

Abstract
Numerous expression systems, engineered strains, and cultivation systems have been developed globally but producing recombinant proteins in the soluble form continues to remain a challenge. Escherichia coli, a preferred host for the recombinant production of biopharmaceuticals and other proteins. Up to 75% of human proteins expressed in E. coli have only 25% in an active soluble form. The proteolytic activity of Lon encoded protease triggers the inclusion bodies leading to heterogenous secreted proteins thereby hampering downstream processing and isolation. Putrescine monooxygenases are versatile with applications in iron acquisition, pathogen control, biotransformation, bio-remediation and redox reaction are still isolated from plant and microbial sources at low yields. As a prerequisite to developing protease knockout E. coli strains, using the Cre-loxP recombination strategy we have built a full-length Lon disruption cassette (5′lon-lox66-cre-KanR-lox71-3′lon) (3368 bp) consisting of upstream and downstream regions of Lon, loxP sites, and Cre gene driven by T7 promoter to the expression of Cre recombinase and a selectable kanamycin resistance gene. Here, after the integration of the knock-out cassette into the host genome, we show the production of homogeneous protein species of recombinant Putrescine monooxygenase by using an E. coli platform strain in which Lon gene is deleted. This Lon knock-out strain secreted more homogeneous protein at a volumetric yield of 60% of the wild-type strain.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12088-023-01056-x.
Keywords: Lon gene disruption cassette, Cre/loxP, Solubility, Marker rescue, Putrescine monooxygenase
Introduction
DNA recombinant technology made possible the expression of several proteins in host organisms and provided an acceptable route when compared to natural sources. E. coli BL21 strain is the workhorse of the expression system for recombinant vaccines and enzymes for functional and structural determinations [1]. Preferred for its well-understood genetics, speedy growth, superior productivity and the low cost [2]. Structures deposited in the PDB database are of the recombinant proteins expressed in E. coli [3]. Several strategies made for efficient production of proteins in E. coli, namely, the use of different mutated host strains, co-production of chaperones and foldases, lowering cultivation temperatures, and addition of a fusion partner [4]. Methods improved the soluble production of recombinant proteins in E. coli, whereas the prediction of robust soluble protein production processes is still a challenge and a necessity [5]. Decrease in productivity is due to the formation of inclusion bodies. This is due to lower proteolytic stability due to degradation at either the N or C terminal ends. The recombinant protein degradations are by ATP-dependent proteases [6]. Therefore, for increased production of proteins, minimal proteolysis and use of the E. coli expression system deficient in proteases is a strategy. It is increased by many methods using a strong promoter's efficient translation.
Previous studies reported that up to 75% of human proteins expressed in E. coli are 25% in an active soluble form using this host system. Steps to avoid the inclusion bodies formation consisted of using low-copy number vectors [7], weak promoters [8], low temperature [9], co-expression of chaperones [10], use of solubilizing partner [11] and fermentation at extreme pH values [12]. The unique strategy used to overcome this problem is the use of host strains carrying mutations in which element production of proteases can sometimes enhance the accumulation of desired recombinant protein by reducing photolytic degradation.
Gene knockout methods were developed in high-performance E. coli strain meant for the production of recombinant proteins. RecA-dependent homologous recombination has been tried in E. coli genome manipulation [13]. Manipulation of genes by Plasmid-based DNA cloning and low-efficiency selection steps for obtaining strain mutants is tried. PCR-mediated direct gene manipulations were verified in S.cerevisiae and P.pastori for an efficient mutant generation. Difficulties in the manipulation of the genome of E. coli are because of the intracellular exo-nucleases which degrade linear DNA [14]. Lambda Red recombination systems with efficient homologous recombination between chromosomal DNA and a linear gene knockout DNA fragment containing an antibiotic resistance gene flanked by two homologous arms [15]. Reports of FLP/FRT from S.cerevisiae and Cre/loxP from bacteriophage P1 are developed to knock out genes [16]. The usage of two plasmids for promoting homologous recombination events and one for removal of resistance marker removal flanked by loxP sites [17] is reported.
Cre/lox system causes genetic modifications introduced into different organisms by recycling a single selectable marker. Towards achieving a marker recycling implementation, there needs introduction of heterologous Cre- expression plasmid and then remove it from the resulting recombinants, which is a time-consuming process. This study aimed to the reduction of the inclusion body formation of putrescine monooxygenase by disruption of the Lon gene. Here, we report the Lon gene knockout of the E. coli K-12 host and its effect on enhancing the solubility of putrescine monooxygenase.PCR methods have used the confirmation of the gene knockout in the host. SDS-PAGE analysis along with enzyme activity was used to analyze the product in the supernatant and compared it with the native host.
