

Abstract
Heterozygous germline mutations in PTEN gene predispose to hamartomas and tumors in different tissues, as well as to neurodevelopmental disorders, and define at genetic level the PTEN Hamartoma Tumor Syndrome (PHTS). The major physiologic role of PTEN protein is the dephosphorylation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), counteracting the pro-oncogenic function of phosphatidylinositol 3-kinase (PI3K), and PTEN mutations in PHTS patients frequently abrogate PTEN PIP3 catalytic activity. PTEN also displays non-canonical PIP3-independent functions, but their involvement in PHTS pathogeny is less understood. We have previously identified and described, at clinical and genetic level, novel PTEN variants of unknown functional significance in PHTS patients. Here, we have performed an extensive functional characterization of these PTEN variants (c.77 C > T, p.(Thr26Ile), T26I; c.284 C > G, p.(Pro95Arg), P95R; c.529 T > A, p.(Tyr177Asn), Y177N; c.781 C > G, p.(Gln261Glu), Q261E; c.829 A > G, p.(Thr277Ala), T277A; and c.929 A > G, p.(Asp310Gly), D310G), including cell expression levels and protein stability, PIP3-phosphatase activity, and subcellular localization. In addition, caspase-3 cleavage analysis in cells has been assessed using a C2-domain caspase-3 cleavage-specific anti-PTEN antibody. We have found complex patterns of functional activity on PTEN variants, ranging from loss of PIP3-phosphatase activity, diminished protein expression and stability, and altered nuclear/cytoplasmic localization, to intact functional properties, when compared with PTEN wild type. Furthermore, we have found that PTEN cleavage at the C2-domain by the pro-apoptotic protease caspase-3 is diminished in specific PTEN PHTS variants. Our findings illustrate the multifaceted molecular features of pathogenic PTEN protein variants, which could account for the complexity in the genotype/phenotype manifestations of PHTS patients.
Subject terms: Molecular biology, Cancer
Introduction
Heterozygous germline mutations in the tumor suppressor gene PTEN (MIM# 601728) define at genetic level the PTEN Hamartoma Tumor Syndrome (PHTS), a complex group of diseases which display highly heterogeneous phenotypes in carrier patients, from severe malformations and high cancer risk to mild neurodevelopmental manifestations. Major syndromes within PHTS include Cowden Syndrome (CS), Bannayan-Riley-Ruvalcaba Syndrome (BRRS), Lhermitte-Duclos Disease (LDD), and Proteus-like Syndrome (MIM# 158350), as well as PTEN autism spectrum disorder (PTEN-ASD) (MIM#605309) [1]. In addition, mutations or absence of the PTEN gene, as well as decrease in PTEN protein expression, are frequent in sporadic tumors from a variety of cancers [2, 3].
PTEN is a unique gene encoding a major PTEN protein of 403 amino acids with phosphatase activity, which counteracts the pro-oncogenic action of class I phosphatidylinositol 3-kinases (PI3K) by converting phosphatidylinositol 3,4,5-trisphosphate (PIP3) in phosphatidylinositol 4,5-bisphosphate (PIP2) [4, 5]. In addition to its PIP3-phosphatase canonical function, PTEN also exerts non-canonical functions as a protein phosphatase and as a catalytically-independent cell homeostasis regulator [6]. A major aspect of PTEN function regulation involves the dynamic changes on its subcellular localization under variable cell conditions, with an active and tightly regulated shuttling between different cellular compartments [7]. In particular, the entry and accumulation of PTEN in the nucleus affects its accessibility to PIP3 substrate at the cell membranes, and makes PTEN competent to perform nuclear phosphatase-independent activities that regulate essential processes including chromosome stability, DNA repair, and gene transcription, among others [8].
Multiple layers of regulation of PTEN biological activity exist, from transcriptional and post-transcriptional regulation of PTEN gene expression to regulation of PTEN translation and reversible post-translational modifications [4, 9, 10]. PTEN is non-reversibly cleaved by caspase-3 during apoptosis. This cleavage targets the PTEN C-terminal tail in a PTEN-phosphorylation-dependent manner, as well as the Asp301 residue at the PTEN C2-domain independently of C-tail phosphorylation [11]. PTEN cleavage regulates PTEN protein half-life and the opening of PTEN to its active conformation competent to bind to membranes [12]. In addition, PTEN caspase cleavage in response to chemotherapy it has been proposed as a potential therapy-resistance mechanism [13]. PTEN intrinsic protein stability is highly dependent on intramolecular interactions in the PTP/C2 domain interface [14], and it is further regulated during cell signaling by phosphorylation, ubiquitination, and protein-interaction events [15–17]. Due to this multi-layered regulation of PTEN protein functions, PTEN missense mutations associated to disease not only target PTEN catalytic residues but also affect residues essential for protein stability, subcellular localization, and protein-protein interactions. This makes difficult, in many cases, the prediction of the functional properties of the PTEN variants present in patients, making necessary to perform dedicated, mutation-specific functional studies.
Using mammalian and yeast cell models as high-throughput platforms, several global analyses of function and protein stability of PTEN variants have been performed, which provide an invaluable guide for experimental research and for the clinics [18, 19]. In addition, dedicated studies on selected panels of mutations associated to disease have unveiled that germline PTEN variants associated to ASD display, in general, higher levels of PIP3-phosphatase activity in cells than PTEN variants associated to tumor syndromes. Alterations in nuclear/cytoplasmic localization, as well as diminished protein stability, have also been found frequently in ASD-associated PTEN variants [20–22]. A multi-model analysis of functional properties of PTEN variants also highlighted the recurrence of decreased PTEN protein stability, although distinct molecular mechanisms of PTEN dysfunction could not be related to distinct pathogenic phenotypes [23]. Other experimental cell and animal models, as well as in silico studies, have been reported that complement the available resources on PTEN mutation-function relationship [24–26]. However, in many cases it is still necessary to analyse individually the functional properties of the distinct PTEN variants found in patients if precision PHTS therapies are aimed to be applied in the next future [27, 28]. We have previously reported the clinical characteristics of a large group of Spanish PHTS patients, including carriers of PTEN mutations encoding novel PTEN variants of unknown functional significance [29]. Here, we present the functional characterization of these PTEN variants using a panel of functional, biochemical, and immunochemical assays. Our results illustrate the complexity of the functional phenotypes associated to some PTEN variants, including the lack of apparent functional alterations using in vitro experimental settings. In addition, we demonstrate for the first time variant-specific alterations in PTEN cleavage at the C2-domain unstructured loop by the pro-apoptotic protease caspase-3.
