

Abstract
Apart from the anti-GD2 antibody, immunotherapy for neuroblastoma has had limited success due to immune evasion mechanisms, coupled with an incomplete understanding of predictors of response. Here, from bulk and single-cell transcriptomic analyses, we identify a subset of neuroblastomas enriched for transcripts associated with immune activation and inhibition and show that these are predominantly characterized by gene expression signatures of the mesenchymal lineage state. By contrast, tumors expressing adrenergic lineage signatures are less immunogenic. The inherent presence or induction of the mesenchymal state through transcriptional reprogramming or therapy resistance is accompanied by innate and adaptive immune gene activation through epigenetic remodeling. Mesenchymal lineage cells promote T cell infiltration by secreting inflammatory cytokines, are efficiently targeted by cytotoxic T and natural killer cells and respond to immune checkpoint blockade. Together, we demonstrate that distinct immunogenic phenotypes define the divergent lineage states of neuroblastoma and highlight the immunogenic potential of the mesenchymal lineage.
Derived from the developing neural crest, neuroblastoma is a common solid tumor of childhood that manifests as extracranial masses arising in the adrenal medulla or sympathetic ganglia. Approximately half of all patients have high-risk features—age >18 months, distant metastases and unfavorable histologic and biological factors including amplification of the MYCN oncogene—which are associated with an adverse prognosis1. Dinutuximab, a monoclonal antibody that promotes immune cell-mediated cytotoxicity against neuroblastoma cells by targeting the disialoganglioside GD2 (ref. 2), has substantially improved survival rates in high-risk patients3. While adoptive natural killer (NK) cells in combination with dinutuximab have shown promising clinical results4, these cells do not provide long-term immunological memory, thereby calling for strategies that lead to more durable responses. However, active immunotherapy approaches involving T cells, including checkpoint inhibition5–7 or adoptive transfer of chimeric antigen receptor (CAR)-modified T cells8,9, have also had limited clinical success.
The major impediments to achieving significant clinical benefit in neuroblastoma include both tumor-intrinsic mechanisms that contribute to immune evasion (IE) and the presence of an immunosuppressive tumor microenvironment (TME)10. These include a low mutational burden leading to a paucity of neoepitopes that can be recognized by cytotoxic T lymphocytes (CTLs)11, downregulation of major histocompatibility complex (MHC) genes and defects in antigen processing and presentation (APP) that render cells resistant to CTLs12, downregulation of cell-surface ligands required for NK cell activation13 and upregulation of checkpoint proteins that protect against NK cell-mediated lysis14 and macrophage-mediated phagocytosis15. Additionally, infiltration of suppressive immune cells, such as T regulatory (Treg) cells16, tumor-associated macrophages17 and myeloid-derived suppressor cells18, and secreted immunosuppressive factors contribute to the generation of a TME that hinders an effective immune response.
Amplification of the MYCN oncogene poses another distinct challenge to immunotherapy against neuroblastoma. This transcription factor is amplified in approximately 50% of high-risk cases and is associated with aggressive disease and a poor clinical outcome19. MYCN-amplified tumors consistently evade immune destruction by downregulating MHC class I molecules12 and are associated with poor infiltration of cytotoxic CD8+ T cells20 and reduced expression of NK cell ligands21. Approximately half of high-risk neuroblastomas do not express amplified MYCN, and their capacity to induce a productive immune response remains unclear.
Thus, although many of the mechanisms of IE in neuroblastoma are known, further understanding of tumor–host interactions will be crucial to enhancing the ability of immunotherapy to target and eliminate tumor cell populations. Especially challenging is the genetic and biologic heterogeneity of neuroblastoma, which makes it difficult to identify factors that indicate the likelihood of an effective immune response and thereby select patients who are most likely to benefit from immunotherapy. Hence, we undertook an unbiased analysis of gene expression signatures across diverse clinical subtypes of primary neuroblastomas and identify the existence of distinct immunogenic phenotypes based on cell lineage state.
Results
A subset of neuroblastomas expresses immune response genes.
To determine whether neuroblastomas are capable of eliciting an immune response, we began by first quantifying tumor-to-tumor gene expression variability in RNA-sequencing (RNA-seq) data from 498 well-annotated primary human tumors representing diverse clinical and genetic subtypes (SEQC-498; GSE49711/GSE62564; Methods). Clustering analysis based on similar tumor transcription profiles yielded four distinct clusters (Fig. 1a, Extended Data Fig. 1a–c and Supplementary Table 1). Cluster 1 (C1, ‘Hi-MYCN’; n = 103, 20%) was enriched for MYCN target genes involved in cell proliferation and biosynthesis (Fig. 1a–c, Extended Data Fig. 1d and Supplementary Table 2), and not surprisingly, segregated primarily with MYCN-amplified tumors (Fig. 1d and Extended Data Fig. 1e). The remaining clusters consisted mainly of MYCN-nonamplified tumors of which cluster 2 (C2, ‘neuronal’; n = 241), making up the largest proportion (48%), was enriched for genes involved in nervous system development (Fig. 1a–c and Extended Data Fig. 1d). Cluster 3 (C3, n = 140), forming 28%, was enriched for tumors that expressed genes involved in immune function—interferon-γ (IFN-γ) response and T cell inflammation and activation—and hence was designated ‘immunogenic’. Cluster 4 (C4, ‘metabolic’; n = 14, 3%), clearly distinct, consisted of International Neuroblastoma Staging System (INSS) stage 4S tumors that expressed fatty acid and cholesterol homeostasis transcripts. The neuronal and metabolic tumors arose predominantly in children <18 months of age with INSS stages 1–3 and 4S disease, while tumors within the immunogenic cluster were associated with age ≥18 months and metastatic disease (n = 66, 47%) (Fig. 1d and Extended Data Fig. 1e). To ensure that these results were not confined to one dataset, we analyzed a 394-neuroblastoma tumor cohort (GSE120572) and similarly noted an immunogenic cluster, again denoting the presence of immune response gene expression in a subset of primary tumors (Extended Data Fig. 1f,g and Methods).
Fig. 1 |. A subset of neuroblastomas expresses signatures of an immune response.
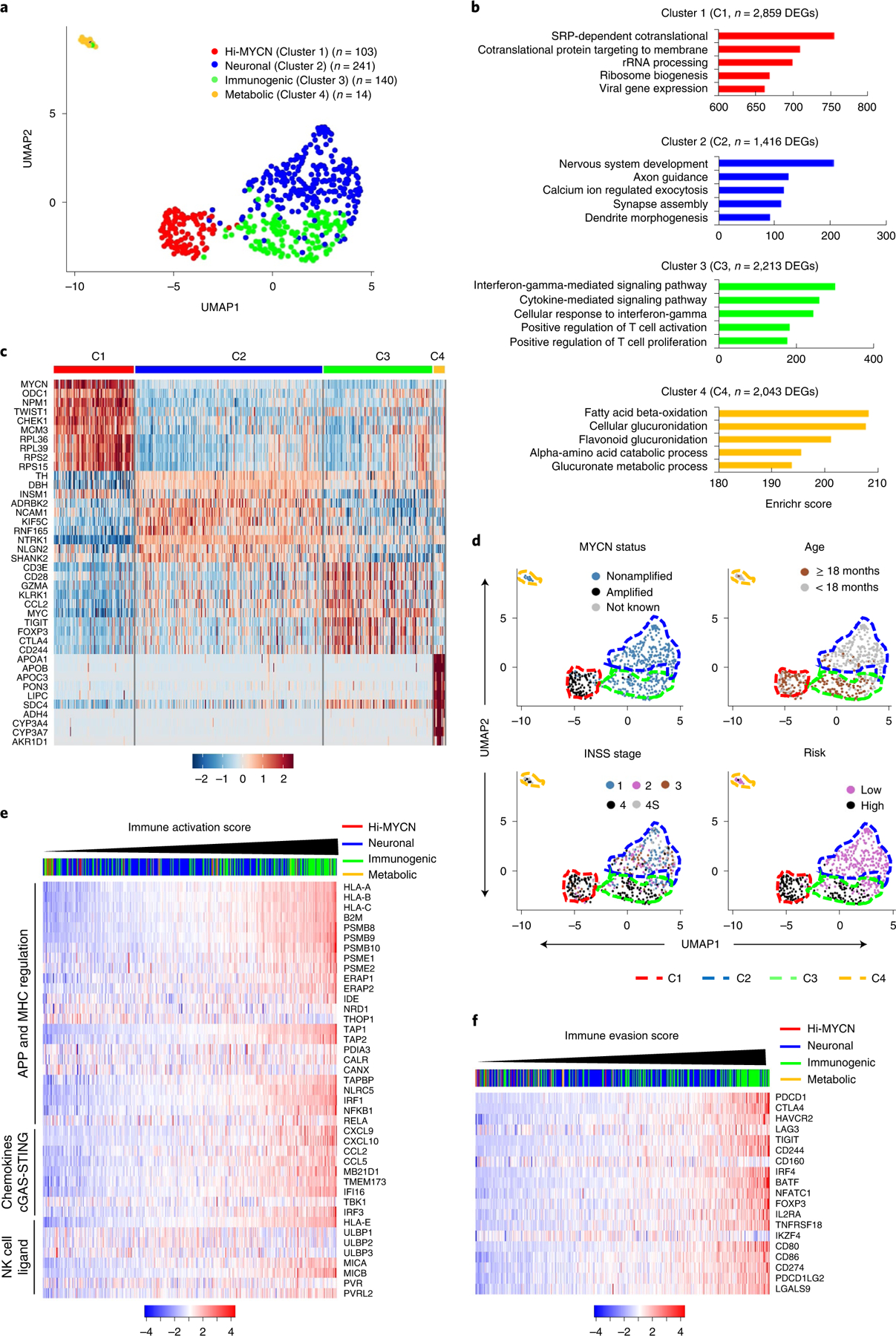
a, Two-dimensional UMAP representations of the gene expression profiles in 498 neuroblastoma (NB) tumors. Each dot represents a tumor. The top 5,000 highly variable genes were selected based on the variance-stabilizing method and 20 significant principle components (PCs) selected and processed to generate clusters representing four NB subtypes. Differentially expressed genes (DEGs) were identified for each cluster using the receiver operating characteristics (ROC) curve to compare one cluster with the other three (log2FC > 0.25). b, GO analysis of DEGs in the four clusters. Significance was assessed by the two-tailed Fisher’s exact test. c, Heat map of expression values of ten representative DEGs within each cluster. Rows are z-score-scaled average expression levels for each gene in all four clusters. d, UMAP visualization of the distribution of the indicated prognostic features among the four clusters. e,f, Heat map of z-score-transformed log2 normalized expression values of IA (e) and IE (f) genes in MYCN-nonamplified NBs (n = 401 tumors). Tumors were ranked based on increasing IA or IE scores. Cluster annotations of the tumors are indicated on the top horizontal bar.
To pursue the immune genes that were differentially enriched in the immunogenic cluster, we generated an immune activation (IA) score based on the relative expression of a curated set of 41 genes with major roles in tumor cell-intrinsic immune function, such as regulation of MHC expression, APP, NK cell recognition and T and NK cell infiltration (Supplementary Table 3). Among the MYCN-nonamplified tumors, a significant number with the highest IA scores fell within the immunogenic and metabolic clusters, while those with intermediate or lower scores were associated with the neuronal and Hi-MYCN clusters, respectively (Fig. 1e, Extended Data Fig. 1h and Supplementary Table 4). Because a cytotoxic immune response is generally accompanied by immune suppression or evasion22,23, we determined whether immune suppression was also represented in these tumors using an IE score based on the relative expression of 19 genes, most of which were markers of T cell dysfunction (Supplementary Table 3). Again, the immunogenic tumor cluster had significantly higher IE scores compared to those of the neuronal and metabolic clusters (Fig. 1f and Extended Data Fig. 1i). We also observed enrichment for IA and IE scores in the immunogenic tumor cluster in the second dataset (GSE120572) (Extended Data Fig. 1j). Consistent with the known poor immunogenicity of MYCN-amplified tumors16,20, tumors within the Hi-MYCN cluster generally had the lowest IA and IE scores, although a small subset (13 of 103 (12.6%)) had scores that were comparable to those within the immunogenic cluster (Extended Data Fig. 1h,i). Therefore, taken together, while the majority of neuroblastomas do not possess an immune response gene signature, a subset, comprising mainly MYCN-nonamplified tumors, have gene expression profiles that are closely linked to immune responsiveness.
Immune signatures are associated with the mesenchymal lineage.
Having identified subsets of neuroblastomas with the potential for immunogenicity, we next sought a biomarker that might consolidate the complex interactions between the immune system and the tumor cells. Therefore, we performed a modular gene coexpression analysis of the 140 tumor transcriptomes within the immunogenic cluster to identify biologically relevant pathways based on similar gene expression patterns24. Of the five modules identified (Extended Data Fig. 2a), M1, with the highest number of coexpressed genes, contained gene sets enriched for epithelial-to-mesenchymal transition, inflammatory response and interferon signaling, suggesting an association between epithelial-to-mesenchymal transition and the preponderance of immune gene expression within the immunogenic cluster (Fig. 2a and Extended Data Fig. 2b). Integration of the coexpression data in module M1 with protein–protein interaction (PPI) data (STRING 11.0 database) identified mesenchymal lineage and immune markers as top regulatory hubs (Extended Data Fig. 2c), leading us to hypothesize that neuroblastoma immunogenicity may be related to lineage state. Two distinct lineage states have been described in neuroblastoma, a differentiated, sympathetic neuron-like adrenergic and a mesenchymal, ‘neural crest cell-like’ phenotype25,26. To test our prediction, we first quantified the tumor adrenergic and mesenchymal identities based on lineage-specific gene expression, assigning adrenergic (A) or mesenchymal (M) scores to each tumor within the four clusters in the SEQC-498 dataset. The tumors in the immunogenic and metabolic clusters had significantly higher mesenchymal scores, whereas those in the Hi-MYCN and neuronal clusters had higher adrenergic scores (Fig. 2b and Extended Data Fig. 2d–e). However, consistent with the known heterogeneity of neuroblastoma, both lineage signatures were observed in each tumor, and the relative mesenchymal lineage enrichment of each tumor (M minus A (M − A) score) showed the strongest correlation with immunogenicity in terms of both IA (Pearson’s correlation coefficient R = 0.71) and IE (R = 0.51) (Extended Data Fig. 2f,g). In agreement, tumors with high M − A scores were represented at a significantly higher proportion within the immunogenic cluster compared with those with low scores, a result that was duplicated in our second dataset (GSE120572) (Extended Data Fig. 3a,b). Finally, a positive relationship between tumor cell-intrinsic immunogenicity and the mesenchymal state was also observed in the subset of MYCN-amplified tumors that expressed immunogenic signatures (Fig. 2c). Together, these results suggest that the relative abundance of a mesenchymal signature is associated with immune gene expression in neuroblastoma.
Fig. 2 |. The mesenchymal cell state is associated with an immunogenic signature.
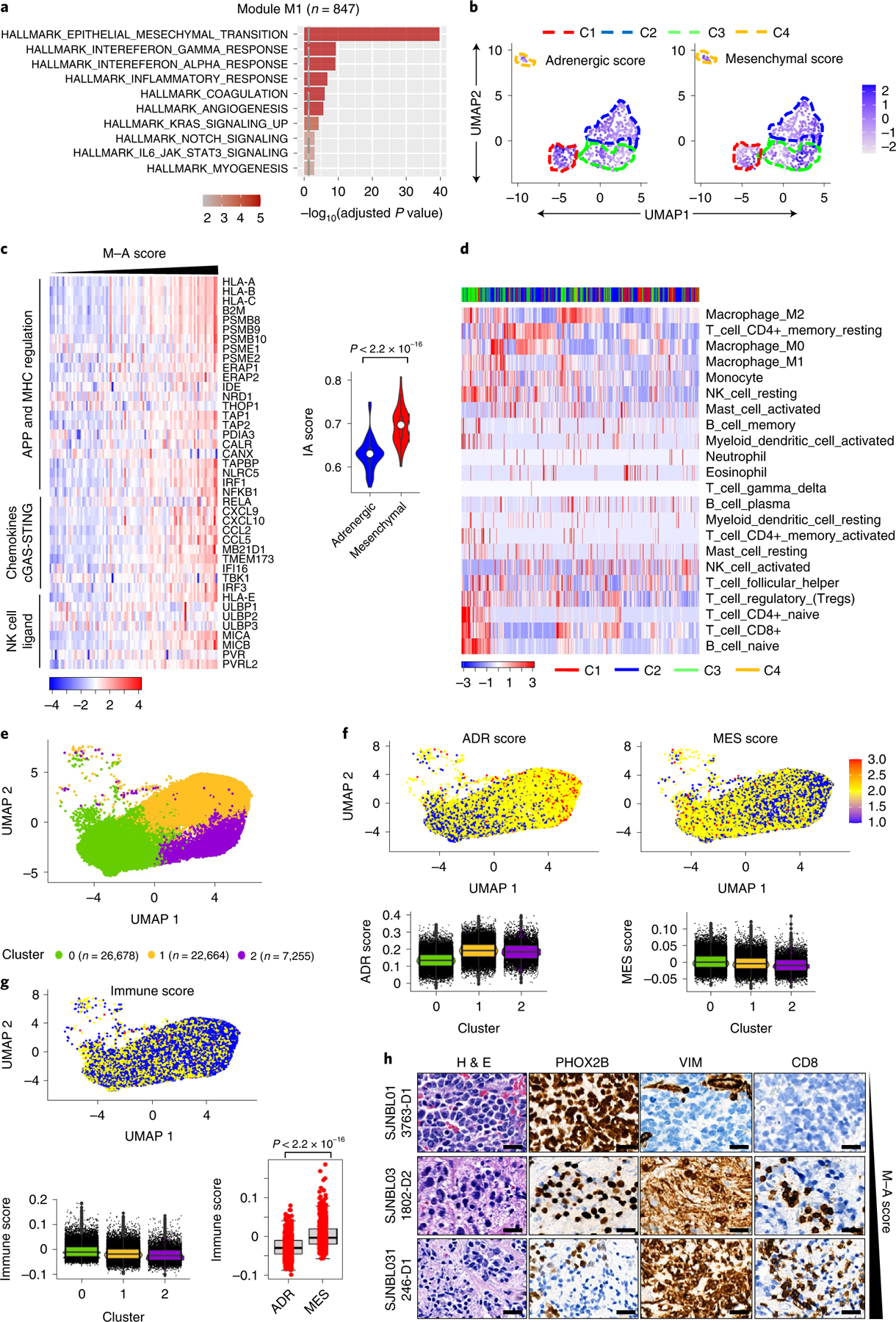
a, GO analysis of coexpressed genes associated with module M1, n = 847 genes. The vertical dashed line indicates the adjusted P value of 0.05. Significance was determined by the two-tailed hypergeometric test. b, UMAP visualization of the distribution of adrenergic and mesenchymal scores among the four tumor clusters. Color bar represents normalized z-scores. Values <2.5 and >2.5 were set to −2.5 and +2.5, respectively, to reduce the effects of extreme outliers (n = 498 tumors). c, Left: heat map representation of tumor cell-intrinsic IA gene expression in MYCN-amplified tumors (n = 92). Samples are ranked by increasing M − A score. The log2 gene expression values were z-score-transformed for visualization. Right: violin plots of tumor immune score distribution, classified as adrenergic or mesenchymal based on the median M − A score. Box plots within the violin plots are defined by center lines (medians), box limits (25th and 75th percentiles) and whiskers (minima and maxima; 1.5 × interquartile range). Significance was determined by the two-tailed Kolmogorov–Smirnov (KS) test. d, Heat map of the hierarchical clustering of z-score-transformed fractional content of the indicated TILs within the four clusters (n = 498 tumors). e,f, UMAP visualization of scRNA-seq data from 56,597 malignant cells from 14 primary NB tumor samples clustered on the basis of transcriptional diversity (e) and the distribution of adrenergic (ADR) and mesenchymal (MES) scores (f, upper). Lower: box plots representing the ADR and MES scores of the malignant cell clusters (cluster 0, n = 26,678; cluster 1, n = 22,664; cluster 2, n = 7,255 cells). g, UMAP visualization (upper) and box plot representation (lower left) of the immune scores of the malignant cell clusters in e. Box plots (lower right) of the immune scores from the upper (MES) and lower (ADR) quartiles of the tumor cells (n = 1,000) ranked according to M − A scores. Significance was determined by the two-tailed KS test. Box plots (f, g) are defined as in c. h, Hematoxylin & eosin (H&E) and IHC analyses of the indicated lineage markers and infiltrating CTLs in human NB tumors arranged based on M − A score. Data are representative of seven tumors. Scale bar, 25 μm.
The preferential overexpression of tumor cell-intrinsic immune genes in the mesenchymal phenotype suggested that these tumors may support increased immune cell infiltration into the TME. To test this prediction, we used two orthogonal approaches. First, we assessed the presence of established signatures of immune cell infiltration27,28 in the tumors arranged according to increasing M − A scores, observing significant enrichment in mesenchymal tumors (Extended Data Fig. 3c–e). Significantly higher expression of not only CTLs and NK cells but also IE markers was seen in high M − A (mesenchymal)-compared with low M − A (adrenergic)-scoring tumors (Extended Data Fig. 3c,e). Second, we used CIBERSORT (Cell type Identification By Estimating Relative Subsets Of known RNA Transcripts)29 as a deconvolution approach to estimate the fraction of tumor-infiltrating leukocytes (TILs) in tumors within the four clusters. Both immune activatory (naïve and memory CD4, cytotoxic CD8, NK) and inhibitory (Treg, M2 macrophage) cell markers were highly enriched in the immunogenic cluster compared with the other clusters (Fig. 2d). Quantification of the average immune cell content in tumors from the upper and lower M − A score quartiles (n = 100 each) revealed significantly higher enrichment of activatory and inhibitory TILs in mesenchymal compared with adrenergic tumors (Extended Data Fig. 3f), indicating that tumor cell-intrinsic immune gene expression in mesenchymal tumors is paralleled by immune cell infiltration into the TME.
