

SUMMARY:
Erdheim-Chester disease is a rare non-Langerhans cell histiocytosis. The disease is widely variable in its severity, ranging from incidental findings in asymptomatic patients to a fatal multisystem illness. CNS involvement occurs in up to one-half of patients, most often leading to diabetes insipidus and cerebellar dysfunction. Imaging findings in neurologic Erdheim-Chester disease are often nonspecific, and the disease is commonly mistaken for close mimickers. Nevertheless, there are many imaging manifestations of Erdheim-Chester disease that are highly suggestive of the disease, which an astute radiologist could use to accurately indicate this diagnosis. This article discusses the imaging appearance, histologic features, clinical manifestations, and management of Erdheim-Chester disease.
A 24-year-old previously healthy man presented to an outside institution with a subacute history of neurologic symptoms that began 2 weeks after a coronavirus disease 2019 (COVID-19) immunization booster, including headaches, slurred speech, and imbalance. MR imaging showed multifocal enhancing abnormalities of his brainstem and spinal cord. CSF analysis detected no oligoclonal bands and 2 white blood cells/high power field (protein = 99 mg/dL, glucose = 67 mg/dL). The patient was given IV methylprednisolone and an oral prednisone taper after testing had ruled out an infection. His symptoms subsequently improved but did not resolve. On examination at our institution, he had an ataxic dysarthria, gait ataxia, and bilateral leg spasticity. After a water-deprivation test, blood and urine analysis revealed elevated prolactin and central diabetes insipidus, with plasma hyperosmolality (302 mOsm/kg) and low urine osmolality (144 mOsm/kg) in the setting of polyuria. Repeat CSF analysis findings were unremarkable except for 8 white cells/high power field (protein = 136 mg/dL and glucose = 49 mg/dL). The patient was then sent for additional MR imaging of his neuraxis.
Imaging
Initial CT images obtained at the outside institution demonstrated patchy areas of hypoattenuation centered around the dentate nuclei of both cerebellar hemispheres. MR imaging demonstrated extensive multifocal T2-hyperintense signal abnormalities and regions of poorly marginated patchy enhancement. These were predominantly located in an almost symmetric distribution in the midbrain, dentate nuclei, and cerebellar peduncles (Fig 1). None of the abnormalities demonstrated restricted diffusion. There was a mild local mass effect associated with these findings, without obstructive hydrocephalus. Lesser involvement was observed in the periatrial white matter and optic chiasm. The pituitary infundibulum was thickened and demonstrated prominent enhancement. In the spine, similar T2-hyperintense enhancing abnormalities were seen throughout the cervical and thoracic cord (Fig 2). A whole-body PET was performed to evaluate a systemic disorder, which revealed FDG uptake in the distal femoral diaphysis with increased intramedullary density on the corresponding CT images.
FIG 1.

MR images of the posterior fossa demonstrate multifocal abnormalities involving both cerebellar hemispheres, particularly located around the dentate nuclei. These regions are bright on T2 and FLAIR (arrows in A and B), lack restricted diffusion (C), are hypointense on T1 (arrows in D), and demonstrate patchy enhancement on postcontrast T1 fat-saturated images (E). Similar patchy enhancement is also noted in the midbrain (dashed oval on F).
FIG 2.

MR imaging of the cervical (A) and thoracic (B and C) spine demonstrates numerous patchy areas of enhancement along the cervicothoracic cord (A and B). Corresponding fat-saturated STIR images of the thoracic spine demonstrate multifocal T2 hyperintensities in these regions (C).
During the patient’s numerous imaging examinations, a long list of potential diagnoses was brought forth. The imaging appearance and temporal relation with the patient’s immunization booster raised the question of a viral encephalomyelitis. Neurosarcoidosis, tuberculous, and primary CNS lymphoma were also considered as possibilities, given the multifocal signal abnormalities. Demyelinating disease was thought unlikely, given the distribution of findings and lumbar puncture results. Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS) was similarly considered unlikely, given the lack of a complete radiologic response to steroids and the size of the enhancing lesions.