Materials and methods
Cells and transfections
Simian kidney COS-7 cells, human breast carcinoma MCF-7 cells, and HEK293 human kidney carcinoma were grown at 37 °C, 5% CO2 in DMEM containing high glucose supplemented with 5% (COS-7) or 10% (MCF-7, HEK293) heat-inactivated fetal bovine serum (FBS), 1 mM L-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Human U87MG glioblastoma cells were grown in DMEM containing high glucose supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1 mM L-glutamine, 1 mM sodium pyruvate, 1% non-essential amino acids, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. The Saccharomyces cerevisiae strain YPH499 (MATa ade2-101 trp1-63 leu2-1 ura3-52 his3-Δ200 lys2-801) was used for heterologous expression of mammalian proteins. YPH499 yeast cells were grown in synthetic complete (SC) medium, containing 0.17% yeast nitrogen base without amino acids, 0.5% ammonium sulfate supplemented with appropriate amino acids and nucleic acid bases, and added 2% glucose (SD), galactose (SG) or raffinose (SR), as required. COS-7 cells were transfected using GenJet reagent (SignaGen, USA) according to the manufacturer instructions, and processed for analysis after 48 h.
Plasmids, mutagenesis, and variant information
The pRK5 PTEN, pYES2 PTEN, pRK5 GST-PTEN, YCpLG myc-p110α-CAAX, and pSG5 AKT1 plasmids have been described [20, 30–32]. The PTEN and GST-PTEN amino acid substitution and truncation variants were made by PCR oligonucleotide site-directed mutagenesis as described [33], and mutations were confirmed by DNA sequencing. Nucleotide and amino acid numbering for PTEN variants correspond to reference sequences from accession numbers NM_000314 and NP_000305, respectively. Nomenclature of variants is according to HGVS. Variants and phenotypes data have been deposited in the LOVD gene variant database (http://www.lovd.nl/3.0/home), with the following accession IDs: 0000352584 (T26I), 0000878862 (P95R), 0000878883 (Y177N), 0000878885 (Q261E), 0000878886 (T277A), 0000878890 (D310G).
Protein stability and phosphatase activity experiments
Whole cell protein extracts from COS-7 cells overexpressing ectopic PTEN variants were prepared by cell lysis in ice-cold M-PERTM lysis buffer (ThermoFisher Scientific) supplemented with PhosSTOP phosphatase inhibitor and cOmplete protease inhibitor cocktails (Roche, Switzerland), followed by centrifugation at 15200 g for 10 min and collection of the supernatant. Proteins (50–100 μg) were resolved in 10% SDS-PAGE under reducing conditions and transferred to PVDF membranes for immunoblot analysis. For experiments of protein stability, cells were treated with 800 μg/mL cycloheximide (CHX) (Sigma Aldrich) or with 10 μM proteasome inhibitor MG132 (Sigma Aldrich) for 6 h before lysis. Immunoblotting was performed using anti-PTEN 6H2.1 monoclonal antibody (mAb) (Merck Millipore, USA) and anti-GAPDH (Santa Cruz Biotechnology, USA) antibody, followed by IRDye-conjugated anti-rabbit or anti-mouse (LI-COR, USA) antibodies. For experiments of PTEN phosphatase activity in mammalian cells, COS-7 cells were co-transfected with the appropriate PTEN variants and HA-AKT1, and lysates were processed for immunoblot using anti-phospho-Ser473-AKT, anti-AKT (Cell Signaling Technologies, USA), and anti-PTEN 6H2.1 antibodies. For determination of phospho-AKT content and PTEN protein stability, bands were visualized and quantified using an Image studioTM software with Odyssey® CLx Imaging System (LI-COR). For experiments of PTEN phosphatase activity in S. cerevisiae, yeasts were transformed by standard procedures, and drop growth assays and GFP-AKT1 distribution analyses were performed as described [30, 34, 35].
mRNA isolation and RT-qPCR
Real time-quantitative PCR (RT-qPCR) was performed using RNA from COS-7 cells transfected with the plasmids of interest. Total RNA was isolated using the IllustraRNAspin mini purification kit (GE Healthcare Life Sciences). 1 μg of total RNA was used for cDNA synthesis using RevertAidTM reverse transcriptase protocol (ThermoFisher Scientific), oligo (dT)18 primers, and RiboLock and RNase inhibitor (all from Fermentas). qPCR was performed as previously described [36], using Agilent AriaMx Real-Time PCR System (Agilent Technologies) and PTEN validated and reference gene (hypoxanthine phosphoribosyltransferase 1 [HPRT1]) QuantiTect primers (Qiagen). Relative quantification was performed using the comparative ΔΔCt method.
Immunofluorescence and microscopy techniques
PTEN subcellular location in COS-7 cells was determined by standard immunofluorescence as previously described, using anti-PTEN 425 A mAb and fluorescein-conjugated anti-mouse antibody [31, 37]. For quantification of PTEN nuclear/cytoplasmic distribution, at least 50 positive cells were scored for each experiment. Cells were rated as showing nuclear staining (N), cytoplasmic staining (C), or staining within both the nucleus and the cytoplasm (N/C). Representative examples of the distinct subcellular localizations determined in our scoring are provided in this section. Nuclei were identified by Hoechst (Sigma-Aldrich) staining. All pictures were taken under a 20X magnification. Measurement of GFP-AKT1 plasma membrane localization in yeast, as an indirect indicator of cellular PIP3 levels, was performed by fluorescence microscopy, as described [30, 34, 35]. ≥100 cells were examined and scored for each condition or experiment for either cytoplasmic or membrane-associated localization.
Caspase-3 cleavage-site experiments
Whole cell protein extracts from COS-7 cells overexpressing ectopic GST-PTEN or PTEN variants were prepared and resolved by 10% or 12% SDS-PAGE as indicated above. In the case of PTEN overexpression, cells were kept untreated or were treated with 50 ng/ml TNF-α (Sigma Aldrich). Immunoblotting was performed to monitor PTEN cleavage at Asp301, using anti-PTEN SP227 mAb (Sigma Aldrich). Anti-PTEN 6H2.1 mAb (Merck Millipore) and anti-GST antibody [38] were used as controls. IRDye-conjugated anti-rabbit or anti-mouse antibodies (LI-COR) were used as secondary antibodies.
