

Graphical abstract

Keywords: HER2 transcriptional downregulator, ELF3-MED23 interaction, Small molecule PPI inhibitor, Hotspot identification, Trastuzumab resistance
Highlights
-
•
ELF3-MED23 hotspot was efficiently deciphered by using customized small molecules.
-
•
ELF3-MED23 PPI depends on specific H-bondings to upregulate HER2 expression.
-
•
PPI inhibitory activity of YK1 was specific to ELF3-MED23.
-
•
YK1-mediated specific H-bond interruption induces promising anticancer activities.
-
•
Anticancer effect of YK1 was even significant against trastuzumab-refractory clones.
-
•
YK1 held highly drug-like properties showing favorable PK profile.
Abstract
Introduction
HER2 overexpression induces cancer aggression and frequent recurrences in many solid tumors. Because HER2 overproduction is generally followed by gene amplification, inhibition of protein–protein interaction (PPI) between transcriptional factor ELF3 and its coactivator MED23 has been considered an effective but challenging strategy.
Objectives
This study aimed to determine the hotspot of ELF3-MED23 PPI and further specify the essential residues and their key interactions in the hotspot which are controllable by small molecules with significant anticancer activity.
Methods
Intensive biological evaluation methods including SEAP, fluorescence polarization, LC-MS/MS-based quantitative, biosensor, GST-pull down assays, and in silico structural analysis were performed to determine hotspot of ELF3-MED23 PPI and to elicit YK1, a novel small molecule PPI inhibitor. The effects of YK1 on possible PPIs of MED23 and the efficacy of trastuzumab were assessed using cell culture and tumor xenograft mouse models.
Results
ELF3-MED23 PPI was found to be specifically dependent on H-bondings between D400, H449 of MED23 and W138, I140 of ELF3 for upregulating HER2 gene transcription. Employing YK1, we confirmed that interruption on these H-bondings significantly attenuated the HER2-mediated oncogenic signaling cascades and exhibited significant in vitro and in vivo anticancer activity against HER2-overexpressing breast and gastric cancers even in their trastuzumab refractory clones.
Conclusion
Our approach to develop specific ELF3-MED23 PPI inhibitor without interfering other PPIs of MED23 can finally lead to successful development of a drug resistance-free compound to interrogate HER2 biology in diverse conditions of cancers overexpressing HER2.
Introduction
Protein–protein interactions (PPIs) are essential phenomena in the human body and are frequently dysregulated in disease states. Despite the essential function of PPIs in humans, efforts to disrupt them have been challenging because PPI interfaces are mostly large and shallow, generally lacking well-defined binding sites. Due to this limitation, while numerous antibody drugs have reached clinical use as prominent PPI inhibitors, only a few of small molecule PPI modulators have been discovered. This is because it is difficult to identify a small-sized ligand capable of binding to the PPI interface with a high affinity [1]. A set of drawbacks related with protein or peptide agents, such as poor cell internalization, low oral bioavailability, and high immunogenicity, have continuously led to various attempts to develop small molecule PPI inhibitors as feasible solution to overcome most of the problems aforementioned [2]. Developing a small molecule modulator for a specific PPI requires identification of ‘hotspot’ for each PPI, representing a small region within the PPI interface that is primarily responsible for the binding affinity between two proteins [3]. Experimental determination of these hotspots is still considered time-consuming and laborious process, despite the various methodological advancements made in this field [4]. Among numerous PPIs of which the hotspot residues are yet unclarified, in this study, we focused on the interaction between E74 Like ETS Transcription Factor 3 (ELF3, also known as ESE-1, ERT, JEN, and epithelium specific Ets transcription factor) and mediator complex subunit 23 (MED23, also known as DRIP130, SUR2, CRSP130, and vitamin D3 receptor interacting protein). ELF3-MED23 interaction is one of the well-known PPIs, which is responsible for regulating the gene expression of Human epidermal growth factor receptor 2 (HER2) (Human gene database (https://www.genecards.org/)).
HER2, a member of the HER family, plays a key role in diverse tumorigenic processes by regulating cell survival, proliferation, and differentiation [5], [6]. HER2 overexpression was first observed in breast cancer and critically assessed as a typical therapeutic marker for breast cancer [7], but nowadays, many reports have also suggested its negative prognostic role in gastrointestinal cancers, especially in gastric cancer, demonstrating its association with cancer aggressiveness and high recurrence rates [8]. For these HER2 overexpressing cancer subtypes, chemotherapy regimens are generally used in combination with trastuzumab (TZMB), an antibody drug that targets the extracellular domain of HER2 protein [9], [10]. Despite clinical successes achieved by TZMB administration, frequently occurring resistance development issues are continuously leading to need for a new, more sustainable therapeutic intervention for HER2-overexpressing cancers. Since overexpression of HER2 is followed by the gene amplification in many solid tumors, which is usually maintained throughout the tumor lifespan [11], [12], [13], we have alternatively investigated on approaches to downregulate HER2 from the gene expression level by specifically inhibiting the interaction between transcription factor (TF), ELF3/ESX/ESE-1/ERT/JEN and its coactivator, MED23/DRIP130/SUR2/CRSP130 [14], [15], [16]. As a necessary TF for HER2 gene expression, ELF3 binds directly to the ETS transcriptional response element of the HER2 promoter and subsequently interacts with MED23 to promote HER2 overproduction [17].
Despite its significance, only a small number of inhibitors have been developed for this PPI due to the lack of concrete structural understanding in the binding interface of ELF3 and MED23 under the limitation that the exact structure of ELF3 has not yet been revealed. This study aimed to determine the hotspot of ELF3-MED23 PPI by specifying the essential residues and their key interactions which are controllable by small molecules.
Materials and methods
Cell viability assay of compounds with diverse cancer cells
Cells were seeded at 104 cells/well in a 96-well cell culture plate for 20 h. After 4 h of starvation with serum-free media, cells were replaced with compound-added medium at designated concentrations and incubated for 72 h at 37 ℃ in a 5 % CO2 incubator. Viability of the cells were finally evaluated by measuring the absorbance at 450 nm, after applying 5 µL of EZ-cytoX to each well. For the measurement, ELISA Microplate Reader (VersaMax, Molecular Devices) was utilized.
Secreted alkaline phosphatase (SEAP) assay
293 T kidney cells were seeded in a 96-well microplate (SPL, Korea) at a density of 5 × 103 cells/well and incubated overnight. Then they were transiently co-transfected with 100 ng of each plasmid and transfection-optimized medium (Welgene, Korea). The co-transfected plasmids were the SEAP reporter gene (pG5IL2SX) and GAL4-ELF3 expression vector (pBJ GAL4-ELF3). Cells were incubated for 3 h with WelFectQ (Welgene, Korea) for transfection, and then the compounds or MED23 protein constructs (pcDNA_MED23 WT, K397A, D400A, H449G, or D400A/H449G) were added. The treated cells were incubated for 12 h. After treatment of compound in a 5 % CO2 incubator for 12 h at 37 ℃, the plate was incubated at 65 ℃ for 3 h to inactivate all enzymes except for SEAP. A mixture of 25 µL of the inactivated medium and 75 µL of double distilled water was added along with 100 µL of the substrate solution, which was made with 1.19 µL of 0.1 M 4-methylumbelliferyl phosphate (Sigma, USA) and 98.81 µL of 2 M diethanolamine (pH 10, Sigma, USA). After incubation overnight at 37 ℃, fluorescence intensity was detected at an excitation wavelength of 360 nm and an emission wavelength of 440 nm using a fluorometer (SPECTRAmax GEMINIEM, Molecular Devices, USA).
Western blot analysis
For the Western blot analyses, cells were seeded in 6-well plates and lysed in 1X RIPA lysis buffer (Cell Signaling, USA) containing 1 % 0.1 M PMSF and 1 % 100X protease inhibitor cocktail solution (Genedepot, USA). The total protein amounts were normalized using the Pierce™ BCA protein assay kit (Thermo Fisher Scientific, USA) and a microplate reader (Tecan Group ltd., Switzerland), equipped at Drug Development Research Core Center. 20 μg of proteins were loaded onto a 10–15 % acrylamide gel for SDS-PAGE and then transferred to a 0.2 μm PVDF membrane (Pall Life Science, USA). 5 % skim milk or 5 % BSA was used for membrane blocking, and then the membranes were incubated with primary antibodies at room temperature (RT) for 3 h or at 4 °C overnight. The blots were washed 3 times with tris-buffered saline-0.1 % Tween20 (1X TBST) and incubated with HRP-conjugated secondary antibodies (GeneTex, USA). The protein bands were detected using ECL solution reagent (GE Healthcare, USA) and LAS-3000 (Fuji Photo Film Co., ltd., Japan). Captured images were evaluated with Multi-Gauge software (Fuji Photo Film Co. ltd.). All indicated primary antibodies were purchased from Thermo Fisher, Cell Signaling, Santa Cruz, Novus, or MBL, and the details are listed in Table S1.