Materials and Methods
Strains, Plasmids, Primers and Media
The E. coli K-12 strain was used for the disruption of the lon gene. The expression plasmid pET151TOPOSpPMO was used for the expression of putrescine monooxygenase. The Cre gene from bacteriophage P5 was codon-optimized for Cre gene expression in E. coli and was obtained from GeneArt, Germany. The plasmid pET28a-(Cre) [18] was used for the cloning of the Cre gene in frame with T7 promoter and was used to generate the cassette needed for the Lon gene disruption in E.coliK-12 strain. The E. coli DH5α strain was used for routine cloning and propagation. Standard cloning procedures were carried out in as described by Sam brook and Russell [19]. The primers were synthesized by Sigma-Aldrich, India. The ΔLonE.coliK-12 was used for the knockout of Lon gene over-expression of pubA gene coding for the putrescine monooxygenase. Prime STAR GXL premix, Emerald Amp GTPCR master mix and DNA ladder (100 bp and 1 Kb) were purchased from Takara Bio., Japan. Nucleospin plasmid isolation, nucleospin gel and PCR cleanup kits were from Macherey – Nagel, Germany, Ni–NTA agarose was from GE-Health Sciences. For the selection of KanR and Amp resistance in E. coli, LB media was supplemented with kanamycin (50 µg/mL) and ampicillin (100 µg/mL).
PCR Mediated Generation of Core and Full-Length Gene Disruption Cassette
The Lon gene sequence of E. coli K-12 strains was retrieved from the NCBI database with the accession numbers (NC_012971.2. and NC_000913.3) respectively. Primers were designed to amplify 250 bp target regions on the 5′ and 3′ ends of the Lon gene from E. coli BL21 (DE3) strain by using the Primer3 software. The terminator codons TAG and TAA were added to the upstream forward primers just before the actual sequence. The sequences of the designed primers are tabulated in Table 1. The sequence confirmed pET28a ( +)-Cre plasmid was used as a template for generating the core cassette, Cre, T7 promoter and kanamycin resistant gene as marker. The forward primer was designed on the 5′ region of the T7 promoter and overlapped with the lox66 sequence. The reverse primer was designed on the Kanamycin resistance gene and overlapped with the lox71 sequence. The forward primer is Lox66T7pFP and the reverse primer is Lox71KanRP.The lox sites (lox71 and lox66) were incorporated in the primers lox66T7pFP and lox71T7pRP used for PCR. The upstream Lon fragment (LonU) of 250 bp was amplified using the primers lonFP and lox66lonRP1and similarly downstream fragment of 250 bp (LonD) was amplified using the primers lox71lonFP1 and LonRP. The amplified 3 fragments namely LonU, LonD and the core cassette were overlapped by using the primers LonFP/LonRP [20]. The PCR product was gel purified and stored at – 20 °C in deep freezer until further use. The amplified product was run on 0.8% agarose gel, purified by gel extraction, dissolved in ultrapure DNase free water, and used in subsequent transformation experiments.
Table 1.
Strains and plasmids used in this study
| Strain/Plasmids | Genotype | Source |
|---|---|---|
| Escherichia coli | ||
| BL21 Star(DE3) | F– ompT hsdSB (rB–, mB–) gal dcm rne131 (DE3) | Invitrogen |
| DH5α | F– φ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK–, mK+) phoA supE44 λ– thi-1 gyrA96 relA1 | Invitrogen |
| K-12 | FΔ(HindIII)::cat (Tra+ Pil+ CamR)/ ung-1 relA1 dut-1 thi-1 spoT1 mcrA | In-house |
| Plasmids | ||
| pET28a(Cre) | The plasmid carrying Cre- Kanr CORE cassette | Our study |
| pColdIV(Cre) | The plasmid carrying Cre- Ampr CORE cassette | Our study |
| pET151TOPOSpPMO | The plasmid carrying PMO gene-Ampr | Our study |
E. coli Competent Cell Preparation and Transformation
The glycerol stock of bacterial culture of E. coli K-12 cells was streaked on LB agar plates grown overnight at 37 °C and a single colony of E. coli was picked from fresh LB plates and inoculated in 10 ml of LB broth and incubated on a shaker at 37 °C for 10 h. 1 ml of starter culture was inoculated to 100 ml LB broth and incubated at 37 °C until the O.D600 reached 0.4–0.6. Immediately the culture was chilled on ice for 20–30 min by swirling occasionally and then centrifuged at 8000 rpm for 8 min at 4 °C and the pellet was re-suspended in 100 ml ice-cold sterile ddH2O and centrifuged at 8000 rpm for 8 min at 4 °C. Repeated the washing step for 2 times and re-suspended the cell pellet in 200 µl of 15% glycerol. Aliquots (50 µl) were pipette into sterile micro-centrifuge tubes stored at -80 °C. For transformation, electro-competent E. coli DH5α cells were mixed with a full-length Lon disruption cassette of 0.6 and 1.2 µg and subjected to pulse at 1800 V, 25 µF, and 200 Ω. The transformed cells were recovered by spreading on LB agar supplemented with kanamycin (50 µg/mL) for the selection of positive transformants.
