

Abstract
Significance:
Cigarette smoke (CS) is a prominent cause of morbidity and death and poses a serious challenge to the current health care system worldwide. Its multifaceted roles have led to cardiovascular, respiratory, immunological, and neoplastic diseases.
Recent Advances:
CS influences both innate and adaptive immunity and regulates immune responses by exacerbating pathogenic immunological responses and/or suppressing defense immunity. There is substantial evidence pointing toward a critical role of CS in vascular immunopathology, but a comprehensive and up-to-date review is lacking.
Critical Issues:
This review aims to synthesize novel conceptual advances on the immunomodulatory action of CS with a focus on the cardiovascular system from the following perspectives: (i) the signaling of danger-associated molecular pattern (DAMP) receptors contributes to CS modulation of inflammation and immunity; (ii) CS reprograms immunometabolism and trained immunity-related metabolic pathways in innate immune cells and T cells, which can be sensed by the cytoplasmic (cytosolic and non-nuclear organelles) reactive oxygen species (ROS) system in vascular cells; (iii) how nuclear ROS drive CS-promoted DNA damage and cell death pathways, thereby amplifying inflammation and immune responses; and (iv) CS induces endothelial cell (EC) dysfunction and vascular inflammation to promote cardiovascular diseases (CVDs).
Future Directions:
Despite significant progress in understanding the cellular and molecular mechanisms linking CS to immunity, further investigations are warranted to elucidate novel mechanisms responsible for CS-mediated immunopathology of CVDs; in particular, the research in redox regulation of immune functions of ECs and their fate affected by CS is still in its infancy.
Keywords: cigarette smoke, morphine, trained immunity, trained tolerance, immunometabolism, cell death
Introduction
Cigarette smoke (CS) accounts for 480,000 adult death annually (1/5 all the deaths/year; 1300 deaths/day) (Warren et al, 2014) and 41,000 deaths from secondhand smoke exposure in the United States (Jamal et al, 2016). Annual economic costs of diseases associated with CS exceed $289 billion (Kohut, 2017). CS contributes significantly to the pathogenesis of several types of major immune-inflammatory diseases including coronary heart disease (CHD) (Centers for Disease Control (CDC), 1989), lung cancers, chronic obstructive pulmonary disease (COPD) (Ishii, 2013), end-stage renal disease (Choi et al, 2019), multiple sclerosis (Alrouji et al, 2019), stroke, and oral inflammatory disease (Table 1).
Table 1.
Cigarette Smoke Promotes Several Types of Major Immunoinflammatory Diseases
| Diseases | Effect of cigarette smoke | PMID |
|---|---|---|
| CVD | Increased neutrophils, lymphocytes, and monocytes Increased inflammatory cytokines and chemokines such as IL-1β and TNF-α Induced endothelial vascular dysfunction and inflammation Increased oxidative stress and ROS production Increased total serum cholesterol, VLDL, LDL, and triglyceride concentrations Increased risk of thrombosis, atherosclerosis, stroke, and CHD Increased cardiovascular morbidity and mortality |
17485580 21285293 15145091 26174518 |
| Respiratory diseases | Increased production of immune mediators and ROS by lung macrophages Increased incidence of COPD and airway inflammation Increased risk of interstitial lung diseases, bronchial asthma, and lung cancer |
22196881 24507834 23631228 |
| CKD | Increased production of proinflammatory mediators Increased EC dysfunction Increased oxidative stress and ROS production Increased cardiovascular morbidity and mortality in CKD patients Increased risk of incident ESRD |
18003763 28339863 22158113 31862942 |
| RA | Increased risk for development and severity of RA Increased rheumatoid nodule formation with multiple joint involvements |
24594022 25479074 |
| MS | Increased incidence and severity of the disease | 32905534 |
| SLE | Increased inflammatory markers associated with decreased anti-inflammatory IL-10 Increased oxidative stress Elevated titers of anti-dsDNA Increased risk of developing SLE |
29724134 |
| IBD | Impaired function of inflammatory cells Increased recruitment of CD4+ and CD8+ T cells, and of CD11b+ DCs Increased proinflammatory chemokines/cytokines (CCR6, CCL20, IL-8) increased in the expression of MHC-II and costimulatory molecules Increased incidence of Crohn's disease Protected against UC by decreased inflammatory mediators and cytokine and decreased gut permeability by nicotine |
20333390 21112082 24691114 21537330 32823518 |
| Oral inflammatory diseases | Impaired function of inflammatory cells Increased inflammatory cytokines Promoted periodontal diseases |
20361572 |
CS increases ROS production, inflammatory cytokine secretion, inflammatory and immune cells, and impairment of inflammatory cells' function. Complex roles of CS have resulted in several diseases, including CVDs, respiratory diseases, CKD, rheumatoid arthritis, multiple sclerosis, systemic lupus erythematosus, and inflammatory bowel disease.
anti-dsDNA, anti-double-stranded DNA; CCL20, chemokine (C-C motif) ligand 20; CCR6, C-C motif chemokine receptor 6; CHD, coronary heart disease; CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; CS, cigarrette smoke; CVD, cardiovascular diseases; DC, dendritic cell; EC, endothelial cell; ESRD, end-stage kidney disease; IBD, inflammatory bowel disease; IL, interleukin; LDL, low-density lipoprotein; MHC-II, MHC class II molecules; MS, multiple sclerosis; RA, rheumatoid arthritis; ROS, reactive oxygen species; SLE, systemic lupus erythematosus; TNF-α, tumor necrosis factor-α; UC, ulcerative colitis; VLDL, very-low-density lipoprotein.
The mechanisms responsible for these effects include increased reactive oxygen species (ROS), membrane receptor (clusters of differentiation)-mediated cell–cell interaction (Horvathova et al, 2009; Xu et al, 2021), noncoding RNA-containing exosome secretion (Maccani and Knopik, 2012; Yang et al, 2017b), and proinflammatory cytokine secretion (Lu et al, 2022; Ni et al, 2021; Zhang et al, 2020a; Zhang et al, 2020b), which lead to both an upregulation in the numbers of leukocytes and an impairment of the function of inflammatory cells. CS induces chronic inflammation in the lungs (Duaso and Duncan, 2012; Wood and Stockley, 2007), which is apparent at the level of tissue damage, and may result in COPD (Jaspers, 2014).
Tobacco smoke (TS) is a carcinogenic and immunomodulatory toxic mixture of more than 5000 chemicals (Talhout et al, 2011). Chronic exposure to CS leads to impaired T cell function (Valiathan et al, 2014), reprogramming of Treg transcriptomes (Shao et al, 2022; Shao et al, 2021b), a heightened level of systemic inflammation (Stämpfli and Anderson, 2009), and depressed antiviral immune responses, particularly in the lungs (Bauer et al, 2013). CS may increase end-organ injury (including atherosclerosis) by activating endothelial cells (ECs), a new innate immune cell type as we proposed (Drummer et al, 2021a; Mai et al, 2013; Shao et al, 2020), and promoting EC–monocyte interactions and increasing leukocyte tissue infiltration. CS induces chronic inflammation by increasing the numbers of neutrophils and macrophages (Jaspers, 2014).
CS administration leads to decreased CD4+ forkhead box P3 (Foxp3)+ T regulatory cells (Tregs, the major anti-inflammation cell type) and reduced expression of anti-inflammatory/immunosuppressive cytokine interleukin (IL)-10 and Treg-specific transcription factor Foxp3. For example, the frequency of Treg cells is lower in CS-exposed mice compared with the control group. More importantly, the frequency of Treg cells is negatively correlated with T helper cell 17 (Th17 cells) and CD8+ IL-17 producing T cells (Tc17 cells) (Duan et al, 2016).
CS is a major reversible risk factor for cardiovascular disease (CVD). CS-induced vascular inflammation and vascular pathology play important roles in the development and progression of subsequent clinical outcomes (Ambrose and Barua, 2004). CS has various cytotoxic effects in a wide range of vascular cell types including vascular ECs and vascular smooth muscle cells (VSMCs), in part, by increasing ROS production, activating nuclear factor kappa B (NF-κB), upregulating adhesion molecules, inducing endothelial activation and endothelial dysfunction, and promoting VSMC phenotypic switching and VSMC matrix degradation. The adhesion of leukocytes on the surface of ECs is an early event in the atherogenesis process and the increased levels of proinflammatory cytokines promote leukocyte-EC adhesion and leukocyte recruitment (Xu et al, 2022; Xu et al, 2021).
CS increases the expression of vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and E-selectin on human umbilical vein endothelial cells (HUVECs), and increased transendothelial migration of monocytes and neutrophils (Bermudez et al, 2002; Shen et al, 1996). In addition, CS induces ferroptosis cell death in VSMCs (Sampilvanjil et al, 2020) and reduces carotid artery VSMC numbers due to increased cell necrosis (Ambalavanan et al, 2001).
Nicotine exposure promotes VSMC migration from the tunica media to atheromatous plaques in the vascular intima and induces VSMC transformation from contractile to synthetic phenotype (VSMC phenotype switching) via nicotinic acetylcholine receptors and G protein-coupled receptors (Yoshiyama et al, 2014). Electronic cigarettes (e-cigarettes), also known as electronic nicotine delivery systems, have become popular as substitutes for conventional tobacco cigarettes. While e-cigarettes contain fewer toxicants than tobacco cigarettes, they still emit detectable levels of volatile organic compounds, aldehydes, and nitrosamines that induce oxidative stress and inflammation (Cheng, 2014). E-cigarette-exposed alveolar macrophages show a significant increase in the production of proinflammatory cytokines/chemokines, apoptosis and necrosis, and ROS production (Scott et al, 2018). Furthermore, e-cigarette exposure significantly increases oxidative stress, oxidized low-density lipoprotein (ox-LDL), and cardiovascular risk (Carnevale et al, 2016; Moheimani et al, 2017).
Chronic e-cigarette exposure induces vascular endothelial dysfunction (Shao et al, 2014), cardiac dysfunction, and atherosclerosis in mice (Espinoza-Derout et al, 2019). In addition, e-cigarette exposure increases the risk for COPD (Bowler et al, 2017) and CVDs (D'Amario et al, 2019).
Signals of Danger-Associated Molecular Pattern Receptors Contribute to the Modulation of Inflammation and Immunity by CS
TS contains several chemicals that are harmful. Among the more than 5000 chemicals identified, at least 250 are reported to be harmful, including hydrogen cyanide, carbon monoxide (CO1), and ammonia (Centers for Disease Control and Prevention et al, 2010; National Center for Chronic Disease Prevention and Health Promotion Office on Smoking and Health, 2014). Based on chemical binding capacities, we tentatively classify them into five groups as follows:
-
(i)
At least 72 chemicals cause cancer (carcinogens) and some of these covalently bind to DNA directly (Goodson et al, 2015) and form DNA adducts. Twenty-eight of the carcinogenic chemicals including polycyclic aromatic hydrocarbons (benzo[a]pyrene), N-nitrosamines, heavy metals (nickel, cadmium, chromium, and arsenic), alkaloids (nicotine and its major metabolite, cotinine), and aromatic amines have immunomodulatory effects on distinct innate and adaptive immune cells and can lead to impaired immunity. These compounds can either downregulate (anti-inflammatory) or upregulate (proinflammatory) the immune response, leading to either an increased susceptibility to the development of cancers or infectious diseases, or immunopotentiation resulting in an increased secretion of inflammatory mediators (Table 2).
-
(ii)
The second group consists of more than 400 odorants. There are distinctive and clinically relevant odorants that evokes a response from 144 odorant receptors (ORs) and 3 trace-amine-associated receptors. The recognition of CS is accomplished by a broad receptor response pattern, and 1-pentanethiol (the odorant most critical for perception of the artificial mimic) is responsible for a small subset of the responsive ORs in this combinatorial code (McClintock et al, 2020). The ratios of >400 odorants over 144 ORs suggest significant degeneracy of CS chemicals in binding to their cellular receptors, about 2.7 odorants/receptors in nonhigh odorant-specific manners.
-
(iii)
Danger-associated molecular pattern (DAMP)/pathogen-associated molecular pattern (PAMP) receptors are the third group. These include toll-like receptors (TLRs), C-type lectin receptors, NOD-like receptors (NLRs including NLRP3), retinoic acid-inducible gene-like receptors, cytosolic DNA sensors, receptor for advanced glycation end products, triggering receptors expressed on myeloid cells, G-protein-coupled receptors, ion channels (Gong et al, 2020), and conditional DAMP receptors (Wang et al, 2016b). Compared with wild-type (WT) mice, CS-induced pulmonary inflammation is unaltered in DAMP/PAMP receptors and TLR2-deficient (Tlr2−/−) (Yang et al, 2008) and TLR4-deficient (Tlr4−/−) mice. CS-induced airway fibrosis, characterized by increased collagen deposition around small airways, is not altered in Tlr2−/− mice, but is attenuated in Tlr4−/− mice compared with CS-exposed WT controls (Haw et al, 2018). In addition, CS amplifies inflammatory reactions and atherogenesis through activation of the H1R-TLR2/4-cyclooxygenase-2 (COX-2) axis (Barua et al, 2015).
-
(iv)
CS inhibits the NLRP3 inflammasome and leads to caspase-1 activation via the TLR4-toll-like receptor adaptor molecule 1 (TICAM1, TRIF)-caspase-8 axis in human macrophages (Buscetta et al, 2020).
-
(v)
The compounds induced by CS and shared with endogenous metabolites (Zhang et al, 2022) as well as chemokines induced by morphine (Rogers, 2020) can use their intrinsic receptors [conditional DAMP receptors as we proposed (Wang et al, 2016b)] to drive or modulate inflammatory signals (Muri and Kopf, 2021; Voss et al, 2021).
Table 2.
