

Abstract
The hematopoietic transcription factor GATA1 induces heme accumulation during erythropoiesis by directly activating genes mediating heme biosynthesis. In addition to its canonical functions as a hemoglobin prosthetic group and enzyme cofactor, heme regulates gene expression in erythroid cells both transcriptionally and post-transcriptionally. Heme binding to the transcriptional repressor BACH1 triggers its proteolytic degradation. In heme-deficient cells, BACH1 accumulates and represses transcription of target genes, including α- and β-like globin genes, preventing the accumulation of cytotoxic free globin chains. A recently described BACH1-independent mechanism of heme-dependent transcriptional regulation is associated with a DNA motif termed heme-regulated motif (HERM), which resides at the majority of loci harboring heme-regulated chromatin accessibility sites. Progress on these problems has led to a paradigm in which cell type-specific transcriptional mechanisms determine the expression of enzymes mediating the synthesis of small molecules, which generate feedback loops, converging upon the transcription factor itself and the genome. This marriage between transcription factors and the small molecules that they control is predicted to be a canonical attribute of regulatory networks governing cell state transitions such as differentiation in the hematopoietic system and more broadly.
Introduction
Heme, the iron-containing protoporphyrin IX, is essential for critical cellular processes, including oxygen transport, electron transfer and catalytic reactions (1). Mechanisms governing heme synthesis, transport and degradation establish and maintain intracellular heme homeostasis. As both excessive and inadequate accumulation of heme are deleterious, heme homeostasis must be tightly regulated as a vital physiological requirement (2, 3)
In mammalian cells, heme is synthesized by multi-step enzymatic reactions that occur in mitochondria and the cytoplasm (3). The first and rate-limiting step of heme biosynthesis is catalyzed by δ-aminolevulinic acid synthase (ALAS), encoded by the ubiquitously expressed ALAS1 and erythroid-specific ALAS2 genes, respectively (4). In erythroid cells, the transcription factor GATA1, which is required for erythropoiesis, activates ALAS2 transcription and heme production via two intronic enhancers (5). Human ALAS2 intron 1 enhancer mutations disrupt heme synthesis and cause X-linked sideroblastic anemia (6, 7). Disruption of the murine intron 1 enhancer abrogates GATA1 occupancy, decreasing Alas2 expression and heme synthesis, thus causing severe anemia and embryonic lethality (8).
In addition to its canonical function as a prosthetic group in metabolic enzymes in all cell types and hemoglobin in erythroid cells, heme regulates gene expression at both transcriptional and translational levels by engaging heme-sensing effector proteins. A variety of heme-sensing proteins have been identified in prokaryotes and eukaryotes that regulate heme homeostasis and fundamental cellular processes (9, 10). In erythroid cells, heme deficiency promotes autophosphorylation and activation of Heme Regulated Inhibitor (HRI), a kinase that phosphorylates and inactivates the translation initiation factor eIF2α (11, 12). Phosphorylation of eIF2α inhibits protein translation, with one important consequence being to restrict α- and β-globin mRNA translation, thus preventing proteotoxicity instigated by the excessive accumulation of heme-free globin chains during iron and heme deficiency (13). Excessive free globin chains form cytotoxic aggregates, analogous to protein aggregation characteristic of certain neurodegenerative disorders. Beyond HRI-mediated translational inhibition, the heme-regulated transcriptional repressor BTB and CNC homology1 (BACH1) provides an additional line of defense to prevent cytotoxic aggregates. In cells with low heme, BACH1 represses globin gene transcription (14, 15) to ensure an appropriate heme and globin stoichiometry. In this review, we discuss existing paradigms and recent progress that has extended and transformed paradigms for how heme utilizes BACH1-dependent and –independent mechanisms to regulate transcription during erythropoiesis and therefore to sustain a vigorous process of erythrocyte generation required for normal physiology and organismal survival.
BACH1-Dependent Transcriptional Regulation by Heme
BACH1 contains an N-terminal BTB domain that mediates protein-protein interactions and a C-terminal basic leucine zipper (bZip) domain mediating sequence-specific DNA binding (Figure 1a). Like other bZip transcription factors, e.g., NF-E2 and NRF2, BACH1 heterodimerizes with small Maf proteins such as MafF, MafG, and MafK. BACH1/Maf heterodimers bind a subset of the Maf recognition elements (MARE) in the genome to repress transcription (16). BACH1 contains 6 cysteine-proline (CP) dipeptide motifs (17) that mediate heme binding within heme regulatory motifs (HRM) of other heme-regulated proteins including ALAS (18, 19) and HRI (20). Biochemical analyses revealed that heme directly binds BACH1 via 4 of the 6 CP motifs (21). Heme binding inhibits BACH1-MafK heterodimer DNA binding and transcriptional regulatory activity without disrupting the BACH1 – MafK protein-protein interaction (17, 22). Heme inhibits BACH1-dependent repression of Hmox1 transcription by preventing BACH1 binding to the MARE motifs in Hmox1 enhancers. In Bach1 knockout mice, Hmox1 expression is elevated in various tissues, providing evidence that BACH1 represses Hmox1 transcription in vivo (23). Similarly, BACH1 inhibits α- and β-like globin gene transcription by binding to MARE motifs within DNaseI hypersensitive sites within the α- and β-like globin gene clusters (24). Treating cells with hemin reduces BACH1 protein levels, elevating α- and β-like globin transcription (14, 15). The BACH1-dependent repression of α- and β-like globin gene transcription ensures a physiological stoichiometry of heme relative to globin chains, thus preventing the cytotoxic accumulation of heme-free globin chains in heme-deficient cells (13).
Figure 1. Heme post-translationally regulates BACH1 function.
