

Abstract
The neural crest is a migratory stem cell population that contributes to various tissues and organs during vertebrate embryonic development. These cells possess remarkable developmental plasticity and give rise to many different cell types, including chondrocytes, osteocytes, peripheral neurons, glia, melanocytes, and smooth muscle cells. Although the genetic mechanisms underlying neural crest development have been extensively studied, many facets of this process remain unexplored. One key aspect of cellular physiology that has gained prominence in the context of embryonic development is metabolic regulation. Recent discoveries in neural crest biology suggest that metabolic regulation may play a central role in the formation, migration, and differentiation of these cells. This possibility is further supported by clinical studies that have demonstrated a high prevalence of neural crest anomalies in babies with congenital metabolic disorders. Here, we examine why neural crest development is prone to metabolic disruption and discuss how carbon metabolism regulates developmental processes like epithelial-to-mesenchymal transition (EMT) and cell migration. Finally, we explore how understanding neural crest metabolism may inform upon the etiology of several congenital birth defects.
Introduction
The neural crest is a migratory stem cell population that contributes to multiple components of the vertebrate body plan, including the craniofacial skeleton, the peripheral and enteric nervous systems, and the pigmentation of the skin (Le Douarin and Kalcheim, 1999). During neurulation, neural crest cells delaminate from the dorsal neural tube (Fig 1) and migrate along stereotypical pathways. Upon reaching the appropriate destinations in the embryo, these cells differentiate into more than thirty distinct cell types such as chondrocytes, melanocytes, neurons, etc. (Bronner and Simoes- Costa, 2016). The neural crest has served as model for the study of multipotency and migration for many decades, and interest in this cell population has also been fueled by its medical importance. Many congenital disabilities, broadly termed as neurocristopathies, result from defects in the formation of the neural crest and its derivatives (Vega-Lopez et al., 2018). While some of these conditions are linked to genetic mutations, they can also be caused by environmental stimuli. The high prevalence of neurocristopathies underscores the complexity of neural crest development and its reliance on the coordination of multiple cellular processes (Vega-Lopez et al., 2018).
Figure 1. Neural crest development and the genesis of metabolic neurocristopathies.
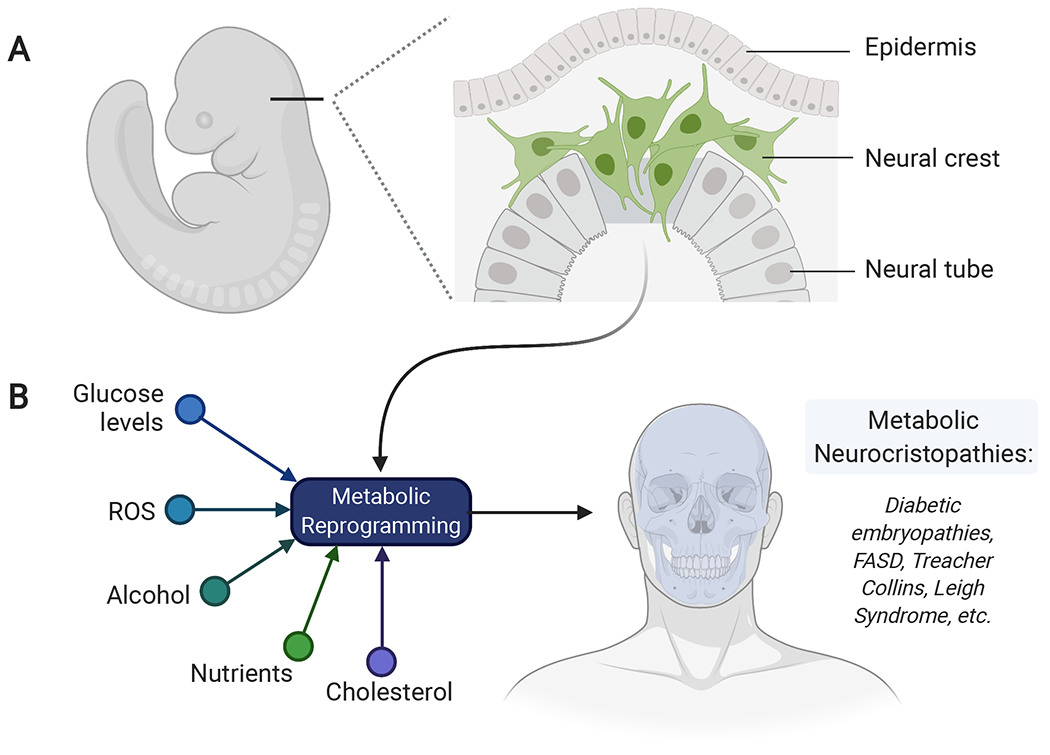
(A) The neural crest is migratory stem cell population that delaminates from the dorsal neural tube and migrates extensively throughout the embryo. Neural crest development requires drastic changes in cellular metabolism for the proper migration and differentiation of these cells. (B) Environmental or genetic factors may disrupt the metabolic transitions observed in neural crest cells, resulting in congenital disorders known as metabolic neurocristopathies. FASD: Fetal alcohol spectrum disorder, ROS: Reactive oxygen species.
The different steps in neural crest formation, which include induction, specification, migration, and differentiation, are governed by an expansive gene regulatory network (GRN) (Simoes-Costa and Bronner, 2015). The neural crest GRN is composed of a large number of genetic interactions between signaling systems, transcription factors, and epigenomic regulators (Hovland et al., 2020; Hu et al., 2014). In the past years, studies in multiple model organisms have allowed us to expand the network’s architecture and gain insight into its logic. Yet, we still have a superficial understanding of how the genetic programs that control cellular behaviors and developmental transitions are coupled with cellular physiology. The complex tasks performed by neural crest cells during embryonic development require the coordination of a number of biochemical processes beyond transcriptional control. One facet of neural crest development that remains relatively unexplored is its metabolic regulation, which recently has taken center stage in many cellular and developmental contexts.
For much of the last century, studies on the links between metabolism and cellular behaviors were bioenergetic in nature, focusing on metabolic changes as a response to cellular needs. An example of this is the Warburg effect (WE), in which cancer cells transition to glycolysis, even in high oxygen environments, to meet the macromolecule demands of persistent proliferation (Hsu and Sabatini, 2008). More recently, numerous studies have suggested that separate from its role in ATP production, metabolism broadly functions as a signaling hub that impinges on other signal transduction pathways as well as on the epigenome and the proteome, to regulate key cellular functions and behaviors (Miyazawa and Aulehla, 2018). Consistent with this idea, we have recently demonstrated that neural crest cells display WE and that this metabolic adaptation is necessary for EMT and migration (Bhattacharya et al., 2020). The importance of this metabolic adaption in neural crest development is further underscored by the etiology of many neurocristopathies. Metabolic syndromes or dietary changes that interfere with glucose metabolism often result in defects in neural crest development (Berio, 2011; Chappell et al., 2009; Smith et al., 2014). Disruption of other bioenergetic pathways such as lipid metabolism and folate synthesis also has shown to have severe consequences on the formation of this stem cell population (See Box 1). This led us to postulate that metabolic regulation is essential for neural crest specification, migration and differentiation by any combination of the following: protecting neural crest cells against oxidative stress, coupling neural crest behavior and physiology, regulating cellular pH, and rapidly producing ATP. In this review, we discuss the diverse roles that carbon metabolism plays in stem cells and throughout embryogenesis, with an emphasis on the neural crest. We also examine why neural crest development is particularly prone to disruptions in metabolism and how these disruptions may result in congenital birth defects (Fig. 1).
Box 1. Other metabolic pathways and metabolites important for neural crest formation.
Apart from carbohydrate metabolism, several other accessory metabolic pathways have essential roles in neural crest development. One such process is the folic acid synthesis pathway, which has been extensively studied in the context of early vertebrate embryogenesis. Folate deficiency during embryogenesis severely affects craniofacial development and causes many of the same phenotypes associated with the neurocristopathies described below. However, only recently have we been able to determine the molecular basis of these conditions. At least three distinct mechanisms have been proposed by which folic acid regulates orofacial development. Firstly, decreased folic acid disrupts methionine metabolism and causes cellular accumulation of the metabolite homocysteine (Boot et al., 2003; Melo et al., 2017). In avian and rat neural tube explants, excess homocysteine promotes neural crest migration but prevents differentiation, particularly into mesenchymal lineages (Boot et al., 2003; Melo et al., 2017). This defect can be fully rescued by adding folic acid to culture media, indicating that the regulation of homocysteine levels is an important function of folate in neural crest cells (Boot et al., 2003).
Secondly, during differentiation, folate metabolism is required for the survival of the neural crest-derived mesenchymal cells. A study by Wahl et al. showed that inhibition of the dihydrofolate reductase enzyme (DHFR) required for folate synthesis, caused increased apoptosis and decreased proliferation of cells in the facial prominences and the developing jaw of the Xenopus embryo (Wahl et al., 2015). The authors further uncovered a novel collaboration between folate and the Retinoic Acid (RA) signaling pathway in neural crest cells, wherein folic acid can rescue the craniofacial defects observed upon RA deficiency in embryos, and the inhibition of DHFR exacerbates the phenotype of RA signaling disruption (Wahl et al., 2015). Whether the folate and RA pathways directly regulate each other or function in parallel remains unresolved. However, this study provides a clinically relevant example of the synergistic interactions between different metabolites that control organ development.
Lastly, being a precursor of S-adenosylmethionine (SAM), folate can control the epigenetic landscape of neural crest cells by impacting histone and DNA methylation. A recent report by Jimenez et al. showed that loss of the neural crest-specific folate transporters, FOLR1 and RFC1, confers a neuronal fate to dorsal neural tube progenitor cells in avian embryos (Jimenez et al., 2018). This is in part due to reduced methylation and de-repression of the neuronal SOX2 gene locus in the neural crest territory, which causes the expansion of the neural plate fate to the dorsal extremities (Jimenez et al., 2018). Taken together, these mechanisms reveal the requirement of folate at every step of neural crest development: from specification to differentiation, and provide clues to how this metabolic pathway is adapted to perform cell specific functions.
