

Abstract
Splice-site variants in cardiac genes may predispose carriers to potentially lethal arrhythmias. To investigate, we screened 1315 probands and first-degree relatives enrolled in the Canadian Hearts in Rhythm Organization (HiRO) registry. 10% (134/1315) of patients in the HiRO registry carry variants within 10 base-pairs of the intron-exon boundary with 78% (104/134) otherwise genotype negative. These 134 probands were carriers of 57 unique variants. For each variant, American College of Medical Genetics and Genomics (ACMG) classification was revisited based on consensus between nine in silico tools. Due in part to the in silico algorithms, seven variants were reclassified from the original report, with the majority (6/7) downgraded. Our analyses predicted 53% (30/57) of variants to be likely/pathogenic. For the 57 variants, an average of 9 tools were able to score variants within splice sites, while 6.5 tools responded for variants outside these sites. With likely/pathogenic classification considered a positive outcome, the ACMG classification was used to calculate sensitivity/specificity of each tool. Among these, Combined Annotation Dependent Depletion (CADD) had good sensitivity (93%) and the highest response rate (131/134, 98%), dbscSNV was also sensitive (97%), and SpliceAI was the most specific (64%) tool. Splice variants remain an important consideration in gene elusive inherited arrhythmia syndromes. Screening for intronic variants, even when restricted to the ±10 positions as performed here may improve genetic testing yield. We compare 9 freely available in silico tools and provide recommendations regarding their predictive capabilities. Moreover, we highlight several novel cardiomyopathy-associated variants which merit further study.
Subject terms: Cardiomyopathies, Gene regulation, Clinical genetics
Introduction
The study of genotype-phenotype relationships has primarily focused on variants of interest in coding regions of DNA. In recent years, next generation sequencing has allowed investigators to study the whole genome, including variants in non-coding regions, with the implication that these variants may have functional consequences causing disease. Some of these variants impact splicing, whereby the primary mRNA sequence is processed to remove non-coding introns.
Variants at exon-intron boundaries are thought to alter spliceosome recognition, especially when they occur in highly conserved splice donor or acceptor sequences [1]. Canonically, the splice donor site at the beginning (5ʹ) of an intron contains a GT dinucleotide and the splice acceptor site at the end (3ʹ) of an intron contains an AG dinucleotide [2]. These sites are central to spliceosome complex recognition and subsequent excision yields mature mRNA; the template for protein translation (Fig. 1). Variants at either dinucleotide site almost invariably alters intron recognition and changes the protein sequence through a variety of mechanisms, with the potential for significant functional consequences. Variants immediately flanking the donor and acceptor sites may also alter splicing, but the association with disease is less clear [3]. Deep intronic variants can create new splice donor or acceptor sites, leading to activation of ‘cryptic’ exons, which may cause various disease states, including inherited heart disease. Approximately 10% of gene-elusive hypertrophic cardiomyopathy (HCM) patients carry deep intronic splice-site variants [4].
Fig. 1. RNA splicing schematic.
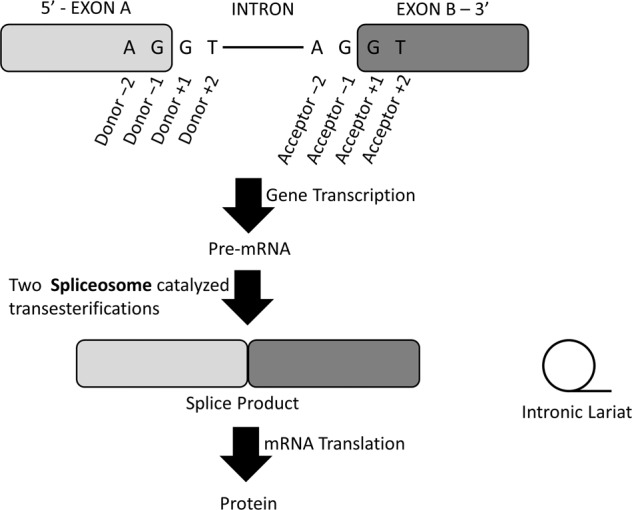
The splice donor/acceptor sites, splicing of the intronic lariat, and protein product are labelled.
It is estimated that 15% of genotype elusive disease cases are due to altered splicing resulting from intronic variations [5]. Changes in the splice sites or introduction of novel splice sites may alter splicing and lead to a frameshift, premature stop codon and truncation, or nonsense mediated decay. Haplo-insufficiency may result, whereby the amount of protein is insufficient to maintain normal function. Alternatively, the poison peptide hypothesis proposes that a mutant protein may have a dominant negative effect [6]. Alternatively, activation of cryptic splice sites can lead to intronic inclusion or cassette exon skipping [7].
Numerous in silico tools aim to predict the deleterious nature of gene variants. For example, SpliceAI is a recently developed, RNA sequencing (RNA-seq) validated tool which uses a 32-layer deep neural network to predict splicing from the pre-mRNA sequence [8]. In silico tools predict splicing impact without the need for traditional in vitro or in vivo validation studies. Each tool has inherent strengths and weaknesses and there is some overlap between the algorithms utilized by each [9]. MaxEntScan (MES), Human Splice Finder (HSF), and Splice Site Prediction by Neural Network (NNSplice) are only optimized for analysis of variants occurring in splice-sites and branch point [10]. MutationTaster makes predictions based on inter-species conservation; it focuses on splice-site alterations and loss of protein function, utilizing features from UniProt [11], Ensembl [12], and the splice-site prediction program NNSplice [13]. It was validated against other applications such as Polymorphism Phenotyping (PolyPhen), which it outperformed [14]. Combined Annotation Dependent Depletion (CADD) derives the deleteriousness of simulated variants through comparison with fixed/nearly fixed alleles. CADD combines 63 distinct annotations, among which the best-performing were the protein-level metrics SIFT and PolyPhen [15]. Deleterious Annotation Using Neural Networks (DANN) is similar to CADD but also uses deep learning to assess variant pathogenicity [16]. dbscSNV’s was developed for single nucleotide variants occurring in the splice consensus sites [9]. SpliceAI utilizes deep learning to automatically extracts sequence features, with the trade-off that it may incorporate irrelevant features into the model [8]. The detailed comparison of these tools is beyond the scope of this work, however, the available selection of tools, accessing each, and output variability between tools makes interpretation difficult and represents hurdles to widespread utility.