Curing of the Transformants and Screening
The kanamycin resistant cells were screened by PCR and to validate the disruption of the Lon locus. PCR was performed at 5′ region using the primers designed as LonChromFP and CreRP. Confirmation of the integration was done by PCR at 3′ region using KanFP and LonChromRP. The PCR was carried out on all the clones to confirm the stability of the integration. After confirmation, the clones were further subjected to induction using IPTG for rescuing the marker. The expression of Cre recombinase supports the recombination event thus making the host kanamycin (30 µg/ml) sensitive. LB agar plates with the absence and presence of kanamycin were used for plating the clones to confirm that the host is cured and becomes kanamycin sensitive. PCR was performed using the LonChromFP and LonChromRP primers designed on the genomic locus for final confirmation. Clones numbered as #7 and #5 were selected for further expression studies.
Heterologous Expression of pubA Gene in Lon Knockout E. coli K-12 Cells
The purified pET151TOPOSpPMO plasmid contained the pubA gene coding for putrescine monoxygenase.1 µl of the plasmid is transformed into competent Lon knockout E.coli K-12 cells and after incubation at 37 °C for 2 h, only 30–50µL of bacterial suspension was plated. Next day, a pure culture of the Lon knockout E.coliK-12 cells containing pET151TOPOSpPMO plasmid was obtained and used to inoculate 10 ml of LB media containing ampicillin (100 µg/ml). The culture is incubated overnight at 37 °C with continuous shaking at 200 rpm. Then sub cultured flask was incubated at 37 °C until the OD 600 nm reached 0.5–0.8. For induction, IPTG (0.5-1 mM) was added to the culture flask. The temperature was set to 37 °C and grown for 4 h at 200 rpm. Un-induced samples were collected at zero hour and induced samples were collected at every hour of induction and were processed for the determination of cell biomass (wet weight) and expressed protein by SDS-PAGE [21]. The percentage of soluble and total expressed protein was estimated from total lysate and the supernatant from the harvested cell pellet of each protein by Bradford method [22]. The assay of SpPMO was determined by a modified Csaky iodine oxidation method [23].
Results and Discussion
The proposed gene knockout strategy involves a site-specific recombination event where the integration of the disruption cassette/knock-out cassette into the host genome takes place, followed by DNA excision catalyzed by the integrated Cre recombinase. For the site-specific integration of the knock-out cassette, it has to be flanked by upstream and downstream homologous regions of the targeted gene. Here, in this study, we have chosen Lon target gene to be knocked out from the E. coli K-12 cells (Table 1). Hence, the target gene sequences were retrieved from the host strains, and 250 bp upstream and downstream target regions of the target genes were identified (Table 2).
Table 2.
Primers used in the study
| Name of Primers | Primer abbreviation | Target region | Sequence (5′-3′) |
|---|---|---|---|
| Lon FP | P1 | 5′ Lon upstream | ATGTAGTAAAATCCTGAGCGTTCTGAACGCA |
| Lox66lLonRP1 | P2 | CATTATACGAACGGTAGCAGTTTCAGCATCTGC | |
| Lox71lonFP1 | P3 | 3′ Lon downstream | ATGCTATACGAACGGTAAATGACCGGTGAGATCAC |
| Lon RP | P4 | CTATTTTGCAGTCACAACCTGCATGCCAGA | |
| Lox66T7pFP | P9 | Core cassette | TACCGTTCGTATAATGTATGCTATACGAAGTTATTAATACGACTCACT |
| Lox71KanRP | P10 | TACCGTTCGTATAGCATACATTATACGAAGTTATATGAGCCATATTCAAC | |
| LonChrom FP | P11 | Lon cassette integration | CGGTCACGGAACATGTCCATC |
| LonChrom RP | P12 | CTTAATAAGGGCAAGCCCGATC | |
| Cre RP | P15 | 5′-region | CGGTCACGGAACATGTCCATC |
| Kan FP | P16 | 3′- region | CAACTCTGGCGCATCGGGCTTC |
Construction of the Lon Gene Disruption Cassette
The strategy for the Lon gene disruption using the Cre/loxP mechanism used in the study is depicted pictorially in Fig. 1. The individual fragments were amplified by PCR and assembled by overlap PCR to form a disruption cassette. The primers were designed in such a way that there was efficient overlap of the fragments. As and when the disruption cassette is transformed to the host, the cassette gets inserted into the genome at the locus of Lon gene by double cross over homologous recombination event. Thereby the Cre gene would fit in the frame with the T7 promoter, upon induction using IPTG, the recombination event takes place and then kanamycin marker gene is removed as seen in the Fig. 1.Due to the unavailability of the genome sequence of the E. coli BL21 Star (DE3) host strain, we have retrieved the lon gene sequence from BL21 (DE3) strain which is the mother strain. The 5′ upstream and 3′ downstream target regions of 250 bp in Lon were identified. Designing primers to amplify approximately 200 to 250 bp sequences upstream and downstream of the fragment was done by taking target regions of Lon gene from E.coli BL21 (DE3) strain (NC_012971.2:427,922–430,276). During the designing of primers a terminator codon TAA is added to upstream forward primers just before the actual sequence. The addition of the terminator codon will avoid any leaky expression even after knockout. Amplification of upstream was done by lonFP/P6RP and downstream fragments using LonRP /P7FP.
