

Abstract
KCNQ1, the major component of the slow-delayed rectifier potassium channel, is responsible for repolarization of cardiac action potential. Mutations in this channel can lead to a variety of diseases, most notably long QT syndrome. It is currently unknown how many of these mutations change channel function and structure on a molecular level. Since tetramerization is key to proper function and structure of the channel, it is likely that mutations modify the stability of KCNQ1 oligomers. Presently, the C-terminal domain of KCNQ1 has been noted as the driving force for oligomer formation. However, truncated versions of this protein lacking the C-terminal domain still tetramerize. Therefore, we explored the role of native cysteine residues in a truncated construct of human KCNQ1, amino acids 100–370, by blocking potential interactions of cysteines with a nitroxide based spin label. Mobility of the spin labels was investigated with continuous wave electron paramagnetic resonance (CW-EPR) spectroscopy. The oligomerization state was examined by gel electrophoresis. The data provide information on tetramerization of human KCNQ1 without the C-terminal domain. Specifically, how blocking the side chains of native cysteines residues reduces oligomerization. A better understanding of tetramer formation could provide improved understanding of the molecular etiology of long QT syndrome and other diseases related to KCNQ1.
Keywords: EPR spectroscopy, KCNQ1, Kv channel, Oligomerization, SDS-PAGE
Introduction
KCNQ1 (Q1, UniProtKB P51787), also known as Kv7.1, is a voltage-gated potassium channel expressed across the body for various physiological purposes such as heart rhythm, metabolite homeostasis, and endocochlear homeostasis [1, 2]. A functional Q1 channel consists of an N-terminal helix (residues 1–121), six transmembrane helices (residues 122–348, S1-S6), and four C-terminal helices (residues 349–676, HA-HD). The transmembrane domain (TMD) consists of the voltage sensing domain (VSD) S1-S4 and the pore domain (PD), S5-S6 [1,3,4]. In vivo, four monomers of Q1 tetramerize to form a fully functional channel [5].
There are several ways that Q1 channel function is regulated in the body. Most notably, Q1 forms a complex with KCNE1 (E1), also known as minK or -subunit, to form the slow-delayed rectifier potassium channel, , which is required for repolarization of the cardiac action potential. In the , E1 modulates Q1 to slow activation kinetics [1,2,6]. Mutations in either protein can cause diseases such as long QT syndrome, Jervell and Lange-Nielsen syndrome, and familial atrial fibrillation [1,3,7]. Calmodulin, another subunit of Q1, regulates channel gating by alleviating inactivation in a Ca2+-dependent manner [4,7,8]. The other main regulator of Q1 channels is the signaling lipid phosphatidylinositol 4,5-bisphosphate (PIP2) which is essential for activation of Q1 homo-tetramers [1,9]. For the IKs to be functional, Q1, E1, calmodulin, and PIP2 all need to be present.
Oligomerization has several important roles within organisms, with the most common being allowing proteins to form large structures without growing the genome [10]. For example, Q1 exhibits channel activity only as a tetramer [5]. There are several point mutations of Q1 that cause long QT syndrome and other diseases; however, it is currently unknown if or how these point mutations change channel structure and function on a molecular level. Several channel diseases could be caused by mutations modifying or disturbing tetramerization of Q1 [5,10,11]. Thus, increasing our understanding of the molecular underpinnings of Q1 tetramerization could indicate if any of these point mutations alter channel formation. Currently, the proximal C-terminal domain, specifically helices HC and HD (Figure 1), has been tied to protein folding, oligomerization, partner specificity, and trafficking to the plasma membrane [3,8,12]. Studies indicate that HD is a four-stranded coiled-coil motif that induces tetramerization within different Kv7 channels [3,12]. This region is similar to the T1 tetramerization domain found in potassium channels outside the Kv7 family [13]. The T1 tetramerization domain and HD have been suggested to enhance channel assembly by increasing proximity of monomeric proteins [13]. Previous studies indicate that isolated Q1 VSD, lacking the pore domain and H domain, is monomeric when expressed in bacteria [14,15]. Nevertheless, there is likely another component of oligomerization within the transmembrane region of the protein [12]. In this study, the possibility of the native cysteines in Q1 TMD, containing both the VSD and PD, to be a major component of tetramerization was explored.