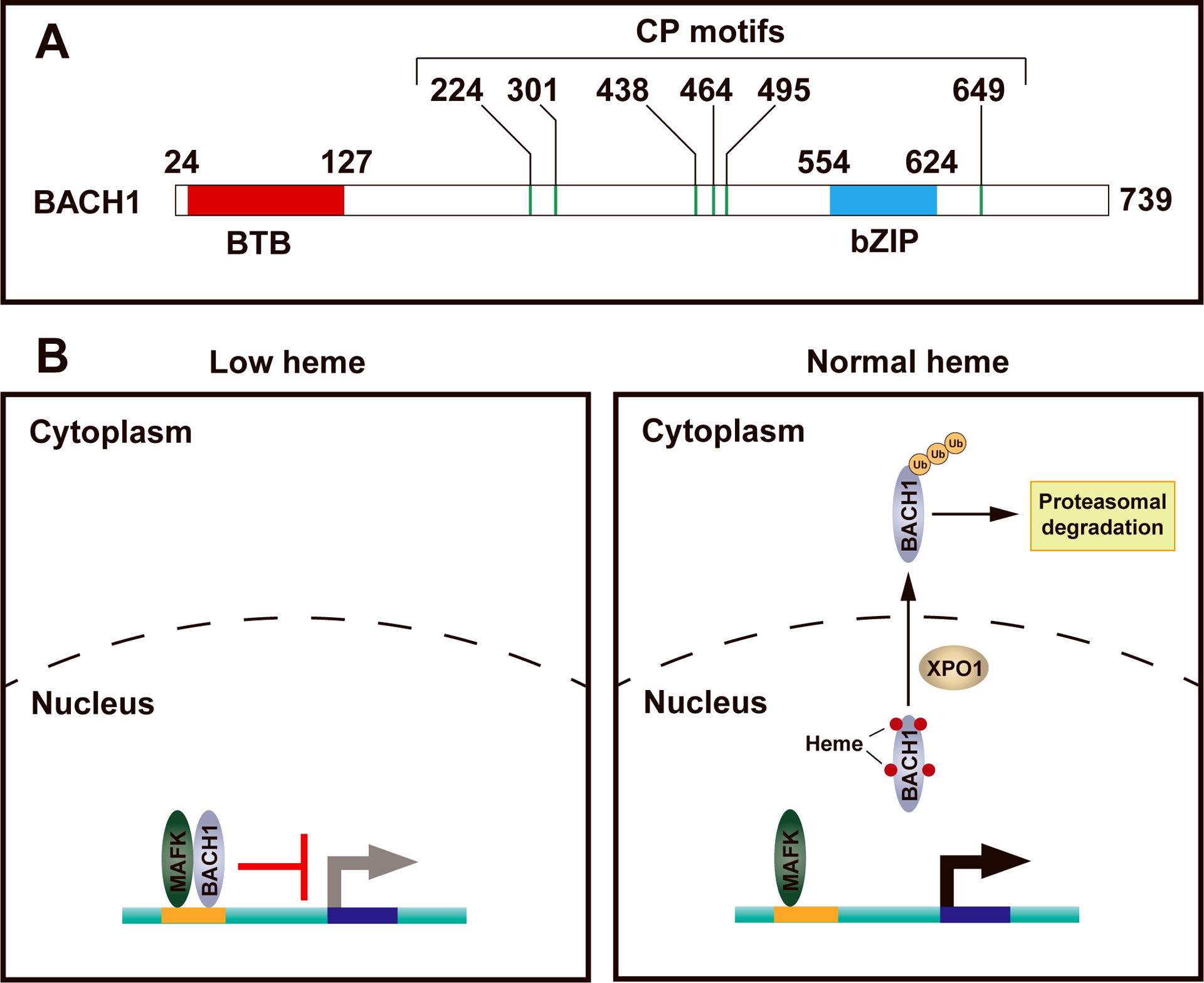
A. Domain structure of murine BACH1 protein. BTB, bric-a-brac–tramtrack–broad complex domain; bZIP, basic leucine zipper domain; CP motif, cysteine-proline dipeptide that mediates heme binding.
B. Heme regulates BACH1 DNA binding, nuclear export and proteasomal degradation. In heme-deficient cells, BACH1 dimerizes with a small Maf protein and inhibits target gene transcription via BACH1 DNA motifs. Upon heme binding to BACH1, BACH1 dissociates from DNA, exits the nucleus into the cytoplasm, and following ubiquitination, it is degraded by the proteasome.
Beyond inhibiting DNA binding, heme regulates BACH1 activity by facilitating BACH1 nuclear export and promoting its proteasomal degradation. Hemin induces the cytoplasmic translocation of BACH1 (25), which is independent of the BTB domain that mediates protein-protein interactions or the cytoplasmic localization signal that mediates cadmium-dependent nuclear export of BACH1 (26). Two of the six CP motifs that mediate heme binding are critical for heme-dependent nuclear export of BACH1. This process requires the broadly utilized nuclear exporter protein XPO1, also known as CRM1 (25).
Zenke-Kawasaki et al. (27) demonstrated that hemin treatment of cells reduces the BACH1 protein level in a dose-dependent manner, and proteasomal inhibition with MG-132 counteracts the downregulation. Using heme-deficient proerythroblast-like G1E-ER-GATA1 cells, Tanimura et al. (5) demonstrated that Bach1 RNA expression is unaffected by heme, while BACH1 protein only accumulates in heme-deficient cells. The E3 ubiquitin ligase HOIL1 (encoded by Rbck1) induces BACH1 polyubiquitination in vitro. HOIL1 is a component of the linear ubiquitin chain assembly complex (LUBAC) (28), and human RBCK1 mutations cause autoimmunity and inflammatory disease (29, 30). Upon co-expression of BACH1 and HOIL1 in NIH 3T3 cells, BACH1 interacts with HOIL1. A dominant-negative HOIL1 mutant delays BACH1 degradation in MEL cells, suggesting that HOIL1 might mediate BACH1 polyubiquitination and degradation (27). Subsequent studies identified FBXL17 (31) or FBXO22 (32) as an E3 ubiquitin ligase that mediates BACH1 degradation in the cervical cancer cell line HeLa or the lung cancer cell lines KP or A549, respectively. Thus, heme-dependent BACH1 degradation might involve tissue-specific E3 ubiquitin ligases that operate in a context-dependent manner (Figure 1b).