Fig. 1.

Schematic work-flow of the knockout of Lon gene in E coli K-12 strain using a Crelox cassette. The Lon 5′ target region (U) and Lon 3′ target region (D) represent sequences (250 bp) upstream and downstream of the Lon ORF respectively. The 34 bp lox sequences (lox66 and lox71) are direct repeats located adjacent to the 250 bp sequence. It flanks the chromosomal Lon gene sequence to be deleted, the T7 promoter, Cre recombinase ORF, the T7 transcription terminator, a marker KanR ORF mediating kanamycin resistance. A double homologous recombination event replaced the Lon ORF in the genome with the crelox cassette. Growth of recombinant cells on IPTG induced the production of the Cre recombinase which recognized the two Lox sites and excised the inner sequence, leaving only one lox site in the genome
The Lon gene upstream and downstream fragments were amplified by PC of sizes 250 bp (Fig. 2a). The CORE cassette was constructed by PCR using primers lox66T7pFP, Lox71KanRP and pET28a( +)Cre plasmid as a template. The restriction sites BamHI and HindIII were incorporated at the 5′ and 3′ ends for cloning of the gene in the same sites into the pET28a vector in frame with the strong T7 promoter. The plasmid pET28a-Cre was used as the template for generating the CORE cassette which contains the lox sites along with Cre and kanamycin in the cassette. The amplified product was 2845 bp (Fig. 2b). The upstream and downstream homologous regions were amplified separately and gel purified. The reverse primer of upstream and forward primer of downstream fragment respectively share 30 bp common nucleotides with the CORE cassette. So, the two homologous fragments overlapped with the CORE cassette by 30-40 bp to allow an efficient fusion. The Lon sites (lox66 and lox71) were incorporated in the primers used for PCR. The forward primer was designed on the 5′ region of the T7 promoter and overlapped with the lon66 sequence. The reverse primer was designed on the kanamycin resistance gene and overlapped with the lox 71 sequence. Using the primers Lox66T7pFP and Lox71KanRP and plasmid pET28a-Cre as the template the amplification of the core cassette (2845 bp) was achieved. The purified core cassette fragment was of high purity with an A260/280 of 1.78 and 140ηg/µL concentration. Thus obtained purified product was used as the template in the construction of a full-length knock-out cassette.
Fig. 2.

a Amplification of the 250 bp target regions of the Lon gene of E. coli K-12 strain. Lane M: 100 bp DNA ladder, Lane 1: PCR product of lon upstream target region (DE3), Lane 2: PCR product of Lon downstream target region. b Amplification of core cassette fragments. Lane M: 1 kb DNA ladder, Lane1: CORE cassette amplicon (lox71-T7promoter-Cre-Kanr-lox66). c Construction of full-length Lon disruption cassette. Lane M: 1 Kb DNA ladder, lanes 1 and 2 show the full-length Lon disruption cassette (5′Lon-lox66-Cre-KanR-lox71-3′Lon) of 3351 bp size after the overlap of 5′Lon upstream fragment, CORE cassette, and 3′Lon downstream fragment
The three fragments of the full length gene disruption cassette are the CORE cassette, Lon gene upstream and Lon gene downstream fragments and were aligned by overlap PCR using LonFP, and LonRP primers to finally get the cassette size of 3351 bp (Fig. 2c). The constructs were sequence verified and were linear before the transformation.
Lon gene Disruption in the E coli k-12 Host
The disruption of Lon gene locus was established by PCR using the primers planned on the genomic Lon locus and an internal primer within the disruption cassette specifically LonChrom FP and Cre RP at the 5′end and on amplification an anticipated product size of DNA fragment of 750 bp was seen (Fig. 3a). Using the primers KanFP and LonChrom RP at the 3′end the PCR showed an amplified product of anticipated product length of 1000 bp (Fig. 3a). Four positive clones were selected and passaged for 50 generations. This confirmed the stability of integration once again by the PCR using the external and internal primers. The clone#5 and Clone#7 were selected and subjected to induction using an inducer IPTG for expression of Cre recombinase and then checked for kanamycin cured host by PCR. During the recombination event, there is excision of Lon gene partially leads to a smaller size fragment compared to the native locus on the wild type host. This confirmation was done using the LonChromFP and LonChrom RP on the ΔlonE.coliK-12 host and the parent E. coliK-12 host. The lon Knockout host amplified the expected product size of 1928 bp and the E. coliK-12 parent amplified the expected size of 2450 bp (Fig. 3). These results validated the disruption of the Lon gene in E. coliK-12 strain.
Fig. 3.