Figure 1:
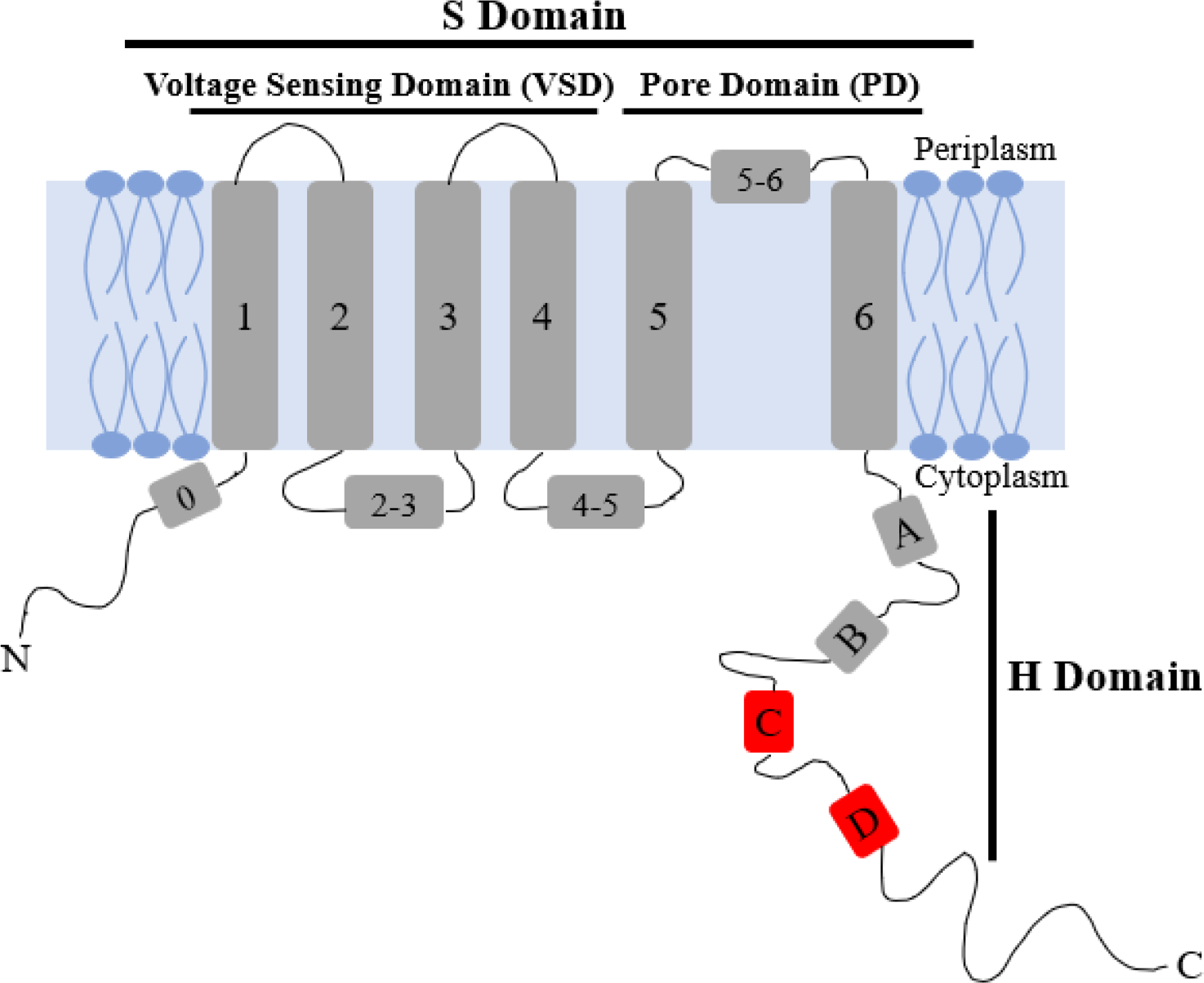
Cartoon depiction of monomeric KCNQ1 containing the voltage sensing domain (VSD), S1-S4; pore domain (PD), S5-S6; and the C-terminal domain, HA-HD. HC and HD are indicated in red.
There are five native cysteines in the TMD of Q1: C122, C136, C180, C214, and C331 (Figure 2). Cryo-EM and X-ray crystallography structures indicate that these cysteines face the hydrophobic membrane and not the aqueous pore [1,4,16]. However, several studies have indicated various adverse functional information from mutated native cysteines [17–20]. For example, Barro-Soria et al., created a C214A/G219C/C331A construct that, when expressed in oocytes, was hyperpolarized ~10 mV compared to the wildtype (WT) [19]. Chung et al., noted that the C136A mutation drastically changes channel function; however, that study did not explore changes in oligomer state [20]. These results suggest that the native cysteines may have a role in channel activity likely through tetramer formation or stability.
Figure 2:
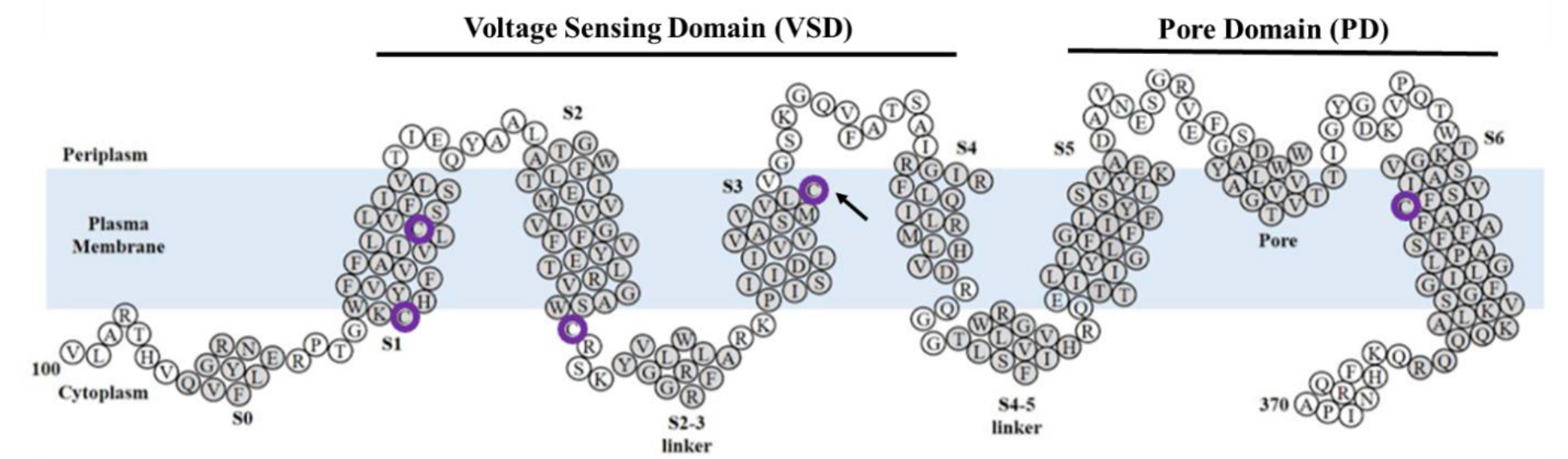
Representation of KCNQ1 used in this study, composed of the transmembrane domain (amino acids 100–370). Native cysteines are circled in purple. For portions of this study, C214 was changed to alanine (arrowhead). Helical regions are shaded in gray and labeled S0-S6. Modified from Dixit et al., 2022 [23].