Results
PTEN germline variants of unknown function
Our previous clinical and genetic analysis from a cohort of PHTS patients revealed several cases carrying PTEN germline variants, in some cases not previously described, which were considered of unknown significance [29]. These included the PTEN variants T26I [c.77 C > T, p.(Thr26Ile)], P95R [c.284 C > G, p.(Pro95Arg)], Y177N [c.529 T > A, p.(Tyr177Asn)], Q261E [c.781 C > G, p.(Gln261Glu)], T277A [c.829 A > G, p.(Thr277Ala)], and D310G [c.929 A > G, p.(Asp310Gly)]. Variants Y177N and Q261E have not been annotated, up to date, in sporadic tumor mutation databases (Table 1). Figure 1 illustrates, in the three-dimensional structure of PTEN protein, the localization of the amino acids targeted by mutations causing these variants. As shown, targeted residues are located on both the PTP- and the C2-domain of PTEN. T26I variant targets the N-terminal nuclear localization signal (NLS) of PTEN, and it has been partially characterized by us [20, 31], whereas P95R variant targets the WPD catalytic loop [14]. The rest of variants do not target amino acids manifestly involved in PTEN catalysis or subcellular localization, although residues Y177 and T277 are located at the PTEN PTP/C2 domain interface, which is important to preserve PTEN functional conformation [14]. The phenotype of the carrier patients is heterogeneous, while they have in common macrocephaly as a referred manifestation, and ASD features were also frequent. Of interest, only one patient (D310G carrier) displayed clear cancer manifestations. This is coincident with the finding of D310G mutation in sporadic tumor samples, as annotated in the databases (Table 1; [29]). The D310G variant targets the end of the unstructured loop (residues 286-309) in the C2-domain of PTEN [14] (Fig. 1), whose physiologic function remains unknown. Since determination of the functional properties of the PTEN proteins encoded by these variants may provide insights into their potential pathogenicity, including risk for cancer, a broad PTEN functional and immunochemical analysis was performed on cells ectopically expressing the variants.
Table 1.
Characteristics of PTEN PHTS variants of unknown significance.
| DNA variant1 | Amino acid substitution1 | Reported in databases2 | Phenotype3 | Age / Sex4 | Reference5 | |||
|---|---|---|---|---|---|---|---|---|
| HGMD | ClinVar | COSMIC | cBioPortal | |||||
| c.77 C > T | p.(Thr26Ile)/T26I | (CM151666) | 3 (189399) | 1, GBM (COSM6936497) | 1, GBM |
Macrocephaly Autism Mental retardation |
11 / M | [50] |
| c.284 C > G | p.(Pro95Arg)/P95R | (CM221495) | - | - | - |
Macrocephaly Polyps, hamartomas |
39 / M | [29] |
| c.529 T > A | p.(Tyr177Asn)/Y177N | (CM221496) | 1 (873327) | - | - |
Macrocephaly Motor delay |
5 / M | [29] |
| c.781 C > G | p.(Gln261Glu)/Q261E | (CM221497) | 1 (873325) | - | - |
Macrocephaly Polyps, lipomas Overgrowth |
34 / M | [29] |
| c.829 A > G | p.(Thr277Ala)/T277A | (CM221498) | 1 (571869) |
1, EC 1, t-all (COSM1349598) |
1, UC |
Macrocephaly Developmental disorder Overgrowth |
9 / M | [29] |
| c.929 A > G | p.(Asp310Gly)/D310G | (CM221499) | 3 (566673) |
2, EC 1, EH 1, LC (COSM1968270) |
1, EC 1, Chol |
Macrocephaly Polyps, hamartomas Several cancers |
29 / M | [29] |
1PTEN DNA germline and protein variants are indicated following HGVS recommended nomenclature. Protein variants are also indicated with the single-letter code amino acid nomenclature. Nucleotide and amino acid numbering corresponds to accessions NM_000314.8 and NP_000305.3, respectively.
2HGMD, Human Gene Mutation Database (Professional) (2022.3); ClinVar (NCBI); COSMIC, Catalogue of Somatic Mutations in Cancer (Wellcome Trust Sanger Institute); cBioPortal, cBioportal for Cancer Genomics (MSK Cancer Center). In the case of HGMD, ClinVar, and COSMIC databases, the ID of the variants is indicated in brackets. Numbers indicate the number of cases with the mutation, followed by the tumor type (GBM glioblastoma, EC endometrium carcinoma, t-all T cell acute lymphoblastic leukemia, EH endometrium hyperplasia, LC liver carcinoma, UC uterine carcinosarcoma, Chol cholangiocarcinoma). All analyzed variants are found with relatively low frequency in patients and in tumors. Note that HGMD database only provides 1 submitted case by variant. - no cases annotated in the database.
3See reference [29] for a more detailed phenotype of each PTEN variant carrier patient.
4Age is indicated in years; M male.
5Original references (to the best of our knowledge) for description of germline DNA variants and patients are provided.
Fig. 1. Depiction of PTEN 3D structure (accession ID5R) and localization of the amino acids targeted by mutations which were analysed in this study (Thr26, Pro95, Tyr177, Gln261, Thr277, and Asp310).
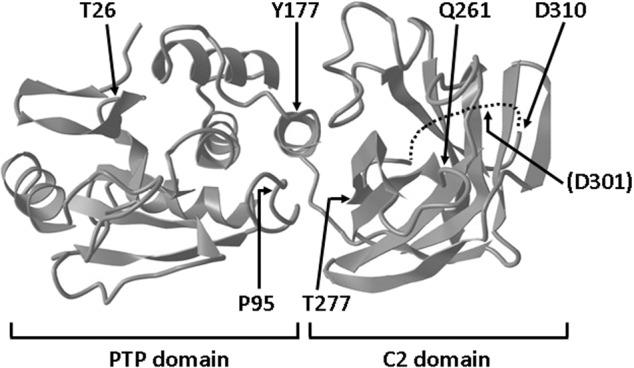
The N-terminal PTP- and C-terminal C2-domain are indicated. Amino acids are indicated in the figure by the single-letter code, and amino acid numbering corresponds to accession NP_000305. The unstructured loop located in the C2 domain (residues 286-309) is depicted as a dotted line, and the Asp301 residue (D301) caspase-3 cleavage site is indicated in brackets.
Expression, protein stability and degradation of PTEN PHTS variants
The cellular expression levels of PTEN protein are determinant for PHTS pathogenesis, and decreased expression or low protein stability of PTEN is relatively frequent in PTEN PHTS variants [22, 23, 30]. The monitoring of the steady-state expression levels of the PTEN variants under scrutiny reflected lower expression of the Y177N and T277A variants compared to PTEN wild type, whereas the rest of variants displayed expression levels similar than PTEN wild type. The PTEN PHTS variant D252G [c.755 A > G; p.(Asp252Gly)], previously reported as unstable [22], also displayed lower steady-state expression levels. This expression pattern was consistently observed in several cell lines, including COS-7 (monkey kidney adenocarcinoma), MCF-7 (human breast carcinoma), U87MG (human glioblastoma), and HEK-293 (human kidney adenocarcinoma) (Fig. 2A, B). To rule out the possibility of distinct expression levels of the mRNAs encoding the distinct PTEN variants in our transfection experiments, we performed parallel qPCR experiments, which showed no significant differences in the mRNA levels of the variants under our experimental conditions (Fig. 2C). These results suggest a shorter half-live for Y177N and T277A PTEN variants than PTEN wild type. PTEN degradation is exerted in part by the ubiquitin-proteasome pathway [39, 40]. Thus, we performed experiments in the presence of cycloheximide or MG132, to test for the abundance of these variants after protein synthesis- or proteasome-inhibition, respectively, in comparison with PTEN wild type or PTEN D252G. As shown, the variants Y177N, T277A, and D252G displayed a decrease in their protein abundance after cycloheximide cell treatment, when compared to PTEN wild type or the other PTEN variants (Fig. 2D). In the presence of the proteasome inhibitor MG132, the Y177N, T277A, and D252G variants, and to a less extent the D310G variant, showed a more prominent relative accumulation than PTEN wild type (Fig. 2E), suggesting that their decrease in expression is due, at least in part, to increased proteasome-mediated degradation. As mentioned, both Y177 and T277 are located at the PTP/C2 domain interface of PTEN, and T277 forms hydrogen bonds with amino acids V191 and G251, which would be lost in the T277A variant (https://www3.cmbi.umcn.nl/hope/input/). Thus, the PTEN PHTS protein variants Y177N and T277A show diminished expression levels in cells, likely due to altered conformation and decreased protein stability, and increased proteasome-mediated degradation.