To determine whether the association between tumor lineage state and immunogenicity observed in bulk RNA-seq data was seen at the single-cell level, we analyzed recently published single-cell RNA-sequencing (scRNA-seq) data from 14 primary neuroblastomas30. Clustering analyses of gene expression was performed on each tumor individually, and the data were visualized using uniform manifold approximation and projection (UMAP). Nonmalignant cells clustered separately from the malignant cells and were excluded from further analysis (Extended Data Fig. 4a). Within individual tumors, the malignant cells clustered as distinct populations expressing both adrenergic and mesenchymal signatures (Extended Data Fig. 4a–c), in keeping with the known intratumoral heterogeneity in neuroblastoma30. Moreover, the intertumoral heterogeneity observed in our bulk RNA-seq analysis was also replicated at the single-cell level (Extended Data Fig. 4d). Finally, a significant correlation was observed between tumor enrichment for mesenchymal lineage signatures and inflammatory gene expression at the single-cell level, similar to our findings in bulk RNA-seq analysis (Fig. 2e–g, Extended Data Fig. 5a–c and Methods).
Next, to determine whether the relationship between lineage state and immune response gene expression was recapitulated histologically, we analyzed T cell infiltration in seven primary neuroblastomas for which matched bulk RNA-seq data were available (https://www.stjude.cloud)31 (Supplementary Table 5). Consistent with the observed intratumoral and intertumoral heterogeneity, these tumors exhibited a wide range of M − A scores (Extended Data Fig. 5d). Tumors with low M − A scores (relatively more adrenergic), represented by sample SJNBL013763_D1, showed abundant expression of the adrenergic lineage marker PHOX2B and sparse staining with the mesenchymal marker vimentin and, importantly, lacked CD8+ T cell infiltration (Fig. 2h). By contrast, samples with moderate and high M − A scores (relatively more mesenchymal), represented by SJNBL031802_D2 and SJNBL031246_ D1, respectively, showed fewer PHOX2B-positive but abundant vimentin-positive tumor cells and were infiltrated by CD8+ T cells. In essence, these histological findings, in concert with gene expression data at both bulk and single-cell levels, suggest that tumor cell lineage state is associated with immunogenicity and reveal that heterogeneity among constituent tumor cells is associated with differential immune cell infiltration into the TME.
Transition to the mesenchymal state promotes immunogenicity.
To identify representative models with which to further evaluate the association of tumor cell-intrinsic immunogenicity with lineage state, we analyzed adrenergic and mesenchymal human neuroblastoma cell line data (GSE28019)25,26, noting a positive correlation between mesenchymal lineage marker and IA gene expression (Fig. 3a,b and Extended Data Fig. 6a,b). We focused on the SH-SY5Y and SH-EP subclones of the MYCN-nonamplified SK-N-SH cell line32 that were determined to be adrenergic and mesenchymal, respectively26 (Fig. 3a,b and Extended Data Fig. 6c). We noted that immune regulatory genes, especially those involved in APP, activation of MHC expression, type-I interferon signaling and encoding ligands for the activating NK cell receptor, NKG2D (NK cell lectin-like receptor, KLRK1), were differentially upregulated in mesenchymal SH-EP compared with adrenergic SH-SY5Y cells (Fig. 3c and Extended Data Fig. 6c,d). We next questioned whether acquisition of the mesenchymal phenotype would be sufficient to render adrenergic cells immunogenic. One of the top overexpressed genes in SH-EP cells is the homeobox transcription factor, PRRX1, a core lineage-specific marker of the mesenchymal state25,26 (Extended Data Fig. 6e). Overexpression of doxycycline (dox)-inducible PRRX1 in adrenergic SH-SY5Y cells, but not that of its DNA-binding mutants (harboring individual deletions of the three α-helices, ΔH1, ΔH2 and ΔH3, within the PRRX1 homeodomain), led to gradual loss of the adrenergic marker PHOX2B, together with increased fibronectin (FN1) and YAP mesenchymal marker expression (Fig. 3d). Concurrent with this phenotypic switch, wild-type (WT) PRRX1 induction led to the increased expression of tumor cell-intrinsic immune genes involved in APP (TAP1, TAP2, LMP2, LMP7) and innate immune recognition of mislocalized DNA (IFI16 and STING/TMEM173) (Fig. 3d,e and Extended Data Fig. 6f), which were also differentially upregulated in mesenchymal SH-EP cells (Fig. 3b,c and Extended Data Fig. 6c). These changes were accompanied by a sustained increase in cell-surface MHC expression (Fig. 3f), and that of MICA and MICB, ligands for the NKG2D receptor, in a minor population of cells (Fig. 3g). These findings suggest that conversion from the adrenergic to the mesenchymal cell state may be sufficient to induce the expression of tumor cell-intrinsic immune genes.
Fig. 3 |. Reprogramming adrenergic cells to the mesenchymal lineage induces immune gene expression.
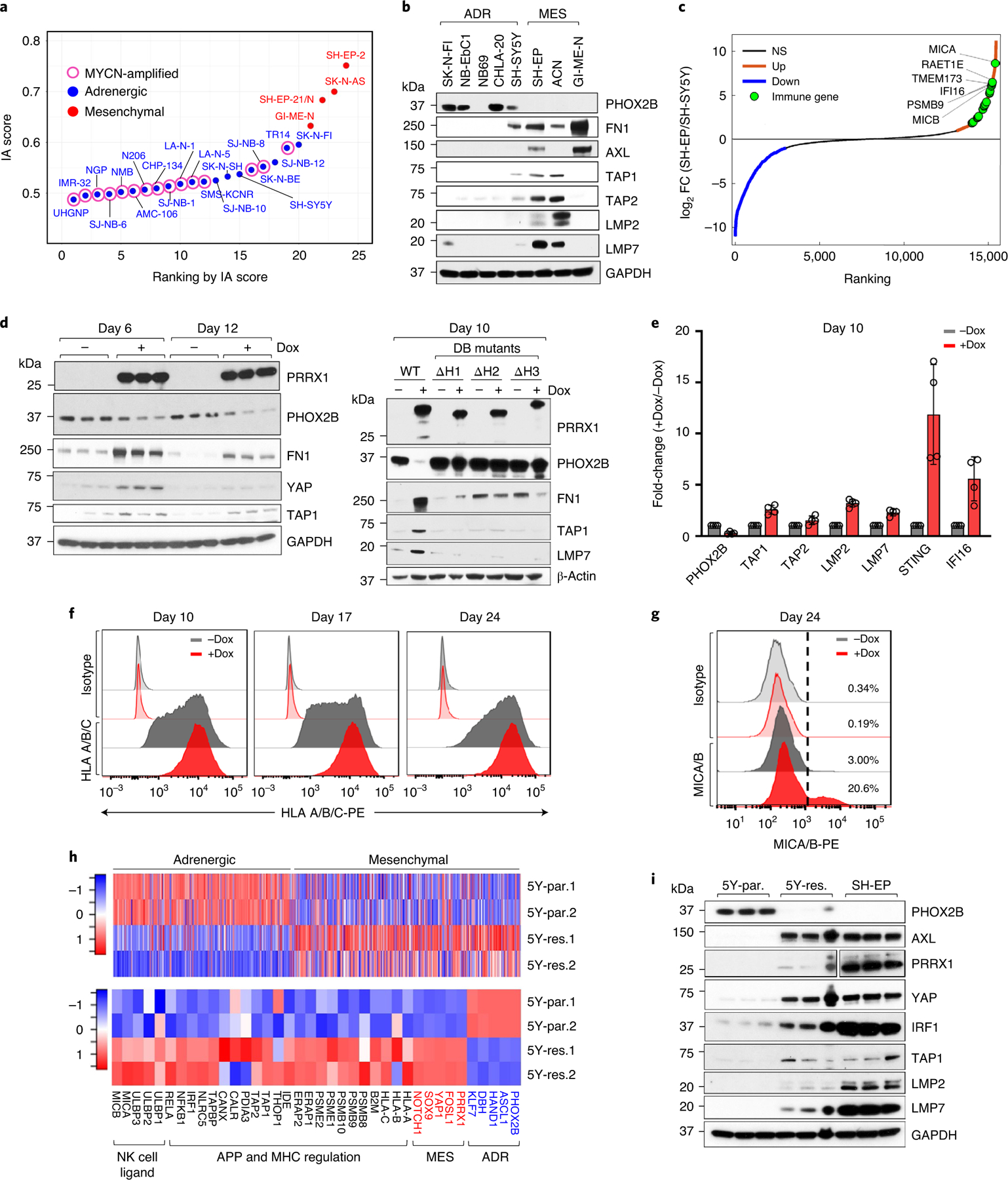
a, Scatter plot of IA scores in human NB cell lines arranged based on increasing scores and designated as adrenergic or mesenchymal based on lineage-specific gene expression. b, Western blot (WB) analysis of lineage markers and APPs in NB cells (n = 2 independent repeats). c, Waterfall plot of the fold-change in RNA expression levels of up- and downregulated genes in SH-EP compared with SH-SY5Y cells. Significance was determined by the two-tailed Wald test. d, Left: WB analysis of the indicated proteins in SH-SY5Y cells expressing dox-inducible PRRX1 in the presence or absence of dox (200 ng ml−1) at the indicated time points (n = 3 independent experiments). Right: WB analysis of the indicated proteins in SH-SY5Y cells expressing WT or DNA-binding (DB) mutants of PRRX1 (n = 1 independent experiment). e, RT–qPCR analysis of immune response genes in the same cells as in d. Data represent mean ± standard deviation (s.d.) (n = 4 independent experiments). f, FACS analysis of HLA expression following PRRX1 overexpression in SH-SY5Y cells at the indicated time points. PRRX1 induction for 10, 16 and 23 days led to a 2.07 ± 0.127 (P = 0.0066), 1.95 ± 0.097 (P = 0.0031) and 1.61 ± 0.021 (P < 0.0001) fold increase in HLA geometric mean fluorescent intensity (gMFI), respectively. Data represent mean ± s.d. (n = 3 independent experiments); two-tailed paired Student’s t-test. g, FACS analysis of MICA/MICB expression after PRRX1 induction for 24 days as in f. Plots are representative of two independent experiments. h, Heat maps of lineage gene signatures (top), and individual lineage marker, APP and NKG2D ligand gene expression (bottom) in 5Y-par. and 5Y-res. cells (n = 2 independent experiments). Rows represent z-scores of log2 expression values. i, WB analysis of lineage marker and APP pathway genes in the indicated cells (n = 3 independent experiments). PRRX1 panels represent two different exposures. GAPDH or β-actin was used as loading control in immunoblots. NS, not significant.
Transition from the adrenergic to the mesenchymal state in neuroblastoma is thought to accompany chemotherapy resistance and relapse26. Whether this transition in the face of treatment pressure might include the acquisition of a proinflammatory signature is unclear, leading us to compare adrenergic neuroblastoma cells that had gained mesenchymal features during the development of treatment resistance against their therapy-sensitive, adrenergic counterparts. We used an isogenic pair of cell lines: adrenergic SH-SY5Y neuroblastoma cells that express the ALKF1174L mutation and are sensitive to the ALK inhibitor ceritinib (LDK378) (5Y-par.; half-maximum inhibitory concentration (IC50) = 215 nM), and their ceritinib-resistant derivatives33 (5Y-res.; IC50 = 1,310 nM) (Extended Data Fig. 6g) that had acquired mesenchymal features during resistance development, including PRRX1 overexpression (Fig. 3h and Extended Data Fig. 6h,i). We noted that the transition to a more mesenchymal state was accompanied by the upregulation of various immune-related pathways—those engaged in MHC expression, APP, NKG2D-mediated NK cell activation and Toll-like receptor 3 (TLR3) signaling (Fig. 3h,i and Extended Data Fig. 6i,j).
We next determined whether such resistance-associated lineage state switching and the accompanying immune gene upregulation observed in cells in culture was also true in primary patient samples. Therefore, we analyzed the transcriptomic profiles of seven matched neuroblastoma tumor pairs obtained at diagnosis and relapse34 (Extended Data Fig. 7a–c and Methods). Two matched pairs had acquired mesenchymal features at relapse (Extended Data Fig. 7d,e) and showed elevated expression of cytotoxic T and NK cell signatures and checkpoint markers, PD1 and CTLA4 (Extended Data Fig. 7f–h). In addition, scRNA-seq analysis of an independent pair of matched diagnosis and relapsed tumors35 revealed a lower adrenergic score in the relapsed tumor (Extended Data Fig. 8a–c) and, consistent with our hypothesis, increased immune cell infiltration compared with the tumor at diagnosis (Extended Data Fig. 8d). Thus, resistance or relapse that is associated with a lineage state switch from an adrenergic to a mesenchymal phenotype is accompanied by increased immune cell infiltration.
Immune genes are epigenetically regulated during lineage switching.
As lineage plasticity in neuroblastoma is epigenetically driven25,26,36,37, we next questioned whether the altered immune response gene expression observed in the individual lineage states could be due to changes in chromatin organization. Indeed, increased active H3K4me3 and loss of repressive H3K27me3 histone binding were seen at immune genes that were upregulated upon PRRX1 induction in adrenergic SH-SY5Y cells (Fig. 4a and Extended Data Fig. 9a). To understand the epigenetic modifications that occur during spontaneous transition between the two lineage states, we compared adrenergic (5Y-par.) cells with those that had acquired mesenchymal characteristics with drug resistance (5Y-res.), using SH-EP cells as a typical example of the mesenchymal state (Fig. 3h). As expected, super-enhancers, identified by H3K27ac occupancy at enhancer regions, were associated with genes that conferred mesenchymal identity in 5Y-res. cells, while those in 5Y-par. cells were at adrenergic lineage genes (Extended Data Fig. 9b). Despite the higher expression of genes associated with immune responsiveness in mesenchymal lineage cells, these genes were not associated with super-enhancers. Rather, significantly higher enrichment of the H3K27ac and H3K4me3 active marks was seen at the promoters of cell-intrinsic immune genes (transcription start site (TSS) ± 2 kilobases (kb)) in 5Y-res. and SH-EP compared with 5Y-par. cells (Fig. 4b,c). On the other hand, adrenergic 5Y-par. cells showed significantly higher occupancies of the H3K27me3 repressive mark at immune gene promoters such as those encoding the NKG2D ligands MICB and ULBP3 (Fig. 4d–f). These immune response genes were also highly occupied by the Polycomb repressive complex 2 (PRC2) subunits EZH2 and SUZ12, which promote H3K27me3 deposition at repressed chromatin38. (Extended Data Fig. 9c), suggesting that immune gene repression is an active phenomenon in this lineage state.
Fig. 4 |. Activation of immune gene expression associated with cell state transition is epigenetically regulated.
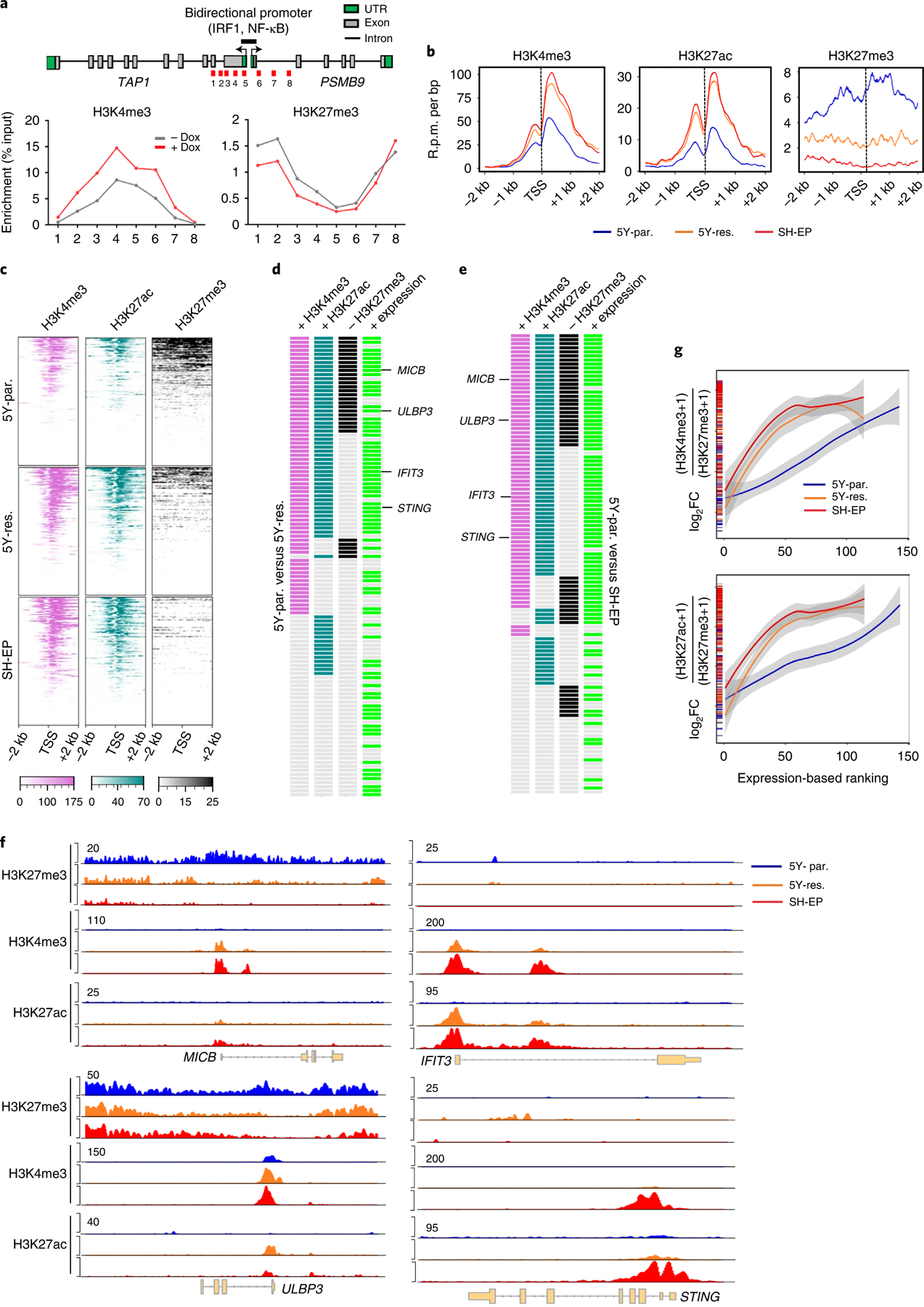
a, Upper: linear representation of TAP1 and PSMB9 gene loci showing the locations of the bidirectional promoter and IRF1 and NF-κB binding sites. Lower: ChIP–qPCR analysis of H3K4me3 and H3K27me3 enrichment at the indicated amplicons (1–8, shown in red in upper) in SH-SY5Y cells expressing dox-inducible PRRX1 in the presence or absence of dox (200 ng ml−1) for 10 days. Data points represent mean of two technical replicates of two independent experiments. b, Metagene representations of average ChIP–seq occupancies of the indicated histone marks at the promoters of tumor cell-intrinsic immune genes (TSS ± 2 kb) in 5Y-par. (n = 143 genes), 5Y-res. (n = 114 genes) and SH-EP cells (n = 114 genes). c, Heat map of histone enrichment at the same immune gene promoters as in b, ranked in decreasing order of occupancy in the indicated cells. Each row represents the normalized densities of histone marks within a ±2 kb window centered on the TSS. d,e, Representation of pair-wise comparisons between 5Y-par. and 5Y-res. cells (d), and 5Y-par. and SH-EP cells (e). The changes (+, gained; −, lost) in occupancies of the active (H3K4me3, H3K27ac) and repressive (H3K27me3) histone marks (log2FC ≥ 0.75, TSS ± 2 kb), together with the corresponding changes in RNA expression (+, overexpressed; log2FC ≥ 1) of each of the tumor cell-intrinsic immune genes analyzed in b, are shown. f, ChIP–seq tracks depicting the gain of active histone binding together with the loss of repressive histone binding at MICB and ULBP3 gene loci (left) or gain of active marks without changes in repressive mark occupancy at IFIT3 and STING loci (right). Signal intensity is given at the top left corner of each track. g, Loess regression analysis of the correlation between the ratios of active to repressive histone binding at the promoters of immune response genes and their RNA expression. Genes are ranked based on increasing expression. Shaded regions represent 95% confidence intervals. UTR, untranslated region.
We next sought to determine whether immune response gene activation during the transition from sensitivity to resistance represented a switch from repressive to active chromatin or a de novo gain of active chromatin marks. Compared with adrenergic 5Y-par. cells, mesenchymal 5Y-res. and SH-EP cells gained significant deposition of the active histone marks at immune response genes, which corresponded with their increased expression in these cells (H3K4me3, 60% (68 of 114) and 62% (71 of 114); H3K27ac, 58% (66 of 114) and 67% (76 of 114), respectively) (Fig. 4d,e and Extended Data Fig. 9d–f). Such gain was accompanied by a concomitant loss of H3K27me3 binding at immune genes in 5Y-res. and SH-EP cells (25% (29 of 114) and 35% (40 of 114), respectively), as represented by the NKG2D ligands MICB and ULBP3 (Fig. 4d–f and Extended Data Fig. 9d–f). At the same time in these cells, several immune genes, including the IFN-regulated factors IFIT3 and STING, gained either one or both active marks without changes in occupancy of the repressive mark (48% (55 of 114) and 41% (47 of 114), respectively). Overall, mesenchymal cells showed a higher ratio of active to repressive histone occupancy at the TSSs of immune-related genes (Extended Data Fig. 9g), which, importantly, also correlated with their increased expression in this cell state (Fig. 4g).