The selected splice tools utilized in this study are readily accessible through online platforms without the need for local program installation. These programs are optimized for analysis, for example in a clinic setting, of a limited number of variants and/or when genetic reports do not contain raw sequencing information as was the case with our data set. Diagnostic pipelines may incorporate other tools which will be further discussed below. The role of further bioinformatic requirements when considering whole exome or whole genome sequencing are not addressed in our manuscript or analyses.
In silico tools often do not indicate the splicing error and are often low specificity [17]. As a result, American College of Medical Genetics and Genomics for Molecular Pathology (ACMG-AMP) criteria associated with in silico tools can be supportive of pathogenicity (PP3) while functional analysis of RNA can serve as strong evidence (PS3) [18]. In this study, we used nine in silico tools to analyze 57 unique splice-site variants to uncover trends, revisit ACMG-AMP classification of each, and assess the utility of these tools.
We have interrogated the Canadian National Hearts in Rhythm Organization (HiRO) registry (NCT04189822) which contains cases from the Cardiac Arrest Survivors with Preserved Ejection Fraction Registry (CASPER) (NCT00292032), Long QT syndrome (LQTS) (NCT02425189), and Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) (NCT01804699) sub-registries. This study surveys the prevalence of splice variants across a large, well characterized population and assesses the utility of in silico prediction tools for rapid characterization of novel splice variants.
Materials and methods
Patient cohort
This project was conducted in compliance with the protocols and principles laid down in the Declaration of Helsinki and approved in full by the Providence Health/University of British Columbia ethics board (REB number H20-00129). A retrospective analysis of the HiRO registry was used to identify probands and first-degree relatives with heritable arrhythmias and/or cardiomyopathies enrolled between March 2004 and January 2020. The strength of applied diagnoses is dependent on the available clinical data based on previously defined criteria [19], whereby patients have possible, probable, or definite disease.
Variant evaluation and in silico workflow
We compiled a list of intronic splice variants, then input these variants into nine unique in silico tools (Table S1). Sequencing data was not generated or analyzed for these patients, rather genetic reports containing the identified variants was utilized. Human Genome Variation Society (HGVS) nomenclature corresponding to the canonical transcript is reported here and was used for interpretation as the majority of genetic test reports did not provide the specific transcript ID. The splice-site is variably defined but likely encompasses the last three base pairs of the exon and first six base pairs of the intron (donor) as well as the last 20 nucleotides of an intron and the first three base pairs of an exon (acceptor). We focused on ±10 base pairs as this covers the majority of these regions, has sufficient sensitivity in detecting most variants, and accounts for variability in test reading frames used by different commercial laboratories. For each individual, panel sequencing was used with the details of screened genes provided in the supplement (Table S2). Blueprint Genetics reports variants within ±20 base pairs, while GeneDx does so for variants “close to the splice junction”, and smaller regional labs do not clearly communicate the reading frame.
The in silico work-flow is detailed here and summarized in Fig. 2. Each variant was initially queried using Varsome version 8.1 [20] which annotates variant allele frequency from the gnomAD v2.1.1 database [21] and splice prediction scores from DANN version 2014 [16], MutationTaster version2021 [14], and dbscSNV v1.1 [22]. MutationTaster predicts the disease-causing potential of a variant based on nucleotide conservation, alterations in the splice site, and potential loss of protein function [14]. DANN and dbscSNV (ADA and RF) use machine learning algorithms to classify the potential impact of a variant [16].
Fig. 2. Work-flow of utilized in silico tools.

The in silico tools and other online tools used in the analysis of splice-site variants are outlined in a stepwise manner. Deleterious Annotation Using Neural Networks (DANN) evaluates genome wide, MutationTaster evaluates genome wide but test data sets were limited to donor (last 3 exonic bases and 6 first intronic bases) and acceptor (last 12 intronic bases and 2 first exonic bases) splice sites., dbscSNV (ADA and RF score) evaluates from −3→ +8 at the 5ʹ and −12→+2 at the 3ʹ end, Splice Site Prediction by Neural Network (NNSplice), Combined Annotation Dependent Depletion (CADD) evaluates genome wide, SpliceAI evaluates genome wide, Human Splice Finder (HSF) evaluates genome wide, MaxEntScan (MES) evaluates −3→+6 at 5’ end and −20→+3 at 3ʹ end.
To compare the wildtype splice-site sequence and variant sequence, were entered into NNSplice 0.9 version [13]. CADD v1.5 [15] requires the entry of chromosome number, position, and nucleotide change; all available through Varsome. CADD scores variants by combining dozens of other scores such as SIFT [23] and PolyPhen [24] which independently attempt to predict the effect of nucleotide substitutions on protein stability and function through examination of cross-species conservation features. Franklin also summarizes numerous resources in one easy-to-use interface which includes a SpliceAI v1.3.1 [8] score. HSF v3.1 [25] and MES version 2.0 [26] scores are accessible through a single online interface. MES, NNSplice, and HSF are integrated into the commercially available software package Alamut (Interactive Biosoftware, Rouen, France).
(ACMG-AMP) criteria
The in silico results were used in part to apply the 2015 ACMG-AMP criteria. Variants within canonical splice regions (±1/2 position) were assigned PVS1 (presumed null effect) when identified in a gene where loss-of-function (LOF) is an established mechanism for disease. For example, based on recent evidence, variants in RyR2 canonical splice dinucleotides were assigned PVS1 [27]. PS3 criteria was assigned to variants where in vitro/vivo studies support the deleterious impact of a variant, whereas BS3 supports no impact.
Output scores from the in silico tools and predefined critical thresholds were used to determined pathogenicity. PP3 or BP4 ratings were applied when multiple computational tools indicated that the variant does/not have a deleterious effect, respectively. When at least 3 tools responded, a BP4/PP3 rating was assigned based on agreement between the majority. The literature from databases such as ClinVar, Pubmed, and Google Scholar were used to assign additional ACMG-AMP criteria [18].
The proportion of variants in which evidence from the in silico prediction tools contributed to a change in variant interpretation was determined. Diagnostic implications of variant position (±1/2 vs. ±3 to ±10) was compared.
