

Abstract
Background
The Human Leukocyte Antigen (HLA) genes are a group of highly polymorphic genes that are located in the Major Histocompatibility Complex (MHC) region on chromosome 6. The HLA genotype affects the presentability of tumour antigens to the immune system. While knowledge of these genotypes is of utmost importance to study differences in immune responses between cancer patients, gold standard, PCR-derived genotypes are rarely available in large Next Generation Sequencing (NGS) datasets. Therefore, a variety of methods for in silico NGS-based HLA genotyping have been developed, bypassing the need to determine these genotypes with separate experiments. However, there is currently no consensus on the best performing tool.
Results
We evaluated 13 MHC class I and/or class II HLA callers that are currently available for free academic use and run on either Whole Exome Sequencing (WES) or RNA sequencing data. Computational resource requirements were highly variable between these tools. Three orthogonal approaches were used to evaluate the accuracy on several large publicly available datasets: a direct benchmark using PCR-derived gold standard HLA calls, a correlation analysis with population-based allele frequencies and an analysis of the concordance between the different tools. The highest MHC-I calling accuracies were found for Optitype (98.0%) and arcasHLA (99.4%) on WES and RNA sequencing data respectively, while for MHC-II HLA-HD was the most accurate tool for both data types (96.2% and 99.4% on WES and RNA data respectively).
Conclusion
The optimal strategy for HLA genotyping from NGS data depends on the availability of either WES or RNA data, the size of the dataset and the available computational resources. If sufficient resources are available, we recommend Optitype and HLA-HD for MHC-I and MHC-II genotype calling respectively.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-023-09351-z.
Keywords: HLA genotyping, Benchmark, Tumour-immune interaction
Background
The human Major Histocompatibility Complex (MHC) is a gene complex located on the p-arm of chromosome 6 that contains two large clusters of genes with antigen processing and presentation functions: the MHC class I and MHC class II regions [1–3].
MHC class I molecules are involved in the presentation of endogenous antigens to cytotoxic T-cells and consist of a heavy chain encoded by one of the MHC class I genes (HLA-A, HLA-B or HLA-C), and a light β2 microglobulin chain [4–6]. Their role in tumour immunity has been established for a long time [7]. Indeed, they can present neoantigens, small mutated peptides, to CD8 + T cells, resulting in an immune response and cancer cell death [8, 9].
The most frequently studied MHC class II genes include HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA1 and HLA-DRB1. They encode alpha/beta heterodimers that form the MHC class II protein complex. The role of these genes in anti-tumour immunity is emerging [10–12]. MHC class II mediated tumour-immune interaction occurs either via an indirect or a direct mechanism. First, cancer cells can secrete neoantigens that are subsequently taken up and presented on the MHC-II of antigen presenting cells infiltrating the tumour [10, 13, 14]. Additionally, some tumours express MHC-II themselves and can directly interact with CD4 + T-cells [10, 14].
The peptide-binding region of HLA molecules is highly polymorphic and specific HLA alleles determine neoantigen binding and presentation to the immune system. Genotype dependent differences in HLA binding affinity could lead to differential responses to immunotherapy, as illustrated by the association that has been described between MHC-I genotypes (e.g., HLA-B62) and survival in immune checkpoint blockade (ICB)-treated advanced melanoma patients [15]. It is currently unclear whether MHC-II genotypes also determine responses to immunotherapy.
Such association studies require knowledge of the HLA genotype. PCR methods are currently the gold standard for this genotyping but datasets with PCR-based HLA calls are rarely available [16–18]. HLA genotyping can also be performed on Next Generation Sequencing (NGS) data. A plethora of tools has been developed for this task. Polysolver and Optitype are often recommended as the best performing tools for MHC-I genotyping [19]. For MHC-II genotyping there is currently no consensus about the best method. Several benchmarks have been performed previously [17, 19–26], but these were either not applied to MHC class II or did not include some recently published tools.
In this study, we compiled a list of 13 tools that predict HLA genotypes from NGS data and benchmarked their performance on both the 1000 Genomes dataset and on an independent cell line dataset [27]. Subsequently, we assessed their performance on 9162 WES and 9761 RNA sequencing files from The Cancer Genome Atlas (TCGA) by comparing the predicted allele frequencies with reference population allele frequencies. Based on these findings, we give recommendations on which tool to use for a given data type and how the outputs of multiple tools can be combined into a consensus prediction.
Results
Selection of 13 HLA genotyping tools with variable computational resource requirements
We identified 22 available HLA genotyping tools from literature (Table 1). Thirteen tools that were free for academic use, applicable on Whole Exome Sequencing (WES), Whole Genome Sequencing (WGS) or RNA-Seq data and ran on Ubuntu 20.04 were included in this study: arcasHLA, HLA-HD, HLA-VBSeq, HLA*LA, HLAforest, HLAminer, HLAscan, Kourami, Optitype, PHLAT, Polysolver, seq2HLA and xHLA (Table S1). These HLA typing algorithms mainly differ in how they map sequencing reads to a panel of reference HLA allele sequences and the strategy they use to subsequently score candidate alleles [28, 29] (Table S2; see Supplementary Note for method-specific details). All 13 tools can make allele predictions for the three MHC class I genes (HLA-A, HLA-B and HLA-C) and 9 tools support additional calling of the MHC class II genes HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1. Two methods support only a subset of the MHC class II genes: xHLA does not support calling HLA-DPA1 and HLA-DQA1, while PHLAT does not support HLA-DPA1 and HLA-DPB1. The tools also differ in which data types they support: 6 of them require WES data, 3 tools require RNA data and the 4 remaining tools support both data types (Table 1).
Table 1.