Another metabolite that is crucial to neural crest formation is cholesterol. Cholesterol is the precursor of all steroid hormones and also plays a critical role during embryogenesis by moderating the Sonic Hedgehog Signaling (Shh) pathway (Lewis et al., 2001; Riobo, 2012). In humans, mutations in enzymes associated with cholesterol transport and biosynthesis cause embryonic defects in neural tube and limb formation and affect neural crest-derived structures such as the facial mesenchyme and cardiac outflow tract (Porter and Herman, 2011). These defects have been primarily attributed to disrupted Shh signaling, which is indispensable for the development of these affected tissues. In the neural crest, Shh signaling is specifically required for the differentiation of progenitor cells into skeletal and non-skeletal mesenchymal cells that form facial structures and the branchial arches (Quintana et al., 2017). Interestingly, despite its important function in neural tube development, Shh primarily promotes mesenchymal fate in neural crest cells and is not necessary for neuronal differentiation. Additionally, Shh signaling functions to pattern the neural crest-derived cardiac outflow tract and is generally required for the survival and proliferation of cranial and cardiac neural crest cells. Indeed, disruption of cholesterol metabolism does not have overt effects on derivates of trunk neural crest cells, suggesting that the relevance of this pathway maybe axial-specific.
Given the ubiquity of cholesterol metabolism, it is important to consider how the tissue/axial-specific function of this pathway is brought about in neural crest cells. A study by Iwata et al. implicates the cranial specific TGF-β signaling system in control of lipid metabolism in neural crest-derived mesenchyme (Iwata et al., 2014). Murine mutants of TGF-β receptors display facial dysmorphisms similar to that observed upon inhibition of cholesterol biosynthesis. By further characterizing palatal mesenchyme cells obtained from these mutants, the authors uncovered that inhibition of TGF-β signaling prevents lipolysis and subsequent lipid turnover in these cells. This, in turn compromises Shh signaling, resulting in decreased cell proliferation and apoptosis. However, these cellular defects can be rescued by administration of Shh protein, confirming that this signaling pathway is epistatic to TGF-β in neural crest-derived facial mesenchyme (Iwata et al., 2014). Together with the studies discussed above, these findings provide an important clue to the function of lipid/cholesterol metabolism as a pivotal node that links distinct signaling pathways to mediate tissue-specific functions in neural crest cells.
Metabolic regulation of neural crest development
Central carbon metabolism, which breaks down glucose, is the primary ATP production mechanism in all living cells. It comprises three sequential pathways: Glycolysis, the Krebs Cycle, and the Electron Transport Chain (ETC) through which, in the presence of oxygen, glucose is completely oxidized to produce CO2, H2O, and 38 ATP molecules (Noor et al., 2010). This combined process is referred to as Oxidative Phosphorylation (OXPHOS) and is the default cellular respiration mechanism for somatic cells under aerobic conditions (Gautheron, 1984; Noor et al., 2010) (Fig. 2). Additionally, glucose can also be incompletely oxidized through glycolysis to produce lactate, which does not enter the subsequent pathways. This process is independent of oxygen availability and is known as aerobic glycolysis when occurring under normoxic conditions (Lunt and Vander Heiden, 2011; Noor et al., 2010) (Fig. 2b). Aerobic glycolysis results in the net gain of only 2 ATP per glucose molecule, and thus increased flux through this pathway necessitates high glucose uptake to meet cellular energy demands. Though not mutually exclusive, aerobic glycolysis and OXPHOS are antagonistic mechanisms of glucose breakdown. The balance between these pathways has been studied extensively in various tissues and distinct biological contexts (Ito and Suda, 2014; Jose et al., 2011; Shyh-Chang and Ng, 2017; Zheng, 2012). In general, most differentiated cells display OXPHOS, while proliferative cells such as stem cells and tumor cells preferentially utilize aerobic glycolysis (Fig 1). The benefits and drawbacks of using one process versus the other are cell type-specific and not only reflect metabolic regulation but also affect cell identity and behavior.
Figure 2. Glucose metabolism in differentiated versus tumor and stem cells.

(A) Oxidative Phosphorylation (OXPHOS) is the default cellular respiration mechanism for somatic cells under aerobic conditions. It results in 38 molecules of ATP per molecule of glucose. In the absence of oxygen, these cells engage in glycolysis to produce only 4 molecules of ATP. (B) Cancer and stem cell populations like the neural crest are highly glycolytic, even in the presence of oxygen. These cells display a metabolic adaptation known as the Warburg Effect and produce approximately 4 molecules of ATP per molecule of glucose.
Congenital diseases caused by disrupted glucose metabolism are often associated with severe defects in neural crest-derived tissues, indicating that this metabolic pathway has a lineage-specific role in neural crest formation (Chappell et al., 2009; Smith et al., 2014). Furthermore, as discussed below, early neural crest cells are susceptible to oxidative stress, a byproduct of excessive OXPHOS, suggesting that a balance between the distinct modes of glucose metabolism may be critical for proper development of the cell type. Despite this, the dynamics of carbon metabolism and its broader functions in neural crest cells have not been resolved. In the 1940s, a study measuring cellular respiration in amphibian neural tube explants reported that, despite aerobic culture conditions, delaminating and migratory neural crest cells display low oxygen uptake rates and that oxygen consumption increases significantly only when the cells begin to differentiate (Flickinger, 1949). This observation was further supported by reports showing that activity of the critical OXPHOS enzyme cytochrome C oxidase is low in undifferentiated migratory quail neural crest cells but is elevated in differentiating cells, particularly in those committed to neuronal lineages (Liu et al., 1990). Additionally, recent studies in murine embryos have revealed that tissue-specific deletion of the mTOR kinase, a critical upstream regulator of OXPHOS, results in apoptosis of only post- migratory neural crest cells (Nie et al., 2018). These observations indicate that OXPHOS plays a more prominent role in later neural crest development.
These findings raised the question of what is the metabolic state of early neural crest cells. Early on, it was hypothesized that undifferentiated neural crest cells maintain a basal state of metabolism (low OXPHOS) and only become energetic (high OXPHOS) during differentiation (Flickinger, 1949, Liu et al.,1990). However, studies from our group and others have recently uncovered that migratory neural crest cells are energetic and that their primary mode of glucose metabolism is aerobic glycolysis (Bhattacharya et al., 2020; Keuls et al., 2020). This discovery underscores a novel feature of neural crest metabolism that has so far been underappreciated. Below, we have described how this metabolic state is specifically regulated in neural crest cells and its possible impacts on the biology of this progenitor population.
Neural crest and aerobic glycolysis
Elevated flux of a metabolic pathway is usually associated with enhanced expression of its rate-limiting enzymes. We thus utilized a time-course series of RNA-seq data in chick neural crest cells to assess the transcriptional dynamics of enzymes involved in glucose breakdown. This analysis revealed that, before undergoing EMT, cranial neural crest cells specifically upregulate the expression of several glycolytic genes, including PFKP, GAPDH, and LDHA. Consistent with these findings, pre-migratory chick neural crest cells display hallmarks of aerobic glycolysis, including increased glucose uptake, lactate production, and low rates of oxygen consumption. Remarkably, pharmacological inhibition of glycolysis, but not OXPHOS, disrupted EMT and inhibited neural crest migration (Bhattacharya et al., 2020).
By examining the mechanism underlying this phenotype, we uncovered that aerobic glycolysis promotes neural crest EMT by activating the YAP/TEAD pathway, a known regulator of cell migration and invasion (Plouffe et al., 2015) (Fig. 3). In neural crest cells, WE stabilizes the interaction between the transcription factor TEAD1 and its activator YAP1, thus promoting the formation of a functional complex that can activate the transcription of downstream target genes. By profiling the genomic occupancy of the active form of YAP1, we identified several direct targets of YAP/TEAD signaling in neural crest cells, which included EMT factors such as SOX9, ZEB2, ETS1, TWIST1 and PRRX2 (Bhattacharya et al., 2020), as well as several genes involved in cell migration and extracellular matrix remodeling (Fig. 3) (Simoes-Costa and Bronner, 2015). Our work directly implicates carbon metabolism in neural crest development for the first time and highlights how bioenergetic shifts impact GRNs to control cell identity and behavior in vivo (Bhattacharya et al., 2020). Notably, another recent study reported that in murine cranial neural crest cells, the switch from the pre-migratory to the migratory state is also accompanied by an increase in glycolytic gene expression and a decrease in the transcription of OXPHOS enzymes (Keuls et al., 2020). These observations support our findings in the chick embryo and suggest that modulation of glucose breakdown is an evolutionarily conserved mechanism of controlling neural crest migration.
Figure 3. Control of neural crest delamination and migration by the Warburg effect.
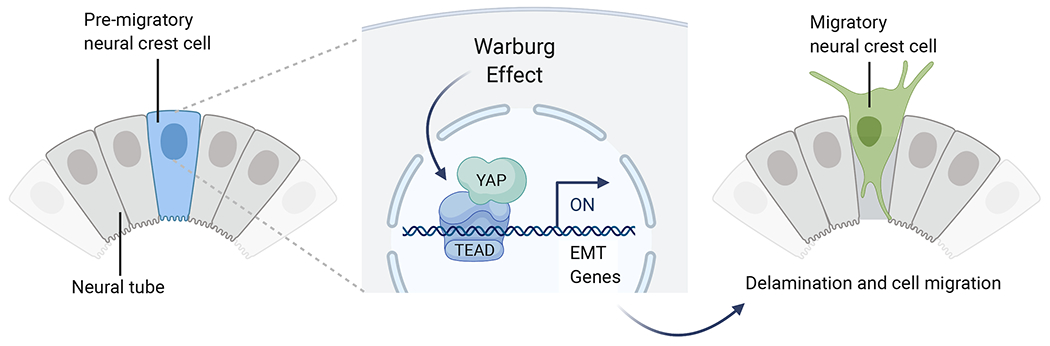
High glycolytic flux in the pre-migratory neural crest promotes the interaction of YAP and TEAD, leading to the activation of genes that promote epithelial to mesenchymal transition (EMT). TAP/TEAD directly interact with tissue-specific enhancers to promote the EMT regulatory program (Adapted from Bhattacharya et al., 2020).