Fig. 2. Steady-state expression and stability of PTEN PHTS variants.

A COS-7 (left panel) or MCF-7 (right panel) cells were transfected with pRK5 plasmids encoding the indicated PTEN variants (EV empty vector, WT wild type), and cell lysates were resolved by SDS-PAGE 10% followed by sequential immunoblot using anti-PTEN 6H2.1 mAb and anti-GAPDH antibody. Top panels show representative experiments, and bottom panels show quantification of the PTEN bands from at least two independent experiments. Data are shown as relative expression with respect to PTEN wild type (wt = 1) ±SD. Statistical significance (Student’s t test P values) of the difference of some variants with respect to wild type is indicated with asterisks: ***p < 0.001, **p < 0.005, *p < 0.05. B U87MG (top panel) or HEK293 (bottom panel) cells were transfected and processed for immunoblot as in A. Representative experiments are shown. C COS-7 cells were transfected and processed for immunoblot as in A (top panel; note that PTEN wild type is duplicated [WT, WT’]) or were processed for RNA isolation and RT-qPCR (bottom panel). ΔΔCt values for PTEN amplification, using HPRT1 as the reference gene, are shown. D, E COS-7 cells were transfected as in A and kept untreated (−) or incubated (+) in the presence of cycloheximide (CHX, 800 μg/ml) or MG132 (10 μM) for 6 h to monitor protein degradation. Top panels show representative experiments, and bottom panels show quantification of the PTEN bands from two independent experiments. Data are shown as relative expression with respect to PTEN wild type under untreated conditions (wt = 1) ±SD. Numbers in the top indicate the ratio non-treate/treated for each variant. Note the inverted trends of variants Y177N and T277A in the presence CHX and MG132, in line with compromised stability.
PIP3 phosphatase activity in cells of PTEN PHTS variants
To test the activity of PTEN in mammalian cells, we determined the phosphorylation of AKT, as a surrogate marker of PTEN PIP3 phosphatase activity, in the presence of the different PTEN variants. COS-7 cells were co-transfected with plasmids encoding the PTEN variants and AKT1, and pAKT content was monitored by immunoblot using an anti-pAKT (Ser473) antibody. PTEN wild type (WT) and the catalytically inactive PTEN C124S [c.371 G > C; p.(Cys124Ser)] mutation were used for comparisons. In this assay, basal pAKT content in cells is detected upon transfection with empy vector (EV), and pAKT levels diminish upon transfection of PTEN WT, but not upon transfection of catalytically defective PTEN variants. As shown, the P95R variant was fully inactive, the T26I and Y177N variants displayed decreased activity, and the Q261E, T277A, and D310G variants displayed similar activity than PTEN wild type (Fig. 3A). Next, we tested the activity of the PTEN variants using our heterologous yeast system, which monitors with high fidelity and sensitivity the PIP3 phosphatase activity of PTEN in eukaryotic cells, by measuring PIP3 levels in the membrane of the yeast upon ectopic expression of mammalian hyperactive PI3K catalytic subunit (p110α-CAAX) and PTEN [30, 34]. The results of PTEN activity from the yeast were concordant with the results from mammalian cells, with a PIP3 phosphatase activity as follows: Q261E,T277A,D310G > T26I,Y177N > P95R (Fig. 3B). We conclude from our determinations that P95R variant lacks PIP3 phosphatase activity in cells, T26I and Y177N variants display compromised PIP3 phosphatase activity, and Q261E, T277A and D310G variants display PIP3 phosphatase activity similar than PTEN wild type (Fig. 3C).This is of interest in the case of T277A, since this variant displayed low steady-state expression levels compared to PTEN wild type (Fig. 2). In this regard, Yang et al. studied the PTEN T277A variant in the context of sporadic brain cancer, reporting a decreased half-live for it, in association with a slight decrease in PIP3 phosphatase activity in cells [41]. Although slight changes in PTEN PIP3 phosphatase activity may account for pathogenicity, the possibility exists that additional deleterious mechanisms of PTEN function exist in variants such as PTEN T277A.
Fig. 3. PIP3-phosphatase functional activity of PTEN PHTS variants in cells.

A. Functional activity of PTEN variants in mammalian cells. COS-7 cells were co-transfected with different combinations of plasmids encoding AKT1 and PTEN variants (WT PTEN wild type, EV empty vector), and the phosphorylation of AKT (as an indirect measurement of PTEN PIP3-phosphatase activity) was monitored by immunoblotting using anti-pAKT (Ser473) antibody. The expression of total AKT, PTEN, and GAPDH (as a loading control) was also monitored using specific antibodies. In the top panel, a representative experiment is shown. In the bottom panel, the pAKT/AKT ratio from each condition is shown, after quantification of the bands from at least three independent experiments. Statistical significance (Student’s t test P values) of the difference of some variants with respect to wild type is indicated with asterisks: *p < 0.05. B Functional activity of PTEN variants in a heterologous yeast S. cerevisiae system by yeast growth drop assay. Cells were transformed with different combinations of plasmids encoding a hyperactive form of the mammalian PI3K p110α catalytic subunit (p110α-CAAX) and PTEN variants, under glucose growth conditions (OFF, no induction of heterologous proteins) or galactose growth conditions (ON induction of heterologous proteins). The growth of yeast cells is inhibited by p110α-CAAX (p110α-CAAX + EV [empty vector]), which converts essential pools of PIP2 into PIP3. This can be prevented by the expression of active PTEN (p110α-CAAX + PTEN wild type [WT]) but not catalytically inactive PTEN mutation (p110α-CAAX + PTEN C124S). −, no phosphatase activity; +, partial phosphatase activity; +++, phosphatase activity similar to PTEN WT. Bars correspond to the mean ± SD. C Functional activity of PTEN variants in a heterologous yeast S. cerevisiae system by microscopy monitoring using a GFP-AKT1 reporter. Cells were co-transformed with plasmids encoding a GFP-AKT1 reporter, which binds to PIP3 at the plasma membrane, and the PTEN variants. Removal of the GFP-AKT1 reporter form the plasma membrane is a read-out of PTEN activity on PI3K-generated PIP3 substrate. Data are the average of three experiments on three different clones (n > 100 cells per clone). Bars correspond to the mean ± SD. In the graphs from A and C, statistical significance (Student’s t test P values) of the difference of some variants with respect to wild type is indicated with asterisks: ***p < 0.001, **p < 0.005, *p < 0.05.
