

Abstract
Background
With millions of victims worldwide, multiple sclerosis is the second most common cause of disability among young adults. Although formidable advancements have been made in understanding the disease, the neurodegeneration associated with multiple sclerosis is only partially counteracted by current treatments, and effective therapy for progressive multiple sclerosis remains an unmet need. Therefore, new approaches are required to delay demyelination and the resulting disability and to restore neural function by promoting remyelination and neuronal repair.
Aims
The article reviews the latest literature in this field.
Materials and methods
The fibroblast growth factor (FGF) signaling pathway is a promising target in progressive multiple sclerosis.
Discussion
FGF signal transduction contributes to establishing the oligodendrocyte lineage, neural stem cell proliferation and differentiation, and myelination of the central nervous system. Furthermore, FGF signaling is implicated in the control of neuroinflammation. In recent years, interventions targeting FGF, and its receptor (FGFR) have been shown to ameliorate autoimmune encephalomyelitis symptoms in multiple sclerosis animal models moderately.
Conclusion
Here, we summarize the recent findings and investigate the role of FGF/FGFR signaling in the onset and progression, discuss the potential therapeutic advances, and offer fresh insights into managing multiple sclerosis.
Keywords: fibroblast growth factor receptors, fibroblast growth factors, inflammation, multiple sclerosis, remyelination
Fibroblast growth factors are involved in the MS development and crucial processes.
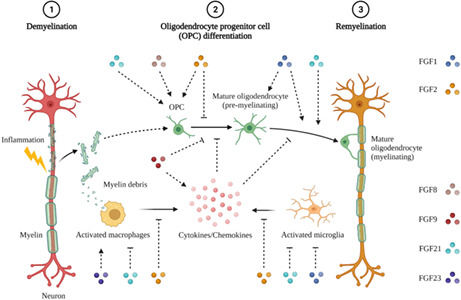
1. INTRODUCTION
Multiple sclerosis (MS) is a chronic demyelinating condition of the central nervous system (CNS) caused by lymphocytic infiltration, damaging axons, and their associated myelin sheath. 1 , 2 It is the leading cause of non‐traumatic neurological disorders in young adults and causes significant disability, and high direct and indirect costs worldwide. Dysregulated immunity, genetic predisposition, and environmental variables interact in a complicated way to cause MS. 1 Vitamin D insufficiency, related to reduced exposure to ultraviolet type B light, is the main environmental factor. The risk of MS is higher for adolescent patients living in northern or southern latitudes than those in equatorial regions. 3 Approximately 85% of MS cases begin with episodic neurological impairment involving the brainstem, optic nerve, and spinal cord, which resolves spontaneously, is termed relapsing–remitting MS (RRMS), and mainly affects individuals in early adulthood. 1 Approximately 20% of patients develop symptoms after age 40, and disability progression in the male population with late onset is faster than in young RRMS patients. 4 In 50% to 60% of RRMS cases, the condition develops into secondary progressive MS (SPMS), characterized by an infrequent or completely terminated relapse after 10–15 years with a slow progression of irreversible disabilities associated with neurodegeneration. 5 , 6 Unidirectional progressive disability from the outset is observed in 15% of MS cases and is referred to as primary progressive MS (PPMS). For this patient cohort, the rapid onset of early neurodegeneration is the best predictor of long‐term progression rates. 6 In addition to experimental autoimmune encephalomyelitis (EAE), the most frequently employed model, 7 several demyelinating models have also been used in MS research. Both clinical and pathological characteristics of human MS are shared by the murine hepatitis virus (MHV) strain A59 that causes demyelination in animal models, 8 and lysophosphatidylcholine has been used to induce inflammatory demyelination, in which the myelin structures, as well as the blood–brain barrier (BBB), are disrupted, but neuronal loss is absent. 9 Moreover, the chronic cuprizone demyelination model leads to consistent demyelination followed by spontaneous remyelination within a short period. 10 Thus, these models are valuable supplements to EAE research and are appropriate for studying the mechanisms of demyelination and therapeutic interventions for MS.
The fibroblast growth factor (FGF) family contains 23 members, including 18 protein ligands (the murine FGF15 and the human FGF19 genes are orthologous) and four fibroblast homologous factors (FHFs). FGF ligands exercise their functions by binding with their high‐affinity receptor family (FGFR1‐4) and are involved in fundamental physiological processes in adults, including wound repair, angiogenesis, and metabolism. 11 , 12 The roles of FGF/FGFR signaling on essential cellular function control indicates the relevance of this axis in the pathogenesis of MS. 13 , 14 Over the past few years, interventions targeting FGF/FGFR have moderately ameliorated symptoms in animal models, and the conditional deletion of FGFR1 and FGFR2 has shown remarkable therapeutic promise. 15 , 16 Furthermore, several FGF family members are strongly associated with the pathogenesis and course of MS. 17 , 18 , 19 Here, we summarize the latest relevant findings, discuss the function of FGF/FGFR signaling in MS pathogenesis, and describe potential therapeutic advances, providing fresh perspectives on MS therapy.