Immunoprecipitation assay
NCI-N87 cells were seeded in a 100-mm cell culture dish and cultured until they reached 60–70 % confluency, cells were transfected with p3Xflag-myc-CMV26-empty or p3Xflag-myc-CMV26-MED23WT plasmid DNAs using Lipofectamine® 2000 (Invitrogen, USA). After 12 h transfection, YK1 (5 μM) was treated for additional 12 h. Cell lysate preparation and total protein quantification were then performed using the same procedure as for the Western blot analyses. Total 750 μg of cell extracts were incubated with 20 μL of FLAG-agarose bead (Sigma Aldrich, USA) slurry at 4 °C for 4 h on a rotator. After the incubation, samples were centrifuged for 1 min at 12000 rpm to precipitate the beads. Supernatants were then removed and the remaining beads were washed 3 times with 200 μL of lysis buffer (RIPA). After removal of final supernatant, the beads were eluted with 2X sample buffer by boiling at 98 °C for 5 min. The immunoprecipitated proteins were loaded and separated by SDS-PAGE and analyzed using western blot analyses.
GST pull down assay
HEK293 cells were seeded in a 100 mm cell culture dish and cultured until they reached 70–80 % confluency, and then the indicated GST-tagged ELF3 construct was transfected using Lipofectamine® 2000 (Invitrogen, USA). Cell lysate preparation was performed using the same procedure as for the western blot analyses. 1000 μg of cell extracts were incubated with 20 μL of Glutathione Sepharose™ beads (GE Healthcare, UK) for 1 h at 4 °C on a rotator. The Sepharose beads were washed 3 times with 200 μL of ice-cold 1X PBS. After removal of final supernatant, the beads were eluted with elution buffer (20 mM Glutathione, 100 mM Tris-HCl (pH 8.0), 120 mM NaCl, 10 % glycerol). The precipitated proteins were loaded and separated by SDS-PAGE and analyzed using western blot analyses.
Luciferase promoter assay
HEK293 cells were plated in 6 well plate and transfected with 1 µg of pNeuLite (Addgene) alone or in combination with 0.5 µg of pcDNA3.1-flag-ELF3WT (provided from Dr. Seung Bae Rho, National Cancer Center, Republic of Korea) and p3Xflag-myc-CMV26-MED23 mutant plasmids as indicated. For the normalization of the luciferase activity of each sample, 0.5 µg of β-galactosidase expression plasmid (provided from Dr. Eun-Sook Hwang, Ewha Womans University, Republic of Korea) was also added in every sample groups. All of the transfections were conducted using Lipofectamine® 2000 Transfection Reagent (Invitrogen, USA). After 24 h, firefly luciferase and β-galactosidase activities were evaluated with the Infinite M200 PRO Microplate reader (Tecan Group ltd., Switzerland) using the Luciferase Assay System (Promega) and Galacto-Light Plus β-Galactosidase Reporter Gene Assay System (Invitrogen), respectively, according to the manufacturers’ protocols.
Fluorescence polarization assay
Various concentrations of (His)6-MED23391–582 and fluorescein isothiocyanate (FITC)-labeled ELF3129–145 peptide (10 nM) were mixed together in assay buffer (20 mM sodium phosphate (pH 8.0) containing 30 mM NaCl, and 5 mM β-mercaptoethanol) to determine the Kd value. The final reaction volume was adjusted to 50 μL, and samples were incubated for 30 min at RT. Determination of the Kd value was conducted with Prism 6.0 (GraphPad Software, USA) using the least-squares non-linear fit method. The FP signals were measured in millipolarization (mP) units using the Infinite F200 PRO microplate reader (Tecan Group ltd., Switzerland) at excitation/emission wavelengths of 485/535 nm. For the displacement assays, graded concentrations of YK1 and unlabeled-ELF3137–144 peptide were mixed together with a mixture of 80 nM (His)6-MED23391–582 and 10 nM FITC-ELF3129–145. Efficacy of compound YK1 was evaluated by measuring the extent of decrease in the FP signals, using unlabeled peptide as positive control. To determine the IC50 of each compound, four-parameter logistic equation was applied through the Table Curve 2D program (SPSS Inc.). Ki values were finally calculated based on the Cheng-Prusoff equation: Ki = IC50/1+([Ligand]/Kd). FITC-labeled ELF3129–145 and unlabeled peptide were synthesized by Peptron Inc., Korea, and (His)6-MED23391–582 was produced as previously described [18], [19]. Experiments were performed in triplicate in flat-bottomed 96-well black plates with a non-binding surface (Thermo Scientific Nunc, USA).
Quantitative Real-Time PCR
FavorPrep™ Tri-RNA reagent (FAVORGEN Biotech Corp., Taiwan) and a PrimeScript™ RT Reagent Kit (Takara Bio Inc., Japan) were used according to the manufacturers’ instructions to extract RNA from cells and synthesize complementary DNA (cDNA), respectively. Quantitative analysis of the demonstrated genes was performed using a SensiFAST™ SYBR No-ROX kit (Bioline, Canada). PCR amplification was conducted using the CFX96TM real-time PCR detection system (Bio-Rad, USA) under the following protocol: polymerase activation at 95 °C for 2 min, followed by 27 cycles of amplifications of 95 °C for 10 sec, 56 °C for 10 sec, and 72 °C for 20 sec. The relative quantity of mRNA was determined using the ΔΔCt method and normalized by GAPDH or ACTIN. The primer sequences used in this study are summarized in Table S2.
Kinase inhibition assay
Kinase inhibitory activity of YK1 at 10 µM and 25 µM was evaluated through the Eurofins kinase profiling services. Its inhibitory effects toward c-RAF, EGFR, ErbB2, ErbB4, and PI3 kinases were analyzed according to the Kinase Profiler Service Assay protocols [20]. The scintillation values were calculated as the percent of kinase inhibition with respect to the control.
Tumor xenografts
NCI-N87 and JIMT-1 cells (5 × 106 cells) in 100 μL of 1X phosphate buffered saline (PBS) were subcutaneously injected into the right flank of 5-week-old female athymic nude mice (Envigo, USA) and 4-week-old female NOD-SCID (Koatech, Pyeongtaek, South Korea) mice, respectively. Mice were randomly separated into each of the designated groups when the tumours reached a volume of 90–100 mm3. After that, drug administration was repeated six times via intravenous route for NCI-N87 xenografts, and eight times through intraperitoneal route for JIMT-1 xenografts, both at 3-day intervals. YK1 and TZMB were equally given at the concentration of 4 mg/kg, while they were separately prepared in DMAC/Tween80/saline (5:10:85) mixture and saline, respectively. Changes in the tumour size were monitored for additional days after drug injection was completed, until the average tumor size of the control group reached 2000–2500 mm3. As shown in the figures, mice were sacrificed 28 days after the first drug injection for NCI-N87 and 46 days for JIMT-1 xenografts. Tumors were then immediately excised from each mouse, and the relative tumor sizes were determined. The tumor length (L) and width (W) were measured with calipers, and the tumor volume was determined according to the formula: (L × W2) / 2.
IHC assay for xenograft mouse model
Tumors obtained from the xenograft mouse model were used to make a paraffin-embedded block section for immunohistochemistry (IHC). All IHC procedures followed general protocols. The indicated HER2 and Ki-67 primary antibodies were incubated at 4 °C overnight and then rinsed 3 times with 1X PBS, incubated with secondary antibody, developed with a Vectastain ABC kit (Vector Laboratories, USA), and stained with DAB solution (Dako, Carpinteria, USA), all following the manufacturers’ protocols. After counterstaining with hematoxylin (USA), IHC staining was evaluated by light microscopy at 200 × magnification. IHC staining was evaluated semi-quantitatively by adopting the IHC score. Final scores were calculated by multiplying the intensity and fraction score (percentage of samples counted at each scale), producing a range from 0 to 300. All imaging and assessments were performed using an Axiophot 2 apparatus (Carl Zeiss MicroImaging Inc., Thornwood, NY, USA), equipped at Drug Development Research Core Center. The details of the applied antibodies and dilution ratios are listed in Table S1.
Cell cycle analysis
5 × 105 NCI-N87 cells were seeded in 60-mm dishes. When 70–80 % confluence was reached, YK1 was treated at different concentrations for 8 h. The cells were trypsinized and then washed with ice-cold PBS (pH 7.4). For the cell cycle analysis, cells were fixed with 70 % ethanol and incubated for 30 min at 4 °C and then stained using FxCycleTM PI/RNase staining solution (InvitrogenTM, USA) according to the manufacturer’s instructions. Cell cycle analyses were performed using a fluorescence-activated cell sorting (FACS) instrument (BD Biosciences, USA), equipped at Drug Development Research Core Center. At least 5,000 cells were measured for each sample.
Annexin V/PI double staining apoptosis assay
Cells were prepared using the same conditions as given above for the cell cycle analysis, except that YK1 was used at various concentrations for 24 h. Cells were trypsinized and washed with 1X PBS, and then each sample was resuspended in 100 μL of 1X Annexin V binding buffer using an FITC Annexin V Apoptosis Detection kit I (BD PharmingenTM) according to the manufacturer’s instructions. The induction of apoptosis was assessed using the FACS instrument (BD Biosciences). 10,000 cells were measured for each sample.
Clonogenic assay
Cells were seeded in 6-well culture plates at a density of 2000 cells/well, followed by incubation with or without treatment of TZMB (10 or 20 μg/mL) and YK1 (0.5 or 1 μM) for 14 days. Cells were then fixed with 100 % methanol for 1 h and stained with 200 μL of crystal violet solution (1 % [w/v] in absolute methanol) per well [21]. Cells were rinsed with tap water and analyzed. The images were taken using ChemiDoc bio-image analyzer (Bio-Rad) and quantified by ImageJ software (NIH, Bethesda, MD, USA). All steps after fixation were performed at room temperature.