Results
1315 participants in the HiRO registry were found to carry 1761 gene variants (Table S2). 134 (10.2%) of these patients carried 57 unique splice-region variants in 23 genes. Mean age at first assessment was 42.7 ± 18.0 years. 49% of patients were females, 83% identified as Caucasian, and 51% were symptomatic, most commonly experiencing palpitations (19%), cardiac arrest (16%), or syncope (12%) (Table 1). The most common diagnosis was ARVC (36%), while 27 probands did not have a clear diagnosis (19.6%) (Tables S3 and S4). Most patients were symptomatic probands (44%) or asymptomatic family members (43%). Among the 57 splice-region variants, 28 were in splice sites, 30 were absent from gnomAD, and 6 had a gnomAD allele frequency less than 0.001%.
Table 1.
Demographic and phenotypic data.
| Variants seen in one individual (n = 41) | Variants seen in more than one individual (n = 93) | Total individuals with a splice variant (n = 134) | |
|---|---|---|---|
| Female gender | 17 (41.5%) | 50 (53.8%) | 67 (50.0%) |
| Caucasian ethnicity | 30 (73.2%) | 81 (87.1%) | 111 (82.8%) |
| Symptomatic | 29 (70.7%) | 39 (41.9%) | 68 (50.7%) |
| Presyncope | 5 (12.2%) | 6 (6.5%) | 11 (8.2%) |
| Syncope | 10 (24.4%) | 6 (6.5%) | 16 (11.9%) |
| Palpitations | 11 (26.8%) | 15 (16.1%) | 26 (19.4%) |
| Cardiac arrest | 14 (34.1%) | 8 (8.6%) | 22 (16.4%) |
| Total ARVC | 9 (22.0%) | 39 (41.9%) | 48 (35.8%) |
| Definite ARVC | 6 (66.7%) | 22 (56.4%) | 28 (58.3%) |
| Probable ARVC | 1 (11.1%) | 9 (23.1%) | 10 (20.8%) |
| Possible ARVC | 2 (22.2%) | 8 (20.5%) | 10 (20.8%) |
| Total LQTS | 7 (17.1%) | 13 (14.0%) | 20 (14.9%) |
| Definite LQTS | 4 (57.1%) | 9 (69.2%) | 13 (65%) |
| Probable LQTS | 1 (14.3%) | 2 (15.4%) | 3 (15%) |
| Possible LQTS | 2 (28.6%) | 2 (15.4%) | 4 (20%) |
| Total HCM | 5 (12.2%) | 1 (1.1%) | 6 (4.5%) |
| Definite HCM | 1 (20%) | 0 | 1 (16.7%) |
| Probable HCM | 4 (80%) | 1 (100%) | 5 (83.3%) |
| Possible HCM | 0 | 0 | 0 |
| Unaffected | 0 (0%) | 5 (5.4%) | 5 (3.7%) |
| Age at first assessment | 44.3 ± 16.6 | 42.0 ± 18.6 | 42.7 ± 18.0 |
| Symptomatic proband | 28 (68.3%) | 31 (33.3%) | 59 (44.0%) |
| Symptomatic family member | 1 (2.4%) | 8 (8.6%) | 9 (6.7%) |
| Asymptomatic primary referral | 6 (14.6%) | 3 (3.2%) | 9 (6.7%) |
| Asymptomatic family member | 6 (14.6%) | 51 (54.8%) | 60 (42.5%) |
LMNA (53%), PKP2 (25%), TTN (21%), and MYBPC3 (10%) stand out as genes where a significant percentage of the variants identified occur in splice-sites. 48/134 (36%) of individuals carrying splice-site variants had a diagnosis of ARVC, 36/48 (75%) of these were otherwise genotype-negative. Of the remaining 25%, 2 were carriers of LP variants with the rest VUS. In HiRO, 10/57 (18%) of the unique splice-site variants were found in PKP2; 7/10 (70%) of these variants received a “pathogenic” rating with the rest being VUS.
57% of the variants occurred in the splice sites. The number of variants identified decreased with prolongation of the reading frame with ±6 positions containing 88% of the identified variants. 8% of variants were missense (Fig. 3).
Fig. 3. Position of identified splice-site variants.
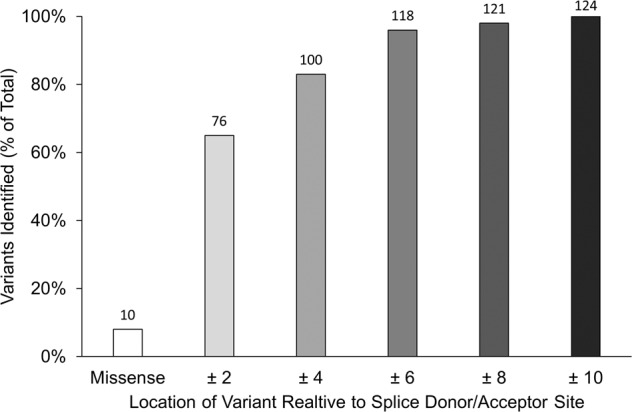
This figure indicates the position of variants relative to the splice donor/acceptor site as a percentage of patient carriers. The label on each column indicates the number of individuals found to carry a variant within the indicated distance from the splice site. 8% of the variants were missense. 57% of variants occurred in the ±2, 85% in ±4, 88% in ±6, 90% in ±8, and 93% in ±10 positions.
Variants identified in a single proband
Forty-one variants were only observed in one individual. The average age of assessment in these patients was 44.3 ± 16.6 years old. 42% of these patients were females, 73% were of Caucasian ethnicity, and ARVC was the most common diagnosis (22%). Most of these patients (68%) were symptomatic probands. A minority (3/41, 7%) also carried a non-splice site variant classified as LP or P, possibly causative of the observed phenotype (Table S3).
PKP2 (NM_001005242.3):c.1034 + 12 C > T (VUS) occurs in the +12 position, however it serves as a variant of interest. This variant presented with different phenotypes in the same family; sudden cardiac death (SCD) in a 19-year-old male with post-mortem findings consistent with ARVC and an asymptomatic 45-year-old female diagnosed with Brugada Syndrome (BrS) following procainamide challenge.
Variants seen in multiple patients
Sixteen unique variants were seen in more than one individual, with 93 patients carrying a variant that was also seen in at least one other individual in the registry. 55% of these patients were asymptomatic first-degree family members (Table 1). Among these patients, only thirteen (13/93, 14%) carried additional non-splice site variants, with eleven patients carrying a VUS, and only 2 patients (2%) with a LP or P variant.