Continuous-wave electron paramagnetic resonance (CW-EPR) spectroscopy is a powerful biophysical spectroscopic technique used to study the structural and dynamic properties of nitroxide-based spin-labeled proteins. One of the most common spin labels, S-(1-oxyl-2,2,5,5,-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methane sulfonothioate (MTSL), attaches to proteins via formation of a disulfide bond with the side chain of cysteine (Figure 3). This inherently blocks any potential interactions cysteine may have in the protein structure. Furthermore, line-shape analysis of the peaks of the CW-EPR spectra can be used to examine the mobility of the spin label attached to the protein [21,22]. Specifically, when the side chain of the spin label is constrained by its environment (e.g. the membrane or other regions of the protein), the CW-EPR spectra broaden, indicating a decrease in spin label mobility [21,22]. Conversely, when the linewidths of the CW-EPR spectra sharpen, the spin label has increased mobility (e.g. less constrained by the environment) [21,22]. In addition to spin label side chain mobility, the motion of the protein backbone can impact observed flexibility [22].
Figure 3:
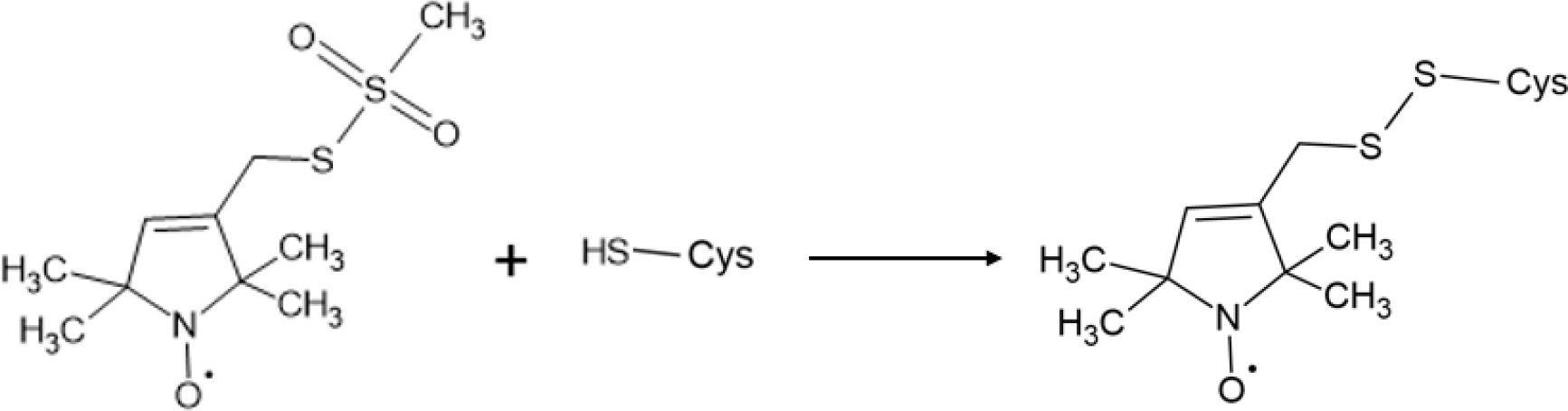
Schematic of the reaction between MTSL spin label and how it attaches to protein.
Previously, it has been shown that a truncated construct of human Q1 containing the transmembrane domain (TMD), which includes the VSD and PD (amino acids 100–370), can be successfully expressed and purified from recombinant E. coli [23]. This construct of Q1 TMD still forms oligomers, even though it is missing the C-terminal domain, suggesting that other interactions play a role in tetramerization [23]. It is hypothesized that the native cysteines promote or stabilize channel tetramerization. In this study, the role of native cysteines in the tetramerization of Q1 TMD was explored by blocking the cysteine side chain with MTSL spin label. Structural and dynamic information of Q1 TMD was obtained using CW-EPR spectroscopy. The oligomerization state was observed via sodium dodecyl sulfate – polyacrylamide gel electrophoresis (SDS-PAGE). Our data show tetramerization changes without the interaction of the native cysteines.
Materials and Methods
Site-Directed Mutagenesis
Primer-based site-directed mutagenesis was performed on the human Q1 TMD in a pET-21b(+) plasmid using PfuUltra II High Fidelity DNA Polymerase (Agilent) according to the manufacturer’s instructions, with the addition of 2% dimethylsulfoxide. Primers were designed using (DNASTAR Lasergene software (Madison, WI)). Purified plasmid DNA was obtained from transformed XL-Gold cells using QIAprep Spin Miniprep Kit (Qiagen). All mutagenesis was confirmed using sanger sequencing (Genewiz).
Over-Expression and Purification
pET-21b(+) containing the gene for human Q1 TMD was transformed into E. coli BL21-CodonPlus (DE3)-RP cells (Agilent). Cells were grown in terrific broth (Fisher Scientific) at 37 C, supplemented with 200 μg/mL ampicillin and 50 μg/mL chloramphenicol with continual rotatory shaking at 250 rpm, until the OD600 reached 0.6–0.8. The cultures were then cooled to 25 C and induced with 1 mM IPTG and incubated overnight with shaking at 250 rpm [23].
The cells were harvested by centrifugation at 11000 ×g for 10 minutes at 4 C. The cells were lysed using a 20 fold excess of lysis buffer to gram of cells (75 mM Tris-base, 300 mM NaCl, 0.2 mM EDTA, pH 7.5), 40x w/v LDR stock (100 mg/mL lysozyme, 10 mg/mL DNase, 10 mg/mL RNase), 200x PMSF stock (20 mg/mL), 5 mM Magnesium acetate, and 2 mM TCEP with tumbling at room temperature for 30 minutes. Cell suspension was then sonicated for 6.5 minutes with pulse 5 sec on/off at 40% amplitude (Fisher Scientific Sonic Dismembrator Model 500). To dissolve all membranes, 3% empigen BB detergent (Millipore Sigma) was added and mixed with shaking overnight at 4 C. All remaining undissolved membranes were removed via centrifugation at 27000 ×g and 4 C for 30 minutes, and the supernatant was retained [23].