Carcinogenic Constituents from Cigarette Smoke Induce Reactive Oxygen Species Production and Oxidative DNA Damage, Inhibit Immune Cell Generation and Proliferation, Modulate Cytokine Secretion, and Suppress Immune Functions
| Carcinogen | Immune cells/tissue | Effects | Proinflammatory (+) Anti-inflammatory (*) | Receptor/pathway | PMID |
|---|---|---|---|---|---|
| Benz[j]aceanthrylene | Macrophages | DNA damage | 9054606 | ||
| Lymphocytes | DNA damage | 9744557 | |||
| Benz[a]anthracene | Lymphocytes | Increased ROS production and oxidative DNA damage | 21888224 | ||
| T cells | Inhibited T cell proliferation Inhibited IL-2 production Induced immunosuppression |
* | Signal transduction mediated by TCR and IL-2R | 8931739 | |
| T cells | Suppressed helper T cell activation | 6219909 | |||
| Benzo[b]fluoranthene | HPBECs | Increased innate and adaptive immune signals Increased expression of oxidative stress genes |
+ | 30090392 | |
| Macrophages | Increased secretion of inflammatory cytokines | + | 18830893 | ||
| Benzo[j]fluoranthene | Mouse skin | A potent tumor initiator with high tumorigenic activity | 3677067 | ||
| B[k]F | Mouse | Reduced T cells in thymus and spleen Reduced CD4+ IL-2+ cells |
* | Immunosuppression through IL-2 production | 16326422 |
| BaP | BMDMs | Impaired proliferation and decreased number of mature cells Increased expression of F4/80 and MHC-II Increased TNF-α and IL-10 |
+ | AhR | 30099064 |
| Mouse/mouse epidermal cells | Increased neutrophil and macrophage accumulation Increased secretion of IL-5, IL-13, IL-33, MCP-1 Increased expression of MHC-II and CD86 expression Increased Th1/Th2 responses and allergic airway inflammation Induced VEGF PI-3K/AP-1 activation |
+ | 26918773 16461351 |
||
| T cells | Increased expression of AhR and CYP1 expression | Activation of AhR/CYP1-metabolizing enzymes | 28461126 24412381 |
||
| BMDMs | Decreased proinflammatory cytokines Increased expression of IL-10, MHC-II, CD14, Fcγ receptor I (FcγRI/CD64) Increased NO production and phagocytosis |
* | AhR activation | 30053493 | |
| DCs and keratinocytes | Increased secretion of cytokines | + | AhR activation | 30748024 | |
| T cells | Inhibited T cell proliferation Decreased production of IFN-γ, IL-2, and IL-4 |
* | Ca2+/CaM/NF-κB and Ca2+/CaM/CaN/NFAT signal transduction pathways | 28290727 | |
| Cyclopenta[c,d]pyrene | Leukocytes | Increased DNA adducts | 8200070 | ||
| 5-Methylchrysene | Mouse skin | Increased DNA adducts | 1643254 | ||
| BD | Mouse | Suppressed cytotoxic T cell generation | 3787617 2401263 |
||
| Benzene | Human and mouse inhalation | Decreased proliferation of B/T cells Suppressed antibody production by of B cells Oxidative stress imbalance Decreased p53 expression |
2941900 29883905 9129168 |
||
| Naphthalene | Macrophages | Increased lipid peroxidation Increased cytochrome c reduction and oxidative stress |
+ | 9667488 | |
| Styrene | Mouse | DNA damage in lymphocytes, liver, bone marrow, and kidney | 9783323 | ||
| Human | Reduced proportion of T helper cells Increased T suppressor cells Increased NK cells |
1630405 | |||
| Human | Increased expression of adhesion molecules on lymphocytes, monocytes, and granulocytes | + | 12141393 | ||
| N-Nitrosodimethylamine | Mouse | Suppressed cell-mediated immunity and humoral immunity | * | 1433375 6716271 3156199 |
|
| PMNs | Increased expression of iNOS, phospho-PI3K, phospho-IκBα and NF-κB, phospho-Akt (T308), phospho-Akt (S473), and phospho-IKKαβ, c-Jun and FosB Increased apoptosis |
PI3K-Akt/PKB pathway | 30722700 23971717 |
||
| N-Nitrosodiethanolamine | Lymphocytes | Increased mutagenicity and genotoxicity | 2808483 3204103 |
||
| Catechol | Human PBMCs | Inhibited production of IL-2 and IL-1β | * | 10932071 | |
| NNN | Rat oral and esophageal mucosa and keratinocytes | Alteration of immune regulation genes (Aire, Ctla4, and CD80) and inflammation (Ephx2 and Inpp5d) | 26785143 | ||
| NNN | Rat oral and esophageal mucosa and keratinocytes | Alteration of immune regulation genes (Aire, Ctla4, and CD80) and inflammation (Ephx2 and Inpp5d) | 26785143 | ||
| NNK | CTLs | Increased expression of adhesion molecule CD62L Impaired expansion capacity of CTLs Reduced memory programming |
23673295 | ||
| Epithelial cells/macrophages | Inhibited secretion of IL-8, IL-6, MCP-1, and TNF | * | 15762874 17096151 14764458 |
||
| Macrophages | Increased release of soluble TNF Decreased IL-10 synthesis |
+ | 11258792 | ||
| o-Anisidine | Hepatocytes/macrophages | Increased proinflammatory cytokines Induced ROS generation |
+ | eIF2- and Nrf2-mediated oxidative stress response | 28089782 |
| Trp-P-1 | DCs | Increased expression of costimulatory receptors (CD80, CD86, and MHC) Inhibited DC maturation Inhibited IL-12 and TNF-α production Attenuated T cell proliferation/activation induced by DCs |
+ | signaling pathways mediated through p38 kinase | 21078543 |
| Macrophages | Inhibited IL-8 expression Inhibited phosphorylation of p38 MAP kinase |
* | Intracellular calcium/p38 MAP kinase-dependent | 18281166 | |
| Benzo[b]furan | Cancer cells | Increased expression of proapoptotic genes (TNFRSF 10A, TNFRSF 10B, CASP8, BAX, BID, NOXA, APAF1) Activated JNK and p38 kinase |
PI3K/Akt/mTOR signaling and mitochondrial-mediated apoptosis | 30465514 27149364 |
|
| FA | Splenocytes | Increased T cell differentiation into regulatory T cells Suppressed Th1-, Th2-, and Th17-related splenic cytokines Suppressed effector T cell activity and decreased T cell-related cytokines |
* | Calcineurin-NFAT signaling | 33046725 |
| Human exposure | Increased CD19+ B cells and CD56+ NK cells Increased IL-10 and IL-4 Decreased IL-8 and IFN-γ |
* | 25157974 | ||
| Acetaldehyde | Splenocytes | Decreased cytokine production Inhibited aerobic glycolysis-related signal in T cells Inhibited NK activity and CTL-mediated lysis |
* | 30121625 2690659 |
|
| Human exposure | Degranulation of human mast cells and histamine release Induced GMCSF production and NF-κB activation Increased allergic inflammation |
+ | 17590989 | ||
| PMN/monocytes | Increased ROS production Decreased PMN phagocytic functions |
+ | |||
| PBMC | Inhibited T cell and B cell proliferation Decreased release of IFN-γ |
8953156 | |||
| Cadmium | Macrophages Mast cells |
Increased ROS production Increased TNF-α and nitrite production Decreased mast cell TNF-α and IgE-mediated histamine release |
+ | 30828855 | |
| Peripheral blood neutrophils and splenocytes | Increased expression of cytochrome P450s enzymes Increased ROS production Induced autophagy and apoptosis in splenocytes |
miR-216a-PI3K/AKT | 32058096 30985881 32563067 | ||
| Cobalt | HMEC-1 Macrophages |
Increased inflammatory markers such as IL-6, IL-8, ICAM-1, and sICAM-1 | + | TLR4 | 27835611 |
| Murine macrophages | Increased oxidative stress and ROS production Decreased OCR and induced mitochondrial dysfunction |
30144138 | |||
| Lead (inorganic) | Macrophages | Increased cell death Increased the antioxidant enzymatic activity of catalase Decreased macrophage phagocytic index, nitric oxide production, endosomal/lysosomal system stability |
16757190 17959351 |
||
| N-nitrosopiperidine | Esophageal epithelium | Increased cell death | 16816872 |
Twenty-eight out of 72 carcinogens in cigarette smoke (PMID: 21324834) have been characterized to affect the activity and function of different innate and adaptive immune cells.
AhR, aryl hydrocarbon receptor; B[k]F, benzo[k]fluoranthene; BaP, benzo[a]pyrene; BD, 1,3-butadiene; BMDMs, bone marrow-derived macrophages; CTLs, CD8+ cytotoxic T lymphocytes; CYP1, cytochrome P450 family 1; FA, formaldehyde; GMCSF, granulocyte/macrophage-colony-stimulating factor; HMEC-1, human microvascular endothelial cell 1; HPBECs, human primary bronchial epithelial cells; ICAM-1, intercellular adhesion molecule 1; IFN-γ, interferon-gamma; JNK, c-Jun N-terminal kinase; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa B; NK, natural killer; NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; NNN, N′-nitrosonornicotine; NO, nitric oxide; Nrf2, nuclear factor erythroid 2-related factor 2; OCR, oxygen consumption rate; PBMCs, peripheral blood mononuclear cells; PHA, phytohemagglutinin; PI3-AKT, phosphoinositide-3-kinase–protein kinase B; PMNs, polymorphonuclear neutrophils; sICAM-1, soluble intercellular adhesion molecule 1; Th, T helper cell; TLR, toll-like receptor; VEGF, vascular endothelial growth factor.
Trained immunity is a relatively new concept where signaling from DAMP receptors can modulate immunity. TLR agonists have been shown to trigger trained immunity through metabolic reprogramming and epigenetic modifications, which drive profound augmentation of antimicrobial functions (Owen et al, 2020). In addition, inflammasome NLRP3 was reported to mediate trained immunity following the use of Western diet (high-fat diet) and could thereby mediate the potentially deleterious effects of trained immunity in inflammatory diseases. Taken together, although we do not know whether signals triggered by all the DAMPs/PAMPs modulate trained immunity, CS activates signaling of TLRs and inflammasomes, which could lead to the establishment of chronic inflammation via trained immunity (Christ et al, 2018).
Cigarette Smoke Reprograms Immunometabolism and Trained Immunity-Related Metabolic Pathways in Innate Immune Cells and T Cells, Which Can Be Sensed by the Cytoplasmic (Cytosolic and Non-Nucleus Organelles) ROS System
CS influences both innate and adaptive immunity and plays dual functions in regulating immunity by promoting antipathogen immune responses or suppressing defensive immunity. Noncarcinogenic DAMPs from CS activate both innate and adaptive immune cells including macrophages, neutrophils, dendritic cells (DCs), natural killer (NK) cells, B cells, CD4+ T helper cells (T helper cell 1, Th1) (Th1/Th2/Th17), CD4+Foxp3+ Tregs, CD8+ T cells, and memory T/B lymphocytes (Table 3).
Table 3.
Noncarcinogenic Danger-Associated Molecular Patterns from Cigarette Smoke Activate Both Innate and Adaptive Immune Cells
| Cell type | Effects | PMID |
|---|---|---|
| Macrophages | Increased cell activation Increased expression of TLR4 Increased secretion of proinflammatory mediators (TNF-α) Increased ROS production Impaired bactericidal and phagocytotic processes Activation of IL-1R-associated kinase, p38, and NF-κB |
12033743 17630319 16004610 16620395 19409098 17947684 |
| Neutrophils | Increased cell number Increased IL-8 production Increased ROS production Enhanced degradation of IκB-α/β proteins and activity of p65 and p50 |
6556892 12960242 |
| DCs | Increased number of immature DCs and decreased number of mature DCs Decreased T cell stimulatory capacity and Th1-cell polarization Increased expression of MHC-II molecules and costimulatory molecules CD40 and CD86 Increased CD4+ cells rather than CD8+ cells |
18337593 25338516 16055867 |
| NK cells | Decreased NK cell numbers and activity Decreased cytotoxic activity and cytokine production Decreased IFN-γ, TNF-α secretions, and perforin expression |
26201093 18055568 |
| B lymphocytes | Increased levels of memory B cells and memory IgG+ B cells Increased levels of circulating IgE Decreased IgG, IgM, and IgA Increased expression of nicotinic receptors |
19909533 24502245 14500745 25011477 |
| T cells | Increased lung CD3+, CD8+, and CD4+ T cells Increased the percentage of CD8+ NKG2D+ cells Enhanced percentage of CD8+ CD69+ cells and cell surface expression of CD69 Increased IFN-γ and CCR6 expression Increased Th17 cells and Th17-related cytokines (IL-17A, IL-6, and IL-23) Increased percentages of Treg cells Increased activation of cytotoxic CD8+ T cells Increased production of IL-1β, IL-6, IL-10, IL-12p70, TNF-α, and IFN-γ Decreased CD4+ CD25+ Treg cells, Foxp3, TGF-β, and IL-10 |
18706444 21763119 20863413 20646637 23044435 23319833 28745532 22070100 |
CS affects both innate and adaptive immunity and plays dual functions in regulating immunity by promoting pathogenic immune responses or suppressing defensive immunity. Innate immune cells affected by CS include macrophages, neutrophils, DCs, and NK cells, while adaptive immune cells affected by CS mainly include T helper cells (Th1/Th2/Th17), CD4+CD25+ regulatory T cells, CD8+ T cells, B cells, and memory T/B lymphocytes.
Foxp3, forkhead box P3; TGF-β, transforming growth factor-β.
Innate immune cells can develop exacerbated long-term immune responses and inflammatory phenotype following brief exposure to endogenous or exogenous DAMPs, which results in a primed and significantly enhanced inflammatory response toward a second challenge after the return to a nonactivated state. This phenomenon is known as innate immune memory or trained immunity (Drummer et al, 2021a; Lu et al, 2019; Zhong et al, 2020). As shown in Figure 1, the establishment of trained immunity involves metabolic reprogramming and epigenetic modification of the innate immune cells, allowing qualitatively and quantitatively adjusted responses of innate immune cells to subsequent time-delayed heterologous stimulations (Netea et al, 2020; Netea et al, 2019; Netea et al, 2016).
FIG. 1.
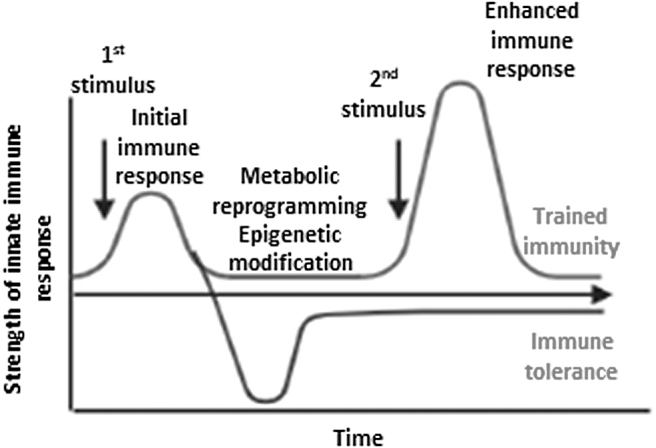
The concept of trained immunity and trained tolerance. Trained immunity involves metabolic reprogramming and epigenetic modification of the innate immune cells, allowing qualitatively and quantitatively adjusted responses of innate immune cells to subsequent time-delayed heterologous stimulation. Inappropriate trained immunity response can contribute to disease progression, resulting in either a chronic hyperinflammatory state or a persistent state of immunological tolerance.
Trained immunity is not only important for host defense and vaccine responses but also for promotion of the pathogenesis of chronic inflammation including metabolic CVDs such as atherosclerosis (Shao et al, 2021a) and synergies among the disease risk factors (Drummer et al, 2021b; Fagenson et al, 2020).
In contrast to the memory function in adaptive immune system with specialized cell subsets to carry out memory function (i.e., memory T cells and memory B cells) (Shen et al, 2019), trained immunity can occur in innate immune cells, including monocytes/macrophages (Lai et al, 2019), DCs (Netea et al, 2020), NK cells, aortic cells (Lu et al, 2022), innate immune functions of T cells, and Treg cells (Lee et al, 2020; Ni et al, 2021; Seyda et al, 2016; Zhang et al, 2020b), and nontraditional immune cells, such as ECs (Lu et al, 2022; Lu et al, 2019; Mai et al, 2013; Shao et al, 2020), VSMCs (Flores-Gomez et al, 2021; Lu et al, 2022; Schnack et al, 2019), fibroblasts (Drummer et al, 2021a), and hepatocytes (Drummer et al, 2021b; Fagenson et al, 2020). Furthermore, after the second challenge, these innate cells can alternatively respond less strongly than to the primary response, and the anti-inflammatory mechanisms are promoted resulting in a state of innate immunological tolerance to prevent tissue damage and limit the inflammatory response (Netea et al, 2020).
It has been reported that T cells play important roles in innate immunity and in antigen nonspecific protection (Berg and Forman, 2006). On the contrary, as adaptive immune cells, T cells are regulated by cytokines and cell–cell communication signals of the innate immune system (Kwesi-Maliepaard et al, 2021; Stäger and Kaye, 2004). Along the same line, we recently reported that CD4+Foxp3+ Treg cells have many active innate immune pathways (Ni et al, 2021; Zhang et al, 2020b), and Tregs can sustain their immunosuppressive functions (Shao et al, 2021b) although Treg cell plasticity in atherosclerosis with a chronic inflammatory environment has been reported (Shao et al, 2021b; Xu et al, 2018).
Our reports indicate that Treg cells and other adaptive immune cells not only respond to antigen stimulation (Yan et al, 2008) but also respond to the stimulation by DAMPs/PAMPs similar to that of innate immune cells (Ke et al, 2008; Lopez-Pastrana et al, 2015; Ni et al, 2021; Yang et al, 2015; Yin et al, 2013). Evidence supporting this new model is that DAMPs/PAMPs sensor inflammasome-dependent release of cytokines, and antigen can activate, shape, or even inhibit adaptive immune responses (Deets and Vance, 2021). TLRs, cytokine receptors, and T cell and B cell antigen receptors have been shown to activate trained immunity-related metabolic pathways, which include phosphoinositide-3-kinase–protein kinase B, mammalian target of rapamycin (Bekkering et al, 2021), liver kinase B1 (also known as serine/threonine kinase 11) (Timilshina et al, 2019), and AMP-activated protein kinase signaling pathways (Cheng et al, 2014; Chou et al, 2022).
CS constituents including nicotine can bind to the intracellular and outer membrane receptors and induce metabolic reprograming and increase disease risk (Fig. 2). CS exposure in several disease conditions, which would include COPD (Pauwels et al, 2011), hyperlipidemia (high-fat diet feeding) (Wu et al, 2018), and CHD (Mao et al, 2021), enhances the innate immune response and promotes disease progression (Table 4). It has been reported that increased energy metabolism and the electron transport chain (Yin and O'Neill, 2021) via (i) glycolysis, (ii) acetyl-CoA generation, (iii) mevalonate synthesis (part of cholesterol biosynthesis), (iv) glutaminolysis (converting glutamine into tricarboxylic acid [TCA] cycle metabolites through the activity of multiple enzymes), and (v) epigenetic modification (Ferreira et al, 2022; Lu et al, 2019) contribute significantly to the establishment of trained immunity. Trained immunity can contribute to disease progression, resulting in a chronic hyperinflammatory state (Shao et al, 2021a).
FIG. 2.