2. UNDERSTANDING THE PATHOGENESIS OF MS
2.1. Inflammation
Multiple sclerosis is a chronic inflammatory demyelinating and degenerative condition of the CNS. Inflammation of the spinal cord and brain is invariably present in all phases of MS and declines with disease progression. 20 A dominant aspect of the early pathology in RRMS patients is active inflammatory demyelinating lesions, which arise through inflammatory infiltrates associated with disrupted BBB. 21 At this stage, lesions of relapsing MS have more plentiful macrophages than any type of progressive MS (PMS) to phagocytose the myelin degradation products. 22 In contrast, chronic lesions predominate in progressive disease. Lymphocyte infiltration is initially blocked in the leptomeninges and blood vessels behind an intact BBB. 23 Here, T cells attract immune cells into the CNS by interacting with major histocompatibility complex class II+ microglia 24 and produce adhesion molecules, chemokines, and a variety of proinflammatory cytokines. 20 Another type of inflammation is densely populated by B cells of the brain's connective tissue spaces adjacent to an intact BBB, where they may form aggregates or tertiary lymph follicles. 21 , 25 Interestingly, the inflammatory state of the CNS might provide a favorable environment for lymphocyte proliferation and expansion. 20 Furthermore, mononuclear phagocytes (MPs), namely resident microglia and the macrophages differentiated from infiltrating monocytes, also act as a significant part in the pathologic mechanisms of PMS. According to experimental and clinical studies, MPs present in demyelinating lesions secrete chemokines that induce lymphocytes to infiltrate the CNS and thus provide an inflammatory environment. They also generate reactive oxygen and nitrogen species, which leads to oligodendrocyte and neuronal cell death. 1 , 26 , 27 In addition, CNS glial cells may initiate an immunological response in MS (particularly in PMS, as it is intimately linked to the chronic activation of the innate immune system). 28 Some studies have pointed out that, at least in certain situations, MS may originate from a primary injury within the CNS, possibly associated with oligodendrocytes, followed by glial activation and ultimately by immune‐mediated inflammatory activation as a secondary response. 25
2.2. Demyelination and neurodegeneration
Demyelination leads to a reduction in axonal integrity and, over time, to neuronal dysfunction. Neurodegeneration is a characteristic of MS and a major contributor to clinical impairment and decreased quality of life. Demyelinated axons become frangible and suffer damage from activated immunological and glial cells releasing cytokines, oxidative products, and free radicals. Even in the predominantly inflammatory demyelinating stage of the disease, transected axons are abundant, demonstrating that axonal loss occurs at disease onset and continues with time. In the initial phases of RRMS, the axonal loss has no immediate substantial clinical impact. As lesions accumulate with time, however, the clinical aspects of MS become driven by axonal loss. Thus, it is believed that the brain's ability to adjust for further axonal loss exhausts before RRMS and SPMS shift. At this stage, MS lesions include remyelination, inflammation resolution without repair, or a “smoldering” state of coexistence of inflammation and myelin degeneration. 29 In PMS, depletion of the myelination capacity by both oligodendrocyte precursor cells (OPCs) and residual oligodendrocytes is critical. Recent studies of MS using human single‐nucleus RNAseq demonstrated that oligodendrocytes respond rapidly to oxidative stress, with the downregulation of homeostasis and myelin synthesis genes. 30 Moreover, when the dynamics of oligodendrocyte generation in MS brain tissue were assessed by 14C methods, it was found that the demyelination is partly caused by the depletion of the myelination ability of the surviving oligodendrocytes rather than by an impairment in OPC differentiation. 31 These results may impact the establishment of disease models and the development of myelin regeneration strategies for PMS. RRMS has become a pharmacologically treatable condition. However, PMS continues to face treatment challenges because of the persistent accumulation of neurological impairments and disabilities.
2.3. Remyelination failure
Demyelination can occur parallel to regeneration processes, which restore some of the destroyed myelin‐generating cells and rebuild the myelin sheath around axons. This is accomplished by the activation, migration, and polarization of resident OPCs and neural stem cells (NSCs) into myelin cells, initiating an oligodendrocyte‐driven repair process known as remyelination. The identity of cells that cause remyelination in the CNS of MS patients has been a subject of debate. OPCs and mature oligodendrocytes that have survived are two potential candidates. This discussion is crucial because therapeutic approaches to enhance remyelination may differ depending on the specific cellular pathways involved. Lineage tracing experiments revealed that newly generated oligodendrocytes derived from OPCs form new myelin sheaths in demyelinated regions. 32 However, it has also been found that surviving oligodendrocytes can expand and remyelinate axons in MS. 31 Moreover, myelin sheaths derived from OPCs are thinner and less functional than those generated by the surviving oligodendrocytes. Myelin cells in the adult CNS can also differentiate from NSCs in the subventricular region. The microenvironment of the demyelinating lesions substantially impacts OPC and NSC homeostasis, in addition to uncontrollable factors such as gender and age and may also be the target of future remyelination treatment strategies. Other glial cells, like microglia, are momentous to remyelination and aid in removing myelin debris and releasing neurotrophic factors that support OPC functions. 33 , 34 Among them, CX3CR1, a fractalkine receptor that is abundantly expressed on microglia, has been shown to affect the ability of these cells to phagocytose. Reduced microglial phagocytosis in cuprizone‐treated CX3CR1‐deficient animals causes a continuous accumulation of myelin debris, inhibiting remyelination due to insufficient OPC recruitment. 35 Additionally, the disequilibrium of pro‐regeneration and inhibitory elements limits the remyelination capacity of OPCs and oligodendrocytes. OPC RNA sequencing revealed that the mTOR pathway plays a substantial role in remyelination failure. This pathway can be manipulated by caloric restriction or by administration of the AMPK‐agonist metformin to reverse the decline in OPC differentiation and restore their ability to remyelinate axons. 5 Moreover, multiple OPC differentiation inhibitors, including PSA‐NCAM, Lingo‐1, Jagged, and Galectin‐4, appear relatively overexpressed. Additionally, IFN‐γ, Gli1, and Sirt1 inhibited the proliferation and differentiation of NSCs in demyelinating lesions. 34 Remyelination failure leads to axonal loss and neurodegenerative changes over time. Therefore, specific targeting of this pathological process is expected to deliver a breakthrough in MS treatment in the future.
3. FGF/FGFR SYSTEM
3.1. FGFs, FGFRs, and co‐receptors
The 18 FGFs cluster into six subfamilies, with the FGF1, FGF4, FGF7, FGF8, and FGF9 subfamilies functioning in a paracrine manner and the FGF19 subfamily members operating as endocrine factors. 11 FGFRs contain a single transmembrane helix (termed TM), three extracellular immunoglobulin‐like domains (termed D1‐3), and two intracellular tyrosine kinase domains (termed TK1‐2). An eight‐residue acid box, a hallmark of FGFRs, is located between D1 and D2 and, together with the D1 loop, plays an autoinhibitory role in receptor activation. 36 The FGF‐binding region is in D2 and interacts with D3 providing specificity. Two alternative splice sites (D3b and D3c) of the D3 protein show distinct FGF binding specificities (Figure 1A). The FGFR D3b isoform is commonly seen in epithelial cells, whereas the FGFR D3c isoform is typically found in mesenchymal cells. Although FGFR1‐3 exhibits frequent alternative splicing, there is no isoform due to the absence of alternative splicing exons in FGFR4. 36 , 37 Depending on the combination with co‐receptors, which include heparin sulfate (HS)/proteoglycans (HSPGs) and Klotho proteins, can FGFs‐FGFRs binding elicit a signal. Most of FGFs feature HS binding domains, and HSPGs are widely distributed in the extracellular matrix. Their different affinities determine whether they work in a paracrine, autocrine, or endocrine way. 11 , 38 Not only can HSPGs tether FGFs and enable them to function in an autocrine or paracrine way, but they also enhance FGFS signaling by forming FGF/FGFR/HSPG complexes. 39 In contrast, the endocrine FGF subclass ligands (FGF19, FGF21, and FGF23), with a weak affinity for HSPGs, utilize Klotho proteins as co‐receptors for binding to their respective FGFRs 37 , 40 (Figure 1B). However, they exhibit a strong affinity for FGFR/Klotho complexes but a limited affinity for individual FGFRs or Klotho proteins. 41 Klotho proteins are a class of transmembrane proteins consisting of the α‐, β‐, and γ‐Klotho subunits. α‐Klotho is necessary for the activity of FGF23, and the biological effects of FGF19 and FGF21 need β‐Klotho (Table 1).