Mesenchymal neuroblastoma cells induce NK cell degranulation.
We next investigated whether the observed association of cell lineage state with tumor immunogenicity was borne out in functional studies of NK cell activity. Since the interaction of an NK cell activating receptor with its cognate ligands on target cells is the first step toward a cytotoxic response (Fig. 5a), we compared the ability of mesenchymal 5Y-res. and SH-EP cells to bind to the recombinant NKG2D receptor fusion (NKG2D-Fc) protein. In keeping with their increased expression of NKG2D ligands (Fig. 5b), increased binding was observed in both cell types, compared with adrenergic 5Y-par. cells (Fig. 5c). Using cell-surface CD107a as a marker of degranulation, we next determined that coculture of 5Y-res. and SH-EP cells with healthy human NK cells resulted in increased NK cell degranulation compared to that with 5Y-par. cells (Fig. 5d). Blockade of NKG2D receptor activity using an anti-NKG2D monoclonal antibody abrogated the increased NK cell degranulation in 5Y-res. and SH-EP cells (Fig. 5e), signifying that the modest but robust increase in degranulation was NKG2D receptor-specific. Coculture with the mesenchymal lineage cells also led to significant downregulation of NK cell-surface-associated NKG2D receptor expression (Fig. 5f), consistent with its internalization and degradation upon interaction with its cognate ligands39. Finally, cytotoxicity assays with human NK cells showed that cell killing in mesenchymal SH-EP cells was reduced with the anti-NKG2D antibody, whereas such inhibition had no effect on adrenergic 5Y-par. cells (Fig. 5g). Such NKG2D-dependent cytotoxicity was seen also in another mesenchymal cell line, GI-ME-N, which serves as an excellent NK cell target owing to its lack of surface MHC expression (Fig. 5g).
Fig. 5 |. Mesenchymal NB cells induce NKG2D-dependent NK activation.
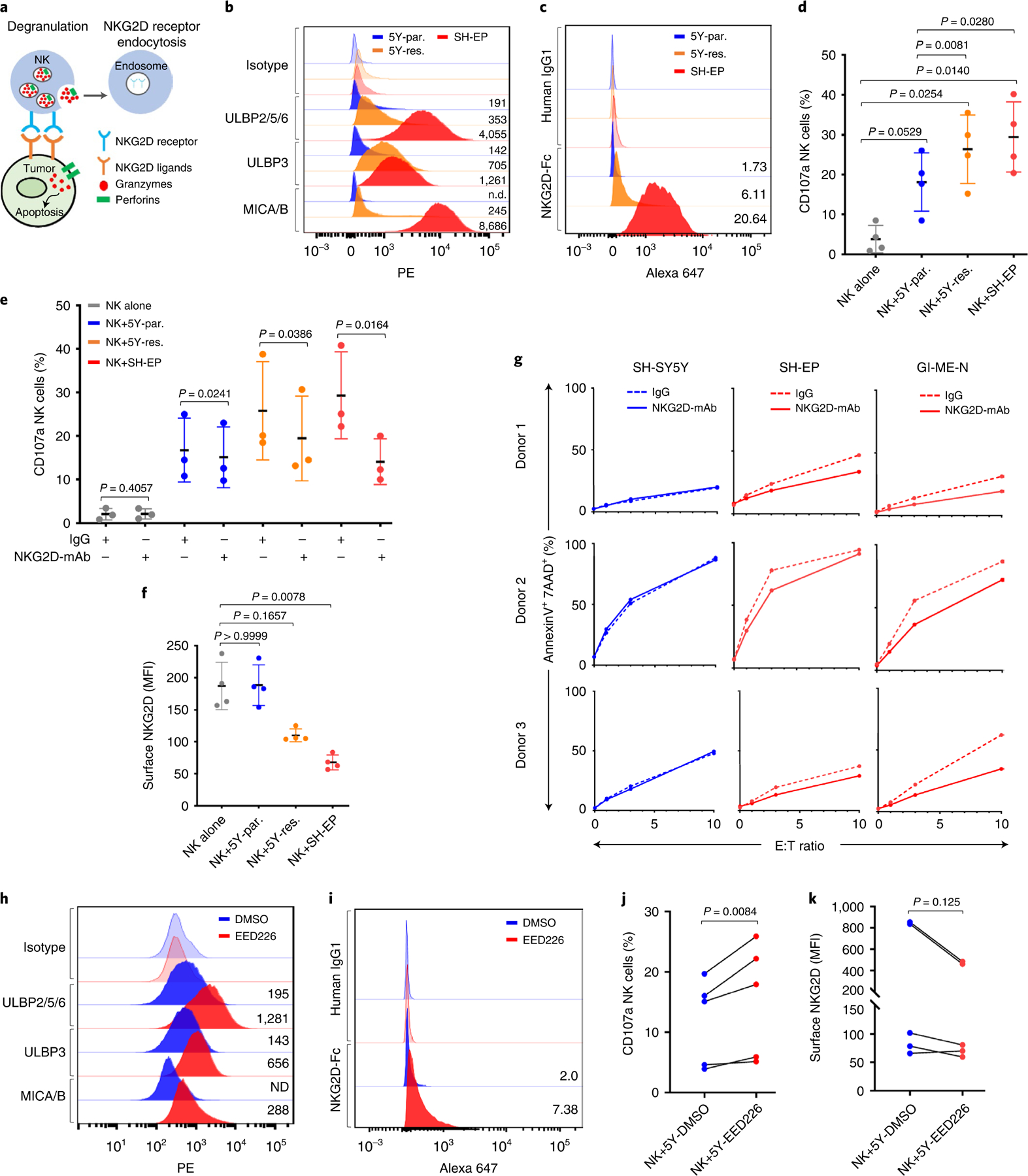
a, Schematic interaction between NKG2D receptors and cognate ligands, leading to NK cell degranulation and receptor endocytosis. b, FACS analysis of NKG2D ligands in the indicated cells. Plots are representative of four independent experiments; numbers indicate isotype-normalized gMFI. c, FACS analysis of recombinant NKG2D-Fc or IgG1 binding in the indicated cells. Numbers denote fold-change in Alexa-647 median fluorescent intensity (MFI) (NKG2D-Fc/IgG1) (n = 3 independent experiments, P = 0.0040, one-way ANOVA followed by test for linear trend). d, Dot plot quantifying degranulation in naïve NK cells (NK alone) or following coculture with indicated targets. Data represent mean ± s.d., four independent experiments. Significance was calculated using repeated measures (RM) one-way ANOVA with the Geisser–Greenhouse correction followed by Sidak’s multiple comparisons test. e, Dot plot quantifying the effect of control IgG1 or NKG2D blocking antibody on NK cell degranulation following coculture with the indicated targets. Data represent mean ± s.d., three independent experiments. Significance was calculated using the two-tailed ratio paired t-test. f, Dot plot quantifying NKG2D MFI in NK cells following coculture with the indicated targets. Data represent means ± s.d., four independent experiments. Significance was calculated using Friedman test followed by Dunn’s multiple comparisons test. g, X–Y plots of NK cell cytotoxicity with control IgG1 or anti-NKG2D antibody against the indicated targets. Data represent three independent experiments. h, FACS analysis of NKG2D ligands in SH-SY5Y cells treated with DMSO or EED226 (5 µM for 8 d). Plots are representative of two independent experiments. Numbers indicate isotype-normalized gMFI. i, FACS analysis of NKG2D-Fc or IgG1 binding in SH-SY5Y cells treated with either DMSO or EED226 as in f. Numbers indicate fold-change in Alexa-647 MFI (NKG2D-Fc/IgG1) (n = 3 independent experiments, P = 0.0060, two-tailed paired t-test). j,k, Before–after plots of degranulation (j) and NKG2D MFI (k) following coculture of naïve NK cells with SH-SY5Y cells treated with DMSO or EED226. Data in j and k represent five independent experiments. Significance in j was derived using the two-tailed ratio paired t-test and in k using the two-tailed Wilcoxon matched-pairs signed rank test. DMSO, dimethylsulfoxide; ND, not determined; PE, phycoerythrin.
The observation that genes encoding NKG2D ligands are repressed by the PRC2 complex in adrenergic 5Y-par. cells (Extended Data Fig. 9c) prompted us to examine whether PRC2 inhibitors could induce the expression of these transcripts and influence NK cell function. Treatment of SH-SY5Y cells with EED226, an allosteric PRC2 complex inhibitor40, led to increased expression and surface localization of the NKG2D ligands which, in turn, led to their increased binding to the NKG2D-Fc protein and a modest increase in NK cell degranulation compared with cells treated with DMSO alone (Fig. 5h–j). Finally, coculture of EED226-treated adrenergic SH-SY5Y cells with primary human NK cells led to loss of surface NKG2D receptor expression (Fig. 5k). Overall, these results suggest that the lineage-specific expression of NKG2D ligands in mesenchymal lineage neuroblastoma has a functional impact on NK cell activity and that pretreatment of adrenergic cells with PRC2 inhibitors could upregulate NKG2D ligand expression and lead to increased NK cell activation.
Mesenchymal neuroblastoma cells can induce a T cell response.
We next investigated the lineage-specific induction of a T cell response using CTLs directed against an HLA-A*02-restricted peptide, SLLQHLIGL, derived from the PRAME cancer testis antigen that functions as an immunogenic target in neuroblastoma41. HLA-A2-expressing, adrenergic (SK-N-DZ, NLF) and mesenchymal (CHP-212, GI-ME-N) neuroblastoma cell lines that express PRAME (Fig. 6a) were utilized for this experiment. As negative controls, we used genotypically HLA-A2-positive IMR-32 cells that lack HLA-A2 and PRAME expression and HLA-A2-negative SMS-KAN cells that express PRAME. We first asked whether IFN signaling was utilized to engage PRAME-specific CTLs. IFN-γ acted in a nondiscriminatory manner to upregulate surface HLA and increase cell kill against both adrenergic and mesenchymal but not control cells (Fig. 6b,c). Owing to the prominence of TLR3 gene signatures in the mesenchymal lineage (Extended Data Fig. 6j) and their role in regulating neuroblastoma growth through both tumor cell-autonomous and immune-mediated mechanisms42, we next analyzed this signaling pathway. Interestingly, while both adrenergic and mesenchymal cells with an HLA-A*02 genotype expressed PRAME, only mesenchymal cells were responsive to the TLR3 ligand poly(I:C), as seen by HLA-A2 and HLA-ABC upregulation (Fig. 6b). In agreement, coculture with PRAME CTLs led to significantly higher cytotoxicity in poly(I:C)-treated mesenchymal cells compared with their adrenergic counterparts or the two negative control cell lines (Fig. 6c). Moreover, CXCL10, a major driver of T cell infiltration in solid tumors, was upregulated in conditioned media from all five mesenchymal neuroblastoma cell lines, compared with only one of five adrenergic cells (Fig. 6d). Mesenchymal cells also showed a differential increase in CCL5, IL4, Eotaxin (CCL11) and MCP1 (CCL2) chemotactic factors which guide T and other immune cells to the tumor site (Fig. 6e). Finally, we measured the ability of Jurkat T cells engineered to express CXCR3, the cognate receptor for CXCLs 9, 10 and 11 (CXCR3-Jurkat), and primary human CD8+ T cells to infiltrate neuroblastoma spheroids following poly(I:C) stimulation, observing significantly increased recruitment of these cells into mesenchymal compared with adrenergic tumor spheroids (Fig. 6f,g). Collectively, these results suggest that evoking the innate immune response through TLR3 stimulation in mesenchymal cells enables antigen recognition and cytotoxicity, in contrast to adrenergic cells that are refractory to such activation.
Fig. 6 |. Mesenchymal NB cells are killed by antigen-specific CTLs.
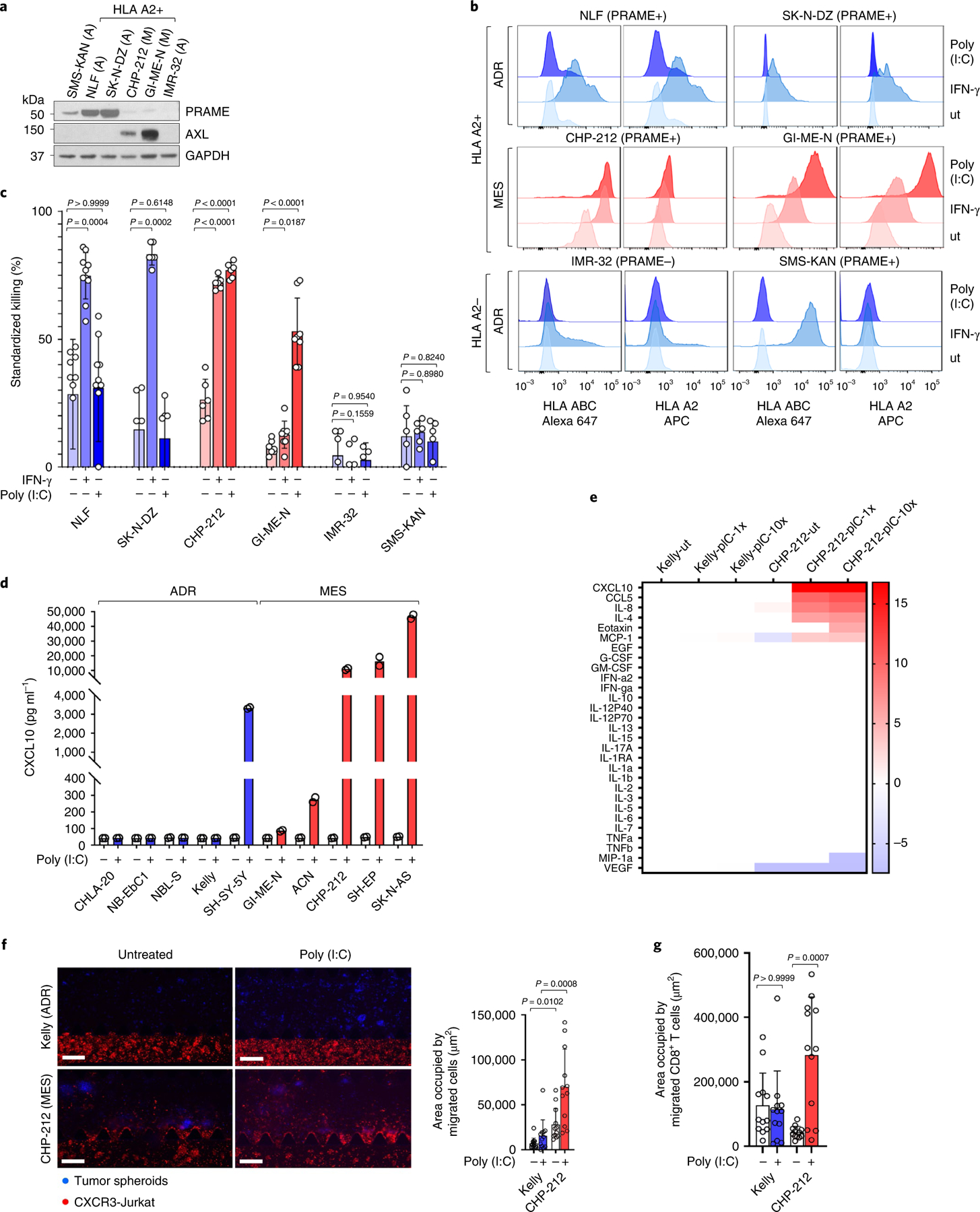
a, WB analysis of PRAME and AXL in the indicated adrenergic (A) or mesenchymal (M) NB cells. GAPDH, loading control. b, FACS analysis of HLA subtypes in the indicated cells, untreated (ut) or following IFN-γ or poly(I:C). Data (a, b) are representative of two independent experiments. c, Bar graphs quantifying cytotoxicity of PRAME-specific CTLs against neuroblastoma cells following IFN-γ (100 U ml−1) or poly(I:C) (10 µg ml−1) × 24 h and coculture at an E:T ratio of 10:1. Data represent mean ± s.d. of n = 10 (NLF), n = 6 (SK-N-DZ, CHP-212,SMS-KAN) and n = 8 (GI-ME-N) independent experiments for all conditions, and n = 6 (untreated and IFN-γ) and n = 4 (poly(I:C)) independent experiments in IMR-32 cells. Significance was derived using the Friedman test followed by Dunn’s multiple comparisons test (NLF); mixed-effects analysis followed by Sidak’s multiple comparisons test (IMR-32); and repeated-measures one-way ANOVA and Geisser–Greenhouse correction followed by Sidak’s multiple comparisons test (SK-N-DZ, CHP-212, GI-ME-N, SMS-KAN). d, ELISA analysis of CXCL10 in conditioned media from the indicated neuroblastoma cells ± poly(I:C) (1 µg ml−1) for 24 h. Data represent mean of two independent experiments, P = 0.0485, Kruskal–Wallis test followed by Dunn’s multiple comparisons test. e, Heat map of log2FCs in the expression of the indicated proteins in conditioned media from adrenergic Kelly or mesenchymal CHP-212 cells, untreated (ut) or treated with 1 or 10 µg ml−1 poly(I:C) (pIC-1x or pIC-10x) × 24 h, and analyzed using multiplex cytokine profiling. Data were normalized to the Kelly-ut sample. f, Left: immunofluorescence images of CXCR3-Jurkat cell migration toward Kelly or CHP-212 spheroids grown in a 3D microfluidic device ± poly(I:C). Scale bar, 200 µm. Right: quantification of area occupied by migrated cells. Data represent mean ± s.d., 12 regions of interest (ROIs) per condition, significance derived using Kruskal–Wallis test followed by Dunn’s multiple comparisons test. g, Quantification of area occupied by CD8+ T cells migrating toward the tumor spheroids in f. Data represent means ± s.d., 12 ROIs per condition. Significance was calculated as in f. Data in f and g are representative of two independent experiments each.
Mesenchymal neuroblastomas are amenable to checkpoint therapy.
We next determined whether the ability of mesenchymal neuroblastoma cells to induce a T cell response translated to in vivo models. To this end, we utilized the murine neuroblastoma cell line NB-9464, derived from tumors arising in the TH-MYCN genetically engineered mouse model43. This model was generated in immunocompetent C57BL/6 mice, and the tumors recapitulate the genetic and immunological features of human neuroblastoma43,44. Interestingly, we observed that NB-9464 cells could be sorted on the basis of surface MHC class I H-2Kb expression into high (H-2Kbhi)- or low (H-2Kblo)-expressing populations (Fig. 7a and Extended Data Fig. 10a,b), both of which expressed transgenic human MYCN (Fig. 7b). Consistent with our hypothesis, H-2Kbhi cells were enriched for mesenchymal markers including Prrx1 (Fig. 7b), and showed enhanced migration and invasion, as might be expected from their neural crest cell-like state (Extended Data Fig. 10c,d). We determined that H-2Kbhi cells, in comparison with H-2Kblo cells, were capable of exogenous antigen presentation using the well-characterized chicken ovalbumin-derived peptide (OVA257–264 or SIINFEKL) (Fig. 7c). Next, we determined whether such antigen presentation enabled H-2Kbhi mesenchymal cells to be recognized by OT-I CD8+ T cells from C57BL/6 mice that express a transgenic T cell receptor (TCR) that specifically recognizes the H-2Kb–SIINFEKL complex. H-2Kbhi cells loaded with the SIINFEKL peptide led to increased OT-I cell activation compared with adrenergic H-2Kblo cells, as determined by the expression of cell surface CD69, an early marker of T cell activation (Fig. 7d). We next investigated whether the ability of mesenchymal H-2Kbhi cells to engage CTLs might license them to respond to immune checkpoint blockade (ICB). Both H-2Kblo and H-2Kbhi cells formed tumors in syngeneic C57BL/6 (H-2Kb haplotype) mice, with persistently higher H-2Kb and Prrx1 expression seen in H-2Kbhi tumors (Fig. 7e and Extended Data Fig. 10e). Treatment with anti-PD1 and anti-CTLA4 antibodies resulted in more pronounced reductions in tumor growth and prolongation of survival in H-2Kbhi compared with H-2Kblo models (Fig. 7f,g and Extended Data Fig. 10f). In agreement, the CD8 to Treg and conventional T cell to Treg ratios, indices of a favorable ICB response, were higher in H-2Kbhi tumors, although this difference did not achieve statistical significance (Extended Data Fig. 10g). Together, these results suggest that the immunogenicity of mesenchymal neuroblastoma cells translates into functional engagement with CTLs and confers responsiveness to checkpoint therapy.
Fig. 7 |. Mesenchymal NB cells functionally engage with cytotoxic T cells and are responsive to ICB.