Operative Report
The decision was made to perform a left cerebellar stereotactic needle biopsy, because it was deemed the safest area with the highest yield for diagnosis. An initial biopsy attempt was aborted because brisk bleeding was encountered after early tissue sampling, pathology was nondiagnostic, and the patient’s postoperative course was uneventful. The second procedure was planned a few weeks later using a slightly different trajectory while targeting the same region. The patient was anesthetized and positioned lateral with the head fixed in a Mayfield head holder. By means of neuronavigation, a linear incision was performed over the planned entry point, and a new burr-hole was obtained. After a small durotomy and corticotomy were performed, a stereotactic biopsy needle was carefully advanced to the target under neuronavigation guidance. Several cylinders of tissue were obtained for biopsy without incident. Frozen pathology was consistent with lesional tissue. The patient awoke at his neurologic baseline and had an uneventful postoperative course.
Pathology
Histopathologic diagnosis in this case was very challenging. The initial cerebellar biopsy was nondiagnostic, showing only the cerebellar cortex with nonspecific Purkinje cell loss and Bergmann gliosis. The subsequent biopsy demonstrated white matter with mild, diffuse hypercellularity (Fig 3). The increased cellularity was due predominantly to foamy macrophages with small nuclei, present in a perivascular distribution and infiltrating the white matter (highlighted by CD68 immunostain). Although cytologically bland, by immunohistochemistry, the foamy macrophages showed apparent expression of BRAF p.V600E, a finding that was confirmed by digital droplet polymerase chain reaction. There was no acute inflammation, multinucleated giant cells, or eosinophils. Stains to evaluate a viral etiology or glial neoplasm were negative. Overall, in the context of the clinical history and imaging, the findings were most consistent with Erdheim-Chester disease (ECD). Subsequently, the patient underwent biopsy of a skin lesion on his back. Histopathologic findings of that biopsy were much more impressive, with numerous, large BRAF-mutant foamy histocytes and Touton giant cells, findings characteristic of ECD.
FIG 3.

H&E-stained sections of the cerebellar biopsy (A) demonstrate mildly hypercellular white matter with increased foamy macrophages (black arrows), highlighted by CD68 immunostain (B). The macrophages were cytologically bland but show expression of BRAF p.V600E (C), supporting the presence of a histiocytic neoplasm. A subsequent skin biopsy (D) shows more characteristic features of ECD, including large, foamy macrophages (black arrows) and Touton giant cells (red arrows), which were positive for BRAF p.V600E (E). Original magnification ×200 for all images.
DISCUSSION
ECD is a rare non-Langerhans cell histiocytosis, in which tissues are infiltrated by lipid-laden histiocytes.1 Affected patients are typically between their fifth and seventh decades of life, and the disease has a predilection for men.2 ECD can affect any tissue, and the clinical manifestations are extremely heterogeneous.3 Because bone, particularly the appendicular skeleton, is almost universally involved, patients may present with nonspecific bone pain; however, bony disease is frequently asymptomatic.4 The severity of ECD ranges from incidental discovery in asymptomatic patients to smoldering focal involvement and multisystem fatal forms.5
CNS involvement occurs in up to 50% of patients with ECD.6,7 The most commonly involved sites are the brain parenchyma, hypothalamic pituitary axis, and meninges.8 Clinical symptoms of neurologic ECD are dependent on the sites of involvement. Diabetes insipidus is the most frequent manifestation; seizures, pontocerebellar dysfunction, and other neuroendocrine abnormalities including panhypopituitarism are also common.9 In rare cases, ECD can present as a neuropsychiatric disorder or cognitive decline.10
The imaging appearance of neurologic ECD is dependent on distribution and extent. Intracranially, ECD often involves the posterior fossa. The most common findings are multifocal FLAIR hyperintensities of the dentate nuclei and brainstem, demonstrating variable enhancement, with or without atrophy of the affected structures.11 Supratentorial structures, too, can be involved, though less often. In other patients, dural-based enhancing masses are seen, sometimes with a stellate appearance.1,12 Sella imaging may demonstrate enlargement of the pituitary gland and/or stalk, sometimes with signal alterations of the affected structures, including absence of the expected hyperintense T1 signal in the posterior pituitary gland. Orbital involvement is frequent and includes infiltrating soft-tissue masses with concurrent exophthalmos. Associated osteosclerosis of the maxillofacial bones may be present.11 Spinal involvement is less common and has variable appearances. Some patients have multifocal T2-hyperintense, enhancing intramedullary lesions.13 Others have extramedullary masses that may compress or even infiltrate the cord (Fig 4).