FIGURE 1.
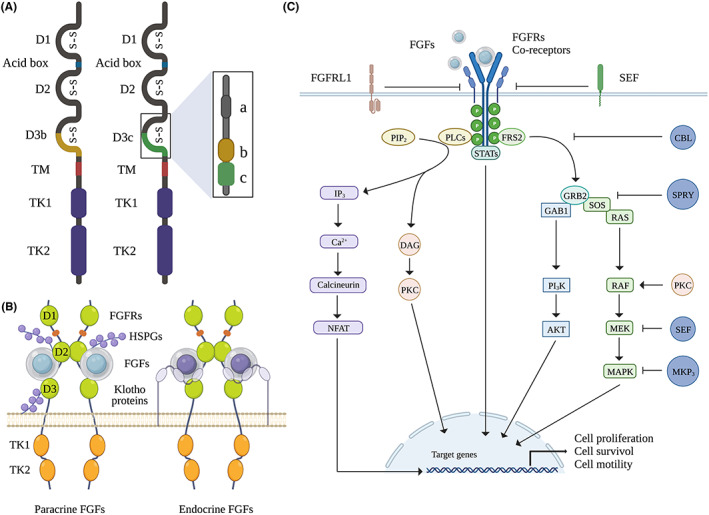
The FGF/FGFR signaling system. (A) FGFR monomer structures: FGFR is a form of the extracellular domains and intracellular catalytic domains linked by a single pass transmembrane domain. Except for FGFR4, the other three FGFR coding genes generate two major splice variants in D3, termed as D3b and D3c, which are essential determinants of ligand binding specificity. (B) The relative orientation of the FGF/FGFR/co‐receptor complex. (C) The downstream pathways of FGF/FGFR signaling: binding of FGFs triggers the dimerization and activation of FGFRs. Activated FGFRs phosphorylate FRS2, which binds to SH2 domain‐containing adaptor GRB2 and GRB2 will subsequently bind to SOS and GAB1 to activate RAS/RAF/MAPKs pathway, including ERK, p38 and JNK, as well as the PI3K/AKT pathway. Independent of the FRS2 binding, FGF signals also activate STATs and PLCγ. Activated PLCγ hydrolyzes PIP2 to DAG and PIP3, which stimulates calcium release from the endoplasmic reticulum and activation of calcium/calmodulin dependent protein kinases. FGFRL1 and SEF are transmembrane proteins and can interact directly with FGFRs to negatively regulate it. Phosphorylation of the MAPK/ERK cascade can be negatively regulated by SEF. SPRY acts at the level of Grb2 to attenuate FGF/FGFR signaling. MKP3 functions as a negative regulator by affecting the phosphorylation of the ERK. AKT, protein kinase B (AKT); DAG, diacylglycerol; ERK, extracellular signal regulated kinase; FRS2, FGFR substrate 2; GAB1, GRB2 associated binding protein 1; JNK, c‐Jun N‐terminal kinase; MAPK, mitogen activated protein kinase; PI3K, phosphatidylinositol 3‐kinase; PKC, protein kinase C; PLCγ, phospholipase Cγ; SOS, son of seven.
TABLE 1.
Classification of FGF ligands and their corresponding receptors.
| Function manner | FGF subfamily | FGFs | FGFRs | Co‐receptors |
|---|---|---|---|---|
| Paracrine FGFs | FGF1 | FGF1 (aFGF) | All FGFRs | HSPGs |
| FGF2 (bFGF) | FGFR1b, FGFR1c, FGFR2c, FGFR3c, FGF4 | |||
| FGF4 | FGF4 | FGFR1c, FGFR2c, FGFR3c, FGF4 | ||
| FGF5 | FGFR1c, FGFR2c | |||
| FGF6 | FGFR1c, FGFR2c, FGFR3c, FGF4 | |||
| FGF7 | FGF3 | FGFR1b, FGFR2b | ||
| FGF7 (KGF) | FGFR1b, FGFR2b | |||
| FGF10 | FGFR1b, FGFR2b | |||
| FGF22 | FGFR1b, FGFR2b | |||
| FGF8 | FGF8 | FGFR1c, FGF2c, FGFR3b, FGFR3c, FGFR4 | ||
| FGF17 | FGFR1c, FGF2c, FGFR3b, FGFR3c, FGFR4 | |||
| FGF18 | FGFR1c, FGF2c, FGFR3b, FGFR3c, FGFR4 | |||
| FGF9 | FGF9 | FGFR1c, FGF2c, FGFR3b, FGFR3c, FGFR4 | ||
| FGF16 | FGFR1c, FGF2c, FGFR3b, FGFR3c, FGFR4 | |||
| FGF20 | FGFR1c, FGF2c, FGFR3b, FGFR3c, FGFR4 | |||
| Endocrine FGFs | FGF19 | FGF15/19 | FGFR1c, FGF2c, FGFR3c, FGFR4 | β‐Klotho |
| FGF21 | FGFR1c, FGFR3c | |||
| FGF23 | FGFR1c, FGFR3c, FGFR4 | α‐Klotho | ||
| Intracellular FGFs | FHFs | FGF11 | ||
| FGF12 | ||||
| FGF13 | ||||
| FGF14 |
Abbreviations: FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; HSPGs, heparin sulfate (HS)/proteoglycans.
3.2. FGF/FGFR signaling
Upon ligand binding, FGFRs dimerize and cause the phosphorylation and activation of intracellular tyrosine kinases. 42 FGFR substrate 2 (FRS2), a key adaptor protein, is phosphorylated by activated FGFRs, which causes the activation of four intracellular pathways: the phospholipase Cγ (PLCγ) signal; signal transducer and activator of transcription (STAT) signal; phosphatidylinositol 3‐kinase (PI3K)/protein kinase B (AKT) signal; and mitogen‐activated protein kinase (MAPK) signal. 43 , 44 , 45 Moreover, FGFR‐like 1 (FGFRL1), 46 SPRY, CBL, SEF, MAPK phosphatase 3 (MKP3), and MKP1 can also negatively regulate FGF/FGFR signaling to varying extents. These regulatory factors modulate intracellular signaling or ligand binding 42 (Figure 1C).