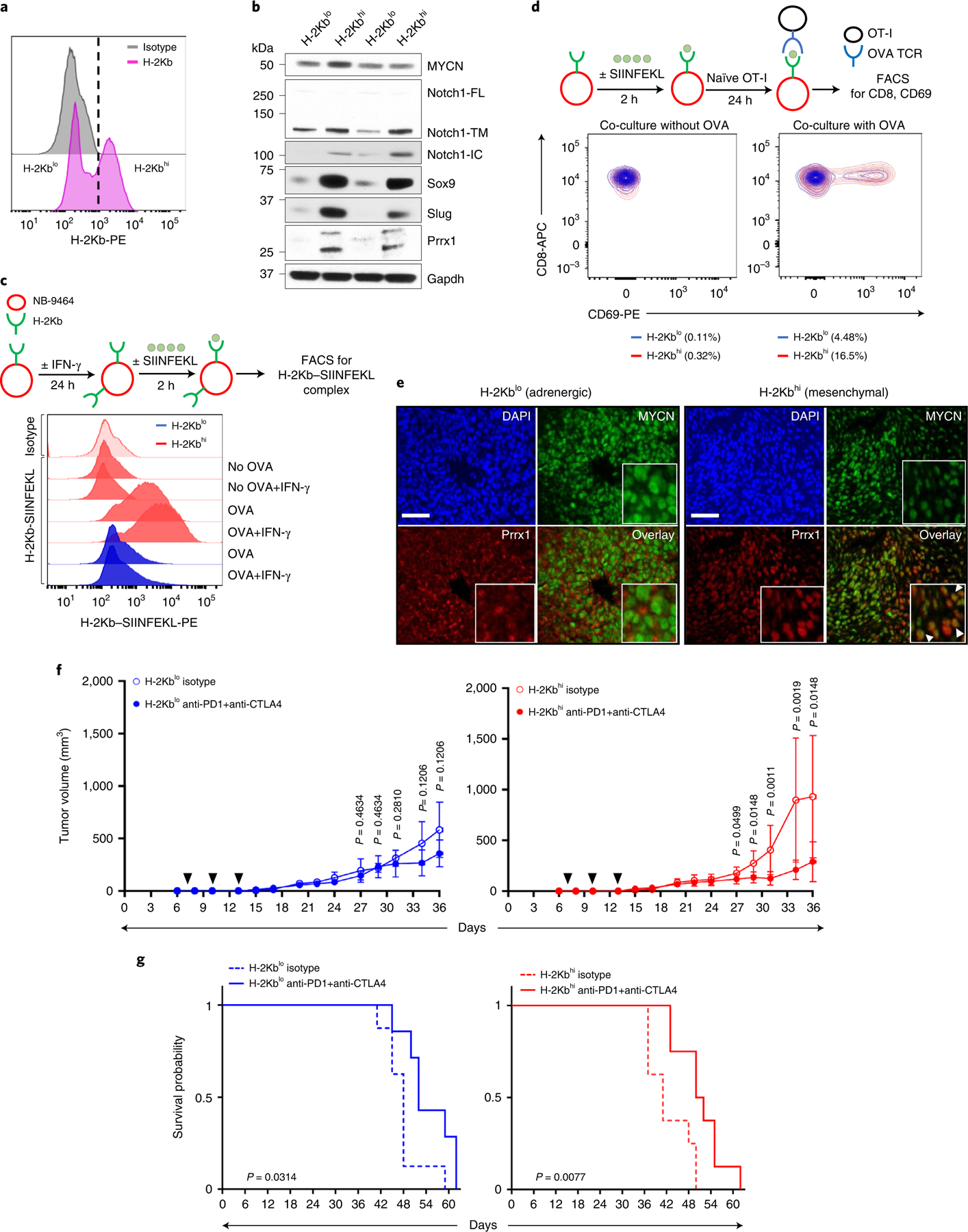
a, FACS analysis of H-2Kb expression in NB-9464 cells. The vertical dashed line denotes the logscale expression value used to gate H-2Kblo and H-2Kbhi populations. Data are representative of two independent experiments. b, WB analysis of lineage markers in H-2Kblo and H-2Kbhi cell populations. n = 2 independent experiments. Notch1-FL, full length; -TM, transmembrane; -IC, intracellular. GAPDH, loading control. c, Upper: schematic of OVA binding assay. Lower: FACS analysis of H-2Kb–SIINFEKL complex in H-2Kbhi and H-2Kblo cells at baseline or following IFN-γ (100 ng ml−1 × 24 h) ± OVA peptide. Data are representative of one independent experiment. d, Upper: schematic of NB-9464-OT-I coculture assay. Lower: contour plots showing the percentage of naïve OT-I cells that were activated (CD8+ CD69+) following coculture with H-2Kblo and H-2Kbhi cells for 24 h with or without the OVA peptide. Data are representative of one independent experiment. e, Immunofluorescence images of MYCN and Prrx1 expression in murine tumors derived from H-2Kblo and H-2Kbhi cells. Nuclei are counterstained with DAPI. Insets: nuclear costaining of MYCN and Prrx1 (arrowheads). Scale bar, 100 µm. Data are representative of n = 3 tumors for each group. f, Tumor volumes in C57BL/6 mice injected subcutaneously with 1 × 106 H-2Kblo or H-2Kbhi cells and treated with anti-PD1 + anti-CTLA4 antibodies or isotype control on days 7, 10 and 13 after tumor inoculation (black arrowheads). Data represent mean ± s.d. n = 8 (H-2Kblo isotype); n = 7 (H-2Kblo anti-PD1 + anti-CTLA4); n = 8 (H-2Kbhi isotype); n = 8 (H-2Kbhi anti-PD1 + anti-CTLA4) mice per group across all time points. Significance was calculated using the two-tailed Mann–Whitney test. g, Kaplan–Meier analysis of mice bearing H-2Kblo or H-2Kbhi tumors treated with isotype or anti-PD1 + anti-CTLA4 antibodies as in f. Significance was determined by the log-rank test.
Discussion
Despite the therapeutic success of dinutuximab in neuroblastoma3, the factors that enable such a response are less clear, as is the extent to which this immunogenic landscape could be leveraged for favorable outcome. Here, we demonstrate that each neuroblastoma lineage state is characterized by distinct cell-intrinsic and -extrinsic immune gene expression, with the mesenchymal lineage state having the capacity to elicit an anti-tumor immune response. The inherent presence or induction of this state in tumor cells is accompanied by the activation of innate and adaptive immune genes through epigenetic remodeling at gene promoters. We demonstrate that mesenchymal cells promote T cell infiltration by secreting inflammatory cytokines, are efficiently targeted by cytotoxic T and NK cells and respond to ICB in an immunocompetent mouse model, thereby signifying the role of tumor cell lineage in shaping immune response in neuroblastoma.
The intimate link between tumor cell lineage and the propensity of eliciting an immune response is substantiated by studies showing that diverse cellular states strongly influence the engagement of innate and adaptive immune pathways45. Inhibition of cancer cell-intrinsic pathways that promote T cell exclusion, for example, sensitizes melanomas to immunotherapy through cell state switching46. In agreement with our findings, recent analyses of bulk gene expression signatures from the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) and Gabriella Miller Kids First (GMKF) neuroblastoma cohorts indicate that transcriptional programs driven by adrenergic transcription factors are enriched in non-T-cell-inflamed tumors without MYCN amplification47. In addition, the recent identification of immunologically cold neuroendocrine and highly inflamed non-neuroendocrine mesenchymal subtypes in small cell lung cancer (SCLC), another neural crest-derived tumor48,49, suggests that coregulation of tumor-intrinsic immunogenicity and mesenchymal gene expression programs may be a conserved feature of neuroendocrine tumors.
Our studies highlight the molecular mechanisms that drive inflammatory gene expression in mesenchymal neuroblastoma cells. First, we observe that lineage-specific transcription factors that constitute the mesenchymal core regulatory circuitry (CRC) regulate immune gene expression. We show that PRRX1, a component of the mesenchymal CRC, leads to transcriptional upregulation of MHC class I and APP genes. Similar to PRRX1, other transcription factors of the mesenchymal CRC, including interferon regulatory factors 1–3 and IFI16, have been shown to function as major drivers of tumor cell-intrinsic innate and adaptive immune responses50,51. The upregulation of DNA damage sensor proteins such as STING in mesenchymal lineage neuroblastomas could also contribute to the increased abundance of TILs in these tumors, as seen in SCLC52. Second, we have established that the changes in immune gene expression that accompany adrenergic-to-mesenchymal transition are epigenetically regulated. Unlike lineage identity genes that are regulated by super-enhancers, tumor cell-intrinsic immune genes involved in diverse immune functions such as the inflammatory response, IFN-γ signaling and NK cell recognition are governed by changes in promoter structure, achieved by either de novo acquisition of permissive chromatin or epigenetic switching from PRC2-mediated repression to a permissive chromatin landscape. Future studies will address how PRRX1 and other mesenchymal transcription factors drive epigenetic changes to support immune gene expression.
We demonstrate that while the mesenchymal state promotes upregulation of MHC class I expression and CTL infiltration, this lineage state is also characterized by the activation of immune checkpoints—Treg cells and exhaustion markers linked to immune suppression. The presence of both IA and suppression markers suggests that patients with mesenchymal tumors may benefit from ICB therapy, as checkpoint inhibitors are likely to be most effective in those with a pre-existing but dampened anti-tumor immune response22,23. In agreement with this notion, our results show that mesenchymal H-2Kbhi tumors are susceptible to PD1 and CTLA4 inhibition, although there are several caveats toward clinical realization of these findings. Our data need to be validated in additional immunocompetent neuroblastoma models. Reduced tumor burden following dual antibody treatment has been observed in Neuro2a and NB3TL models53,54, although the extent to which they represent the intratumoral heterogeneity of human neuroblastomas and TH-MYCN tumors is not known. It may be important to consider the status of the Food and Drug Administration (FDA)-approved predictive biomarkers of ICB response while enrolling patients with mesenchymal tumors to ICB trials, although neuroblastomas in general are known to lack these indices55. The small numbers of neuroblastoma patients included in early-phase ICB trials render analysis of tumor cell lineage in responders versus nonresponders challenging. However, the recent demonstration of the predictive power of mesenchymal lineage-specific responsiveness in a retrospective analysis of over 400 SCLC patients treated with ICB48 supports our conclusions. Additionally, our observation that relapsed neuroblastomas with mesenchymal features exhibit increased immunogenicity, including elevated expression of checkpoint markers, suggests that ICB therapy may be more effective at relapse. Indeed, it is noteworthy that combination therapy with dinutuximab and the anti-PD1 antibody, nivolumab, resulted in partial remission in two recently reported cases of relapsed neuroblastoma56.
A pertinent point highlighted by our study is the concept that triggering an innate immune response in mesenchymal neuroblastoma cells enables CTL activity. This is important because simultaneous targeting of innate and adaptive immune regulators in neuroblastoma results in complete remission and anti-tumor memory57. Specifically, we demonstrate that TLR3 activation in mesenchymal cells induces proinflammatory chemokines, promotes T cell migration and accentuates killing by PRAME-specific CTLs. Together with a recent study that also highlights a lineage-specific function of TLR3 in neuroblastoma58, it is plausible that TLR3 activation in mesenchymal tumor cells, alone or in combination with adoptively transferred CTLs or checkpoint inhibition, could lead to immune-mediated tumor regression.
Our data also suggest that lineage-restricted immune gene expression is epigenetically regulated, thus adding to the growing body of evidence for tumor cell-autonomous function of PRC2 as a barrier to anti-tumor immunity59. Similar to the reports demonstrating Polycomb-mediated suppression of MHC class I gene expression in neuroblastoma60, our finding of PRC2 deposition at genes that encode activating NK cell receptor ligands in adrenergic neuroblastoma cells suggests the possibility of utilizing PRC2 inhibitors to augment NK cell responses against these cells. That said, it is possible that such reactivation of immune genes could be accompanied by the acquisition of other mesenchymal features such as therapy resistance or invasiveness. However, the critical effector function of NK cells in anti-tumor immunity against neuroblastoma44, coupled with the promising responses in patients with other solid tumors to NKG2D-directed CAR NK cell therapy, imply that PRC2 inhibition in combination with NK cell therapy may be beneficial. In conclusion, our study uncovers an unappreciated link between immunogenicity and tumor lineage state in neuroblastoma and rationalizes future interrogations into avenues through which the vulnerability of mesenchymal cells to immune-mediated targeting could be harnessed clinically.
Methods
Ethical approvals.
Human NK cells were isolated from blood collars of healthy donors obtained under Institutional Review Board-approved protocols of the Dana-Farber Cancer Institute (DFCI) and Boston Children’s Hospital. All human tumor specimens were obtained under an Institutional Review Board-approved protocol of St Jude Children’s Research Hospital, and informed consent was obtained from all subjects. All mouse experiments were performed with approval from the DFCI Institutional Animal Care and Use Committee (IACUC).
Cell culture.
Human neuroblastoma cell lines (Kelly, NBL-S, CHP-212, SH-SY5Y, SH-EP, CHLA-20, NB69, SK-N-FI, SK-N-BE2, SMS-KAN, IMR-32) were obtained from the Children’s Oncology Group biobank. ACN, GI-ME-N, NB-EbC1, SK-N-DZ and NLF were gifts from A. Thomas Look and Kimberly Stegmaier at DFCI, and murine NB-9464 cells were from To-Ha Thai at Beth Israel Deaconess Medical Center, Boston, MA, USA. Cell lines were authenticated through short tandem repeat analyses and were routinely tested for mycoplasma. Neuroblastoma cells were grown in RPMI-1640 medium (Invitrogen) supplemented with 10% FBS (Invitrogen) and 1% penicillin/streptomycin (Life Technologies). HEK293T cells obtained from the American Type Culture Collection were grown in DMEM (Invitrogen) with 10% FBS and 1% penicillin/streptomycin. SH-SY5Y cells resistant to ceritinib (LDK378) were described previously33 and were grown in complete RPMI-1640 containing 1.5 µM LDK378.
Compounds.
Ceritinib (LDK378) and EED226 were purchased from Selleck Chemicals, and dox and dimethylsulfoxide from Sigma-Aldrich.
Generation of PRRX1-inducible cells.
Complementary DNAs encoding WT and DNA-binding mutants of human PRRX1A were cloned into the pInducer20 lentiviral plasmid (gift from Stephen Elledge, Addgene no. 44012). The DNA-binding mutants harbor individual deletions of the three α-helices within the homeodomain: ΔH1 (aa 103–116); ΔH2 (aa 121–131); ΔH3 (aa 135–151). The lentivirus was packaged by cotransfection of pInducer20 plasmid with the helper plasmids, pCMV-deltaR8.91 and pMD2.G-VSV-G, into HEK293T cells using the TransIT-LT1 Transfection Reagent (Mirus Bio) and supernatants collected 48 hours after transfection. SH-SY5Y cells were transduced with the supernatant and selected using neomycin (5 µg ml−1). Gene expression was induced with dox (200 ng ml−1) in RPMI-1640 medium supplemented with 10% tetracycline-negative FBS and 1% penicillin/streptomycin.
Derivation of NB-9464 cell lines.
NB-9464 cells were sorted based on H-2Kb expression using fluorescence-activated cell sorting (FACS) analysis. A threshold logscale expression value of 103 was used to gate H-2Kblo (<103) and H-2Kbhi (≥103) cell populations which were then expanded to NB-9464-H-2Kblo and NB-9464-H-2Kbhi cell lines.
FACS analysis.
Adherent cells were disassociated using Accutase (Invitrogen). For each staining reaction, 1 × 106 live cells were placed in a 12 × 75-mm2 polystyrene round-bottom tube (Falcon), resuspended in 100 µl of 1 × PBS and stained with Zombie near-infrared (Zombie NIR) viability dye (BioLegend) at 1:1,000 dilution for 15 min at room temperature. Cells were then washed once in FACS buffer (0.5% BSA in 1 × PBS), resuspended in 100 µl of FACS buffer and incubated in 5 µl of Human TruStain FcX (Fc receptor blocking solution, BioLegend) for 10 min at room temperature. Next, appropriate volumes of conjugated fluorescent primary antibodies at predetermined optimum concentrations (Supplementary Table 7) were added and incubated on ice for 20 min. Cells were then washed once in 2 ml of FACS buffer by centrifugation at 435g for 5 min. All samples were analyzed on an LSRFortessa or FACSCanto II cytometer (Becton Dickinson) using FACSDiva and FlowJo software (Becton Dickinson). A minimum of 10,000 events were counted per sample. The histograms for the isotype controls in Fig. 3g are the same as those in the day 24 panel in Fig. 3f, as MICA/B and HLA-A/B/C data were obtained from the same experiment.
Cell viability assay.
Cells were seeded into 96-well plates at 2 × 103 cells per well. After 24 hours, cells were treated with increasing concentrations of LDK378 (1 nM to 10 μM) in dimethylsulfoxide. Dimethylsulfoxide solvent served as a negative control. After 72 hours, cell viability was analyzed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). Drug concentrations that inhibited cell growth by 50% (IC50) were determined using a nonlinear regression curve fit with GraphPad Prism 8 software.
Cell migration and invasion assays.
NB-9464-H-2Kblo or NB-9464-H-2Kbhi cells in serum-free medium (0.5 × 106 cells per ml) were added to the upper chamber of Transwell chambers (Falcon), and inserts (8 μm) were placed in the lower chamber containing medium with 10% FBS. Following incubation at 37 °C for 8 hours, cells in the lower chamber were fixed with methanol and stained with crystal violet (Sigma-Aldrich), then photographed with a light microscope at ×100 magnification and migration quantified as the number of cells per high-power field. Cell invasion was measured using the fluorometric QCM ECMatrix Cell Invasion Assay (Millipore) according to the manufacturer’s instructions.
RNA extraction and RT–qPCR.
Total RNA was isolated using the RNeasy Mini kit (Qiagen) and reverse transcribed using Superscript IV VILO master mix (Thermo Fisher Scientific) following the manufacturer’s protocol. Quantitative polymerase chain reaction with reverse transcription (RT–qPCR) was performed using conventional methods on a ViiA 7 (Thermo Fisher Scientific) or StepOnePlus (Applied Biosystems) real-time PCR system61. Primer sequences are listed in Supplementary Table 6.
Synthetic RNA spike-in and RNA-seq.
Biological duplicates (5 × 106 cells per replicate) were homogenized in 1 ml of TRIzol Reagent (Invitrogen) and purified using the mirVANA miRNA isolation kit (Ambion) following the manufacturer’s instructions. Total RNA was treated with DNA-free DNase I (Ambion), spiked-in with ERCC RNA Spike-In Mix (Ambion) and analyzed on an Agilent 2100 Bioanalyzer (Agilent Technologies) for integrity. Sequencing libraries were prepared using LP-KAPA mRNA Hyper Prep and sequenced using Illumina HiSeq for 40 bases.
Western blotting.
Cells were homogenized in NP40 lysis buffer (Life Technologies) containing 1 × complete EDTA-free protease inhibitor cocktail and 1 × PhosSTOP (Roche). Protein concentration was measured using the DC Protein Assay (Bio-Rad). Total protein was denatured in LDS sample buffer (Invitrogen), and immunoblotting performed as previously described61. Antibodies are listed in Supplementary Table 7.
ChIP–qPCR analysis.
Soluble chromatin was prepared from SH-SY5Y cells without or with dox-inducible PRRX1 expression (200 ng ml−1 dox for 10 days) as previously described61,62. Chromatin immunoprecipitation (ChIP) was performed using various antibodies (Supplementary Table 7), followed by qPCR (ChIP–qPCR) on purified ChIP DNA for the genomic regions of interest (ROIs). Relative enrichment was quantified using the percentage input method. Primer sequences are listed in Supplementary Table 6.
ChIP–seq analysis.
Soluble chromatin was prepared from approximately 4 × 107 cells as previously described61,62. Chromatin equivalent of 1 × 107 cells was used in each immunoprecipitation reaction. Antibodies are listed in Supplementary Table 7. Purified ChIP DNA was used to prepare Illumina multiplexed sequencing libraries using the NEBNext Ultra II DNA Library Prep kit and the NEBNext Multiplex Oligos for Illumina (New England Biolabs) according to the manufacturer’s protocols. Libraries with distinct indices were multiplexed and run together on the Illumina NextSeq 500 (SY-415–1001, Illumina) for 75 base pairs.
NKG2D binding assay.
Cells were stained with the Zombie NIR (or Zombie Violet) viability dye and incubated with 10 µg ml−1 recombinant human NKG2D-Fc chimeric protein (R&D Systems) or an equivalent concentration of human IgG (BioLegend) for 1 hour on ice, following which they were washed and stained with an AlexaFluor 647-conjugated anti-human IgG (Supplementary Table 7) for 30 min and analyzed by flow cytometry as described above.
NK cell degranulation assay.
Human peripheral blood NK cells were isolated from blood collars using a RosetteSep human NK cell enrichment cocktail (STEMCELL Technologies) followed by Ficoll density gradient centrifugation (20 min, 800g). NK cells were cultured overnight in RPMI-1640 supplemented with 10% FBS, 1% penicillin/streptomycin and 2.5 ng ml−1 IL-15 (R&D Systems) and cocultured for 4 hours with confluent monolayers of neuroblastoma cells at a 1:2 effector to target (E:T) ratio with CD107a PerCP-Cy5.5 antibody. At the endpoint, NK cells were collected; stained with Zombie Yellow (BioLegend), CD56-FITC (or NKp46-AlexaFluor 647) and NKG2D-PE (or mouse IgG1-PE) antibodies; and fixed with 1% paraformaldehyde (Polysciences). For the NKG2D blocking assay, NK cells were incubated with purified anti-NKG2D antibody or mouse IgG1 isotype control for 30 min at 37 °C, following which the degranulation assay was performed. NK cells were analyzed by flow cytometry as described above. Gating strategy is shown in Supplementary Data Fig. 1a. Antibodies are detailed in Supplementary Table 7.