Split luciferase complementation biosensor
All biosensors were constructed using an In-Fusion® HD Cloning Kit with firefly (Photinus pyralis) luciferase from the pGL3-basic vector (Promega) template. Luciferase was split into two fragments (Nluc and Cluc). Full-length ELF3 (ELF3WT) and MED23 variants were cloned into the HindIII and Kpnl sites and the Notl and Kpnl sites, respectively, of p3Xflag-myc-CMV26. Full-length cDNA for human ELF3 (accession number NM_001114309.1) was fused with the N-terminal fragments of the split luciferase (Nluc), and full-length cDNA of MED23 (accession number NM_001270521.1) was fused with the C-terminal fragments of the split luciferase (Cluc). Both inserts were cloned into the p3Xflag-myc-CMV26 vector. Nluc-empty and Cluc-empty constructs were prepared afterward using the generated full-length ELF3 and MED23 constructs as a template. KOD-Plus-Mutagenesis kit was used. The primer sequences for these experiments are summarized in Table S2.
In silico docking study
Docking studies were performed using a protein–protein docking model for the ELF3–MED23 interaction obtained through the ClusPro webserver. The best pose was selected according to previous experimental evidence [22], [23]. Prior to docking, amino acids 137–144 of ELF3 (ELF3137-144) were extracted as an internal ligand. After removing the ELF3 protein from the model, protomol was generated using ELF3137-144. The 3D structure of YK1 was generated by the SKETCH module implemented in the SYBYL program (Tripos Inc., St. Louis, USA). Energy-minimization was conducted through the Tripos force field using Gasteiger-Huckel [24] charges. All the molecular docking was performed through Surflex-Dock interfaced with Sybyl-X 2.3. (Tripos International, 2012). The resulting poses were additionally visualized through PoseView [25] to identify important contacts between ELF3 and MED23.
Quantifying Protein-Small molecule interaction using LC-MS/MS
HEK293 cells were seeded in 100 mm3 dish and incubated until it reached 70–80 % confluency. Cells were then transfected with p3Xflag-myc-CMV26-MED23WT and p3Xflag-myc-CMV26-MED23D400A/H449G plasmids for 24 h and lysed to obtain protein extracts. To specifically isolate the MED23-bound YK1, the whole lysates were incubated with YK1 for 4 h and finally pulled-down using FLAG-agarose beads (Sigma Aldrich, USA). Non-bound compounds were completely removed by washing the precipitated beads for at least 3 times. To separate YK1 out from precipitated MED23, he samples (50 μL) were extracted with 1 mL of acetonitrile and centrifuged for 10 min at 4 °C with 13,000 rpm. Each sample was aliquoted from the upper layer (1000 μL) to a new tube and evaporated to dryness under nitrogen gas at 37 °C. For the analysis of YK1, the residue was dissolved in 50 μL of ACN and vortexed for 10 min. The sample was then mixed with 1 μL of the internal standard (Compound 18, hereafter YK2; structurally similar to YK1, but lacking binding affinity to MED23; Table 1), 5000 μg/mL in ACN) and extracted with 450 μL of ACN by sonicating and vortex-mixing. MED23-bound YK1 was finally analyzed using Agilent 6460 triple quadrupole mass spectrometry (Agilent Technologies, USA) with an electrospray interface, equipped at Drug Development Research Core Center. The separation was achieved on the column of XSelect® HSS T3 2.5 μm (2.1 × 100 mm; 2.5 μm particle size; Island Waters). The supernatants of each sample and standard solutions were injected into the LC-MS/MS system. The column temperature was maintained at 37 °C and the injection volume was set to 1 μL. The mobile phase (A) contained 0.1 % formic acid in water. The mobile phase (B) contained 0.1 % formic acid in ACN. The flow rate was 0.2 mL/min in gradient mode: 0 min, 70 % (B); 0–3 min 100 % (B); 3–5 min 100 % (B); 5.1–12 min, 70 % (B). The total run time was 12 min.
Table 1.
Direct ELF3-MED23 PPI inhibitory activity (%) of 25 compounds and the MED23 residues that created interactions with each compound.
| Group |
Structure |
Compounds |
Interacting residuesaby in silico study |
SEAP assay |
|||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 |
Inhibition (%)b |
IC50 (μM)c |
||||
| – | – | – | – | – |
Peptide (8 a.a.) |
D400, E405, H449 | 53 | – | |
| – | – | – | – | – | Gefitinib | D400, H449 | 85 | 3.29 ± 0.14 | |
| 1 | 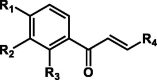 |
H | OH | H | 2-Furanyl | 1 | D400, H449 | 86 | 2.13 ± 0.04 |
| H | OH | H | p-Methoxyphenyl | 2 | H449 | 41 | – | ||
| H | OH | H | p-Methoxynaphthalenyl | 3 | D400, H449 | 70 | 1.72 ± 0.02 | ||
| H | OH | H | p-Chlorophenyl | 4 | D400, H449 | 80 | 2.97 ± 0.04 | ||
| H | OH | H | p-Hydroxyphenyl | 5 | F399, K406, H449 | 60 | 3.96 ± 0.04 | ||
| H | H | OH | p-Methoxyphenyl | 6 | D400, H449 | 90 | 1.98 ± 0.34 | ||
| H | H | OH | p-Methoxynaphthalenyl | 7 | D400 | 80 | 2.39 ± 0.36 | ||
| H | H | OH | 2-Furanyl | 8 | H449 | 50 | – | ||
| H | H | OH | p-Hydroxyphenyl | 9 | F399, Y403, E405, H449 | 50 | – | ||
| H | H | OH | p-Chlorophenyl | 10 | D400 | 80 | 3.93 ± 0.74 | ||
| H | H | OH | 2-Thiophenyl | 11 | H449 | 50 | – | ||
| 2 | 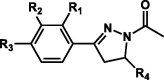 |
H | OH | H | 2-Furanyl | 12 | H449 | 58 | – |
| H | OH | H | p-Methoxyphenyl | 13 | H449 | 58 | – | ||
| H | OH | H | p-Methoxynaphthalenyl | 14 | F399, W429 | 14 | – | ||
| H | OH | H | p-Chlorophenyl | 15 | D400, H449 | 95 | 1.18 ± 0.34 | ||
| H | OH | H | p-Hydroxyphenyl | 16 | F399, W429, P446 | 37 | – | ||
| OH | H | H | 2-Furanyl | 17 | – | 23 | – | ||
| OH | H | H | 1-Methoxynaphthalenyl | 18 | – | 0 | – | ||
| OH | H | H | p-Hydroxyphenyl | 19 | F399, Y403 | 39 | – | ||
| OH | H | H | p-Chlorophenyl | 20 | H449 | 45 | – | ||
| OH | H | H | 2-Thiophenyl | 21 | H449 | 57 | – | ||
| OH | H | H | p-Methoxyphenyl | 22 | D400 | 63 | – | ||
| 3 | 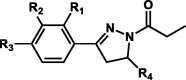 |
H | OH | H | p-Methoxyphenyl | 23 | D400, H449 | 87 | 5.01 ± 1.16 |
| H | OH | H | 2-Thiophenyl | 24 | – | 34 | – | ||
| H | OH | H | p-Chlorophenyl | 25 | F399, H449 | 42 | – | ||
Residues of MED23 which interacted with each of the indicated compounds through H-bonding.
SEAP inhibitory activity (%) of each compound at 10 μM representing the ELF3-MED23 interaction inhibitory activity.
IC50 value of each compounds against SEAP inhibitory activity.
Quantification and statistical analysis
All experiments were performed at least three times, and the mean ± standard deviation is expressed for all data. Statistics were calculated by one-way analysis of variance (ANOVA) or Student’s t-test with Prism 6.0 (GraphPad Software, USA), and the differences between two values were considered statistically significant when P values (described using single, double or triple asterisks) were < 0.05, <0.01, and < 0.001.
Ethics experiment
All experiments involving animals were conducted according to the ethical policies and procedures approved by the Institutional Animal Care and Use Committee (IACUC) at the Ewha Womans University (No. 16–052).