4. ROLE OF FGF/FGFR SIGNALING IN CNS
FGF/FGFR signaling not only plays a crucial role in CNS formation during development but also has a broad function in the adult CNS. A prototype member, FGF1, is expressed by neurons in adult neural tissue and acts as a mitogen in neurodevelopmental processes. 47 , 48 , 49 FGF1 stimulated NSC cell expansion and neurite outgrowth in neurons. 50 , 51 In vitro and in vivo, FGF2 controls NSC proliferation. Under the induction of FGF2, it has been discovered that undifferentiated precursor cells in the adult mouse proliferated and differentiated into various CNS cells, such as neurons, oligodendrocytes, and astrocytes. 52 Several studies have shown that FGF2 is also crucial for NSC proliferation in vivo. Granule precursor neuron proliferation was four times higher after subcutaneous injection of FGF2 and had a 250% increase in the subventricular zone of the lateral ventricles. Furthermore, a 68% and 50% increase in DNA synthesis in hippocampal and whole cerebellar homogenates were observed following in vivo FGF2 treatment. 53 Moreover, the generation and dendritic development of new dentate granule cells was also enhanced after intracerebroventricular FGF2 infusion. 54 However, the positive regulatory effect of FGF2 on neuronal proliferation was reversed after treatment with FGF2‐specific neutralizing antibodies. 55 These results indicate that FGF2 is an important neurogenic factor that directly acts on the mitosis of NSCs to promote their proliferation. Conversely, according to multiple studies, FGF2 is not necessary to proliferate neuronal precursors, as FGF2‐deficient mice show normal neural progenitor proliferation during development. However, these mice exhibited partial cerebral cortex loss, impaired neural stem cell differentiation, and increased CNS cell apoptosis, suggesting that FGF2 triggers neural progenitors to migrate and differentiate. 56 , 57 In addition, other FGFs, such as FGF4, FGF8, FGF9, FGF10, and FGF21, regulate neuronal fate. 58 , 59 , 60 , 61 , 62
The CNS myelin‐producing cells, oligodendrocytes, play a central role in generating and preserving the pace and the power of axonal electrical impulses. Neurological deficits in MS result from myelin damage or insufficient remyelination. Understanding the signals involved in developing oligodendrocyte‐driven myelination may shed light on demyelinating disease prevention and treatment. OPCs migrate to various brain regions during development, transforming into myelin‐producing cells. 63 , 64 Several growth factors, including FGFs, control the development of oligodendrocytes. 65 However, the effect of FGF/FGFR signaling on oligodendrocyte development is regulated by the differential expression of FGFRs. Early and late OPCs express FGFR3, FGFR2 is expressed in mature oligodendrocytes, and both express FGFR1 but not FGFR4. 66 , 67 Each FGFR has different roles and focuses during the development and maturation of oligodendrocytes. FGFR1 may transduce signals that stimulate early OPCs proliferation and migration, while FGFR3 signaling primarily controls the late OPCs transformation to oligodendrocytes, and FGFR2 is mainly involved in modulating oligodendrocyte differentiation and myelination. 65 FGFR1 and FGFR2 in CNS myelination are crucial since they initiate myelination, regulate myelin thickness independently of oligodendrocyte differentiation, and contribute to the remyelination of chronically demyelinated lesions. 68 Insufficient myelin protein production by oligodendrocytes in FGFR1/FGFR2‐double knockout mice prevents myelination from reaching average levels of myelin thickness. 68 , 69
Furthermore, ERK1/2 and PI3K/AKT/mTOR, the downstream mediators of these FGFR signaling pathways, sequentially regulate myelination by affecting distinct stages of the oligodendrocyte lineage. ERK1/2 signaling regulates both the transition from early to late OPCs and subsequent immature oligodendrocyte stages. The mTOR signaling pathway is necessary to transform from immature to mature oligodendrocytes. 67 , 68 There are many members of the FGF family, each with distinct roles in myelination. FGF2 promotes OPC migration and proliferation but prevents them from differentiating at the terminal stage. FGF2 also affects mature, post‐mitotic oligodendrocytes and causes increased process elongation through FGFR2 stimulation and decreased re‐entry into the cell cycle and myelin proteins via FGFR1. 65 , 70 FGF8 and its related subfamily member FGF17 target OPCs and selectively activate FGFR3 to inhibit their differentiation. However, FGF9 stimulates myelination through the specific activation of FGFR2 in differentiated oligodendrocytes. 71 FGF18 exerts corresponding functions by activating FGFR3 and FGFR2 in OPCs and differentiated oligodendrocytes, respectively. 70 Circulating FGF21 has been shown to enhance OPC proliferation in both in vivo and in vitro studies, and this was reliant on β‐Klotho presence. 9 , 62 However, under normal circumstances, FGF21 transport into the CNS is limited, as the amount of FGF21 in healthy individuals' cerebrospinal fluid (CSF) is approximately 60% lower than that in the peripheral circulation. 72 Nonetheless, when the BBB is damaged, FGF21 penetrates the CNS to directly promote myelination. Moreover, FGF21 also regulates the expression of the VEGF2 receptor, which modulates OPC migration and indirectly affects oligodendrocyte development and remyelination. 9
Immunity is continually regulated by FGF signaling, which is, in turn, modified by immune cells during inflammation and tissue healing. FGF signaling cascades are important in CNS inflammatory responses by modulating immune homeostasis and host defense. FGF1 reduces the inflammatory response associated with neuropathic pain by inducing the production of the Th2 cytokine IL‐4, upregulating arginase‐I (Arg‐I), and suppressing the activation of microglia and astrocytes. 73 In a study conducted by Forouzanfar et al., 73 FGF1 treatment reduced the ratios of Bcl2, cleaved caspase 3, MMP‐2, IL‐1β, and Iba1 in model animals of chronic sciatic nerve contractile injury, and modulated apoptosis and neuroinflammation during treatment of neuropathic pain. Both the production of FGF2 protein and the phosphorylation of downstream molecule ERK were restrained in a neuroinflammation‐induced depression model. However, exogenous infusion of FGF2 not only prevented the reduction in ERK1/2 phosphorylation in neuroinflammatory states but also inhibited the expression of proinflammatory cytokines like IL‐1β, IL‐6, and TNFα while increasing the amount of the anti‐inflammatory cytokine IL‐10. These responsive changes reversed depressive‐like behaviors and neuroinflammation‐induced impairment of hippocampal neurogenesis and were blocked by FGFR inhibitors. 74 , 75 Notably, FGF2 signaling has demonstrated immune regulatory actions in the aging brain by re‐establishing the balance of proinflammatory cytokines. 74 Furthermore, FGF2 supplementation attenuated various inflammatory parameters in spontaneous epileptic lesions, with IL‐1β, whose expression was almost entirely blocked, appearing to have the greatest effect. 76 FGF9, another member of the FGF family, recruits macrophages. Studies have elucidated that FGF9 signaling promoted inflammation and neuronal apoptosis by affecting the stimulation of M1‐type macrophages and the ERK signaling pathway. In contrast, knockdown of the FGF9 gene inhibited macrophage recruitment, thereby attenuating nerve damage. 77 Controlling inflammatory responses is thought to be a viable therapeutic approach for stroke. Like other FGFs, after a stroke, recombinant human FGF21 has anti‐inflammatory properties that attenuate inflammatory cell polarization and the infiltration of peripheral immune cells, showing its potential as an anti‐inflammatory agent in stroke. 78 Taken together, FGF/FGFR signaling presumably exerts a pivotal role in neural tissue regeneration, remyelination, and neuroinflammation.