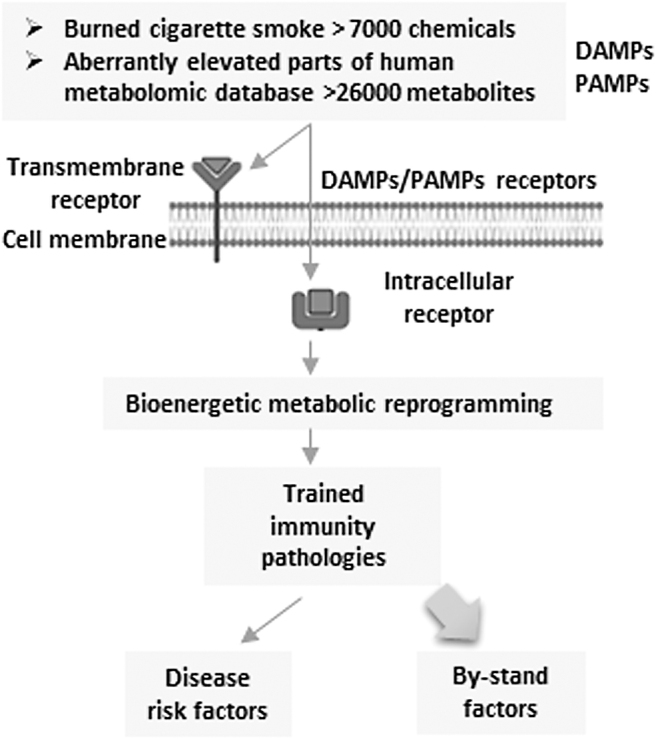
Several DAMP receptor pathways induce innate immune memory (trained immunity) and sustained inflammation, tissue remodeling, and damage. CS promotes disease risk factors by binding to its receptors and induces bioenergetic metabolic reprogramming to induce trained immunity pathology. CS, cigarette smoke; DAMP, danger-associated molecular pattern.
Table 4.
Cigarette Smoke Exacerbates Inflammatory and Immune Responses
| Smoke component | Cell type/tissue/disease condition | Effect | Receptor/pathway | PMID |
|---|---|---|---|---|
| CS-exposed patients | Lung tissue of COPD patients | Increased expression of IL-1α and IL-1β Promoted inflammation and activated NLRP3-related pyroptosis pathway |
IL-1α and Nlrp3/caspase-1/IL-1β | 21622588 |
| Nicotine | HFD-fed ApoE−/− mice | Increased inflammatory cytokine secretion Increased cleavage of caspase-1, IL-1β, and IL-18 secretion Increased atherosclerotic plaques Increased activation of NLRP3-ASC inflammasome Increased pyroptosis in HAECs |
ROS-NLRP3-mediated EC pyroptosis | 29416034 |
| Nicotine | BMDM/ECs from CHD patients | Increased plasma IL-1β and IL-18 Increased TXNIP expression Increased mitochondrial ROS production Increased activation of NLRP3 and caspase-1 inflammasome Increased GSDMD expression Increased monocyte/macrophage dysfunction |
TXNIP/NLRP3-mediated pyroptotic pathway | 33626512 |
| Nicotine | Macrophages from HFD-fed ApoE−/− mice | Increased expression of cleaved caspase1, NLRP3, IL-1β, IL-18 Increased LDH release Induced pyroptosis |
HDAC6/NF-κB/NLRP3 signaling pathway | 33321327 |
| Smoke exposure | Pseudomonas aeruginosa infected mouse lung | Delayed rate of bacterial clearance Increased neutrophils and mononuclear cells infiltration Increased proinflammatory cytokines and chemokines |
15317669 | |
| CS | Angiotensin-II induced hypertension in mice | Induced mitochondrial oxidative stress and endothelial dysfunction Induced oxidation of cardiolipin (mitochondrial oxidative stress biomarker) Increased blood pressure |
30608177 | |
| Chronic nicotine exposure | Renal IRI in mice | Exacerbated renal IRI and oxidative stress-induced injury Aggravate acute renal ischemia- or oxidative stress-induced stress kinase signaling |
21511693 23892062 22552933 |
|
| CSE | Human bronchial epithelial cell | Increased expression of TLR4 Increased binding of LPS Increased release of IL-8 by LPS stimulated cells Increased neutrophil chemotaxis Amplification of inflammation |
Activation of ERK pathway | 18217953 |
| CS | Mice with poly (I:C) administration | Increased infiltration of macrophages, neutrophils, and eosinophils in the bronchoalveolar lavage Induced inflammatory response |
28468623 | |
| CS | NTHI | Exacerbated NTHI-mediated chronic respiratory inflammation Increased proinflammatory IL-1β, IL-6, and TNF-α Decreased IFN-γ and IL-4 secretion |
24752444 |
CSE, cigarette smoke extract; GSDMD, gasdermin D; HAECs, human aortic endothelial cells; HDAC6, histone deacetylase 6; HFD, high-fat diet; IRI, ischemia–reperfusion injury; LDH, lactate-dehydrogenase; LPS, lipopolysaccharide; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; NTHI, nontypeable Haemophilus influenzae; TXNIP, thioredoxin-interacting protein.
We previously reported that the anti-inflammatory cytokine IL-35 is an inflammation-induced cytokine (Li et al, 2018; Li et al, 2012), in contrast to housekeeping anti-inflammatory cytokines such as transforming growth factor-β (Li et al, 2012). We reported that anti-inflammatory cytokines IL-35 and IL-10 can inhibit EC activation and vascular inflammation but spare the modulation of trained immunity gene expression (Li et al, 2020). Similarly, concomitant to a proinflammatory response, anti-inflammatory mechanisms such as trained innate immune tolerance, with a feature of upregulated immune responsive gene 1 (IRG1) itaconate synthesis enzyme (Domínguez-Andrés et al, 2019), and homeostasis molecular patterns (Wang et al, 2016b) are induced to prevent excessive inflammation and tissue damage and to limit the inflammatory response in time.
Overall, CS has the capacity to induce opposing effects on the immune system, with chronic exposure increasing inflammation on the one hand, and potentially reducing inflammation by trained immune tolerance on the other (Table 5).
Table 5.
Cigarette Smoke Can Induce Trained Innate Immune Tolerance
| Cell type/disease | Effect | Receptor/pathway | PMID | |
|---|---|---|---|---|
| CSE | MDMs and THP-1 cells | Inhibited LPS- and LPS plus ATP-induced IL-1β and IL-18 release and pro-IL-1β, pro-IL-18, and NLRP3 expression Decreased LPS-induced lactate release and basal glycolytic flux and impaired glycolytic burst |
NLRP3-independent and TLR4-TRIF-caspase-8-dependent pathway | 31914643 |
| A single injection of nicotine | Renal I/R injury in rats | Attenuated renal dysfunction and tubular necrosis induced by I/R injury Ameliorated acute tubular damage following renal I/R injury Reduced TNF-α protein expression and leukocyte infiltration of the kidney |
α7nAChR | 18401335 |
| A single injection of nicotine | Renal I/R injury in rats | Reduced the α7nAChR protein after I/R injury Inhibited renal I/R-induced STAT3 activation |
α7nAChR | 18614620 |
| Nicotine (500 μM) | LPS-stimulated primary human macrophages | Inhibited TNF release from LPS-stimulated macrophages | 12508119 | |
| Nicotine | IL-18-stimulated PBMC | Inhibited IL-18-enhanced expression of ICAM-1 and CD40 Inhibited production of IL-12, TNF-α, and IFN-γ by IL-18-stimulated cells |
α7nAChR | 16966384 |
| Pretreatment with CSE | LPS stimulated bronchial epithelial cells | Inhibited LPS-induced GM-CSF and IL-8 protein release Suppressed neutrophil chemotaxis induced by LPS Downregulated the activity of LPS-induced transcription factor AP-1 Attenuated inflammatory response induced by LPS |
Suppression of AP-1 activation | 15356167 |
| Pretreatment with CSE | Anti-CD3 and PMA stimulated PBMCs | Suppressed production of IL-1β, TNF-α, IL-2, and IFN-γ | 10932071 | |
| CS | Lung macrophages of Apoe−/− mice | Increased itaconate metabolite Increased expression of IRG1 Prevents overshooting macrophage activation in the lungs May counteract the oxidative challenge from the activated immune system as well as directly from CS exposure. |
32419906 |
CS increases the expression of anti-inflammatory cytokines and decreases the expression of proinflammatory cytokines and chemokines.
α7nAChR, α7 nicotinic acetylcholine receptor; IRG1, immune responsive gene 1; MDMs, monocyte-derived macrophages; PMA, phorbol myristate acetate; STAT3, signal transducer and activator of transcription 3.
Trained innate immune tolerance leads to a persistent state of innate immunological tolerance, a new mechanism that dampens the inflammatory response of the host to maintain homeostasis and prevent tissue damage and organ failure, but with the subsequent risk of secondary infections and other diseases related to decreased activity of the immune system (Netea et al, 2020). Itaconate accumulation has been shown to inhibit the expression of proinflammatory mediators such as IL-6, IL-1β, and IL-12p70 in lipopolysaccharide (LPS)-stimulated macrophages (Lampropoulou et al, 2016).
As shown in Figure 3, itaconate is upregulated in macrophages activated by LPS via increased cis-aconitate decarboxylase/IRG1 transcription. Overproduction of itaconate activates the antioxidant transcription factor nuclear factor erythroid 2–related factor 2 (Nrf2) pathway by alkylation of Kelch-like ECH-associated protein 1, which induces the transcription of various Nrf2-dependent antioxidant and anti-inflammatory genes. This would include heme oxygenase-1, glutamate cysteine synthase, and glutathione synthetase (for glutathione biosynthesis). Itaconate can also inhibit succinate dehydrogenase and reduce ROS generation and IL-1β secretion. Itaconate promotes the transcription of activating transcription factor 3, which directly inhibits the inhibitor of nuclear factor kappa B zeta (IκBζ) expression and leads to decreased expression of IL-6. In addition, itaconate directly alkylates the cysteine residues of glyceraldehyde-3-phosphate dehydrogenase, aldolase, fructose-bisphosphate A (leading to reduced glycolysis), and downregulates IL-1β secretion, thereby depressing the inflammatory response (O'Neill and Artyomov, 2019).
FIG. 3.

The concept of trained immune tolerance. Upon LPS stimulation, itaconate production is increased by CAD/IRG1 transcription. Overproduction of itaconate activates the antioxidant transcription factor Nrf2 by alkylation Keap1, which induces the transcription of various Nrf2-dependent antioxidant and anti-inflammatory genes. Itaconate can also inhibit SDH and reduce ROS generation and IL-1β secretion. Itaconate promotes the transcription of ATF3, which directly inhibits the IκBζ expression to reduce IL-6 secretion. In addition, itaconate directly alkylates the cysteine residue 22 of GAPDH and ALDOA to inhibit glycolysis, and reduces IL-1β secretion, thereby alleviating the inflammatory response. CS exposure can significantly increase the expression of IRG1 and the abundance of itaconate metabolites. ALDOA, aldolase, fructose-bisphosphate A; ATF3, activating transcription factor 3; CAD, cis-aconitate decarboxylase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IκBζ, inhibitor of nuclear factor-kappa B zeta; IL, interleukin; IRG1, immune responsive gene 1; LPS, lipopolysaccharide; Keap1, kelch-like ECH-associated protein 1; Nrf2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; SDH, succinate dehydrogenase.
CS can increase the anti-inflammatory gene expression and decreases the expression of proinflammatory cytokines and chemokines, leading to an immunosuppressive effect. CS exposure significantly increases the abundance of itaconate metabolites and the expression of IRG1 (Table 5) (Titz et al, 2020).
Recent progress on trained innate immune tolerance may lead to the development of a new anti-inflammatory therapy such as ItaCORMs (Krause et al, 2021). Although it is unclear whether CS promotes functionally trained immunity or trained immune tolerance, CS has divergent effects on innate immunity in promoting inflammatory bowel disease but inhibiting ulcerative colitis (AlQasrawi et al, 2020). These findings demonstrate that innate immune cells and adaptive immune cells are equipped with innate immune signaling pathways, presumably including both innate immune memory (trained immunity) and innate immune tolerance, in response to stimulation by various DAMPs, PAMPs, and other environmental stimuli derived from CS.
Based on extensive analysis, we recently proposed that ROS systems are a new integrated network for sensing homeostasis and alarming stress in organelle metabolic processes (Sun et al, 2020). In addition to critical functions in signaling (Li et al, 2016a; Li et al, 2013) and damaging DNAs (nuclear ROS), ROS contribute to a large number of human diseases (Hybertson et al, 2011) and aging (Kowalska et al, 2020). Thus, cytoplasmic ROS and reactive nitric species (RNS) (Michel and Vanhoutte, 2010) can be used as the indicators of metabolic reprogramming. CS contributes to metabolic dysregulation and a shift of metabolism in innate immune cells (Table 6). CS inhibits alveolar macrophage mitochondrial respiration while inducing glycolysis and ROS (Aridgides et al, 2019).
Table 6.
Metabolic Reprogramming Induced by Cigarette Smoke May Be the Novel Mechanisms Underlying the Establishment of Trained Innate Immunity and Trained Tolerance
| Smoke component | Cell type/tissue/species | Effect | PMID |
|---|---|---|---|
| Chronic CS | Lung cells | Increased expression of TCA cycle enzymes Increased expression of enzymes involved in glutamine metabolism, fatty acid degradation, and lactate synthesis Induced mitochondrial metabolic reprogramming |
29042306 |
| CS and electronic cigarette vaping | Increased sphingolipid metabolites Decreased TCA cycle metabolites |
34072305 | |
| Short-term CS | Alveolar type-II cells | Increased glycolysis Increased mitochondria β-oxidation Decreases levels of phosphatidylcholine |
24625219 |
| Short-term CS | Mouse lung (mitochondria) | Increased pentose phosphate pathway Increased expression and activity of complexes II, III, IV, and V Upregulation of genes involved in glycolysis, TCA cycle, mitochondrial fatty acid oxidation, and redox regulation |
23064950 |
| CSE | Pulmonary microvascular ECs | Increased glycolysis Decreased mitochondrial respiration Decreased fatty acid oxidation |
31555131 |
| CS | Alveolar macrophages | Increased glycolysis Decreased mitochondrial respiration Increased ROS and metabolic dysfunction |
31270372 |
| Chewing tobacco | Epithelial cells | Increased oxidative phosphorylation Decreased expression of enzymes involved in the glycolytic pathway Increased expression of mitochondrial proteins involved in the electron transport chain and enzymes of the TCA cycle |
31438645 |
| CS | Human lymphocytes | Decreased mitochondrial complex IV activity Increased lipid peroxidation |
10383908 |
| CSE | Monocyte-derived macrophages and THP-1 cells | Decreased basal glycolytic flux Impaired glycolytic burst in response to LPS |
31914643 |
CS induces metabolic reprogramming depending on many variables, including the type of tobacco and duration of use, and type of exposed cells.
TCA, tricarboxylic acid.
Furthermore, short-term (4 and 8 weeks) exposure of A/J mice to CS (a model for lung tumorigenesis caused by TS) (Witschi, 2005) induces upregulation of genes encoding glycolysis, TCA cycle, mitochondrial fatty acid oxidation pathway, and redox regulation (Agarwal et al, 2014). Increased expression of nuclear genes in the glycolytic pathway and decreased levels of mitochondrial genes following exposure to either the main stream extract or side stream smoke extract support the notion that CS significantly contributes to the transformation of nonmalignant esophageal epithelial cells into a tumorigenic phenotype via metabolic reprogramming (Kim et al, 2010). In addition, acute CS exposure leads to a reversible airspace enlargement in A/J mice, indicative of alveolar damage. A decrease in respiration rates while metabolizing glucose is observed in the CS-exposed group, indicating altered glycolysis that is compensated by an increase in fatty acid palmitate utilization.
Fatty acid palmitate utilization is accompanied by an increase in the expression of CD36 and carnitine palmitoyl transferase 1 in type II alveolar cells for the transport of palmitate into the cells and into mitochondria, respectively (Agarwal et al, 2014). As we and others reported, fatty acid oxidation is also essential for inflammasome activation in proinflammatory type 1 macrophages (M1) (Batista-Gonzalez et al, 2019; Lai et al, 2019).
Epigenetic alteration, including DNA methylation (Jamaluddin et al, 2007) and histone posttranslational modifications (Shao et al, 2016), has been shown to play a significant role in the progression of CS-related diseases (Table 7). DNA methylation is catalyzed by a protein family known as DNA methyltransferases (DNMTs), which transfer methyl groups from S-adenosyl-l-methionine to the 5-carbon position of cytosine residues in DNA. Several studies demonstrate the associations between smoking and altered DNA methylation at multiple cytosine-phosphateguanine (CpG) sites (Breitling et al, 2011; Shenker et al, 2013; Wan et al, 2012). Some DNA methylation sites associated with tobacco smoking have also been localized to genes related to CHD (Breitling et al, 2012) and pulmonary disease (Wauters et al, 2015). Some studies have found different DNA methylation associated CpGs in smokers versus nonsmokers (Zeilinger et al, 2013). CS increases DNA methylation and inflammation in macrophages (Yu et al, 2016) and CS-exposed individuals (Siedlinski et al, 2012). DNMTs are significantly overexpressed in the lung tissues of the smokers compared with the nonsmokers (Kwon et al, 2007). Histone acetylation, methylation, phosphorylation, and ubiquitination are the most broadly studied and extensively characterized histone posttranslational modifications in terms of the regulation of chromatin structure and transcriptional activity (Li et al, 2018; Shao et al, 2016).