Most splice variants (14/16, 88%) were associated with a single diagnosis across multiple carriers. With respect to the variants associated with more than one diagnosis, LMNA (NM_170708.4):c.1609-3 C > G was seen in ten unaffected individuals, one patient had unclassified cardiomyopathy, and one patient had AV block/Torsades. NEXN (NM_144573.4):c.1053 + 1 G > A was seen in two individuals with LV non-compaction and one patient with BrS (Table S4).
ACMG-AMP classification of variant pathogenicity
Among the 27 PVS1 variants, 25 (92.6%) received a LP or P designation, whereas of the 30 variants that did not receive the PVS1 rating, only 4 (13%) received a LP/P designation (ChiSquare = 44.00, p = <0.0001).
44 variants were classified as PP3, 10 as BP4, and 3 were not classified as less than 3 tools reported a prediction. Of the 28 variants in splice sites, 23 (82%) were classified as pathogenic, with the other 5 variants classified as VUS, despite the PVS1 and PP3 criteria assigned and ultimately due to a lack of supporting data (Tables 2 and S5).
Table 2.
In silico analysis and ACMG classification.
| Gene Variant | In silico prediction | BP4/PP3 | ACMG criteria | Classification |
|---|---|---|---|---|
| ACTN2 (NM_001103.4):c.1255 + 2dupT | 1/1 | PM2 | VUS | |
| CACNB2 (NM_201571.4):c.1122 + 3 A > T | 0/9 | BP4 | BS1, BP4, BP6 | LB |
| CASQ2 (NM_001232.4):c.606 + 1 G > C | 9/9 | PP3 | PVS1, PM2, PP3, PP5 | P |
| DSC2 (NM_024422.6):c.631-2 A > G | 9/9 | PP3 | PVS1, PS3, PP3 | P |
| DSC2 (NM_024422.6):c.943-1 G > A | 9/9 | PP3 | PVS1, PM2, PP3, PP5 | P |
| DSG2 (NM_001943.5):c.523 + 1 G > C | 9/9 | PP3 | PP3 | VUS |
| DSG2 (NM_001943.5):c.1880-2 A > G | 9/9 | PP3 | PS3, PM2, PP3 | P |
| DSP (NM_004415.4):c.939 + 1 G > A | 9/9 | PP3 | PVS1, PS3, PM2, PP3 | P |
| EMD (NM_000117.3):c.265 + 2 T > A | 9/9 | PP3 | PM4, PM2, PP3, PP5 | LP |
| FLNC (NM_001458.5):c.7385-1 G > A | 7/9 | PP3 | PVS1, PM2, PP3 | P |
| GLB1 (NM_000404.4):c.1480-4 G > T | 0/8 | BP4 | BP4 | VUS |
| GLB1 (NM_001317040.1):c.1879-5_1879-4dup | 0/1 | BS1 | VUS | |
| JUP (NM_002230.4):c.1653 + 1 G > A | 9/9 | PP3 | PVS1, PM2, PP3, BP2 | P |
| KCNH2 (NM_000238.4):c.472 + 1 G > C | 9/9 | PP3 | PVS1, PM2, PP3 | P |
| KCNH2 (NM_000238.4):c.1128 G > A‡ | 5/6 | PP3 | PVS1, PM2, PP3, PP5 | P |
| KCNH2 (NM_000238.4):c.2145 G > A‡ | 6/8 | PP3 | PVS1, PM2, PP3, PP5 | P |
| KCNH2 (NM_000238.4):c.2692 + 8 G > A | 2/8 | BP4 | BS1, BS4, BP4 | LB |
| KCNQ1 (NM_000218.3):c.387-5 T > A | 5/7 | PP3 | PM2, PP3 | VUS |
| KCNQ1 (NM_000218.3):c.477 + 1 G > A | 9/9 | PP3 | PVS1, PS3, PP3, PP5, BP2 | P |
| KCNQ1 (NM_000218.3):c.477 + 5 G > A | 5/8 | PP3 | PS3, PP3 | LP |
| KCNQ1 (NM_000218.3):c.921 G > A‡ | 4/8 | PP3 | PS3, PM5, PP3 | LP |
| KCNQ1 (NM_000218.3):c.1033-3 C > T | 0/8 | BP4 | BP4, BP6 | LB |
| KCNQ1 (NM_000218.3):c.1686-1 G > T | 9/9 | PP3 | PVS1, PM2, PP3 | P |
| KRAS (NM_001369786.1):c.*5-3 C > T | 0/5 | BP4 | PM1, PM2, BP4 | VUS |
| LMNA (NM_001282626.2):c.937-1 G > C | 7/9 | PP3 | PVS1, PM2, PP3 | P |
| LMNA (NM_170708.4):c.1609-3 C > G | 6/7 | PP3 | PM2, PP3, PP5, BS4, BP1 | VUS |
| MIB1 (NM_020774.4):c.2050-4 T > C | 0/8 | BP4 | PM2, BP4 | VUS |
| MYBPC3 (NM_000256.3):c.1227-2 A > G | 6/9 | PP3 | PVS1, PM2, PM5, PP3, PP5 | P |
| MYBPC3 (NM_000256.3):c.1624G > C‡ | 7/9 | PP3 | PVS1, PS3, PM5, PP3 | P |
| MYBPC3 (NM_000256.3):c.1897 + 1 G > A | 9/9 | PP3 | PVS1, PM2, PP3, PP5 | P |
| MYBPC3 (NM_000256.3):c.2737 + 1 G > C | 9/9 | PP3 | PVS1, PM2, PP3, PP5 | P |
| MYH7 (NM_000257.4):c.639 + 5 G > T | 5/8 | PP3 | PP3 | VUS |
| NEXN (NM_144573.4):c.1053 + 1 G > A | 9/9 | PP3 | PVS1, PP3, BS4, BP5 | VUS |
| PKP2 (NM_001005242.3):c.224-3 C > G | 5/7 | PP3 | PM2, PP3 | VUS |
| PKP2 (NM_001005242.3):c.337-2 A > T | 9/9 | PP3 | PVS1, PM2, PP3, PP5 | P |
| PKP2 (NM_001005242.3):c.1034 + 1 G > A | 9/9 | PP3 | PVS1, PP3, PP5 | P |
| PKP2 (NM_004572.4): c.1510 + 5 G > A | 4/6 | PP3 | PP3 | VUS |
| PKP2 (NM_004572.4): c.1584 G > A | 1/5 | BP4 | BP4, BP6, BP7 | VUS |
| PKP2 (NM_004572.4):c.1689-1 G > C | 8/9 | PP3 | PVS1, PP3, PP5 | P |
| PKP2 (NM_004572.4):c.1688 + 1 G > A | 9/9 | PP3 | PVS1, PP3, PP5 | P |
| PKP2 (NM_004572.4):c.2146-1 G > C | 8/8 | PP3 | PVS1, PS3, PP3 | P |
| PKP2 (NM_004572.4):c.2299 + 1 G > A | 9/9 | PP3 | PVS1, PM2, PP3, PP5 | P |
| PKP2 (NM_001005242.3):c.2357 + 1 G > A | 9/9 | PP3 | PVS1, PP3, PP5 | P |
| RYR2 (NM_001035.3):c.6022 + 5 G > A | 5/8 | PP3 | PP3, BS1 | VUS |
| RYR2 (NM_001035.3):c.8715-2 A > G | 9/9 | PP3 | PVS1, PM2, PP3, BP2 | P |