5. EXPLORATION OF FGF/FGFR IN MS
5.1. FGF1 subfamily
In MS, FGF1 is predominantly expressed in remyelinated lesions, with its production being lower in the demyelinated lesion core than in the remyelinated rim. 79 In cerebellar slice cultures, FGF1 promoted remyelination and directly accelerated myelination. Furthermore, by inducing the upregulation of the leukemia inhibitory factor (LIF) and the chemokine CXCL8 in human astrocytes, which are involved in the recruitment of oligodendrocytes, FGF1 indirectly promoted the induction of remyelination. 79 LIF has been demonstrated to support oligodendrocyte development and survival, 80 , 81 , 82 in addition to promoting myelination. 83 In EAE, it has also been proven to prevent oligodendrocyte death 84 and promote remyelination. 85 Astrocytes at the edges of active MS lesions secrete high levels of CXCL8, thereby recruiting OPCs into the lesions and participating in their regeneration. 86 , 87
How FGF2 affects oligodendrocyte responses during demyelination and remyelination in MS is debatable. On one hand, FGF2 is regarded as a neuroprotective agent that promotes remyelination in MS. 17 FGF2 has mainly been detected in microglial and macrophages of active, chronic‐active, and chronic‐inactive lesions in MS. 88 An increase in serum FGF2 levels was also found in gadolinium‐enhanced lesions in RRMS and disability progression of SPMS. 89 However, FGF2 peaked in the initial stage of remyelination. 90 Consistently, ciliary neurotrophic factor (CNTF) expression was induced around remyelination lesions of MHV‐A59‐infected model mice, and FGF2 and its receptor were induced in spinal cord astrocytes after CNTF injection, suggesting that CNTF acts through the FGF2/FGFR pathway. 91 Endogenous FGF2 activity was tested in glial cultures purified from demyelinated lesions of MHV‐A59‐infected mice, and these experiments demonstrated its ability to act as an effective mitogen related to the OPC proliferative response in demyelinating and remyelinating tissues. 92 FGF2 null EAE presented intensive clinical symptoms compared to normal EAE, 17 and FGF2 gene therapy reverted this phenomenon 93 and significantly reduced the infiltration of myelotoxic cells into the CNS. Moreover, mice overexpressing the FGF2 gene showed increased OPCs and myelin‐forming numbers in demyelinating regions. 93 Indeed, FGF2 null mice had noticeably more degenerative nerve fibers and axonal loss. Concurrently, there was a considerable reduction in the quantity of remyelinated axons. 17 On the other hand, it has been suggested that FGF2 is a negative factor in both the myelination processes and remyelination failure. In the cuprizone‐induced mouse model, FGF2 expression levels were increased, 94 and high FGF2 levels induced marked destruction of mature oligodendrocytes and severe myelin loss in the CNS. 95 FGF2 knockout promoted oligodendrocyte regeneration by spontaneously enhancing the proliferation of OPCs in vivo during the lesion recovery phase, thereby promoting remyelination 94 and reducing axonal atrophy in demyelinating lesions. 96 Stimulation of lesion‐derived glial cells in MHV‐A59‐infected mice showed that high concentrations of FGF2 promoted OPC proliferation. In contrast, OPC development into oligodendrocytes was favored in vitro by attenuating endogenous FGF2 via neutralizing antibody. 97 Indeed, these outcomes align with the improved OPC differentiation observed in FGF2−/− animals. The role of FGF2 is variable since chronically high FGF2 levels in the CSF reverse the positive benefits, and the secretion of FGF2 by immune cells increases with disease progression in MS/EAE.
5.2. FGF8 subfamily
FGF8, FGF17, and FGF18 belong to the FGF8 subfamily. They share 60 to 80 percent of amino acid sequence similarity and have comparable receptor binding features. 98 FGF8 is an original factor that, consistent with FGF2 roles, induces OPC activation, migration, and proliferation but does not hamper differentiation. In the FGF8‐treated medium, a remarkable increase in the number of OPCs was detected, and they displayed both immature and mature oligodendrocyte markers. Therefore, it appears that FGF8 stimulates OPC proliferation without inhibiting differentiation, ultimately producing more mature oligodendrocytes. It was observed that FGF8 bound to FGFRs could attract OPCs and induce their migration, which was also recapitulated in animal models for demyelination. 99 When a demyelinating lesion arises, OPCs that are present throughout life in the CNS are prepared to differentiate into mature oligodendrocytes. 100 However, dysfunction of OPC activation in MS affects remyelination processes. 101 A reduced capacity for myelination may be caused in part by the attenuation of OPC migration and differentiation, positioning FGF8 as a potential therapeutic target.