FIG 4.
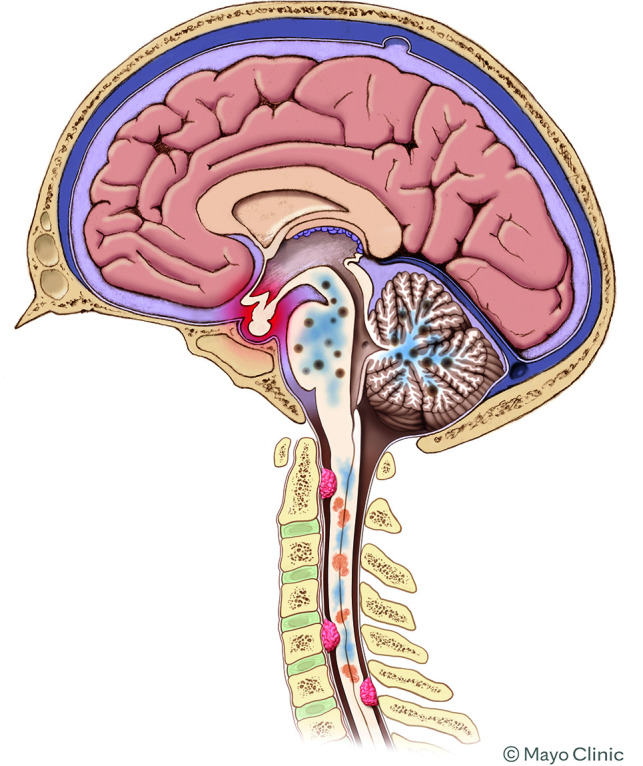
Schematic showing the various types of involvement of ECD in the CNS. Intracranially, the most common findings are focal areas of enhancement in the brainstem and/or cerebellum, with surrounding edema. The pituitary infundibulum is commonly enlarged. In the spine, both intramedullary lesions and extramedullary lesions are observed, which are often associated with adjacent edema.
The imaging findings of CNS ECD are protean and location-dependent; as a result, the differential considerations are highly variable. The patchy brainstem enhancement seen in many patients can mimic CLIPPERS or CNS lymphoma but tends to be more confluent than CLIPPERS and does not respond to steroids like these 2 entities.14 Supratentorial involvement of white matter lesions, though rare, may raise concern for MS. Dural-based masses can share the appearance of meningiomas, though this is more common in Rosai-Dorfman disease (RDD).15,16 In the spine, extramedullary masses along nerve roots have been mistaken for schwannomas.17
Other histiocytic disorders can also involve the CNS. RDD is characterized by histiocyte proliferation that leads to lymphatic sinus dilation. Although patients often present with painless cervical lymphadenopathy, extranodal disease is not uncommon.18 As stated above, intracranial manifestations of RDD mimic meningiomas: The disease typically presents as dural-based masses, often around the cavernous sinuses and sella region.19
Langerhans cell histiocytosis (LCH) is characterized by accumulations of epidermal dendritic cells. CNS involvement is relatively rare, usually presenting as a pituitary dysfunction such as diabetes insipidus. On imaging, the most common findings are thickening of the pituitary stalk, an enhancing suprasellar mass, and loss of normal T1 signal in the posterior pituitary gland.15 Neurodegenerative changes can also be seen, presenting as symmetric signal abnormalities in the dentate nuclei, pons, and basal ganglia, as well as focal or diffuse parenchymal atrophy.20 Patients also commonly have skeletal involvement, with lytic lesions in the skull and vertebra plana in the spine (Table).