Potential functions of aerobic glycolysis in migratory neural crest cells
Our study indicates that WE promotes the activation of the YAP/TEAD pathway, promoting EMT and cell migration in neural crest cells. However, given the pleiotropic function of this metabolic adaptation in different contexts, it is likely WE impacts other aspects of neural crest physiology. One such role of WE could be regulating Reactive Oxygen Species (ROS) levels in the migrating cells. Not only ROS can function to mediate cellular signaling (Finkel, 2011), but neural crest cells are especially sensitive to ROS, which can cause oxidative stress and severely affect the development of this cell type (Laforgia et al., 2018). In respiring cells, most ROS are generated as byproducts of mitochondrial electron transport (Murphy, 2009). However, WE can significantly reduce cellular ROS production by decreasing reliance on mitochondrial respiration (Brand and Hermfisse, 1997; Mullarky and Cantley, 2015). Additionally, pathways promoting redox homeostasis, such as the pentose phosphate pathway (PPP), are often upregulated in cells undergoing aerobic glycolysis. Increased flux through PPP produces the reducing intermediate NADPH, which alongside glutathione, function to quench cellular ROS (Cho et al., 2018; Patra and Hay, 2014). Downstream of WE, these mechanisms may also be essential for preventing oxidative stress in migratory neural crest cells.
Another function of WE could be modulating extracellular matrix proteins to make the microenvironment conducive to neural crest migration. In metastatic tumors, glycolytic cells secrete excess lactate, which renders the environment acidic (Estrella et al., 2013). This shift in pH activates metalloproteases and cathepsins and promotes the degradation of extracellular matrix proteins to facilitate cell migration (Li et al., 2016). Furthermore, metabolic intermediates of aerobic glycolysis and enzymes of this pathway can function as cytokines that promote cell invasion through autocrine signaling (Kathagen- Buhmann et al., 2018). Neural crest migration and cancer invasion implicate many of the same proteases and chemokine signaling pathways ((Acloque et al., 2009)). Thus, in principle, WE could function through some or all of the above mechanisms to promote neural crest migration.
Apart from their canonical function in glucose metabolism, glycolytic enzymes can also directly control cellular signaling systems. A recent study by Figueiredo et al. reported that the enzyme PFKFB4 is an important regulator of the AKT pathway in neural crest cells of Xenopus embryos. By performing stage-specific knockdown of the enzyme, the authors were able to show that PFKFB4 function is essential both for specification and migration of neural crest cells. Interestingly, the role of this protein during specification was found to be independent of glycolysis and is mediated solely through the activation of the AKT pathway. However, during neural crest migration, PFKFB4 function was dependent both on glycolysis and AKT signaling (Figueiredo et al., 2017). This hints at yet another potential mechanism by which glucose metabolism co-operates with a signaling system to drive neural crest migration. As discussed below, AKT as well as the broader FGF pathway has been implicated in the regulation of WE in other developmental contexts. Thus, it is possible that PFKFB4 mediated activation of AKT pathway during neural crest specification may play a causal role in the upregulation of glycolysis during neural crest migration.
Lastly, aerobic glycolysis may also affect the epigenetic landscape of neural crest cells. As discussed below, a link between glycolytic metabolism and histone acetylation is well documented (Cluntun et al., 2015; Liu et al., 2015; Lu and Thompson, 2012). Removal of glucose causes a global reduction in chromatin acetylation, decreasing the transcription of several genes, including those of glycolytic enzymes. Conversely, histone deacetylation is also sensitive to cellular glycolytic flux (Wellen and Thompson, 2012). The activity of histone deacetylases, such as sirtuins, is modulated by the cytoplasmic NAD+/NADH ratio, an increase in which accentuates these enzymes’ function (Cluntun et al., 2015; Wellen and Thompson, 2012). Importantly, delaminating neural crest cells also display a high NAD+/NADH ratio (Bhattacharya et al., 2020), indicating that aerobic glycolysis may be essential for maintaining a balance between histone acetylation and deacetylation in this progenitor population. Additionally, a recent study reported the association of WE with another novel epigenetic modification: lactate-derived lactylation of histone lysine residues (Zhang et al., 2019). The authors discovered that lactylation is a glycolysis-regulated widespread histone mark that stimulates gene expression. Histone lactylation is, in fact, more specific than acetylation and is only observed in cells displaying high glycolytic flux (Zhang et al., 2019). Thus, the identification of this epigenetic mark provides another clue regarding the non-metabolic functions of WE, which could be relevant to neural crest biology. In summary, a number of developmental processes intrinsic to neural crest cells may be facilitated by WE. We hypothesize that, in coordination with its neural crest specific role as a regulator of the YAP/TEAD pathway, these downstream functions of WE ensure that the neural crest EMT program is initiated only in metabolically primed cells that are equipped to modify the microenvironment.
Warburg Effect in other developmental contexts
While the physiological role of WE remains controversial, its proposed functions in cancer cells include rapid ATP production, promoting flux through biosynthetic pathways, modulating the tumor microenvironment, and impacting cellular signaling through ROS and chromatin modulation (Liberti and Locasale, 2016). By regulating these distinct aspects of cellular physiology, WE promotes cell survival, proliferation, and migration, making this phenomenon indispensable for tumor growth and metastasis (Liberti and Locasale, 2016). Extensive cell proliferation and migration also underlie embryonic morphogenesis, which suggested to researchers early on that WE may be similarly relevant during normal development (Gardner and Leese, 1987, 1990). The concept of WE as a developmental phenomenon has gained traction and studies over the past decade have provided elegant examples of how aerobic glycolysis regulates cell identity in developmental systems. These findings summarized below, indicate that WE is a tightly-regulated developmental mechanism rather than an anomaly of cancer cells.
Lessons in carbon metabolism from embryonic stem cells
The most well-studied function of aerobic glycolysis in development is its critical role in regulating pluripotency and proliferation of Embryonic Stem Cells (ESCs) (Ito and Suda, 2014; Shyh-Chang and Ng, 2017). Like cancer cells, ESCs in culture proliferate rapidly and have shorter cell cycle than differentiated cells, prompting researchers to investigate their energetic and biosynthetic demands. Studies in both human and mouse primed ESCs revealed that these cells are almost exclusively glycolytic and display increased activity of the PPP pathway under normoxic conditions (Gu et al., 2016; Sperber et al., 2015; Zhou et al., 2012). Together, these metabolic pathways regulate the stemness of ESCs, protect them from oxidative stress, and produce biomolecules (such as ribose and NADPH) required for nucleotide synthesis and DNA replication (Shyh-Chang and Ng, 2017). Notably, differentiation of ESCs to adult cell types is associated with a metabolic shift to OXPHOS (Varum et al., 2011). Conversely, nuclear reprogramming of somatic cells to induced Pluripotent Stem Cells (iPSCs) results in a metabolic shift to aerobic glycolysis (Folmes et al., 2011). These observations indicate that WE is intricately associated with pluripotency. Indeed, a seminal study by Moussaief and colleagues reported that increased glycolysis in ESCs increases the production of acetyl-CoA and promotes histone acetylation (Moussaieff et al., 2015). The authors further demonstrated that a shift to OXPHOS results in the loss of these acetylation marks within the first hour of differentiation. This can be prevented by adding exogenous acetate, which sustains ESC pluripotency (Moussaieff et al., 2015). Apart from acetylation in ESCs, WE regulate other epigenetic marks such as histone methylation and O-N-acetylglycolysation, which either enhances the transcription of pluripotency factors or silences the expression of differentiation genes in ESCs (Hanover et al., 2012; Jang et al., 2012; Shi et al., 2013). Moreover, as a feed-forward mechanism, key glycolytic enzymes, including HK2 and PKM2 and glucose transporter GLUT1, are direct transcriptional targets of core pluripotency factors c-MYC, SOX2, and OCT4 (Kim et al., 2015; Yu et al., 2019). Another important ESC factor, the RNA binding protein LIN28A/B also inhibits mitochondrial respiration and directly alters the cellular proteome to promote glycolysis (Zhang et al., 2016).This complex crosstalk illustrates that stem cell identity and cellular metabolism are reciprocally regulated in ESCs and emphasizes the importance of metabolism in the regulation of developmental plasticity.
The above observations obtained from cultured ESCs in vitro also hold true for pluripotent stem cells that compose the Inner Cell Mass (ICM) of human and murine blastocysts in vivo. Analysis of metabolic flux in early murine embryos revealed a transition from OXPHOS to aerobic glycolysis as embryos progress from a single-cell zygote to morula and blastula stages (Kaneko, 2016; Martin and Leese, 1995). This metabolic shift is driven by an increased expression of glycolytic genes and decreased mitochondrial potential of the ICM (Houghton, 2006; Kaneko, 2016). Post implantation, despite a surge in oxygen availability, the murine ICM continues to elevate its glycolytic flux until the beginning of germ layer differentiation (Houghton, 2006; Kaneko, 2016; reviewed by Intlekofer and Finley, 2019). These findings reiterate the association between WE and pluripotency and show that complementary metabolic transitions drive ICM formation and germ layer differentiation in vivo.
Notably, the role of WE in the regulation of stemness extends beyond pluripotent cells and is also relevant in several tissue- specific multipotent progenitor cells such as Neural Stem/Progenitor Cells (NSPCs) and Haemopoietic Stem Cells (HSCs) (reviewed by Candelario et al., 2013; Ito and Suda, 2014). Both NSPCs and HSCs are quiescent stem cells that preferentially utilize glycolysis. In fact, in their respective tissues, these cells exist within a hypoxic niche that promotes WE downstream of HIF1a-mediated upregulation of glycolytic genes (De Filippis and Delia, 2011; Mohyeldin et al., 2010; Suda et al., 2011). The dependence of both NSPCs and HSCs on WE suggests that, rather than rapid ATP production, the primary function of glycolysis in stem cells is the regulation of developmental potential. Thus, it is likely that the mechanisms by which WE regulates stemness is cell type-specific and dependent upon the functional and environmental context of stem cell populations.
In this context, the development of the vertebrate retina provides another interesting example of how aerobic glycolysis sustains progenitor identity in vivo. This is exemplified in a study by Agathocleous and colleagues, which reported that proliferating progenitor cells within the retina of Xenopus and Zebrafish embryos are glycolytic (Agathocleous et al., 2012). Unlike their differentiated counterparts, these cells have reduced sensitivity to OXPHOS inhibition. The authors showed that WE is essential for the survival and proliferation of these progenitor cells and that glycolysis mediates this function by directly regulating the cell cycle. Lastly, as observed in several other contexts, the differentiation of retinal progenitors in the Xenopus embryo was accompanied by a transition to OXPHOS and reduced dependence on glycolysis (Agathocleous et al., 2012). Taken together, these findings echo a common theme: the balance between aerobic glycolysis and OXPHOS regulates cell state transitions during embryogenesis. Since the neural crest shares many regulatory features with embryonic stem cells, it is likely that many of the mechanisms described here are shared between these cell populations. Comparative studies in metabolic regulation of cell identity will be vital to obtaining a holistic perspective on the role of WE in stem cell populations in the developing embryo.