| RYR (NM_001035.3):c.9129-10 T > A | 0/7 | BP4 | PM2, BP4 | VUS |
| SCN5A (NM_000335.5):c.392 + 3 A > G | 6/7 | PP3 | PM2, PP3 | VUS |
| SCN5A (NM_000335.5):c.483 G > A‡ | 4/8 | PP3 | PM2, PP3 | VUS |
| SCN5A (NM_198056.3):c.4437 + 5 G > A | 2/5 | BP4 | PP4, BP4 | VUS |
| TRDN (NM_006073.4):c.1322-10 T > A | 6/6 | PP3 | PP3 | VUS |
| TPM1 (NM_001018005.2):c.375-3 C > T | 0/8 | BP4 | BS1, BP4, BP6 | LB |
| TTN (NM_001267550.2):c.7330 + 5 G > C | 7/7 | PP3 | PM2, PP3, BP1 | VUS |
| TTN (NM_001267550.2):c.33340 G > T‡ | 7/9 | PP3 | BP1, PP3 | VUS |
| TTN (NM_001267550.2):c.33911-6_33911-5insG | 2/2 | BS1 | VUS | |
| TTN (NM_001267550.2):c.35875 + 1 G > C | 9/9 | PP3 | PVS1, PP3, BP2 | VUS |
| TTN (NM_001267550.2):c.51739 + 1 G > C | 7/9 | PP3 | PM2, PM4, PP3, PP5, BS4 | LP |
| TTN (NM_133378.4):c.51923-1 G > C | 9/9 | PP3 | PVS1, PM2, PP3 | P |
‡Located at the last base pair position of the exon.
In silico prediction column indicates the number of tools which predicted a pathogenic variant/the number of tools which responded ie. 0/9 indicates that none of the tools predicted a damaging effect out of nine which scored the variant.
After completion of in silico analyses, we reclassified 7 variants based on the ACMG-AMP criteria; 6 were downgraded and 1 upgraded (Table S5). In silico analysis contributed to 3 of these reclassifications; with CACNB2 (NM_201571.4):c.1122 + 3 A > T reclassified from VUS to LB, FLNC (NM_001458.5):c.7385-1 G > A reclassified from LP to P, and KCNQ1 (NM_000218.3):c.477 + 5 G > A reclassified from LP/P to P.
Functional data correlated well with the in silico tests as all 8 of the variants with a PS3 rating also received a PP3 rating; and 5 of these variants were classified as “pathogenic” (62.5%).
76 patients carried variants in the splice donor/acceptor sites, among these 33 (43%) were symptomatic with 31 (41%) having a definite diagnosis. In contrast, of the 58 patients carrying variants outside these sites 23 (40%) were symptomatic (ChiSquare = 0.19, p = 0.66) and 23 (40%) had a definite diagnosis (ChiSquare = 0.018, p = 0.89) (Tables S3 and S4). There was no significant difference between variants within or adjacent to the splice sites with respect to symptomaticity.
An average of 8.96/9 tools scored the 28 variants in the ±1/2 positions and there was good consensus with 8.64/9 tools on average predicting pathogenicity. In contrast, 29 variants outside these positions were scored by an average of 6.5/9 tools with an average of 3.2/9 tools predicting pathogenicity. Insertions and deletions were rarely classified by in silico tools; for the two duplications and one insertion which we screened, a total of 4 tools responded.
In silico tool sensitivity and specificity
DANN and CADD responded most frequently when queried with the variants we analyzed (54/57, 95%). MutationTaster (32/57, 56%) and MES (35/57, 61%) responded least frequently. To calculate sensitivity and specificity, we considered PP3 as a positive and BP4 as a negative prediction. The final classification of LP/P was deemed to be a positive and a LB/B/VUS rating a negative result. For example, a BP4 output and a P rating would be deemed a false negative, while a PP3 and a LP would be a true positive. Every tool was found to have high sensitivity (>80%) in predicting pathogenicity with MutationTaster and MES having a sensitivity of 100%, however MES had a specificity of 0% as it failed to detect any true negatives. For MutationTaster, these values are similar to the sensitivity (89%) and specificity (93%) reported by the developers [14]. Among the other tools, dbscSNV (ADA/RF) (97%), HSF (96%), CADD (93%), and DANN (93%) were sensitive. Specificity was lower for all tools, with SpliceAI (64%), DANN (63%), and CADD (54%) predicting the greatest number of true negatives. These results are summarized in Fig. 4.
Fig. 4. Predictive capability of in silico tools.

The sensitivity (white) and specificity (black) for each tool is displayed as a percentage. Sensitivity was determined from the number of true positives as a ratio of true positives and false negatives. Specificity was determined from the number of true negatives as a ratio of true negatives and false positives. The final classification of LP or P was considered a positive result and any other ACMG-AMP classification considered negative for each prediction. The table lists the false negatives, false positives, true negatives, and true positives that were used to calculate the sensitivity and specificity for each tool.