NK cell killing assay.
Human NK cells were cultured overnight in RPMI-1640 supplemented with 10% FBS, 1% penicillin/streptomycin and 1 ng ml−1 recombinant human IL-15 (Miltenyi Biotec) and then incubated with purified anti-NKG2D antibody or isotype control as described above. Target cells were labeled with 5 µM CellTrace Violet (Thermo Fisher Scientific) in PBS for 20 min at 37 °C. Antibody-treated NK and CellTrace Violet-labeled target cells were washed twice with RPMI-1640 and cocultured at the indicated E:T ratios for 18–24 hours at 37 °C. At the endpoint, cells were spun down, trypsinized and stained with Annexin V-PE and 7-AAD (both BD Biosciences) in Annexin V binding buffer (BioLegend) for 15 min, and cytotoxicity was analyzed by flow cytometry. Gating strategy is shown in Supplementary Data Fig. 1b. Antibodies are detailed in Supplementary Table 7.
T cell cytotoxicity assay.
Human peripheral blood CD8+ T cells were isolated using Ficoll isopaque density gradient centrifugation followed by CD8 selection using magnetic bead separation (Miltenyi Biotech) and lentivirally transduced with a TCR specific for the PRAME425–433 peptide, SLLQHLIGL63, as previously described41,64. PRAME-TCR-positive cells were sorted based on Vβ1 expression using a FACSAria II (Becton Dickinson) and were stimulated once every other week using a feeder cell mixture comprising irradiated allogenic peripheral blood mononuclear cells and lymphoblastoid cell lines in Glutamax-supplemented RPMI media (Gibco) containing 10% pooled human serum, 1% penicillin/streptomycin, 50 U ml−1 IL-2 and 1 μg ml−1 phytohemagglutinin (both Miltenyi Biotec). Target cells were untreated or cultured overnight in the presence of 100 U ml−1 IFN-γ (R&D Systems) or 10 µg ml−1 poly (I:C) (Thermo Fisher Scientific), then MHC class I and HLA-A2 expression was assessed and then cells were washed and cocultured overnight with PRAME-specific T cells at an E:T ratio of 10:1. At the endpoint, cells were spun down, trypsinized and stained with fixable viability dye (eFluor780, Thermo Fisher Scientific). Cytotoxicity was analyzed using the high-throughput sampler of the LSRFortessa. Standardized killing was calculated by normalizing target cytotoxicity to target cell control. Gating strategy is shown in Supplementary Data Fig. 1c. Antibodies are detailed in Supplementary Table 7.
ELISA.
Adrenergic and mesenchymal cells (1 × 106 each per well) were plated into six-well plates and, 24 hours later, treated with or without 1 µg ml−1 poly(I:C) for 24 h. Cell-free supernatants were stored at −20 °C until further use. CXCL10 in cell-free supernatants was measured using a human CXCL10 ELISA kit (R&D Systems, DIP100) per the manufacturer’s instructions and concentration (pgml−1) analyzed using linear regression. Samples above the limit of detection were diluted 40-fold and re-analyzed.
Multiplexed cytokine profiling.
Multiplexed cytokine profiling of cell-free supernatants treated with or without poly(I:C) (1 or 10 µg ml−1) was performed using the Human Cytokine/Chemokine Magnetic Bead Panel (Millipore, HCYTMAG-60K-PX30) on a Luminex MAGPIX system (Merck Millipore). The concentration of each protein (pg ml−1) in conditioned media was derived using a five-parameter curve-fitting model. Fold-changes relative to the corresponding control were calculated and plotted as log2 fold-change (log2FC). Lower and upper limits of quantitation were imputed from standard curves for cytokines/chemokines above or below the limit of detection.
Three-dimensional T cell migration assay.
The T cell migration assay was performed as previously described65. Briefly, cells were plated at a density of 5 × 105 cells per well of a six-well plate and transfected with 1 µg ml−1 poly(I:C) (Invitrogen) using the X-tremeGENE HP DNA Transfection Reagent (Roche) for 24 hours. Spheroids were generated by seeding 5 × 105 poly(I:C)-transfected or control cells on an ultra-low attachment dish (Corning) for 24 hours and labeled with eFluor 450 (Invitrogen) per the manufacturer’s instructions. Spheroids were pelleted and resuspended in type-I rat tail collagen (Corning) at a final concentration of 2.5 mg ml−1 following the addition of 10 × PBS containing phenol red on ice. The pH of the resulting spheroid suspension was adjusted to 7.0–7.5 using NaOH and confirmed using PANPEHA Whatman paper (Sigma-Aldrich). The spheroid–collagen suspension was then introduced into the central channel of the three-dimensional (3D) microfluidic cell culture chamber (AIM Biotech and ref. 66). Collagen hydrogels containing cancer cell spheroids were incubated for 40 min at 37 °C in humidity chambers, following which RPMI-1640 medium containing either (1) 6 × 104 CXCR3-overexpressing Jurkat cells65 or (2) 6 × 104 primary human CD8+ T cells supplemented with IL-2 (10 ng ml−1), at an E:T ratio of 2:1 was perfused through one of the side channels located next to the central channel. CD8+ T cells were isolated from peripheral blood mononuclear cells using manufacturer protocols (EasySep Human T Cell Isolation Kit, STEMCELL Technologies) and expanded using T Cell Activation/Expansion Kit (Miltenyi Biotech, purity >90%). Both CXCR3-overexpressing Jurkat and primary human CD8+ T cells were labeled with Cell Tracker Red (Thermo Fisher Scientific) before coculture. The cancer cell spheroids and T cells were cocultured for 3 days, following which T cell migration into the collagen hydrogel was visualized through images captured on a Nikon Eclipse 80i fluorescence microscope equipped with Z-stack (Prior) and a CoolSNAP CCD camera (Roper Scientific), and analyzed using NIS-Elements AR software package. Quantification of T cell infiltration into the central channel was performed by measuring the total area occupied by the Cell Tracker Red-positive cells located in ROIs (at least 6 ROIs per microfluidic cell culture chamber).
Antigen presentation assay.
Approximately 1 × 106 NB-9464-H-2Kblo or NB-9464-H-2Kbhi cells were seeded onto 10-cm plates, treated with recombinant mouse IFN-γ (BioLegend) (100 ng ml−1) for 24 h and pulsed with 100 ng ml−1 SIINFEKL peptide at 37 °C for 2 hours. Cells were subsequently washed with 1 × PBS to remove unbound peptide and processed for FACS analysis of H-2Kb–SIINFEKL complex (antibodies in Supplementary Table 7).
T cell activation assays.
OT-I TCR transgenic mice were purchased from the Jackson Laboratory. Splenocytes were collected and T cells isolated from the mononuclear layer using Ficoll separation and directly used in coculture assays. Successful enrichment of CD8+ T cells was confirmed by FACS analysis. Adherent NB-9464-H-2Kblo and NB-9464-H-2Kbhi cells were pulsed with 100 ng ml−1 SIINFEKL at 37 °C for 2 h, washed with 1 × PBS and cocultured with unstimulated OT-1 T cells at a ratio of 1:1 for 24 h. OT-I cells were then collected, sequentially stained with the Zombie NIR viability dye and CD8-FITC and CD69-PE antibodies, fixed with 1% paraformaldehyde (Polysciences) and analyzed by flow cytometry. Gating strategy is shown in Supplementary Data Fig. 1d. Antibodies are detailed in Supplementary Table 7.
In vivo experiments.
For the pilot study, approximately 1 × 106 NB-9464-H-2Kblo or NB-9464-H-2Kbhi cells were resuspended in growth factor-reduced Matrigel and injected subcutaneously in a 100-μl volume into the right flanks of 6–9-week-old female C57BL/6 mice (Charles River Laboratories; n = 7 mice per group). Mouse weights and tumor volumes were measured every 2–3 days, beginning on day 6 after injection. Tumor measurements were obtained using electronic calipers and volumes calculated using the modified ellipsoid formula: ½ (length × width)67. Animals were euthanized when tumors reached 2 cm in any dimension based on IACUC criteria for maximum tumor burden. Tumors were collected, snap-frozen in liquid nitrogen and stored at −80 °C for downstream analysis. For the immunotherapy studies, subcutaneous NB-9464-H-2Kblo and NB-9464-H-2Kbhi tumors were generated as above except that the cells were resuspended in PBS before injection. Tumor-bearing mice (n = 15 per group) were treated with either (1) 100 μg each of InVivoMAb anti-PD1 (BioXCell, clone 29F.1A12) and InVivoMAb anti-CTLA4 (BioXCell, clone 9H10) or (2) 100 μg each of isotype-matched control antibodies (InVivoMAb Rat IgG2a (BioXCell 0089) and InVivoMAb Syrian hamster polyclonal IgG (BioXCell 0087)) via intraperitoneal administration on days 7, 10 and 13 after inoculation. Tumor volumes were monitored and measured as above. Tumors from five mice per group were collected on day 36 for FACS analysis, and the remaining ten mice per group monitored for tumor growth and survival until endpoint. The following animals from the remaining ten mice per group were omitted from the final analysis because they (1) did not form palpable tumors (n = 1, H-2Kblo anti-PD1 + anti-CTLA4), (2) formed palpable tumors that were not measurable when alive (found to be growing inwards when euthanized for body condition score and weight-loss endpoints; n = 1, H-2Kblo anti-PD1 + anti-CTLA4; n = 1, H-2Kbhi isotype), (3) formed tumors that became ulcerated before reaching the volume endpoint (n = 2, H-2Kblo isotype; n = 1, H-2Kbhi isotype) or (4) had tumors that exhibited substantially different growth characteristics compared with the other animals in the cohort (n = 1, H-2Kblo anti-PD1 + anti-CTLA4; n = 2, H-2Kbhi anti-PD1 + anti-CTLA4). The data in Fig. 7f,g and Extended Data Fig. 10f reflect these events.
FACS analysis of tumor-infiltrating immune cells.
Tumors from five mice per group were collected on day 36 after injection. Resected tumors were weighed (15–4,840 mg) and sequentially treated with GentleMACS and Collagenase I (Worthington Biochemical) at 2 mg ml−1 in 1 × DPBS containing calcium and magnesium, followed again by GentleMACS treatment. The resulting single-cell suspensions were filtered through a 70-µm filter, supplemented with ice-cold PBS containing 2% FBS and centrifuged at 350g for 5 min at 4 °C. TILs were isolated from the cell pellets using CD45+ MACS positive selection. CD45+ cells were blocked with MACS buffer containing 2% anti-mouse CD16/32 (BioLegend) and 5% mouse and rat sera, and surface stained with antibodies (Supplementary Table 7) in Brilliant Stain Buffer (BD Biosciences) for 30 min at 4 °C. Dead cells were excluded using NIR live dead cell stain (1:1,000, Invitrogen). For intracellular staining, all samples were fixed using eBioscience Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher) for 20 min at 4 °C, washed with 1 × MACS buffer and left overnight at 4 °C, followed by blocking as above and Foxp3 staining. Spherotech AccuCount Fluorescent particles were added to all samples for cell quantification followed by FACS analysis. Single-color compensation controls and fluorescence-minus-one thresholds were used to set gate margins. Absolute cell counts were normalized to the total tumor weight before calculating ratios between distinct immune cell populations.
Immunofluorescence (IF) and immunohistochemistry (IHC) analyses are included in the Supplementary Information.
Analysis of RNA-seq data.
RNA-seq data processing and identification of differentially expressed genes.
Single-end RNA-seq samples with 40-base-pair read lengths were mapped to the human genome (GRCh38) and External RNA Controls Consortium (ERCC) spike-in sequences. Reads were mapped to the genome using Bowtie2 (v.2.3.4.3) and default parameters. Reads that overlapped with the genomic locations for exonic regions were used to calculate gene counts with the FeatureCounts package (Subread package of v.1.6.3). Spike-in read counts for each sample were used to normalize library sizes. These read counts were used to calculate the sample-specific size factor by using the function estimateSizeFactors (DESeq2) available in R. Normalized sample coverage profiles were then created from previously determined size factors by using bamCoverage (DeepTools v.3.1.1) and parameters ‘--scaleFactor --skipNonCoveredRegions’. To check the reproducibility of biological replicates for each condition, principal component analysis (PCA) and correlation (Spearman’s rank coefficient) were assessed from the sample coverage profiles at genome-wide scale and visualized using scatterplots and heat maps. Because these analyses showed a high correlation of sample coverage profiles between replicates, replicates were merged using samtools merge and processed again as described for the individual replicates. Next, differential gene expression analysis was performed using the DESeq2 in R. To detect differentially expressed genes (DEGs) in each sample, raw read counts from RNA-seq data were imported to the DESeq2 and the size factors calculated using the estimateSizeFactors function. A transcript with an absolute log2FC ≥ 1.5 and an adjusted P ≤ 0.01 was considered significant.
Identification of tumor clusters using UMAP.
The log-transformed reads per million from RNA-seq data from the 498 neuroblastomas were used to identify tumor subtypes. For clustering, 5,000 highly variable genes were identified using the FindVariableFeatures function from the Seurat R package (v.4.1.0), with a selection method ‘vst’ (variance-stabilizing transformation)68, and dimensionality reduction carried out on the variable genes using PCA. Next, significant principal components (PCs) were identified with the ElbowPlot function in the Seurat R package, and 20 PCs (from PC1 to PC20) were selected as significant components for clustering where the percentage change in consecutive PCs was <0.05%. FindNeighbors and FindClusters functions with a parameter resolution of 0.4 were used to perform clustering in Seurat. Distinct clusters were identified with the Louvain graph-based clustering algorithm69 and visualized using UMAP70. Next, to characterize each cluster, DEGs unique to each cluster were identified. A standard receiver operating characteristics classifier test was run with the function FindMarkers from the Seurat R package to identify DEGs in the different clusters, where each cluster was compared with the other three clusters and DEGs selected only if the average log2FC difference was ≥0.25. Gene ontology (GO) analysis of DEGs was performed with the enrichR package (https://amp.pharm.mssm.edu/Enrichr/). The clinical annotations were overlaid onto the clusters using the AddMetaData function and the results visualized using UMAP in the Seurat R package.
Enrichment analysis.
All GO terms were ranked based on the enrichR combined score, calculated by multiplying the adjusted P value with the z-score using the Fisher’s exact test. This test was also used to determine significant overlaps between the queried gene sets and other publicly available datasets. Enrichment was considered significant for an adjusted P ≤ 0.01, unless stated otherwise.
Gene coexpression analysis.
Gene coexpression analysis was performed on the log2 normalized expression data from the tumors within the immunogenic cluster using the CEMiTool (v.1.10.2) (coexpression modules identification tool) R package24. Expression data were processed after filtering out low-expressed genes by using default parameters. Coexpressed genes were identified as modules. To identify GO terms enriched within each module, overrepresentation analysis was performed via the clusterProfiler (v.3.14.3) R package71 using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The hypergeometric test was performed between the coexpressed genes in the modules and the gene sets in the KEGG database; significant GO terms were listed along with the −log10 adjusted P value. To identify regulatory genes (hubs) within the M1 module, PPIs were integrated with coexpression data within this module. Next, coexpressed M1 module genes were searched for within the STRING 11.0 PPI database and all possible interactions identified. The gene network was created by the ‘plot_interaction’ function within CEMiTool and the genes with high degrees of connectivity highlighted as hub genes within both the coexpression module and the PPI network.
Derivation of gene signature scores.
IA and IE scores were calculated from a curated set of genes (Supplementary Table 3) into a single value for all MYCN-nonamplified tumors. All genes were ranked based on their expression levels in each tumor. The rank order was established for all genes, and the percentiles were calculated based on the full list of genes. The average percentile value was calculated for all IA and IE signature genes in a tumor, and, thus, an IA/IE score was assigned to each tumor. Similarly, adrenergic (A) and mesenchymal (M) scores were calculated for each tumor from the list of gene signatures detailed by Groningen et al.26. A total of 6 IA genes were removed from the 485-gene mesenchymal signature genes to ensure that the IA signature genes did not overlap with predefined adrenergic and mesenchymal signatures. To further assign a tumor as either adrenergic or mesenchymal, the M-score was subtracted from the A score (M − A score) and tumors were arranged according to increasing M − A scores. IA, A and M scores were calculated for 24 neuroblastoma cell lines in the same manner.
IA/IE and lineage score correlation.
IA, IE, A, M and M − A score correlation was assessed using the R (v.4.2.0) corrplot package. Pearson’s correlation coefficients were calculated for all pair-wise correlations among all parameters.
Estimation of immune cell content in neuroblastoma tumors.
Cell type identification by estimating relative subsets (CIBERSORT)29 was used to evaluate immune cell fractions from gene expression data using the R package ‘immunedeconv’ (v.2.0.0). RNA transcript estimations were generated for all 498 neuroblastoma tumors using the LM22 signature matrix available for 22 immune cell types. CIBERSORT was run in ‘Absolute mode’ with quantile normalization disabled as recommended for tumor RNA-seq data and the overall immune content produced by the algorithm compared among tumors.
sc/single-nucleus (sn)RNA-seq data analysis.
Gene expression count matrices were downloaded from GSM4186961, GSM4186962 and EGAS00001004388 sc/snRNA-seq datasets. Quality control was carried out for each sample dataset by removing doublet72 cells or nuclei with <500 genes and 1,000 unique molecular identifiers, 10% of which were from mitochondrial genes, using the Seurat R package (v.4.1.0). The top 4,000 highly variable genes were identified using the FindVariableFeatures function with a selection method ‘vst’68 for each tumor. Next, the variable genes were used to perform PCA, and significant PCs identified with the ElbowPlot function. Twenty significant PCs for snRNA-seq (n = 14) and 50 PCs for scRNA-seq (n = 2) data were used as input with the FindNeighbors and FindClusters functions in the Louvain graph-based algorithm69, and clusters were visualized using UMAP70. Next, to annotate the clusters based on cell type, previously identified gene expression signatures30,35 and SingleR73 were used. Malignant cells within each sample were identified by inferring chromosomal copy number alterations from gene expression data using inferCNV (https://github.com/broadinstitute/inferCNV). The nonmalignant immune and endothelial cells were used as references to estimate copy number alterations in the malignant cells. Next, the snRNA-seq data from the 14 samples30 were integrated using Harmony (v.0.1.0)74. In total, 4,000 variable genes were identified across all 14 tumors, and the first 20 PCs analyzed with the RunHarmony function. FindClusters and RunUMAP functions were used to carry out dimension reduction and cluster visualization, respectively. Additionally, to determine the adrenergic, mesenchymal and immune scores (IA and IFN-response signatures) of each malignant cell, the AddModuleScore function (Seurat R v.4.1.0) was applied using previously published signatures26.
Identification of IFN-response signatures.
IFN-response signatures were retrieved from the 498-tumor cohort and 5Y-par., 5Y-res. and SH-EP cells. DEGs in the immunogenic cluster were selected from the UMAP clustering analysis if the average log2FC difference in expression value was ≥0.25 compared with the other clusters. GO enrichment analysis of the DEGs was performed with the enrichR package in R and IFN-response-related GO terms chosen if the adjusted P value was <0.01. In parallel, genes whose expression was consistently upregulated in the three cell lines were identified. DEGs were selected through pair-wise comparisons between adrenergic (5Y-par.) and mesenchymal (5Y-res. and SH-EP) cells using the DESeq2 package in R. Genes upregulated by 0.5-fold in the mesenchymal compared with adrenergic cells and associated with IFN-response-specific GO terms were identified. Finally, the overlapping IFN-response genes in both tumors and cell lines were selected to derive NB-specific IFN-response signatures.
Analysis of matched diagnosis and relapse samples.
Microarray data from seven matched diagnosis and relapsed tumor pairs, and four tumors at diagnosis (total = 18 samples)34, were log2 transformed. Using the Loess regression model from the Giotto package (v.1.0.3)75, 5,264 variable genes were selected for clustering. These variable genes were used to perform PCA, and PC1 and PC2 that showed the highest transcriptomic variability were used to cluster tumors using the Leiden algorithm76 and clusters were visualized using UMAP.
ChIP–seq analysis.
Data processing.
All ChIP–seq raw datasets were processed as previously described61 and using the Fastqc tool (v.0.11.7) to identify possible sequencing errors and biases. Reads were aligned to the human genome (build hg19, GRCh37.75) using the Bowtie (v.2.3.4.3) mapper with default parameters. Unique and nonduplicate reads that mapped to the reference genome were further processed using Samtools (v.1.9) and the MarkDuplicates (v.2.1.1) command of Picard tools. Next, antibody enrichment in each replicate compared with input samples was verified using the PlotFingerprint command of deepTools (v.3.1.1). Peak caller MACS2 (2.1.1) was used to identity peaks. Bigwig files were created for visualization with bedGraphToBigWig.
Identification of super-enhancer regions.
Super-enhancers were identified using the ROSE algorithm (https://bitbucket.org/young_computation/rose/src/master/) as previously described61.
Analysis of histone binding between lineage states.