Results
In Silico-based structural analysis to predict hotspot of ELF3-MED23 complex
To gain in-depth structural insights into the binding module of ELF3 and MED23, homology model of ELF3 protein was built using Phyre2 server (https://www.sbg.bio.ic.ac.uk/phyre2; web portal for protein modelling, prediction, and analysis) [26], in which 69 % of the residues were modelled at > 90 % confidence. Together with the known structure of MED23 (PDB ID: 6H02) [27], we then implemented the automated protein–protein docking server ClusPro [28], [29] to predict the mode of interaction (Fig. 1A) using the balanced coefficient weighting method. Total 27 poses were estimated here. Final model was selected upon the experimental evidence from our earlier reports explaining the interaction between the 391–582 residues of MED23 (highlighted in green) and the TAD domain (129–145, in magenta) of ELF3 (Fig. 1A). Specifically, the yellow-highlighted ELF3 residues of S137 to E144 (8 a.a) were previously suggested as residues required for the interaction [18], [23]. For detailed validation of the model, we prepared ELF3137-144 peptide as a ligand and performed docking studies on the binding interface from the predicted complex (Fig. 1B, S1A-B). Consistent with previous results, 5 hydrophobic residues (W138, I139, I140, L142, and L143) of ELF3 were in direct hydrophobic contact with K406, D400, E405, H449, P446, and M396 of MED23 (Fig. 1B, S1A). Hydrophilic amino acids S137, E141, and E144 of ELF3 were positioned to protrude outward without making direct interactions with MED23 (Fig. S1B). Since all features demonstrated by the model were in exact agreement with the prior experimental evidence, further analyses were conducted based upon this model. Among the 5 key hydrophobic residues of ELF3, the indole moiety of W138 formed a specific H-bond with D400 and fairly strong π-π interactions with F399 of MED23. The I139 and I140 residues of ELF3 were also predicted to create additional H-bonds with E405 and H449 in MED23, respectively. Besides, several hydrophobic contacts between ELF3 and the D400, H449, E405, M396, P446 and K406 residues of MED23 were also suggested to consolidate the interaction as well (Fig. 1B, S1A). To elucidate the residues of MED23 that are critically involved in the interaction with ELF3, we performed docking studies using adamanolol, wrenchnolol, canertinib, and gefitinib which were previously reported to exhibit ELF3-MED23 PPI inhibitory activity [14], [15], [18], [23]. We found that all the tested compounds and ELF3137-144 formed H-bonds with D400 and H449 of MED23 in common (Fig. 1C-E). ELF3137-144 and canertinib, which showed superior ELF3-MED23 PPI inhibitory activity than gefitinib [14] created additional π-contacts with F399 of MED23 (Fig. S1C). Thus, we presumed D400, H449 and F399 residues of MED23 served as hotspot for ELF3-MED23 PPI by forming H-bonds (D400, H449) and π-stacking (F399) with ELF3.
Fig. 1.

In silico-based structural analysis to predict hotspot of ELF3-MED23 complex. (A) Protein binding model of MED23WT and ELF3WT prepared using the Phyre2 server. The important residues responsible for ELF3–MED23 binding are depicted in different colors. (B) 3D docking view of the ELF3137-144 peptide on MED23 protein (PDB: 6H02). The key residues of MED23 and ELF3 involved in the interaction were labelled in green and black, respectively. The detailed 2D interaction diagrams between ELF3137-144 and MED23 is depicted in Figure S1A. (C) Docking pose of wrenchnolol, adamanolol, gefitinib, and canertinib that were previously identified to have ELF3-MED23 PPI inhibitory activity. (D) 2D interaction diagrams of the gefitinib docking generated by PoseView (https://proteins.plus). Gefitinib has been shown to have H-bondings to the D400 and H449 residues of MED23 with additional hydrophobic contacts via M396, F399, D400, E405, L443, P446, and P448 but no π-π interaction with F399. (E) Summary of H-bonding residues and -stacking of MED23 involved in the interaction with each of the indicated compounds. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Specification of key residues and interactions in the hotspot of ELF3-MED23 PPI
To confirm whether the D400, H449 residues are indeed key contributors for ELF3-MED23 PPI, we utilized our previously developed SEAP assay system [14] and additionally transfected several point mutants of full-length MED23 construct. MED23WT induced an increase in SEAP activity but not the MED23D400A, MED23H449G, and MED23D400A/H449G mutants which lack the ability to form H-bonds with ELF3 (Fig. 2A). Introduction of alanine in K397 residue (MED23K397A), which was predicted as a non-contributing residue for ELF3-MED23 PPI (Fig. 1), produced no significant changes compared to MED23WT (Fig. 2A). For further evaluation, we prepared biosensors using Nluc-ELF3 and various Cluc-MED23391-582 constructs and measured the bioluminescence intensity complementally generated by ELF3-MED23 binding (Fig. 2B). Co-transfection of Nluc-ELF3 and Cluc-MED23391-582 WT or K397A distinctly increased the luciferase activity [5.2- (for WT) and 4.8-fold (for K397A) changes relative to control, respectively], but Cluc-MED23391-582D400A and H449G constructs did not produce such changes (Fig. 2C-D). Same trend was also observed in GST-pull down assay using point mutants of full-length MED23. The interaction between ELF3 and MED23 was significantly lost in the single mutants of D400A and H449G, but not in K397A (Fig. 2E). Through successful interaction of MED23 with ELF3 (WT and K397A), HER2 expression levels were upregulated with a direct increase in the transcriptional activity of ELF3 (Fig. 2F-G). Consistent with the results so far, D400A and H449G variants of MED23 failed to increase the HER2 level. This verifies that direct H-bonding to ELF3 via D400 and H449 residues of MED23 is crucial in the ELF3-MED23 PPI to activate HER2 gene transcription.
Fig. 2.

Specification of key residues and required interaction in the hotspot of ELF3-MED23 PPI. (A) To verify D400 and H449 of MED23 as key residues for ELF3-MED23 PPI, the impact of each MED23 mutant on the binding degree between gal4-ELF3 and MED23 was measured by SEAP activity. The results are expressed in bar graph (n = 5, mean ± S.D.). ANOVA (* and ***P < 0.05 and 0.001, respectively, ns = non-significant). MED23WT and MED23K397A increased the SEAP activity but D400A and H449G single mutants and D400A/H449G double mutant did not. (B) Graphical scheme of designed biosensor. The N-terminal fragment of luciferase was tagged to ELF3, and the C-terminal was linked to the MED23 constructs. (C, D) The degree of protein binding between ELF3 and different MED23391-582 fragments was evaluated by co-transfecting the generated constructs to HEK293 cells for 24 h. Representative image (C) and the quantification result (D) was demonstrated in a bar graph (n = 4, mean ± S.D.). ANOVA (***P < 0.001, ns = non-significant). (E) The extent of direct protein interaction between several full-length MED23 mutants and ELF3 was assessed by GST pull down assay. (F, G) Changes in the transcriptional activity (F) and HER2 expression level (G) were determined along with co-expression of ELF3 with different full-length MED23 constructs.
Small Molecule-aided confirmation of key residues of MED23 in ELF3-MED23 PPI
To concretize our findings, we utilized the results of an in silico study of the binding module of gefitinib at the ELF3-MED23 interface to design and synthesize compounds with minimal essential structural elements. According to the 3D and 2D pose views of ELF3137-144 (Fig. 1B, S1A-B) and gefitinib (Fig. 1C-D) in the binding site, D400 and H449 of MED23 served as H-bond acceptors and donors, respectively. In addition, the 3-chloro-4-fluorophenyl group of gefitinib and the indole ring of W138 in ELF3137-144 both formed an important lipophilic contact within the hydrophobic pocket near F399 and D400. The F399 residue served as a π-stacking provider, contributing to stabilize the binding of each ligand at the interface. Based on these observations, we aimed to remove the non-essential side chains from gefitinib and to design structures that have both H-bonding donor and acceptor moieties for the D400 and H449 residues of MED23, respectively (Fig. S2). Besides maintaining the key interaction core, we attempted to enhance the binding favorability by incorporating potential π-stacking interaction moieties into the structure, allowing compounds to fit tightly into the hydrophobic pocket. Based upon this strategy, we adopted chalcone and pyrazoline as skeletal cores for potential modulators of ELF3-MED23 interaction and finally prepared focused series of 25 compounds (11 chalcones and 14 pyrazolines) (Fig. S2 and Scheme S1).
ELF3-MED23 PPI inhibitory activity of all the prepared compounds was measured through SEAP assay [14]. Compounds that showed over 65 % inhibition of the ELF3-MED23 interaction (black highlighted in the bar graph in Fig. S3A), were further evaluated in diverse concentrations (3, 5, and 10 µM) to verify the initial screening data (Fig. S3B). By measuring cell viability in parallel under the same experimental conditions as the reporter gene assay, we confirmed the compound-induced specific disruption of the ELF3-MED23 interaction and not due to the non-specific cytotoxicity of the compound (Fig. S3). We then performed in silico docking studies of all the prepared compounds against the identified hotspot of MED23, and evaluated the correlation between the actual PPI inhibitory efficacy and the predicted structural requirements. Compounds that were predicted to form H-bonds with both D400 and H449 residues showed better ELF3-MED23 PPI inhibitory activity (Table 1 and Fig. S4). For further evaluation, we selected compound 15 (hereinafter referred to as YK1) showing the strongest cellular ELF3-MED23 PPI inhibitory activity [95 % inhibition at 10 μM, IC50 (μM) = 1.18 ± 0.34]. Superimposing the final docking poses of gefitinib and YK1 showed that both compounds exhibited similar binding orientation within the hotspot of ELF3-MED23 PPI (Fig. 3A). Instead of the nitrogen atom in the quinazoline ring of gefitinib, the oxygen atom of the methoxy group on the pyrazoline ring of YK1 served as H-bond acceptors for H449 of MED23. The hydrogen atom of hydroxyl group on the phenyl ring of YK1 served as the H-bond donor for D400 of MED23, equivalent to the quinazoline ring of gefitinib. Unlike gefitinib, YK1 formed an H-bond with the oxygen in the backbone of the D400 residue, which readily created additional π-contacts with F399 of MED23, enhancing overall binding. Moreover, similar to ELF3137-144 peptide, YK1 was also predicted to form hydrophobic interactions with MED23 residues, F399, E405 and P446 (Fig. 3B). The ability of YK1 to directly bind to MED23 via forming H-bonds with D400 and H449 residues was validated using LC-MS/MS-based quantitative analysis method, as illustrated in Fig. S5. YK1 was able to bind to the FLAG-tagged MED23WT, but not to the double mutant MED23D400A/H449G (Fig. 3C). Also, among the different fragments of MED23, YK1 specifically interacted with the MED23391-625, the fragment containing D400 and H449 residues (Fig. 3D). To ensure whether YK1 could indeed inhibit the ELF3-MED23 PPI, we measured changes in the bioluminescence intensity in the absence and presence of YK1 using Nluc-ELF3 and Cluc-MED23391-462 constructs as illustrated in Fig. 2B. Luciferase activity markedly elevated by Nluc-ELF3WT and Cluc-MED23391-462 (6-fold change vs control) was significantly reduced by co-treatment of YK1 in a dose-dependent manner (Fig. 3E-F), verifying that YK1 specifically bound to this MED23391-462 region and interfered with the ELF3-MED23 PPI.