Discussion
This study of the national HiRO registry has identified variants which may effect the splicing of cardiac genes leading to disease. Trends in prevalence of splice-site variants, genotype-phenotype relationships, variable expressivity, and differential phenotypic presentations are highlighted here. We have also compiled a selection of available in silico tools to aid clinicians in determining the pathogenic potential of rare variants. Among 1315 participants and 1761 gene variants, 134 patients (10.2%) carried intronic splice-region variants. 57 unique variants were identified and this was the sole variant identified in 78% (104/134) of patients. Splice variants were felt to be either likely pathogenic or pathogenic in the majority of cases (30/57, 53%) and correlated well with observed clinical phenotypes (27/57, 47%). In silico analyses aided in reclassification of 7 (12%) variants, with pathogenicity downgraded in the majority, correlating well with the observed clinical findings.
Six new splice acceptor and 2 new donor sites were predicted by HSF/MES; findings which require further in vitro validation (Table S5). One of the splice variants identified was upgraded from LP to P, and five variants were downgraded from: LP/P to LP, VUS to LB, P to VUS, LP/P to VUS, LP to VUS, and P to VUS. For those downgraded, the available functional data were insufficient to support a LP/P rating.
The design of in silico tools is predicated on several assumptions and each has inherent strengths and weaknesses. Newly developed tools tend to be validated against older tools and often improve upon the predictive potential [9]. MutationTaster and MES had the highest sensitivity (~100%); yet responded less frequently than other tools.
A result was provided most frequently by the tools CADD/DANN (54/57, 95%), SpliceAI (52/57, 91%), and dbscSNV (51/57, 89%). Conversely, MutationTaster (32/57, 56%), MES (35/57, 61%), and HSF (36/57, 63%) provided a score least frequently, which was likely due to several factors. Tools such as CADD, SpliceAI, dbscSNV, and DANN do not pick up insertions, deletions, or duplications as they exclusively respond to single nucleotide variants (SNV). NNSplice, HSF, and MES are optimized for variants occurring at splice sites and branch points [10]. The frame of evaluation is limited in some tools such as MES and to a lesser extent dbscSNV (Table S1). Similarly, MutationTaster was originally optimized on data containing variants adjacent to the splice site [14]. Lastly, missense mutations were generally poorly analyzed.
MES had poor specificity (0%) as it failed to detect any true negatives and in fact did not provide a response when queried with any of the variants which we eventually labelled as unlikely to be LP/P. The reasons for this are discussed above and include training on variants adjacent to the splice sites (-3→+6 at the 5’ end and -20→+3 at the 3’ end) and lack of response to silent mutations. With our data set, MES responded the least frequently (35/57, 61%) among the tools tested. Others have shown MES to have a high false positive rate, particularly when analyzing deep intronic variants. We also found this to be the case with 29% (10/35) false positive rate [28].
MES is optimized for detection of variants occurring at splice sites and branch points and at these foci it appears accurate [28]. Other groups have shown this tool to be highly accurate when analyzing splice donor sites [29] and Jian et al. found it both highly sensitive (86%) and specific (92%) [17]. This tool was originally developed and optimized on a relatively small number of ~1800 transcripts [26].
In contrast, HSF and dbscSNV responded with greater frequency (63% and 89%, respectively) and had similarly high sensitivity (~97%) in predicting pathogenicity. CADD had high sensitivity in predicting true positives (93%). This deep learning tool combines 63 annotation scores to predict splicing impact [16]. Specificity was generally poor for all nine tools; SpliceAI (64%), DANN (63%), and CADD (54%) were most accurate at ruling out variant pathogenicity.
A recent publication used functional assessments of 249 VUS to test the predictive capabilities of nine in silico algorithms including: CADD, Splicing Clinically Applicable Pathogenicity Prediction (S-CAP), Modular Modeling of Splicing (MMSplice), and MES [30]. Consistent with our results, they report that SpliceAI outperformed the others. A consensus score (using 4/8 algorithms to support splicing disruption) outperformed each individual algorithms but was marginally worse than SpliceAI, whereas weighted scoring of the algorithms provided only marginal gains compared to SpliceAI [30].
Overall, if selecting a limited number of tools to use clinically, we would recommend the recently developed SpliceAI which uses deep learning; a more flexible methodology compared to most of the other tools. SpliceAI was well rounded: responding for a large number of the variants while being specific/sensitive in predicting the nature of each. CADD and dbscSNV also performed well and may be used to complete a triad of clinically applicable in silico tools. CADD in particular has been shown by other comprehensive studies to perform particularly well in predicting variant pathogenicity [31]. When tested separately, these three tools alone had the same accuracy (93%) as the entire toolset (Fig. 4).
The clinical relevance of variants adjacent to the splice sites is debatable [1, 3]. 57% of our identified variants were found in the canonical sites (Fig. 3), yet a significant number (58/134, 43%) occurred outside these sites. The need for evaluating flanking regions is highlighted by KCNQ1 (NM_000218.3):c.387-5 T > A, identified in a proband and her two children. All three presented with LQTS and 5/7 tools predicted an effect on splicing leading to a PP3 rating. Evidence suggests pathogenicity is due to out-of-frame skipping of exon 2, with the mutant encoding the majority (~85%) of transcripts. Members of two families with LQTS were previously reported as carriers of this variant [32]. RNA analysis in heterozygous carriers showed that as little as 10% functional protein is protective against congenital deafness occurring in Jervell and Lange-Nielsen Syndrome, but is insufficient to maintain normal QT duration [32, 33].
A variant in the +12 position, initially excluded from our analysis due to it’s location, is an interesting example of different phenotypes within a family. It also highlights that it may be worthwhile to extend analysis into deep intronic regions in genotype negative families. This family was identified after the tragic SCD of a 19-year-old male. Post-mortem examination was suggestive of ARVC and the patient was a carrier of PKP2 (NM_001005242.3):c.1034 + 12 C > T. The patient’s mother was asymptomatic on presentation and without clinical evidence of ARVC. She carried the same variant and a positive procainamide challenge yielded a diagnosis of BrS. The relationship between PKP2 variants and BrS has only recently been uncovered, with studies pointing to an overlap in the pathophysiology of BrS and ARVC [34, 35]. This variant highlights the limitations of in silico tools, particularly when considering variants located outside the splice regions. Only 5/9 tools responded when queried, and none indicated an impact on splicing.