Subcellular localization of PTEN PHTS variants
PTEN protein is known to actively shuttle between cytosolic and nuclear compartments by different mechanisms, which constitutes an important determinant of PTEN biological functions [31, 42]. This prompted us to examine the subcellular localization of the PTEN variants by immunofluorescence analysis, using anti-PTEN antibodies, both in a PTEN wild type background (which mostly locates in the cytoplasm) and on a PTEN 1-375 background (which accumulates in the nucleus) [43]. The PTEN variants analysed did not show major differences in the subcellular localization with respect to PTEN wild type, except for the P95R, Y177N, and T277A variants, which displayed a slight shift towards more marked cytoplasmic localization (Fig. 4A). Remarkably, the nuclear accumulation of PTEN 1-375 was partially prevented in the Y177N, and T277A variants, whereas the nuclear accumulation of PTEN 1-375 T26I, Q261E, and D310G variants was unaffected. PTEN 1-375 P95R displayed an intermediate nuclear and nuclear/cytoplasmic localization (Fig. 4B, C). Prevention of nuclear accumulation of PTEN T277A variant in a background of non-C-terminal phosphorylated PTEN-GFP has also been reported [41]. Together, these findings attribute differential changes in subcellular localization, affected by the PTEN amino acid sequence background, to the P95R, Y177N, and T277A PTEN PHTS variants.
Fig. 4. Subcellular localization of PTEN PHTS variants.

COS-7 cells were transfected with pRK5 plasmids encoding the indicated PTEN variants (WT wild type), in a background of PTEN (panels A and C), or PTEN 1-375 (panels B and C), and cells were analyzed by standard immunofluorescence using anti-PTEN 425 mAb. The quantification of percentages of cells with nuclear (N), cytoplasmic (C), or nuclear/cytoplasmic (N/C) localization is shown in panels A and B. Data are shown as percentage of cells ± SD from at least two independent experiments. Statistical significance (Student’s t test P values) of the difference of the cytoplasmic (A) or nuclear (B) localization of some variants with respect to wild type is indicated with asterisks: *p < 0.05. Examples of representative images of cells transfected with plasmids encoding the indicated PTEN or PTEN 1-375 variants are shown in panel C, with indication of cells with nuclear (N), nuclear/cytoplasmic (N/C), or cytoplasmic (C) localization.
C2-domain caspase-3 cleavage of PTEN PHTS variants
PTEN is cleaved by caspase-3 at residue Asp301 (D301) in the unstructured loop from PTEN C2-domain [11] (Figs. 1 and 5A). This can be readily monitored using the anti-PTEN SP227 mAb, which recognizes the PTEN truncation 1-301 but not PTEN wild type or other PTEN truncations (Fig. 5B). As the use of this mAb for academic purposes has not been reported yet, we aimed to characterize its epitope specificity for its application in the monitoring of PTEN cleavage at Asp301, an aspect of PTEN biology with potential functional implications that remains unexplored. Mutation to Ala of residues Gln298 [p.(Gln298Ala), Q298A], Glu299 [p.(Glu299Ala), E299A], Ile300 [p.(Ile300Ala), I300A], or Asp301 [p.(Asp301Ala), D301A], but not mutation to Ala of residue Asp297 [p.(Asp297Ala), D297A), abrogated the reactivity of SP227 mAb (Fig. 5B). Furthermore, a GST-fusion protein containing at its C-terminus PTEN residues (298)QEID(301) (GST-QEID) was recognized by SP227 mAb, but not a GST-fusion containing PTEN residues (299)EID(301) (GST-EID) (Fig. 5C). Thus, SP227 mAb recognizes the C-terminal motif -QEID generated on PTEN upon caspase-3 cleavage. PTEN cleavage at Asp301 is enhanced upon apoptotic stimulation, such as TNF-α cell treatment [11], making possible a PTEN functional relevance for this specific modification. However, how PTEN PHTS mutations may affect Asp301 PTEN cleavage is unknown. Next, we monitored the effect of the analysed PTEN mutations on PTEN Asp301 cleavage. As shown, mutations T26I, P95R, Y177N, and T277A displayed diminished PTEN Asp301 cleavage, although it should be mentioned that the diminished basal expression of the Y177N and T277A variants hampers the interpretation of the results with these mutations. The catalytically inactive C124S variant also displayed a slight diminished Asp301 cleavage, especially manifested in the presence of TNF-α. On the other hand, Asp301 cleavage of mutations Q261E and D310G was achieved efficiently, both in the absence and in the presence of TNF-α (Fig. 5D). We conclude that specific PTEN PHTS variants may have compromised their C2-domain-loop cleavage in cells by caspase-3.
Fig. 5. Cleavage of PTEN PHTS variants at Asp301 as determined using the anti-PTEN SP227 mAb.

A Schematic of PTEN showing the caspase-3 cleavage motif (amino acids 297-301) and the Asp301 (D301) cleavage site at the unstructured loop at PTEN C2 domain. The arrow indicates the cleavage site. B Reactivity of the anti-PTEN SP227 mAb with GST-PTEN (N-terminal tagging) truncations. COS-7 cells were transfected with pRK5 plasmids encoding the indicated GST-PTEN truncations (EV empty vector, WT wild type), and cell lysates were analyzed by immunoblot using anti-PTEN SP227 or anti-GST (as a control of expression). In the right panel, the indicated amino acid substitutions, targeting the caspase-3 cleavage motif, were analysed in the background of GST-PTEN 1-301. C Reactivity of the anti-PTEN SP227 mAb with GST containing amino acids from the caspase-3 cleavage motif at PTEN C2 domain. COS-7 cells were transfected with pRK5 plasmids encoding the indicated GST fusion proteins (EV, empty vector), and cell lysates were analyzed as in B. D Cleavage of PTEN variants at Asp301. COS-7 cells were transfected with pRK5 plasmids encoding the indicated PTEN variants (EV empty vector, WT wild type), and then were kept untreated or were treated with TNF-α (50 ng/ml) for 6 h, followed by immunoblot using anti-PTEN SP227 or anti-PTEN 6H2.1 antibodies. The left panel shows a representative experiment (arrows indicate migration of PTEN [left upper panel] or PTEN 1-301 [left bottom panel]). The right panel shows the relative detection of PTEN bands from the PTEN variants as compared with PTEN wild type (WT) under untreated conditions (PTEN full-length and PTEN 1-301 normalized to 1), after quantification of the bands from three independent experiments. Results are shown as the ratio of the PTEN 1-301/PTEN bands (note that, for normalization, this ratio is = 1 in PTEN WT under untreated conditions). Statistical significance (Student’s t test P values) of the difference of some variants with respect to wild type is indicated with asterisks: ***p < 0.001, **p < 0.005.