Fig. 3.

Small molecule-aided confirmation of key residues of MED23 in ELF3-MED23 PPI and discovery of YK1 as a specific tool for blocking ELF3-MED23 PPI. (A) Binding orientations of gefitinib and YK1 were compared by overlaying the final docking poses of gefitinib and YK1. MOLCAD hydrogen bonding surface was shown to visualize the distribution of hydrogen bond donors (red) and acceptors (blue). (B) 2D interaction diagrams of the docking model of YK1 was generated through PoseView (https://proteins.plus). Strong π-contacts with F399 along with specific H-bonding to D400 and H449 of MED23 were observed. (C) LC-MS/MS analysis result confirmed direct binding between MED23WT and YK1 via H-bonds with D400 and H449 residues. Binding was significantly lost with MED23 D400A/H449G construct. (D) LC-MS/MS analysis assessed YK1 binding affinity toward various MED23 fragments. YK1 specifically interacted with the 391–625 a.a. region of MED23. (E, F) Representative image (E) and quantification result (n = 3) (F) of the luciferase biosensor assay conducted using Nluc-ELF3WT and Cluc-MED23391-462 fragment. YK1 was co-administered to the ELF3-MED23391-462 interacting system to confirm its PPI inhibitory activity. (G) The extent of ELF3-MED23 PPI inhibition was assessed by the decreased fluorescence polarization (mP) values due to the release of FITC-labeled ELF3 peptide complexed with MED23391-582 protein by dose-dependent treatment of YK1 or unlabeled ELF3 peptide. YK2 was used as a negative control. (H) The IC50 and Ki values of YK1 were calculated from the fluorescence polarization assay results. (n = 3, mean ± S.D.). (I) PPI inhibitory activity of YK1 was evaluated against MED23 and its diverse binding partners using immunoprecipitation assay. YK1 specifically blocked the ELF3-MED23 PPI. (J) mRNA levels of HER2 were evaluated by dose-dependent treatment of YK1 (n = 3, mean ± S.D., GAPDH was used as a control for normalization), ANOVA, ***P < 0.001 vs CON. (K) Protein levels of HER2 and HER2-related downstream signal molecules were evaluated with dose-dependent treatment of YK1. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
YK1 as a direct regulator for HER2 by selectively inhibiting ELF3-MED23 PPI
Through in vitro fluorescence polarization (FP) assay, YK1 was reconfirmed as a direct PPI inhibitor of ELF3-MED23, as it markedly decreased the FP signal through inducing the release of FITC-labeled ELF3129-145 peptide from MED23391-582 (Fig. 3G-H) [18], whereas YK2 made no such changes (Fig. 3G). Using the Kd value calculated from the titration curve of (His)6-MED23391–582 and FITC-labeled ELF3129–145 peptide (Fig. S6), we additionally calculated the Ki value of each compound as explained in the method section. Inhibitory activity of YK1 exceeded that of the unlabeled ELF3137-144 peptide (Ki value; YK1 vs peptide; 0.78 ± 0.05 μM vs 4.27 ± 0.25 μM; 5.5-fold; Fig. 3H). To determine whether the PPI inhibitory activity of YK1 is specific to ELF3-MED23, we also evaluated its effect on some other PPIs between MED23 and its known partner transcription factors, such as ELK1 and IRF7 [27], [30], [31]. Both ELK1- and IRF7-MED23 PPIs were not disturbed by YK1 (Fig. 3I). YK1, an ELF3-MED23 PPI-specific inhibitor, induced significant downregulation of the HER2 gene and protein expression levels in cells in dose-dependent manner (Fig. 3J-K). However, unlike gefitinib [20], YK1 did not exhibit significant inhibitory activity against any of HER family kinases (EGFR, ErbB2, and ErbB4) or AKT/MAPK pathway-related kinases (PI3 kinase and c-RAF) (Table S3). It reflects that YK1-induced transcriptional downregulation of HER2 was specifically triggered by ELF3-MED23 PPI inhibition, resulting in attenuation of the HER2-related signaling cascade. Further evaluation of the antiproliferative effect of YK1 on several gastric cancer cells revealed that YK1 more potently inhibited the cell growth of NCI-N87, which highly expresses HER2 (IC50 = 1.56 ± 0.14 μM), but almost non-cytotoxic in normal mammalian cells (at least 44-fold less toxic compared to NCI-N87 cells, see Fig. S7), indicating that the antiproliferative activity of YK1 is dependent on HER2 overexpression.
Direct application of small molecule PPI inhibitor as an anticancer drug targeting HER2-overexpressing gastric cancer
We further evaluated whether YK1-mediated ELF3-MED23 PPI blockage could be linked to significant tumor suppression against HER2-positive gastric cancers, the cancer subtype in which HER2 is generally known as a negative prognostic factor [8]. Kaplan-Meier survival analysis using publicly available dataset of 1,065 gastric cancer patients (https://www.kmplot.com/gastric/) revealed a significant negative correlation between HER2 mRNA level and the overall survival (OS) or progression-free survival (PFS) (Fig. 4A-B). Intravenous (IV) administration of 4 mg/kg of YK1 to NCI-N87 xenograft mice facilitated significant tumor growth retardation with significant reduction in tumor volume (Fig. 4C-D). IHC analysis confirmed that this outcome was due to the significant reduction (1.8-fold compared to untreated control) of HER2 level in the tumor (Fig. 4E, upper panel), which was accompanied by remarkable decrease (4.5-fold compared to untreated control) in the proliferation marker, Ki67 (Fig. 4E, lower panel). Consistent with previous studies showing that HER2 overexpression and gene amplification are closely related to G1/S phase cell-cycle dysregulation [32], [33], time- and dose-dependent treatment of YK1 resulted in a significant increase in the cell fraction of the G1 phase as well as a decrease in the S and G2/M phases, overall promoting G1 arrest (Fig. 4F-G). YK1 also promoted apoptosis markedly in a dose-dependent manner (Fig. 4H), inducing an increase in pro-apoptotic markers, c-PARP and c-caspase 7, but a decrease in anti-apoptotic marker, bcl-2 (Fig. 4I). Prominent anticancer activity of YK1 was also supported by favorable physicochemical properties and safety profiles; reasonable solubility (103.2 ± 0.6 μg/ml at pH 7), log P value (2.98), and permeability (Fig. S8A) with no association of genotoxicity or cardiotoxicity (Figs. S8B-C). This indicates that its properties generally meet the criteria of drug-likeness. The PK profile of YK1 also demonstrated modest bioavailability (Ft, 25.1 %; Fig. S8D) with a long half-life [T1/2 (IV), 9.74 ± 2.76 h, T1/2 (PO), 10.9 ± 11.00 h], presumably indicating that the distribution rate predominates over the elimination rate. Throughout the animal experiments, no safety issues were observed; the animals’ food and water consumption rates were normal, and no significant changes in body weight occurred (data not shown).
Fig. 4.

Clinical relevance of HER2 overexpression and promising in vitro and in vivo anticancer activities of YK1 based upon significant downregulation of HER2. (A, B) Overall survival (log rank test, P = 0.00088) (A) and progression-free survival (log rank test, P = 0.019) (B) of 1,065 gastric cancer patients were analyzed, and the Kaplan-Meier plots were generated by the Kaplan-Meier Plotter (http://www.kmplot.com). (C) Tumor regression was markedly promoted by YK1 in an NCI-N87-xenograft mouse model (n = 5 per group; intravenous (IV) injection of YK1 (4 mg/kg every 3 days). (D) Photograph of the tumors collected from the vehicle- and YK1–treated mice (upper panel). Tumor volumes were evaluated at the indicated time points by measuring the length and width of the tumor with callipers using the equation (length × width2) / 2 (mean ± S.E.M.) (lower panel). Student’s t-test, ***P < 0.001 vs CON. (E) IHC stains of HER2 and Ki67 (proliferation marker) in the tumors. IHC score quantification was conducted using Image J (10 independent fields per sample were assessed, mean ± S.D.). Student’s t test, ***P < 0.001 vs CON. (F, G) G1 arrest was induced by YK1 with time-dependency (10 μM at each time point; n = 3) (F) concentration-dependency (24 h treatment at the indicated concentrations, n = 3) (G). ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001 vs CON. (H) Proportions of apoptotic cell fractions were remarkably increased with treatment of YK1 in a dose-dependent manner (24 h treatment at indicated concentrations). (I) YK1–mediated induction of apoptosis was confirmed by increased pro-apoptotic (e.g., c-PARP and c-casp7) and decreased anti-apoptotic (e.g., bcl-2) markers (24 h treatment at indicated concentrations).