5.3. FGF9 subfamily
The cascade of pathogenic events in MS eventually leads to the loss of neurons and axons, which can be measured by reduced brain volume on volumetric magnetic resonance imaging (MRI) in vivo. Between 0.5% and 1.5% of MS patients develop brain atrophy each year, and during the progressive stages of the disease, the deep gray matter structures display a higher rate of degeneration. 2 It was shown that plasma FGF9 levels are strongly associated with brain volume loss in MS patients, and the annual percentage of brain volume change was inversely related to these levels. 102 FGF9 was high in early active lesions and was upregulated in ongoing lesions of MS patients with longstanding progressive disease. However, FGF9 levels were remarkably lower in healthy white matter and almost nonexistent in chronically demyelinated inactive lesions. 18 Furthermore, GFAP+ astrocytes and OLIG2+ and NOGO‐A+ oligodendrocytes were shown to produce FGF9. This suggests that FGF9 was induced by a localized glial response toward ongoing tissue damage in MS. Although FGF9 was discovered to prevent OPCs from developing into mature oligodendrocytes, the authors did not agree that this direct effect was responsible for the inhibition of remyelination. Rather, FGF9 still serves as an OPC stimulant and contributes to the generation of proteolipid protein+ (PLP+) oligodendrocytes in the complex cellular environment. 18 However, this proliferation is of little importance since FGF9 inhibited the differentiation of precursor oligodendrocytes into mature myelination‐competent oligodendrocytes through an astrocyte‐dependent mechanism. Furthermore, FGF9 also enhanced the expression of chemokines CCL2 and CCL7, 18 which are known to be expressed in MS lesions and recruit macrophages and microglia to initiate the inflammation of MS. The dual pathogenic role of FGF9 in MS, which involves both triggering a proinflammatory response and inhibiting remyelination, has significant implications for the etiology of the disease.
5.4. FGF19 subfamily
The endocrine FGF subclass ligands (FGF19, FGF21, and FGF23) are also known as the FGF19 subfamily. FGF21, mainly released by the skeletal muscle, pancreas, liver, kidney, and adipose tissue, exerts pleiotropic effects in regulating glucose, lipid, and energy homeostasis. 103 Apart from metabolic regulation, it has been recently discovered that FGF21 is secreted by neurons and exhibits neuroprotective functions. 104 FGF21 was dramatically downregulated after cerebral ischemia, and the upregulation could restore brain function by reducing cerebral infarction and ameliorating neuronal cell death. 105 Moreover, FGF21 promoted remyelination after traumatic brain injury. 103 In the lysophosphatidylcholine‐induced demyelination model, treatment with a neutralizing antibody against FGF21 or gene knockout abolished these positive effects on OPCs. 9 The effect of FGF21 on OPCs seems to be limited to promoting proliferation without affecting their differentiation or apoptosis. Furthermore, FGF21 did not modulate the cell fate of astrocytes, and Kuroda et al. detected no significant proliferation of OPCs cultured in astrocyte supernatant with FGF21 pre‐treatment, indicating that FGF21 acts directly as an OPC mitogen. Interestingly, FGF21‐mediated proliferation of human OPCs in autopsy samples from MS patients has been observed. 9 Therefore, we can infer that FGF21‐mediated OPC proliferation and consequent remyelination are conserved in CNS demyelinating models.
FGF23 was first identified in the ventrolateral thalamic nucleus of the mouse brain, and its physiological role has recently attracted significant attention. 106 The FGF23 protein is present in three distinct forms in the bloodstream: a full‐length mature form and two inactive (C‐ and N‐terminal) fragments. 107 It is generally accepted that intact FGF23 is a bioactive factor that controls the metabolism of phosphate and vitamin D. In contrast, high levels of the inactive FGF23 forms have been demonstrated to inhibit these effects. 108 Considered a bone‐derived hormone, FGF23 is primarily released by osteocytes and osteoblasts in the skeleton. It is a component of the novel hormonal bone–parathyroid–kidney axis, which interacts to form hormonal homeostasis. 109 , 110 Several studies have shown that serum FGF23 concentrations were elevated in RRMS patients. However, calcitriol levels were reduced, indicating that elevated FGF23 levels in MS may disrupt the FGF23‐PTH‐vitamin D axis, resulting in pathological effects. 19 , 111 However, in a study that measured plasma levels of FGF23 in 91 MS patients and 92 healthy controls, no difference was observed. 112 Similar results were obtained in a study by Alagha et al. 113 In addition, they also found that the secretion of FGF23 in the CSF of MS patients was comparable to that of the healthy population. There are several reasons for these diverging results: (1) The study of elevated FGF23 was conducted in RRMS, and the latter two studies were the result of a mixture of all clinical subtypes of MS. Furthermore, the pathogenesis of MS subtypes differs, which can lead to different outcomes; (2) As mentioned earlier, FGF23 exists in two forms, an intact active form, and an incomplete form. Of the studies that concluded that FGF23 was elevated, intact FGF23 was measure, while the other studies measured all forms of FGF23, which may have biased the results; (3) The experimental samples and methods used in these studies differed slightly, which may also cause inconsistent results. The elevated FGF23 in MS patients inhibited 1‐α‐hydroxylase and upregulated 24‐α‐hydroxylase to eliminate 1,25‐(OH)2D3 levels. 111 Previous studies have demonstrated that vitamin D has an immunomodulatory effect and promotes the multiplication of NSCs and their differentiation into mature neurons and oligodendrocytes. 114 FGF23 might be involved in immune responses via suppression of vitamin D production and direct interaction with immune cells such as macrophages and dendritic cells (DCs). Under the stimulation of LPS/IFN‐γ, activated DCs and macrophages contribute to the increased serum FGF23 levels, mediated by the nuclear factor‐κB and the JAK/STAT1 signaling pathways. 115 Compelling studies suggest that FGF23 targets macrophages, which express FGFR1 along with DCs, and exhibit increased α‐Klotho expression upon inflammatory stimulation. 116 The interaction of FGF23 with the FGFR1/α‐Klotho complex affects the polarization of macrophages to the M1 phenotype, blocks the transition to the M2 phenotype and induces M1‐type macrophages to secrete TNFα and suppress Arg‐I expression in M2 macrophages, resulting in sustained inflammation. 115 , 116 However, 1,25‐(OH)2D3 has been reported to have the opposite effect on cytokine secretion of macrophages. 116 FGF23 has a proinflammatory function and counters the regulatory action of 1,25‐(OH)2D3 on immune responses. Accordingly, it is conceivable that elevated FGF23 levels contribute to MS by promoting inflammation.