Discussion
Our functional analysis of the panel of PTEN PHTS mutations reflects the functional heterogeneity attributed to PTEN variants found in association with disease, with most of the variants analyzed (T26I, P95R, Y177N, and T277A) manifesting several alterations in our assays (Table 2). This is in accordance with previous reports analysing in parallel other sets of PTEN germline mutations using complementary methodologies [20, 22, 23, 44], as well as with the comparative analysis of the tumor spectrum phenotype from several Pten knock-in mice [45]. In line with the importance of PTEN protein expression levels for appropriate PTEN function, we have found two PTEN variants (Y177N, T277A) displaying low steady-state expression and a trend of increased proteasome-mediated degradation. Dedicated studies are required to ascertain the contribution of altered phosphorylation and ubiquitination of these variants in their altered protein stability. In this regard, high-frequency PHTS mutations targeting the Arg173 PTEN residue, which cause diminished protein stability, display increased ubiquitination at specific PTEN Lys residues [46, 47]. In addition to the frequent impairment of PIP3 phosphatase activity in cells, changes in the PTEN cytoplasmic/nuclear ratio stands as one of the more common alterations in PHTS patients. In our analysis, the P95R, Y177N, and T277A variants showed diminished nuclear accumulation in the background of PTEN 1-375, a recombinant proteoform that mimics one of the PTEN C-terminal caspase-3 protein products [11]. When comparing PTEN (major cytoplasmic localization) with PTEN 1-375 (major nuclear localization; ref. [31]), the variants P95R, Y177N, and T277A displayed enhanced cytoplasmic localization. We speculate that different regulatory mechanisms of PTEN subcellular localization could be affected by distinct PTEN PHTS mutations.
Table 2.
Functional analysis of PTEN PHTS variants of unknown significance.
| Variant1 | Phosphatase activity2 | Expression3 | Stability3 | Localization PTEN4 | Localization PTEN 1-3754 | Asp301- cleavage5 |
|---|---|---|---|---|---|---|
| WT | +++ | +++ | +++ | C | N | + |
| T26I c.77 C > T | + | +++ | +++ | C | N | - |
| P95R c.284 C > G | - | +++ | +++ | C | N/C, N | - |
| Y177N c.529 T > A | + | + | + | C | N/C | (?) |
| Q261E c.781 C > G | +++ | +++ | +++ | C | N | + |
| T277A c.829 A > G | +++ | + | + | C | N/C | (?) |
| D310G c.929 A > G | +++ | +++ | +++ | C | N | + |
1PTEN protein variants are indicated with the single-letter code amino acid nomenclature. DNA germline variants are indicated following HGVS recommended nomenclature. Nucleotide and amino acid numbering corresponds to accessions NM_000314.8 and NP_000305.3, respectively.
2As determined both in mammalian and yeast cells.; -lack of activity; +, diminished activity. +++, similar activity to PTEN WT
3+, diminished compared to PTEN WT+++, similar to PTEN WT;.
4Major subcellular localization in PTEN or PTEN 1-375 background. N nuclear, N/C nuclear/cytoplasmic, C cytoplasmic
5As determined using the anti-PTEN SP227 antibody. Determination of cleavage on Y177N and T277A variants is hampered by their low expression and it is indicated as (?).
We did not detect significant alterations in the variants Q261E and D310G targeting the PTEN C2-domain, suggesting the existence of uncovered regulatory mechanisms of PTEN function. The Q261E mutation incorporates a negative charge in the CBR3 loop at PTEN C2 domain, which has been involved in binding to membranes [14]. This makes possible altered membrane binding properties for the PTEN Q261E variant. On the other hand, the D310G mutation removes a negative charge near the unstructured loop at the C2 domain, whose functions are unknown. The implementation of alternative PTEN function experimental assays will be central to unveil the potential pathogenicity of these types of mutations. In this regard, the T26I and P96R variants displayed diminished caspase-3 cleavage at Asp301 at the C2-domain unstructured loop, as monitored using a specific anti-PTEN Asp301-cleaved antibody. This could be a consequence of impaired catalytic activity [11], but it could also be the result of subtle alterations in protein-protein interactions and subcellular localization. The regulation and functional consequences of Asp301 PTEN cleavage are unknown, which deserves dedicated studies. In this regard, the PTEN Asp301 caspase-3 cleavage is not regulated by PTEN C-terminal tail phosphorylation by CK2, distinct to the PTEN caspase-3 cleavage at the PTEN C-tail [11]. PTEN structure and intrinsic phosphatase activity is maintained in the absence of the C2-domain unstructured loop (residues 286-309) containing the Asp301 residue [14], suggesting a non-essential role for this region in PTEN enzymatic activity. However, amino acid substitution variants targeting PTEN residues 286-309 are found in PHTS patients and in tumors, suggesting an active regulatory role for the C2-domain unstructured loop linked to pathogenesis. In this regard, this loop contains the Lys289 residue, which is targeted by mutation in PHTS and whose ubiquitination is important for facilitating PTEN nuclear import [48], an event that has been related with chemoresistance to temozolomide in glioblastoma cells [49]. Also of interest, PTEN caspase cleavage has been associated with resistance to cisplatin treatment in the A2780 ovarian cancer cell line [13]. Further work is necessary to elucidate the potential role of alterations in PTEN caspase-3 cleavage or PTEN nuclear import in PHTS pathogenesis and resistance to anti-cancer chemotherapy.
Acknowledgements
We thank Gustavo Pérez‐Nanclares and Ana Belén de la Hoz (Genetics-Genomics Core facility, Biocruces Bizkaia Health Research Institute) for their expert assistance with DNA sequencing., and Javier Díez García (Microscopy core facility, Biocruces Bizkaia Health Research Institute) for expert microscopy technical support.
Author contributions
LT, JM, IR-E, TF-A, and SL designed and performed experiments, and performed data analysis. CEN-X designed experiments, supervised the work and performed data analysis. JIL and MM supervised the work. FM, MC, and MU shared information and provided feedback. VJC and RP designed experiments, performed data analysis and wrote the manuscript. All authors revised the manuscript.
Funding
This work has been supported in part by grant BBH-19-001 (to RP) from PTEN Research Foundation (United Kingdom); grants SAF2016-79847-R (to RP and JIL), and PID2019-105342GB-I00 (to VJC and MM) from Ministerio de Economía y Competitividad (Spain and The European Regional Development Fund); and grant S2017/BMD‐3691(InGEMICS‐CM) from Comunidad de Madrid and European Structural and Investment Funds (to VJC and MM). LT has been the recipient of a predoctoral fellowship from Asociación Española Contra el Cáncer (AECC, Junta Provincial de Bizkaia, Spain). JM has been the recipient of a predoctoral fellowship (PRE_2014_1_285) from Gobierno Vasco, Departamento de Educación (Basque Country, Spain). CN-X is the recipient of a Miguel Servet Research Contract from Instituto de Salud Carlos III (grant number CP20/00008).