Table 7.
Cigarette Smoke Induces Epigenetic Innate Immune Memory in the Form of Histone Acetylation and Methylation and Provides a Foundation for Sustained Inflammation, in Which Acetyl and Methyl Donations Are Generated by CS-Induced Metabolic Reprogramming
| HATs/HDACs | Smoking | Tissues/cells/species | Changes by CS | PMID |
|---|---|---|---|---|
| I: DNA methylation | ||||
| DNMT1 | CS | Macrophages | Increased DNMT1 expression Increased proinflammatory cytokine production Decreased PPAR-γ |
17053888 27530451 |
| JAK3 KRT1 RUNX3 |
CS | CS-exposed individuals | Hypermethylation of RUNX3, JAK3, and KRT1 genes associated with CRP in COPD increased inflammation | 22617718 |
| II: Histone posttranslational modification: HATs and HDACs | ||||
| HAT, CBP/p300 | CSE | Bronchial epithelial cells | Increased expression of HAT, p300/CBP Increase acetylation of histones (H3/H4) and NF-κB in COPD Increased inflammation |
27925185 19811389 |
| HDAC1 | CSE/CS | Rat lung tissue and macrophages | Decreased expression of HDAC1 Increased MCP-1, IL-8, TNF-α, and MMP9 expressions increased level of acetylated H3K9, Increased inflammation |
26033389 |
| HDAC2 | CS | Lung tissue of COPD smokers | Decreased cytoplasmic expression of HDAC2 Increased acetylation of histones H3 and H4 Increased expression of IL-12p40 Decreased IκBα expression Increased inflammation |
30659954 16574938 |
| HDAC3 | CSE | Bronchial epithelial cells Alveolar macrophages |
Decreased SIRT1, HDAC2, HDAC3, and FoxO3 Increased TLR4 and survivin, LPS binding, and ERK ½ activation Increased IL8 and IL1β production Increased inflammation |
30659954 22613758 |
| HDAC4 | Chronic CS | Mouse lung and lung tissue of COPD smokers | Decreased expression of HDAC4 Repressed c-Jun and IL-17A in COPD |
27793800 |
| HDAC5/8 | CS | Lung tissue and alveolar macrophages of COPD smokers | Decreased HDAC5 expression Increased IL-8 expression Increased histone-4 acetylation |
15888697 |
| HDAC6 | CSE | Mouse epithelial cells and lung microvascular ECs/chronic smokers with COPD | Increased HDAC6 expression | 24200693 26452072 |
| HDAC7 | CS | Human exposure | Decreased HDAC7 expression Increased HIF1α |
22172637 |
| HDAC10 | CS | Lung tissues of smokers with COPD | Decreased HDAC10 expression | 30214182 |
| SIRT1 | Smoker with COPD | PBMCs | Decreased expression and activity of SIRT1 | 29861836 27167200 |
| SIRT3 | CS | WT mice | Decreased sirtuin-3 expression Increased mitochondrial oxidative stress Induced endothelial dysfunction |
30608177 |
| SIRT4 | CS | Human pulmonary microvascular ECs | Decreased SIRT4 expression Induced mononuclear cell adhesion Induced VCAM-1, E-selectin, and NF-κB activation Increased proinflammatory cytokines IL-1β, TNF-α, and IL-6 |
24603126 |
| SIRT5 | CSE | Lung epithelial cells | Increased SIRT5 expression and FOXO3 acetylation | 25981116 |
| SIRT6 | CSE | Human bronchial epithelial cells | Decreased SIRT6 expression Induced cellular senescence |
24367027 |
CS induces hyperacetylation and increases H3 and H4acetylation by increasing HATs and decreasing HDACs as well as modification of DNA methylation.
CS, cigarette smoke; DNMT, DNA methyltransferase; HATs, histone acetyltransferases; VCAM-1, vascular cell adhesion molecule 1; WT, wild type.
Histone posttranslational modifications play a critical role in gene expression by adding or removing the acetyl or methyl group, which is catalyzed by various enzymes (Bannister and Kouzarides, 2011). Also, lysine residues in histones can be oxidized by the lysyl oxidase family of proteins (Serra-Bardenys and Peiroó, 2021). Furthermore, epigenetic modification in the form of histone acetylation and methylation, which are catalyzed by histone acetyltransferases (HATs), histone demethylases, and histone deacetylases (HDACs), is associated with trained immunity. For example, there are increases inmarkers of open chromatin such as histone 3 lysine 4 trimethylation (H3K4me3), H3K4me1, and histone 3 lysine 27 acetylation (H3K27ac). Simultaneously, there is a decrease in histone markers depicting closed chromatin such as histone 3 lysine 9 dimethylation (H3K9me2). Epigenetic alterations include histone methylation and acetylation that work in concert to regulate gene transcription in a heritable manner.
The enzymes that regulate these epigenetic modifications can be activated by smoking, which further mediates the expression of multiple inflammatory genes (van der Heijden et al, 2018). Finally, CS has been shown to increase histone acetylation by increasing the expression of HATs and decreasing the expression of HDACs (Ito et al, 2005; Sundar et al, 2014).
Transgenerational epigenetic inheritance occurs when developmental programming is transmitted across generations that were not exposed to the initial environment, which triggered the change. The transgenerational programming has been described for metabolic and CVD risk (Krauss-Etschmann et al, 2015). A previous study showed significant hypomethylation in the placentas of babies born to mothers who smoked during pregnancy compared with that of nonsmoking mothers. This hypomethylation was found to correlate with increased placental cytochrome P450 family 1 subfamily A member 1 (CYP1A1) expression, which may have implications for xenobiotic metabolism in the offspring (Suter et al, 2011). Moreover, hypermethylation of the brain-derived neurotrophic factor may be responsible for its decreased expression with subsequent behavioral consequences in infants, children, and adolescents exposed in utero to maternal cigarette smoke (Knopik et al, 2012).
Taken together, these results have demonstrated that CS may modulate trained immunity and trained tolerance pathways via regulating energy metabolisms and epigenetic modifications. Since CS has divergent pro- and anti-inflammatory activities (AlQasrawi et al, 2020), we suggest that CS modulates inflammation and immunity via a linked trained innate immunity and trained innate immune tolerance process.
Five Nucleus-Localized ROS Activate CS-Promoted DNA Damage and Cell Death Pathways to Potentially Amplify Inflammation and Immune Responses via Releasing Alarmins
The toxicity of CS is largely attributed to its ability to generate ROS. CS contains a number of highly unstable free radicals, and these free radicals enhance ROS and RNS production leading to oxidative/nitrosative stress. Increased oxidative/nitrosative stress plays an important role in the pathogenesis of several diseases such as diabetes, lung cancer, atherosclerosis, and COPD. Accumulating evidence suggests a key role for smoke-induced ROS and the resulting oxidative stress in inflammation and cancer. CS is also known to stimulate ROS production by activating ROS sources, such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria (Fetterman et al, 2017; Kim et al, 2014; Li et al, 2016a).
Five ROS are localized in the nucleus including superoxide (O2−), nitrogen dioxide (NO2−), hydroxyl radical (OH−), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl) (Lu et al, 2021; Sun et al, 2020), and it has been reported that CS extract produces superoxide by reacting with bicarbonate (Park et al, 2021). Nitrogen oxides (NOx), most notably nitrogen dioxide (NO2−), are among the most hazardous forms of air pollution. TS is a major indoor source of NOx. Substantial increases in NOx concentrations are found when smoking only one cigarette (Braun et al, 2021), suggesting that NOx exposure occurs for secondhand smokers following smoking in indoor rooms. In addition, cigarette tar in aqueous solution contains a quinone–hydroquinone–semiquinone complex that can reduce oxygen to produce superoxide and hence hydrogen peroxide and the hydroxyl radical.
The cigarette tar radical can penetrate viable cells, bind to DNA, and induce nicks (Pryor, 1997; Zeng et al, 2018). Moreover, nicotine mediates hypochlorous acid-induced nuclear protein damage in mammalian cells (Salama et al, 2014). Chronic CS exposure results in increased oxidative stress leading to diaphragm muscle dysfunction (Barreiro et al, 2012), and increased expression of pro-oxidant genes and proinflammatory markers (Khanna et al, 2013). CS also increases intracellular ROS, cytokine expression, basal mitochondrial ROS, and oxidative stress in alveolar macrophages (Table 8) (Lugg et al, 2022).
Table 8.
Cigarette Smoke Induces Reactive Oxygen Species and Oxidative Stress, Which Promote Inflammation, Inflammatory Cell Death (Pyroptosis), Oxidative DNA Damage, and Cancers
| Cell type/tissue/species | Effect | PMID | |
|---|---|---|---|
| CS-exposed humans, mice, and pig | Skeletal muscles Diaphragm |
Induced oxidative stress and inflammation leading to muscle dysfunction | 20413628 22349133 |
| CS-exposed rat | Serum/brain | Increased expression of pro-oxidant genes (iNOS, NOX4, dual oxidase 1, and p22[phox]) leading to oxidative stress Increased expression of proinflammatory markers (IFN-γ, TNF-α, IL-1α, IL-1β, IL-23, IL-6, IL-23, IL-17, IL-10, TGF-β, T-bet, and FoxP3) and increased inflammation |
23031832 |
| CSE | Type II alveolar epithelial cells | Decreased cell viability Decreased expression of VEGF Increased ROS production and oxidative stress |
25607112 |
| CSE | Mouse β cells | Increased β cell endoplasmic reticulum and oxidative stress Reduced insulin secretion Impaired β cell survival |
32283079 |
| CS | Mouse lung | Increased MDA and MPO activities Decreased SOD activity Induced oxidative stress Reduced the expression of the mitochondrial fusion protein OPA1 and fission protein MFF Induced mitochondrial dysfunction |
34221235 |
| Acute CS exposure | Human alveolar epithelial cell | Increased cellular oxidative stress and autophagy | 31016633 |
| Electronic cigarette | Rats | Induced oxidative stress | 34515107 |
| CS exposure | Alveolar macrophages | Increased intracellular ROS Increased IL-8 cytokine production Increased basal mitochondrial ROS Increased oxidative stress |
33986144 |
| Nicotine | Mouse VSMCs/Apoe−/− mice | Induced autophagy and VSMC phenotype switching Increased ROS production and NF-κB signaling pathway Increased aortic atherosclerotic plaque |
30856513 |
iNOS, inducible nitric oxide synthase; MDA, malondialdehyde; MFF, mitochondrial fission factor; MPO, myeloperoxidase; OPA1, optic atrophy protein 1; SOD, superoxide dismutase; VSMCs, vascular smooth muscle cells.
Several new cell death pathways (Xiong et al, 2009; Yan et al, 2008; Yang et al, 2005; Yang et al, 2002; Yang et al, 1997) have been recently reported including intrinsic apoptosis, extrinsic apoptosis, mitochondrial permeability transition-driven necrosis, necroptosis, ferroptosis, pyroptosis (inflammatory cell death), parthanatos (excessive oxidative damage to DNA leading to overactivation of poly(ADP-ribose), polymerase [PARP]), entotic cell death, neutrophil extracellular trap (NETotic) cell death, lysosome-dependent cell death, autophagy-dependent cell death, immunogenic cell death, cellular senescence, mitotic catastrophe (Galluzzi et al, 2018; Shao et al, 2021a; Wang et al, 2019), and alkaliptosis and oxelptosis (Tang et al, 2019).
Among the 16 cell death types, most cell death types are involved in inflammation (Galluzzi et al, 2018; Wang et al, 2019; Yan et al, 2008; Yang et al, 2021). Moreover, a new pathway for proinflammatory programmed cell death, PANoptosis, has been described, which is controlled by a recently identified cytoplasmic multimeric protein complex named the PANoptosome. The PANoptosome can engage, in parallel, three key modes of programmed cell death—pyroptosis, apoptosis, and necroptosis (Samir et al, 2020). The cells undergoing death emit numerous signals that interact with the host to dictate the immunological signature of cellular stress and death. In the absence of reactive (nontolerated) antigenic determinants (which is generally the case for healthy cells), such signals may drive inflammation but cannot engage adaptive immunity.
Conversely, when cells exhibit sufficient reactive nontolerated antigenicity, as in the case of infected or malignant cells with tumor antigens (Yang and Yang, 2005; Yang et al, 2006), their death can culminate with adaptive immune responses that are executed by cytotoxic T lymphocytes and elicit immunological memory (Kroemer et al, 2022; Shen et al, 2019).
In addition to carcinogenic factors of CS exposure, oxidative stress plays significant roles in mediating CS-triggered cell death (Aoshiba and Nagai, 2003). CS exposure induces cellular senescence and seven different types of cell death including apoptosis, necrosis, necroptosis, ferroptosis, pyroptosis (Lopez-Pastrana et al, 2015; Wang et al, 2016a; Yin et al, 2015), parthanatos pathway of cell death, and autophagic cell death (Table 9) (Liu et al, 2021). CS exposure induces mitochondria-to-nuclear translocation of apoptosis-inducing factor and endonuclease G within the first 3 h characteristic of the parthanatos pathway.
Table 9.
Cigarette Smoke Induces Seven Types of Inflammation-Related Cell Death and Senescence
| Smoke components | Model/cell type | Effects | PMID |
|---|---|---|---|
| CSE | Bronchial epithelial cells | Induced cell death via repressing PRMT6/AKT signaling | 33260152 |
| CS | Bronchial epithelial cells | Activated parthanatos pathway of cell death | 31396404 |
| CSS | Olfactory epithelium | Induced apoptotic cell death and TNF expression | 29950987 |
| CS | Alveolar epithelial cells | Enhanced oxidative stress-induced apoptosis and/or necrosis | 19570263 |
| TS | Human premonocytic | Induced apoptosis and necrosis | 9755110 |
| CS | Human bronchial epithelial cell | Induced necroptotic cell death and release of DAMP and proinflammatory cytokines | 26719146 |
| CS | Human umbilical vein ECs | Induced prolonged endoplasmic reticulum stress and autophagic cell death | 21676957 |
| CSE | Bronchial epithelial cells | Reduced cell proliferation Increased β-galactosidase activity Increased cellular senescence |
27237816 |
| CSE | Mice and human bronchial epithelial cells | Increased expression of NLRP3 Increased caspase-1 activity and cleaved GSDMD Increased release of IL-1β and IL-18 Induced pyroptosis |
33465393 |
| CSE | Human bronchial epithelial cells | Induced ferroptosis cell death | 31316058 |
| Acute cigarette smoke exposure | Normal and COPD human bronchial epithelial cells | Induced apoptosis and CXCL8/IL8 production Induced epithelial to mesenchymal transition |
28468623 |
| CSE | Human bronchial epithelial cells/bronchoalveolar lavage fluid of CS-exposed mice | Induced necroptotic cell death Induced release of DAMPs Induced release of proinflammatory cytokines via MyD88 signaling |
26719146 |
| CS | Bovine pulmonary artery ECs and rat lung microvascular ECs/mice | Increased EC apoptosis | 24853558 |
| CSE | Rat VSMCs | Induced ferroptosis and cell death Increased inflammatory responses |
31975626 |
CS induces seven types of newly characterized cell death pathways in epithelial cells and ECs including apoptosis, necrosis, ferroptosis, pyroptosis, necroptosis, parthanatos pathway cell death, autophagic cell death, and cellular senescence (PMID:29362479; 31355136).
CSS, cigarette smoke solution; CXCL8, C-X-C motif chemokine ligand 8; DAMP, danger-associated molecular pattern; PRMT6, protein arginine methyltransferase 6; TS, tobacco smoke.
These diseases cause oxidative stress in neurons that also produce peroxynitrite (ONOO−, also a nuclear ROS) from the reaction of superoxide anions and nitric oxide causing DNA damage with subsequent PARP-1 activation (Sun et al, 2020). The use of a specific PARP-1 inhibitor, BMN673, abrogates the effect of smoke-induced activation of the parthanatos pathway. Smoke-mediated activation of the parthanatos pathway is increased in human bronchial epithelial cells obtained from chronic smokers (Künzi and Holt, 2019).
Inflammatory cell death can amplify inflammation via at least three pathways: (i) caspase-1-caspase-4 (human)/caspase-11 (mouse) facilitated N-gasdermin D membrane pore to release big proinflammatory secretomes as we previously reported (Lu et al, 2022; Lu et al, 2021; Ni et al, 2021; Xu et al, 2021); (ii) release of alarmins (DAMPs) from dying cells (Yang et al, 2017a); and (iii) NETotic cell death (Delgado-Rizo et al, 2017).