Possibility of small molecule ELF3-MED23 PPI inhibitor as a strategy to overcome trastuzumab resistance in HER2-overexpressing cancers
In various HER2-positive cancer types, where HER2 overexpression is considered a clinically significant negative prognostic factor, trastuzumab is applied as first line therapy [9], [10] despite the high risk of resistance development within 1 year of medication [32], [34]. Thus, we evaluated whether YK1 could be utilized to overcome trastuzumab-resistance using three HER2-positive TZMB-resistant cancer cell lines prepared according to the previous method; JIMT-1 (ER-/PR-/HER2+ subtype, from a TZMB-receiving patient showing innate resistance), the TZMB-refractory BT474 (BT-TR; ER+/PR+/HER2+ subtype, acquired resistance), and the TZMB-refractory NCI-N87 (NCI-N87 TR; HER2+ subtype, acquired resistance [35]. YK1 treatment reliably inhibited the growth of both TZMB-sensitive (BT474 and NCI-N87) and -resistant (BT474 TR, NCI-N87 TR, and JIMT-1) cancer cells, whereas the activity of TZMB was only effective in the sensitive cells (Fig. 5A-C). Treatment of YK1 effectively downregulated the HER2 expression and its downstream signals such as phosphorylated AKT and MAPK in both TZMB-sensitive (BT474 and NCI-N87) and -resistant (BT474 TR, NCI-N87 TR, and JIMT-1) cancer cells. In contrast, TZMB treatment failed to make such changes in BT474 TR, NCI-N87 TR, and JIMT-1 cells (Fig. 5D-F). This trend was further confirmed by clonogenic assay in which YK1 effectively inhibited the survival and proliferation of TZMB-resistant BT474 TR, NCI-N87 TR, and JIMT-1 cancer cells, whereas TZMB did not (Fig. 5G-I). The efficacy of YK1 to overcome TZMB resistance was once again validated in vivo by administering 4 mg/kg of YK1 and TZMB, respectively, to JIMT-1 xenograft mice. TZMB dose determination was referenced to the clinically-applied dosing regimen [36]. YK1 treatment significantly reduced tumor growth and volume (Fig. 5J-L), along with a remarkable decrease in HER2 levels (Fig. 5M, 1.6-fold and 1.4-fold decrease compared to untreated control and TZMB-treated group, respectively) and the proliferation marker, Ki67 (Fig. 5N, 2.5-fold and 2.9-fold decrease compared to untreated control and TZMB-treated group, respectively). TZMB failed to demonstrate these significant changes. Meanwhile, body weight of the mice was generally similar throughout the experiment (Fig. S9).
Fig. 5.


Possibility of YK1 to overcome trastuzumab resistance in HER2-overexpressing cancers. (A-C) Dose-dependent treatment of YK1 and TZMB-induced growth inhibitory effect on parent cells (BT474 and NCI-N87) and TZMB-resistance acquired cells (BT474 TR, NCI-N87 TR, and JIMT-1) (n = 5). ANOVA, ns=non-significant, **P < 0.01, ***P < 0.001 versus the parent cells. (D-F) Dose-dependent treatment of YK1 and TZMB-induced effect on protein levels of HER2 and HER2-related major downstream signal molecules in TZMB-sensitive and TZMB-resistant cells. (G-I) Survival and proliferation of TZMB-sensitive and TZMB-resistant cells after dose-dependently treated with YK1 and TZMB, respectively, through clonogenic assay. (J-L) (J) Tumor regression was markedly promoted only by YK1 in JIMT-1-xenograft mouse model (n = 6 per group; 4 mg/kg IV injection every 3 days of YK1 and TZMB, respectively). (K) Photograph of the tumors collected from the vehicle- and YK1 or TZMB –treated mice. (L) Tumor volumes were evaluated at the indicated time points by measuring the length and width of the tumor with callipers using the equation (length x width2 ) / 2 (mean ± S.E.M.). Student’s t-test, ***P < 0.001 vs. CON, ###P < 0.001 vs. TZMB. (M, N) IHC stains of HER2 and Ki67 in the tumors. IHC score quantification was conducted using Image J (6 independent fields per sample were assessed, mean ± S.D.). Student’s t test, ***P < 0.001 vs. CON, ###P < 0.01 vs. TZMB.
Discussion
Various PPIs are typically required events in the human body for cells to regulate specific cellular signaling pathways, such as Wnt/β-Catenin and TGF-β pathways. The highly complex and interconnected network makes PPI an essential event to maintain homeostatic function under normal conditions. However, when exaggerated, it becomes a factor to cause tumorigenesis and/or metastasis [1], [37]. Therefore, ongoing efforts have been made to identify inhibitors for a single specific PPI in response to the needs to regulate specific biological pathways with the aim of reducing side effects and off-target effects, and understanding the exact function of a certain protein that may vary with binding partners in context-dependent manner [38]. A good example in this case is the ELF3-MED23 PPI which is essential for HER2 expression. Overexpression of HER2 is frequent in diverse tumors, such as breast, gastric, ovarian, and prostate cancers. Rates of HER2 overexpression in each cancer type were reported to be ∼ 25 % of all breast cancers, ∼6–35 % of all gastric cancers, ∼9–32 % of all ovarian cancers, and 25 % of untreated prostate cancers [50]. Although the main mechanism of HER2 overexpression is HER2 gene amplification, it also has been found that high transcription rates of HER2 per gene copy is involved [51]. Enhanced HER2 gene transcription is induced even in HER2-low or HER2-negative breast cancer after radiation therapy or endocrine therapy, which leads to resistance in treatment [52]. HER2-targeted therapies such as TZMB and pertuzumab have been recommended in combination with chemotherapy for HER2-overexpressing breast and gastric cancer patients [9], [10], but frequently emerging resistance has created a need for alternative strategies. Thus, instead of targeting already overexpressed HER2, several experimental attempts have been made to identify small molecules that transcriptionally inhibit HER2 expression [53], [54], [55], [56], [57], [58], but nothing has yet reached clinical trials. HER2 expression-related PPIs occur between various TFs and coactivators as follows: TFs are ELF3 [17], [22], activator protein-2 (AP-2) [39], SP1 [40], [41], signal transducer and activator of transcription 3 (STAT3) [42], Y-box binding protein-1 (YBX1 or YB1) [43], and EGR2 [44], [45], and coactivators are MED23, CITED2 (Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 2) [44], [45], and Yin Yang 1 (YY1) [46], [47]. Among them, the binding interface of ELF3 and MED23 is the most attractive target for HER2 modulation, since unlike other TFs, HER2 and ELF3 form a specific reciprocal regulatory relationship [48], [49]. It is that downregulation of HER2 by selectively inhibiting the ELF3-MED23 interaction could induce reduction in ELF3 and lead to persistent attenuation of HER2 signaling.
In this study, we systematically utilized molecular modelling and intensive biological evaluation methods to elucidate for the first time that ELF3-MED23 PPI specifically requires H-bonding with the D400 and H449 residues of MED23 to upregulate HER2 gene transcription. To be more precise at structural analysis, we started from the full-length protein structures of ELF3 (371 a.a.) and MED23 (1368 a.a.) and narrowed down to essential short peptide of ELF3 (8 a.a) in the PPI interface. Thus, it can be said that the model utilized is very close to the actual tertiary structure with minimal chance to have wrongly taken a different pose. Rather than directly applying mutation studies in this structural model, we first performed in silico study utilizing several compounds previously identified as possessing ELF3-MED23 PPI inhibitory activity to specifically define the critical residues for MED23- ELF3 PPI. This directly led to the idea that D400 and H449 of MED23 are the key residues for the interaction. Among the four compounds, binding mode of gefitinib was finally utilized as the starting point for the design of additional small molecule series. Though canertinib showed the most potent ELF3-MED23 PPI inhibitory activity, however, due to its severe cytotoxicity, gefitinib was utilized, which showed the second-highest efficacy with negligible nonspecific cytotoxicity. Since chalcones generally have two aromatic rings linked by a conjugated ketone core that can act as a hydrogen acceptor, we designed either R1 or R2 substituent to be hydroxyl group to act as a potential hydrogen donor. Using these chalcones, we also prepared pyrazolines with abidingly restricted spatial location of the intramolecular hydrogen acceptor and donor moieties, in order to promote more stable binding of compounds within the ELF3-MED23 interface.
Deciphering PPI interface in the molecular level and developing specific small molecule inhibitors for a specific single PPI has long been considered a difficult task, because the binding interfaces of PPI are relatively flat and large compared to receptor binding sites or enzyme active sites and the process to determine the hotspot residues and their position within the interface is time-consuming and tedious [1], [3], [59]. Nevertheless, mutational analysis of PPI interfaces revealed that small 'hotspots', but not all residues of the PPI interface, accounted for most of the binding energy of PPIs. This concept of hotspot, defined as being hydrophobic with conformational adaptivity and being centered in the PPI interface that can be covered by a small molecule, has led to successful PPI inhibitor discovery, some of which have undergone clinical trials [59]. Birinapant (also known as TL32711), a peptidomimetic inhibitor of second mitochondrial activator of caspases (SMAC) by interrupting PPI between SMAC and cellular inhibitors of apoptosis 1 has recently achieved FDA approval for the treatment of metastatic RET fusion-positive non-small cell lung cancer [60], [61]. Thus, it is very encouraging to discover that transcriptional regulation of HER2 expression can be facilitated simply by regulating only two hotspot residues (D400 and H449) out of 1368 amino acids of MED23. However, considering the number of attempts to develop various types of PPI inhibitors, those drugs that have reached the clinical use are still in a very limited proportion, as most of them exhibit relatively high molecular weight, low cell permeability, and poor oral bioavailability [3], [62]. This implicates that the customized small molecule to the PPI interface on the basis of structural insights, not only can serve as a useful tool for deciphering PPI hotspots, but can also hold a great developmental value as a druggable therapeutic agent. Therefore, these findings will provide a useful groundwork for future studies using a rational approach to develop drugs targeting PPIs and will open a new therapeutic landscape for HER2-overexpressing cancers, including gastric and breast cancers.