a PCR verification of the double homologous recombination of crelox cassette in the Lon locus of Kanamycin resistant Lon knockout hosts (Clone#5 and Clone#7) at the 5′ end and 3′end regions confirms the integration of the cassette at the correct locus. The 5′end, 3′end and full-length PCR as done using the primers: LonChromFP and CreRP, KanFP and LonChromRP and LonFP and Lon RP respectively. b Lane M: DNA ladder, Lane 2: Amplified product size of 2450 bp Lane 4:750 bp Lane 6:1000 bp. Lane 1, 3 and 5 do not show bands for the integration of the cassette in Clone#5
Sensitivity towards Kanamycin was tested for Kanamycin sensitivity by plating the Clones on LB plate without and with Kanamycin. There was no removal of the antibiotic cassette and no transformation with the Cre recombinase expression plasmid as there is inbuilt Cre recombinase within the cassette. During the curing of the kanamycin-resistant transformants were selected and grown with antibiotic and without antibiotic at 30 °C. After culturing at 30 °C, a large majority of transformants lost the lox flanked antibiotic gene and thus, were fully antibiotic susceptible. The resulting strains were tested by PCR using primers flanking the deleted region. There was no growth of the clones in the plates containing kanamycin which confirmed that the KanR gene is lost with the deletion of cassette and lead to the sensitivity. Two clones namely Clone #7 and Clone #5 were selected and used for further growth studies. The growth characteristic of Clone #7 was determined in comparison with the parent strain to confirm the effect of the gene knockout on the growth indicators. A growth curve was plotted and found that the knockout had no change on the growth as depicted in Fig. 4. The growth studies of the ΔLonE.coliK-12 host in comparison with E. coliK-12 showed analogous growth characteristics. The knockout of the Lon gene does not have any unfavorable effect on the characteristics of the cells during their growth for 12 h and similar results are seen during the growth studies of P. pastoris strain GS115 (ATCC 20,864) after disruption of KEX1 gene [10].
Fig. 4.
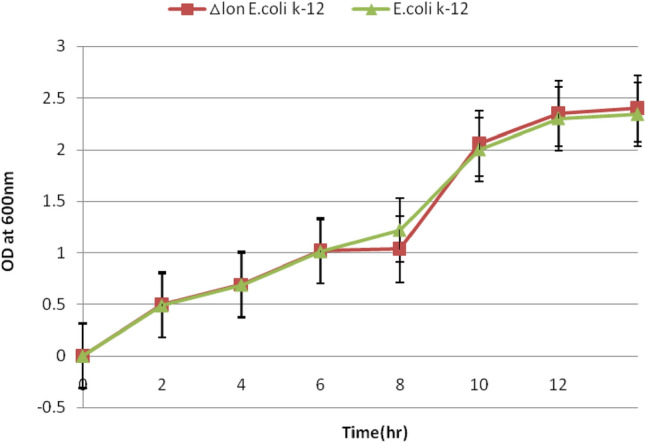
The growth studies of the ΔLon E.coli K-12 host in comparison with parent E. coli K12 strain shows analogous growth characteristics. The knockout of the Lon gene does not have any unfavorable effect on the characteristics of the cells during their growth for 12 h
Expression of the pubA Gene Coding Putrescine Monooxygenase
The photo plasmid of Putrescine monooxygenase was not liberalized and was transformed into E. coli K-12 cells. ΔlonE.coliK-12 host by electroporation as described previously. The transformations were selected on LB media. Clones that were resistant to ampicillin were selected and subjected to induction using IPTG. The expression of putrescine monooxygenase was analyzed by SDS PAGE and out of the eighteen clones, Clone#18 consistently gave higher expression productivity of putrescine monooxygenase and was chosen for over expression.
The earlier usage of E. coli BL21 (DE3) cells as expression host produced initial minimum insoluble SpPMO after overnight incubation at 20℃ in LB medium while incubator at 37℃ resulted in large amounts of SpPMO as completely insoluble inclusion bodies [23]. Similar results were seen during the expression of MbsG gene in the pVP55A vector [24]. Figure 5 shows the improved solubility of SpPMO expressed in the cell line ΔlonE.coli K-12 with 90% soluble protein and total activity was reported to be 120.110 ηnmol/min compared to the total activity expressed in cell free extract of 85.110ηmol/min. The specific activity remained unchanged and showed similar to 9.15 0.5µmolmin−1 mg−1.The comparison of the solubility percentage of expressed SpPMO in ΔLon E.coli K-12 and native cells is summarized in Table 3. The best results in terms of solubility of SpPMO were achieved with ΔlonE.coli K-12 cells at 37℃. There was no need of co-expression with chaperone GroES/EL plasmids needed for proper folding. The error rate for the determination of protein expressed in soluble form and the total expressed form was ± 5%. There was a 16% increase in the solubility at 37℃ and a 33% increase in solubility at 20℃. Densitometry analysis of expressed bands in SDS-PAGE gel showed SpPMO band from Δlon E.coli K-12 to be 15% denser than SpPMO band isolated from wild-type E. coli K-12 cells.