High-mobility group box protein 1 (HMGB1), a highly conserved nonhistone nuclear protein, binds to DNA to regulate the structure of chromosomes and maintain transcription, replication, DNA repair, and nucleosome assembly. HMGB1 is actively or passively released into the extracellular space during cell activation or necrosis. Extracellular HMGB1 as an alarmin can initiate immune responses alone or combined with other substances such as nucleic acid. It has been reported that HMGB1 is involved in various inflammatory responses, the inflammatory-reparative response (Foglio et al, 2022), and autoimmunity (Dong et al, 2022). In addition to HMGB1, alarmins also include a surprising number of chromatin-binding moieties, such as HMGN1, IL-1α, and IL-33, as well as heat shock proteins, S100 proteins, ATP, and uric acid crystals (Yang et al, 2017a). It has been reported that CS can silence innate lymphoid cell function and facilitates an exacerbated type I IL33-dependent inflammatory response to infection (Kearley et al, 2015).
Taken together, five nucleus-localized ROS can activate CS-promoted DNA damage (Zeng et al, 2018) and potentially trigger cell death. CS-induced cell death can amplify inflammation and immune responses. In the future, more detailed work is needed to determine whether combinations of several cell death pathways mediate trained immunity-promoted CS-triggered cell death.
Cigarette Smoke Increases ROS and EC Activation/Dysfunction to Induce Vascular Inflammation and Subsequently Promote CVDs
Vascular endothelium plays a fundamental role in the regulation of vascular tone, inflammatory response, vascular growth, and thrombotic balance by producing important vasodilators with antiatherosclerotic and antiaggregatory properties such as nitric oxide (NO) and prostacyclin (Vane et al, 1990). Endothelium is among the first line of the body's defense system. The normal vascular endothelium is taken as a gatekeeper of cardiovascular health, whereas abnormality of vascular endothelium is a major contributor to CVDs, such as atherosclerosis, aging, hypertension, obesity, and diabetes. Based on the capacities of ECs in carrying out 11 important innate immune functions that are originally considered to be carried out by prototypic innate immune cell-type macrophages, in 2013, we proposed a novel concept that ECs are innate immune cells (Drummer et al, 2021a; Mai et al, 2013; Shao et al, 2021a; Shao et al, 2020).
To consolidate this new concept, we reported that ECs are equipped with innate immune sensing function including canonical inflammasome and caspase-1 (Li et al, 2017; Li et al, 2016b; Lopez-Pastrana et al, 2015; Lu et al, 2021; Wang et al, 2016a; Yin et al, 2015), immune checkpoint receptor upregulation in response to proinflammatory cytokine stimulation (Shen et al, 2019), upregulation of six types of canonical and noncanonical secretomes (Lu et al, 2022), upregulation of a list of new CDs (clusters of differentiation) (Xu et al, 2021), and upregulation of innate immune regulatome and myelopoiesis factor (Shao et al, 2021a).
The five cardinal signs of inflammation are redness, swelling, heat, pain, and loss of function. ECs play a major role in the initiation of inflammatory process. Endothelial activation encompasses a range of endothelial responses to inflammatory signals including changes in thromboresistance, altered vasomotor tone, and loss of barrier function. Once activated, the EC facilitates cellular trafficking. Leukocyte activation and transmigration are important for innate and adaptive immunity. Endothelial dysfunction is widely used to describe the nonphysiologic activity of ECs. The pathophysiology of endothelial dysfunction is complex and multifactorial (Shao et al, 2014). These include imbalanced vasodilation and vasoconstriction, decreased bioavailability of NO, reduced activity of endothelial nitric oxide synthase (eNOS), and increased production of ROS, increased proinflammatory factors. The resulting oxidative stress caused by these factors within the vascular wall contributes significantly to the pathophysiology of endothelial dysfunction and, subsequently, vascular inflammation and CVDs (Shao et al, 2014).
Endothelial dysfunction is a central early event in the pathogenesis of most CVDs and provides an important link between diseases such as hypertension, chronic renal failure, and diabetes. Previous reports showed that inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interact with ECs or VSMCs to induce endothelial nitric oxide dysfunction, ROS production, and VSMC proliferation, resulting in endothelial dysfunction and promotion of CVD. Indications regarding endothelial function are mainly assessed using flow-mediated dilation (FMD). FMD is a noninvasive measurement of brachial artery diameter changes following an increase in shear stress induced by reactive hyperemia (Corretti et al, 2002). Brachial FMD showed a significant predictive value for cardiovascular events (Ras et al, 2013). CS is the most important modifiable risk factor and plays a critical role in the pathogenesis and development of atherosclerotic CVDs by, at least in part, endothelial dysfunction.
CS causes increased oxidative stress by several mechanisms, including (i) direct damage by radical species and (ii) the inflammatory response caused by CS. CS-induced oxidative stress and ROS induce expression of cell adhesion molecules to promote leukocytes—EC adhesion and VSMC proliferation (Table 10) (Teasdale et al, 2014). CS, e-cigarette, and nicotine administration have all been shown to reduce NO bioavailability, which is the central mechanism in the pathophysiology of endothelial dysfunction (DiGiacomo et al, 2018; He et al, 2017).
Table 10.
Cigarette Smoke Induces Endothelial Cell Activation/Dysfunction and Vascular Inflammation via Increasing Reactive Oxygen Species Production, Activating Nuclear Factor Kappa B, and Upregulating Adhesion Molecules
| Smoke components | Model/cell type | Effects | PMID |
|---|---|---|---|
| CSE | Mouse cerebral microvascular ECs | Decreased cell viability Increased paracellular permeability Increased proinflammatory cytokines Increased ROS production |
31666540 |
| CSE | Pulmonary ECs | Increased endothelial monolayer permeability Induced EC activation and promoted lung inflammation |
29351435 |
| CSE | HMVEC-L | Increased PAF which promotes transendothelial migration Increased cell surface expression of adhesion molecules such as P-selectin, E-selectin, ICAM-1, and VCAM-1 Increased polymorphonuclear leukocytes adherence |
21984569 |
| CSE | Carotid arteries of rats | Impaired vascular relaxations and induced endothelial dysfunction Increased ROS production Increased expression of adhesion molecules (ICAM-1, VCAM-1, and E-selectin) and monocyte adhesion. Increased expression of inflammatory markers (iNOS, TNF-α, IL-1β, and IL-6) Induced activation of NF-κB |
17213480 |
| CSC | HUVEC | Increased expression of ICAM-1 and VCAM-1 Induced activation of NF-κB Increased transendothelial migration of monocytes and neutrophils Induced activation of protein kinase C |
8928867 21651795 |
| CSE | HLMVECs/mouse lung tissue cells | Increased expression of CXCR3 Increased responsiveness to EMAP II and IP-10 to induce apoptosis |
22936405 |
| CS and e-cigarette | Male C57BL/6 mice | Increases superoxide radical and NADPH oxidase in mouse aorta Decreases eNOS, p-eNOS Ser1177, and BH4 expression in aorta Impaired ACh-induced endothelium-dependent relaxation in mouse aortic segments |
32412791 35089811 |
| CSE | HAECs | Decrease in eNOS expression Increase in expression of iNOS, NLRP3, caspase-1p20, and IL-1β |
34768128 |
| CS and e-cigarette | Healthy smokers and nonsmoker adults | Increased soluble NOX2-derived peptide and oxidative stress Decrease in NO bioavailability and FMD |
27108682 |
Ach, acetylcholine; CSC, cigarette smoke condensate; CXCR3, C-X-C motif chemokine receptor 3; EMAP II, endothelial monocyte-activating polypeptide II; eNOS, endothelial nitric oxide synthase; FMD, flow-mediated dilation; HLMVECs, human lung microvascular endothelial cells; HuAoSMCs, human aorta primary smooth muscle cells; HUVEC, human umbilical vein endothelial cells; IP-10, interferon-inducible protein–10; NADPH, nicotinamide adenine dinucleotide phosphate; PAF, platelet-activating factor.
eNOS is an enzyme that is responsible to produce NO in ECs. Exposure to CS in EC can reduce the expression of eNOS genes and proteins, resulting EC dysfunction (He et al, 2017; Su et al, 1998). A decrease of eNOS and NO levels will increase vascular tone, increase expression of adhesion molecules, trigger coagulation cascade and inflammation (Kaur et al, 2018), and increases the risk of atherosclerotic CVDs (Fig. 4). Measurement of FMD represents a useful tool to assess the impacts of smoking on the vascular wall and the efficacy of treatment options for early smoking-induced proatherogenic changes in the vasculature. CS showed a significant decrease in FMD (Esen et al, 2004). A possible mechanism for lowering FMD induced by CS is reduced production of NO and increased expression of adhesion molecules, leading to impaired endothelial function.
FIG. 4.

The schematic figure showed that CS and nicotine induce endothelial dysfunction by binding to different types of DAMP receptors and causing direct damage to endothelial cells, decreasing eNOS and NO bioavailability leading to increased oxidative stress and adhesion molecules, therefore increasing inflammatory response and vascular inflammation to promote initiation and progression of cardiovascular diseases. eNOS, endothelial nitric oxide synthase; NO, nitric oxide.
Conclusion
Cigarette smoke (CS) has a major impact on health issues worldwide. Many of the health care consequences of cigarette smoking could be related to its ability to depress the immune system although the mechanisms by which CS alters immunity are not completely understood. The toxic chemicals present in CS have broad immunomodulatory consequences that include altered innate and adaptive immunity and interrupt immunological homeostasis. This leads to several diseases and exerts paradoxical effects on inflammation. We published one such inflammation paradox between proinflammatory cytokines and microRNAs (Liu et al, 2020). We observed increased ROS and oxidative stress generation, increased DNA damage and cell death, as well as metabolic reprogramming and epigenetic modification. In particularly, CS acts as a double-edged sword that either exacerbates pathological immune responses or attenuates the defensive function of the immune system, possibly due to the complexities and functional diversities of CS components.
Even though CS exerts diverse effects on immune responses, the net effect is deleterious rather than beneficial. We have reviewed experimental evidence that showed (i) the signals of DAMP receptors contribute to CS modulation of inflammation and immunity; (ii) CS reprograms immunometabolism and trained immunity-/trained innate immune tolerance-related metabolic pathways in innate immune cells and T cells; (iii) five nucleus-localized ROS activate CS-promoted DNA damage and cell death pathways to potentially amplify inflammation and immune responses; and (iv) CS increases ROS and EC activation/dysfunction to induce vascular inflammation and subsequently promotes CVDs.
Cigarette smoking attributes significantly to CHD deaths and doubles the risk of ischemic stroke. Smoking acts synergistically with other disease risk factors, substantially increasing the risk of CHD, peripheral vascular disease, cancer, chronic lung disease, and many other chronic diseases (Shinton and Beevers, 1989). In the presence of a high-fat diet (hyperlipidemia), a known trained immunity inducer (Christ et al, 2018), nicotine exposure significantly increased vascular inflammation and atherosclerosis in apolipoprotein E-deficient (ApoE−/−) mice (Wu et al, 2018). CS in angiotensin II-induced hypertension increased mitochondrial oxidative stress, which contributes to endothelial dysfunction and CVDs (Dikalov et al, 2019). Furthermore, smoking substantially increases the risk of CVD in patients with type 2 diabetes (Yang et al, 2022).
Cigarette smoking is known to both increase susceptibility to infection and drives inflammation. Exposure to CS impaired host defense response and immune suppressive effects by suppressing multiple host defense mechanisms such as epithelial cell responses and recruitment and activation of innate immune cells including monocytes/macrophages, neutrophils, and NK cells. Recently, smokers have been found to be at a higher risk of developing severe forms of coronavirus disease 2019 (COVID-19) (Masso-Silva et al, 2021). E-cigarette aerosol inhalation (vaping) has been associated with several inflammatory lung disorders. Previous study demonstrated that mice exposed to e-cigarette aerosol showed a marked reduction in IgA immunoglobulins and CD4 T cells associated with increased neutrophil activation, indicating that e-cigarette vapers may be at a higher risk of increasing the immune response and developing inflammatory disorders of the lungs (Masso-Silva et al, 2021). E-cigarette-exposed alveolar macrophages show a significant increase in the production of proinflammatory cytokines/chemokines, apoptosis and necrosis, and ROS production (Scott et al, 2018), as well as oxidative stress, ox-LDL, and cardiovascular risk (Carnevale et al, 2016; Moheimani et al, 2017).
Chronic e-cigarette exposure induces vascular endothelial dysfunction, cardiac dysfunction, and atherosclerosis in mice (El-Mahdy et al, 2021; Espinoza-Derout et al, 2019) and increases risk for COPD (Bowler et al, 2017) and CVDs (D'Amario et al, 2019). On the contrary, another clinical study has found that CS or vaping e-cigarettes showed a reduction in the expression of immune-related genes. Also, vaping e-cigarettes was associated with suppression of a large number of unique genes and e-cigarette users showed greater suppression of genes common with those changed in cigarette smokers.
Furthermore, vaping e-cigarettes suppressed the expression of transcription factors, such as early growth response 1, which was functionally associated with decreased expression of 5 target genes in cigarette smokers and 18 target genes in e-cigarette users (Martin et al, 2016).
CS modulates the immune functions of ECs. CS exposure increases pulmonary endothelial barrier permeability and causes endothelial activation/dysfunction. CS decreased eNOS and inhibited eNOS activity. In addition, CS increased ROS production, and oxidative stress-mediated reduction in eNOS-NO signaling is a significant contributor to CS-induced vascular endothelial dysfunction. CS stimulates the expression of P-selectin, E-selectin, ICAM-1, and VCAM-1, as well as cytokines and chemokines, including TNF-α, IL-6, and IL-1β, via NADPH oxidase-dependent NF-κB transcriptional activation. CS also increases adherence of monocytes to the endothelium and transendothelial migration, as well as neutrophil transmigration across HUVECs and induced upregulation of the CXCR3 receptor in ECs. CS synergizes with the inflammatory cytokine IL-1β to increase vascular permeability and endothelial dysfunction via ROS/p38/phosphatase and tensin homolog-mediated tyrosinephosphorylation of vascular endothelial cadherin and β-catenin and subsequent β-catenin nuclear translocation and expression of inflammatory genes, such as COX-2 in cardiac ECs.
In our working model (Fig. 5), we showed that after an initial exposure to the first stimulus, innate immune cells with “memory” traits respond more rapidly and with a high magnitude to secondary stimulation. After the secondary stimulus, cells can respond by increasing the proinflammatory mediator, mediated by (i) metabolic reprogramming involving increased aerobic glycolysis, increased acetyl CoA generation, increased mevalonate synthesis, glutaminolysis, and increased production of lactate and fumarate; and (ii) epigenetic modification including increased H3K27ac and H3K4me3. Also, cell can respond with decreasing the magnitude of immune response (innate immune tolerance) characterized by decreased proinflammatory and increased anti-inflammatory mediators mediated by metabolic reprogramming including decreased aerobic glycolysis, increased production of itaconate metabolites, as well as epigenetic rewiring including increased H3K9me3.
FIG. 5.

Our working model. Representative model of innate immune memory and immune tolerance response. After initial exposure to the first stimulus, innate immune cells with “memory” traits respond rapidly with a high magnitude of immune response to the secondary stimulation, increased proinflammatory mediators mediated by metabolic reprogramming such as increased aerobic glycolysis, increased acetyl CoA generation, increased mevalonate synthesis, glutaminolysis, and increased production of fumarate and lactate as well as epigenetic modification such as increased H3K27ac and H3K4me3. Also, after exposure to the first stimulus, cells respond with a decreased magnitude of innate immune response (tolerance) characterized by decreased proinflammatory and increased anti-inflammatory mediators mediated by metabolic reprogramming such as decreased aerobic glycolysis, increased production of itaconate metabolites, as well as epigenetic rewiring such as increased and H3K9me3. H3K4me3, histone 3 lysine 4 trimethylations; H3K9me3, histone 3 lysine 9 trimethylation; H3K27ac, histone 3 lysine 27 acetylations.
The carcinogenic and immunomodulatory toxins in CS bind to several receptors on the cell membrane, cytosol, and nucleus leading to increases in the cytosolic ROS and five nuclear ROS expression. This leads to the induction of seven types of cell death and subsequent alarmins and NET release, which amplify inflammation and induced trained immunity. Metabolic reprogramming induced by CS can result in the generation of acetyl and methyl groups and induce histone remodeling and exacerbate immune responses to induce trained immunity. CS exposure significantly upregulates the IRG1 expression and increases the abundance of itaconate metabolites (Titz et al, 2020) resulting in immunosuppression.