Data availability
The datasets generated during and/or analysed during the current study are available in the LOVD gene variant database (http://www.lovd.nl/3.0/home), with the following accession ID: 0000352584 (T26I), 0000878862 (P95R), 0000878883 (Y177N), 0000878885 (Q261E), 0000878886 (T277A), 0000878890 (D310G).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Yehia L, Keel E, Eng C. The Clinical Spectrum of PTEN Mutations. Annu Rev Med. 2020;71:103–16. doi: 10.1146/annurev-med-052218-125823. [DOI] [PubMed] [Google Scholar]
- 2.Ngeow J, Eng C. PTEN in Hereditary and Sporadic Cancer. Cold Spring Harbor Perspect Med. 2020;1:a036087. [DOI] [PMC free article] [PubMed]
- 3.Pulido R, Mingo J, Gaafar A, Nunes-Xavier CE, Luna S, Torices L, et al. Precise Immunodetection of PTEN Protein in Human Neoplasia. Cold Spring Harb Perspect Med. 2019;9:a036293. doi: 10.1101/cshperspect.a036293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol cell Biol. 2018;19:547–62. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 5.Pulido R. PTEN: a yin-yang master regulator protein in health and disease. Methods. 2015;77-78:3–10. doi: 10.1016/j.ymeth.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 6.A Papa, PP Pandolfi. Phosphatase-independent functions of the tumor suppressor PTEN. In: Neel B, Tonks N, editors. Protein Tyrosine Phosphatases in Cancer. New York, NY: Springer; 2016;247–60.
- 7.Bononi A, Pinton P. Study of PTEN subcellular localization. Methods. 2015;77-78:92–103. doi: 10.1016/j.ymeth.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Misra S, Ghosh G, Chowdhury SG, Karmakar P. Non-canonical function of nuclear PTEN and its implication on tumorigenesis. DNA Repair (Amst) 2021;107:103197. doi: 10.1016/j.dnarep.2021.103197. [DOI] [PubMed] [Google Scholar]
- 9.Leslie NR, Kriplani N, Hermida MA, Alvarez-Garcia V, Wise HM. The PTEN protein: cellular localization and post-translational regulation. Biochemical Soc Trans. 2016;44:273–8. doi: 10.1042/BST20150224. [DOI] [PubMed] [Google Scholar]
- 10.Sellars E, Gabra M, Salmena L. The Complex Landscape of PTEN mRNA Regulation. Cold Spring Harb Perspect Med. 2020;10:a036236. doi: 10.1101/cshperspect.a036236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres J, Rodriguez J, Myers MP, Valiente M, Graves JD, Tonks NK, et al. Phosphorylation-regulated cleavage of the tumor suppressor PTEN by caspase-3: implications for the control of protein stability and PTEN-protein interactions. J Biol Chem. 2003;278:30652–60. doi: 10.1074/jbc.M212610200. [DOI] [PubMed] [Google Scholar]
- 12.Gil A, Andrés-Pons A, Pulido R. Nuclear PTEN: a tale of many tails. Cell death Differ. 2007;14:395–9. doi: 10.1038/sj.cdd.4402073. [DOI] [PubMed] [Google Scholar]
- 13.Singh M, Chaudhry P, Fabi F, Asselin E. Cisplatin-induced caspase activation mediates PTEN cleavage in ovarian cancer cells: a potential mechanism of chemoresistance. BMC Cancer. 2013;13:233. doi: 10.1186/1471-2407-13-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–34. doi: 10.1016/S0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 15.Fragoso R, Barata JT. Kinases, tails and more: Regulation of PTEN function by phosphorylation. Methods. 2014. [DOI] [PubMed]
- 16.Sotelo NS, Schepens JT, Valiente M, Hendriks WJ, Pulido R. PTEN-PDZ domain interactions: binding of PTEN to PDZ domains of PTPN13. Methods. 2015;77-78:147–56. doi: 10.1016/j.ymeth.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 17.Wang K, Liu J, Li YL, Li JP, Zhang R. Ubiquitination/de-ubiquitination: A promising therapeutic target for PTEN reactivation in cancer. Biochim Biophys Acta Rev Cancer. 2022;1877:188723. doi: 10.1016/j.bbcan.2022.188723. [DOI] [PubMed] [Google Scholar]
- 18.Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet. 2018;50:874–82. doi: 10.1038/s41588-018-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mighell TL, Evans-Dutson S, O’Roak BJ. A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am J Hum Genet. 2018;102:943–55. doi: 10.1016/j.ajhg.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mingo J, Rodriguez-Escudero I, Luna S, Fernandez-Acero T, Amo L, Jonasson AR, et al. A pathogenic role for germline PTEN variants which accumulate into the nucleus. Eur J Hum Genet: EJHG. 2018;26:1180–7. doi: 10.1038/s41431-018-0155-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodríguez-Escudero I, Oliver MD, Andrés-Pons A, Molina M, Cid VJ, Pulido R. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet. 2011;20:4132–42. doi: 10.1093/hmg/ddr337. [DOI] [PubMed] [Google Scholar]
- 22.Spinelli L, Black FM, Berg JN, Eickholt BJ, Leslie NR. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet. 2015;52:128–34. doi: 10.1136/jmedgenet-2014-102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Post KL, Belmadani M, Ganguly P, Meili F, Dingwall R, McDiarmid TA, et al. Multi-model functionalization of disease-associated PTEN missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat Commun. 2020;11:2073. doi: 10.1038/s41467-020-15943-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chao JT, Hollman R, Meyers WM, Meili F, Matreyek KA, Dean P, et al. A Premalignant Cell-Based Model for Functionalization and Classification of PTEN Variants. Cancer Res. 2020;80:2775–89. doi: 10.1158/0008-5472.CAN-19-3278. [DOI] [PubMed] [Google Scholar]
- 25.Ganguly P, Madonsela L, Chao JT, Loewen CJR, O’Connor TP, Verheyen EM, et al. A scalable Drosophila assay for clinical interpretation of human PTEN variants in suppression of PI3K/AKT induced cellular proliferation. PLoS Genet. 2021;17:e1009774. doi: 10.1371/journal.pgen.1009774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith IN, Thacker S, Seyfi M, Cheng F, Eng C. Conformational Dynamics and Allosteric Regulation Landscapes of Germline PTEN Mutations Associated with Autism Compared to Those Associated with Cancer. Am J Hum Genet. 2019;104:861–78. doi: 10.1016/j.ajhg.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cid VJ, Rodríguez-Escudero I, Andrés-Pons A, Romá-Mateo C, Gil A, den Hertog J, et al. Assessment of PTEN tumor suppressor activity in nonmammalian models: the year of the yeast. Oncogene. 2008;27:5431–42. doi: 10.1038/onc.2008.240. [DOI] [PubMed] [Google Scholar]