To purify Q1 TMD, the supernatant was incubated with 3 mL pre-equilibrated Ni(II)-NTA superflow resin (Qiagen) for 30 minutes at 4 C. The resin-supernatant mixture was then centrifuged at 2700 ×g and 4 C for 10 minutes to separate the protein-bound resin fraction based on density and then transferred to a column. The protein-bound resin was washed with cold buffer A (40 mM HEPES and 300 mM NaCl, pH 7.5), 3% empigen BB detergent, 50 mM imidazole, and 2 mM TCEP. Treatment of the column with cold buffer A, 0.3% n-Dodecylphosphocholine (DPC), also known as Fos-Choline 12, (Anatrace), and 2 mM TCEP resulted in the exchange of detergent from empigen BB to DPC. Q1 TMD was eluted with 250 mM imidazole, 0.5% DPC, and 2 mM TCEP. To concentrate, the eluent was placed on an ultracel regenerated cellulose membrane, 3 kDa molecular cut off (Millipore Sigma) and centrifuged at 4300 ×g at 4 C for 30 minutes. Finally, buffer exchange was conducted using an ultracel regenerated cellulose membrane, 3 kDa molecular cut-off, with 50 mM phosphate buffer, pH 7, and 0.05% DPC at 4300 ×g at 4 C for 30 minutes, 3 times [23].
MTSL spin-labeling
250 mM stock MTSL (Toronto Research Chemicals) was added directly to concentrated Q1 protein in 50 mM phosphate buffer, pH 7.0, and 0.05% DPC at a 30:1 MTSL: protein molar ratio. The solution incubated overnight at room temperature with vigorous shaking. The mixture was then placed on an ultracel regenerated cellulose membrane, 3 kDa molecular cut-off, for centrifugal filtering with 50 mM phosphate buffer, pH 7, and 0.05% DPC at 4300 ×g at 4 C for 30 minutes, 3 times. To remove any excess spin label, the solution was incubated with 3 mL pre-equilibrated Ni(II)-NTA superflow resin overnight at 4 C. The resin was washed with 300 mL/column 50 mM phosphate buffer, pH 7.0, and 0.05% DPC. Finally, the protein was eluted with 4 mL/column 50 mM phosphate buffer, pH 7.0, 250 mM imidazole, and 0.5% DPC.
Circular Dichroism (CD) Spectroscopy
All CD experiments were performed using an Aviv Circular Dichroism Spectrometer model 435. Twenty scans were collected and signal averaged from 185 nm to 260 nm, at 25 C. The averaging time was 3.0 sec, and the settling time was 0.333 sec. Protein samples were at a concentration of 0.090 mg/mL in 50 mM phosphate buffer, pH 7.0, and 0.05% DPC. To account for background absorbance, spectra of 50 mM phosphate buffer, pH 7.0, and 0.05% DPC were collected and subtracted from all protein samples. The spectra were analyzed according to the following formula derived from Chen, 1972 [24].
| Equation 1 |
is the mean residue ellipticity value at is the value of structures and random coils at is the value of helices at .[24]
CW-EPR Spectroscopy
CW-EPR spectroscopy experiments were conducted on a Bruker EMX X-band spectrometer. EPR spectra were recorded over a scan width of 100 G with a field modulation of 1 G at a modulation frequency of 100 kHz. Q1 protein samples were placed in a glass capillary tube in a volume of 100 μL and at a concentration of 35 μM. Every sample was scanned 20 times with a microwave intensity of 10 mW at 25 C.
SDS-PAGE and Silver Staining
1 μg of each protein sample were subjected to separation by SDS-PAGE through 12.5% polyacrylamide gels. Samples were mixed with sample solubilizing buffer (50 mM Tris pH 6.8, 0.1 M DTT, 2.5% SDS, 15% glycerol, and 0.05% bromophenol blue) prior to loading the gel. After electrophoresis, the gel was silver stained according to the protocol developed by Chavellet, Luche, and Rabilloud [25]. The gel was imaging using a Bio-Rad GelDoc Go System (Supplemental Figure 1).
Results and Discussion
Circular dichroism (CD) spectroscopy was used to determine the nature of the secondary structure of Q1 TMD and Q1 C214A TMD in the presence and absence of bound MTSL spin label (Figure 4) to determine if MTSL binding has an effect on secondary structure. Under our assay conditions, all variants display clear signs of alpha helical components with a positive peak at 195 nm and negative peaks at 208 nm and 222 nm. Additionally, all variants are approximately 25% ± 5% alpha helix, as calculated by Equation 1; therefore, it is reasonable to conclude that a mutation of C214 to alanine (C214A) and labeling the remaining cysteines with MTSL does not significantly alter the secondary structure of KCNQ1. Furthermore, the tertiary structure of the protein was inferred to not be disrupted by the mutagenesis or the MTSL labeling.