Overview of evaluated tools for NGS-based HLA genotyping. Checkmarks and crosses indicate which NGS methods (WES and/or RNA-Seq) and input file types (FASTQ and/or BAM) are supported and for which genes predictions can be made. The tools in the upper part of the table are benchmarked in this study. Tools in the lower part of the table did not fulfil our inclusion criteria and were not further considered. * Works preferentially with WGS instead of WES data
| Data type | Input filetype | HLA loci | Version | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WES | RNA | BAM | FASTQ | A | B | C | DPA1 | DPB1 | DQA1 | DQB1 | DRB1 | |||
| Included | arcasHLA [18] | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 0.2.0 |
| HLA-HD [30] | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 1.3.0 | |
| HLA-VBSeq [31] | ✓* | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 2 | |
| HLA*LA [32] | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 1.0.1 | |
| HLAforest [33] | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 1 | |
| HLAminer [34] | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 1.4 | |
| HLAscan [35] | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 2.1.4 | |
| Kourami [36] | ✓* | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 0.9.6 | |
| Optitype [16] | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | 1.3.5 | |
| PHLAT [37] | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | 1.1 | |
| Polysolver [8] | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | 4 | |
| seq2HLA [38] | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | 2.3 | |
| xHLA [39] | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | 0.0.0 | |
| Not included | ALPHLARD-NT [40] | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| ATHLATES [41] | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✓ | ✓ | ||
| HLAProfiler [42] | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| HLAreporter [43] | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| HLAssign [44] | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| OncoHLA [45] | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| PolyPheMe [46] | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✓ | ✓ | ||
| SNP2HLA [47] | ✗ | ✗ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| SOAP-HLA [48] | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
Firstly, the computing time and memory usage of the thirteen selected tools were measured on a random subset of 10 WES and 10 RNA sequencing files from the TCGA project (Fig. 1).
Fig. 1.

Computational resource consumption of the 13 selected tools. (A-B) Boxplots compare the resources needed by the different tools to analyse one sequencing file on a system with a single CPU core. Each tool was applied on WES and/or RNA sequencing files (n = 10), as indicated at the top of the figure. Different tools are represented with a different colour of the boxplot, as indicated in the legend. The y-axes are displayed on a logarithmic scale. (A) Time consumption per sample. (B) Maximal memory consumption per sample
Among the 10 WES-supporting methods Optitype (median 2.48 h) and HLA*LA (median 1.84 h) require the largest computing time. The remaining WES tools take less than 1 h per file, with HLAminer, Kourami and PHLAT being the fastest (97 s, 225 s and 253 s respectively). Apart from being computationally intensive, HLA*LA is also the most memory demanding WES tool (median 36.3 GiB per file). Other WES tools with a median memory consumption higher than 5 GiB are xHLA (median 22.9 GiB), Kourami (median 9.3 GiB) and HLA-HD (median 6.7 GiB). The relatively low memory usage of Polysolver makes it feasible to compensate for its long running time by processing multiple samples in parallel.
Among the 7 RNA-supporting methods, HLA-HD has the longest computing time per sample (median 15.0 h). At the other end of the spectrum, the sole pseudoalignment-based tool arcasHLA takes only 38s per file. The most memory intensive tool is HLA-HD (median memory peaks of 103.1 GiB), followed by Optitype (median 34.1 GiB). The other RNA tools have a memory usage lower than 10 GiB. Remarkably, HLAminer, PHLAT and HLA-HD, which are compatible with both WES and RNA data take a longer time on RNA data (median computing time per sample is 29.4, 8.9, 6.8 times longer for HLA-HD, PHLAT and HLAminer respectively).
HLA*LA and HLA-HD are the best performing MHC class II genotyping tools on WES data
The 10 selected algorithms that are compatible with WES data were benchmarked using data from the 1000 Genomes Project [49] (average HLA gene read depth = 40x +/- 16.7). Predictions were made for HLA-A (n = 1012), HLA-B (n = 1011), HLA-C (n = 1010), HLA-DQB1 (n = 1008), HLA-DRB1 (n = 1000) and HLA-DQA1 (n = 68) (Fig. 2). HLA-DPA1 and HLA-DPB1 were not benchmarked due to the lack of available gold standard calls. For MHC-I genes (HLA-A, HLA-B, HLA-C), the best accuracy was obtained with Optitype (98.0%), followed by Polysolver and HLA*LA (94.9% and 94.4% respectively). For MHC-II genes (HLA-DQA1, HLA-DQB1 and HLA-DRB1), the best allele predictions were made using HLA-HD and HLA*LA (96.2% and 95.7% accuracy respectively). These were the only two methods to reach an accuracy of 90% on all tested MHC-II genes. HLAscan (74.2%), HLA-VBSeq (60.2%) and HLAminer (53.8%) performed considerably worse than the other tools.
Fig. 2.

HLA allele prediction accuracies. Radar plots of HLA allele prediction accuracies on samples from the 1000 Genomes Project. Coloured lines represent different genes, as indicated in the legend below the plots. Corners of the radar plots correspond to the tools that were evaluated for that data type. The Meta tool corresponds to the 4-tool consensus metaclassifier
We observed large variabilities in calling accuracies between MHC class II genes (Fig. 2). Overall, HLA-DQB1 was the hardest MHC-II gene to call. Except for PHLAT, all tools obtained their worst MHC-II call accuracy on this gene. HLA-DQA1, on the other hand, was the gene with the highest calling accuracy for all tools that support it, except for HLAminer and Kourami.
Incorrect calls are either caused by wrong allele calls or a failure to make an allele call. HLA-VBSeq and HLAminer had both a high rate of wrong and failed calls (Figures S1-S2). When HLAscan or Kourami were able to make a call, their predictions were mostly reliable (Figure S1), but these tools regularly failed to produce an output (Figure S2). Miscalled samples had a significantly lower average read depth in the HLA genes than correctly called samples for most tools (Figure S3). Notably, large differences in coverage sensitivity were observed between the different tools, with Kourami and HLA-VBSeq being the most sensitive and Optitype being the least affected (Figure S4). An in silico analysis that simulated the effect of lowering coverage (to 50%, 10%, 5% and 1%) for the best performing tools suggested that the minimal average read depth to get 90% accuracy is 12.2x and 17.4x for MHC-I with Optitype and MHC-II with HLA-HD respectively (Figure S5).
Subsequently, we performed an independent benchmark using the smaller NCI-60 cell line dataset (n = 58, average HLA gene read depth = 37x +/- 25.8), which largely confirmed our results (Figure S6). Additionally, this analysis indicated that the best performing MHC class II supporting tools also performed well on HLA-DPB1.