Aerobic glycolysis in organogenesis
Our understanding of the role of carbon metabolism in neural crest development can benefit from an examination of the metabolic transitions that take place in other embryonic cell types. As discussed above, germ layer differentiation and organogenesis in vertebrate embryos are usually associated with a shift to oxidative metabolism. However, as in the case of neural crest, specific cell types within the developing embryo can transition back to a glycolytic state to engage in distinct biological processes. In particular, migratory progenitor cells commonly upregulate glycolysis to drive organ morphogenesis. A classic example of this is the “tip” and “stalk” Endothelial Cells (EC) that mediate blood vessel formation in the embryo (De Bock et al., 2013). The tip ECs are migratory and promote vessel sprouting while the proliferative stalk ECs function to elongate the blood vessel (Potente et al., 2011). By measuring the metabolic flux of cultured ECs in vitro, De Bock and colleagues uncovered that these cells display very high glycolytic flux comparable to that observed in tumor cells (De Bock et al.,2013). This metabolic alternation in ECs is mediated by the action of sprouting signals such as FGF and VEGF, that increase the expression of glycolysis activator enzyme PFKFB3. Depletion of PFKFB3 in ECs expectedly reduces the glycolytic flux and inhibits sprout formation and EC migration in vitro and embryonic angiogenesis in vivo. This demonstrates that high glycolytic flux is required for proper tip cell positioning in the developing blood vessel, and that PFKFB3 expression is necessary and sufficient to promote the formation and activity of tip cells in vitro and in vivo. Interestingly, this “pro-tip” function of aerobic glycolysis counters the “pro-stalk” activity of Notch signaling. This balancing act is essential for maintaining the proper ratio of tip:stalk cells in the growing vessel (De Bock et al., 2013). This elegant study thus shows how aerobic glycolysis can mediate the crosstalk between different signaling pathways (such as FGF and Notch) to regulate cellular identity and function during organogenesis.
A series of recent studies characterizing the metabolic transitions that control somite formation also support the premise that WE functions as a mediator of developmental signals in specific cellular contexts. By studying paraxial mesoderm segmentation during somitogenesis, Oginuma and colleagues, and Bulusu and colleagues independently observed that a glycolytic gradient in the presomitic mesoderm is required for the posterior elongation of the embryonic axis (Bulusu et al., 2017; Oginuma et al., 2017). Furthermore, Oginuma and colleagues showed that this gradient is established downstream of FGF signaling, which increases the expression of glycolytic enzymes in the developing tail bud. In a follow-up study, they reported that aerobic glycolysis-mediated acidosis elevates the intracellular pH of NMPs, leading to the activation of Wnt signaling through acetylation and stabilization of the effector protein β-catenin (Oginuma et al., 2020). This enhanced Wnt activity establishes a mesodermal identity in multipotent NMPs and may also promote their migration towards the forming somite by regulating cell motility. Additionally, Wnt signaling transcriptionally controls the expression of FGF ligands and receptors in the paraxial mesoderm (Oginuma et al., 2020). As discussed above, elevated FGF signaling activates aerobic glycolysis and consequentially the Wnt pathway, thus feeding into a positive feedback loop that ensures tight coordination between Wnt and FGF signaling during somitogenesis.
Crosstalk between aerobic glycolysis and FGF appears to be a recurring theme during organogenesis. A recent study by Kantarci and colleagues indicates that FGF and aerobic glycolysis can reciprocally regulate each other and that this mechanism is broadly relevant in the formation of multiple tissues within the embryo (Kantarci et al., 2020). By harnessing the power of reverse genetics, the authors of this study disrupted the glycolytic enzyme phosphoglycerate kinase-1(PGK1) in zebrafish embryos and observed defective neuronal development in the otic vesicle and central/peripheral nervous system, as well as disruption of hair cell formation. Interestingly, both neuron and hair cell development are controlled by FGF signaling, prompting the authors to investigate how aerobic glycolysis impacts this signaling pathway. Through a series of functional experiments, the study found that lactate elevates the basal levels of FGF effectors MAPK and ETV5B in glycolytic cells, which primes them to respond efficiently to changes in this signaling pathway (Kantarci et al., 2020). Taken together, the above studies shed light on how signaling systems work in concert with metabolic regulation to drive changes in cell identity.
Effects of metabolic disorders in neural crest development
Our examination of carbon metabolism in neural crest cells indicates that their metabolic states are precisely regulated during development. Prior to delamination, these cells display basal cellular metabolism (Fig. 4). However, as they become primed for migration, there is a striking upregulation of glycolytic enzymes, and the cells become highly glycolytic, exhibiting the hallmarks of the Warburg effect. Glycolytic flux increases during migration, but once neural crest cells reach their final destinations they begin differentiating. As observed in many other developmental cell types, differentiation is accompanied by an increase in the expression of OXPHOS enzymes and reduction in levels of aerobic glycolysis. Thus, there are two major bioenergetic shifts during neural crest development: the first during delamination and the second at the onset of differentiation (Fig. 4). We propose that disruptions in these metabolic transitions may affect the migration and differentiation of neural crest cells, leading to metabolic neurocristopathies (Fig. 1).
Figure 4. Metabolic transitions during neural crest development.
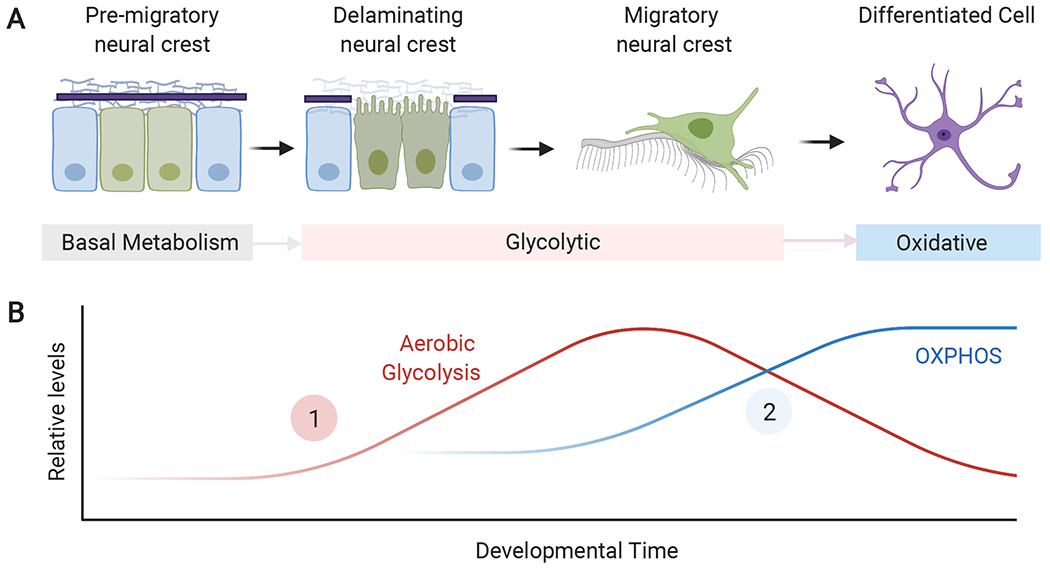
(A) Early pre-migratory neural crest cells initially exhibit low levels of glycolysis and oxidative phosphorylation. As the cells become primed for epithelial to mesenchymal transition, they transition to a highly glycolytic state and undergo delamination and migration. After migration, neural crest cells differentiate and engage in oxidative phosphorylation (OXPHOS). (B) Estimated levels of glycolysis and oxidative phosphorylation during neural crest development. Neural crest cells undergo two main developmental transitions, when they transition from quiescent to glycolytic (1), and when they differentiate and display high levels of mitochondrial respiration (2). OXPHOS: Oxidative phosphorylation.
Neurocristopathies are a set of congenital disabilities that arise from defects in the formation of the neural crest and its derivatives (Vega-Lopez et al., 2018). These pathologies are often associated with exposure of the fetus to abnormal levels of different metabolites, including high glucose (Chappell et al., 2009) and alcohol (Smith et al., 2014; Zhang et al., 2018). Furthermore, mutations in genes that affect the cellular response to metabolic stress can cause neurocristopathies such as Treacher Collins syndrome (Sakai et al., 2016). Other congenital diseases caused by mutations in mitochondrial proteins, such as Leigh syndrome, also have severe craniofacial phenotypes indicative of improper neural crest development (Berio, 2011). This is consistent with the view that neural crest cells are specifically sensitive to metabolic abnormalities, underscoring the importance of energy homeostasis in the formation of this cell population. Below, we describe the phenotypes associated with some metabolic neurocristopathies and discuss their etiology.
Diabetic Embryopathies
Maternal diabetes is strongly associated with increased craniofacial malformations such as cleft palate, cardiac outflow tract defects, and holoprosencephaly (Castori, 2013). Though maternal diabetes has been linked to embryopathies since the 1930s, the specific diabetic teratogens causing these malformations remain to be determined. Initial studies in animal models suggested that hyperglycemia is the crucial factor, since exposing mouse and rat embryos to diabetic glucose levels recapitulates the malformations observed in human neonates (Fine et al., 1999). Community-based approaches to monitor and manage maternal blood glucose levels during gestation were also found to be effective in moderating birth defects (Kitzmiller et al., 1991). However, more recent reports implicate maternal hyperketonemia, disrupted fetal arachidonic acid metabolism, and increased oxidative and cellular stress in diabetic embryopathy (Eriksson and Wentzel, 2016). Given the pleiotropic effect of diabetes on adult tissue (DeFronzo et al., 2015), it is likely that the cause behind maternal diabetes embryopathy is multifactorial. Furthermore, since neural crest cells are significantly affected by high levels of extracellular glucose and oxidative stress, maternal diabetes may impinge on neural crest development at multiple levels and via distinct mechanisms.