Fig. 5.
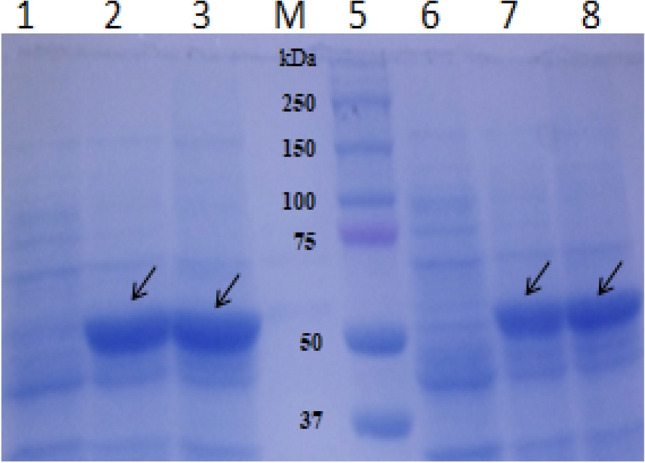
Comparison of Putrescine monooxygenase expression level in Lon knockout E. coliK-12 and wild type E. coliK-12 cells containing plasmid pET151TOPOSpPMO Lane1: Un-induced knockout transformant, Lane2: Induced knockout transformant for 3 h at 37℃, Lane3: Induced knockout transformant for 4 h at 37℃, Lane M: Dual colour precision plus protein standards, Lane5: Un-induced wild-type transformant, Lane6: Induced wild-type transformant for 3 h at 37℃, Lane7: Induced wild-type transformant for 4 h at 37℃
Table 3.
Comparison of the solubility percentages of determined SpPMO in knockout cells and wild-type E. coli K-12 grown at three different temperatures after IPTG induction
| Cell line | Solubility % | |||
|---|---|---|---|---|
| 37℃ | 25℃ | 16℃ | ||
| Δlon E. coli K-12 | Determined SpPMOT | 69 | 70 | 69 |
| Soluble SpPMOS | 58 | 60 | 62 | |
| E.coli K-12 | Determined SpPMOT | 67 | 68 | 70 |
| Soluble SpPMOS | 25 | 35 | 38 | |
T: % of the total SpPMO determined with respect to total determined protein at the time of
maximum folding S: % of the total SpPMO determined with respect to total SpPMO
Bioavailability of iron is a major challenge faced by saprophytic microorganisms. In order to tackle this, they produce iron-chelating molecules called siderophores. N-hydroxylating monooxygenases (NMO) are one of the principle enzymes involved in the production of hydroxamate siderophores [24]. In our previous study, the putrescine monooxygenase enzyme was expressed in E coli BL21 Star DE3 cells and was present as inclusion bodies. Secondly, chaperones were co-expressed to assist the heterologous expression of putrescine monooxygenase from Shewanella putrefaciens 95 [25]. The solubility increased to around 10%. Earlier a novel knockout strategy to enhance recombinant protein expression in E coli was created for blocking the cellular stress response and high levels of recombinant asparginase [26]. Researchers cloned and disrupted the Lon gene and studied the regulation of its expression in Streptomyces lividans. Lon mutant grew more slowly than the wild type and spore germination was impaired at high temperatures.
Previously described reports/reviews suggest creating protease deficient strain either by ultraviolet-causing mutagenesis or by site-directed mutagenesis [27]. The studies using UVM and SDM techniques allow the gene to be mutated but no maximum abolishment of proteolytic activity are observed. Therefore, in such situations, knock out of the genes encoding Lon and Ompt proteases in E. coli by Cre/lox mechanism was adapted here. We have also built a full-length Lon disruption cassette (5′Ompt-lox66-cre-KanR-lox71-3′Ompt) of size 3368 bp consisting of upstream and downstream regions of Ompt, loxP sites, and Cre gene driven by T7 promoter to the expression of Cre recombinase and a selectable Kanamycin resistance gene (Supplementary data Fig.S3). Double knock out combinations to be created from the best performing single knock outs leading to a further enhancement in expression levels. Using the Cre/lox mechanism, total inactivation of the proteolytic enzyme is achieved without hampering the growth of the host organism [28]. The mechanism recognized the lox P sites and catalyzed the recombination between the two loxP sites. We knocked out the targeted protease gene and marker gene (used for selection) using the Cre lox mechanism.