Future directions
Future studies may explore the unique predictive capabilities of a myriad of available in silico tools in comparison with those utilized here. MutPred Splice uses machine learning to predict the potential consequences of exonic changes on pre-mRNA splicing [36]. Splicing Prediction in Consensus Elements (SPiCE), is a downloadable program that builds on SpliceSiteFinder-like [1] and MES to predict the impact of spliceogenic variants with high sensitivity and specificity [37]. S-CAP builds on seven tools including CADD and MutPred Splice to develop its own unique features; offering significant sensitivity in detecting splice-disrupting VUS [38]. MMSplice uses neural networks trained on large genomic datasets to predict the effect of variants on splicing and pathogenicity [39]. The Super-QUick Information content Random-forest Learning of Splice variants (SQURILS) algorithm uses interpretable wild-type and variant sequence features for machine learning-based prediction, doing so with high accuracy comparable to CADD and SpliceAI [40]. These programs require raw sequencing data in variant call format (vcf); a limitation which precluded comparison in this study with the nine selected tools. MutPred Splice, SQURILS, and SPiCE can also take single variant genomic position as input. When queried, SPiCE performed well, responding to 93% of the variants, with a sensitivity of 86.2%, and a specificity of 45.8% (Table S6). Despite their unique strengths, these tools do not necessarily have a predictive advantage over those tested here. For example, Rowlands et al. found SpliceAI to outperform both MMSplice and S-CAP [30] while Rentzsch et al. showed that S-CAP outperforms CADD on canonical splice site variants and intronic SNVs [41].
In vitro and in vivo studies on human tissue can define and measure the consequences of genetic variants on splicing, and report effects on RNA metabolism and cell physiology. RNA extracted from venous blood samples can be used to investigate splice variants for select cardiac genes, however may not always replicate or reflect tissue level expression profiles [42]. Human induced pluripotent stem cells (hiPSC) can be derived directly from patients with cardiac disease and subsequently differentiated into hiPSC-derived cardiomyocytes. Transcriptome analysis of hiPSC can be used to discover pathogenic splice variants [43]. Leveraging transcriptomics can create a clearer genotype-to-phenotype picture [44, 45]. By pairing RNA-seq, bioinformatics, and proteomics, a more accurate categorization of a patient’s splicing patterns and gene expression can be obtained [46].
Limitations
Published reports were used to study the impact of variants, however the majority of the variants presented here have not been studied at the RNA or protein level to allow for correlation with clinical data. We limited our study to variants within ±10 base pairs of the splice site to incorporate variants reported by different clinical labs as we lacked access to the sequencing source data. Distant splice regulatory elements are of some importance but were not capture here [4, 8, 47–49]. In addition, in silico tool do not capture aspects of splicing which are actively being studied, for example the role of RNA secondary structures [50]. The patients included in this study were sequenced by different labs at different times.
Conclusion
A significant proportion of patients with inherited arrhythmia and cardiomyopathy syndromes carry clinically relevant variants in the non-coding segments of the DNA: 10% of patients in the HiRO database were found to carry splice-site variants accounting for 8% of all the variants in the database. We tested nine in silico tools, that can predict the impact of these intronic variants. These tools responded most frequently when queried with variants in the canonical splice sites. A number of tools were found to have high sensitivity and moderate specificity in predicting pathogenicity. To allow for rapid risk stratification, without sacrificing predictive accuracy in the clinical setting, we recommend an abridged version of the toolkit comprising of three in silico tools: CADD, SpliceAI, and dbscSNV. Contingent on clinician comfort, consensus between two or three of these tools may be used to guide further investigations or subsequent treatment course. The novel variants explored here, particularly those where in silico tools suggest an impact on splicing and for which functional data is lacking, are an excellent starting point for in vitro characterization.
Supplementary information
Author contributions
Experimental design: KR, BD, RDB, ADK, ZWML. Data acquisition: KR, BD. Data analysis and interpretation: KR, BD, RDB, ADK, ZWML. Manuscript preparation: KR, BD, MC, DC, RDB, ADK, ZWML. Manuscript editting: KR, BD, MC, DC, JDR, RT, MSG, JSH, CSS, SS, CS, CM, PA, HD, RH, LA, RL, CS, AF, JA, SK, BM, WA, JC-T, JJ, MG, MT, RDB, ADK, ZWML.
Funding
This work was supported by the Faculty of Medicine Summer Student Research Program at the University of British Columbia provided to KR. ZL was funded by the Michael Smith Foundation for Health Research and the Cardiology Academic Practice Plan at the University of British Columbia. The study was supported by the Heart in Rhythm Organization (Dr. Krahn, Principal Investigator) that receives support from the Canadian Institute of Health Research (RN380020 – 406814).
Data availability
The data underlying this article are available within the text, appendices, and online supplementary material.
Competing interests
The authors declare no competing interests.
Ethical approval
This project was conducted in compliance with the protocols and principles laid down in the Declaration of Helsinki and approved in full by the Providence Health/University of British Columbia ethics board (REB number H20-00129).
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41431-022-01193-9.
