q-VD-Oph | Selleck Chem | Cat #S7311 |
| Human TNF-α Neutralizing Rabbit mAb | Cell Signaling | Cat #7321 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Invitrogen | Cat #13778150 |
| Recombinant IFN-β | Peprotech | Cat #300-02BC |
| Doxycycline | Fisher Scientific | Cat #AC446060050 |
| Cycloheximide | Selleck Chem | Cat#S7418 |
| Calf intestinal phosphatase (CIP) | New England Biolabs | Cat #M0525S |
| 2’-3’-cGAMP | Invivogen | Cat #tlrl-nacga23 |
| Tetraethylthiuram disulfide (Disulfiram) | Sigma-Aldrich | Cat #86720 |
| Necrosulfonamide | Sigma-Aldrich | Cat #480073 |
| Ruxolitinib | Selleck Chem | Cat #S1378 |
| BX-795 | Selleck Chem | Cat #S1274 |
| ACHP | Fisher Scientific | Cat#45-471-0 |
| Bengamide B | Fisher Scientific | Cat#52-731-00U |
| Pierce™ Sodium meta-periodate | Thermo Fisher | Cat #20504 |
| Zeba desalting columns | Thermo Fisher | Cat #89882 |
| Alt-R® S.p. Cas9 Nuclease | IDT | Cat #1081058 |
| Neon™ Transfection System | Invitrogen | Cat # MPK5000 |
| Neon™ Transfection System 10 μL Kit | Invitrogen | Cat #MPK1025 |
| Lipofectamine 3000 | Invitrogen | Cat #L3000001 |
| Digitonin | Calbiochem | Cat #CAS 11024-24-1 |
| Amersham ECL Prime Western Blotting Detection Reagent | Cytiva Life Sciences | Cat#RPN2236 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher | Cat #34094 |
| 4–15% Mini-PROTEAN® TGX™ Precast Protein Gels, 15-well | Bio-Rad | Cat #4561086 |
| TRI reagent (Trizol) | Zymo Research | Cat #R2050-1-200 |
| RNA Clean & Concentrator | Zymo Research | Cat #R1017 |
| High-Capacity cDNA reverse transcription kit | Applied Biosystems | Cat #4368813 |
| RNase Inhibitor, Human Placenta | New England Biolabs | Cat#M0307L |
| SYBR Green Master Mix | Applied Biosystems | Cat #4309155 |
| DMEM, high glucose, pyruvate | Cellgro | Cat #10-013-CV |
| DMEM, no glucose, no glutamine, no phenol red | Gibco | Cat #A1443001 |
| Airway epithelial cell basal medium | ATCC | Cat #PCS-300-030 |
| Bronchial epithelial cell growth kit | ATCC | Cat #PCS-300-040™ |
| Direct-zol RNA Miniprep kit | Zymo Research | Cat #R2052 |
| OPTI-MEM | Gibco | Cat #31985070 |
| Critical commercial assays | ||
| LumiKine™ Xpress hIFN-β 2.0 | Invivogen | Cat code luex-hifnbv2 |
| Human CCL5/RANTES Quantikine ELISA Kit | R&D systems | Cat #DRN00B |
| Human IL-6 Quantikine ELISA Kit | R&D systems | Cat #D6050 |
| Human TNF-alpha Quantikine ELISA Kit | R&D systems | Cat #DTA00D |
| Live/Dead ™ cell imaging kit | Thermo Fisher | Cat #R37601 |
| Sytox Green Nucleic Acid stain | Thermo Fisher | Cat #S7020 |
| Deposited Data | ||
| Raw data from RNA-seq | This study | GSE173953 |
| Raw imaging data | This study | 10.17632/ndwddpcyzm.1 |
| Experimental models: Cell lines | ||
| U2OS (WT, ΔG3BPs, ΔPKR, ΔUBAP2L) | Laboratory of Dr. Paul J. Anderson | Paul Anderson lab (Aulas et al., 2017; Kedersha et al., 2016; Sanders et al., 2020) |
| U2OS (ΔMAVS, ΔRIG-I, ΔG3BPs ΔRNase L, ΔPKR, ΔRNase L, ΔG3BPs ΔIRF3 and ΔG3BPs ΔPKR) | This study | N/A |
| A549 WT | Laboratory of Dr. Susan Weiss | Susan Weiss lab (Li et al., 2017) |
| A549 (ΔG3BPs, ΔG3BP ΔMAVS and ΔG3BPs ΔRNase L) | This study | N/A |
| HBEC3-KT | ATCC | CRL-4051 |
| HBEC3ΔG3BPs | This study | N/A |
| HeLa WT | Laboratory of Dr. Gracjan Michlewski | Gracjan Michlewski lab (Choudhury et al., 2014) |
| HeLaΔG3BPs | This study | N/A |
| HEK293T | ATCC | CRL-11268 |
| Oligonucleotides | ||
| 112 or 162 bp dsRNA | In-house | (Cadena et al., 2019) |
| Cy5-Monohydrazide | Cytiva | Cat #PA15121 |
| Poly I:C | Invivogen | Cat #tlrl-pic |
| ON-TARGETplus Human ADAR1 (103) siRNA Set | Horizon Discovery | Cat #LQ-008630-00-0020 |
| ON-TARGETplus Non-targeting Pool | Horizon Discovery | Cat #001810-10-20 |
| Primer sequences | See table S1 | N/A |
| Recombinant DNA | ||
| plentiCRISPRv2 | Addgene | Cat #98290 |
| psPAX2 | In-house | James DeCaprio lab (Cheng et al., 2017) |
| pMD2.G VSV-G | In-house | James DeCaprio lab (Cheng et al., 2017) |
| pInducer20 | In-house | Hidde Ploegh Lab (Ashour et al., 2015) |
| pInducer20-hG3BP1-IRES-hG3BP2 | This study | N/A |
| pFLAG-CMV4 empty vector | In-house | Ahmad et al., 2018 |
| pFLAG-CMV4-MDA5 (R337G) | In-house | Ahmad et al., 2018 |
| pFLAG-CMV4-MDA5 (G495R) | In-house | Ahmad et al., 2018 |