HLA-HD, PHLAT and arcasHLA are the best performing MHC class II genotyping tools on RNA data
We then evaluated the 7 selected methods that support HLA calling on RNA sequencing data from the 1000 Genomes Project [50] (average HLA gene read depth = 2807x +/- 1300). Predictions were made for HLA-A (n = 373), HLA-B (n = 372), HLA-C (n = 372), HLA-DQB1 (n = 371), HLA-DRB1 (n = 362) and HLA-DQA1 (n = 53) (Fig. 2).
ArcasHLA and Optitype had the best MHC-I allele predictions (99.4% and 99.2% accuracy, respectively), followed by HLA-HD (98.0%), seq2HLA (95.9%) and PHLAT (95.4%). Similar accuracies were found for MHC-II allele predictions, with HLA-HD, PHLAT and arcasHLA performing the best (99.4%, 98.9% and 98.1%, respectively). Contrary to its good prediction of MHC class I alleles, seq2HLA has a lower accuracy for MHC class II (87.8%). RNA-based tools were generally less affected by coverage differences than DNA-based tools, which is likely related to the higher absolute coverage of RNA-Seq as compared to WES data (Figures S3-S5). The high MHC-I accuracies of arcasHLA and Optitype were confirmed on the independent NCI-60 dataset (91.8% and 90.0%, respectively; n = 58, average HLA gene read depth = 578x +/- 837). The accuracy of HLA-HD, PHLAT and seq2HLA was worse on the cell lines than in the benchmark on the 1000 Genomes data (86.6%, 83.3% and 82.3%, respectively). As MHC-II is generally not expressed in cell lines, this benchmark was not performed for those genes.
Correlation and concordance analyses on large independent datasets confirm the benchmarking results
Being one of the few large sequencing datasets for which gold standard HLA genotypes for both MHC classes are available, many algorithms included in our benchmark were developed, optimized and validated using files from the 1000 Genomes Project, introducing a potential bias. Additionally, no evaluation was possible for HLA-DPA1 and HLA-DPB1, due to the lack of gold standard HLA calls. Therefore, we performed an indirect and independent evaluation on a large NGS dataset obtained from TCGA (WES: n = 9162 with an average HLA read depth = 66x +/- 28.6; RNA: n = 9761 with an average HLA read depth = 3076x +/- 2775).
We first compared the observed allele frequencies for each tool with the expected population frequencies. We calculated how often each of the alleles was predicted by a certain tool to obtain an observed allele frequency, stratifying for Caucasian American (n = 7935) and African American (n = 938) ethnicities. By comparing these frequencies to the expected allele frequencies, as derived from Allele Frequency Net [51], strong significant correlations were found for the WES-based tools HLA-HD (minimal Pearson’s r = 0.970; P = 1.5*10− 5), HLA*LA (min. r = 0.968; P = 7.6*10− 5), Optitype (min. r = 0.978; P = 5.5*10− 108), Polysolver (min. r = 0.976; P = 4.7*10− 58) and xHLA (min. r = 0.978; P = 4.4*10− 115) and for the RNA-based tools Optitype (min. r = 0.972; P = 6.2*10− 47), arcasHLA (min. r = 0.939; P = 1.2*10− 19) and PHLAT (min. r = 0.937; P = 2.1*10− 5). The correlations were considerably worse for HLA‑VBSeq (min. r = 0.867; P = 4.1*10− 23), HLAminer (min. r = 0.557; P = 1.2*10− 8 and r = 0.593; P = 6*10− 7, for WES and RNA respectively) and HLAforest (min. r = 0.423; P = 1.8*10− 3) than for the other tools (Fig. 3). These findings largely confirm the results of the benchmark on the 1000 Genomes data. Notably, among the well performing tools, arcasHLA had a worse correlation for HLA-DRB1 in African Americans (r = 0.939; P = 1.2*10− 19), which is mainly due to the discrepancy between the observed and predicted frequency of HLA-DRB1*14:02 in this population (Figure S7).
Fig. 3.

Correlations between observed and expected allele frequencies. Heatmap of correlations between observed allele frequencies and frequencies expected in an African American and in a Caucasian American population. Vertical axis indicates the tools, with different colours representing the data type (WES or RNA) on which the tool was applied. Rows were sorted according to the mean correlation of the tool. Size of the circles indicates the P value of the correlation test as indicated in legend. Absent circles indicate that the tool could not be evaluated on that gene
We then calculated for each pair of tools how often their predictions are concordant (Figures S8-S11). Tools that performed poorly in the previous analyses (e.g., HLAminer, HLA-VBSeq and HLAforest) consistently have a low concordance with all other tools. In contrary, tools that scored high in the previous analyses (such as Optitype, HLA*LA, arcasHLA and HLA-HD) made predictions that are consistent with each other. Noteworthy, this is also the case for HLA-DPA1 and HLA-DPB1, two genes for which no gold standard data was available, suggesting that predictions for these genes are reliable as well.
A consensus metaclassifier improves HLA predictions for WES data
We noted that only for a very small fraction of the samples the genotypes are wrongly typed by all tools simultaneously (median 0.79% for WES and 0.68% for RNA; Figures S12-S13). This complementarity of the tools’ allele predictions opens the possibility to combine predictions of different HLA callers into a consensus prediction. We first applied a majority voting algorithm to the output of all tools, with the predicted allele pair being the one with the most votes. On the WES data, this approach outperforms the predictions of each individual tool for all genes. This is best illustrated by the HLA-DQB1 gene, where the accuracies increased from 93.2% with the best performing tool (HLA*LA) to 96.3% when the voting metaclassifier was used. On RNA data, where the best tools already attain accuracies over 99% by themselves, only minor improvements were made by combining the results (Figure S14).
Based on these results, we determined the minimal number of tools that must be included in the WES-based metaclassifier to produce reliable results (See Methods; Fig. 4). For the WES data, including 4 tools in the model led to a considerable improvement for all genes for both MHC classes. The best accuracies were observed when Optitype, HLA*LA, Kourami and Polysolver were combined for MHC-I predictions (99.0% accuracy) and with HLA*LA, HLA-HD, PHLAT and xHLA for MHC-II predictions (98.4% accuracy). Raising the number of tools further only resulted in marginal gains. Strikingly, the accuracy of the HLA-DQB1 allele predictions even decreases when more tools were included in the model. Therefore, we suggest combining the output of 4 tools for both MHC classes.
Fig. 4.