While studies in diverse cell types have analyzed BACH1-linked biochemical and genetic mechanisms, only recently have insights emerged on how BACH1 negotiates the numerous MAREs in the genome to control genome function. In addition to the BACH1 ChIP-seq analysis in human K562 erythroleukemia cells and H1 human ES cells from the ENCODE project (33), Warnatz et al. (34) combined BACH1 ChIP-seq and microarray in HEK293 cells with BACH1 knockdown and identified BACH1 target genes linked to cell cycle regulation and apoptosis. Matsumoto et al. (35) analyzed BACH1 function in immortalized mouse embryonic fibroblasts (iMEF) and discovered multiple BACH1 target genes involved in lipid metabolism, including Pparg, encoding the master regulator of adipocyte differentiation PPARγ. Bach1 deficiency promotes the differentiation of iMEFs to adipocytes in the presence of PPARγ ligands, suggesting that BACH1 regulates adipocyte differentiation through PPARγ. Ebina-Shibuya et al. (36) analyzed BACH1 and BACH2 targeting in the M1 murine myeloid leukemia cell line and demonstrated that BACH1 and BACH2 regulate genes involved in the inflammatory response. Unlike BACH1, which is expressed in erythroid and myeloid cells, BACH2 is expressed primarily in lymphoid cells and regulates B- and T-lymphocyte differentiation and function (37). BACH2 has 5 CP motifs and binds heme via an intrinsically disordered region (38). Heme binding inhibits BACH2 DNA binding in vitro and promotes its degradation in B cells (39). Analyses of Bach1 and Bach2 knockout mice revealed that both transcription factors promote erythropoiesis, while suppressing myelopoiesis by reducing expression of C/EBPβ and its target genes in myeloid progenitor cells (40). Although the studies highlighted above identified BACH1 target genes in non-erythroid cells, how heme regulates the expression of these genes remains unclear.
BACH1-Independent Transcriptional Regulation by Heme
To systematically analyze heme-dependent transcriptional mechanisms during erythropoiesis, Tanimura et al. (5) utilized the murine G1E-ER-GATA1 cell genetic complementation system (41, 42). These GATA1-null proerythroblast-like cells stably express a conditionally active allele of GATA1 fused to the ligand binding domain of the estrogen receptor. In the absence of β-estradiol, ER-GATA1 resides in an inactive state, and β-estradiol activates ER-GATA1, instigating an erythroid gene expression program and erythroid maturation (43, 44). CRISPR-Cas9 was used to engineer a mutant G1E-ER-GATA1 cell line, in which the WGATAR motifs within two intronic enhancers of Alas2 were mutated (5). The mutant cells contain 30-fold lower heme in comparison to wild-type cells after 48 h of β-estradiol treatment. Supplementing the mutant cells with 5-aminolevulinic acid (5-ALA), the small molecule product of ALAS enzymatic activity, restored heme biosynthesis. RNA-seq analyses of β-estradiol-induced wild-type, mutant, and 5-ALA-treated mutant cells revealed heme-regulated genes during erythroid maturation. While 1340 genes are heme-regulated (differentially expressed between wild-type and mutant cells), 1033 of which are also GATA1-regulated; 94 are rescued by 5-ALA. Of the 94 GATA1/heme/5-ALA-regulated genes, 66 are up-regulated by both GATA1 and heme, suggesting that heme facilitates GATA1 activity in these contexts (5).
Depleting Bach1 with shRNA in Alas2 enhancer mutant, heme-deficient cells restores the expression of select heme-regulated genes, raising the possibility that additional mechanisms, potentially not involving Bach1, contribute to dysregulated gene expression in this system (5). The BACH1-sensitive genes include established BACH1 targets, such as Hba-a1 (14), Hbb-b1 (15), and Slc7a11 (34), and genes not known to be BACH1 targets, including Tbcel and Fbxo30. By contrast, downregulating BACH1 did not affect expression of the heme-regulated genes Sqstm1 and Slc30a1. Since SQSTM1 is upregulated upon BACH1 knockdown in HEK293 cells (34), a given gene might be BACH1-regulated in certain, but not all, cellular contexts. The GATA1- and heme-activated gene Slc30a1 encodes a zinc exporter that regulates intracellular zinc in erythroblasts. Zinc chelation, which reduces intracellular zinc, induces cell death in immature erythroblasts, while Slc30a1 knockdown, which increases intracellular zinc, promotes terminal differentiation (45). Thus, the heme-dependent, BACH1-independent mechanism regulates the expression of genes critical for erythropoiesis.
To elucidate the mechanism of heme-dependent genomic regulation, Liao et al. utilized the assay for transposase-accessible chromatin sequencing (ATAC-seq) (46) to establish a genome-wide atlas of heme-regulated chromatin with wild-type and mutant (heme-deficient) G1E-ER-GATA1 cells. This study identified 11,340 GATA1-regulated ATAC-seq peaks and 1,122 heme-regulated peaks; 526 peaks are co-regulated by GATA1 and heme. The majority of the 526 GATA1/heme-co-regulated peaks are co-activated or co-repressed by GATA1 and heme, suggesting that heme amplification of GATA1 activity to regulate chromatin accessibility is the most common mechanism (47).