Demographics and most frequently observed CNS imaging features of ECD, RDD, and LCH
| ECD | LCH | RDD | |
|---|---|---|---|
| Demographics | M>>F, 50–70 yrs | M > F, 1–3 yrs | M > F, young adults |
| Presentation | Commonly asymptomatic; CNS involvement leads to diabetes insipidus, pontocerebellar symptoms, and seizures | Nonspecific, including lethargy, fever, bone pain, diabetes insipidus | Lymphadenopathy, night sweats, weight loss |
| CNS imaging | Patchy enhancement and T2 hyperintensity in the posterior fossa; enlargement and enhancement of the pituitary stalk; loss of posterior pituitary T1 signal; intra- and extra medullary spinal lesions | Enlargement and enhancement of the pituitary stalk; loss of posterior pituitary T1 signal; neurodegenerative changes in the basal ganglia, pons, and dentate nuclei; parenchymal atrophy | Enlarged cervical lymph nodes; enhancing dural-based masses that mimic meningiomas |
Note:—M indicates male; F, female.
Fortunately, involvement of extra-CNS organs can provide further diagnostic clues. The most common finding is sclerotic osseous lesions symmetrically involving the diaphysis of the long bones. On technetium Tc99m hydroxymethylene diphosphonate bone scans, these will demonstrate strong uptake, particularly in the distal metaphyseal regions of the femur and proximal epiphyseal regions of the tibia (the so-called “hot knees” sign, which is pathognomonic of ECD).21,22 Perinephric fibrosis related to fat infiltration leads to the appearance of “hairy kidneys,” while retroperitoneal fibrosis can encase the descending aorta (“coated aorta”).23,24
Histologically, ECD is characterized by infiltrates of foamy, lipid-laden histocytes with small nuclei. Multinucleated Touton giant cells are frequently present. By immunohistochemistry, the neoplastic cells are highlighted by the histiocytic markers CD68 and CD163 but are negative for CD1a and langerin, differentiating ECD from LCH, and are negative or weakly positive for S-100, differentiating ECD from RDD.25 The histologic findings in CNS ECD may be subtle relative to other organ systems, and confirmation of a mitogen-activated protein kinase (MAPK) alteration is often necessary to establish a definitive diagnosis. The mutation-specific BRAF p.V600E antibody can identify cases having this alteration. Identification of other MAPK alterations requires sequencing studies. MAPK alterations are not specific to ECD and may be observed in LCH as well as gliomas and other tumor types. Therefore, diagnosis of ECD requires integration of clinical, histologic, and molecular features.
Current treatment of these disorders relies on identification of pathogenic mutations in the affected tissue and treatment with pathway inhibitors. Patients with CNS involvement invariably require treatment, whereas isolated single-system disease such as pulmonary disease can often be monitored. Vemurafenib, a BRAF inhibitor, was the first FDA-approved therapy for histiocytic disorders for patients with the BRAF p.V600E mutation.26 Cobimetinib is a mitogen-activated ERK kinase (MEK) inhibitor, which was approved in 2022 for treatment of ECD, regardless of mutation status. For patients not responding to these agents who do not have another targetable mutation, cytotoxic therapies such as cladribine or methotrexate are typically used.