Accuracies of meta-prediction models with an increasing number of included tools. Tools were added one by one to the consensus metaclassifier model. At each step, the prediction accuracies of the best performing metaclassifier model for a given number of tools were plotted at the top of the figure. Unfilled markers are placed at the smallest number of tools where the maximal accuracy was obtained for that gene. Black lines indicate the average accuracy of the consensus predictions for the two MHC classes (averaged over all genes of that class). The table below the plot indicates which tools were selected in each model for a given number of tools
To evaluate whether the good performance of this approach is generalizable to other datasets, we assessed the correlation between the expected allele frequencies and the allele frequencies observed using the 4-tool WES consensus predictions on the TCGA dataset and compared the results with our previous findings. The allele frequencies predicted by the metaclassifier correlated better with the expected allele frequencies (Fig. 3) than was the case for the individual tools that supported all genes of interest.
Discussion
Rapid technological advancements in NGS have resulted in the generation of numerous publicly available DNA and RNA sequencing datasets. These data have been critical for understanding the genomic basis of human carcinogenesis [52]. In the field of immuno-oncology, genomic data have also been used to study immune selection [53, 54] and, additionally, the availability of corresponding clinical data opens possibilities for studying HLA-dependent cancer susceptibility or even differences in clinical ICB responses between cancer patients [15, 55–57]. However, this requires that the HLA genotype for each subject can be accurately determined. An ever-increasing number of NGS-based HLA typing software applications have been developed. In this study, we benchmarked the performance of 13 publicly available tools. To our knowledge, this is the most extensive benchmark of MHC genotyping tools that has been performed so far (Table S3).
First, we evaluated the tools by comparing their output to genotypes derived from a PCR-based approach. While PCR methods are the gold standard for HLA typing, they have limitations that could lead to ambiguous typing results [58]. Furthermore, inconsistencies have been reported across PCR-based HLA typing datasets that are available for the 1000 Genomes samples [59] which could have affected our benchmarking results. Therefore, we also used 2 other, indirect approaches to assess the performance of the different tools.
Both a concordance analysis between the tools’ predictions and a correlation analysis between predicted and expected allele frequencies confirmed our benchmarking results. To avoid biasing the results of this correlation analysis, we disabled ethnicity-specific allele frequencies for the algorithms that support this (i.e., arcasHLA and Polysolver). However, in the case of arcasHLA, when no specific ethnicity is specified, it uses prior frequencies that depend on the prevalence of the alleles in the entire human population, possibly hindering its ability to call alleles that are uncommon in the specified population. This is illustrated by the worse correlation between observed and expected allele frequencies of arcasHLA for HLA-DRB1 in the African American population, due to an overestimation of the frequency of the rare HLA-DRB1*14:02 allele.
The benchmarking of DNA-based tools was limited to WES data in our study. This likely explains the worse performance and strong coverage sensitivity of both Kourami and HLA-VBSeq, which are algorithms that were primarily developed to be applied on (high-coverage) Whole Genome Sequencing data [36, 60].
We found that Optitype, Polysolver, HLA-HD, HLA*LA and xHLA are all solid choices for WES-based MHC genotyping, while Optitype, HLA-HD, arcasHLA and PHLAT are the better performing tools for RNA data. On the other hand, HLAminer, HLA-VBSeq and HLAscan performed rather poorly in our benchmark. Similar trends were observed in previous independent benchmarking studies [17, 20, 22–26] that focused on a subset of tools and/or genes (Table S3), with the exception of xHLA where we obtained considerably higher accuracies on WES data than reported in a study by Chen et al. [22].
The optimal strategy for HLA genotyping depends on a few factors: the availability of WES or RNA data, the size of the dataset that needs to be analysed and the available computational resources. Additionally, MHC class II typing based on RNA data is only feasible on sequencing data derived from MHC-II expressing cells. For WES data, Optitype and HLA-HD are the best performing individual tools for MHC class I and MHC class II typing, respectively. For RNA data, the same tools are recommended when sufficient computational resources are available. However, the large resource and time consumption of HLA-HD on RNA data makes its usage rather impractical on large datasets. As an alternative, arcasHLA is recommended, which is both the fastest and more accurate tool for RNA that supports all 5 MHC class II genes. Finally, we have demonstrated that the accuracy of the WES-based HLA genotype predictions can be improved further by combining the output of Optitype, HLA*LA, Kourami and Polysolver for MHC-I typing and combining HLA*LA, HLA-HD, PHLAT and xHLA for MHC-II typing using a majority voting rule. The drawback of this metaclassifier approach is that it vastly increases the computational requirements, implying it is only a realistic option if sufficient resources are available or the sample size is relatively small. For RNA data a similar metaclassifier approach did not lead to a further improvement of the prediction accuracies.
Conclusions
Our extensive benchmark demonstrated that the optimal strategy for HLA genotyping from NGS data depends on the availability of either DNA or RNA sequencing data, the size of the dataset and the available computational resources. If sufficient resources are available, we recommend Optitype and HLA-HD for MHC-I and MHC-II genotype calling respectively.
Methods
Selection of tools
A list of existing HLA genotyping tools for NGS data was compiled from literature between October and December 2020. The tools that fulfilled the following criteria were selected for further analysis: the tool should be free for academic use, support WES and/or RNA sequencing data, should not require enrichment of the HLA region before sequencing and should be a Linux command line tool that we could successfully run on our system. When the authors provided instructions on how to update the IPD-IMGT/HLA database used by their tool, this database was updated to version 3.43. This was the case for three tools: HLA-HD, HLAminer and Kourami.