Construction of two new Lon/OmpT gene disruption cassettes with Kanr marker genes and Cre/loxP recombination system of bacteriophage P1 is carried out. Cre/loxP system has been integrated into the E. coli genome for further use in marker removal. PCR amplification of the upstream and downstream regions of Lon and OmpT target regions from E. coli BL21 Star (DE3) and Origami 2(DE3) was achieved. E. coli codon-optimized gene sequence of Cre recombinase deposited to NCBI GenBank with an accession no. (MK033600). E. coli codon-optimized Cre gene was cloned into the pColdIV vector and Cre gene expression studies failed to show that the Cold Shock promoter A (CspA) is compatible with obtaining a highly soluble recombinant Cre recombinase. In comparison to the pColdIV vector, the T7 promoter-based pET28a( +) vector system enhanced the production of soluble Cre recombinase in E. coli BL21 Star (DE3) host cells. In-vitro cleavage assay showed that the purified Cre recombinase had comparable activity and stability profile with the commercial sources. We constructed the core cassette (2777 bp) containing T7 promoter, Cre gene and kanamycin resistance gene sequence, derived from pET28a ( +) Cre plasmid.
E. coli has become one of the most important model organisms in biological sciences and the main workhorse in biotechnology. Thus remain a few challenges in obtaining the recombinant enzymes in the soluble form. Protease deletion from the E. coli genome system allowed the recombinant proteins to bypass the host defense mechanism and express the foreign genes in an active soluble form. Lon-encoded protease is an essential component of the protein quality-control systems that have evolved in all cells to protect against the harmful effects of unfolded proteins. OmpT protease is a subtype of the family of Ompt in proteases, which are found on some gram-negative species of bacteria and encode housekeeping protease that degrades foreign peptide material that the bacteria encounter. Deletion/ disruption of these protease genes from the E. coli system allows the recombinant proteins to bypass the host defense mechanism and expresses the foreign genes in active soluble form [9]. Pre-requisite to knocking out these specific proteases from the E. coli host genome, is to build a disruption knockout cassette/ recombination cassette (Fig. 1).
The genetic tools that we constructed allow the application of the Cre/loxP system in E coli to generate marker-free small deletions. However, obtaining clones where in the second event of homologous recombination may require several rounds of passaging in non-selective media. Occasionally, chromogenic screening is hampered by the natural pigmentation of the colonies in some strains. Use of the counter-selectable markers often implies the use of a mutant strain [9].The steps based on homologous recombination are always associated with the possibility of selecting or screening the desired events by antibiotic resistance, a simple and robust procedure. The excision of the Lon gene using the Cre/loxP system resulted in a very high (> 95%) excision frequency [29], so there is no need for selection of the desired mutant (supplementary data). Later to the excision, we see a small scar of 33 bp and no exogenous DNA is retained in the final strain.
The findings of this study represent a step toward developing a protease knock-out E. coli created by Cre-lox mechanism. Expression of Cre-recombinase catalyzed Cre-lox technology would delete the mentioned protease genes from the genome of two E. coli strains along with the rescue of the marker gene. Understanding this technology gets us closer to the minimal genome concept. Eliminates the certain protease genes to enhance the capacity of E.coli to synthesize more soluble recombinant proteins. Taking together all the research findings towards synthetic genomes of E.coli leads us to a path of bacterial molecular genetics that gains importance in many fields of biotechnology such as the production of molecules/ drugs for human well-being.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Funding by Vision Group for Science and Technology, Govt. of Karnataka, (GRD No.869)
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Demain AL, Preeti V. Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv. 2009;27(3):297–306. doi: 10.1016/j.biotechadv.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Gupta SK, Shukla P. Advanced technologies for improved expression of recombinant proteins in bacteria: perspectives and applications. Crit Rev Biotechnol. 2016;36(6):1089–1098. doi: 10.3109/07388551.2015.1084264. [DOI] [PubMed] [Google Scholar]
- 3.Bird LE. High throughput construction and small scale expression screening of multi-tag vectors in Escherichia coli. Methods. 2011;55:29–37. doi: 10.1016/j.ymeth.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbial Biotechnol. 2003;60:523–553. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 5.Jana S, Deb JK. Strategies for efficient production of heterologous proteins in Escherichia coli. Appl Microbiol Biotechnol. 2005;67:289–298. doi: 10.1007/s00253-004-1814-0. [DOI] [PubMed] [Google Scholar]
- 6.Rozkov A, Schweda T, Veide A. Dynamics of proteolysis band and it s influence and on the accumulation of intracellular recombination proteins. Enzyme Microb Technol. 2000;27(10):743–748. doi: 10.1016/s0141-0229(00)00294-5. [DOI] [PubMed] [Google Scholar]