Studies in chick, mouse, and rat models have revealed that maternal diabetes induces apoptosis of cranial and cardiac neural crest cells. A study by Wang and colleagues shows that chick embryos developed in the presence of glucose display facial bone defects caused by apoptosis of early neural crest cells (Wang et al., 2015). The authors determined that the hyperglycemic environment activates the PI3K/ERK signaling pathway promoting autophagy, the excess of which results in neural crest apoptosis in a p53-dependent manner (Wang et al., 2015). Consistent with this conclusion, pharmacological inhibition of autophagy rescues glucose-induced cranial neural crest apoptosis, highlighting the therapeutic relevance of this pathway (Wang et al., 2015).
Similar observations were also made in an earlier study by Morgan and colleagues, which demonstrated that hyperglycemia and oxidative stress directly disrupt cardiac neural crest induction (Morgan et al., 2008). In this study, the authors showed that embryos of both diabetic and transiently hyperglycemic mice have increased apoptosis of PAX3+ neural crest cells and subsequently display cardiac outflow defects. Interestingly, these phenotypes could be rescued by administering antioxidants to hyperglycemic pregnant mice, leading the authors to conclude that oxidative stress is the primary cause of diabetic embryopathy in cardiac neural crest cells (Morgan et al., 2008). In support of this hypothesis, a transient increase in oxidative stress by subcutaneous injection with actinomycin in pregnant mice recapitulated the cardiac neural crest defects, further indicating that oxidative stress is a downstream effect of hyperglycemia (Morgan et al., 2008).
A separate study by Wentzel and colleagues provides insights into a possible mechanism by which high glucose levels increase oxidative stress in neural crest cells (Wentzel and Eriksson, 2011). By assessing the gene expression profiles of cranial and trunk neural crest cells isolated from rat embryos, the authors showed that when cultured in the presence of high glucose, cranial neural crest cells specifically downregulate the expression of antioxidant genes such as NRF2, Superoxide dismutase, Catalase, and PARP. (Wentzel and Eriksson, 2011). This suggests that, in addition to altering the cells’ metabolic profile, hyperglycemia can impinge on their transcriptional program, reducing the ability of neural crest cells to respond to oxidative stress. The specificity of this transcriptional alteration to the cranial subpopulation further indicates an axial-specific susceptibility of neural crest cells to hyperglycemia and oxidative stress.
Taken together, these studies describe two distinct mechanisms: (1) upregulation of autophagy and (2) increase in oxidative stress, by which maternal diabetes can promote apoptosis of neural crest cells. However, since an important effect of oxidative stress is the upregulation of autophagy (Filomeni et al., 2015; Yun et al., 2020), these two mechanisms likely merge into a single signaling cascade where, downstream of hyperglycemia, cellular oxidative stress upregulates autophagy to cause neural crest apoptosis in a p53-dependent manner. If substantiated, this mechanism emphasizes the potential of dietary antioxidant supplementation to pregnant diabetic mothers as a possible intervention for diabetic embryopathy.
Fetal Alcohol Spectrum Disorder
Fetal Alcohol Spectrum Disorder (FASD) is a neurocristopathy that results from prenatal alcohol exposure (Smith et al., 2014). It can be diagnosed by characteristic craniofacial dysmorphia, including flattened midface and mouth area, thin upper lip, and micrognathia. Infants suffering from FASD may also display sensory and language disorders, neurological abnormalities, and growth defects (Smith et al., 2014; Wozniak et al., 2019). Prenatal alcohol exposure impacts multiple stages of neural crest development. During early gastrulation, high maternal blood alcohol levels disrupt neuroectoderm development and decrease expression of crucial neural crest markers genes in the embryo (Cartwright and Smith, 1995). Alcohol also suppresses cranial neural crest migration (Cartwright and Smith, 1995; Rovasio and Battiato, 1995). In vivo studies in the chick embryo have revealed that upon exposure to ethanol, neural crest cells fail to emerge from the neural tube, indicative of defects in EMT. The few cells that do migrate initially follow appropriate migratory routes but do not arrive at the correct locations in the embryo, further suggesting disruption of signaling cues (Cartwright and Smith, 1995; Rovasio and Battiato, 1995). A recent study in zebrafish embryos demonstrated that upon ethanol challenge, the left-right symmetry of migratory cranial neural crest cells is lost, and the cells display an uncharacteristic retrograde motion that prevents travel over longer distances (Boric et al., 2013). Intriguingly, complementary in vitro studies using neural crest explants revealed that the migration defects observed upon ethanol exposure are specific to cranial neural crest cells and are significantly moderate in its trunk counterparts (Czarnobaj et al., 2014; Rovasio and Battiato, 2002), thus affirming the axial differences in the neural crest’s ability to respond to metabolic stress.
The above defects in cranial neural crest formation and migration are partly attributed to increased apoptosis of these cells upon alcohol exposure (Debelak-Kragtorp et al., 2003; Flentke et al., 2011; Rovasio and Battiato, 1995; Smith et al., 2014). Even low doses of ethanol induce cell death in up to 50% of delaminating neural crest cells, and inhibition of apoptosis can rescue the facial anomalies observed in FASD (Cartwright et al., 1998; Dunty et al., 2001; Rovasio and Battiato, 2002). At least two distinct mechanisms have been described by which alcohol promotes cell death in the neural crest. The first mechanism involves the interaction of ethanol with a yet unidentified G-protein coupled receptor, which elevates the levels of inositol triphosphate (IP3) and causes a massive increase in intracellular Ca2+ levels (Debelak-Kragtorp et al., 2003; Garic-Stankovic et al., 2005). This transient calcium wave activates the calcium/calmodulin-dependent protein kinase II (CaMKII), specifically in the dorsal neural folds (Garic et al., 2011). CaMKII, in turn phosphorylates and destabilizes β-catenin, thus effectively inhibiting the Wnt signaling pathway (Flentke et al., 2011). This disrupts neural crest specification and migration, leading to apoptosis of this cell population (Flentke et al., 2011).
An alternate mechanism implicates oxidative stress and depletion of intracellular metabolites as the primary cause of ethanol-induced neural crest apoptosis (Chen and Sulik, 1996; Chen et al., 2013; Davis et al., 1990). Alcohol is known to induce oxidative stress by generating excess ROS, which neural crest cells are particularly sensitive to due to their low levels of endogenous antioxidants (Davis et al., 1990). The mechanism through which ethanol metabolism generates ROS in neural crest cells remains unknown. Despite this, several antioxidants, including astaxanthin, vitamin C, vitamin E, and beta-carotene have effectively prevented FASD in animal models (Zhang et al., 2018). Interestingly, neural crest cells increase expression of NRF2, a transcription factor and master regulator of the antioxidant response after ethanol exposure, likely to compensate for the elevated oxidative stress (Chen et al., 2013). While this modest increase in NRF2 transcription is not sufficient to reduce apoptosis, Chen and colleagues observed that overexpression of NRF2 or treatment with its chemical agonists increased the expression of antioxidant factors such as SOD1, catalase, GPX3, and NQO; and decreased ROS generation and cell death in the neural crest (Chen et al., 2013). Ethanol additionally impacts cellular metabolism by decreasing the availability of cholesterol esters and depleting polyamines (Poodeh et al., 2014; Li et al., 2007). The former results in suppression of the Sonic Hedgehog Signaling pathway and has severe consequences on neurogenesis and possibly neural crest development (Li et al., 2007). Reduced availability of polyamines such as putrescine and spermidine negatively affect DNA replication, which may inhibit the proliferation of pre-migratory neural crest cells, resulting in apoptosis (Poodeh et al., 2014).
Prenatal alcohol exposure can also directly impact the migration of neural crest cells. Apart from increased apoptosis, the profound migratory defects described above result from improper EMT of cranial neural crest cells and defects in filopodia formation and substrate adhesion (Hassler and Moran, 1986; Oyedele and Kramer, 2013; Rovasio and Battiato, 2002). Intriguingly, these phenotypes mirror those observed upon inhibition of aerobic glycolysis during the migration of cranial neural crest cells. As discussed above, aerobic glycolysis necessitates increased uptake of glucose by delaminating neural crest cells (Bhattacharya et al., 2020). However, a well-characterized feature of FASD is significantly decreased fetal blood glucose level and reduced glucose uptake by the embryo. This could prevent aerobic glycolysis in the neural crest due to the unavailability of glucose, which may be an additional mechanism by which prenatal alcohol exposure disrupts delamination and migration of this cell type. Future studies aimed at understanding how ethanol challenge impacts central glucose metabolism could verify this possibility and provide insights on mechanisms by which ethanol impacts OXPHOS to promote ROS production.
Treacher Collins Syndrome
Neurocristopathies can also be caused by an inadequate or insufficient response to metabolic stress. An example of this is Treacher Collins syndrome (TCS), characterized by jawbone and cheekbone hypoplasia, external ear anomalies, cleft palate, and periorbital abnormalities (Aljerian and Gilardino, 2019). Of the four causative genes, autosomal dominant mutations in TCOF1 is the most common and is observed in 90% of TCS patients (Group, 1996). TCS-associated mutations in TCOF1 result in reduced expression of the nucleolar protein Treacle, which causes impaired ribosome biogenesis (Valdez et al., 2004), increased nucleolar stress (Rubbi and Milner, 2003), and apoptosis of neural crest cells (Jones et al., 2008). Historically, defects in ribosome function were considered to be the primary cause of neural crest apoptosis in TCS patients. However, recent reports indicate that haploinsufficiency of TCOF1 dampens the oxidative stress response in neuroepithelium, resulting in cell apoptosis in a p53-dependent manner (de Peralta et al., 2016; Sakai et al., 2016).
A study by Sakai and colleagues demonstrated that a key function of Treacle is to interact with the DNA damage sensor MRNM complex (Mre11-Rad51-NBS1-MDC1) and localize at the sites of oxidative stress-induced DNA strand breaks (Sakai et al., 2016). At these damaged loci, Treacle recruits the DNA repair protein BRCA1 to fix the strand breaks and restore cell cycle progression following oxidative stress. The authors further demonstrated that in early embryos, the neuroepithelium exists in a highly oxidative state with elevated ROS levels, which explains why TCOF1 loss of function has a severe effect on neural crest development. Importantly, by injecting the antioxidant N-acetyl cysteine into TCOF1 +/− pregnant mice, the authors could rescue the craniofacial deformations associated with TCS. This suggests that antioxidant treatments can potentially alleviate and even prevent pathogenesis in TCOF1 +/−embryos (Sakai et al., 2016).