De novo motif finding in heme-regulated ATAC-seq peaks identified a DNA motif that resembles the BACH1 binding motif (35). Surprisingly, the BACH1 motif resides at less than 30% of the heme-regulated peaks. The application of de novo motif finding with peaks lacking BACH1 motifs revealed an AG-rich motif, which is deemed Heme-Regulated Motif (HERM). HERM resides at greater than 70% of heme-regulated peaks. Statistical analyses demonstrated that HERM is enriched in heme-regulated ATAC-seq peaks relative to random genomic regions or non-heme-regulated peaks (47).
Amalgamating ATAC-seq-detected accessible chromatin sites with RNA-seq data (5) revealed 169 genes regulated by heme at the RNA and chromatin levels (47). These genes conform to six regulatory modes based on the upregulation or downregulation of their expression by heme and GATA1, and whether a given gene contains BACH1 motif or HERM. Each regulatory mode contains genes with BACH1 motif or HERM. Thus, the BACH1 motif and HERM do not appear to predict the regulatory mode. Depleting BACH1 with shRNA in heme-deficient cells rescued the expression of BACH1-motif-containing, but not HERM-containing genes, in the group of GATA1- and heme-co-activated genes. These results are consistent with a model in which HERM and BACH1 function in parallel to facilitate GATA1-mediated transcriptional activation (Fig. 2).
Figure 2. Context-dependent heme amplification of GATA1 activity: parallel BACH1-dependent and independent mechanisms.
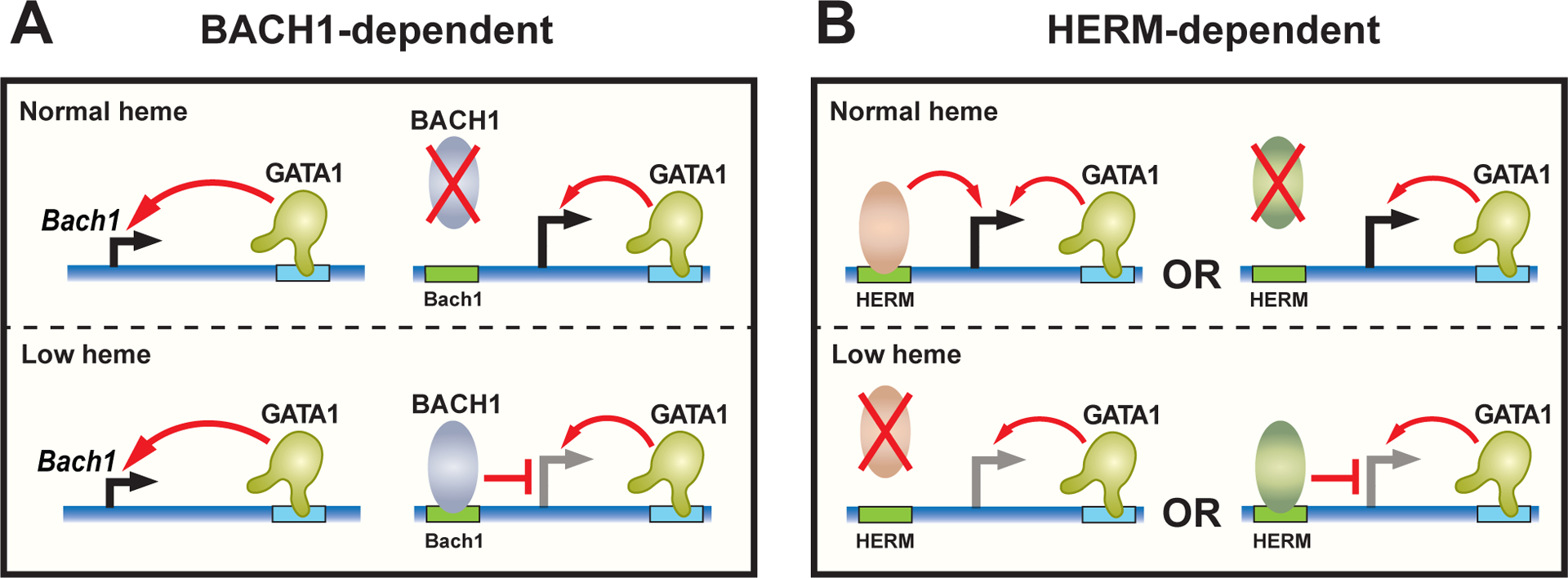
A. Heme facilitates GATA1 activity via BACH1. GATA1 activates Bach1 transcription. In heme-deficient cells, BACH1 protein accumulates, binds BACH1 DNA motifs, and opposes GATA1 activity to activate target gene transcription. In a normal heme environment, BACH1 protein proteolysis dominates over mechanisms that promote BACH1 synthesis, thus negating the mechanism that antagonizes GATA1.
B. Heme facilitates GATA1 activity via HERM. The presence of a HERM-binding transcriptional activator or the absence of a HERM-binding repressor in a physiological heme environment facilitates GATA1-mediated activation of target gene transcription.
The study described above provided evidence that HERM can mediate heme-dependent transcriptional repression that counteracts GATA1-mediated transcriptional activation (47). Pklr encodes the liver- and erythroid-specific pyruvate kinase essential for glycolysis (48), and PKLR mutations cause pyruvate kinase deficiency and nonspherocytic hemolytic anemia (49). Pklr expression is upregulated by GATA1, but further upregulated (by ~100 fold) in heme-deficient cells. shRNA-mediated BACH1 depletion does not affect Pklr expression, suggesting that Pklr is regulated by a BACH1-independent mechanism. Pklr contains an intronic heme-regulated ATAC-seq peak, which harbors two HERMs that are 29 bp apart. Disrupting the HERMs by CRISPR-Cas9 reduced Pklr expression in heme-deficient cells, but did not elevate Pklr expression in cells with normal heme, suggesting that HERM mediates the upregulated Pklr expression in a heme-deficient cell (Fig. 3a). Comparison of HERM to known motifs revealed similarity to a motif implicated in ZNF263 binding, which was identified from a ChIP-seq analysis in K562 cells (50). However, whether heme regulates binding of ZNF263 or another transcription factor to HERM requires future studies.