To analyze the changes in occupancies of active (H3K27ac and H3K4me3) and repressive (H3K27me3) histone marks during the transition from adrenergic (5Y-par.) to mesenchymal (5Y-res., SH-EP) states, the peaks were compared for histone marks identified by MACS2 at the promoter regions. The promoter regions (TSS ± 2 kb) of all annotated protein coding genes were identified and the peaks of the three histone marks retrieved. To analyze the differential binding of each histone mark between 5Y-par. and 5Y-res. cells, all significant peaks that overlapped by at least 50% at the promoter regions were first combined. The 50% threshold was used to avoid merging peaks that had clear and distinct summits. Next, the normalized active or repressive histone read densities were calculated for each region and a ratio of (log2 (5Y-res./5Y-par.)) derived. Peak regions with ≥0.75 or <0.75 fold-change of the ratio value were categorized as 5Y-res.- or 5Y-par.-cell-specific peaks, respectively, while other regions were denoted as ‘shared’ peaks. Shared peaks had a similar enrichment of either active or repressive histone marks in both the cell types. Similar comparisons were performed for active or repressive histone marks between 5Y-par. and SH-EP cells. To further compare changes in all histone marks at the promoters of immune genes, gain of significant H3K27ac and H3K4me3 and loss of H3K27me3 signals were identified in mesenchymal compared with adrenergic cells.
Integrated analysis of histone binding and gene expression.
Cell type-specific differential enrichment of H3K27me3 and H3K4me3 binding in 5Y-par., 5Y-res. and SH-EP cells was determined by calculating the log2 (H3K4me3 + 1/H3K27me3 + 1) ratios at the promoters of immune genes. To examine the association between gene expression and differential enrichment of H3K27me3 and H3K4me3 binding in immune genes in these cells, genes were ranked based on their expression values and plotted against the calculated ratios between the histone marks. For each cell line, a loess regression curve was computed for the ratio values. Similarly, cell-specific differential enrichment of H3K27me3 and H3K27ac binding in the cells was determined and a loess regression curve fitted for the ratios of log2 (H3K27ac + 1/H3K27me3 + 1) and gene expression values at immune gene promoters.
Statistics and reproducibility.
No statistical method was used to predetermine sample sizes, but our sample sizes were similar to those reported in previous publications23,60,61. All experiments (except where noted in figure legends) were performed in at least two independent replicates with reproducible results. Data exclusions for the in vivo studies are explained in the Reporting Summary and in the Methods section. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. Statistical analyses were performed in GraphPad Prism (v.8.4.3) or R (v.4.2.0). For all experiments, normality and equal variances were formally tested using Shapiro–Wilk test and F-test or Brown–Forsythe test, respectively. In general, comparisons of non-normal data points between groups were carried out with nonparametric approaches. Normally distributed samples were analyzed using Student’s t-test or analysis of variance (ANOVA).
Extended Data
Extended Data Fig. 1 |. Tumors within the immunogenic cluster express both IA and IE markers.
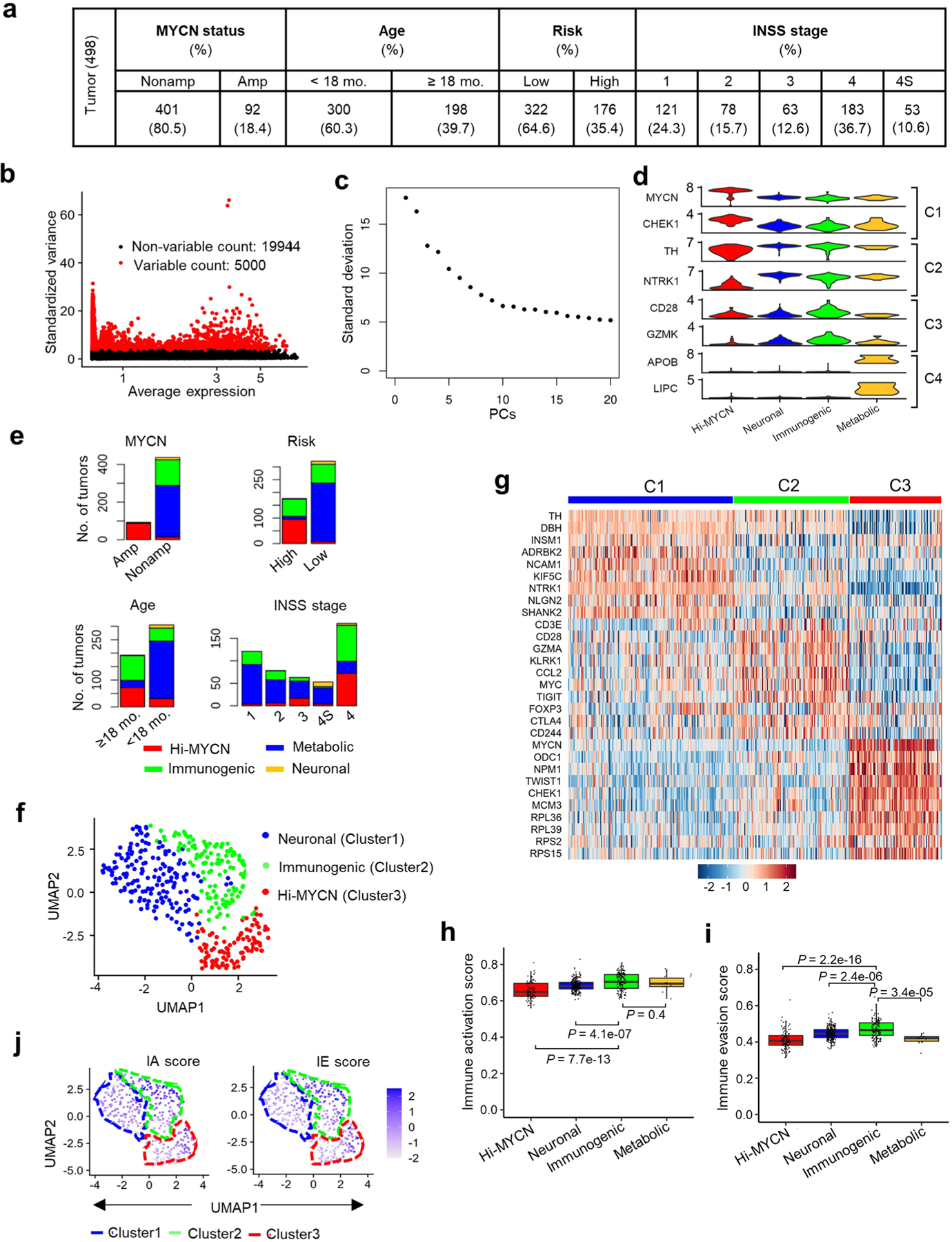
a, Distribution of the 498 primary NB tumors in the data set (SEQC-498; GSE49711) within the indicated prognostic categories. Risk stratification was based on the Children’s Oncology Group risk classification. INSS, International Neuroblastoma Staging System32. b, Scatter plot of the standardized variance in expression of all protein coding genes within the 498 tumors. Red dots indicate the top 5000 variably expressed genes. c, Elbow plot representing the percentage variance for the top 20 principal components, PCs (n = 20). d, Violin plots showing the expression of representative marker genes across the four clusters in 498 tumors. e, Stacked bar plots showing the distribution of tumors within the defined prognostic features within each cluster. Amp, amplified; Nonamp, nonamplified. f, Two-dimensional UMAP representations of the gene expression profiles in 394 NB tumors (GSE120572). Each dot represents a tumor. The top 3000 highly variable genes were selected based on the variance-stabilizing method33 and 20 significant principal components (PCs) selected and processed in UMAP to generate three clusters representing three NB subtypes. The DEGs were identified for each cluster using the receiver operating characteristics (ROC) curve to compare one cluster with others (log2 FC > 0.25). g, Heat map of expression values of 10 representative DEGs within each cluster in (f). Rows are z-score scaled average expression levels for each gene in all three clusters. h, i, Box plots comparing IA (h) and IE (i) scores within the four clusters (n = 103 tumors in Hi-MYCN, 241 in neuronal, 140 in Immunogenic and 14 in metabolic) from the SEQC-498 tumor data set. All box plots are defined by center lines (medians), box limits (25th and 75th percentiles) and whiskers (minima and maxima; the smallest and largest data range). Significance was determined by the two-tailed Wilcoxon rank-sum test. j, UMAP visualization of the distribution of IA and IE scores among the three tumor clusters derived from the 394 NBs in the GSE120572 dataset. Color bar represents normalized z-scores. Values <2.5 and >2.5 were set to −2.5 and +2.5, respectively, to reduce the effects of extreme outliers.
Extended Data Fig. 2 |. Cell lineage is significantly associated with immune gene signatures in NB.
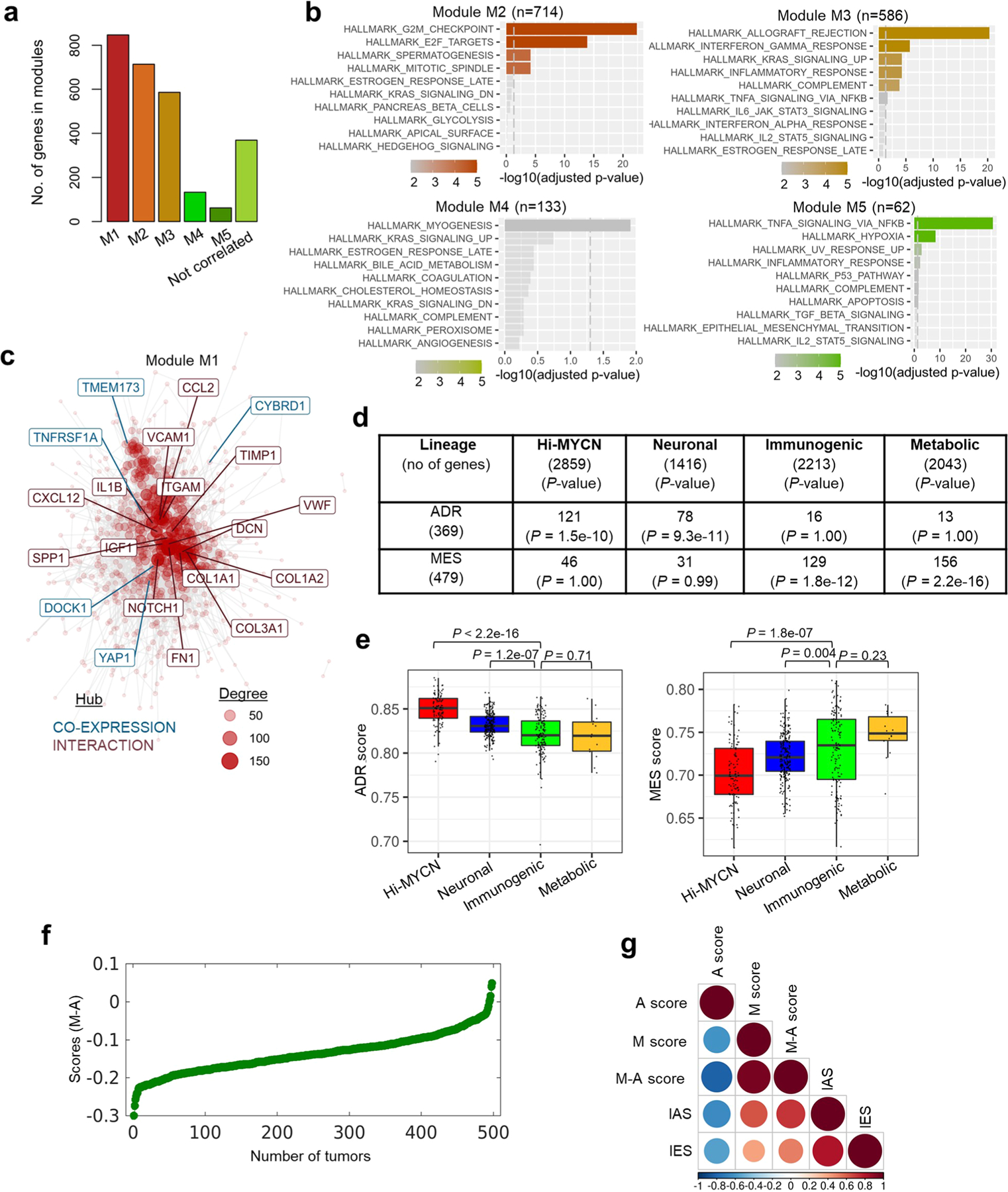
a, Bar plots representing the numbers of co-expressed genes within each module. b, GO analysis of co-expressed genes associated with indicated modules (KEGG database). The vertical dashed lines indicate the adjusted P-value of 0.05, two-tailed hypergeometric test. c, Gene network representing all possible interactions in module M1. The topmost connected genes (hubs) are indicated. Hubs derived from module M1 are colored blue (co-expression) and those from the STRING database are indicated in red (interaction). The size of each node corresponds to the degree of interaction. d, Summary of the overlap between the DEGs associated with the four tumor clusters and the adrenergic or mesenchymal signature genes as per Groningen et al., 201738. Significance determined by two-tailed Fisher’s exact test. e, Box plots comparing the expression of adrenergic and mesenchymal scores within the four clusters (n = 498 tumors [Hi-MYCN (n = 103), neuronal (241), immunogenic (140), metabolic (14)] derived from the SEQC-498 tumor data set. All box plots are defined by center lines (medians), box limits (25th and 75th percentiles) and whiskers (minima and maxima; the smallest and largest data range). The immunogenic cluster was compared with the other three clusters and significance determined by the two-tailed Wilcoxon rank-sum test. f, Scatter plot of the 498 primary NB tumors ranked based on increasing M-A scores. g, Pearson correlation matrix showing pairwise correlation values among the indicated parameters. The colors and sizes of the circles indicate the correlation coefficient values, with the least (smaller blue circles) to the most (larger brown circles) degree of association between the parameters shown.
Extended Data Fig. 3 |. The relative mesenchymal score (M-A score) is positively correlated with an immunogenic signature.
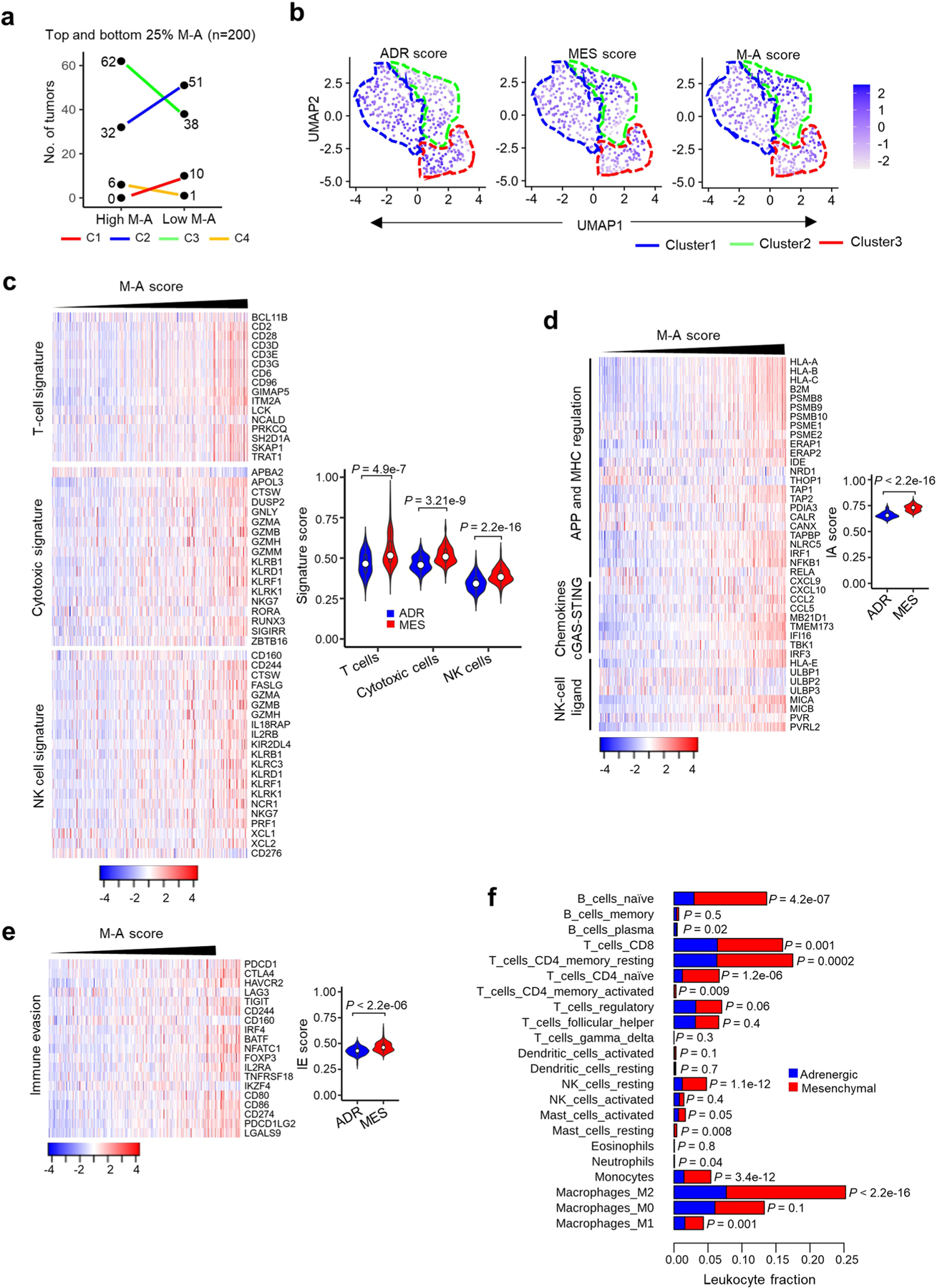
a, Dot plots showing the distribution of MYCN-nonamplified tumors (n = 400) within each of the clusters based on ranked M-A scores. Tumors from the upper (high M-A) and lower (low M-A) M-A score quartiles are shown (n = 200 tumors; P = 0.0011 for C3). Statistical significance assessed by the two-tailed Fisher’s exact test. b, UMAP visualization of the distribution of adrenergic, mesenchymal, and M-A scores among the three tumor clusters derived from 394 NBs in the GSE120572 dataset. Color bar represents normalized z-scores. Values <2.5 and >2.5 were set to −2.5 and +2.5, respectively, to reduce the effects of extreme outliers. c, Left, Heat map of the indicated immune cell signatures in MYCN-nonamplified tumors (n = 401), ranked by increasing M-A scores. Log2 gene expression values were z-score transformed for heat map visualization. Right, violin plots comparing the quantitative scores of the indicated immune cell signatures in 100 tumors each from the upper (mesenchymal) and lower (adrenergic) quartiles of the tumor M-A scores using the two-sided K-S test. Box plots within the violin plots are defined by center lines (medians), box limits (25th and 75th percentiles) and whiskers (minima and maxima; 1.5X the interquartile range). d, e, Heat maps depicting IA (d) and IE (e) signatures in MYCN-nonamplified tumors, ranked by increasing M-A score. Log2 gene expression values were z-score transformed for visualization. Violin plots comparing the distribution of IA (d) and IE (e) signatures in 100 tumors each from upper (mesenchymal) and lower (adrenergic) quartiles of the tumor M-A scores are shown next to the heat maps. Significance determined by the two-sided K-S test. Box plots within the violin plots are defined as in (c). f, Bar diagram comparing the CIBERSORT-estimated fractional content of the indicated tumor-infiltrating leukocytes between MYCN-nonamplified adrenergic and mesenchymal tumors. Adrenergic and mesenchymal tumors were assigned as in (c). Data represent the mean, n = 100 tumors, P determined by the two-tailed Welch’s t-test.
Extended Data Fig. 4 |. Cellular heterogeneity predicts immune gene expression in NB tumors.