The anti-inflammatory effect of CS is counteracted by the proinflammatory effect of other CS constituents, resulting in reduced immunosuppression effects and increased inflammation and trained immunity in a phenomenon known as inflammation paradox (Liu et al, 2020; Virtue et al, 2017) or second inflammation wave as we proposed (Johnson et al, 2020; Liu et al, 2020) (Fig. 6).
FIG. 6.

Our working model. The carcinogenic and immunomodulatory toxins in CS bind to different receptors on the cell membrane, cytosol, and nucleus leading to increased cytoplasmic (cytosolic and non-nucleus organelle) ROS, which further induces five different nuclear ROS. Increased ROS production results in the induction of seven types of cell death and subsequent alarmins and neutrophil extracellular trap release, which amplify inflammation and induce trained immunity. Metabolic reprogramming induced by CS can induce epigenetic remodeling and exacerbate immune repose to induce trained immunity. CS exposure significantly upregulates the IRG1 expression and increases the abundance of itaconate metabolites resulting in immunosuppression. The anti-inflammatory effect of CS is counteracted by the proinflammatory effect of other CS constituents resulting in reduced immunosuppression effects and increased inflammation and trained immunity in a phenomenon known as inflammation paradox1 or second inflammation wave. AChR, acetylcholine receptor; CLRs, C-type lectin receptors; GPCRs, G-protein-coupled receptors; NLRs, NOD-like receptors; PAMPs, pathogen-associated molecular patterns; RAGE, receptor for advanced glycation end products; RLRs, retinoic acid-inducible gene-like receptors; TREMs, triggering receptors expressed on myeloid cells; TLRs, toll-like receptors.
In conclusion, the effects of CS on immune responses are wide-ranging and complex, and both potentiation (proinflammatory/trained immunity) and attenuation (anti-inflammatory/trained tolerance) may be induced. The net effect of CS on the immune response and inflammation depends on many variables, including the dose and duration of CS use, cell types, tissue types, and contexture, and the presence of other factors at the time of immune cell stimulation. The recognition of specific mechanisms by which CS affects immune response is an important step toward elucidating mechanisms of CS-induced diseases and may identify novel therapeutic approaches for the management of diseases that afflict smokers.
Acknowledgments
F.S. carried out the primary literature search and drafted the article. Others provided material input and helped in revising the article. X.F.Y. conceived the study and provided field expertise. All the authors read and approved the final article.
Abbreviations Used
- α7nAChR
α7 nicotinic acetylcholine receptor
- Ach
acetylcholine
- AChR
acetylcholine receptor
- AhR
aryl hydrocarbon receptor
- ALDOA
aldolase, fructose-bisphosphate A
- ApoE−/−
apolipoprotein E deficient
- ATF3
activating transcription factor 3
- B[k]F
benzo[k]fluoranthene
- BaP
benzo[a]pyrene
- BD
1,3-butadiene
- BMDMs
bone marrow-derived macrophages
- CAD
cis-aconitate decarboxylase
- CCL20
chemokine (C-C motif) ligand 20
- CCR6
C-C motif chemokine receptor 6
- CDs
clusters of differentiation
- CHD
coronary heart disease
- CKD
chronic kidney diseases
- CLRs
C-type lectin receptors
- COPD
chronic obstructive pulmonary disease
- COVID-19
coronavirus disease 2019
- COX-2
cyclooxygenase-2
- CpG
cytosine-phosphate-guanine
- CS
cigarette smoke
- CSC
cigarette smoke condensate
- CSE
cigarette smoke extract
- CTLs
CD8+ cytotoxic T lymphocytes
- CVD
cardiovascular disease
- CXCL8
C-X-C motif chemokine ligand 8
- CXCR3
C-X-C motif chemokine receptor 3
- CYP1A1
cytochrome P450 family 1 subfamily A member 1
- DAMPs
danger-associated molecular pattern
- DCs
dendritic cells
- DNMTs
DNA methyltransferases
- dsDNA
double-stranded DNA
- e-cigarettes
electronic cigarettes
- ECs
endothelial cells
- EMAP II
endothelial monocyte-activating polypeptide II
- eNOS
endothelial nitric oxide synthase
- ESRD
end-stage kidney disease
- FA
formaldehyde
- FMD
flow-mediated dilation
- Foxp3
forkhead box P3
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GMCSF
granulocyte/macrophage-colony-stimulating factor
- GPCRs
G-protein-coupled receptors
- GSDMD
gasdermin D
- H1R
histamine receptor 1
- H2O2
hydrogen peroxide
- H3K27ac
histone 3 lysine 27 acetylation
- H3K4me3
histone 3 lysine 4 trimethylation
- H3K9me2
histone 3 lysine 9 dimethylation
- H3K9me3
histone 3 lysine 9 trimethylation
- HAECs
human aortic endothelial cells
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- HFD
high-fat diet
- HLMVECs
human lung microvascular endothelial cells
- HMEC-1
human microvascular endothelial cell 1
- HMGB1
high-mobility group box protein 1
- HMGN1
high-mobility group nucleosome binding domain 1
- HOCl
hypochlorous acid
- HPBECs
human primary bronchial epithelial cells
- HuAoSMCs
human aorta primary smooth muscle cells
- HUVECs
human umbilical vein endothelial cells
- IBD
inflammatory bowel disease
- ICAM-1
intercellular adhesion molecule 1
- IFN-γ
interferon-gamma
- IκBζ
inhibitor of nuclear factor-kappa B zeta
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IP-10
interferon-inducible protein–10
- IRG1
immune responsive gene 1
- IRI
ischemia–reperfusion injury
- JNK
c-Jun N-terminal kinase
- Keap1
kelch-like ECH-associated protein 1
- LDH
lactate-dehydrogenase
- LDL
low-density lipoprotein
- LPS
lipopolysaccharide
- MDA
malondialdehyde
- MDMs
monocyte-derived macrophages
- MFF
mitochondrial fission factor
- MHC-II
MHC class II molecules
- MPO
myeloperoxidase
- MS
multiple sclerosis
- mTOR
mammalian target of rapamycin
- NADPH
nicotinamide adenine dinucleotide phosphate
- NETs
neutrophil extracellular traps
- NF-κB
nuclear factor kappa B
- NK
natural killer
- NLRP3
NOD-, LRR-, and pyrin domain-containing protein 3
- NLRs
NOD-like receptors
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNN
N′-nitrosonornicotine
- NO
nitric oxide
- NO2−
nitrogen dioxide
- NOx
nitrogen oxides
- Nrf2
nuclear factor erythroid 2-related factor 2
- NTHI
nontypeable Haemophilus influenzae
- O2−
superoxide
- OCR
oxygen consumption rate
- OH−
hydroxyl radical
- OPA1
optic atrophy protein 1
- ORs
odorant receptors
- ox-LDL
oxidized low-density lipoprotein
- PAF
platelet-activating factor
- PAMPs
pathogen-associated molecular patterns
- PARP
poly(ADP-ribose) polymerase
- PBMCs
peripheral blood mononuclear cells
- PI3-AKT
phosphoinositide-3-kinase–protein kinase B
- PMA
phorbol myristate acetate
- PMA
phorbol myristate acetate
- PMNs
polymorphonuclear neutrophils
- PRMT6
protein arginine methyltransferase 6
- RA
rheumatoid arthritis
- RAGE
receptor for advanced glycation end products
- RLRs
retinoic acid-inducible gene-like receptors
- RNS
reactive nitric species
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- sICAM-1
soluble intercellular adhesion molecule 1
- SLE
systemic lupus erythematosus
- SOD
superoxide dismutase
- STAT3
signal transducer and activator of transcription 3
- TCA
tricarboxylic acid
- TGF-β
transforming growth factor-β
- Th
T helper cell
- TICAM1
toll-like receptor adaptor molecule 1
- TLRs
toll-like receptors
- TNF-α
tumor necrosis factor-α
- Tregs
T regulatory cells
- TREMs
triggering receptors expressed on myeloid cells
- TS
tobacco smoke
- TXNIP
thioredoxin-interacting protein
- UC
ulcerative colitis
- VCAM-1
vascular cell adhesion molecule 1
- VEGF
vascular endothelial growth factor
- VLDL
very-low-density lipoprotein
- VSMCs
vascular smooth muscle cells
- WT
wild type
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Our research activities are supported by grants from the National Institutes of Health (NIH)/(HL131460, HL132399, HL138749, HL147565, HL130233, DK104116, DK113775, P30 DA13429, RO1 DA040619, and RO1 DA049745). The content in this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- Agarwal AR, Yin F, Cadenas E. Short-term cigarette smoke exposure leads to metabolic alterations in lung alveolar cells. Am J Respir Cell Mol Biol 2014;51(2):284–293; doi: 10.1165/rcmb.2013-0523OC [DOI] [PubMed] [Google Scholar]
- AlQasrawi D, Qasem A, Naser SA. Divergent effect of cigarette smoke on innate immunity in inflammatory bowel disease: A nicotine-infection interaction. Int J Mol Sci 2020;21(16):E5801; doi: 10.3390/ijms21165801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alrouji M, Manouchehrinia A, Gran B, et al. Effects of cigarette smoke on immunity, neuroinflammation and multiple sclerosis. J Neuroimmunol 2019;329:24–34; doi: 10.1016/j.jneuroim.2018.10.004 [DOI] [PubMed] [Google Scholar]
- Ambalavanan N, Carlo WF, Bulger A, et al. Effect of cigarette smoke extract on neonatal porcine vascular smooth muscle cells. Toxicol Appl Pharmacol 2001;170(2):130–136; doi: 10.1006/taap.2000.9094 [DOI] [PubMed] [Google Scholar]
- Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J Am Coll Cardiol 2004;43(10):1731–1737; doi: 10.1016/j.jacc.2003.12.047 [DOI] [PubMed] [Google Scholar]
- Aoshiba K, Nagai A. Oxidative stress, cell death, and other damage to alveolar epithelial cells induced by cigarette smoke. Tob Induc Dis 2003;1(3):219–226; doi: 10.1186/1617-9625-1-3-219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridgides DS, Mellinger DL, Armstrong DA, et al. Functional and metabolic impairment in cigarette smoke-exposed macrophages is tied to oxidative stress. Sci Rep 2019;9(1):9624; doi: 10.1038/s41598-019-46045-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011;21(3):381–395; doi: 10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro E, del Puerto-Nevado L, Puig-Vilanova E, et al. Cigarette smoke-induced oxidative stress in skeletal muscles of mice. Respir Physiol Neurobiol 2012;182(1):9–17; doi: 10.1016/j.resp.2012.02.001 [DOI] [PubMed] [Google Scholar]
- Barua RS, Sharma M, Dileepan KN. Cigarette smoke amplifies inflammatory response and atherosclerosis progression through activation of the H1R-TLR2/4-COX2 axis. Front Immunol 2015;6:572; doi: 10.3389/fimmu.2015.00572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Gonzalez A, Vidal R, Criollo A, et al. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol 2019;10:2993; doi: 10.3389/fimmu.2019.02993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CMT, Morissette MC, Stämpfli MR. The influence of cigarette smoking on viral infections: Translating bench science to impact COPD pathogenesis and acute exacerbations of COPD clinically. Chest 2013;143(1):196–206; doi: 10.1378/chest.12-0930 [DOI] [PubMed] [Google Scholar]
- Bekkering S, Domínguez-Andrés J, Joosten LAB, et al. Trained immunity: Reprogramming innate immunity in health and disease. Annu Rev Immunol 2021;39:667–693; doi: 10.1146/annurev-immunol-102119-073855 [DOI] [PubMed] [Google Scholar]
- Berg RE, Forman J. The role of CD8 T cells in innate immunity and in antigen non-specific protection. Curr Opin Immunol 2006;18(3):338–343; doi: 10.1016/j.coi.2006.03.010 [DOI] [PubMed] [Google Scholar]
- Bermudez EA, Rifai N, Buring JE, et al. Relation between markers of systemic vascular inflammation and smoking in women. Am J Cardiol 2002;89(9):1117–1119; doi: 10.1016/s0002-9149(02)02284-1 [DOI] [PubMed] [Google Scholar]
- Bowler RP, Hansel NN, Jacobson S, et al. Electronic cigarette use in US adults at risk for or with COPD: Analysis from two observational cohorts. J Gen Intern Med 2017;32(12):1315–1322; doi: 10.1007/s11606-017-4150-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, Klingelhöfer D, Müller R, et al. The impact of second-hand smoke on nitrogen oxides concentrations in a small interior. Sci Rep 2021;11(1):11703; doi: 10.1038/s41598-021-90994-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling LP, Salzmann K, Rothenbacher D, et al. Smoking, F2RL3 methylation, and prognosis in stable coronary heart disease. Eur Heart J 2012;33(22):2841–2848; doi: 10.1093/eurheartj/ehs091 [DOI] [PubMed] [Google Scholar]
- Breitling LP, Yang R, Korn B, et al. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011;88(4):450–457; doi: 10.1016/j.ajhg.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscetta M, Di Vincenzo S, Miele M, et al. Cigarette smoke inhibits the NLRP3 inflammasome and leads to caspase-1 activation via the TLR4-TRIF-caspase-8 axis in human macrophages. FASEB J 2020;34(1):1819–1832; doi: 10.1096/fj.201901239R [DOI] [PubMed] [Google Scholar]
- Carnevale R, Sciarretta S, Violi F, et al. Acute impact of tobacco vs electronic cigarette smoking on oxidative stress and vascular function. Chest 2016;150(3):606–612; doi: 10.1016/j.chest.2016.04.012 [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control (CDC). The Surgeon General's 1989 Report on Reducing the Health Consequences of Smoking: 25 Years of Progress. MMWR Suppl 1989;38(2):1–32. [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (U.S.), National Center for Chronic Disease Prevention and Health Promotion (U.S.), and Office on Smoking and Health (US). How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General. Publications and Reports of the Surgeon General. Centers for Disease Control and Prevention: Atlanta, GA; 2010. [PubMed] [Google Scholar]
- Cheng S-C, Quintin J, Cramer RA, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014;345(6204):1250684; doi: 10.1126/science.1250684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T. Chemical evaluation of electronic cigarettes. Tob Control 2014;23(Suppl 2):ii11–ii17; doi: 10.1136/tobaccocontrol-2013-051482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Han K-D, Oh TR, et al. Smoking and risk of incident end-stage kidney disease in general population: A nationwide population-based cohort study from Korea. Sci Rep 2019;9(1):19511; doi: 10.1038/s41598-019-56113-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou W-C, Rampanelli E, Li X, et al. Impact of intracellular innate immune receptors on immunometabolism. Cell Mol Immunol 2022;19(3):337–351; doi: 10.1038/s41423-021-00780-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ A, Günther P, Lauterbach MAR, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 2018;172(1–2):162–175.e14; doi: 10.1016/j.cell.2017.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corretti MC, Anderson TJ, Benjamin EJ, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: A report of the international brachial artery reactivity task force. J Am Coll Cardiol 2002;39(2):257–265; doi: 10.1016/s0735-1097(01)01746-6 [DOI] [PubMed] [Google Scholar]
- D'Amario D, Migliaro S, Borovac JA, et al. Electronic cigarettes and cardiovascular risk: Caution waiting for evidence. Eur Cardiol 2019;14(3):151–158; doi: 10.15420/ecr.2019.16.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deets KA, Vance RE. Inflammasomes and adaptive immune responses. Nat Immunol 2021;22(4):412–422; doi: 10.1038/s41590-021-00869-6 [DOI] [PubMed] [Google Scholar]
- Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, et al. Neutrophil extracellular traps and its implications in inflammation: An overview. Front Immunol 2017;8:81; doi: 10.3389/fimmu.2017.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiacomo SI, Jazayeri M-A, Barua RS, et al. Environmental tobacco smoke and cardiovascular disease. Int J Environ Res Public Health 2018;16(1):E96; doi: 10.3390/ijerph16010096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S, Itani H, Richmond B, et al. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am J Physiol Heart Circ Physiol 2019;316(3):H639–H646; doi: 10.1152/ajpheart.00595.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez-Andrés J, Novakovic B, Li Y, et al. The itaconate pathway is a central regulatory node linking innate immune tolerance and trained immunity. Cell Metab 2019;29(1):211–220.e5; doi: 10.1016/j.cmet.2018.09.003 [DOI] [PubMed] [Google Scholar]
- Dong Y, Ming B, Dong L. The role of HMGB1 in rheumatic diseases. Front Immunol 2022;13:815257; doi: 10.3389/fimmu.2022.815257 [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Drummer C, Saaoud F, Shao (
 ) Y, et al. Trained immunity and reactivity of macrophages and endothelial cells. Arterioscler Thromb Vasc Biol
2021a;41(3):1032–1046; doi: 10.1161/ATVBAHA.120.315452 [DOI] [PMC free article] [PubMed] [Google Scholar]
) Y, et al. Trained immunity and reactivity of macrophages and endothelial cells. Arterioscler Thromb Vasc Biol
2021a;41(3):1032–1046; doi: 10.1161/ATVBAHA.120.315452 [DOI] [PMC free article] [PubMed] [Google Scholar] - Drummer CIV, Saaoud F, Sun Y, et al. Hyperlipidemia may synergize with hypomethylation in establishing trained immunity and promoting inflammation in NASH and NAFLD. J Immunol Res 2021b;2021:3928323; doi: 10.1155/2021/3928323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan M-C, Zhang J-Q, Liang Y, et al. Infiltration of IL-17-Producing T cells and Treg cells in a mouse model of smoke-induced emphysema. Inflammation 2016;39(4):1334–1344; doi: 10.1007/s10753-016-0365-8 [DOI] [PubMed] [Google Scholar]