Next-generation sequencing datasets for benchmark
Slices of the 1012 CRAM files of WES data from the 1000 Genomes on GRCh38 dataset [49] that were used for the benchmark on WES data were obtained from the International Genome Sample Resource using the samtools view command (version 1.12). The following contigs were included in the download: the MHC region on the primary assembly (chr6:28,509,970–33,480,727), all 525 contigs starting with HLA- and all unmapped reads. The sliced BAM files for the RNA benchmark were obtained from the Geuvadis [50] RNA-Seq dataset (part of the 1000 Genomes Project) via ArrayExpress (accession number E-GEUV-1). All reads mapped to the MHC region and the unmapped reads were included in the download. Sequencing data from NCI-60 cell lines [24] were obtained from the Sequence Read Archive with accession numbers SRP150855 (WES) [27] and SRP133178 (RNA) [61]. The NCI-60 sequencing data were realigned according to the same alignment pipeline used by the 1000 Genomes on GRCh38 dataset [49]: reads were aligned to the complete GRCh38 reference genome, including alternate (ALT) contigs and HLA sequences, using an alternative scaffold-aware version of BWA-MEM. As done in the same 1000 Genomes alignment pipeline, PCR-introduced duplicates were marked using the markduplicates function in BioBamBam (version 2.0.182). Aligned sequences of Whole Exome Sequencing (WES) and RNA sequencing experiments from The Cancer Genome Atlas (TCGA) were downloaded in BAM format from the Genomic Data Commons (GDC) portal. All 9162 available BAM files of blood-derived normal WES samples were selected. For RNA-Seq, all 9762 RNA-Seq samples that were derived from primary tumours and were aligned using the “STAR 2-Pass” workflow, were selected. Reads mapped to the MHC region of chromosome 6 (chr6:28,509,970–33,480,727) and unmapped reads were extracted from the BAM files and downloaded following the instructions that are described in the GDC API. For the RNA-Seq samples one file failed to download after multiple attempts. The resulting dataset consists of 9162 blood-derived normal WES samples and 9761 primary tumour RNA-Seq samples from 33 available cancer types. The most resource intensive RNA tools were applied on a subset of the TCGA dataset. Optitype was applied on 2226 RNA files, HLAforest on 2900 files and HLA-HD was not applied on the TCGA data.
Calculating the coverage of sequencing data and assessing its influence on accuracy
For all downloaded whole-exome and RNA sequencing files, the average read depth in each of the exons of the HLA genes (HLA-A, HLA-B, HLA-C, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1) was determined using Mosdepth (version 0.2.9) [62]. To assess the influence of coverage on the HLA typing accuracy, we first calculated the average HLA read depth by averaging the read depth in the most polymorphic region of the HLA genes (i.e., the exons encoding the peptide binding region: exons 2 and 3 for MHC-I and exon 2 for MHC-II). The average HLA read depths for genes and samples that were correctly predicted (both alleles correct) were then compared with the average HLA read depths that correspond to incorrect predictions using a Wilcoxon rank sum test. Subsequently, a logistic regression model was fitted that relates the average HLA read depths for a gene and sample with the correctness of the corresponding allele pair prediction. Then, we performed an in silico analysis to simulate the effect of lowering coverage. 100 WES and 100 RNA sequencing files were randomly selected. From each of these files, subsampled BAM files were derived that contain respectively 100%, 50%, 10%, 5%, 1% of the reads of the original file (using the samtools view command, version 1.12). To obtain an absolute read depth for these samples, we multiplied the average HLA read depth by the fraction of the reads that was retained. The minimum read depth required to obtain an accuracy of 90% was then calculated by linearly interpolating the results of this analysis.
Gold standard HLA typing data
Gold standard PCR-based HLA calls for the samples from the 1000 Genomes on GRCh38 dataset were provided by three earlier studies [26–28]. The HLA genotypes from these datasets were merged. Where the calls did not agree, the calls by Gourraud et al. [63] were preferred. For the NCI-60 cell lines, PCR-based HLA genotypes were provided in a study by Adams et al. [29]. For both reference datasets alleles were mapped to the corresponding G-groups, as defined by IPD-IMGT (http://hla.alleles.org/alleles/g_groups.html), and trimmed to second-field resolution.
HLA allele predictions
All 13 selected tools were run on the sliced BAM files following the guidelines of the authors. For tools requiring FASTQ input files, a FASTQ file was extracted from the sliced BAM files using samtools fastq. For HLAscan, which supports input files in either file format, the input was provided in BAM format. For tools that allowed to specify a list of loci that should be called HLA-A, HLA-B, HLA-C, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1 were chosen. Kourami was run with the -a (additional loci) parameter to call the HLA-DPA1 and HLA-DPB1 genes. In rare cases, this led to a crash of the tool and Kourami was run again without the -a parameter. For HLAminer only the HPRA mode was evaluated. xHLA, Polysolver and HLA-VBSeq were not compatible with BAM files that are aligned to a reference genome build that includes ALT contigs. For these tools, an additional realignment step was performed before the tool was executed. Input data for xHLA and Polysolver were realigned to a GRCh38 build that excludes ALT contigs. The input data for HLA-VBSeq was realigned to GRCh37. All allele predictions were mapped to the corresponding G-groups and trimmed at second-field resolution.
Measuring the resource consumption
The running time and memory consumption required by the tools were measured for a random subset of 10 WES and 10 RNA sequencing files from the TCGA project. Each tool was executed in a separate Docker container (version 19.03.3) that was allocated a single CPU core. When the package provided a parameter to specify the number of threads, this was set to 1. Per file, the memory usage of the Docker container was monitored using the docker stats command. The running time was calculated as the time interval between the start and the end of the tool, excluding the time to start the Docker container. Pre-processing steps related to realignment to a different genome build (as required for xHLA, Polysolver and HLA-VBSeq) were not included in the resource consumption assessment. For HLA-HD the analysis of a single sample did not complete successfully as the required amount of memory exceeded what we have available on our system.
Performance metric
For each sample, two allele predictions were made. An allele prediction was labelled “correct” when it was listed as one of the two alleles in the gold standard for that patient. When a tool made a homozygous prediction, while the gold standard was heterozygous, at most one of the two predictions was labelled “correct” for that sample. The accuracy of the predictions is then defined as the proportion of all correctly predicted alleles divided by twice the number of samples. Samples where the gold standard was missing for a particular gene were ignored for that gene.
Population frequency data
Lists of expected HLA allele frequencies for an African American and for a Caucasian American population were constructed based on 18 different studies in the Allele Frequency Net [51] database (Table S4). The studies were selected based on the following criteria. First, we required that the study was conducted on a Black or Caucasoid population from the United States. This was not possible for HLA-DPA1 where no HLA allele frequencies were available for these ethnicities. As a substitute, the allele frequencies of three European populations (French, Swedish and Basques) were used to approximate the allele frequencies for this gene in Caucasian Americans. As a second requirement, the HLA calls should be determined by a PCR-based method. Thirdly, the Allele Frequency Net database should have assigned a gold label (i.e., allele frequency sums to 1, sample size of study > 50, and at least 2-field resolution) to the study for the gene of interest. Lastly, it was required that the subjects included in the selected studies were healthy subjects (i.e., selected for an anthropological study, blood donors, bone marrow registry or controls for a disease study). Allele frequencies from different studies were combined by taking the average frequency, weighted according to the study’s sample size. All alleles were mapped to the corresponding G-groups and trimmed at second-field resolution.