5.5. Fibroblast growth factor receptors
To characterize the role of FGFRs in oligodendrocytes in vitro, researchers used the multi‐kinase inhibitor and FGFR1‐3 inhibitor to block FGFR signaling. The application of inhibitors increased the expression of the myelin‐associated proteins PLP and 2′,3′‐cyclic‐nucleotide 3′‐phosphodiesterase. Furthermore, upregulation of neurotrophic factor BDNF/TrkB signaling and decreased expression of the remyelination inhibitor semaphorin 3A were observed. These effects may be related to the reduction in downstream molecules of the FGF/FGFR signaling pathway, such as ERK and AKT phosphorylation, in oligodendrocytes. 117 These findings have been confirmed in vivo. Conditional ablation of FGFR1 or FGFR2 in oligodendrocytes alleviated the symptoms of motor deficits in MOG35‐55‐induced EAE. 14 , 16 , 118 In the spinal cord of FGFR1ind−/− and FGFR2ind−/− animals, myelin, axonal loss, and the infiltration of inflammatory cells were decreased in the chronic phase of EAE without causing any alterations in the acute phase. The protective effects of the oligodendrocyte‐specific deletion of FGFR1 and FGFR2 on EAE are mainly manifested in two aspects. First, the knockdown of FGFRs can regulate the inflammatory environment in the CNS. T cells and B cells, as well as macrophages and activated microglia, were significantly reduced in the spinal cord and cerebellum of FGFR1ind−/− mice, along with the decreased expression of the proinflammatory cytokines TNFα, IL‐1β, and IL‐6, and the chemokine CX3Cl1 and its receptor CXC3R1. 14 , 118 Furthermore, FGFR2ind−/− mice showed a similar phenotype, 16 and this low‐inflammatory environment facilitates remyelination and tissue repair of EAE lesions. Second, the deletion of FGFR1 and FGFR2 exhibited robust pro‐remyelination activity. Although the depletion of FGFR1 and FGFR2 did not affect the numbers of cells from the oligodendrocyte lineage, the high expression of pERK, pAKT, BDNF, and TrkB, and the low level of Lingo‐1 in the spinal cord was caused by specific knockdown of FGFR1. 14 , 118 The phenotype of FGFR2ind−/− mice varied slightly, with an increase in PLP‐positive cells in the spinal cord and a decrease in the expression of SEMA3A. At the same time, there were no significant increases in pERK or TrkB, 16 indicating that the two FGFR receptors in the EAE model mediate different effects (Figure 2). In the cuprizone demyelination model, acute‐phase demyelination and myelin recovery were not affected by oligodendrocyte‐specific deletions of FGFR1 and FGFR2. However, in the chronic period, double knockout of FGFR1 and FGFR2 caused remyelination failure. 119 Additionally, single deletion of FGFR1 promoted remyelination and functional recovery in the chronic phase. 15 Therefore, these data suggest that FGFR signaling inhibits regenerative processes, particularly chronic demyelination.
FIGURE 2.
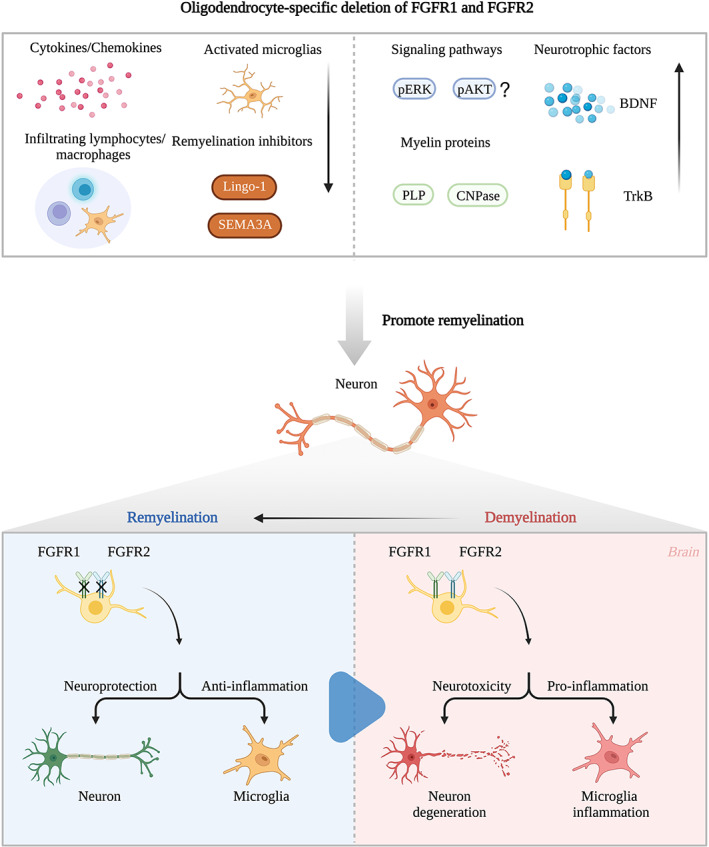
Role of oligodendrocyte‐specific deletion of FGFR1 and FGFR2. Pro‐remyelination and anti‐inflammatory mechanisms of oligodendrocyte‐specific deletion of FGFR1 and FGFR2 in EAE. BDNF, brain derived neurotrophic factor; CNPase, 2′,3′‐cyclic‐nucleotide 3′‐phosphodiesterase; EAE, experimental autoimmune encephalomyelitis; PLP, Proteolipid protein; SEMA3A, Semaphorin 3A.
6. SUMMARY
Current MS treatments have achieved near complete control over RRMS and focal neuroinflammation. However, no effective treatment strategies are available concerning neurodegeneration and disability. FGF signal transduction is involved in the development of the CNS, enhances the proliferation and axonal growth of NSCs in adults, and promotes their differentiation into various CNS cells, such as neurons and oligodendrocytes. Furthermore, the development of oligodendrocytes is inseparably linked to FGFs, and distinct FGFRs are expressed at different stages of their development; therefore, FGFs play pivotal roles at each stage. Surprisingly, several studies have shown that neuroinflammation is regulated partly by FGF signaling, as FGF1, FGF2, and FGF21 have anti‐inflammatory functions, while FGF9 induces neuroinflammation. These results all emphasize the importance of FGF/FGFR signaling in the pathogenesis of MS. Some FGFs are mainly expressed in acute demyelinating lesions and mediate disease progression. Others display high concentrations in remyelinating lesions in the chronic phase of MS and promote the life cycle of OPCs and the maturation of oligodendrocytes, which helps to enhance tissue repair and restore motor function (Table 2). However, due to the large size of the FGF family and their far‐spanning actions, coupled with their ambiguous roles in MS and their evolution through disease progression, it is difficult to explore their current therapeutic utility for MS. Furthermore, before FGFs become relevant biomarkers for monitoring MS disease course and disease identification, various conditions need to be met: the role of other members of the FGF family in MS and the correlation between FGFs and clinical data, including MS subtypes, clinical features, and disease stages, require further investigation. 120 In addition, more research is needed to detect the sensitivity, specificity, and stability of FGFs as biomarkers and they must be compared with existing oligoclonal bands and IgG to clarify their characteristics. 121
TABLE 2.