Figure 4:
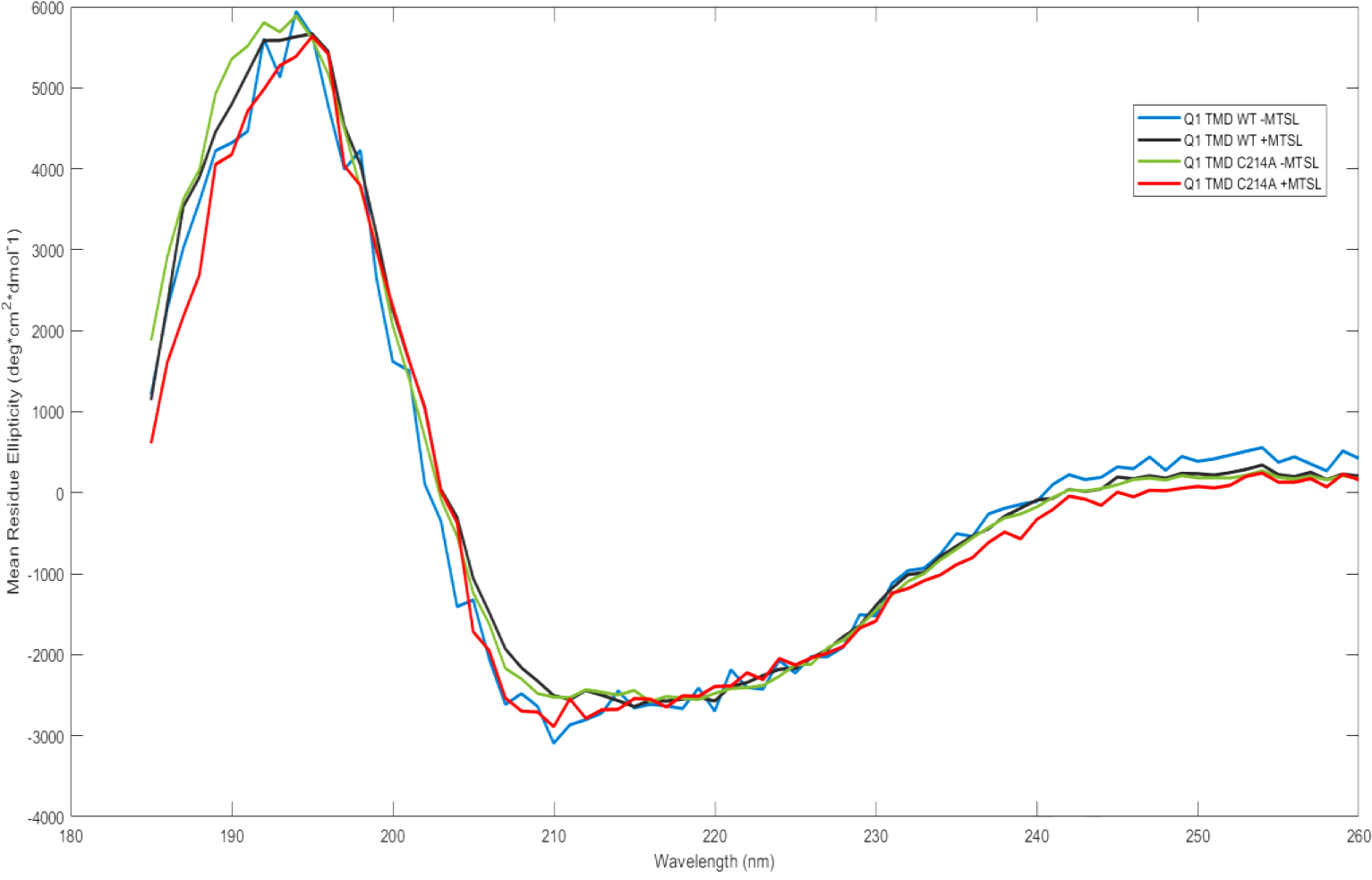
Circular Dichroism spectra of all Q1 TMD variants in (0.09 mg/mL) 0.05% DPC and 50 mM phosphate buffer, pH 7.0. Q1 TMD WT −MTSL (blue), Q1 TMD WT +MTSL (black), Q1 TMD C214A −MTSL (green), and Q1 TMD C214A +MTSL (red).
Continuous-wave (CW) EPR spectroscopy was used to compare the relative dynamic properties of the WT and Q1 C214A TMD. EPR line-shape analysis of the Q1 C214A TMD spectrum indicates that it is broader than the WT Q1 TMD spectra, indicating less spin label mobility in the C214A protein (Figure 5). This is likely due to the bound MTSL spin labels disrupting the stability of WT Q1 tetramers; thereby, reducing the amount of tetramers. The spin labels are likely less constrained in the Q1 monomer compared to Q1 oligomers