Correlation between expected and observed allele frequencies.
For all tools and for each supported data type, the number of times that each allele was called was counted. This count was divided by the total number of samples to obtain the “observed allele frequency”. The Pearson correlation was calculated between observed allele frequencies and the allele frequencies that were expected based on the Allele Frequency Net database.
Concordance of predictions among different tools
Per gene, the concordance of the predictions between each pair of tools was assessed by counting the number of allele pair predictions made by the first tool that were also made by the second tool (for the same sample and gene). Samples where one of both tools did not make a prediction were not considered. This analysis was performed on the 1000 Genomes and TCGA dataset.
Consensus HLA predictions
A majority voting rule was used to determine the most likely HLA genotype for each sample. For each gene of interest, we selected the pair of alleles that has been predicted the most frequently for that sample (i.e., outputted by the highest number of tools). When ties occurred (i.e., multiple allele pairs had equal numbers of predictions), priority was given to the allele pair that was predicted by the tool with the best individual performance for that gene.
Selecting a minimum number of tools to make consensus HLA predictions
The minimal set of tools that must be included in the majority voting scheme to make reliable consensus predictions was determined using an iterative procedure. Initially, two tools were selected for the model: the tool that performed the best in the benchmark on the 1000 Genomes data and the one that best complements that tool. The latter tool was defined as the tool that most often made a correct prediction (for both alleles) on the samples that were wrongly predicted by the best performing tool. Additional tools were added to this initial model with k = 2 tools in a stepwise manner. At each step, a model with 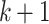 tools was obtained by adding one additional tool to the model with
tools was obtained by adding one additional tool to the model with 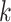 tools. To determine which additional tool would be the most suitable choice, we evaluated all unselected tools and added the tool to the model that led to the largest increase (or the smallest decrease) in accuracy. This procedure was repeated until we obtained a model where all tools were selected.
tools. To determine which additional tool would be the most suitable choice, we evaluated all unselected tools and added the tool to the model that led to the largest increase (or the smallest decrease) in accuracy. This procedure was repeated until we obtained a model where all tools were selected.
Hardware and software environment
Analyses were performed on Ubuntu 20.04 on a Dell EMC PowerEdge R940xa server with 4 Intel Xeon Gold 6240 CPUs (2.60 GHz), each with 18 physical CPU cores, and 376 GiB RAM installed.
Data processing and statistical analysis
Data processing and statistical analyses were performed using R (version 4.0).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. This publication was supported by the University Foundation of Belgium (Universitaire Stichting van België).
Authors’ contributions
J.V.d.E conceptualized the study. A.C., J.S. and P.M. acquired the data and software. A.C., J.S., P.M. and J.V.d.E. performed the analyses. A.C., J.V.d.E. and K.M. contributed to writing and revising the manuscript. All authors read and approved the manuscript.
Funding
This work was supported by the Ghent University Special Research Fund Starting Grant (JVdE, BOF.STG.2019.0073.01, https://www.ugent.be/en/research/funding/bof) and Kom op tegen Kanker (Stand up to Cancer), the Flemish cancer society (AC; STI.VLK.2022.0013.01). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data Availability
The study is based based on publicly available data from the 1000 Genomes Project (https://www.internationalgenome.org/) and The Cancer Genome Atlas (https://www.cancer.gov/ccg/research/genome-sequencing/tcga/using-tcga-data). The code used to produce the results described in this manuscript is available at https://github.com/CCGGlab/mhc_genotyping.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Trowsdale J. Genomic structure and function in the MHC. Trends Genet. 1993;9:117–22. doi: 10.1016/0168-9525(93)90205-V. [DOI] [PubMed] [Google Scholar]
- 2.Beck S, Geraghty D, Inoko H, Rowen L, Aguado B, Bahram S et al. Complete sequence and gene map of a human major histocompatibility complex. Nature 1999 401:6756. 1999;401:921–3. [DOI] [PubMed]
- 3.Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, et al. Gene map of the extended human MHC. Nat Rev Genet. 2004;2004 5:12. doi: 10.1038/nrg1489. [DOI] [PubMed] [Google Scholar]
- 4.Halenius A, Gerke C, Hengel H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: so many targets—but how many arrows in the quiver? Cellular & Molecular Immunology 2015 12:2. 2014;12:139–53. [DOI] [PMC free article] [PubMed]
- 5.Allen RL, Hogan L. Non-classical MHC, class I Molecules (MHC-Ib) eLS. 2013 doi: 10.1002/9780470015902.A0024246. [DOI] [Google Scholar]
- 6.Hewitt EW. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology. 2003;110:163. doi: 10.1046/j.1365-2567.2003.01738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philipps C, McMillan M, Flood PM, Murphy DB, Forman J, Lancki D, et al. Identification of a unique tumor-specific antigen as a novel class I major histocompatibility molecule. Proc Natl Acad Sci U S A. 1985;82:5140–4. doi: 10.1073/pnas.82.15.5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (1979). 2015;348:69–74. [DOI] [PubMed]
- 10.Axelrod ML, Cook RS, Johnson DB, Balko JM. Biological consequences of MHC-II expression by tumor cells in cancer. Clin Cancer Res. 2019;25:2392–402. doi: 10.1158/1078-0432.CCR-18-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696–701. doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Z, Chen F, Meng F, Wei J, Liu B. MHC class II restricted neoantigen: a promising target in tumor immunotherapy. Cancer Lett. 2017;392:17–25. doi: 10.1016/j.canlet.2016.12.039. [DOI] [PubMed] [Google Scholar]