Conclusion
PPI is no longer a new concept but still of a great interest. This is because the general signal cascade can be selectively regulated by modulating specific PPIs destined to propagate the target signal of interest. Deciphering PPI interfaces and identification of their modulators are commonly considered challenging, and especially applying PPI modulators to therapeutics is fastidious. Developing selective PPI regulators becomes easier under circumstances where hotspots are clearly defined, but these cases are generally infrequent due to the lack of precise 3D structural insights into the PPI interface. Thus, in many cases, drug development relies on a conventional HTS system, which often leads to identification of compounds with non-specific PPI inhibitory activities and complicated structure. This, in turn, requires additional efforts to optimize the compound through an intensive structural modification process. Based upon these facts, the significance of this study resides in disclosing an efficient and powerful strategy for solving the specific PPI interface under the conditions where the target protein has multiple binding partners, and the 3D crystal structure is available for only one of the target proteins. Representatively, we focused on the ELF3-MED23 interaction, which is known to increase transcription of HER2, a well-known oncogene. Through iterative complementary molecular modelling studies with intensive biological evaluation methods, we finally identified YK1 with excellent in vitro, and in vivo anticancer activity against various HER2-overexpressing cancers, even in trastuzumab-refractory clones. Our findings serve as a representative case of showing how a small molecule can be utilized to define exact hotspot residues of a specific PPI and can also be directly applied as an anticancer agent with superior druggability.
Compliance with ethics requirements
All Institutional and National Guidelines for the care and use of animals (fisheries) were followed.
CRediT authorship contribution statement
Soo-Yeon Hwang: Conceptualization, Methodology, Formal analysis, Investigation, Writing – original draft. Seojeong Park: Methodology, Validation. Hyunji Jo: Methodology, Validation. Seung Hee Seo: Methodology, Validation. Kyung-Hwa Jeon: Methodology, Validation. Seojeong Kim: Methodology, Validation. Ah-Reum Jung: Methodology, Validation. Chanju Song: Methodology, Validation. Misun Ahn: . Soo Yeon Kwak: Methodology, Validation. Hwa-Jong Lee: Methodology, Validation. Motonari Uesugi: Conceptualization, Writing – review & editing. Younghwa Na: Conceptualization, Methodology, Supervision. Youngjoo Kwon: Conceptualization, Writing – review & editing, Supervision, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
This work was inspired by the international environments of JSPS CORE-to-CORE Program, “Asian Chemical Biology Initiative”. This work was supported by the National Research Foundation of Korea (NRF) grant (2018R1A5A2025286, 2021M3E5E7024855) and Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Ministry of Education (2021R1A6C101A442).
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2022.08.003.
Contributor Information
Younghwa Na, Email: yna7315@cha.ac.kr.
Youngjoo Kwon, Email: ykwon@ewha.ac.kr.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Bojadzic D., Buchwald P. Toward Small-Molecule Inhibition of Protein-Protein Interactions: General Aspects and Recent Progress in Targeting Costimulatory and Coinhibitory (Immune Checkpoint) Interactions. Curr Top Med Chem. 2018;18(8):674–699. doi: 10.2174/1568026618666180531092503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leader B., Baca Q.J., Golan D.E. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7(1):21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 3.Bakail M., Ochsenbein F. Targeting protein–protein interactions, a wide open field for drug design. C R Chim. 2016;19(1-2):19–27. [Google Scholar]
- 4.Kenneth Morrow J., Zhang S. Computational prediction of protein hot spot residues. Curr Pharm Des. 2012;18(9):1255–1265. doi: 10.2174/138161212799436412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgess A.W., Cho H.-S., Eigenbrot C., Ferguson K.M., Garrett T.P.J., Leahy D.J., et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12(3):541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 6.Prenzel N., Fischer O.M., Streit S., Hart S., Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8(1):11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- 7.Slamon D.J., Leyland-Jones B., Shak S., Fuchs H., Paton V., Bajamonde A., et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. The New England journal of medicine. 2001;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 8.Abrahao-Machado L.F., Scapulatempo-Neto C. HER2 testing in gastric cancer: An update. World J Gastroenterol. 2016;22(19):4619–4625. doi: 10.3748/wjg.v22.i19.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.NCCN. NCCN GUIDELINES FOR PATIENTS®. Available at: https://wwwnccnorg/patients/guidelines/content/PDF/stomach-patientpdf. 2019.
- 10.Gradishar W.J., Anderson B.O., Abraham J., Aft R., Agnese D., Allison K.H., et al. Breast Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18(4):452–478. doi: 10.6004/jnccn.2020.0016. [DOI] [PubMed] [Google Scholar]
- 11.Slamon D.J., Godolphin W., Jones L.A., Holt J.A., Wong S.G., Keith D.E., et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science (New York, NY) 1989;244(4905):707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 12.Koeppen H.K.W., Wright B.D., Burt A.D., Quirke P., McNicol A.M., Dybdal N.O., et al. Overexpression of HER2/neu in solid tumours: an immunohistochemical survey. Histopathology. 2001;38(2):96–104. doi: 10.1046/j.1365-2559.2001.01084.x. [DOI] [PubMed] [Google Scholar]
- 13.Cresti N., Lee J., Rourke E., Televantou D., Jamieson D., Verrill M., et al. Genetic variants in the HER2 gene: Influence on HER2 overexpression and loss of heterozygosity in breast cancer. European journal of cancer (Oxford. England. 2016;55:27–37. doi: 10.1016/j.ejca.2015.10.066. [DOI] [PubMed] [Google Scholar]
- 14.Kim H.-L., Jeon K.-H., Jun K.-Y., Choi Y., Kim D.-K., Na Y., et al. A-62176, a potent topoisomerase inhibitor, inhibits the expression of human epidermal growth factor receptor 2. Cancer Lett. 2012;325(1):72–79. doi: 10.1016/j.canlet.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Nam J.M., Jeon K.-H., Kwon H., Lee E., Jun K.-Y., Jin Y.B., et al. Dithiiranylmethyloxy azaxanthone shows potent anti-tumor activity via suppression of HER2 expression and HER2-mediated signals in HER2-overexpressing breast cancer cells. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2013;50(2):181–190. doi: 10.1016/j.ejps.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Jun K.-Y., Kwon Y. Proposal of Dual Inhibitor Targeting ATPase Domains of Topoisomerase II and Heat Shock Protein 90. Biomol Ther (Seoul) 2016;24(5):453–468. doi: 10.4062/biomolther.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang C.-H., Scott G.K., Kuo W.-L., Xiong X., Suzdaltseva Y., Park J.W., et al. ESX: a structurally unique Ets overexpressed early during human breast tumorigenesis. Oncogene. 1997;14(13):1617–1622. doi: 10.1038/sj.onc.1200978. [DOI] [PubMed] [Google Scholar]
- 18.Asada S., Choi Y., Uesugi M. A Gene-Expression Inhibitor that Targets an α-Helix-Mediated Protein Interaction. J Am Chem Soc. 2003;125(17):4992–4993. doi: 10.1021/ja0292703. [DOI] [PubMed] [Google Scholar]
- 19.Jeon K.-H., Jun K.-Y., Kim E.Y., Kwon Y. Expression and purification of a soluble ESX-binding core domain of SUR2. Prep Biochem Biotech. 2013;43(4):364–375. doi: 10.1080/10826068.2012.738273. [DOI] [PubMed] [Google Scholar]
- 20.Segovia-Mendoza M., Gonzalez-Gonzalez M.E., Barrera D., Diaz L., Garcia-Becerra R. Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: preclinical and clinical evidence. Am J Cancer Res. 2015;5(9):2531–2561. [PMC free article] [PubMed] [Google Scholar]
- 21.Franken N.A.P., Rodermond H.M., Stap J., Haveman J., van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 22.Asada S., Choi Y., Yamada M., Wang S.-C., Hung M.-C., Qin J., et al. External control of Her2 expression and cancer cell growth by targeting a Ras-linked coactivator. Proc Natl Acad Sci U S A. 2002;99(20):12747–12752. doi: 10.1073/pnas.202162199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimogawa H., Kwon Y., Mao Q., Kawazoe Y., Choi Y., Asada S., et al. A wrench-shaped synthetic molecule that modulates a transcription factor-coactivator interaction. J Am Chem Soc. 2004;126(11):3461–3471. doi: 10.1021/ja038855+. [DOI] [PubMed] [Google Scholar]
- 24.Gasteiger J., Marsili M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron. 1980;36(22):3219–3228. [Google Scholar]
- 25.Stierand K., Maass P.C., Rarey M. Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinformatics (Oxford, England) 2006;22(14):1710–1716. doi: 10.1093/bioinformatics/btl150. [DOI] [PubMed] [Google Scholar]