corresponding to more mobility in the monomer as indicated in Figure 5. In addition, C214A likely destabilizes oligomers due to the lack of interaction of this amino acid position with the other native cysteines blocked by MTSL. This is further verified by SDS-PAGE in Figure 6.
Figure 5:
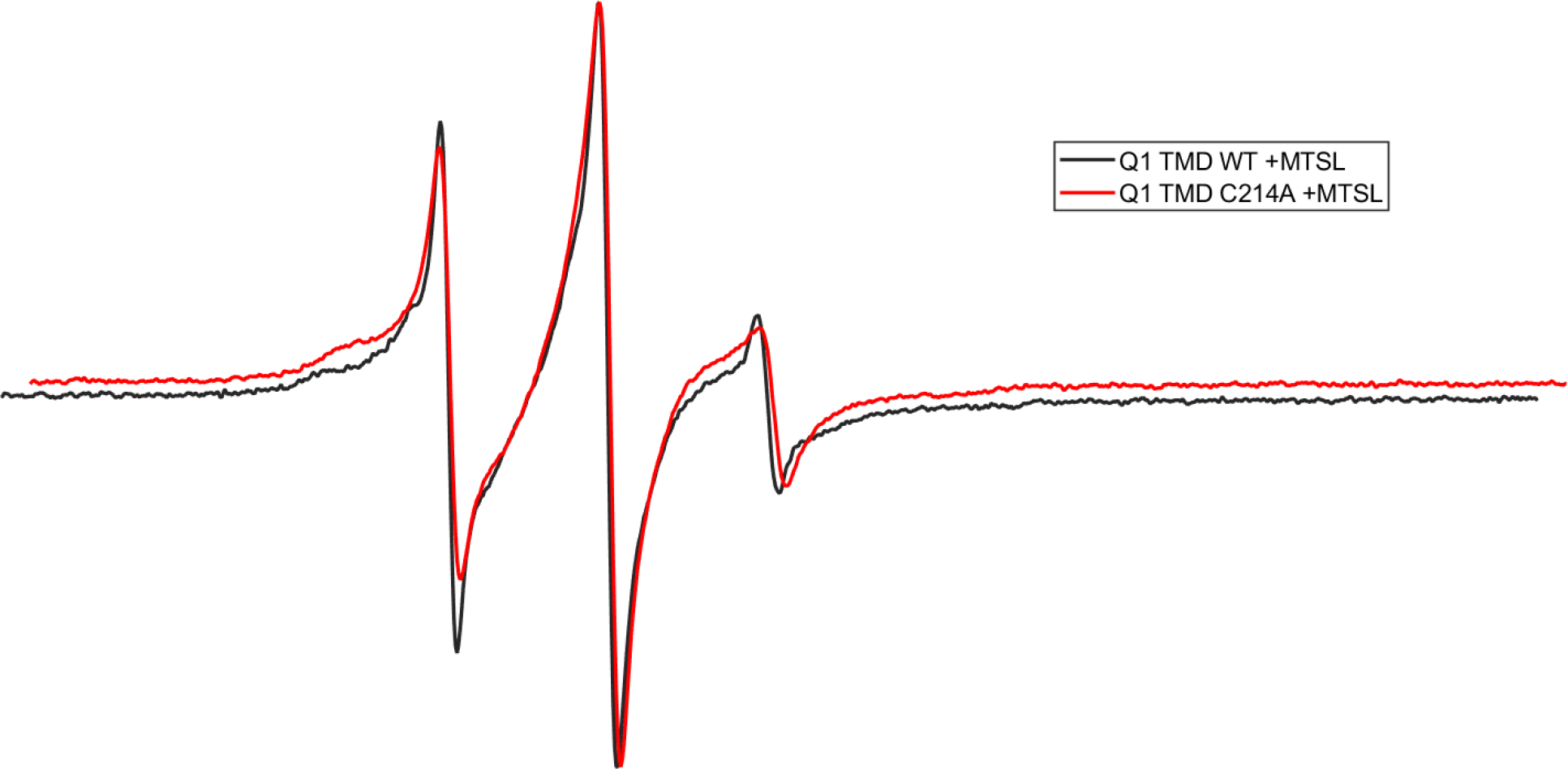
CW-EPR spectra of Q1 TMD WT (black) and Q1 TMD C214A (red) labeled with MTSL in 0.05% DPC and 50 mM phosphate buffer, pH 7.0.
Figure 6:
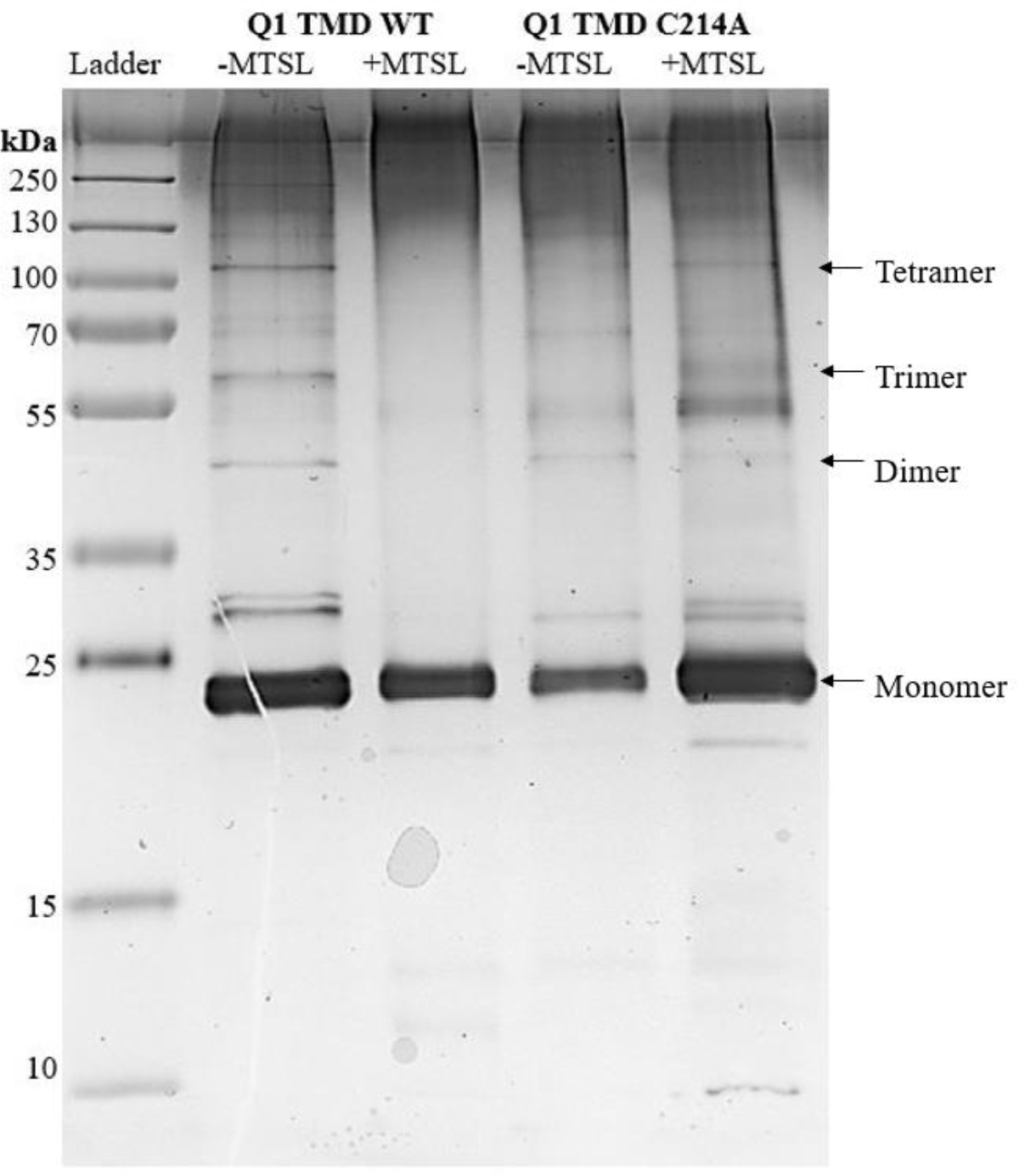
Silver Stained SDS-PAGE gel of all Q1 TMD variants. Lane 1: Page plus protein ladder (Fisher), Lane 2: Q1 TMD WT −MTSL, Lane 3: Q1 TMD WT +MTSL, Lane 4: Q1 TMD C214A −MTSL, Lane 5: Q1 TMD C214A +MTSL. All lanes have approximately 1 μg of protein. Monomeric Q1 TMD is approximately 24 kDa.