- 13.Corthay A, Skovseth DK, Lundin KU, Røsjø E, Omholt H, Hofgaard PO, et al. Primary antitumor immune response mediated by CD4 + T cells. Immunity. 2005;22:371–83. doi: 10.1016/j.immuni.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Haabeth OAW, Fauskanger M, Manzke M, Lundin KU, Corthay A, Bogen B, et al. CD4 + T-cell–mediated rejection of MHC class II–Positive tumor cells is dependent on Antigen Secretion and Indirect Presentation on host APCs. Cancer Res. 2018;78:4573–85. doi: 10.1158/0008-5472.CAN-17-2426. [DOI] [PubMed] [Google Scholar]
- 15.Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (1979). 2018;359:582–7. [DOI] [PMC free article] [PubMed]
- 16.B AS, C S, M MMS, O K OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–6. doi: 10.1093/bioinformatics/btu548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bauer DC, Zadoorian A, Wilson LOW, Alliance MGH, Thorne NP. Evaluation of computational programs to predict HLA genotypes from genomic sequencing data. Brief Bioinform. 2018;19:179–87. doi: 10.1093/bib/bbw097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orenbuch R, Filip I, Comito D, Shaman J, Pe’Er I, Rabadan R. arcasHLA: high-resolution HLA typing from RNAseq. Bioinformatics. 2020;36:33–40. doi: 10.1093/bioinformatics/btz474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matey-Hernandez ML, Brunak S, Izarzugaza JMG. Benchmarking the HLA typing performance of Polysolver and Optitype in 50 danish parental trios. BMC Bioinformatics. 2018;19:1–12. doi: 10.1186/s12859-018-2239-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee M, Seo JH, Song S, Song IH, Kim SY, Kim YA, et al. A New Human Leukocyte Antigen typing Algorithm Combined with currently available genotyping tools based on next-generation sequencing data and guidelines to select the most likely human leukocyte Antigen genotype. Front Immunol. 2021;12:4080. doi: 10.3389/fimmu.2021.688183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Zhou C, Chen K, Huang B, Liu Q, Ye H. Benchmarking HLA genotyping and clarifying HLA impact on survival in tumor immunotherapy. Mol Oncol. 2021;15:1764–82. doi: 10.1002/1878-0261.12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Madireddi S, Nagarkar D, Migdal M, vander Heiden J, Chang D, et al. In silico tools for accurate HLA and KIR inference from clinical sequencing data empower immunogenetics on individual-patient and population scales. Brief Bioinform. 2021;22:1–11. doi: 10.1093/bib/bbaa223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y, Wang K, Fahira A, Yang Q, Sun R, Li Z, et al. Systematic comparative study of computational methods for HLA typing from next-generation sequencing. HLA. 2021;97:481–92. doi: 10.1111/tan.14244. [DOI] [PubMed] [Google Scholar]
- 24.Kiyotani K, Mai TH, Nakamura Y. Comparison of exome-based HLA class I genotyping tools: identification of platform-specific genotyping errors. J Hum Genet 2017. 2016;62:3. doi: 10.1038/jhg.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu P, Yao M, Gong Y, Song Y, Chen Y, Ye Y, et al. Benchmarking the Human Leukocyte Antigen typing performance of three assays and seven next-generation sequencing-based algorithms. Front Immunol. 2021;12:840. doi: 10.3389/fimmu.2021.652258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yi J, Chen L, Xiao Y, Zhao Z, Su X. Investigations of sequencing data and sample type on HLA class Ia typing with different computational tools. Brief Bioinform. 2021;22:1–6. doi: 10.1093/bib/bbaa143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–82. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai Y, Wang D, Fury W. PHLAT: inference of high-resolution HLA types from RNA and whole exome sequencing. Methods Mol Biol. 2018;1802:193–201. doi: 10.1007/978-1-4939-8546-3_13. [DOI] [PubMed] [Google Scholar]
- 29.Klasberg S, Surendranath V, Lange V, Schöfl G. Bioinformatics Strategies, Challenges, and Opportunities for Next Generation sequencing-based HLA genotyping. Transfus Med Hemotherapy. 2019;46:312–25. doi: 10.1159/000502487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawaguchi S, Higasa K, Shimizu M, Yamada R, Matsuda F. HLA-HD: an accurate HLA typing algorithm for next-generation sequencing data. Hum Mutat. 2017;38:788–97. doi: 10.1002/humu.23230. [DOI] [PubMed] [Google Scholar]
- 31.Wang YY, Mimori T, Khor SS, Gervais O, Kawai Y, Hitomi Y, et al. HLA-VBSeq v2: improved HLA calling accuracy with full-length japanese class-I panel. Hum Genome Variation 2019. 2019;6(1):1–5. doi: 10.1038/s41439-019-0061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dilthey AT, Mentzer AJ, Carapito R, Cutland C, Cereb N, Madhi SA, et al. HLA*LA—HLA typing from linearly projected graph alignments. Bioinformatics. 2019;35:4394–6. doi: 10.1093/bioinformatics/btz235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HJ, Pourmand N. HLA Haplotyping from RNA-seq data using Hierarchical Read Weighting. PLoS ONE. 2013;8:e67885. doi: 10.1371/journal.pone.0067885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warren RL, Choe G, Freeman DJ, Castellarin M, Munro S, Moore R, et al. Derivation of HLA types from shotgun sequence datasets. Genome Med. 2012;4:1–8. doi: 10.1186/gm396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ka S, Lee S, Hong J, Cho Y, Sung J, Kim HN, et al. HLAscan: genotyping of the HLA region using next-generation sequencing data. BMC Bioinformatics. 2017;18:1–11. doi: 10.1186/s12859-017-1671-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H, Kingsford C, Kourami Graph-guided assembly for novel human leukocyte antigen allele discovery. Genome Biol. 2018;19:1–16. doi: 10.1186/s13059-018-1388-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bai Y, Ni M, Cooper B, Wei Y, Fury W. Inference of high resolution HLA types using genome-wide RNA or DNA sequencing reads. BMC Genomics. 2014;15:1–16. doi: 10.1186/1471-2164-15-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boegel S, Löwer M, Schäfer M, Bukur T, de Graaf J, Boisguérin V, et al. HLA typing from RNA-Seq sequence reads. Genome Med. 2012;4:1–12. doi: 10.1186/gm403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie C, Yeo ZX, Wong M, Piper J, Long T, Kirkness EF et al. Fast and accurate HLA typing from short-read next-generation sequence data with xHLA. Proceedings of the National Academy of Sciences. 2017;114:8059–64. [DOI] [PMC free article] [PubMed]