References
- 1.Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic acids Res. 1987;15:7155–74. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang M. Statistical features of human exons and their flanking regions. Hum Mol Genet. 1998;7:919–32. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
- 3.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. doi: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagnall RD, Ingles J, Dinger ME, Cowley MJ, Ross SB, Minoche AE, et al. Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2018;72:419–29. doi: 10.1016/j.jacc.2018.04.078. [DOI] [PubMed] [Google Scholar]
- 5.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 6.Veitia RA, Birchler JA. Dominance and gene dosage balance in health and disease: why levels matter! J Pathol: A J Pathological Soc Gt Br Irel. 2010;220:174–85. doi: 10.1002/path.2623. [DOI] [PubMed] [Google Scholar]
- 7.Moon H, Jang HN, Liu Y, Choi N, Oh J, Ha J, et al. Activation of cryptic 3’ splice-sites by SRSF2 contributes to cassette exon skipping. Cells. 2019;8:696. doi: 10.3390/cells8070696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaganathan K, Panagiotopoulou SK, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–48. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 9.Jian X, Boerwinkle E, Liu X. In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genet Med. 2014;16:497–503. doi: 10.1038/gim.2013.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baralle D, Lucassen A, Buratti E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. EMBO Rep. 2009;10:810–6. doi: 10.1038/embor.2009.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic acids Res. 2003;31:3784–8. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hubbard TJ, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, et al. Ensembl 2009. Nucleic acids Res. 2009;37:D690–D7. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J computational Biol. 1997;4:311–23. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 14.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat methods. 2010;7:575–6. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 15.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:761–3. doi: 10.1093/bioinformatics/btu703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic acids Res. 2014;42:13534–44. doi: 10.1093/nar/gku1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies B, Roberts JD, Tadros R, Green MS, Healey JS, Simpson CS, et al. The hearts in rhythm organization: a Canadian national cardiogenetics network. CJC open. 2020;2:652–62. doi: 10.1016/j.cjco.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Aguilera MA, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35:1978. doi: 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–9. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 24.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc in Hum Genet. 2013:7.20:1–7. [DOI] [PMC free article] [PubMed]
- 25.Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids Res. 2009;37:e67–e. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J computational Biol. 2004;11:377–94. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 27.Sun B, Yao J, Ni M, Wei J, Zhong X, Guo W, et al. Cardiac ryanodine receptor calcium release deficiency syndrome. Sci Transl Med. 2021;13:eaba7287. doi: 10.1126/scitranslmed.aba7287. [DOI] [PubMed] [Google Scholar]
- 28.Desmet F-O, Hamroun D, Collod-Béroud G, Claustres M, Béroud C. Bioinformatics identification of splice site signals and prediction of mutation effects. Global Research Network Publishers; 2010, pp. 1–14.
- 29.Houdayer C, Caux‐Moncoutier V, Krieger S, Barrois M, Bonnet F, Bourdon V, et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum Mutat. 2012;33:1228–38. doi: 10.1002/humu.22101. [DOI] [PubMed] [Google Scholar]
- 30.Rowlands C, Thomas HB, Lord J, Wai HA, Arno G, Beaman G, et al. Comparison of in silico strategies to prioritize rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. Sci Rep. 2021;11:1–11. doi: 10.1038/s41598-021-99747-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson D, Lassmann T. A phenotype centric benchmark of variant prioritisation tools. NPJ Genom Med. 2018;3:1–9. doi: 10.1038/s41525-018-0044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhuiyan ZA, Momenah TS, Amin AS, Al-Khadra AS, Alders M, Wilde AA, et al. An intronic mutation leading to incomplete skipping of exon-2 in KCNQ1 rescues hearing in Jervell and Lange-Nielsen syndrome. Prog biophysics Mol Biol. 2008;98:319–27. doi: 10.1016/j.pbiomolbio.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation of the gene for I sK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of long-QT syndrome. Circulation. 1998;97:142–6. doi: 10.1161/01.CIR.97.2.142. [DOI] [PubMed] [Google Scholar]
- 34.Campuzano O, Fernández-Falgueras A, Iglesias A, Brugada R Brugada Syndrome and PKP2: Evidences and uncertainties. Elsevier; 2016. [DOI] [PubMed]
- 35.Ben-Haim Y, Asimaki A, Behr ER Brugada syndrome and arrhythmogenic cardiomyopathy: overlapping disorders of the connexome? EP Europace. 2020. [DOI] [PubMed]
- 36.Mort M, Sterne-Weiler T, Li B, Ball EV, Cooper DN, Radivojac P, et al. MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol. 2014;15:R19. doi: 10.1186/gb-2014-15-1-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leman R, Gaildrat P, Le Gac G, Ka C, Fichou Y, Audrezet MP, et al. Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in silico/in vitro studies: an international collaborative effort. Nucleic acids Res. 2018;46:7913–23. doi: 10.1093/nar/gky372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jagadeesh KA, Paggi JM, Ye JS, Stenson PD, Cooper DN. S-CAP extends pathogenicity prediction to genetic variants that affect RNA splicing. 2019;51:755–63. [DOI] [PubMed]
- 39.Cheng J, Nguyen TYD, Cygan KJ, Çelik MH, Fairbrother WG, Avsec Ž, et al. MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. 2019;20:48. [DOI] [PMC free article] [PubMed]
- 40.Danis D, Jacobsen JOB, Carmody LC, Gargano MA, McMurry JA, Hegde A, et al. Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Am J Hum Genet. 2021;108:1564–77. doi: 10.1016/j.ajhg.2021.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice—improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021;13:31. doi: 10.1186/s13073-021-00835-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singer ES, Ingles J, Semsarian C, Bagnall RD. Key value of RNA analysis of MYBPC3 splice-site variants in hypertrophic cardiomyopathy. Circ Genom Precis Med. 2019;12:e002368. doi: 10.1161/CIRCGEN.118.002368. [DOI] [PubMed] [Google Scholar]
- 43.Holliday M, Singer ES, Ross SB, Lim S, Lal S, Ingles J, et al. Transcriptome sequencing of patients with hypertrophic cardiomyopathy reveals novel splice-altering variants in MYBPC3. Circulation: Genomic and Precision Medicine. 2021. [DOI] [PMC free article] [PubMed]
- 44.Heinig M, Adriaens ME, Schafer S, van Deutekom HW, Lodder EM, Ware JS, et al. Natural genetic variation of the cardiac transcriptome in non-diseased donors and patients with dilated cardiomyopathy. Genome Biol. 2017;18:1–21. doi: 10.1186/s13059-017-1286-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaudhry F, Isherwood J, Bawa T, Patel D, Gurdziel K, Lanfear DE, et al. Single-cell RNA sequencing of the cardiovascular system: new looks for old diseases. Front cardiovascular Med. 2019;6:173. doi: 10.3389/fcvm.2019.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 48.Ito K, Patel PN, Gorham JM, McDonough B, DePalma SR, Adler EE, et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc Natl Acad Sci. 2017;114:7689–94. doi: 10.1073/pnas.1707741114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Pathophysiological mechanisms of dominant and recessive KVLQT1 K+ channel mutations found in inherited cardiac arrhythmias. Hum Mol Genet. 1997;6:1943–9. doi: 10.1093/hmg/6.11.1943. [DOI] [PubMed] [Google Scholar]
- 50.Ule J, Blencowe BJ. Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol cell. 2019;76:329–45. doi: 10.1016/j.molcel.2019.09.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available within the text, appendices, and online supplementary material.