A recent study utilizing a zebrafish model of TCS also confirmed the utility of antioxidants in reducing craniofacial abnormalities in TCS embryos and identified an additional mechanism by which Treacle protects cells from oxidative stress (de Peralta et al., 2016). In this study, Peralta and colleagues showed that Treacle stabilizes ROS protective CNBP protein (Cellular nucleic acid-binding protein), which is crucial for craniofacial development (de Peralta et al., 2016). Though the mechanism by which CNBP promotes antioxidant response is not understood, the authors showed that overexpression of this protein was sufficient to reduce elevated ROS levels and rescue the craniofacial phenotypes of TCS embryos (de Peralta et al., 2016). Altogether, the above studies aimed at delineating the etiology of TCS reiterate the importance of antioxidant response in neural crest development and inform upon therapeutic strategies for managing birth defects arising from the disruption of this protective pathway.
Mitochondrial Diseases
The congenital diseases described above are classical neurocristopathies resulting from defective neural crest metabolism. Intriguingly, mutations in crucial metabolic enzymes also result in birth defects with phenotypes very similar to those observed in FASD and diabetic embryopathy. In this regard, diseases caused by mutations in mitochondrial proteins are particularly relevant as they are often associated with gross craniofacial deformities (Berio, 2011). For example, patients with Leigh syndrome, a life-threatening birth defect caused by mutations in OXPHOS genes, often display craniofacial dysmorphisms, such as maxillary hypoplasia, upturned nose, micrognathia, and smooth philtrum (Berio and Piazzi, 2007). Other OXPHOS-related diseases, such as Kearns-Sayre syndrome and congenital pyruvate dehydrogenase deficiency, are also associated with similar facial abnormalities, albeit with varying severity (Berio, 2011; Finsterer et al., 2020). While these diseases are not traditionally categorized as neurocristopathies, likely due to severe comorbidities in other organs, their phenotypes strongly suggest that neural crest development is particularly vulnerable to mitochondrial dysfunction. The molecular basis of this vulnerability, and indeed the role of OXPHOS in neural crest development, remains uncharacterized. However, this is increasingly becoming a relevant area of research in neural crest and developmental biology.
Conclusions
In the last decades, we have learned much about the connections between metabolism and cellular processes, such as signal transduction and gene regulation. Studies on metabolic diseases, such as diabetes, led to the idea that carbon metabolism has a pervasive role in cellular function (DeFronzo et al., 2015). Since then, this framework has become critical to our understanding of cancer, aging, and neurodegeneration (Hsu and Sabatini, 2008; Procaccini et al.,2016; Barzilai et al., 2012). Yet, we are only now gaining insight on how the metabolic state of embryonic cells contributes to their behavior and identity during embryonic development (Miyazawa and Aulehla, 2018). The reciprocal relationships between metabolism and cellular behaviors are evident in the development of the neural crest. A key conclusion emerging from the existing body of research is that the metabolic state of the neural crest is highly dynamic and involves drastic bioenergetic shifts that are consequential to its development.
At least two metabolic reprogramming events define neural crest development: (i) the transition of delaminating cells from basal to a glycolytic state, which is essential for EMT, and (ii) the transition of post migratory neural crest cells to a high OXPHOS state that is required for differentiation (Fig. 3). The importance of these metabolic transitions was initially determined through experiments in model organisms, and their relevance for human health is supported by studies on neurocristopathies linked to metabolic diseases. Notably, these conditions specifically affect either neural crest migration or differentiation and sometimes disrupt both processes. This indicates that these two steps in neural crest development are the most sensitive to metabolic disruptions. Additional studies on the molecular pathways that work in concert with carbon metabolism are required for the identification of specific mechanisms that are disrupted in metabolic neurocristopathies.
Diseases caused by metabolite imbalance, such as maternal diabetes and FASD, primarily increase cellular ROS levels in delaminating neural crest cells, resulting in their apoptosis and failure to delaminate. We propose that the dysregulation of extracellular glucose levels inherent to these diseases prevents aerobic glycolysis, which can prematurely upregulate OXPHOS and elevate cellular ROS levels. In contrast, congenital diseases caused by mutations in mitochondrial genes disproportionately affect differentiation of neural crest cells, particularly mesenchymal lineages. This is likely because inhibition of mitochondrial respiration prevents the second transition of post migratory neural crest cells to a high OXPHOS state, which appears to be especially critical for craniofacial development.
The phenotypes associated with neurocristopathies also highlight the metabolic heterogeneity of different neural crest populations. As mentioned above, susceptibility of these progenitors to metabolic disruptions appears to be dependent on their location along the embryonic anterior-posterior axis. Neural crest cells derived from the cranial regions appear to be more vulnerable than their trunk counterparts. This observation indicates that the bioenergetic profile and the characteristic metabolic modes of neural crest cells may vary depending on their axial level. As evidenced by recent studies on the metabolic regulation of neuro-mesodermal progenitors, the differentiation of embryonic cells to more mesodermal lineages tend to be significantly reliant on cellular metabolism (Oginuma et al., 2017). This raises the intriguing possibility that the developmental potential of neural crest subpopulations is connected with their bioenergetic state.
Lastly, the above discussion also engenders several outstanding questions, especially regarding the mechanisms controlling the bioenergetic shifts throughout neural crest formation. Our understanding of how metabolic reprogramming is mediated in other developmental contexts could provide essential clues in this regard. In particular, the regulation of aerobic glycolysis in ESCs likely has direct parallels with how this metabolic adaptation is implemented in migratory neural crest cells. Indeed, key transcription factors like HIF1a and c-MYC, which are master regulators of WE in ESCs (Kim et al., 2015; Varum et al., 2011), are also highly expressed in pre-migratory and migratory neural crest cells in a tissue-specific manner in multiple vertebrate models (Barriga et al., 2013; Kerosuo and Bronner, 2016). The depletion of these factors in delaminating neural crest recapitulate the phenotypes observed upon inhibition of WE, suggesting that these regulators may have a conserved role in pluripotent ESCs and multipotent neural crest cells during embryogenesis (Barriga et al., 2013). Similarly, mechanisms that inhibit WE during ESC differentiation may also be active in post-migratory neural crest cells to mediate their transition from a glycolytic to a high OXPHOS state prior to cell fate commitment. Identifying the key players would provide further clues on how the crosstalk between the neural crest GRN and metabolic pathways guide the formation of this cell type.
Acknowledgements
This study was supported by NIH grant R01DE028576 to M.S.-C. We would like to thank the members of the Simoes-Costa Lab for critical reading of the manuscript and valuable suggestions. The figures in this article were created with Biorender.
References
- Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA, 2009. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. The Journal of clinical investigation 119, 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agathocleous M, Love NK, Randlett O, Harris JJ, Liu J, Murray AJ, Harris WA, 2012. Metabolic differentiation in the embryonic retina. Nat Cell Biol 14, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alata Jimenez N, Torres Perez SA, Sanchez-Vasquez E, Fernandino JI, Strobl-Mazzulla PH, 2018. Folate deficiency prevents neural crest fate by disturbing the epigenetic Sox2 repression on the dorsal neural tube. Dev Biol 444 Suppl 1, S193–S201. [DOI] [PubMed] [Google Scholar]
- Aljerian A, Gilardino MS, 2019. Treacher Collins Syndrome. Clin Plast Surg 46, 197–205. [DOI] [PubMed] [Google Scholar]
- Barriga EH, Maxwell PH, Reyes AE, Mayor R, 2013. The hypoxia factor Hif-1alpha controls neural crest chemotaxis and epithelial to mesenchymal transition. The Journal of cell biology 201, 759–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Huffman DM, Muzumdar RH, Bartke A, 2012. The critical role of metabolic pathways in aging. Diabetes 61, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berio A, 2011. Metabolic syndromes and neural crest development. Journal of Biological Research. [Google Scholar]
- Berio A, Piazzi A, 2007. [Leigh syndrome with facial abnormalities: a neurocristopathy]. Pediatr Med Chir 29, 50–54. [PubMed] [Google Scholar]
- Bhattacharya D, Azambuja AP, Simoes-Costa M, 2020. Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev Cell 53, 199–211 e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boot MJ, Steegers-Theunissen RP, Poelmann RE, Van Iperen L, Lindemans J, Gittenberger-de Groot AC, 2003. Folic acid and homocysteine affect neural crest and neuroepithelial cell outgrowth and differentiation in vitro. Dev Dyn 227, 301–308. [DOI] [PubMed] [Google Scholar]
- Boric K, Orio P, Vieville T, Whitlock K, 2013. Quantitative analysis of cell migration using optical flow. PLoS One 8, e69574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand KA, Hermfisse U, 1997. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J 11, 388–395. [DOI] [PubMed] [Google Scholar]
- Bronner ME, Simoes-Costa M, 2016. The Neural Crest Migrating into the Twenty-First Century. Curr Top Dev Biol 116, 115–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulusu V, Prior N, Snaebjornsson MT, Kuehne A, Sonnen KF, Kress J, Stein F, Schultz C, Sauer U, Aulehla A, 2017. Spatiotemporal Analysis of a Glycolytic Activity Gradient Linked to Mouse Embryo Mesoderm Development. Developmental cell 40, 331– 341 e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario KM, Shuttleworth CW, Cunningham LA, 2013. Neural stem/progenitor cells display a low requirement for oxidative metabolism independent of hypoxia-inducible factor- 1alpha expression. J Neurochem 125, 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright MM, Smith SM, 1995. Stage-dependent effects of ethanol on cranial neural crest cell development: partial basis for the phenotypic variations observed in fetal alcohol syndrome. Alcohol Clin Exp Res 19, 1454–1462. [DOI] [PubMed] [Google Scholar]