Figure 3. Context-dependent heme antagonism of GATA1.
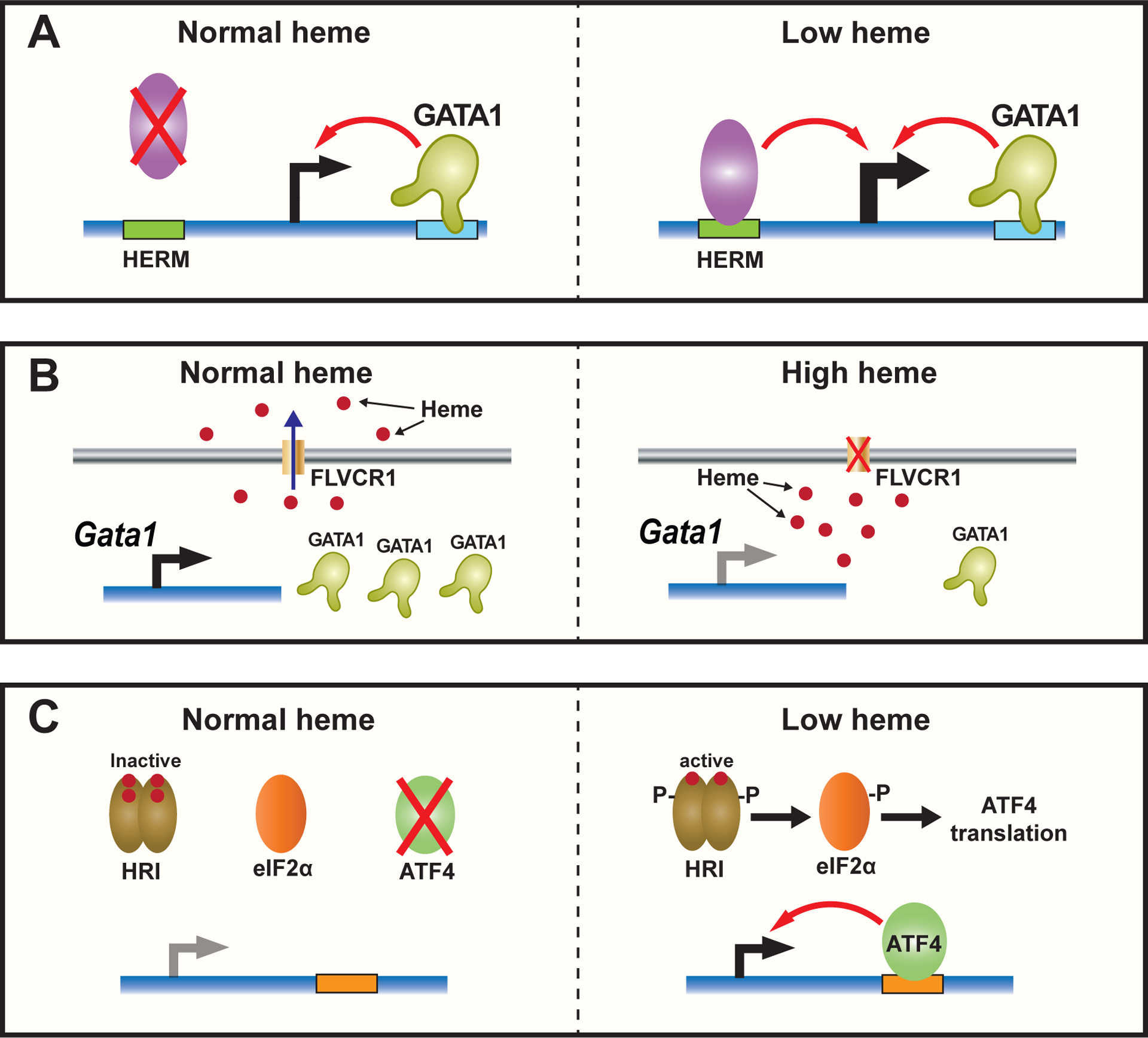
A. Heme antagonism of GATA1 via HERM. Hypothetical mechanism of HERM-binding transcriptional activator that elevates GATA1 target gene transcription in heme-deficient cells, with heme opposing this mechanism.
B. Heme accumulation inhibits GATA1 expression. Depletion of Flvcr1, the plasma membrane heme exporter, increases intracellular heme, which inhibits Gata1 RNA and GATA1 protein expression.
C. HRI-dependent induction of ATF4 activates transcription in heme-deficient cells. In a low-heme environment, HRI is activated and phosphorylates the translational elongation factor, eIF2α, which increases ATF4 translation, enabling ATF4 to activate target genes transcription.
Heme exporters can contribute to intracellular heme content. Feline leukemia virus subgroup C receptor (FLVCR1), a member of the major facilitator superfamily of transporter proteins, is a human heme exporter (51). Depleting Flvcr1 in mice elevates intracellular heme and is embryonic lethal, while neonatal depletion of Flvcr1 causes macrocytic anemia (52, 53). Single-cell RNA-seq analysis in wild-type and Flvcr1 knockout mice revealed that ribosomal protein gene expression is elevated in Flvcr1-deleted early erythroid cells. Gata1 and its target genes are repressed in late erythroid cells lacking Flvcr1 (54). Treating primary human bone marrow cells or K562 cells with 5-ALA, which elevates intracellular heme, reduces GATA1 RNA and protein expression (54). Thus, GATA1 and heme operate in a feedback loop in which GATA1 elevates heme synthesis, and as heme accumulates, it initially facilitates GATA1 activity during early erythropoiesis (5, 45, 47). As heme levels increase further, this is associated with reduced GATA1 expression during terminal differentiation (54) (Figure 3b).