SDS-PAGE was used to analyze the oligomerization state of the Q1 TMD variants (Figure 6). Comparing WT −MTSL (Lane 2) to WT +MTSL (Lane 3), the quantity of dimers and higher order oligomers, as observed by the number and density of the bands, decreases upon adding the spin label. Additionally, comparing C214A −MTSL (Lane 4) to C214A +MTSL (Lane 5), there are fewer oligomers upon the addition of spin label as evidenced by less prominant bands at molecular weights that correlate to Q1 TMD oligomers. This suggests that blocking potential sites of interactions between cysteines during oligomer formation inhibits this association and may drive nonnative interactions as evidenced by the increase in the number and density of bands at molecular weights that are not predicted to be Q1 TMD oligomers as well as the increase of aggregate protein at the top of the lanes. C214A mutation −MTSL (Lane 4) reduced the formation of tetramers compared to the WT −MTSL(Lane 2), even though this residue is not predicted to be in the interface of Q1 association [1]. We expect that C214 interacts with the polar region of the membrane to stabilize oligomers. Previous studies indicate that S217 is just outside of the membrane [26]. Thus, it is reasonable to predict that a residue close by would be associated with the polar region of the membrane.
In this study, the role of cysteine residues in the oligomerization of Q1 TMD was explored in detergent. Detergent is a common membrane mimetic system that encapsulates membrane proteins in micelles [27,28]. However, recent studies suggest that secondary and tertiary structures of the same protein can be quite distinct when comparing in vitro to in vivo structures, due to the complex environment of the cell [26–31]. Because the secondary and tertiary structures are different, the assumption is that the native quaternary structure of proteins also changes depending on the surrounding environment [27,28]. In the context of homo-oligomers, such as KCNQ1, the macromolecular crowding of the cell would drive and stabilize oligomer formation; thus, leading to more contacts that cannot be achieved with membrane mimetics [29].
In conclusion, the proximal C-terminal domain of KCNQ1 is not the only region that drives or stabilizes the formation of oligomers. Our data suggest that C214 play a role in the formation or stability of Q1 tetramers; specifically, by likely stabilizing the tetramer to the polar region of the membrane. Future studies are needed to explore how the other native cysteines, C122, C136, C180, and C331, play a role in oligomerization due to the loss of higher order species upon native cysteine blockage by the spin label MTSL. Additionally, there may still be another component of Q1 tetramerization that can only be detected in cells rather than detergents.
Supplementary Material
Highlights.
Exploration of oligomerization of Kv channels without C-terminal domain
Nitroxide based spin labeling of native cysteine residues alters oligomerization
C214 plays a role in the stability and or formation of KCNQ1 tetramers
Acknowledgements
We would like to thank Emma Gordan with assistance with MatLab. The authors thank the members of the Lorigan and Dabney-Smith research groups for their valuable suggestions with this study. We also thank Charles Sanders and members of his research group for the pET21b+ plasmid with human KCNQ1 100–370 gene.
Funding
This work was generously supported by a NIGMS/NIH Maximizing Investigator’s Research Award (MIRA) R35 GM126935.
Abbreviations
- CD
Circular dichroism spectroscopy
- CW-EPR
Continuous-wave electron paramagnetic resonance spectroscopy
- DPC
Dodecylphosphocholine or Fos-Choline 12
- E1
KCNE1
- IKs
slow-delayed rectifier potassium channel
- Q1
KCNQ1
- MRE
mean residue ellipticity
- MTSL
S-(1-oxyl-2,2,5,5,-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methane sulfonothioate
- PD
Pore domain
- PIP2
phosphatidylinositol 4,5-bisphosphate
- SDS-PAGE
sodium dodecyl sulfate – polyacrylamide gel electrophoresis
- TMD
Transmembrane domain
- VSD
Voltage sensing domain
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sun J, MacKinnon R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell. 2020;180(2):340–347.e9. doi: 10.1016/j.cell.2019.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixit G, Dabney-Smith C, Lorigan GA. The membrane protein KCNQ1 potassium ion channel: Functional diversity and current structural insights. Biochim Biophys Acta - Biomembr. 2020;1862(5):183148. doi: 10.1016/j.bbamem.2019.183148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewer KR, Kuenze G, Vanoye CG, George AL, Meiler J, Sanders CR. Structures Illuminate Cardiac Ion Channel Functions in Health and in Long QT Syndrome. Front Pharmacol. 2020;11:550. doi: 10.3389/fphar.2020.00550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun J, MacKinnon R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell. 2017;169(6):1042–1050.e9. doi: 10.1016/j.cell.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jalily Hasani H, Ganesan A, Ahmed M, Barakat KH. Effects of protein-protein interactions and ligand binding on the ion permeation in KCNQ1 potassium channel. van Veen HW, ed. PLoS One. 2018;13(2):e0191905. doi: 10.1371/journal.pone.0191905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Y, Wang Y, Meng XY, et al. Building KCNQ1/KCNE1 channel models and probing their interactions by molecular-dynamics simulations. Biophys J. 2013;105(11):2461–2473. doi: 10.1016/j.bpj.2013.09.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Eldstrom J, Fedida D. Gating and Regulation of KCNQ1 and KCNQ1 + KCNE1 Channel Complexes. Front Physiol. 2020;11:504. doi: 10.3389/fphys.2020.00504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiener R, Haitin Y, Shamgar L, et al. The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J Biol Chem. 2008;283(9):5815–5830. doi: 10.1074/jbc.M707541200 [DOI] [PubMed] [Google Scholar]