- Cartwright MM, Tessmer LL, Smith SM, 1998. Ethanol-induced neural crest apoptosis is coincident with their endogenous death but is mechanistically distinct. Alcohol Clin Exp Res 22, 142–149. [PubMed] [Google Scholar]
- Castori M, 2013. Diabetic embryopathy: a developmental perspective from fertilization to adulthood. Mol Syndromol 4, 74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JH Jr., Wang XD, Loeken MR, 2009. Diabetes and apoptosis: neural crest cells and neural tube. Apoptosis 14, 1472–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Sulik KK, 1996. Free radicals and ethanol-induced cytotoxicity in neural crest cells. Alcohol Clin Exp Res 20, 1071–1076. [DOI] [PubMed] [Google Scholar]
- Chen X, Liu J, Chen SY, 2013. Over-expression of Nrf2 diminishes ethanol-induced oxidative stress and apoptosis in neural crest cells by inducing an antioxidant response. Reprod Toxicol 42, 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho ES, Cha YH, Kim HS, Kim NH, Yook JI, 2018. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol Ther (Seoul) 26, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluntun AA, Huang H, Dai L, Liu X, Zhao Y, Locasale JW, 2015. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnobaj J, Bagnall KM, Bamforth JS, Milos NC, 2014. The different effects on cranial and trunk neural crest cell behaviour following exposure to a low concentration of alcohol in vitro. Arch Oral Biol 59, 500–512. [DOI] [PubMed] [Google Scholar]
- Davis WL, Crawford LA, Cooper OJ, Farmer GR, Thomas DL, Freeman BL, 1990. Ethanol induces the generation of reactive free radicals by neural crest cells in vitro. J Craniofac Genet Dev Biol 10, 277–293. [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Carmeliet P, 2013. Role of endothelial cell metabolism in vessel sprouting. Cell Metab 18, 634–647. [DOI] [PubMed] [Google Scholar]
- De Filippis L, Delia D, 2011. Hypoxia in the regulation of neural stem cells. Cell Mol Life Sci 68, 2831–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Peralta MS, Mouguelar VS, Sdrigotti MA, Ishiy FA, Fanganiello RD, Passos-Bueno MR, Coux G, Calcaterra NB, 2016. Cnbp ameliorates Treacher Collins Syndrome craniofacial anomalies through a pathway that involves redox-responsive genes. Cell Death Dis 7, e2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelak-Kragtorp KA, Armant DR, Smith SM, 2003. Ethanol-induced cephalic apoptosis requires phospholipase C-dependent intracellular calcium signaling. Alcohol Clin Exp Res 27, 515–523. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, Hu FB, Kahn CR, Raz I, Shulman GI, Simonson DC, Testa MA, Weiss R, 2015. Type 2 diabetes mellitus. Nat Rev Dis Primers 1, 15019. [DOI] [PubMed] [Google Scholar]
- Dunty WC Jr., Chen SY, Zucker RM, Dehart DB, Sulik KK, 2001. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol Clin Exp Res 25, 1523–1535. [PubMed] [Google Scholar]
- Eriksson UJ, Wentzel P, 2016. The status of diabetic embryopathy. Ups J Med Sci 121, 96– 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Johnson J, Gatenby RA, Gillies RJ, 2013. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res 73, 1524–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo AL, Maczkowiak F, Borday C, Pla P, Sittewelle M, Pegoraro C, Monsoro-Burq AH, 2017. PFKFB4 control of AKT signaling is essential for premigratory and migratory neural crest formation. Development 144, 4183–4194. [DOI] [PubMed] [Google Scholar]
- Filomeni G, De Zio D, Cecconi F, 2015. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine EL, Horal M, Chang TI, Fortin G, Loeken MR, 1999. Evidence that elevated glucose causes altered gene expression, apoptosis, and neural tube defects in a mouse model of diabetic pregnancy. Diabetes 48, 2454–2462. [DOI] [PubMed] [Google Scholar]
- Finkel T, 2011. Signal transduction by reactive oxygen species. J Cell Biol 194, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DK, Leese HJ, 1987. Assessment of embryo viability prior to transfer by the noninvasive measurement of glucose uptake. J Exp Zool 242, 103–105 [DOI] [PubMed] [Google Scholar]
- Finsterer J, Winklehner M, Stollberger C, Hummel T, 2020. Unusual Phenotype and Disease Trajectory in Kearns-Sayre Syndrome. Case Rep Neurol Med 2020, 7368527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flentke GR, Garic A, Amberger E, Hernandez M, Smith SM, 2011. Calcium-mediated repression of beta-catenin and its transcriptional signaling mediates neural crest cell death in an avian model of fetal alcohol syndrome. Birth Defects Res A Clin Mol Teratol 91, 591– 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flickinger RA Jr., 1949. A study of the metabolism of amphibian neural crest cells during their migration and pigmentation in vitro. J Exp Zool 112, 465–484. [DOI] [PubMed] [Google Scholar]
- Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A, 2011. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14, 264– 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garic A, Flentke GR, Amberger E, Hernandez M, Smith SM, 2011. CaMKII activation is a novel effector of alcohol’s neurotoxicity in neural crest stem/progenitor cells. J Neurochem 118, 646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garic-Stankovic A, Hernandez MR, Chiang PJ, Debelak-Kragtorp KA, Flentke GR, Armant DR, Smith SM, 2005. Ethanol triggers neural crest apoptosis through the selective activation of a pertussis toxin-sensitive G protein and a phospholipase Cbeta- dependent Ca2+ transient. Alcohol Clin Exp Res 29, 1237–1246. [DOI] [PubMed] [Google Scholar]
- Gautheron DC, 1984. Mitochondrial oxidative phosphorylation and respiratory chain: review. J Inherit Metab Dis 7 Suppl 1, 57–61. [DOI] [PubMed] [Google Scholar]
- Gardner DK, Leese HJ, 1990. Concentrations of nutrients in mouse oviduct fluid and their effects on embryo development and metabolism in vitro. J Reprod Fertil 88, 361–368. [DOI] [PubMed] [Google Scholar]
- Group TTCSC, 1996. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. The Treacher Collins Syndrome Collaborative Group. Nat Genet 12, 130–136. [DOI] [PubMed] [Google Scholar]
- Gu W, Gaeta X, Sahakyan A, Chan AB, Hong CS, Kim R, Braas D, Plath K, Lowry WE, Christofk HR, 2016. Glycolytic Metabolism Plays a Functional Role in Regulating Human Pluripotent Stem Cell State. Cell Stem Cell 19, 476–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi Poodeh S, Alhonen L, Salonurmi T, Savolainen MJ, 2014. Ethanol-induced impairment of polyamine homeostasis--a potential cause of neural tube defect and intrauterine growth restriction in fetal alcohol syndrome. Biochem Biophys Res Commun 446, 173–178. [DOI] [PubMed] [Google Scholar]
- Hanover JA, Krause MW, Love DC, 2012. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol 13, 312–321. [DOI] [PubMed] [Google Scholar]
- Hassler JA, Moran DJ, 1986. Effects of ethanol on the cytoskeleton of migrating and differentiating neural crest cells: possible role in teratogenesis. Journal of craniofacial genetics and developmental biology. Supplement 2, 129–136. [PubMed] [Google Scholar]
- Houghton FD, 2006. Energy metabolism of the inner cell mass and trophectoderm of the mouse blastocyst. Differentiation 74, 11–18. [DOI] [PubMed] [Google Scholar]
- Hovland AS, Rothstein M, Simoes-Costa M, 2020. Network architecture and regulatory logic in neural crest development. Wiley Interdiscip Rev Syst Biol Med 12, e1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PP, Sabatini DM, 2008. Cancer cell metabolism: Warburg and beyond. Cell 134, 703– 707. [DOI] [PubMed] [Google Scholar]
- Hu N, Strobl-Mazzulla PH, Bronner ME, 2014. Epigenetic regulation in neural crest development. Dev Biol 396, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Finley LWS, 2019. Metabolic signatures of cancer cells and stem cells. Nat Metab 1, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Suda T, 2014. Metabolic requirements for the maintenance of self-renewing stem cells.Nat Rev Mol Cell Biol 15, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J, Suzuki A, Pelikan RC, Ho TV, Sanchez-Lara PA, Chai Y, 2014. Modulation of lipid metabolic defects rescues cleft palate in Tgfbr2 mutant mice. Hum Mol Genet 23, 182– 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, Kim TW, Yoon S, Choi SY, Kang TW, Kim SY, Kwon YW, Cho EJ, Youn HD, 2012. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell 11, 62–74. [DOI] [PubMed] [Google Scholar]
- Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, Dixon MJ, Trainor PA, 2008. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nature medicine 14, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose C, Bellance N, Rossignol R, 2011. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta 1807, 552–561. [DOI] [PubMed] [Google Scholar]
- Kaneko KJ, 2016. Metabolism of Preimplantation Embryo Development: A Bystander or an Active Participant? Curr Top Dev Biol 120, 259–310. [DOI] [PubMed] [Google Scholar]
- Kantarci H, Gou Y, Riley BB, 2020. The Warburg Effect and lactate signaling augment Fgf- MAPK to promote sensory-neural development in the otic vesicle. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathagen-Buhmann A, Maire CL, Weller J, Schulte A, Matschke J, Holz M, Ligon KL, Glatzel M, Westphal M, Lamszus K, 2018. The secreted glycolytic enzyme GPI/AMF stimulates glioblastoma cell migration and invasion in an autocrine fashion but can have anti-proliferative effects. Neuro Oncol 20, 1594–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerosuo L, Bronner ME, 2016. cMyc Regulates the Size of the Premigratory Neural Crest Stem Cell Pool. Cell reports 17, 2648–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keuls RA, Kojima K, Lozzi B, Steele JW, Chen Q, Gross SS, Finnell RH, Parchem RJ, 2020. MiR-302 Regulates Glycolysis to Control Cell-Cycle during Neural Tube Closure. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Jang H, Kim TW, Kang BH, Lee SE, Jeon YK, Chung DH, Choi J, Shin J, Cho EJ, Youn HD, 2015. Core Pluripotency Factors Directly Regulate Metabolism in Embryonic Stem Cell to Maintain Pluripotency. Stem Cells 33, 2699–2711. [DOI] [PubMed] [Google Scholar]
- Kitzmiller JL, Gavin LA, Gin GD, Jovanovic-Peterson L, Main EK, Zigrang WD, 1991.Preconception care of diabetes. Glycemic control prevents congenital anomalies. JAMA 265, 731–736. [PubMed] [Google Scholar]