The heme-dependent eIF2α kinase, HRI, which inhibits the translation of globin mRNAs via eIF2α phosphorylation, induces ATF4 (55, 56), a basic leucine zipper (bZip) protein that mediates stress responses (57). RNA-seq analysis in wild-type and Eif2ak1 (encoding HRI) knockout mice during iron deficiency revealed that the majority of genes elevated in wild-type mice with iron deficiency appear to be ATF4 targets involved in amino acid metabolism and synthesis, and these genes are not elevated in Eif2ak1 mutant mice (58). Thus, the HRI-dependent increase of ATF4 in heme-deficient cells may mediate heme-dependent transcriptional repression (Figure 3c). ATF4 shares similar DNA binding motifs with other bZip transcription factors (59, 60). Interestingly, MafK, NFE2, BACH2, and AP-1 motifs, which involve related sequences, also reside within sequences encompassing ATF4 ChIP-seq peaks (60). In principle, ATF4 may function through Bach motifs in chromatin. Thus, since BACH1 is a transcriptional repressor that mediates heme-dependent gene activation, the heme-repressed genes might be regulated by ATF4, or perhaps a related bZip factor through BACH1 motifs in heme-deficient cells.
Conclusion
Heme exerts critical functions during erythropoiesis beyond its canonical role as a hemoglobin component. These functions include facilitating or antagonizing GATA1 function to establish and/or maintain the erythroblast transcriptome via BACH1-dependent and independent mechanisms (5, 45, 47). GATA1 upregulates heme synthesis and expression of the transcriptional repressor BACH1. Heme binding inhibits DNA binding, promotes nuclear export, and induces proteasomal degradation of BACH1, thus enhancing GATA1-dependent activation of genes, including globins and Hmox1 (17, 23, 25, 27). Furthermore, heme can antagonize GATA1 activity by inhibiting GATA1 expression (54) or, in principle, by targeting the genome via the DNA motif HERM (47). HERM mediates heme-dependent activation or repression of genes that are essential for erythropoiesis, including the zinc exporter, Slc30a1 and the metabolic enzyme, Pklr (45, 47). Major unsolved problems include whether HERM-dependent genome targeting involves sequence-specific transcription factor binding, how HERM-dependent and BACH1-dependent mechanisms interface, and how these transcriptional mechanisms are interlinked with heme-dependent post-transcriptional and post-translational mechanisms. As GATA1 activates Alas2 and other genes encoding heme biosynthetic enzymes that generate heme, and heme functions in feedback loops to control GATA1 and the genome, we envision that diverse small molecules, including metals and metabolites, instigate such loops, which are likely to be vital components of GATA factor mechanisms that control cellular functional states, including differentiation (Figure 4).
Figure 4. Feedback loops involving GATA factor, genome, and small molecules control cellular differentiation.
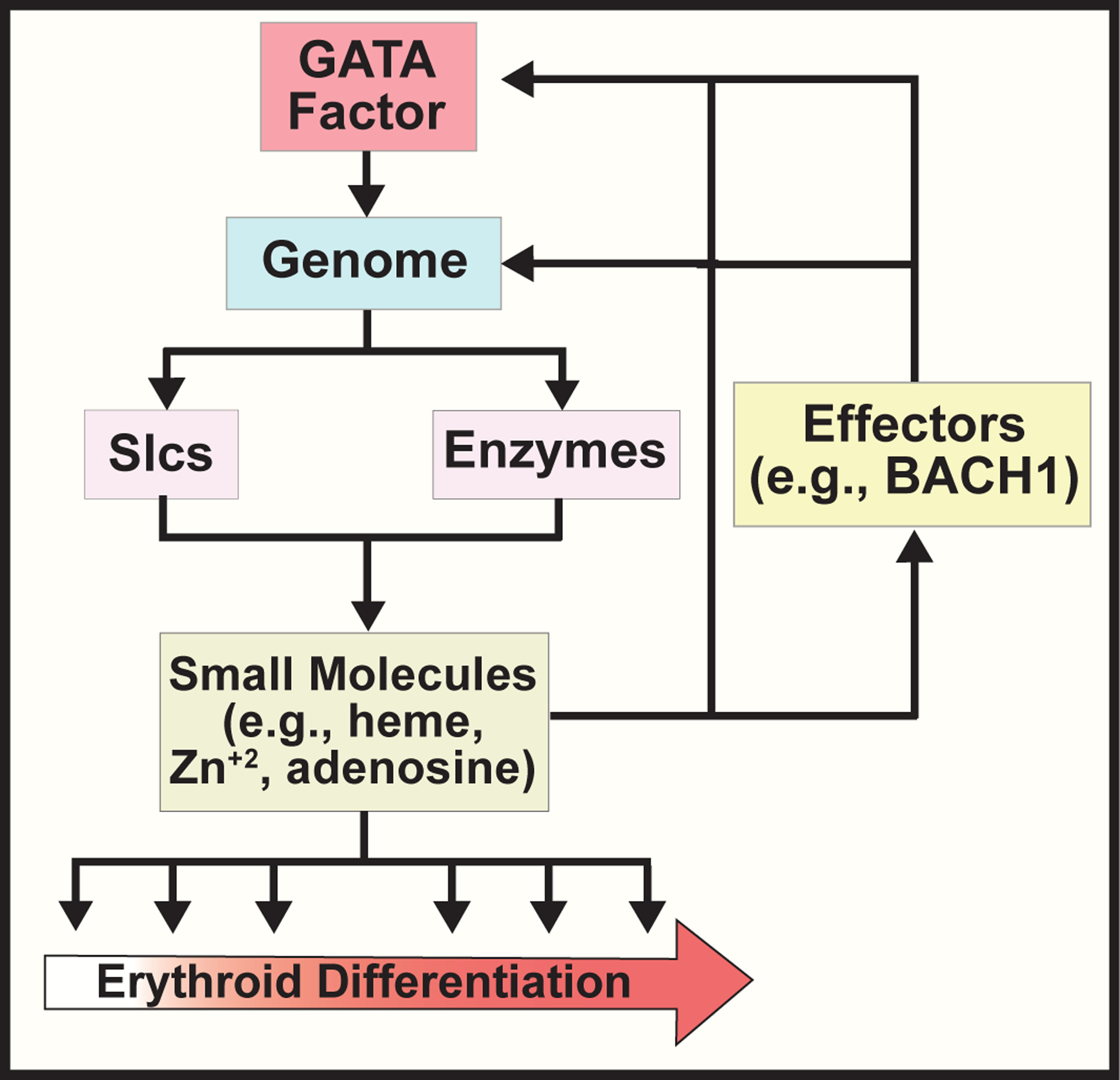
GATA factor dependent genome regulation controls the expression of transporters (SLCs) and biosynthetic enzymes for small molecules. The small molecules then function in a feedback loop to control GATA factor and genome function via protein effectors, e.g. BACH1.