- 26.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monté D., Clantin B., Dewitte F., Lens Z., Rucktooa P., Pardon E., et al. Crystal structure of human Mediator subunit MED23. Nat Commun. 2018;9(1) doi: 10.1038/s41467-018-05967-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vajda S., Yueh C., Beglov D., Bohnuud T., Mottarella S.E., Xia B., et al. New additions to the ClusPro server motivated by CAPRI. Proteins. 2017;85(3):435–444. doi: 10.1002/prot.25219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozakov D., Hall D.R., Xia B., Porter K.A., Padhorny D., Yueh C., et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevens J.L., Cantin G.T., Wang G., Shevchenko A., Shevchenko A., Berk A.J. Transcription Control by E1A and MAP Kinase Pathway via Sur2 Mediator Subunit. Science (New York, NY) 2002;296(5568):755–758. doi: 10.1126/science.1068943. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths SJ, Koegl M, Boutell C, Zenner HL, Crump CM, Pica F, et al. A systematic analysis of host factors reveals a Med23-interferon-λ regulatory axis against herpes simplex virus type 1 replication. PLoS Pathog. 2013;9(8):e1003514. [DOI] [PMC free article] [PubMed]
- 32.Nahta R., Esteva F.J. Trastuzumab: triumphs and tribulations. Oncogene. 2007;26(25):3637–3643. doi: 10.1038/sj.onc.1210379. [DOI] [PubMed] [Google Scholar]
- 33.Eladdadi A., Isaacson D. A mathematical model for the effects of HER2 over-expression on cell cycle progression in breast cancer. Bull Math Biol. 2011;73(12):2865–2887. doi: 10.1007/s11538-011-9663-3. [DOI] [PubMed] [Google Scholar]
- 34.Okines A.F.C., Cunningham D. Trastuzumab: a novel standard option for patients with HER-2-positive advanced gastric or gastro-oesophageal junction cancer. Therapeutic advances in gastroenterology. 2012;5(5):301–318. doi: 10.1177/1756283X12450246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang S.-Y., Choi S.-K., Seo S.H., Jo H., Shin J.-H., Na Y., et al. Specific Roles of HSP27 S15 Phosphorylation Augmenting the Nuclear Function of HER2 to Promote Trastuzumab Resistance. Cancers. 2020;12(6):1540. doi: 10.3390/cancers12061540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waller C.F., Möbius J., Fuentes-Alburo A. Intravenous and subcutaneous formulations of trastuzumab, and trastuzumab biosimilars: implications for clinical practice. Br J Cancer. 2021;124(8):1346–1352. doi: 10.1038/s41416-020-01255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pawson T., Nash P. Protein–protein interactions define specificity in signal transduction. Genes Dev. 2000;14(9):1027–1047. [PubMed] [Google Scholar]
- 38.Mullard A. Protein-protein interaction inhibitors get into the groove. Nat Rev Drug Discov. 2012;11(3):173–175. doi: 10.1038/nrd3680. [DOI] [PubMed] [Google Scholar]
- 39.Vernimmen D., Begon D., Salvador C., Gofflot S., Grooteclaes M., Winkler R. Identification of HTF (HER2 transcription factor) as an AP-2 (activator protein-2) transcription factor and contribution of the HTF binding site to ERBB2 gene overexpression. Biochem J. 2003;370(Pt 1):323–329. doi: 10.1042/BJ20021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y., Gill G.N. Positive and negative regulatory elements in the human erbB-2 gene promoter. Oncogene. 1994;9(8):2269–2276. [PubMed] [Google Scholar]
- 41.Jiang W., Jin Z., Zhou F., Cui J., Wang L., Wang L. High co-expression of Sp1 and HER-2 is correlated with poor prognosis of gastric cancer patients. Surg Oncol. 2015;24(3):220–225. doi: 10.1016/j.suronc.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 42.Qian L.u., Chen L., Shi M., Yu M., Jin B., Hu M., et al. A novel cis-acting element in Her2 promoter regulated by Stat3 in mammary cancer cells. Biochem Biophys Res Commun. 2006;345(2):660–668. doi: 10.1016/j.bbrc.2006.04.153. [DOI] [PubMed] [Google Scholar]
- 43.Kuwano M., Shibata T., Watari K., Ono M. Oncogenic Y-box binding protein-1 as an effective therapeutic target in drug-resistant cancer. Cancer Sci. 2019;110(5):1536–1543. doi: 10.1111/cas.14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dillon R.L., Brown S.T., Ling C., Shioda T., Muller W.J. An EGR2/CITED1 transcription factor complex and the 14-3-3sigma tumor suppressor are involved in regulating ErbB2 expression in a transgenic-mouse model of human breast cancer. Mol Cell Biol. 2007;27(24):8648–8657. doi: 10.1128/MCB.00866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minemura H., Takagi K., Sato A., Takahashi H., Miki Y., Shibahara Y., et al. CITED2 in breast carcinoma as a potent prognostic predictor associated with proliferation, migration and chemoresistance. Cancer Sci. 2016;107(12):1898–1908. doi: 10.1111/cas.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Powe D.G., Akhtar G., Habashy H.O., Abdel-Fatah T., Rakha E.A., Green A.R., et al. Investigating AP-2 and YY1 protein expression as a cause of high HER2 gene transcription in breast cancers with discordant HER2 gene amplification. Breast Cancer Res. 2009;11(6) doi: 10.1186/bcr2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schiano C., Franzese M., Pane K., Garbino N., Soricelli A., Salvatore M., et al. Hybrid (18)F-FDG-PET/MRI Measurement of Standardized Uptake Value Coupled with Yin Yang 1 Signature in Metastatic Breast Cancer. A Preliminary Study. Cancers. 2019;11(10) doi: 10.3390/cancers11101444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neve R.M., Ylstra B., Chang C.-H., Albertson D.G., Benz C.C. ErbB2 activation of ESX gene expression. Oncogene. 2002;21(24):3934–3938. doi: 10.1038/sj.onc.1205503. [DOI] [PubMed] [Google Scholar]
- 49.Eckel K.L., Tentler J.J., Cappetta G.J., Diamond S.E., Gutierrez-Hartmann A. The epithelial-specific ETS transcription factor ESX/ESE-1/Elf-3 modulates breast cancer-associated gene expression. DNA Cell Biol. 2003;22(2):79–94. doi: 10.1089/104454903321515896. [DOI] [PubMed] [Google Scholar]
- 50.Tai W., Mahato R., Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146(3):264–275. doi: 10.1016/j.jconrel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mungamuri S., Murk W., Grumolato L., Bernstein E., Aaronson S. Chromatin modifications sequentially enhance ErbB2 expression in ErbB2-positive breast cancers. Cell Rep. 2013;5(2):302–313. doi: 10.1016/j.celrep.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Q., Kulak M.V., Borcherding N., Maina P.K., Zhang W., Weigel R.J., et al. A novel HER2 gene body enhancer contributes to HER2 expression. Oncogene. 2018;37(5):687–694. doi: 10.1038/onc.2017.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hollywood D.P., Hurst H.C. Targeting gene transcription: a new strategy to down-regulate c-erbB-2 expression in mammary carcinoma. Br J Cancer. 1995;71(4):753–757. doi: 10.1038/bjc.1995.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ebbinghaus S.W., Fortinberry H., Gamper H.B., Jr. Inhibition of transcription elongation in the HER-2/neu coding sequence by triplex-directed covalent modification of the template strand. Biochemistry. 1999;38(2):619–628. doi: 10.1021/bi980981g. [DOI] [PubMed] [Google Scholar]
- 55.Chiang S.-Y., Bürli R.W., Benz C.C., Gawron L., Scott G.K., Dervan P.B., et al. Targeting the ets binding site of the HER2/neu promoter with pyrrole-imidazole polyamides. The Journal of biological chemistry. 2000;275(32):24246–24254. doi: 10.1074/jbc.M000820200. [DOI] [PubMed] [Google Scholar]
- 56.Costamagna A., Rossi Sebastiano M., Natalini D., Simoni M., Valabrega G., Defilippi P., et al. Modeling ErbB2-p130Cas interaction to design new potential anticancer agents. Sci Rep. 2019;9(1) doi: 10.1038/s41598-019-39510-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee L.W., Taylor C.E.C., Desaulniers J.-P., Zhang M., Højfeldt J.W., Pan Q., et al. Inhibition of ErbB2(Her2) expression with small molecule transcription factor mimics. Bioorg Med Chem Lett. 2009;19(21):6233–6236. doi: 10.1016/j.bmcl.2009.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujimoto M., Kito H., Kajikuri J., Ohya S. Transcriptional repression of human epidermal growth factor receptor 2 by ClC-3 Cl(-) /H(+) transporter inhibition in human breast cancer cells. Cancer Sci. 2018;109(9):2781–2791. doi: 10.1111/cas.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arkin M., Tang Y., Wells J. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2014;21(9):1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.FDA approves pralsetinib for lung cancer with RET gene fusions [press release]. September, 04 2020.
- 61.Srivastava AK, Jaganathan S, Stephen L, Hollingshead MG, Layhee A, Damour E, et al. Effect of a Smac Mimetic (TL32711, Birinapant) on the Apoptotic Program and Apoptosis Biomarkers Examined with Validated Multiplex Immunoassays Fit for Clinical Use. Clin Cancer Res. 2016;22(4):1000-10. [DOI] [PMC free article] [PubMed]
- 62.Ran X.u., Gestwicki J.E. Inhibitors of protein-protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Curr Opin Chem Biol. 2018;44:75–86. doi: 10.1016/j.cbpa.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.