- 7.Pacheco B, Crombet L, Loppnau P, Cossar D. A Screening strategy for heterologous protein expression in Escherichia coli with the highest return of investment. Protein Expr Purif. 2012;81:33–41. doi: 10.1016/j.pep.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 8.Studier FW, Daegelen P, Lenski RE, Maslov S, Kim JF. Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E coli B and K-12 genomes. J Mol Biol. 2009;394(4):653–80. doi: 10.1016/j.jmb.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 9.Hui CY, Guo Y, He QS, Peng L, Wu SC, Cao H, Huang SH. Escherichia coli outer membrane protease OmpT confers resistance to urinary cationic peptides. Microbiol Immunol. 2010;54(8):452–459. doi: 10.1111/j.1348-0421.2010.00238.x. [DOI] [PubMed] [Google Scholar]
- 10.Sreenivas S, Krishnaiah SM, Anil HS, Mallikarjun N, Govindappa N, Chatterjee A, Kedarnath NS. Disruption of KEX1 gene reduces the proteolytic degradation of secreted two-chain Insulin glargine in Pichia pastoris. Protein Expr Purif. 2016;118:1–9. doi: 10.1016/j.pep.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Yan X, Yu H, Hong Q, Li S. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl Environ Microbiol. 2008;74(17):5556–5562. doi: 10.1128/AEM.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian W, Song H, Liu Y, Zhang C, Niu Z, Wang H, Qiu B. Improved gene disruption method and Cre-loxP mutant system for multiple gene disruptions in Hansenula polymorpha. J Microbiol Methods. 2009;79:253–259. doi: 10.1016/j.mimet.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Dutra BE, Sutera VA, Lovett ST. RecA-independent recombination is efficient but limited by exonucleases. PNAS. 2007;104(1):216–221. doi: 10.1073/pnas.0608293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werten MW, Van den Bosch TJ, Wind RD, Mooibroek H, DeWolf FA. High yield secretion of recombinant gelatins by Pichia pastoris. Yeast. 1999;15:1077–1096. doi: 10.1002/(SICI)1097-0061(199908)15:11. [DOI] [PubMed] [Google Scholar]
- 15.Serra-Moreno R, Acosta S, Hernalsteens JP. Use of the lambda Red recombinase system to produce recombinant prophages carrying antibiotic resistance genes. BMC Molecular Biol. 2006;7:31–37. doi: 10.1186/1471-2199-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos LDF, Caraty-Philippe L, Darbon E, Pernodet J-L. Marker-free genome engineering in amycolatopsis using the pSAM2 site-specific recombination system. Microorganisms. 2022;10:828. doi: 10.3390/microorganisms10040828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma MS, Mukherjee AK. Genome engineering for improved recombinant protein expression in Escherichia coli. Microb Cell Fact. 2014;19(13):177. doi: 10.1186/s12934-014-0177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srividya D, Anil HS, Saroja NR. Expression and purification of codon-optimized Cre recombinase in E. coli. Protein Expr Purif. 2020;167:105546. doi: 10.1016/j.pe.2019.105546. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook, J, Russell, DW (2001) Molecular cloning: a laboratory manual; cold spring harbor laboratory press: cold spring harbor, NY, USA, 2001; ISBN 0879695773
- 20.Nikolai A, Shevchuk AV, Bryksin YA, Nusinovich F, Cabello C, Margaret S, Stephan L. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucl Acids Res. 2004;32(2):16–19. doi: 10.1093/nar/gnh014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laemmli UK. Cleavage of structural proteins during the assembly of the head of Bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Bradford MM. A rapid and sensitive method for the quantization of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 23.Reeder R, Sobrado P. Substrate binding modulates the activity of Mycobacterium smegmatis G, a flavin-dependent monooxygenase involved in the biosynthesis of hydroxamate-containing siderophores. Biochemistry. 2011;508:489–8496. doi: 10.1021/bi200933h. [DOI] [PubMed] [Google Scholar]
- 24.Visser MB, Majumdar S, Hani E, Sokol PA. Importance of the ornibactin and pyochelin siderophore transport systems in Burkholderia cenocepacia lung infections. Infect Immun. 2004;72(5):2850–2857. doi: 10.1128/iai.72.5.2850-2857.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saroja NR, Anil HS, Srividya D, Supreetha K (2019) Chaperone-assisted expression and purification of putrescine monooxygenase from Shewanella putrefaciens-95 Protein Expr Purif, 157:9–16. doi: 10.1016/j.pep.2019.01.006. [DOI] [PubMed]
- 26.Khushoo A, Pal Y, Mukherjee KJ. Optimization of extracellular production of recombinant asparaginase in Escherichia coli in shake-flask and bioreactor. Appl Microbiol Biotechnol. 2005;68(2):189–197. doi: 10.1007/s00253-004-1867-0. [DOI] [PubMed] [Google Scholar]
- 27.Ohta T, Sutton MD, Guzzo A, Cole S, Ferentz AE, Walker GC. Mutations affecting the ability of the Escherichia coli UmuD′ protein to participate in SOS mutagenesis. J Bacteriol. 1999;181(1):177–185. doi: 10.1128/jb.181.1.177-185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma AK, Shukla E, Janoti DS, Mukherjee KJ, Shiloach J. A novel knock out strategy to enhance recombinant protein expression in Escherichia coli. Microb Cell Fact. 2020;19:148. doi: 10.1186/s12934-020-01407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan X, Yu H, Hong Q, Li S. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl Environ Microbiol. 2008;74:5556–5562. doi: 10.1128/AEM.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.