References
- 1.Dutt S, Hamza I, Bartnikas TB. Molecular Mechanisms of Iron and Heme Metabolism. Annu Rev Nutr. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dailey HA, Meissner PN. Erythroid heme biosynthesis and its disorders. Cold Spring Harb Perspect Med. 2013;3(4):a011676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006;1763(7):723–36. [DOI] [PubMed] [Google Scholar]
- 4.Bishop DF, Henderson AS, Astrin KH. Human delta-aminolevulinate synthase: assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the X chromosome. Genomics. 1990;7(2):207–14. [DOI] [PubMed] [Google Scholar]
- 5.Tanimura N, Miller E, Igarashi K, Yang D, Burstyn JN, Dewey CN, et al. Mechanism governing heme synthesis reveals a GATA factor/heme circuit that controls differentiation. EMBO Rep. 2016;17(2):249–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campagna DR, de Bie CI, Schmitz-Abe K, Sweeney M, Sendamarai AK, Schmidt PJ, et al. X-linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA-binding site mutations. Am J Hematol. 2014;89(3):315–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaneko K, Furuyama K, Fujiwara T, Kobayashi R, Ishida H, Harigae H, et al. Identification of a novel erythroid-specific enhancer for the ALAS2 gene and its loss-of-function mutation which is associated with congenital sideroblastic anemia. Haematologica. 2014;99(2):252–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Zhang J, An W, Wan Y, Ma S, Yin J, et al. Intron 1 GATA site enhances ALAS2 expression indispensably during erythroid differentiation. Nucleic Acids Res. 2017;45(2):657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kruger A, Keppel M, Sharma V, Frunzke J. The diversity of heme sensor systems - heme-responsive transcriptional regulation mediated by transient heme protein interactions. FEMS Microbiol Rev. 2022;46(3). [DOI] [PubMed] [Google Scholar]
- 10.Shimizu T, Lengalova A, Martinek V, Martinkova M. Heme: emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem Soc Rev. 2019;48(24):5624–57. [DOI] [PubMed] [Google Scholar]
- 11.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109(7):2693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen JJ, Zhang S. Translational control by heme-regulated elF2alpha kinase during erythropoiesis. Curr Opin Hematol. 2022;29(3):103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20(23):6909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tahara T, Sun J, Igarashi K, Taketani S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun. 2004;324(1):77–85. [DOI] [PubMed] [Google Scholar]
- 15.Tahara T, Sun J, Nakanishi K, Yamamoto M, Mori H, Saito T, et al. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem. 2004;279(7):5480–7. [DOI] [PubMed] [Google Scholar]
- 16.Oyake T, Itoh K, Motohashi H. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol. 1996;16:6083–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001;20(11):2835–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lathrop JT, Timko MP. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science. 1993;259(5094):522–5. [DOI] [PubMed] [Google Scholar]
- 19.Munakata H, Sun JY, Yoshida K, Nakatani T, Honda E, Hayakawa S, et al. Role of the heme regulatory motif in the heme-mediated inhibition of mitochondrial import of 5-aminolevulinate synthase. J Biochem. 2004;136(2):233–8. [DOI] [PubMed] [Google Scholar]
- 20.Igarashi J, Murase M, Iizuka A, Pichierri F, Martinkova M, Shimizu T. Elucidation of the heme binding site of heme-regulated eukaryotic initiation factor 2alpha kinase and the role of the regulatory motif in heme sensing by spectroscopic and catalytic studies of mutant proteins. J Biol Chem. 2008;283(27):18782–91. [DOI] [PubMed] [Google Scholar]
- 21.Hira S, Tomita T, Matsui T, Igarashi K, Ikeda-Saito M. Bach1, a heme-dependent transcription factor, reveals presence of multiple heme binding sites with distinct coordination structure. IUBMB Life. 2007;59(8–9):542–51. [DOI] [PubMed] [Google Scholar]
- 22.Segawa K, Igarashi K, Murayama K. The Cys-Pro motifs in the intrinsically disordered regions of the transcription factor BACH1 mediate distinct and overlapping functions upon heme binding. FEBS Lett. 2022. [DOI] [PubMed] [Google Scholar]
- 23.Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21(19):5216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci U S A. 2004;101(6):1461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki H, Tashiro S, Hira S, Sun J, Yamazaki C, Zenke Y, et al. Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J. 2004;23(13):2544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki H, Tashiro S, Sun J, Doi H, Satomi S, Igarashi K. Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J Biol Chem. 2003;278(49):49246–53. [DOI] [PubMed] [Google Scholar]