- Duaso M, Duncan D. Health impact of smoking and smoking cessation strategies: Current evidence. Br J Community Nurs 2012;17(8):356–363; doi: 10.12968/bjcn.2012.17.8.356 [DOI] [PubMed] [Google Scholar]
- El-Mahdy MA, Mahgoup EM, Ewees MG, et al. Long-term electronic cigarette exposure induces cardiovascular dysfunction similar to tobacco cigarettes: Role of nicotine and exposure duration. Am J Physiol Heart Circ Physiol 2021;320(5):H2112–H2129; doi: 10.1152/ajpheart.00997.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esen AM, Barutcu I, Acar M, et al. Effect of smoking on endothelial function and wall thickness of brachial artery. Circ J 2004;68(12):1123–1126; doi: 10.1253/circj.68.1123 [DOI] [PubMed] [Google Scholar]
- Espinoza-Derout J, Hasan KM, Shao XM, et al. Chronic intermittent electronic cigarette exposure induces cardiac dysfunction and atherosclerosis in apolipoprotein-E knockout mice. Am J Physiol Heart Circ Physiol 2019;317(2):H445–H459; doi: 10.1152/ajpheart.00738.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagenson AM, Xu K, Saaoud F, et al. Liver ischemia reperfusion injury, enhanced by trained immunity, is attenuated in caspase 1/caspase 11 double gene knockout mice. Pathog Basel Switz 2020;9(11):E879; doi: 10.3390/pathogens9110879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira AV, Domiguéz-Andrés J, Netea MG. The role of cell metabolism in innate immune memory. J Innate Immun 2022;14(1):42–50; doi: 10.1159/000512280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetterman JL, Sammy MJ, Ballinger SW. Mitochondrial toxicity of tobacco smoke and air pollution. Toxicology 2017;391:18–33; doi: 10.1016/j.tox.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Gomez D, Bekkering S, Netea MG, et al. Trained immunity in atherosclerotic cardiovascular disease. Arterioscler Thromb Vasc Biol 2021;41(1):62–69; doi: 10.1161/ATVBAHA.120.314216 [DOI] [PubMed] [Google Scholar]
- Foglio E, Pellegrini L, Russo MA, et al. HMGB1-mediated activation of the inflammatory-reparative response following myocardial infarction. Cells 2022;11(2):216; doi: 10.3390/cells11020216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 2018;25(3):486–541; doi: 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong T, Liu L, Jiang W, et al. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 2020;20(2):95–112; doi: 10.1038/s41577-019-0215-7 [DOI] [PubMed] [Google Scholar]
- Goodson WH, Lowe L, Carpenter DO, et al. Assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment: The challenge ahead. Carcinogenesis 2015;36(Suppl 1):S254–S296; doi: 10.1093/carcin/bgv039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haw TJ, Starkey MR, Pavlidis S, et al. Toll-like receptor 2 and 4 have opposing roles in the pathogenesis of cigarette smoke-induced chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 2018;314(2):L298–L317; doi: 10.1152/ajplung.00154.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Chen Y, Hou C, et al. Cigarette smoke extract changes expression of endothelial nitric oxide synthase (ENOS) and P16(INK4a) and is related to endothelial progenitor cell dysfunction. Med Sci Monit Int Med J Exp Clin Res 2017;23:3224–3231; doi: 10.12659/msm.902746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvathova M, Jahnova E, Szabova M, et al. The relationship between cell surface markers, cytokines, ageing, and cigarette smoking. Bratisl Lek Listy 2009;110(7):394–400. [PubMed] [Google Scholar]
- Hybertson BM, Gao B, Bose SK, et al. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol Aspects Med 2011;32(4–6):234–246; doi: 10.1016/j.mam.2011.10.006 [DOI] [PubMed] [Google Scholar]
- Ishii Y. Smoking and respiratory diseases [in Japanese]. Nihon Rinsho Jpn J Clin Med 2013;71(3):416–420. [PubMed] [Google Scholar]
- Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 2005;352(19):1967–1976; doi: 10.1056/NEJMoa041892 [DOI] [PubMed] [Google Scholar]
- Jamal A, King BA, Neff LJ, et al. Current cigarette smoking among adults—United States, 2005–2015. MMWR Morb Mortal Wkly Rep 2016;65(44):1205–1211; doi: 10.15585/mmwr.mm6544a2 [DOI] [PubMed] [Google Scholar]
- Jamaluddin MDS, Chen I, Yang F, et al. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood 2007;110(10):3648–3655; doi: 10.1182/blood-2007-06-096701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers I. Cigarette smoke effects on innate immune mechanisms in the nasal mucosa. potential effects on the microbiome. Ann Am Thorac Soc 2014;11(Suppl 1):S38–S42; doi: 10.1513/AnnalsATS.201306-154MG [DOI] [PubMed] [Google Scholar]
- Johnson C, Drummer Iv C, Shan H, et al. A novel subset of CD95+ pro-inflammatory macrophages overcome miR155 deficiency and may serve as a switch from metabolically healthy obesity to metabolically unhealthy obesity. Front Immunol 2020;11:619951; doi: 10.3389/fimmu.2020.619951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc Diabetol 2018;17(1):121; doi: 10.1186/s12933-018-0763-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke X, Wang J, Li L, et al. Roles of CD4+CD25(High) FOXP3+ Tregs in lymphomas and tumors are complex. Front Biosci 2008;13:3986–4001; doi: 10.2741/2986 [DOI] [PubMed] [Google Scholar]
- Kearley J, Silver JS, Sanden C, et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 2015;42(3):566–579; doi: 10.1016/j.immuni.2015.02.011 [DOI] [PubMed] [Google Scholar]
- Khanna A, Guo M, Mehra M, et al. Inflammation and oxidative stress induced by cigarette smoke in lewis rat brains. J Neuroimmunol 2013;254(1–2):69–75; doi: 10.1016/j.jneuroim.2012.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Han C-H, Lee M-Y. NADPH oxidase and the cardiovascular toxicity associated with smoking. Toxicol Res 2014;30(3):149–157; doi: 10.5487/TR.2014.30.3.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Huang Y, Lee J, et al. Cellular transformation by cigarette smoke extract involves alteration of glycolysis and mitochondrial function in esophageal epithelial cells. Int J Cancer 2010;127(2):269–281; doi: 10.1002/ijc.25057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopik VS, Maccani MA, Francazio S, et al. The epigenetics of maternal cigarette smoking during pregnancy and effects on child development. Dev Psychopathol 2012;24(4):1377–1390; doi: 10.1017/S0954579412000776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohut SJ. Interactions between nicotine and drugs of abuse: A review of preclinical findings. Am J Drug Alcohol Abuse 2017;43(2):155–170; doi: 10.1080/00952990.2016.1209513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska M, Piekut T, Prendecki M, et al. Mitochondrial and nuclear DNA oxidative damage in physiological and pathological aging. DNA Cell Biol 2020;39(8):1410–1420; doi: 10.1089/dna.2019.5347 [DOI] [PubMed] [Google Scholar]
- Krause BM, Bauer B, Neudörfl J-M, et al. ItaCORMs: Conjugation with a CO-releasing unit greatly enhances the anti-inflammatory activity of itaconates. RSC Med Chem 2021;12(12):2053–2059; doi: 10.1039/d1md00163a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss-Etschmann S, Meyer KF, Dehmel S, et al. Inter- and transgenerational epigenetic inheritance: Evidence in asthma and COPD? Clin Epigenetics 2015;7(1):53; doi: 10.1186/s13148-015-0085-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galassi C, Zitvogel L, et al. Immunogenic cell stress and death. Nat Immunol 2022;23(4):487–500; doi: 10.1038/s41590-022-01132-2 [DOI] [PubMed] [Google Scholar]
- Künzi L, Holt GE. Cigarette smoke activates the parthanatos pathway of cell death in human bronchial epithelial cells. Cell Death Discov 2019;5:127; doi: 10.1038/s41420-019-0205-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwesi-Maliepaard EM, Jacobs H, van Leeuwen F. Signals for antigen-independent differentiation of Memory CD8+ T cells. Cell Mol Life Sci 2021;78(19–20):6395–6408; doi: 10.1007/s00018-021-03912-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y-M, Park JH, Kim H, et al. Different susceptibility of increased DNMT1 expression by exposure to tobacco smoke according to histology in primary non-small cell lung cancer. J Cancer Res Clin Oncol 2007;133(4):219–226; doi: 10.1007/s00432-006-0160-2 [DOI] [PubMed] [Google Scholar]
- Lai B, Wang J, Fagenson A, et al. Twenty novel disease group-specific and 12 new shared macrophage pathways in eight groups of 34 diseases including 24 inflammatory organ diseases and 10 types of tumors. Front Immunol 2019;10:2612; doi: 10.3389/fimmu.2019.02612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampropoulou V, Sergushichev A, Bambouskova M, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 2016;24(1):158–166; doi: 10.1016/j.cmet.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-G, Cho M-Z, Choi J-M. Bystander CD4+ T cells: Crossroads between innate and adaptive immunity. Exp Mol Med 2020;52(8):1255–1263; doi: 10.1038/s12276-020-00486-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Fang P, Li Y, et al. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler Thromb Vasc Biol 2016a;36(6):1090–1100; doi: 10.1161/ATVBAHA.115.306964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Fang P, Mai J, et al. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol OncolJ Hematol Oncol 2013;6:19; doi: 10.1186/1756-8722-6-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Fang P, Sun Y, et al. Anti-inflammatory cytokines IL-35 and IL-10 block atherogenic lysophosphatidylcholine-induced, mitochondrial ROS-mediated innate immune activation, but spare innate immune memory signature in endothelial cells. Redox Biol 2020;28:101373; doi: 10.1016/j.redox.2019.101373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Mai J, Virtue A, et al. IL-35 is a novel responsive anti-inflammatory cytokine—A new system of categorizing anti-inflammatory cytokines. PLoS One 2012;7(3):e33628; doi: 10.1371/journal.pone.0033628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Shao Y, Sha X, et al. IL-35 (interleukin-35) suppresses endothelial cell activation by inhibiting mitochondrial reactive oxygen species-mediated site-specific acetylation of H3K14 (histone 3 lysine 14). Arterioscler Thromb Vasc Biol 2018;38(3):599–609; doi: 10.1161/ATVBAHA.117.310626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y-F, Huang X, Li X, et al. Caspase-1 mediates hyperlipidemia-weakened progenitor cell vessel repair. Front Biosci Landmark Ed 2016b;21(1):178–191; doi: 10.2741/4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y-F, Nanayakkara G, Sun Y, et al. Analyses of caspase-1-regulated transcriptomes in various tissues lead to identification of novel IL-1β-, IL-18- and Sirtuin-1-independent pathways. J Hematol Oncol 2017;10(1):40; doi: 10.1186/s13045-017-0406-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Saredy J, Zhang R, et al. Approaching inflammation paradoxes-proinflammatory cytokine blockages induce inflammatory regulators. Front Immunol 2020;11:554301; doi: 10.3389/fimmu.2020.554301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wu N, Xu K, et al. Organelle crosstalk regulators are regulated in diseases, tumors, and regulatory T cells: Novel classification of organelle crosstalk regulators. Front Cardiovasc Med 2021;8:713170; doi: 10.3389/fcvm.2021.713170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pastrana J, Ferrer LM, Li Y-F, et al. Inhibition of caspase-1 activation in endothelial cells improves angiogenesis: A novel therapeutic potential for ischemia. J Biol Chem 2015;290(28):17485–17494; doi: 10.1074/jbc.M115.641191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Nanayakkara G, Sun Y, et al. Procaspase-1 patrolled to the nucleus of proatherogenic lipid LPC-activated human aortic endothelial cells induces ROS promoter CYP1B1 and strong inflammation. Redox Biol 2021;47:102142; doi: 10.1016/j.redox.2021.102142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Sun Y, Drummer C, et al. Increased acetylation of H3K14 in the genomic regions that encode trained immunity enzymes in lysophosphatidylcholine-activated human aortic endothelial cells—Novel qualification markers for chronic disease risk factors and conditional DAMPs. Redox Biol 2019;24:101221; doi: 10.1016/j.redox.2019.101221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Sun Y, Xu K, et al. Aorta in pathologies may function as an immune organ by upregulating secretomes for immune and vascular cell activation, differentiation and trans-differentiation-early secretomes may serve as drivers for trained immunity. Front Immunol 2022;13:858256; doi: 10.3389/fimmu.2022.858256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugg ST, Scott A, Parekh D, et al. Cigarette smoke exposure and alveolar macrophages: Mechanisms for lung disease. Thorax 2022;77(1):94–101; doi: 10.1136/thoraxjnl-2020-216296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccani MA, Knopik VS. Cigarette smoke exposure-associated alterations to non-coding RNA. Front Genet 2012;3:53; doi: 10.3389/fgene.2012.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai J, Virtue A, Shen J, et al. An evolving new paradigm: endothelial cells—Conditional innate immune cells. J Hematol Oncol 2013;6:61; doi: 10.1186/1756-8722-6-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Li D, Zhou E, et al. Nicotine exacerbates atherosclerosis through a macrophage-mediated endothelial injury pathway. Aging 2021;13(5):7627–7643; doi: 10.18632/aging.202660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EM, Clapp PW, Rebuli ME, et al. E-cigarette use results in suppression of immune and inflammatory-response genes in nasal epithelial cells similar to cigarette smoke. Am J Physiol Lung Cell Mol Physiol 2016;311(1):L135–L144; doi: 10.1152/ajplung.00170.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masso-Silva JA, Moshensky A, Shin J, et al. Chronic e-cigarette aerosol inhalation alters the immune state of the lungs and increases ACE2 expression, raising concern for altered response and susceptibility to SARS-CoV-2. Front Physiol 2021;12:649604; doi: 10.3389/fphys.2021.649604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock TS, Khan N, Alimova Y, et al. Encoding the odor of cigarette smoke. J Neurosci 2020;40(37):7043–7053; doi: 10.1523/JNEUROSCI.1144-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel T, Vanhoutte PM. Cellular signaling and NO production. Pflugers Arch 2010;459(6):807–816; doi: 10.1007/s00424-009-0765-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moheimani RS, Bhetraratana M, Yin F, et al. Increased cardiac sympathetic activity and oxidative stress in habitual electronic cigarette users: Implications for cardiovascular risk. JAMA Cardiol 2017;2(3):278–284; doi: 10.1001/jamacardio.2016.5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muri J, Kopf M. Redox regulation of immunometabolism. Nat Rev Immunol 2021;21(6):363–381; doi: 10.1038/s41577-020-00478-8 [DOI] [PubMed] [Google Scholar]
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. Reports of the Surgeon General. Centers for Disease Control and Prevention (US): Atlanta, GA; 2014. [PubMed] [Google Scholar]
- Netea MG, Domínguez-Andrés J, Barreiro LB, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol 2020;20(6):375–388; doi: 10.1038/s41577-020-0285-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Joosten LAB, Latz E, et al. Trained immunity: A program of innate immune memory in health and disease. Science 2016;352(6284):aaf1098; doi: 10.1126/science.aaf1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Schlitzer A, Placek K, et al. Innate and adaptive immune memory: An evolutionary continuum in the host's response to pathogens. Cell Host Microbe 2019;25(1):13–26; doi: 10.1016/j.chom.2018.12.006 [DOI] [PubMed] [Google Scholar]
- Ni D, Tang T, Lu Y, et al. Canonical secretomes, innate immune caspase-1-, 4/11-gasdermin D non-canonical secretomes and exosomes may contribute to maintain Treg-Ness for Treg immunosuppression, tissue repair and modulate anti-tumor immunity via ROS pathways. Front Immunol 2021;12:678201; doi: 10.3389/fimmu.2021.678201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LAJ, Artyomov MN. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol 2019;19(5):273–281; doi: 10.1038/s41577-019-0128-5 [DOI] [PubMed] [Google Scholar]
- Owen AM, Fults JB, Patil NK, et al. TLR agonists as mediators of trained immunity: mechanistic insight and immunotherapeutic potential to combat infection. Front Immunol 2020;11:622614; doi: 10.3389/fimmu.2020.622614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-M, Jeong H, Seo Y-S, et al. Cigarette smoke extract produces superoxide in aqueous media by reacting with bicarbonate. Toxics 2021;9(11):316; doi: 10.3390/toxics9110316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels NS, Bracke KR, Dupont LL, et al. Role of IL-1α and the Nlrp3/Caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J 2011;38(5):1019–1028; doi: 10.1183/09031936.00158110 [DOI] [PubMed] [Google Scholar]