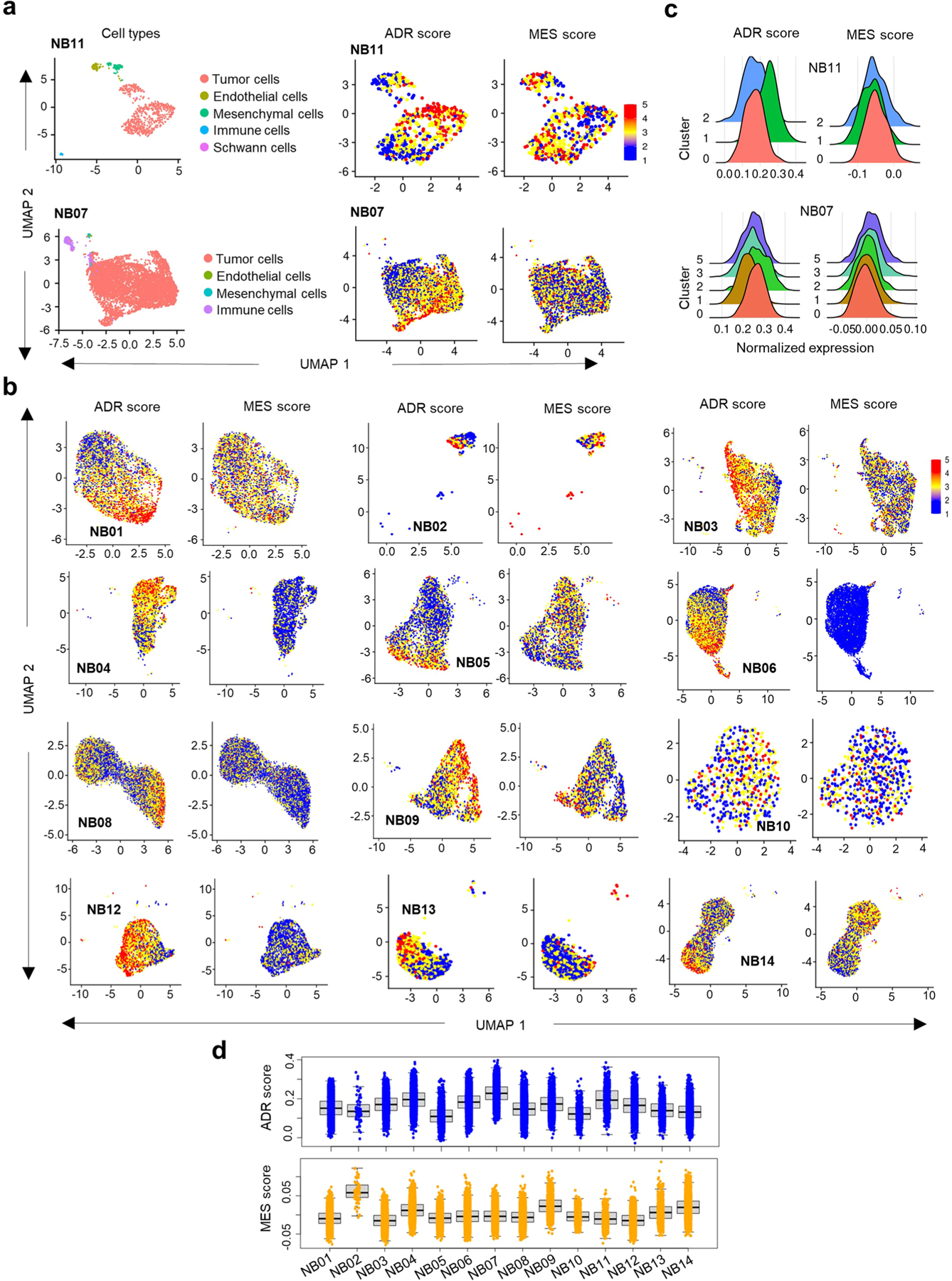
a, UMAP embedding of 691 (top left) and 6498 (bottom left) cell signatures derived from two representative MYCN-amplified and nonamplified tumors (NB11 and NB07), respectively. Clusters of transcriptionally similar cells are colored by cell type. UMAP representation of 573 (top right) and 6299 (bottom right) malignant cell signatures from the same samples as in left panel. b, UMAP representation of the distribution of malignant cells in 12 NB tumors. Cells are colored based on the score calculated from the average expression of genes per cell38. c, Ridge plot representing the distributions of the ADR and MES scores of the malignant cells within the indicated clusters. d, Box plot representing ADR and MES scores of 56,597 malignant cells from 14 primary NB tumor samples based on transcriptional diversity (n = 4543 cells in NB01, 66 in NB02, 4429 in NB03, 3914 in NB04, 3769 in NB05, 7782 in NB06, 6047 in NB07, 10675 in NB08, 2353 in NB09, 587 in NB10, 568 in NB11, 3596 in NB12, 1024 in NB13 and 7244 in NB14). Box plots are defined by center lines (medians), box limits (25th and 75th percentiles) and whiskers (minima and maxima; 1.5X the interquartile range).
Extended Data Fig. 5 |. NB tumors are highly heterogeneous at the inter- and intra-tumoral level.
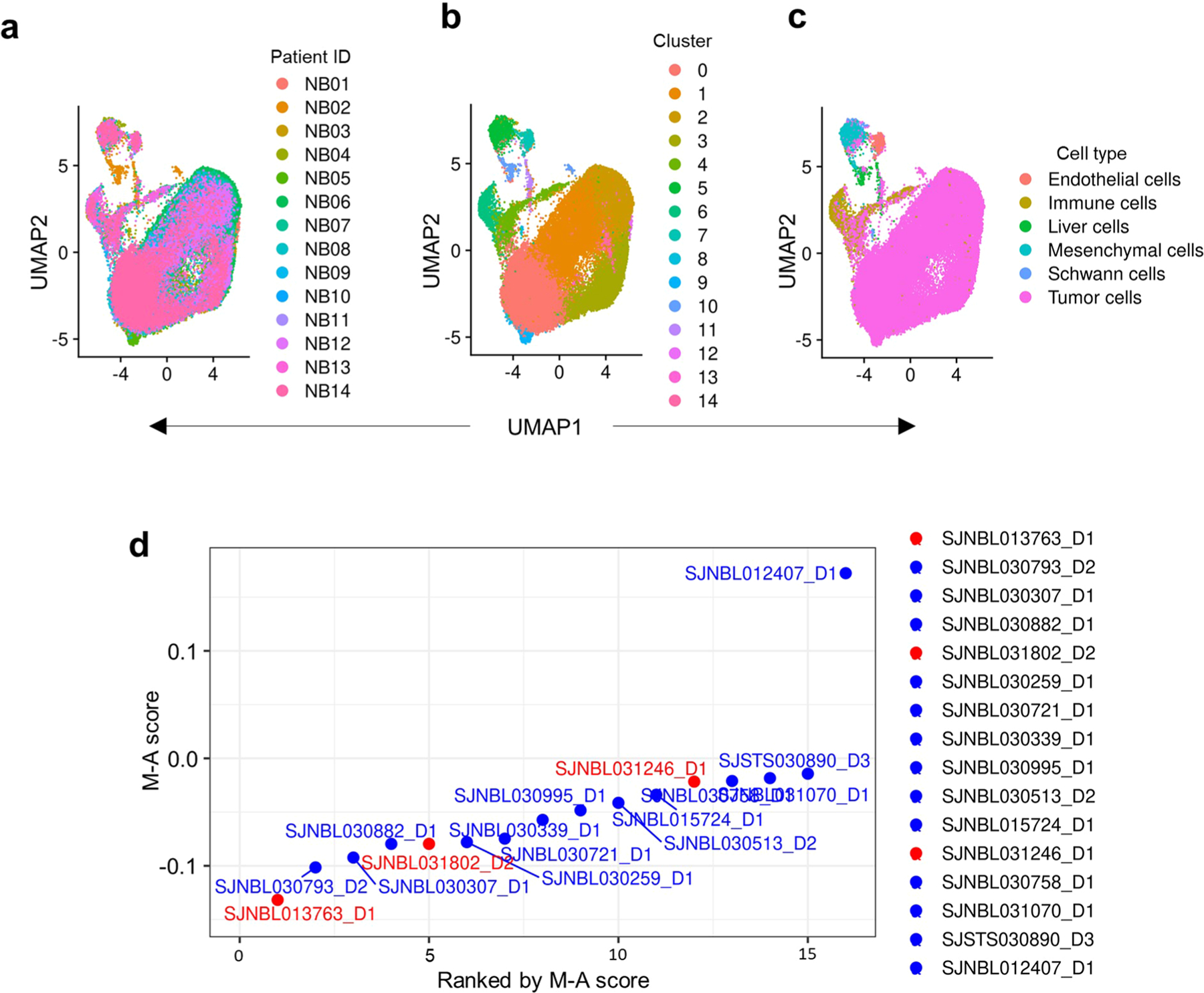
a, b, c, UMAP visualization of 63,291 cells from 14 NB tumors42, colored by samples (a), cells (b) and cell type (c). d, Scatter plot of M-A scores derived from bulk RNA-seq data from 16 NB tumors (St Jude Cloud43). Samples are arranged based on increasing M-A scores. The samples analyzed by IHC are colored in red.
Extended Data Fig. 6 |. Tumor cell-intrinsic immune marker genes are upregulated in mesenchymal NBs.
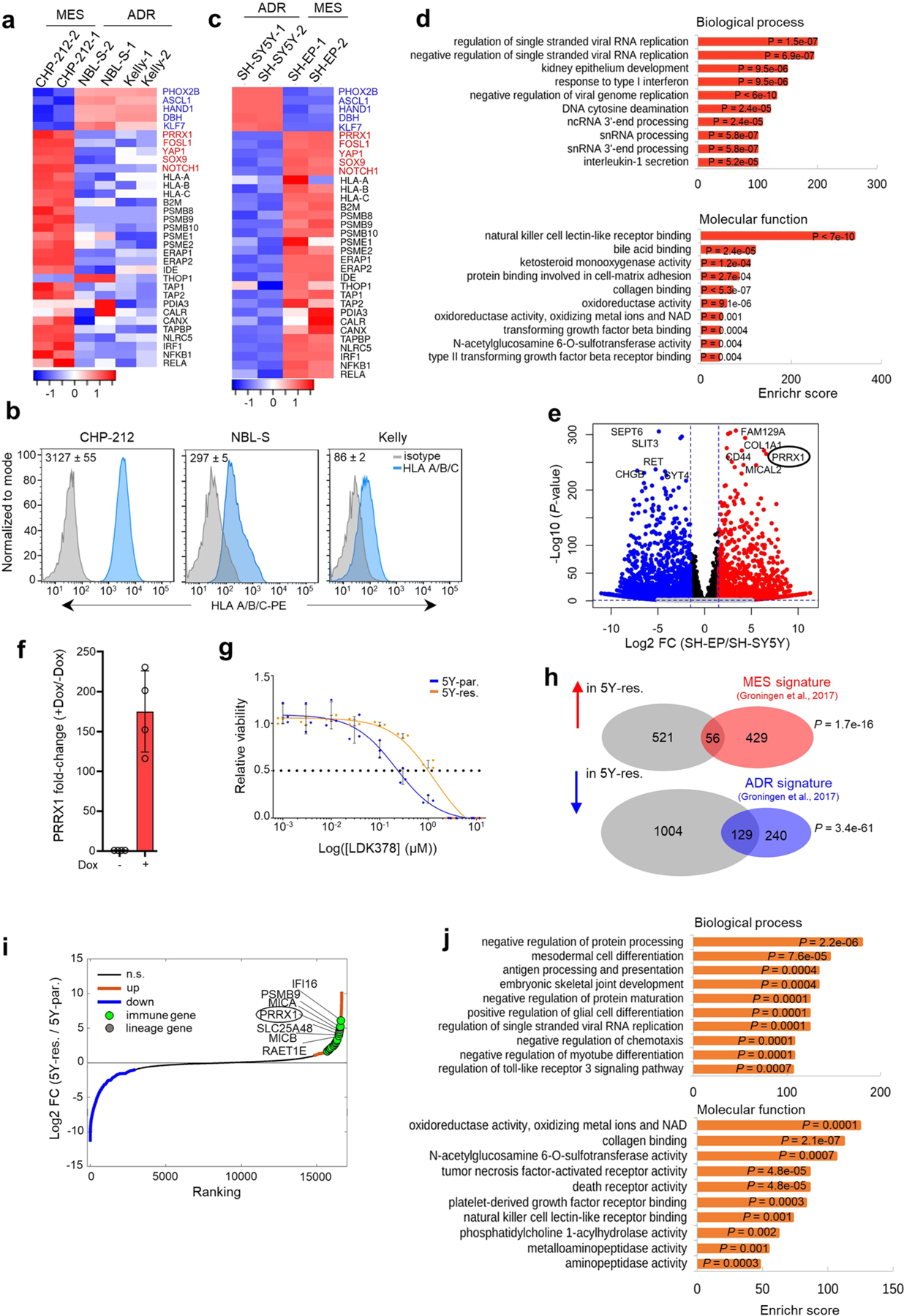
a, Heat maps of lineage marker (blue, adrenergic; red, mesenchymal) and MHC and APM gene (black) expression in the indicated adrenergic and mesenchymal cells (n = 2 independent experiments). Rows are z-scores calculated for each transcript in each cell type. b, FACS analysis of cell surface HLA expression in the cells depicted in a. Numbers on bottom right indicate isotype-normalized HLA gMFI values expressed as mean ± SD, n = 3 independent experiments. c, Heat map of lineage marker (blue, adrenergic; red, mesenchymal), MHC and APM gene (black) expression in SH-SY5Y and SH-EP adrenergic and mesenchymal cells, respectively (n = 2 independent experiments). Rows are z-scores calculated for each transcript in each cell type. d, GO analysis of differentially upregulated genes in mesenchymal SH-EP and adrenergic SH-SY5Y cells. Significance determined by the Fisher’s exact test. e, Volcano plot showing the gene expression changes between mesenchymal SH-EP and adrenergic SH-SY5Y cells. The top ten lineage marker genes are highlighted. The fold changes are represented in log2 scale (X-axis) and the -log10 of the P-values depicted on Y-axis (FDR < 0.1 and log2FC > 1.5). Significance was determined by two-tailed Wald test. f, RT-qPCR analysis of PRRX1 expression in SH-SY5Y cells expressing dox-inducible PRRX1 ± dox for 10 days. Data represent mean ± SD, n = 4 independent experiments. g, Dose-response curves of ceritinib-sensitive (5Y-par.) and -resistant (5Y-res.) cells treated with increasing concentrations of LDK378 for 72 h. Data show mean ± SD, n = 3 technical replicates and is representative of n = 3 independent experiments. h, Venn diagrams depicting the overlap between the DEGs in 5Y-res. cells (compared to 5Y-par. cells) and mesenchymal or adrenergic signatures. P-values were determined by two-tailed Fisher’s exact test. i, Waterfall plot of the fold-change in RNA expression levels of up- and down-regulated genes in 5Y-res. cells compared to 5Y-par. cells; selected lineage and immune genes are highlighted. j, GO analysis of differentially upregulated genes in 5Y-res. compared to 5Y-par. cells. Significance determined by the two-tailed Fisher’s exact test.
Extended Data Fig. 7 |. NBs that acquire mesenchymal phenotypes at relapse show increased immune gene expression.
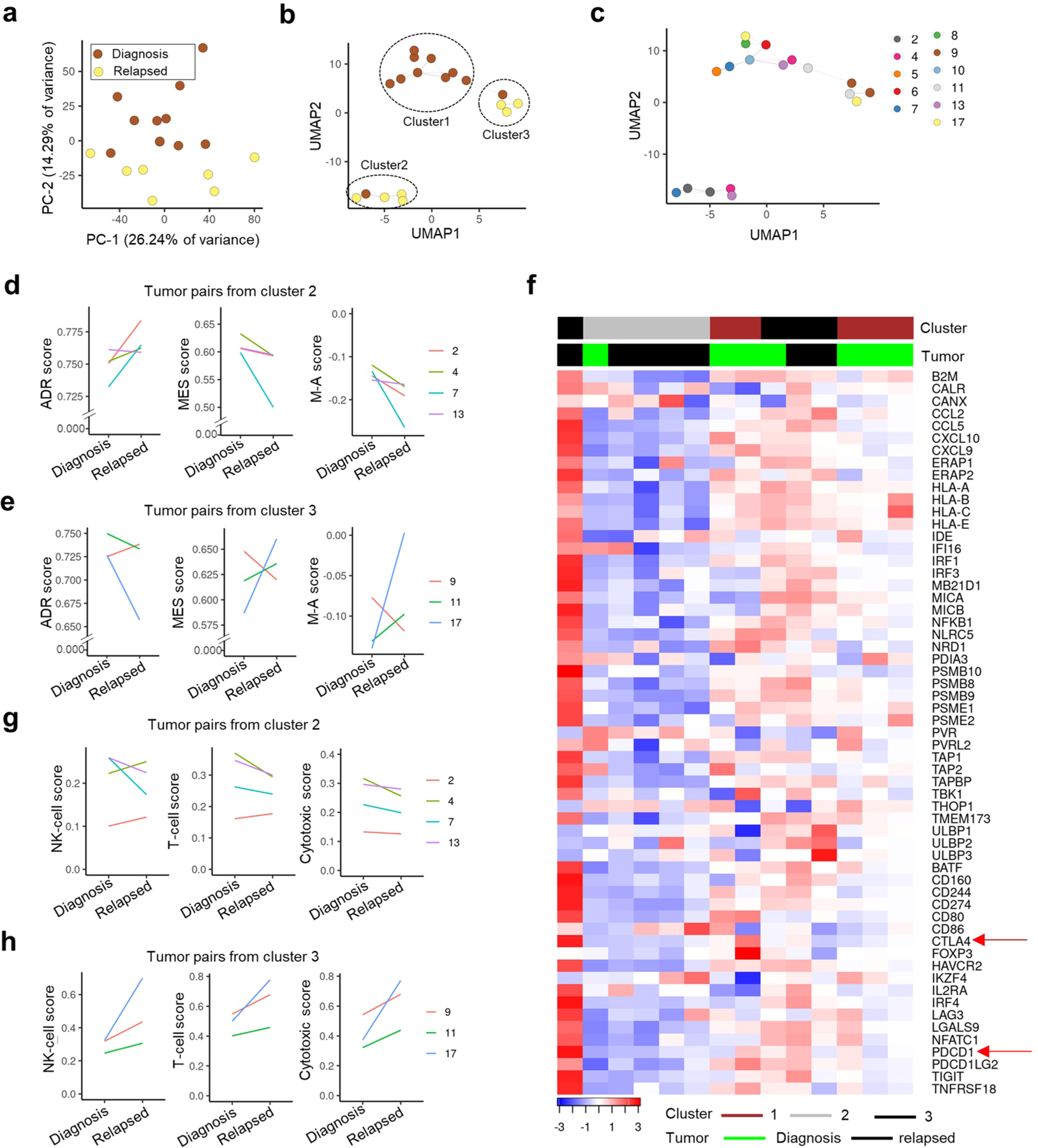
a, PCA plot showing the segregation of the depicted tumors using the 1st and 2nd PCs in 7 matched pairs and 4 single primary tumors. b, c, UMAP plot showing the segregation of primary and relapsed tumors (b) overlaid with the sample numbers (c). d, e, Line plots depicting changes in ADR, MES and M-A (MES-ADR) scores between primary and relapsed tumor pairs in clusters 2 (d) and 3 (e). f, Heat map of z-score-transformed log2 normalized expression values of tumor cell-intrinsic IA and IE genes in the 7 matched tumor pairs (n = 14 tumor samples). g, h, Line plots depicting the changes in the indicated immune cell scores in primary and relapsed tumor pairs in clusters 2 (g) and 3 (h).
Extended Data Fig. 8 |. Enrichment of immune cells is seen at relapse in a matched diagnosis-relapsed NB tumor pair.
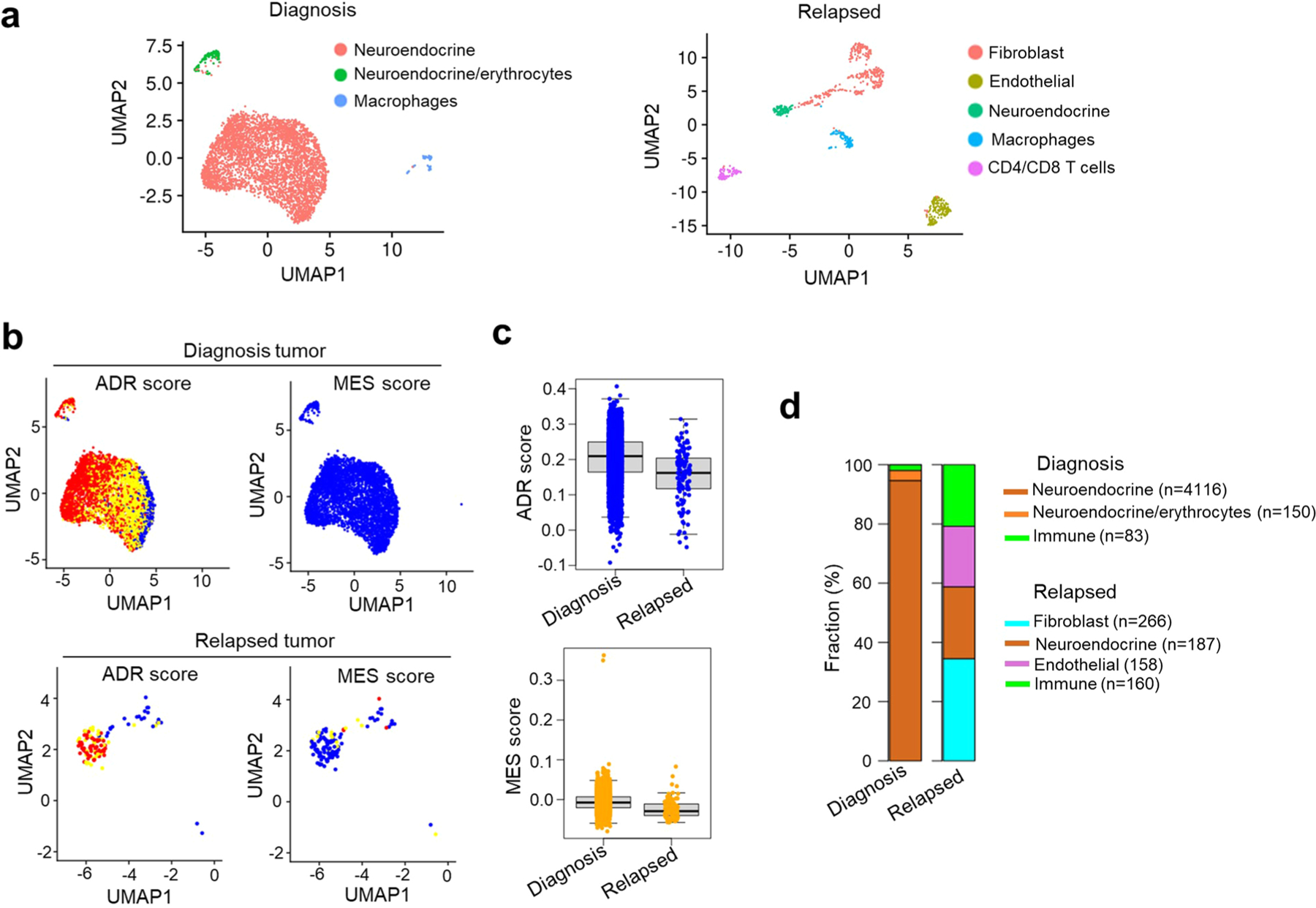
a, UMAP embedding of single cell profiles (dots) from diagnosis (left; n = 4349 cells) and relapsed (right; n = 771 cells) samples, colored by cell subset signatures. b, UMAP visualization of the distribution of ADR and MES scores of malignant cells at diagnosis (top; n = 4266 cells) and relapse (bottom; n = 113 cells). c, Box plot depicting the ADR (top) and MES (bottom) scores of the malignant cells in each sample [diagnosis (n = 4266), relapse (n = 113)]. Box plots are defined by center lines (medians), box limits (25th and 75th percentiles) and whiskers (minima and maxima; the smallest and largest data range). d, Bar plot representing the proportion of neuroendocrine cells (tumor), fibroblasts, endothelial cells and immune cells in each sample.