The treatment of ECD has evolved with time.27 Surgery, radiation therapy, steroids, cytotoxic drugs, and interferon-α have all been used with variable efficacy.28,29 Then, multiple studies found that more than one-half of ECD tissue samples demonstrated BRAF p.V600E mutations.30,31 This discovery led to the use of vemurafenib, a BRAF p.V600E inhibitor, which has shown remarkable efficacy in early reports.32,33 It was subsequently found that patients lacking BRAF p.V600E mutations often have activating mutations elsewhere in the MAPK pathway.34 This finding led to other treatment options, including MEK and rapidly accelerated fibrosarcoma inhibitors.35 Still, no standardized treatment regimen exists; management is usually based on disease severity, the results of genomic analyses, and clinicians’ preferences.
The prognosis of patients with ECD is highly variable and is predominantly influenced by specific organ involvement. CNS disease tends to respond rapidly to BRAF/MEK inhibition if enhancing lesions are present, though in patients with nonenhancing, presumably chronic lesions, residual deficits are typical. A 2011 study of 53 patients found that the 5-year survival was 68%, though these results were published before recent treatment advances.36 Both increased age and CNS involvement are independent predictors of poor prognoses.36,37 In patients with BRAF p.V600E–mutated disease, almost all stabilize or respond to therapy if they tolerate the medication.5,38 Follow-up imaging is typically performed every 3–6 months early on in the treatment course.
After initiation of vemurafenib, this patient had a substantial improvement in gait ataxia within 1 week, going from walking with a walker to no gait aid. At 3-month follow up, he was walking without aid, had returned to work, and had a persistent moderate ataxic dysarthria. MR imaging of the brain at that time demonstrated a reduction in the degree of enhancement within his dentate nuclei.
Case Summary
CNS involvement of ECD is variable. Characteristic findings include patchy T2 hyperintensities and/or enhancement in the brainstem and cerebellum, thickening of the pituitary infundibulum, and dural-based extra-axial masses.
Diabetes insipidus is the most common clinical manifestation of neurologic ECD and can precede the diagnosis by up to a decade. Other neurologic symptoms are dependent on the sites of involvement.
Diagnosis is best made by biopsy, with molecular genetic testing to evaluate causative mutations.
Identification of the biopsy target using vertex-to-toes PET/CT is an essential component of evaluation of these disorders.
Histopathologic findings of CNS ECD can be very subtle relative to other organ systems. In the proper clinical context, molecular testing for BRAF or other MAPK pathway alterations can aid in establishing a definitive diagnosis.
Treatment strategies have evolved during recent years. Targeted therapeutic regimens are now often based on genomic analyses of tissue samples.
ABBREVIATIONS:
- CLIPPERS
chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids
- ECD
Erdheim-Chester disease
- LCH
Langerhans cell histiocytosis
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated ERK kinase
- RDD
Rosai-Dorfman disease
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- 1.Sedrak P, Ketonen L, Hou P, et al. Erdheim-Chester disease of the central nervous system: new manifestations of a rare disease. AJNR Am J Neuroradiol 2011;32:2126–31 10.3174/ajnr.A2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oliveira M, Monteiro S, Dos Santos J, et al. Erdheim-Chester disease: a rare clinical entity. Eur J Case Rep Intern Med 2020;7:001630 10.12890/2020_001630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Todisco A, Cavaliere C, Vaglio A, et al. Erdheim-Chester disease: a challenging diagnosis for an effective therapy. Clin Neurol Neurosurg 2020;194:105841 10.1016/j.clineuro.2020.105841 [DOI] [PubMed] [Google Scholar]