- 40.HayashiShuto MoriyamaTakuya, YamaguchiRui MizunoShinichi. KomuraMitsuhiro, MiyanoSatoru, ALPHLARD-NT: Bayesian Method for Human Leukocyte Antigen Genotyping and Mutation Calling through Simultaneous Analysis of Normal and Tumor Whole-Genome Sequence Data. https://home.liebertpub.com/cmb. 2019;26:923–37. [DOI] [PMC free article] [PubMed]
- 41.Liu C, Yang X, Duffy B, Mohanakumar T, Mitra RD, Zody MC, et al. ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res. 2013;41:e142–2. doi: 10.1093/nar/gkt481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchkovich ML, Brown CC, Robasky K, Chai S, Westfall S, Vincent BG, et al. HLAProfiler utilizes k-mer profiles to improve HLA calling accuracy for rare and common alleles in RNA-seq data. Genome Med. 2017;9:1–15. doi: 10.1186/s13073-017-0473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Y, Yang J, Ying D, Zhang Y, Shotelersuk V, Hirankarn N, et al. HLAreporter: a tool for HLA typing from next generation sequencing data. Genome Med. 2015;7:1–12. doi: 10.1186/s13073-015-0145-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wittig M, Anmarkrud JA, Kässens JC, Koch S, Forster M, Ellinghaus E, et al. Development of a high-resolution NGS-based HLA-typing and analysis pipeline. Nucleic Acids Res. 2015;43:e70. doi: 10.1093/nar/gkv184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sverchkova A, Anzar I, Stratford R, Clancy T. Improved HLA typing of class I and Class II alleles from next-generation sequencing data. HLA. 2019;94:504–13. doi: 10.1111/tan.13685. [DOI] [PubMed] [Google Scholar]
- 46.Abi-Rached L, Gouret P, Yeh JH, di Cristofaro J, Pontarotti P, Picard C, et al. Immune diversity sheds light on missing variation in worldwide genetic diversity panels. PLoS ONE. 2018;13:e0206512. doi: 10.1371/journal.pone.0206512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE. 2013;8:e64683. doi: 10.1371/journal.pone.0064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao H, Wu J, Wang Y, Jiang H, Zhang T, Liu X et al. An Integrated Tool to Study MHC Region: Accurate SNV Detection and HLA Genes Typing in Human MHC Region Using Targeted High-Throughput Sequencing. PLoS ONE. 2013;8. [DOI] [PMC free article] [PubMed]
- 49.Zheng-Bradley X, Streeter I, Fairley S, Richardson D, Clarke L, Flicek P, et al. Alignment of 1000 Genomes Project reads to reference assembly GRCh38. Gigascience. 2017;6:1. doi: 10.1093/gigascience/gix038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lappalainen T, Sammeth M, Friedländer MR, Hoen ’T, Monlong PAC, Rivas J et al. MA,. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013 501:7468. 2013;501:506–11. [DOI] [PMC free article] [PubMed]
- 51.Gonzalez-Galarza FF, McCabe A, Santos EJM dos, Jones J, Takeshita L, Ortega-Rivera ND et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2020;48:D783–8. [DOI] [PMC free article] [PubMed]
- 52.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science (1979). 2013;340:1546–58. [DOI] [PMC free article] [PubMed]
- 53.Claeys A, Luijts T, Marchal K, van den Eynden J. Low immunogenicity of common cancer hot spot mutations resulting in false immunogenic selection signals. PLoS Genet. 2021;17:e1009368. doi: 10.1371/journal.pgen.1009368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van den Eynden J, Jiménez-Sánchez A, Miller ML, Larsson E. Lack of detectable neoantigen depletion signals in the untreated cancer genome. Nat Genet. 2019;51:1741–8. doi: 10.1038/s41588-019-0532-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 2019. 2019;25:12. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naranbhai V, Viard M, Dean M, Groha S, Braun DA, Labaki C, et al. HLA-A*03 and response to immune checkpoint blockade in cancer: an epidemiological biomarker study. Lancet Oncol. 2022;23:172–84. doi: 10.1016/S1470-2045(21)00582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adams SD, Barracchini KC, Chen D, Robbins F, Wang L, Larsen P et al. Ambiguous allele combinations in HLA Class I and Class II sequence-based typing: when precise nucleotide sequencing leads to imprecise allele identification. 2004. 10.1186/1479-5876-2-30. [DOI] [PMC free article] [PubMed]
- 59.Bauer-Mehren A, Bundschus M, Rautschka M, Mayer MA, Sanz F, Furlong LI. Gene-disease network analysis reveals functional modules in mendelian, complex and environmental diseases. PLoS ONE. 2011;6:e20284. doi: 10.1371/journal.pone.0020284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nariai N, Kojima K, Saito S, Mimori T, Sato Y, Kawai Y, et al. HLA-VBSeq: Accurate HLA typing at full resolution from whole-genome sequencing data. BMC Genomics. 2015;16:1–6. doi: 10.1186/1471-2164-16-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reinhold WC, Varma S, Sunshine M, Elloumi F, Ofori-Atta K, Lee S, et al. RNA sequencing of the NCI-60: integration into CellMiner and CellMiner CDB. Cancer Res. 2019;79:3514–24. doi: 10.1158/0008-5472.CAN-18-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pedersen BS, Quinlan AR. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2018;34:867–8. doi: 10.1093/bioinformatics/btx699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gourraud PA, Khankhanian P, Cereb N, Yang SY, Feolo M, Maiers M, et al. HLA diversity in the 1000 genomes dataset. PLoS ONE. 2014;9:e97282. doi: 10.1371/journal.pone.0097282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adams S, Robbins FM, Chen D, Wagage D, Holbeck SL, Morse HC, et al. HLA class I and II genotype of the NCI-60 cell lines. J Transl Med. 2005;3:11. doi: 10.1186/1479-5876-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study is based based on publicly available data from the 1000 Genomes Project (https://www.internationalgenome.org/) and The Cancer Genome Atlas (https://www.cancer.gov/ccg/research/genome-sequencing/tcga/using-tcga-data). The code used to produce the results described in this manuscript is available at https://github.com/CCGGlab/mhc_genotyping.