- 27.Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, et al. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol. 2007;27(19):6962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25(20):4877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13(12):1178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zinngrebe J, Rieser E, Taraborrelli L, Peltzer N, Hartwig T, Ren H, et al. --LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J Exp Med. 2016;213(12):2671–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan MK, Lim HJ, Bennett EJ, Shi Y, Harper JW. Parallel SCF adaptor capture proteomics reveals a role for SCFFBXL17 in NRF2 activation via BACH1 repressor turnover. Mol Cell. 2013;52(1):9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell. 2019;178(2):316–29 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouse EC, Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, et al. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 2012;13(8):418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warnatz HJ, Schmidt D, Manke T, Piccini I, Sultan M, Borodina T, et al. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J Biol Chem. 2011;286(26):23521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumoto M, Kondo K, Shiraki T, Brydun A, Funayama R, Nakayama K, et al. Genomewide approaches for BACH1 target genes in mouse embryonic fibroblasts showed BACH1-Pparg pathway in adipogenesis. Genes Cells. 2016;21(6):553–67. [DOI] [PubMed] [Google Scholar]
- 36.Ebina-Shibuya R, Watanabe-Matsui M, Matsumoto M, Itoh-Nakadai A, Funayama R, Nakayama K, et al. The double knockout of Bach1 and Bach2 in mice reveals shared compensatory mechanisms in regulating alveolar macrophage function and lung surfactant homeostasis. J Biochem. 2016;160(6):333–44. [DOI] [PubMed] [Google Scholar]
- 37.Igarashi K, Itoh-Nakadai A. Orchestration of B lymphoid cells and their inner myeloid by Bach. Curr Opin Immunol. 2016;39:136–42. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe-Matsui M, Matsumoto T, Matsui T, Ikeda-Saito M, Muto A, Murayama K, et al. Heme binds to an intrinsically disordered region of Bach2 and alters its conformation. Arch Biochem Biophys. 2015;565:25–31. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe-Matsui M, Muto A, Matsui T, Itoh-Nakadai A, Nakajima O, Murayama K, et al. Heme regulates B-cell differentiation, antibody class switch, and heme oxygenase-1 expression in B cells as a ligand of Bach2. Blood. 2011;117(20):5438–48. [DOI] [PubMed] [Google Scholar]
- 40.Kato H, Itoh-Nakadai A, Matsumoto M, Ishii Y, Watanabe-Matsui M, Ikeda M, et al. Infection perturbs Bach2- and Bach1-dependent erythroid lineage ‘choice’ to cause anemia. Nat Immunol. 2018;19(10):1059–70. [DOI] [PubMed] [Google Scholar]
- 41.Weiss MJ, Yu C, Orkin SH. Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol Cell Biol. 1997;17(3):1642–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gregory T, Yu C, Ma A, Orkin SH, Blobel GA, Weiss MJ. GATA-1 and erythropoietin cooperate to promoter erythroid cell survival by regulating bcl-xl expression. Blood. 1999;94(1):87–96. [PubMed] [Google Scholar]
- 43.Grass JA, Boyer ME, Pal S, Wu J, Weiss MJ, Bresnick EH. GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc Natl Acad Sci U S A. 2003;100:8811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, et al. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood. 2004;104:3136–47. [DOI] [PubMed] [Google Scholar]
- 45.Tanimura N, Liao R, Wilson GM, Dent MR, Cao M, Burstyn JN, et al. GATA/Heme Multi-omics Reveals a Trace Metal-Dependent Cellular Differentiation Mechanism. Dev Cell. 2018;46(5):581–94 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liao R, Zheng Y, Liu X, Zhang Y, Seim G, Tanimura N, et al. Discovering How Heme Controls Genome Function Through Heme-omics. Cell Rep. 2020;31(13):107832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Predicting therapeutic efficacy for sickle cell anemia. Prog Clin Biol Res. 1987;251:497–505. [PubMed] [Google Scholar]
- 49.Baronciani L, Beutler E. Analysis of pyruvate kinase-deficiency mutations that produce nonspherocytic hemolytic anemia. Proc Natl Acad Sci U S A. 1993;90(9):4324–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frietze S, O’Geen H, Blahnik KR, Jin VX, Farnham PJ. ZNF274 recruits the histone methyltransferase SETDB1 to the 3’ ends of ZNF genes. PLoS One. 2010;5(12):e15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, et al. Identification of a human heme exporter that is essential for erythropoiesis. Cell. 2004;118(6):757–66. [DOI] [PubMed] [Google Scholar]
- 52.Doty RT, Phelps SR, Shadle C, Sanchez-Bonilla M, Keel SB, Abkowitz JL. Coordinate expression of heme and globin is essential for effective erythropoiesis. J Clin Invest. 2015;125(12):4681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319(5864):825–8. [DOI] [PubMed] [Google Scholar]
- 54.Doty RT, Yan X, Lausted C, Munday AD, Yang Z, Yi D, et al. Single-cell analyses demonstrate that a heme-GATA1 feedback loop regulates red cell differentiation. Blood. 2019;133(5):457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suragani RN, Zachariah RS, Velazquez JG, Liu S, Sun CW, Townes TM, et al. Heme-regulated eIF2alpha kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood. 2012;119(22):5276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang S, Macias-Garcia A, Velazquez J, Paltrinieri E, Kaufman RJ, Chen JJ. HRI coordinates translation by eIF2alphaP and mTORC1 to mitigate ineffective erythropoiesis in mice during iron deficiency. Blood. 2018;131(4):450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol Metab. 2017;28(11):794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang S, Macias-Garcia A, Ulirsch JC, Velazquez J, Butty VL, Levine SS, et al. HRI coordinates translation necessary for protein homeostasis and mitochondrial function in erythropoiesis. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pathak SS, Liu D, Li T, de Zavalia N, Zhu L, Li J, et al. The eIF2alpha Kinase GCN2 Modulates Period and Rhythmicity of the Circadian Clock by Translational Control of Atf4. Neuron. 2019;104(4):724–35 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]