- 4.Sánchez-Villalobos JM, Jimeno-Almazán A, López-Peña C, et al. Erdheim-Chester disease mimicking multiple sclerosis or a new association? Mult Scler Relat Disord 2019;30:94–97 10.1016/j.msard.2019.02.008 [DOI] [PubMed] [Google Scholar]
- 5.Pegoraro F, Papo M, Maniscalco V, et al. Erdheim-Chester disease: a rapidly evolving disease model. Leukemia 2020;34:2840–57 10.1038/s41375-020-0944-4 [DOI] [PubMed] [Google Scholar]
- 6.Cohen Aubart F, Idbaih A, Galanaud D, et al. Central nervous system involvement in Erdheim-Chester disease: an observational cohort study. Neurology 2020;95:e2746–54 10.1212/WNL.0000000000010748 [DOI] [PubMed] [Google Scholar]
- 7.Garg N, Lavi ES. Clinical and neuroimaging manifestations of Erdheim-Chester disease: a review. J Neuroimaging 2021;31:35–44 10.1111/jon.12785 [DOI] [PubMed] [Google Scholar]
- 8.Abdelfattah AM, Arnaout K, Tabbara IA. Erdheim-Chester disease: a comprehensive review. Anticancer Res 2014;34:3257–61 [PubMed] [Google Scholar]
- 9.Manaka K, Sato J, Makita N. Neuroendocrine manifestations of Erdheim-Chester disease. Handb Clin Neurol 2021;181:137–47 10.1016/B978-0-12-820683-6.00010-5 [DOI] [PubMed] [Google Scholar]
- 10.Cives M, Simone V, Rizzo FM, et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol 2015;95:1–11 10.1016/j.critrevonc.2015.02.004 [DOI] [PubMed] [Google Scholar]
- 11.Kumar P, Singh A, Gamanagatti S, et al. Imaging findings in Erdheim-Chester disease: what every radiologist needs to know. Pol J Radiol 2018;83:e54–62 10.5114/pjr.2018.73290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caparros-Lefebvre D, Pruvo JP, Rémy M, et al. Neuroradiologic aspects of Chester-Erdheim disease. AJNR Am J Neuroradiol 1995;16:735–40 [PMC free article] [PubMed] [Google Scholar]
- 13.Jeon I, Choi JH. Isolated thoracic intramedullary Erdheim-Chester disease presenting with paraplegia: a case report and literature review. BMC Musculoskelet Disord 2021;22:270 10.1186/s12891-021-04061-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berkman J, Ford C, Johnson E, et al. Misdiagnosis: CNS Erdheim–Chester disease mimicking CLIPPERS. Neuroradiol J 2018;31:399–402 10.1177/1971400917710251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Camelo-Piragua S, Abdullah A, et al. Neuroimaging features of CNS histiocytosis syndromes. Clin Imaging 2020;60:131–40 10.1016/j.clinimag.2019.10.001 [DOI] [PubMed] [Google Scholar]
- 16.Johnson MD, Aulino JP, Jagasia M, et al. Erdheim-Chester disease mimicking multiple meningiomas syndrome. AJNR Am J Neuroradiol 2004;25:134–37 [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Z, Li S, Hong J, et al. Erdheim-Chester disease mimicking lumbar nerve schwannoma: case report and literature review. Spinal Cord Ser Cases 2019;5:90 10.1038/s41394-019-0234-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unadkat BS, Kashikar SV, Bhansali PJ, et al. Intracranial Rosai-Dorfman disease: a case to remember. Cureus 2022;14:e32605 10.7759/cureus.32605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng X, Cheng JL, Gao AK. A study on clinical characteristics and magnetic resonance imaging manifestations on systemic Rosai-Dorfman disease. Chin Med J (Engl) 2018;131:440–47 10.4103/0366-6999.225053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prosch H, Grois N, Wnorowski M, et al. Long-term MR imaging course of neurodegenerative Langerhans cell histiocytosis. AJNR Am J Neuroradiol 2007;28:1022–28 10.3174/ajnr.A0509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerudelli E, Gazzilli M, Bertoli M, et al. Erdheim-Chester disease: the power of nuclear medicine imaging. Rev Esp Med Nucl Imagen Mol (Engl Ed) 2020;39:323–24 10.1016/j.remn.2020.01.003 [DOI] [PubMed] [Google Scholar]