- 28.Leslie NR, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Developmental Biol. 2016;52:30–8. doi: 10.1016/j.semcdb.2016.01.030. [DOI] [PubMed] [Google Scholar]
- 29.Pena-Couso L, Ercibengoa M, Mercadillo F, Gomez-Sanchez D, Inglada-Perez L, Santos M, et al. Considerations on diagnosis and surveillance measures of PTEN hamartoma tumor syndrome: clinical and genetic study in a series of Spanish patients. Orphanet J Rare Dis. 2022;17:85. doi: 10.1186/s13023-021-02079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrés-Pons A, Rodríguez-Escudero I, Gil A, Blanco A, Vega A, Molina M, et al. In vivo functional analysis of the counterbalance of hyperactive phosphatidylinositol 3-kinase p110 catalytic oncoproteins by the tumor suppressor PTEN. Cancer Res. 2007;67:9731–9. doi: 10.1158/0008-5472.CAN-07-1278. [DOI] [PubMed] [Google Scholar]
- 31.Gil A, Andrés-Pons A, Fernández E, Valiente M, Torres J, Cervera J, et al. Nuclear localization of PTEN by a Ran-dependent mechanism enhances apoptosis: Involvement of an N-terminal nuclear localization domain and multiple nuclear exclusion motifs. Mol Biol Cell. 2006;17:4002–13. doi: 10.1091/mbc.e06-05-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrés-Pons A, Gil A, Oliver MD, Sotelo NS, Pulido R. Cytoplasmic p27Kip1 counteracts the pro-apoptotic function of the open conformation of PTEN by retention and destabilization of PTEN outside of the nucleus. Cell Signal. 2012;24:577–87. doi: 10.1016/j.cellsig.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Mingo J, Erramuzpe A, Luna S, Aurtenetxe O, Amo L, Diez I, et al. One-Tube-Only Standardized Site-Directed Mutagenesis: An Alternative Approach to Generate Amino Acid Substitution Collections. PloS One. 2016;11:e0160972. doi: 10.1371/journal.pone.0160972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez-Escudero I, Fernandez-Acero T, Bravo I, Leslie NR, Pulido R, Molina M, et al. Yeast-based methods to assess PTEN phosphoinositide phosphatase activity in vivo. Methods. 2015;77-78:172–9. doi: 10.1016/j.ymeth.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Rodríguez-Escudero I, Roelants FM, Thorner J, Nombela C, Molina M, Cid VJ. Reconstitution of the mammalian PI3K/PTEN/Akt pathway in yeast. Biochemical J. 2005;390:613–23. doi: 10.1042/BJ20050574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nunes-Xavier CE, Pulido R. Global RT-PCR and RT-qPCR Analysis of the mRNA Expression of the Human PTPome. Methods Mol Biol. 2016;1447:25–37. doi: 10.1007/978-1-4939-3746-2_2. [DOI] [PubMed] [Google Scholar]
- 37.Andrés-Pons A, Valiente M, Torres J, Gil A, Roglá I, Ripoll F, et al. Functional definition of relevant epitopes on the tumor suppressor PTEN protein. Cancer Lett. 2005;223:303–12. doi: 10.1016/j.canlet.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 38.Sotelo NS, Valiente M, Gil A, Pulido R. A functional network of the tumor suppressors APC, hDlg, and PTEN, that relies on recognition of specific PDZ-domains. J Cell Biochem. 2012;113:2661–70. doi: 10.1002/jcb.24141. [DOI] [PubMed] [Google Scholar]
- 39.Tolkacheva T, Boddapati M, Sanfiz A, Tsuchida K, Kimmelman AC, Chan AM. Regulation of PTEN binding to MAGI-2 by two putative phosphorylation sites at threonine 382 and 383. Cancer Res. 2001;61:4985–9. [PubMed] [Google Scholar]
- 40.Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. The. J Biol Chem. 2001;276:993–8. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 41.Yang JM, Schiapparelli P, Nguyen HN, Igarashi A, Zhang Q, Abbadi S, et al. Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization. Oncogene. 2017;36:3673–85. doi: 10.1038/onc.2016.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ho J, Cruise ES, Dowling RJO, Stambolic V. PTEN Nuclear Functions. Cold Spring Harb Perspect Med. 2020;10:a036079. doi: 10.1101/cshperspect.a036079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gil A, Rodriguez-Escudero I, Stumpf M, Molina M, Cid VJ, Pulido R. A functional dissection of PTEN N-terminus: implications in PTEN subcellular targeting and tumor suppressor activity. PloS one. 2015;10:e0119287. doi: 10.1371/journal.pone.0119287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong CW, Wang Y, Liu T, Li L, Cheung SKK, Or PM, et al. Autism-associated PTEN missense mutation leads to enhanced nuclear localization and neurite outgrowth in an induced pluripotent stem cell line. The. FEBS J. 2020;287:4848–61. doi: 10.1111/febs.15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H, Karikomi M, Naidu S, Rajmohan R, Caserta E, Chen HZ, et al. Allele-specific tumor spectrum in pten knockin mice. Proc Natl Acad Sci USA. 2010;107:5142–7. doi: 10.1073/pnas.0912524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo Y, He J, Zhang H, Chen R, Li L, Liu X, et al. Linear ubiquitination of PTEN impairs its function to promote prostate cancer progression. Oncogene. 2022;41:4877–92. doi: 10.1038/s41388-022-02485-6. [DOI] [PubMed] [Google Scholar]
- 47.Pearce W, Kessaris N, Leslie NR, Vanhaesebroeck B, Tibarewal P, Classen G, et al. Investigation of PTEN genotype-phenotype correlations in the PTEN hamartoma tumor syndrome (PHTS) using in vitro and in vivo approaches. Mol cancer Res: MCR. 2020;18:B22. doi: 10.1158/1557-3125.PI3K-mTOR18-B22. [DOI] [Google Scholar]
- 48.Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–56. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong L, Li Y, Liu L, Meng X, Li S, Han D, et al. Smurf1 Suppression Enhances Temozolomide Chemosensitivity in Glioblastoma by Facilitating PTEN Nuclear Translocation. Cells. 2022;11:3302. doi: 10.3390/cells11203302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Busa T, Milh M, Degardin N, Girard N, Sigaudy S, Longy M, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol: EJPN: Off J Eur Paediatr Neurol Soc. 2015;19:188–92. doi: 10.1016/j.ejpn.2014.11.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analysed during the current study are available in the LOVD gene variant database (http://www.lovd.nl/3.0/home), with the following accession ID: 0000352584 (T26I), 0000878862 (P95R), 0000878883 (Y177N), 0000878885 (Q261E), 0000878886 (T277A), 0000878890 (D310G).