Extended Data Fig. 9 |. Immune gene activation associated with the mesenchymal state is epigenetically regulated.
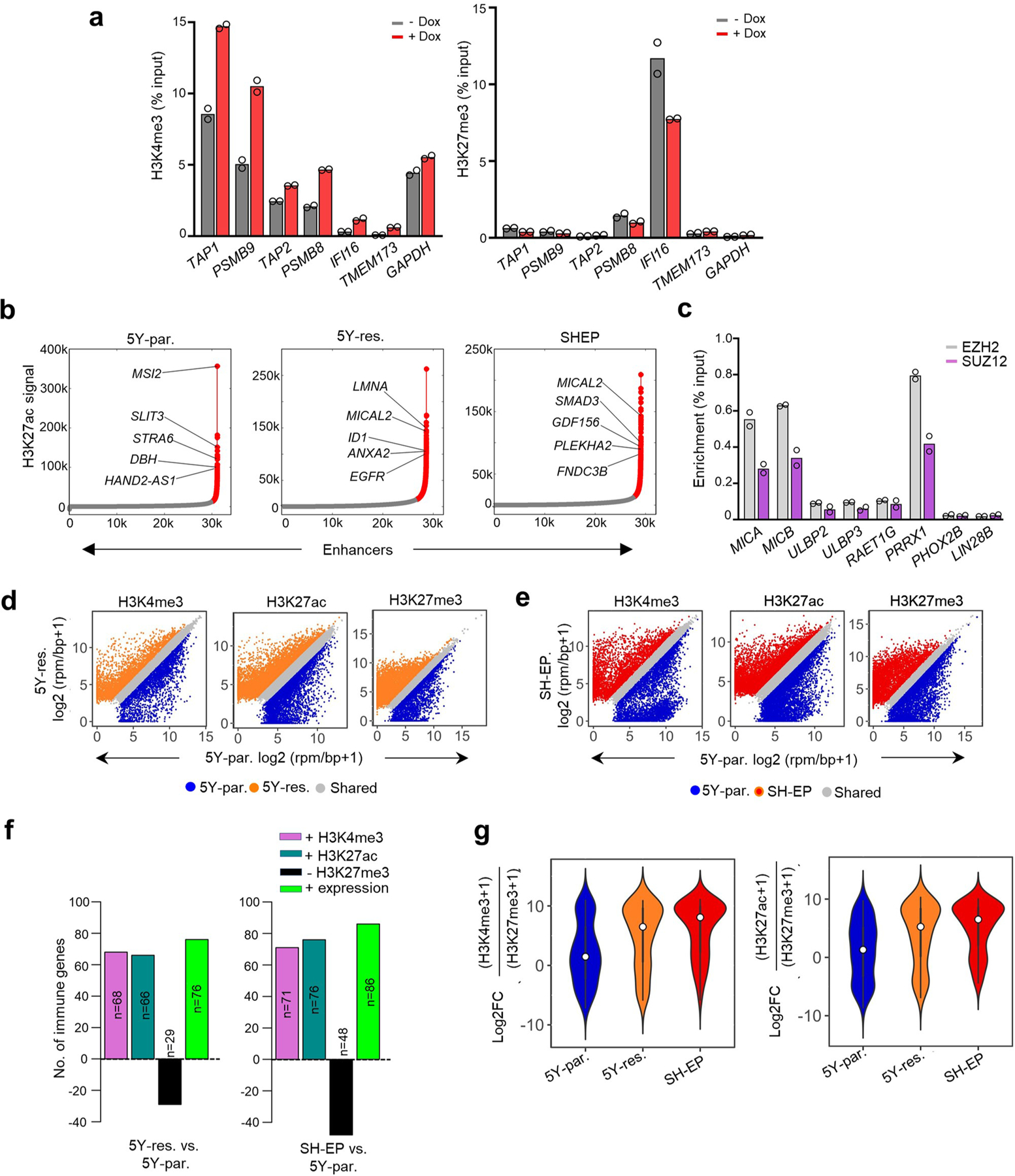
a, ChIP-qPCR analysis of the indicated histone marks at the indicated immune gene promoters in SH-SY5Y cells expressing dox-inducible PRRX1 ± dox. Data points represent mean of 2 technical replicates of 2 independent experiments. Enrichments at TAP1 and PSMB9 loci represent the data corresponding to amplicons 4 and 6, respectively, in Fig. 4a. b, Enhancer regions in 5Y-par., 5Y-res. and SH-EP cells. H3K27ac bound regions identified as significant peaks were stitched together if they were within 12.5 kb of each other (typical enhancers; grey). SEs (red) were defined as stitched enhancers surpassing the threshold signal based on the inclination point in all cell types. In 5Y-par., 5Y-res. and SH-EP cells, 2.94% (915/31116), 6.56% (1880/28635) and 4.18% (1215/29057) of the enhancers were classified as SEs, respectively. The top five SE-associated lineage-specific genes are highlighted. c, ChIP-qPCR analysis of EZH2 and SUZ12 enrichment at the indicated immune genes in 5Y-par. cells. Negative control loci, PHOX2B and LIN28B. Data represent mean of n = 2 independent experiments. d, e, Scatter plots representing the differential binding of the indicated histone marks at the promoter regions of all protein coding genes between 5Y-par and 5Y-res. (d), and 5Y-par. and SH-EP cells (e). rpm/bp, reads per million per base pair. A ≥ 0.75 log2FC threshold was used to identify unique peaks for each histone mark. f, Bar plots representing the numbers of immune genes with increased promoter deposition of H3K4me3 and H3K27ac (log2 FC ≥ 0.75) and loss of H3K27me3 (log2 FC ≥ 0.75) marks, together with increased RNA expression (log2 FC ≥ 1) in 5Y-res. (left) or SH-EP (right) compared to 5Y-par. cells. g, Violin plots of the ratios of active to repressive histone marks surrounding immune gene promoters in the indicated cells. Significance determined by the two-tailed Wilcoxon rank-sum test. Box plots within the violin plots defined by center lines (medians), box limits (25th and 75th percentiles) and whiskers (minima and maxima; 1.5X the interquartile range).
Extended Data Fig. 10 |. Tumors derived from NB-9464 H-2Kbhi cells show mesenchymal properties.
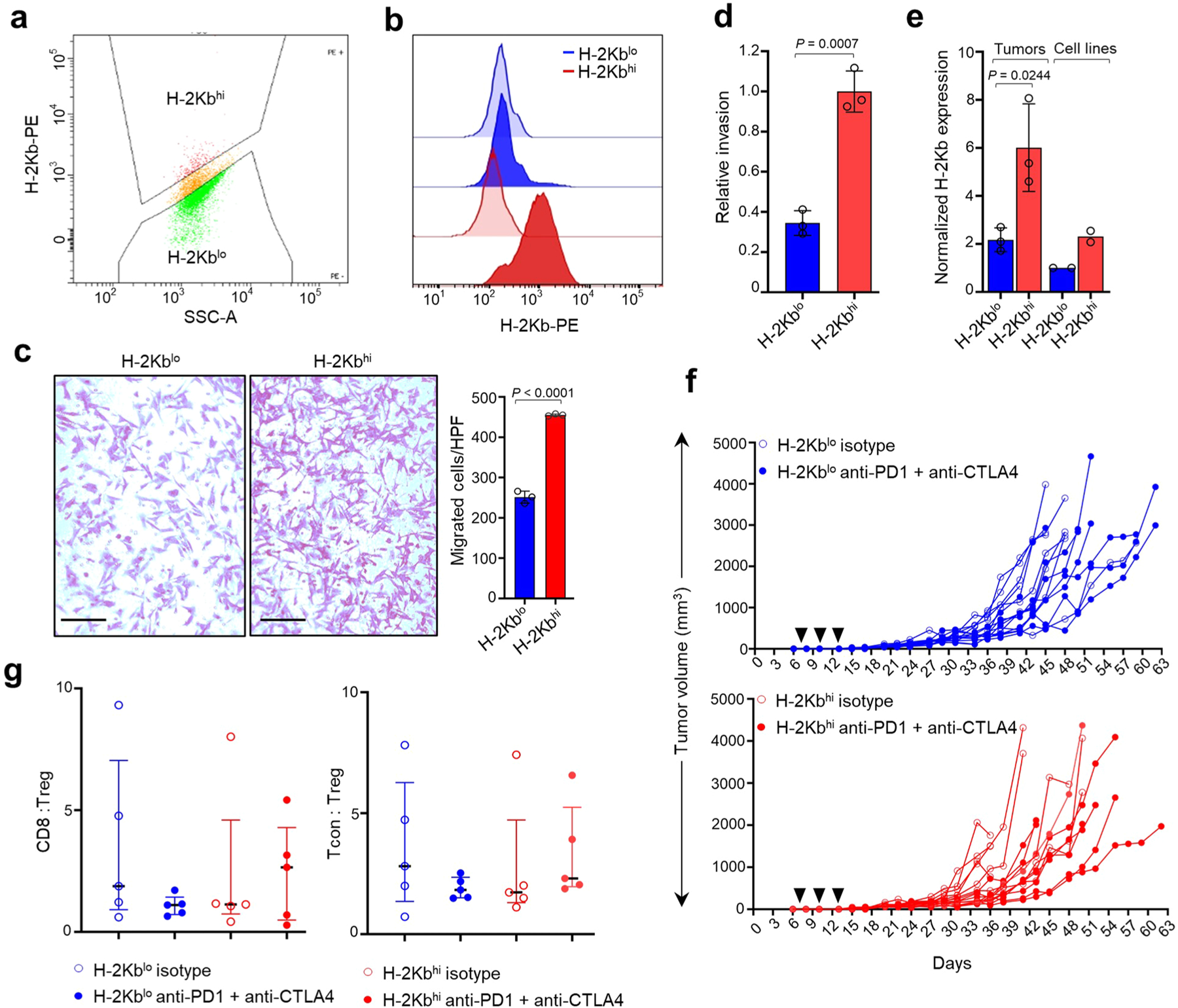
a, FACS plot showing gating conditions for sorting NB-9464 cells into H-2Kbhi and H-2Kblo populations. X-axis, side scatter; Y axis, H-2Kb fluorescence intensity. A logscale expression value of 103 was used to gate H-2Kbhi (≥ 103) and H-2Kblo (<103) populations. b, FACS analyses of H-2Kb expression (dark colored histograms) in H-2Kblo and H-2Kbhi cells compared to isotype controls (light colored histograms). Plots representative of 2 independent experiments. c, Left, Bright field images of crystal violet-stained H-2Kblo and H-2Kbhi cells in transwell migration assays. Scale bar, 100 µm. Right, Quantification of migrating cells per high-power field (HPF). Data represent mean ± SD of cells from three HPFs per cell line, significance derived using unpaired two-tailed Student’s t-test. Data representative of n = 3 independent experiments. d, Quantification of the relative invasiveness of H-2Kblo and H-2Kbhi cells. Data represent mean ± SD from three technical replicates, significance derived using unpaired two-tailed Student’s t-test. Data representative of 2 independent experiments. e, RT-qPCR analysis of H-2Kb expression in H-2Kblo and H-2Kbhi tumors and the cell lines from which they were derived. Data were normalized to H-2Kb expression in H-2Kblo cells and represent mean ± SD, n = 3 tumors per group, P determined by the unpaired two-tailed Student’s t-test. f, Tumor volumes in C57BL/6 mice injected subcutaneously with 1 ×106 H-2Kblo or H-2Kbhi cells and treated with anti-PD1 + anti-CTLA4 antibodies or isotype control (black arrowheads) on days 7, 10, and 13 after tumor inoculation. Data represent growth of individual mouse tumors, n = 8, H-2Kblo isotype; n = 7, H-2Kblo anti-PD1 + anti-CTLA4; n = 8, H-2Kbhi isotype; n = 8, H-2Kbhi anti-PD1 + anti-CTLA4. g, FACS analysis of the ratios between tumor infiltrating CD8 + T or conventional T (Tcon) and Treg cells (CD8:Treg and Tcon:Treg, respectively) in H-2Kblo and H-2Kbhi tumors treated with isotype control or anti-PD1 + anti-CTLA4 antibodies. Data represent medians with interquartile ranges, n = 5 tumors per group.
Supplementary Material
Acknowledgements
We thank C. Cardoso and M. Harlow from the George laboratory and S. Sen Santara, Y. Zhang and Z. Zhang from the Lieberman laboratory for helpful discussions. We thank M. Zimmerman, A. T. Look and K. Stegmaier for sharing cell lines. We thank the following members of the former Haining laboratory at DFCI for sharing resources and experimental advice: U. Gerdemann, D. Comstock, K. Yates, A. Word and A. Long. We thank F. Westermann’s group for sharing the Seurat object containing the count matrix derived from the snRNA-seq dataset. We thank H. Tillman and the St. Jude Veterinary Pathology Core for their assistance with IHC staining. The results shown here are in part based on data curated by the R2: Genomics Analysis and Visualization Platform: http://r2.amc.nl/. This work was supported by a St. Baldrick’s Foundation Childhood Cancer Research Grant, grant no. DOD CA191000 (R.E.G. and R.J.); NIH grant no. R01-CA197336 (R.E.G); and the Cookies for Kids’ Cancer Foundation (R.E.G); the Ted and Eileen Pasquarello Research Fund (R.R.); and Villa Joep Foundation, grant no. IWOV-Actief.51391.180034 (S.N.). S. Spranger is a recipient of a Pew Stewart Scholarship. S. Sengupta and M.K. were supported by the Rally Foundation for Childhood Cancer Research and Infinite Love for Kids Fighting Cancer, M. Campisi by an AIRC Fellowship for Abroad, B.C.M. by the National Center for Advancing Translational Sciences/NIH Award no. KL2 TR002542 and D.N.D. by an Alex’s Lemonade Stand Foundation Young Investigator Fellowship.
Footnotes
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s43018-022-00427-5.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43018-022-00427-5.
Peer review information Nature Cancer thanks William A. Weiss and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
The RNA-seq and ChIP–seq datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO) repository under accession number GSE165750. Source data are provided with this paper. The authors declare that all other data supporting the findings of this study are available within the paper and its Supplementary Information files or are available from the corresponding author on reasonable request.
References
- 1.Cheung NK & Dyer MA Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 13, 397–411 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker E et al. Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res 51, 144–149 (1991). [PubMed] [Google Scholar]
- 3.Yu AL et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med 363, 1324–1334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Federico SM et al. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin. Cancer Res 23, 6441–6449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merchant MS et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin. Cancer Res 22, 1364–1370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis KL et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol 21, 541–550 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geoerger B et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol 21, 121–133 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Louis CU et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JR et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther 15, 825–833 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Casey DL & Cheung NV Immunotherapy of pediatric solid tumors: treatments at a crossroads, with an emphasis on antibodies. Cancer Immunol. Res 8, 161–166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pugh TJ et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet 45, 279–284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernards R, Dessain SK & Weinberg RA N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell 47, 667–674 (1986). [DOI] [PubMed] [Google Scholar]
- 13.Raffaghello L et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia 6, 558–568 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castriconi R et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc. Natl Acad. Sci. USA 101, 12640–12645 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Theruvath J et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med 28, 333–344 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei JS et al. Clinically relevant cytotoxic immune cell signatures and clonal expansion of T-cell receptors in high-risk MYCN-not-amplified human neuroblastoma. Clin. Cancer Res 24, 5673–5684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asgharzadeh S et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol 30, 3525–3532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao Y et al. Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin. Cancer Res 22, 3849–3859 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Brodeur GM, Seeger RC, Schwab M, Varmus HE & Bishop JM Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224, 1121–1124 (1984). [DOI] [PubMed] [Google Scholar]
- 20.Layer JP et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology 6, e1320626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brandetti E et al. MYCN is an immunosuppressive oncogene dampening the expression of ligands for NK-cell-activating receptors in human high-risk neuroblastoma. Oncoimmunology 6, e1316439 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spranger S et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci. Transl. Med 5, 200ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spranger S, Bao R & Gajewski TF Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Russo PST et al. CEMiTool: a Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinf 19, 56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boeva V et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet 49, 1408–1413 (2017). [DOI] [PubMed] [Google Scholar]
- 26.van Groningen T et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet 49, 1261–1266 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Bindea G et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Cursons J et al. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol. Res 7, 1162–1174 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Newman AM et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jansky S et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat. Genet 53, 683–693 (2021). [DOI] [PubMed] [Google Scholar]
- 31.McLeod C et al. St. Jude Cloud: a pediatric cancer genomic data-sharing ecosystem. Cancer Discov 11, 1082–1099 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross RA, Spengler BA & Biedler JL Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J. Natl Cancer Inst 71, 741–747 (1983). [PubMed] [Google Scholar]
- 33.Debruyne DN et al. ALK inhibitor resistance in ALKF1174L-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 35, 3681–3691 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schramm A et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet 47, 872–877 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Slyper M et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med 26, 792–802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chipumuro E et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durbin AD et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet 50, 1240–1246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Margueron R & Reinberg D The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Molfetta R et al. Regulation of NKG2D expression and signaling by endocytosis. Trends Immunol 37, 790–802 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Qi W et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol 13, 381–388 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Spel L et al. Natural killer cells facilitate PRAME-specific T-cell reactivity against neuroblastoma. Oncotarget 6, 35770–35781 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boes M & Meyer-Wentrup F TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett 361, 49–56 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG & Bishop JM Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J 16, 2985–2995 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kroesen M et al. A transplantable TH-MYCN transgenic tumor model in C57Bl/6 mice for preclinical immunological studies in neuroblastoma. Int. J. Cancer 134, 1335–1345 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Malladi S et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 165, 45–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jerby-Arnon L et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997 e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao R et al. Immunogenomic determinants of tumor microenvironment correlate with superior survival in high-risk neuroblastoma. J. Immunother. Cancer 9, e002417 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gay CM et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 39, 346–360 e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahadevan NR et al. Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov 11, 1952–1969 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang CH, Hammer J, Loh JE, Fodor WL & Flavell RA The activation of major histocompatibility complex class I genes by interferon regulatory factor-1 (IRF-1). Immunogenetics 35, 378–384 (1992). [DOI] [PubMed] [Google Scholar]
- 51.Unterholzner L et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol 11, 997–1004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sen T et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 9, 646–661 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shirinbak S et al. Combined immune checkpoint blockade increases CD8+CD28+PD-1+ effector T cells and provides a therapeutic strategy for patients with neuroblastoma. Oncoimmunology 10, 1838140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srinivasan P, Wu X, Basu M, Rossi C & Sandler AD PD-L1 checkpoint inhibition and anti-CTLA-4 whole tumor cell vaccination counter adaptive immune resistance: a mouse neuroblastoma model that mimics human disease. PLoS Med 15, e1002497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wienke J et al. The immune landscape of neuroblastoma: challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur. J. Cancer 144, 123–150 (2021). [DOI] [PubMed] [Google Scholar]
- 56.Ehlert K et al. Nivolumab and dinutuximab beta in two patients with refractory neuroblastoma. J. Immunother. Cancer 8, e000540 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Voeller J et al. Combined innate and adaptive immunotherapy overcomes resistance of immunologically cold syngeneic murine neuroblastoma to checkpoint inhibition. J. Immunother. Cancer 7, 344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wolpaw AJ et al. Epigenetic state determines inflammatory sensing in neuroblastoma. Proc. Natl Acad. Sci. USA 119, e2102358119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peng D et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burr ML et al. An evolutionarily conserved function of Polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 36, 385–401.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Debruyne DN et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 572, 676–680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang H et al. Extracellular domain shedding of the ALK receptor mediates neuroblastoma cell migration. Cell Rep 36, 109363 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amir AL et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin. Cancer Res 17, 5615–5625 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Lo Presti V et al. Efficient lentiviral transduction method to gene modify cord blood CD8+ T cells for cancer therapy applications. Mol. Ther. Methods Clin. Dev 21, 357–368 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kitajima S et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov 9, 34–45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aref AR et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 18, 3129–3143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomayko MM & Reynolds CP Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol 24, 148–154 (1989). [DOI] [PubMed] [Google Scholar]
- 68.Durbin BP, Hardin JS, Hawkins DM & Rocke DM A variance-stabilizing transformation for gene-expression microarray data. Bioinformatics 18, S105–S110 (2002). [DOI] [PubMed] [Google Scholar]
- 69.Zhu X et al. Single-cell clustering based on shared nearest neighbor and graph partitioning. Interdiscip. Sci 12, 117–130 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McGinnis CS, Murrow LM & Gartner ZJ DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst 8, 329–337 e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aran D et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dries R et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol 22, 78 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and ChIP–seq datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO) repository under accession number GSE165750. Source data are provided with this paper. The authors declare that all other data supporting the findings of this study are available within the paper and its Supplementary Information files or are available from the corresponding author on reasonable request.