- Laforgia N, Di Mauro A, Favia Guarnieri G, Varvara D, De Cosmo L, Panza R, Capozza M, Baldassarre ME, Resta N, 2018. The Role of Oxidative Stress in the Pathomechanism of Congenital Malformations. Oxid Med Cell Longev 2018, 7404082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin N, Kalcheim C, 1999. The neural crest, 2nd ed. Cambridge University Press, Cambridge, UK ; New York, NY, USA. [Google Scholar]
- Lewis PM, Dunn MP, McMahon JA, Logan M, Martin JF, St-Jacques B, McMahon AP, 2001. Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell 105, 599–612. [DOI] [PubMed] [Google Scholar]
- Li X, Yu X, Dai D, Song X, Xu W, 2016. The altered glucose metabolism in tumor and a tumor acidic microenvironment associated with extracellular matrix metalloproteinase inducer and monocarboxylate transporters. Oncotarget 7, 23141–23155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Yang HT, Zdanowicz M, Sicklick JK, Qi Y, Camp TJ, Diehl AM, 2007. Fetal alcohol exposure impairs Hedgehog cholesterol modification and signaling. Lab Invest 87, 231–240. [DOI] [PubMed] [Google Scholar]
- Liberti MV, Locasale JW, 2016. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wilcox DA, Sieber-Blum M, Wong-Riley M, 1990. Developing neural crest cells in culture: correlation of cytochrome oxidase activity with SSEA-1 and dopamine-beta-hydroxylase immunoreactivity. Brain Res 535, 271–280. [DOI] [PubMed] [Google Scholar]
- Liu XS, Little JB, Yuan ZM, 2015. Glycolytic metabolism influences global chromatin structure. Oncotarget 6, 4214–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Thompson CB, 2012. Metabolic regulation of epigenetics. Cell Metab 16, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG, 2011. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27, 441–464. [DOI] [PubMed] [Google Scholar]
- Martin KL, Leese HJ, 1995. Role of glucose in mouse preimplantation embryo development. Mol Reprod Dev 40, 436–443. [DOI] [PubMed] [Google Scholar]
- Melo FR, Bressan RB, Costa-Silva B, Trentin AG, 2017. Effects of Folic Acid and Homocysteine on the Morphogenesis of Mouse Cephalic Neural Crest Cells In Vitro. Cell Mol Neurobiol 37, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa H, Aulehla A, 2018. Revisiting the role of metabolism during development. Development 145. [DOI] [PubMed] [Google Scholar]
- Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A, 2010. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7, 150–161. [DOI] [PubMed] [Google Scholar]
- Morgan SC, Relaix F, Sandell LL, Loeken MR, 2008. Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res A Clin Mol Teratol 82, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, Bomze D, Elena-Herrmann B, Scherf T, Nissim- Rafinia M, Kempa S, Itskovitz-Eldor J, Meshorer E, Aberdam D, Nahmias Y, 2015. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab 21, 392–402. [DOI] [PubMed] [Google Scholar]
- Mullarky E, Cantley LC, 2015. Diverting Glycolysis to Combat Oxidative Stress, in: Nakao K, Minato N, Uemoto S (Eds.), Innovative Medicine: Basic Research and Development, Tokyo, pp. 3–23. [Google Scholar]
- Murphy MP, 2009. How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, Zheng J, Ricupero CL, He L, Jiao K, Mao JJ, 2018. mTOR acts as a pivotal signaling hub for neural crest cells during craniofacial development. PLoS Genet 14, e1007491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Uyeda K, 1995. Purification and Characterization of a Novel Xylulose 5-Phosphate-activated Protein Phosphatase Catalyzing Dephosphorylation of Frutcose-6-phosphate, 2-kinase:Fructose-2,6-bisphosphatase. J Biol Chem 270, 26341–26346. [DOI] [PubMed] [Google Scholar]
- Noor E, Eden E, Milo R, Alon U, 2010. Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol Cell 39, 809–820. [DOI] [PubMed] [Google Scholar]
- Oginuma M, Harima Y, Tarazona OA, Diaz-Cuadros M, Michaut A, Ishitani T, Xiong F, Pourquie O, 2020. Intracellular pH controls WNT downstream of glycolysis in amniote embryos. Nature 584, 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oginuma M, Moncuquet P, Xiong F, Karoly E, Chal J, Guevorkian K, Pourquie O, 2017. A Gradient of Glycolytic Activity Coordinates FGF and Wnt Signaling during Elongation of the Body Axis in Amniote Embryos. Dev Cell 40, 342–353 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyedele OO, Kramer B, 2013. Nuanced but significant: how ethanol perturbs avian cranial neural crest cell actin cytoskeleton, migration and proliferation. Alcohol 47, 417–426. [DOI] [PubMed] [Google Scholar]
- Patra KC, Hay N, 2014. The pentose phosphate pathway and cancer. Trends Biochem Sci 39, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe SW, Hong AW, Guan K, 2015. Disease Implication of the Hippo/YAP pathway. Trends Mol Med 21, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter FD, Herman GE, 2011. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res 52, 6–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, Carmeliet P, 2011. Basic and therapeutic aspects of angiogenesis.Cell 146, 873–887. [DOI] [PubMed] [Google Scholar]
- Procaccini C, Santopaolo M, Faicchia D, Colamatteo A, Formisano L, de Candia P, Galgani M, De Rosa V, Matarese G, 2016. Role of metabolism in neurodegenerative disorders. Metabolism 65, 1376–1390. [DOI] [PubMed] [Google Scholar]
- Quintana AM, Hernandez JA, Gonzalez CG, 2017. Functional analysis of the zebrafish ortholog of HMGCS1 reveals independent functions for cholesterol and isoprenoids in craniofacial development. PLoS One 12, e0180856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riobo NA, 2012. Cholesterol and its derivatives in Sonic Hedgehog signaling and cancer. Curr Opin Pharmacol 12, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovasio RA, Battiato NL, 1995. Role of early migratory neural crest cells in developmental anomalies induced by ethanol. Int J Dev Biol 39, 421–422. [PubMed] [Google Scholar]
- Rovasio RA, Battiato NL, 2002. Ethanol induces morphological and dynamic changes on in vivo and in vitro neural crest cells. Alcohol Clin Exp Res 26, 1286–1298. [DOI] [PubMed] [Google Scholar]
- Rubbi CP, Milner J, 2003. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. The EMBO journal 22, 6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai D, Dixon J, Achilleos A, Dixon M, Trainor PA, 2016. Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nat Commun 7, 10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi FT, Kim H, Lu W, He Q, Liu D, Goodell MA, Wan M, Songyang Z, 2013. Ten- eleven translocation 1 (Tet1) is regulated by O-linked N-acetylglucosamine transferase (Ogt) for target gene repression in mouse embryonic stem cells. J Biol Chem 288, 20776– 20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Ng HH, 2017. The metabolic programming of stem cells. Genes Dev 31, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes-Costa M, Bronner ME, 2015. Establishing neural crest identity: a gene regulatory recipe. Development 142, 242–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Garic A, Flentke GR, Berres ME, 2014. Neural crest development in fetal alcohol syndrome. Birth Defects Res C Embryo Today 102, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, Fischer KA, Devi A, Detraux D, Gu H, Battle SL, Showalter M, Valensisi C, Bielas JH, Ericson NG, Margaretha L, Robitaille AM, Margineantu D, Fiehn O, Hockenbery D, Blau CA, Raftery D, Margolin AA, Hawkins RD, Moon RT, Ware CB, Ruohola-Baker H, 2015. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol 17, 1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K, Semenza GL, 2011. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9, 298–310. [DOI] [PubMed] [Google Scholar]
- Valdez BC, Henning D, So RB, Dixon J, Dixon MJ, 2004. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proceedings of the National Academy of Sciences of the United States of America 101, 10709–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley C.A.t., Ramalho-Santos J, Van Houten B, Schatten G, 2011. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 6, e20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Lopez GA, Cerrizuela S, Tribulo C, Aybar MJ, 2018. Neurocristopathies: New insights 150 years after the neural crest discovery. Dev Biol 444 Suppl 1, S110–S143. [DOI] [PubMed] [Google Scholar]
- Wahl SE, Kennedy AE, Wyatt BH, Moore AD, Pridgen DE, Cherry AM, Mavila CB, Dickinson AJ, 2015. The role of folate metabolism in orofacial development and clefting. Dev Biol 405, 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XY, Li S, Wang G, Ma ZL, Chuai M, Cao L, Yang X, 2015. High glucose environment inhibits cranial neural crest survival by activating excessive autophagy in the chick embryo. Sci Rep 5, 18321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB, 2012. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol 13, 270–276. [DOI] [PubMed] [Google Scholar]
- Wentzel P, Eriksson UJ, 2011. Altered gene expression in rat cranial neural crest cells exposed to a teratogenic glucose concentration in vitro: paradoxical downregulation of antioxidative defense genes. Birth Defects Res B Dev Reprod Toxicol 92, 487–497. [DOI] [PubMed] [Google Scholar]
- Wozniak JR, Riley EP, Charness ME, 2019. Clinical presentation, diagnosis, and management of fetal alcohol spectrum disorder. Lancet Neurol 18, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Ji KY, Zhang J, Xu Y, Ying Y, Mai T, Xu S, Zhang QB, Yao KT, Xu Y, 2019. Core pluripotency factors promote glycolysis of human embryonic stem cells by activating GLUT1 enhancer. Protein Cell 10, 668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun HR, Jo YH, Kim J, Shin Y, Kim SS, Choi TG, 2020. Roles of Autophagy in Oxidative Stress. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, Ding J, Czyz D, Hu R, Ye Z, He M, Zheng YG, Shuman HA, Dai L, Ren B, Roeder RG, Becker L, Zhao Y, 2019. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ratanasirintrawoot S, Chandrasekaran S, Wu Z, Ficarro SB, Yu C, Ross CA, Cacchiarelli D, Xia Q, Seligson M, Shinoda G, Xie W, Cahan P, Wang L, Ng SC, Tintara S, Trapnell C, Onder T, Loh YH, Mikkelsen T, Sliz P, Teitell MA, Asara JM, Marto JA, Li H, Collins JJ, Daley GQ, 2016. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell stem cell 19, 66–80. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li Y, Peng Y, 2018. A review of interventions against fetal alcohol spectrum disorder targeting oxidative stress. Int J Dev Neurosci 71, 140–145. [DOI] [PubMed] [Google Scholar]
- Zheng J, 2012. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett 4, 1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, Ruohola-Baker H, 2012. HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J 31, 2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]