- 22.Fasulo S, Alkomos MF, Pjetergjoka R, et al. Erdheim-Chester disease presenting at the central nervous system. Autops Case Rep 2021;11:e2021321 10.4322/acr.2021.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu SZ, Zhou X, Song A, et al. Exophthalmos and coated aorta in Erdheim–Chester disease. Rheumatology (Oxford) 2020;59:2651–52 10.1093/rheumatology/keaa022 [DOI] [PubMed] [Google Scholar]
- 24.Hazim AZ, Acosta-Medina AA, Young JR, et al. ; Mayo Clinic-University of Alabama at Birmingham Histiocytosis Working Group. Classical and non-classical phenotypes of Erdheim-Chester disease: correlating clinical, radiographic and genotypic findings. Br J Haematol 2022;199:454–57 10.1111/bjh.18422 [DOI] [PubMed] [Google Scholar]
- 25.Emile JF, Tirabosco R, Demicco, Elizabeth. WHO Classification of Tumours Editorial Board. Central Nervous System Tumours. Vol 6. 5th ed. 2021 [Google Scholar]
- 26.Go RS, Jacobsen E, Baiocchi R, et al. Histiocytic Neoplasms, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2021;19:1277–303 10.6004/jnccn.2021.0053 [DOI] [PubMed] [Google Scholar]
- 27.Munoz J, Janku F, Cohen PR, et al. Erdheim-Chester disease: characteristics and management. Mayo Clin Proc 2014;89:985–96 10.1016/j.mayocp.2014.01.023 [DOI] [PubMed] [Google Scholar]
- 28.Pan Z, Kleinschmidt-DeMasters BK. CNS Erdheim-Chester disease: a challenge to diagnose. J Neuropathol Exp Neurol 2017;76:986–96 10.1093/jnen/nlx095 [DOI] [PubMed] [Google Scholar]
- 29.Haque A, Pérez CA, Reddy TA, et al. Erdheim-Chester disease with isolated CNS involvement: a systematic review of the literature. Neurol Int 2022;14:716–26 10.3390/neurolint14030060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Badalian-Very G. A common progenitor cell in LCH and ECD. Blood 2014;124:991–92 10.1182/blood-2014-06-581736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytosis. Blood 2012;120:2700–03 10.1182/blood-2012-05-430140 [DOI] [PubMed] [Google Scholar]
- 32.Tzoulis C, Schwarzlmüller T, Gjerde IO, et al. Excellent response of intramedullary Erdheim-Chester disease to vemurafenib: a case report. BMC Res Notes 2015;8:171 10.1186/s13104-015-1135-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costa S, Julião MJ, Silva S, et al. Erdheim-Chester disease: a rare non-Langerhans histiocytosis. BMJ Case Rep 2021;14:e24114 10.1136/bcr-2020-241143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starkebaum G, Hendrie P. Erdheim-Chester disease. Best Pract Res Clin Rheumatol 2020;34:101510 10.1016/j.berh.2020.101510 [DOI] [PubMed] [Google Scholar]
- 35.Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov 2016;6:154–65 10.1158/2159-8290.CD-15-0913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnaud L, Hervier B, Néel A, et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood 2011;117:2778–82 10.1182/blood-2010-06-294108 [DOI] [PubMed] [Google Scholar]
- 37.Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim-Chester disease: clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996;75:157–69 10.1097/00005792-199605000-00005 [DOI] [PubMed] [Google Scholar]
- 38.Diamond EL, Subbiah V, Lockhart AC, et al. Vemurafenib for BRAF V600-mutant Erdheim-Chester disease and Langerhans cell histiocytosis: analysis of data from the histology-independent, Phase 2, open-label VE-BASKET study. JAMA Oncol 2018;4:384–88 10.1001/jamaoncol.2017.5029 [DOI] [PMC free article] [PubMed] [Google Scholar]