- Pryor WA. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ Health Perspect 1997;105(Suppl 4):875–882; doi: 10.1289/ehp.97105s4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ras RT, Streppel MT, Draijer R, et al. Flow-mediated dilation and cardiovascular risk prediction: A systematic review with meta-analysis. Int J Cardiol 2013;168(1):344–351; doi: 10.1016/j.ijcard.2012.09.047 [DOI] [PubMed] [Google Scholar]
- Rogers TJ. Bidirectional regulation of opioid and chemokine function. Front Immunol 2020;11:94; doi: 10.3389/fimmu.2020.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama SA, Arab HH, Omar HA, et al. Nicotine mediates hypochlorous acid-induced nuclear protein damage in mammalian cells. Inflammation 2014;37(3):785–792; doi: 10.1007/s10753-013-9797-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samir P, Malireddi RKS, Kanneganti T-D. The PANoptosome: A deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol 2020;10:238; doi: 10.3389/fcimb.2020.00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampilvanjil A, Karasawa T, Yamada N, et al. Cigarette smoke extract induces ferroptosis in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 2020;318(3):H508–H518; doi: 10.1152/ajpheart.00559.2019 [DOI] [PubMed] [Google Scholar]
- Schnack L, Sohrabi Y, Lagache SMM, et al. Mechanisms of trained innate immunity in OxLDL primed human coronary smooth muscle cells. Front Immunol 2019;10:13; doi: 10.3389/fimmu.2019.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A, Lugg ST, Aldridge K, et al. Pro-inflammatory effects of e-cigarette vapour condensate on human alveolar macrophages. Thorax 2018;73(12):1161–1169; doi: 10.1136/thoraxjnl-2018-211663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Bardenys G, Peiró S. Enzymatic lysine oxidation as a posttranslational modification. FEBS J 2021. [Epub ahead of print]; doi: 10.1111/febs.16205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyda M, Elkhal A, Quante M, et al. T cells going innate. Trends Immunol 2016;37(8):546–556; doi: 10.1016/j.it.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Cheng Z, Li X, et al. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction—A novel mechanism for maintaining vascular function. J Hematol Oncol 2014;7:80; doi: 10.1186/s13045-014-0080-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Chernaya V, Johnson C, et al. Metabolic diseases downregulate the majority of histone modification enzymes, making a few upregulated enzymes novel therapeutic targets—“Sand Out and Gold Stays”. J Cardiovasc Transl Res 2016;9(1):49–66; doi: 10.1007/s12265-015-9664-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Cornwell W, Xu K, et al. Chronic exposure to the combination of cigarette smoke and morphine decreases CD4+ regulatory T cell numbers by reprogramming the Treg cell transcriptome. Front Immunol 2022;13:887681; doi: 10.3389/fimmu.2022.887681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Saredy J, Xu K, et al. Endothelial immunity trained by coronavirus infections, DAMP stimulations and regulated by anti-oxidant NRF2 may contribute to inflammations, myelopoiesis, COVID-19 cytokine storms and thromboembolism. Front Immunol 2021a;12:653110; doi: 10.3389/fimmu.2021.653110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Saredy J, Yang WY, et al. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol 2020;40(6):e138–e152; doi: 10.1161/ATVBAHA.120.314330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Yang WY, Saaoud F, et al. IL-35 promotes CD4+Foxp3+ Tregs and inhibits atherosclerosis via maintaining CCR5-amplified Treg-suppressive mechanisms. JCI Insight 2021b;6(19):e152511; doi: 10.1172/jci.insight.152511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Wu N, Nanayakkara G, et al. Co-signaling receptors regulate T-cell plasticity and immune tolerance. Front Biosci Landmark Ed 2019;24(1):96–132; doi: 10.2741/4710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Rattan V, Sultana C, et al. Cigarette smoke condensate-induced adhesion molecule expression and transendothelial migration of monocytes. Am J Physiol 1996;270(5 Pt 2):H1624–H1633; doi: 10.1152/ajpheart.1996.270.5.H1624 [DOI] [PubMed] [Google Scholar]
- Shenker NS, Ueland PM, Polidoro S, et al. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiol Camb Mass 2013;24(5):712–716; doi: 10.1097/EDE.0b013e31829d5cb3 [DOI] [PubMed] [Google Scholar]
- Shinton R, Beevers G. Meta-analysis of relation between cigarette smoking and stroke. BMJ 1989;298(6676):789–794; doi: 10.1136/bmj.298.6676.789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siedlinski M, Klanderman B, Sandhaus RA, et al. Association of cigarette smoking and CRP levels with DNA methylation in α-1 antitrypsin deficiency. Epigenetics 2012;7(7):720–728; doi: 10.4161/epi.20319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stäger S, Kaye PM. CD8+ T-cell priming regulated by cytokines of the innate immune system. Trends Mol Med 2004;10(8):366–371; doi: 10.1016/j.molmed.2004.06.003 [DOI] [PubMed] [Google Scholar]
- Stämpfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol 2009;9(5):377–384; doi: 10.1038/nri2530 [DOI] [PubMed] [Google Scholar]
- Su Y, Han W, Giraldo C, et al. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol 1998;19(5):819–825; doi: 10.1165/ajrcmb.19.5.3091 [DOI] [PubMed] [Google Scholar]
- Sun Y, Lu Y, Saredy J, et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol 2020;37:101696; doi: 10.1016/j.redox.2020.101696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar IK, Nevid MZ, Friedman AE, et al. Cigarette smoke induces distinct histone modifications in lung cells: Implications for the pathogenesis of COPD and lung cancer. J Proteome Res 2014;13(2):982–996; doi: 10.1021/pr400998n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter M, Ma J, Harris A, et al. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics 2011;6(11):1284–1294; doi: 10.4161/epi.6.11.17819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhout R, Schulz T, Florek E, et al. Hazardous compounds in tobacco smoke. Int J Environ Res Public Health 2011;8(2):613–628; doi: 10.3390/ijerph8020613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res 2019;29(5):347–364; doi: 10.1038/s41422-019-0164-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teasdale JE, Hazell G, Newby AC, et al. Paradoxical effects of cigarette smoke extract and high laminar flow on tumour necrosis factor-alpha induced VCAM-1 up-regulation—Implications for endothelial dysfunction. Atherosclerosis 2014;237(2):e13–e14; doi: 10.1016/j.atherosclerosis.2014.10.074 [DOI] [Google Scholar]
- Timilshina M, You Z, Lacher SM, et al. Activation of mevalonate pathway via LKB1 is essential for stability of Treg cells. Cell Rep 2019;27(10):2948–2961.e7; doi: 10.1016/j.celrep.2019.05.020 [DOI] [PubMed] [Google Scholar]
- Titz B, Szostak J, Sewer A, et al. Multi-omics systems toxicology study of mouse lung assessing the effects of aerosols from two heat-not-burn tobacco products and cigarette smoke. Comput Struct Biotechnol J 2020;18:1056–1073; doi: 10.1016/j.csbj.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiathan R, Miguez MJ, Patel B, et al. Tobacco smoking increases immune activation and impairs T-cell function in HIV infected patients on antiretrovirals: A cross-sectional pilot study. PLoS One 2014;9(5):e97698; doi: 10.1371/journal.pone.0097698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden CDCC, Noz MP, Joosten LAB, et al. Epigenetics and trained immunity. Antioxid Redox Signal 2018;29(11):1023–1040; doi: 10.1089/ars.2017.7310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR, Anggård EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med 1990;323(1):27–36; doi: 10.1056/NEJM199007053230106 [DOI] [PubMed] [Google Scholar]
- Virtue A, Johnson C, Lopez-Pastraña J, et al. MicroRNA-155 deficiency leads to decreased atherosclerosis, increased white adipose tissue obesity, and non-alcoholic fatty liver disease: A novel mouse model of obesity paradox. J Biol Chem 2017;292(4):1267–1287; doi: 10.1074/jbc.M116.739839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss K, Hong HS, Bader JE, et al. A guide to interrogating immunometabolism. Nat Rev Immunol 2021;21(10):637–652; doi: 10.1038/s41577-021-00529-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan ES, Qiu W, Baccarelli A, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet 2012;21(13):3073–3082; doi: 10.1093/hmg/dds135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lai B, Nanayakkara G, et al. Experimental data-mining analyses reveal new roles of low-intensity ultrasound in differentiating cell death regulatome in cancer and non-cancer cells via potential modulation of chromatin long-range interactions. Front Oncol 2019;9:600; doi: 10.3389/fonc.2019.00600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Fu H, Nanayakkara G, et al. Novel extracellular and nuclear caspase-1 and inflammasomes propagate inflammation and regulate gene expression: A comprehensive database mining study. J Hematol Oncol 2016a;9(1):122; doi: 10.1186/s13045-016-0351-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y-F, Nanayakkara G, et al. Lysophospholipid receptors, as novel conditional danger receptors and homeostatic receptors modulate inflammation-novel paradigm and therapeutic potential. J Cardiovasc Transl Res 2016b;9(4):343–359; doi: 10.1007/s12265-016-9700-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren GW, Alberg AJ, Kraft AS, et al. The 2014 Surgeon General's Report: “The Health Consequences of Smoking—50 Years of Progress”: A paradigm shift in cancer care. Cancer 2014;120(13):1914–1916; doi: 10.1002/cncr.28695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauters E, Janssens W, Vansteenkiste J, et al. DNA methylation profiling of non-small cell lung cancer reveals a COPD-driven immune-related signature. Thorax 2015;70(12):1113–1122; doi: 10.1136/thoraxjnl-2015-207288 [DOI] [PubMed] [Google Scholar]
- Witschi H. A/J mouse as a model for lung tumorigenesis caused by tobacco smoke: Strengths and weaknesses. Exp Lung Res 2005;31(1):3–18; doi: 10.1080/01902140490494959 [DOI] [PubMed] [Google Scholar]
- Wood AM, Stockley RA. Alpha one antitrypsin deficiency: From gene to treatment. Respiration 2007;74(5):481–492; doi: 10.1159/000105536 [DOI] [PubMed] [Google Scholar]
- Wu X, Zhang H, Qi W, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis 2018;9(2):171; doi: 10.1038/s41419-017-0257-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z, Yan Y, Song J, et al. Expression of TCTP antisense in CD25(High) regulatory T cells aggravates cuff-injured vascular inflammation. Atherosclerosis 2009;203(2):401–408; doi: 10.1016/j.atherosclerosis.2008.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Saaoud F, Yu S, et al. Monocyte adhesion assays for detecting endothelial cell activation in vascular inflammation and atherosclerosis. Methods Mol Biol 2022;2419:169–182; doi: 10.1007/978-1-0716-1924-7_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Shao Y, Saaoud F, et al. Novel knowledge-based transcriptomic profiling of lipid lysophosphatidylinositol-induced endothelial cell activation. Front Cardiovasc Med 2021;8:773473; doi: 10.3389/fcvm.2021.773473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Yang WY, Nanayakkara GK, et al. GATA3, HDAC6, and BCL6 regulate FOXP3+ Treg plasticity and determine Treg conversion into either novel antigen-presenting cell-like Treg or Th1-Treg. Front Immunol 2018;9:45; doi: 10.3389/fimmu.2018.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Xiong Z, Zhang S, et al. CD25high T cells with a prolonged survival inhibit development of diabetes. Int J Immunopathol Pharmacol 2008;21(4):767–780; doi: 10.1177/039463200802100401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Han Z, Oppenheim JJ. Alarmins and immunity. Immunol Rev 2017a;280(1):41–56; doi: 10.1111/imr.12577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Chen IH, Xiong Z, et al. Model of stimulation-responsive splicing and strategies in identification of immunogenic isoforms of tumor antigens and autoantigens. Clin Immunol 2006;121(2):121–133; doi: 10.1016/j.clim.2006.06.007 [DOI] [PubMed] [Google Scholar]
- Yang F, Yang X-F. New concepts in tumor antigens: Their significance in future immunotherapies for tumors. Cell Mol Immunol 2005;2(5):331–341 [PubMed] [Google Scholar]
- Yang Q, Nanayakkara GK, Drummer C, et al. Low-intensity ultrasound-induced anti-inflammatory effects are mediated by several new mechanisms including gene induction, immunosuppressor cell promotion, and enhancement of exosome biogenesis and docking. Front Physiol 2017b;8:818; doi: 10.3389/fphys.2017.00818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Zhang R, Tang P, et al. Ultrasound may suppress tumor growth, inhibit inflammation, and establish tolerogenesis by remodeling innatome via pathways of ROS, immune checkpoints, cytokines, and trained immunity/tolerance. J Immunol Res 2021;2021:6664453; doi: 10.1155/2021/6664453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WY, Shao Y, Lopez-Pastrana J, et al. Pathological conditions re-shape physiological Tregs into pathological Tregs. Burns Trauma 2015;3(1):1; doi: 10.1186/s41038-015-0001-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XF, Weber GF, Cantor H. A novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity 1997;7(5):629–639; doi: 10.1016/s1074-7613(00)80384-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X-F, Ye Q, Press B, et al. Analysis of the complex genomic structure of Bcl-x and its relationship to Bcl-x(Gamma) expression after CD28-dependent costimulation. Mol Immunol 2002;39(1–2):45–55; doi: 10.1016/s0161-5890(02)00049-4 [DOI] [PubMed] [Google Scholar]
- Yang X-F, Yin Y, Wang H. Vascular inflammation and atherogenesis are activated via receptors for PAMPs and suppressed by regulatory T cells. Drug Discov Today 2008;5(2):125–142; doi: 10.1016/j.ddstr.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Peng N, Chen G, et al. Interaction between smoking and diabetes in relation to subsequent risk of cardiovascular events. Cardiovasc Diabetol 2022;21(1):14; doi: 10.1186/s12933-022-01447-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Xiong Z, Zhang S, et al. Bcl-XL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem J 2005;392(Pt 1):135–143; doi: 10.1042/BJ20050698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, O'Neill LAJ. The role of the electron transport chain in immunity. FASEB J 2021;35(12):e21974; doi: 10.1096/fj.202101161R [DOI] [PubMed] [Google Scholar]
- Yin Y, Li X, Sha X, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol 2015;35(4):804–816; doi: 10.1161/ATVBAHA.115.305282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Pastrana JL, Li X, et al. Inflammasomes: Sensors of metabolic stresses for vascular inflammation. Front Biosci Landmark Ed 2013;18(2):638–649; doi: 10.2741/4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama S, Chen Z, Okagaki T, et al. Nicotine exposure alters human vascular smooth muscle cell phenotype from a contractile to a synthetic type. Atherosclerosis 2014;237(2):464–470; doi: 10.1016/j.atherosclerosis.2014.10.019 [DOI] [PubMed] [Google Scholar]
- Yu J, Qiu Y, Yang J, et al. DNMT1-PPARγ pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci Rep 2016;6:30053; doi: 10.1038/srep30053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilinger S, Kühnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One 2013;8(5):e63812; doi: 10.1371/journal.pone.0063812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Nanayakkara GK, Shao Y, et al. DNA checkpoint and repair factors are nuclear sensors for intracellular organelle stresses-inflammations and cancers can have high genomic risks. Front Physiol 2018;9:516; doi: 10.3389/fphys.2018.00516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Saredy J, Shao Y, et al. End-stage renal disease is different from chronic kidney disease in upregulating ROS-modulated proinflammatory secretome in PBMCs—A novel multiple-hit model for disease progression. Redox Biol 2020a;34:101460; doi: 10.1016/j.redox.2020.101460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Xu K, Shao Y, et al. Tissue Treg secretomes and transcription factors shared with stem cells contribute to a treg niche to maintain Treg-Ness with 80% innate immune pathways, and functions of immunosuppression and tissue repair. Front Immunol 2020b;11:632239; doi: 10.3389/fimmu.2020.632239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Liang Y, Li L, et al. Succinate: A novel mediator to promote atherosclerotic lesion progression. DNA Cell Biol 2022;41(3):285–291; doi: 10.1089/dna.2021.0345 [DOI] [PubMed] [Google Scholar]
- Zhong C, Yang X, Feng Y, et al. Trained immunity: An underlying driver of inflammatory atherosclerosis. Front Immunol 2020;11:284; doi: 10.3389/fimmu.2020.00284 [DOI] [PMC free article] [PubMed] [Google Scholar]