- 9.Zaydman MA, Silva JR, Delaloye K, et al. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci U S A. 2013;110(32):13180–13185. doi: 10.1073/pnas.1305167110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashimoto K, Panchenko AR. Mechanisms of protein oligomerization, the critical role of insertions and deletions in maintaining different oligomeric states. Proc Natl Acad Sci U S A. 2010;107(47):20352–20357. doi: 10.1073/PNAS.1012999107/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang H, Kuenze G, Smith JA, et al. Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci Adv. 2018;4(3). doi: 10.1126/sciadv.aar2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haitin Y, Attali B. The C-terminus of Kv7 channels: a multifunctional module. J Physiol. 2008;586(Pt 7):1803. doi: 10.1113/JPHYSIOL.2007.149187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.N Z, YN J, LY J. An artificial tetramerization domain restores efficient assembly of functional Shaker channels lacking T1. Proc Natl Acad Sci U S A. 2000;97(7):3591–3595. doi: 10.1073/PNAS.060016797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423(6935):42–48. doi: 10.1038/nature01581 [DOI] [PubMed] [Google Scholar]
- 15.Peng D, Kim JH, Kroncke BM, et al. Purification and structural study of the voltage-sensor domain of the human KCNQ1 potassium ion channel. Biochemistry. 2014;53(12):2032–2042. doi: 10.1021/bi500102w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sachyani D, Dvir M, Strulovich R, et al. Structural Basis of a Kv7.1 Potassium Channel Gating Module: Studies of the Intracellular C-Terminal Domain in Complex with Calmodulin. Structure. 2014;22(11):1582–1594. doi: 10.1016/j.str.2014.07.016 [DOI] [PubMed] [Google Scholar]
- 17.Nakajo K, Kubo Y. KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J Gen Physiol. 2007;130(3):269–281. doi: 10.1085/jgp.200709805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osteen JD, Barro-Soria R, Robey S, Sampson KJ, Kass RS, Larsson HP. Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc Natl Acad Sci U S A. 2012;109(18):7103–7108. doi: 10.1073/pnas.1201582109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barro-Soria R, Rebolledo S, Liin SI, et al. KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat Commun. 2014;5(1):1–12. doi: 10.1038/ncomms4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung DY, Chan PJ, Bankston JR, et al. Location of KCNE1 relative to KCNQ1 in the IKS potassium channel by disulfide cross-linking of substituted cysteines. Proc Natl Acad Sci U S A. 2009;106(3):743–748. doi: 10.1073/pnas.0811897106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahu ID, Lorigan GA. Site-Directed Spin Labeling EPR for Studying Membrane Proteins. Biomed Res Int. 2018;2018(1). doi: 10.1155/2018/3248289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klug CS, Feix JB. Methods and Applications of Site-Directed Spin Labeling EPR Spectroscopy. Methods Cell Biol. 2008;84:617–658. doi: 10.1016/S0091-679X(07)84020-9 [DOI] [PubMed] [Google Scholar]
- 23.Dixit G, Stowe RB, Bates A, et al. Purification and membrane interactions of human KCNQ1100–370 potassium ion channel. Biochim Biophys Acta - Biomembr. 2022;1864(11):184010. doi: 10.1016/J.BBAMEM.2022.184010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y-H, Tsi Yang J, Martinez HM. Determination of the Secondary Structures of Proteins by Circular Dichroism and Optical Rotatory Dispersion. Biochemistry. 1972;11(22):4120–4131. [DOI] [PubMed] [Google Scholar]
- 25.Chevallet M, Luche S, Rabilloud T. Silver staining of proteins in polyacrylamide gels. Nat Protoc 2006 14. 2006;1(4):1852–1858. doi: 10.1038/nprot.2006.288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dixit G, Sahu ID, Reynolds WD, et al. Probing the Dynamics and Structural Topology of the Reconstituted Human KCNQ1 Voltage Sensor Domain (Q1-VSD) in Lipid Bilayers Using Electron Paramagnetic Resonance Spectroscopy. Biochemistry. Published online 2019. doi: 10.1021/acs.biochem.8b01042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou HX, Cross TA. Influences of Membrane Mimetic Environments on Membrane Protein Structures. http://dx.doi.org/101146/annurev-biophys-083012-130326. 2013;42(1):361–392.doi: 10.1146/ANNUREV-BIOPHYS-083012-130326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majeed S, Ahmad AB, Sehar U, Georgieva ER. Lipid Membrane Mimetics in Functional and Structural Studies of Integral Membrane Proteins. Membranes (Basel). 2021;11(9). doi: 10.3390/MEMBRANES11090685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuznetsova IM, Turoverov KK, Uversky VN. What Macromolecular Crowding Can Do to a Protein. Int J Mol Sci 2014, Vol 15, Pages 23090–23140. 2014;15(12):23090–23140. doi: 10.3390/IJMS151223090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonucci A, Ouari O, Guigliarelli B, Belle V, Mileo E. In-Cell EPR: Progress towards Structural Studies Inside Cells. ChemBioChem. 2020;21(4):451–460. doi: 10.1002/CBIC.201900291 [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Mills JD, Periasamy A. Protein localization in living cells and tissues using FRET and FLIM. Differentiation. 2003;71(9–10):528–541. doi: 10.1111/J.1432-0436.2003.07109007.X [DOI] [PubMed] [Google Scholar]
- 32.Luchinat E, Banci L. A unique tool for cellular structural biology: In-cell NMR. J Biol Chem. 2016;291(8):3776–3784. doi: 10.1074/JBC.R115.643247/ATTACHMENT/FBEF8D7A-6A32-42B5-8DEE-BB261961A110/MMC2.PDF [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.