Variety actions of FGFs in MS.
| FGFs | Concentration in MS | Roles in remyelination | Roles in neuroinflammation |
|---|---|---|---|
| FGF1 | Predominantly expressed in remyelinated lesions; lower in the demyelinated lesion core compared to the remyelinated rim | Upregulates expression of LIF and CXCL8 in astrocytes to support oligodendrocyte maturation, differentiation, and survival | Reduces the inflammatory response via inducing the expression of IL‐4, Arg‐I and inhibiting the activation of microglia and astrocytes |
| FGF2 | Mainly detected in active lesions and ambitus of chronic‐active as well as chronic‐inactive lesions | Enhances the proliferation and survival of OPCs in the early stage, and inhibits their differentiation into oligodendrocytes after prolonged stimulation | Prevents the expression of proinflammatory cytokines such as IL‐1β, IL‐6, and TNFα and increases the anti‐inflammatory cytokine IL‐10 levels |
| FGF8 | Not detected | Induces OPCs' activation and migration | Not yet identified |
| FGF9 | Highly expressed in early active lesions, and upregulated in ongoing lesions; strongly associated to loss of brain volume in MS patients | Inhibits the differentiation of isolated OPC into mature oligodendrocytes | Enhances the expression of proinflammatory genes, such as chemokines CCL2 and CCL7 |
| FGF21 | Not detected | Mediates OPCs proliferation and leads remyelination | Attenuates inflammatory cell polarization and infiltration of peripheral immune cells into CNS |
| FGF23 | Serum concentration elevates; no significant increasement in CSF | Not yet identified | Counters the regulatory action of 1,25‐(OH)2D3 on immune responses; affects the polarization of macrophages to the M1 phenotype and induces the secretion of TNFα, blocks the transition to M2 macrophages and the expression of Arg‐I |
Abbreviations: CSF, cerebrospinal fluid; FGF, fibroblast growth factor; LIF, leukemia inhibitory factor; MS, Multiple sclerosis; OPC, oligodendrocyte precursor cell.
In contrast, FGFRs have shown promising therapeutic potential. FGFRs are widely expressed in CNS and immune cells (Table 3) and may be involved in many aspects of MS pathogenesis; therefore, therapies targeting FGFRs can exert multiple effects. Furthermore, conditional deletion of FGFR1 and FGFR2 has improved EAE symptoms, especially in the chronic phase, leading to amelioration of the inflammatory microenvironment, less demyelination, and greater axonal density. The underlying mechanisms may be changes in the levels of ERK and AKT phosphorylation, and the expression of BDNF and several remyelination inhibitors. The increase in BDNF expression is particularly interesting since glatiramer acetate and fingolimod, both currently available MS treatments, have been reported to upregulate BDNF expression; this upregulation may be associated with their therapeutic efficacy. 122 Based on the findings described here, targeting FGFRs is a promising strategy for treating MS patients.
TABLE 3.
Cell expression of FGFRs.
| FGFRs | Cell type | References | |
|---|---|---|---|
| FGFR1 |
Macrophage T lymphocyte B lymphocyte NK cell |
Neuron Microglia Astrocyte OPC Oligodendrocyte |
118, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132 |
| FGFR2 |
Macrophage NK cell |
Neuron Microglia Astrocyte OPC Oligodendrocyte |
123, 129, 132, 133, 134 |
| FGFR3 |
Macrophage B lymphocyte |
Neuron Microglia Astrocyte OPC Oligodendrocyte |
90, 123, 132, 135, 136, 137 |
| FGFR4 | Macrophage |
Neuron Microglia Astrocyte OPC |
123, 132, 138 |
Abbreviations: FGFR, fibroblast growth factor receptor; OPC, oligodendrocyte precursor cell.
AUTHOR CONTRIBUTIONS
Zhiguo Chen and Tao Jin designed the study. Qingxiang Zhang wrote the main text, produced the tables and figures. Kaili Zhang was responsible for literature review and data collection. Jie Zhu and Tao Jin critically revised the whole manuscript. All authors read and approved the final manuscript.
FUNDING INFORMATION
This work was supported by grants from the General Program of the National Natural Science Foundation of China (No. 82171337).
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
ACKNOWLEDGMENTS
We would like to thank Editage (www.editage.cn) for English language editing.
Zhang Q, Chen Z, Zhang K, Zhu J, Jin T. FGF/FGFR system in the central nervous system demyelinating disease: Recent progress and implications for multiple sclerosis. CNS Neurosci Ther. 2023;29:1497‐1511. doi: 10.1111/cns.14176
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378(2):169‐180. doi: 10.1056/NEJMra1401483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622‐1636. doi: 10.1016/S0140-6736(18)30481-1 [DOI] [PubMed] [Google Scholar]
- 3. Sintzel MB, Rametta M, Reder AT. Vitamin D and multiple sclerosis: a comprehensive review. Neurol Ther. 2018;7(1):59‐85. doi: 10.1007/s40120-017-0086-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D'Amico E, Patti F, Zanghi A, Chisari CG, Lo Fermo S, Zappia M. Late‐onset and young‐onset relapsing‐remitting multiple sclerosis: evidence from a retrospective long‐term follow‐up study. Eur J Neurol. 2018;25(12):1425‐1431. doi: 10.1111/ene.13745 [DOI] [PubMed] [Google Scholar]
- 5. Cunniffe N, Coles A. Promoting remyelination in multiple sclerosis. J Neurol. 2021;268(1):30‐44. doi: 10.1007/s00415-019-09421-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olek MJ. Multiple Sclerosis. Ann Intern Med. 2021;174(6):ITC81‐ITC96. doi: 10.7326/AITC202106150 [DOI] [PubMed] [Google Scholar]
- 7. Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079‐1106. doi: 10.1111/j.1476-5381.2011.01302.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kishore A, Kanaujia A, Nag S, et al. Different mechanisms of inflammation induced in virus and autoimmune‐mediated models of multiple sclerosis in C57BL6 mice. Biomed Res Int. 2013;2013:589048. doi: 10.1155/2013/589048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuroda M, Muramatsu R, Maedera N, et al. Peripherally derived FGF21 promotes remyelination in the central nervous system. J Clin Invest. 2017;127(9):3496‐3509. doi: 10.1172/JCI94337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Torkildsen O, Brunborg LA, Myhr KM, Bo L. The cuprizone model for demyelination. Acta Neurol Scand Suppl. 2008;188:72‐76. doi: 10.1111/j.1600-0404.2008.01036.x [DOI] [PubMed] [Google Scholar]
- 11. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235‐253. doi: 10.1038/nrd2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang L, Zhou F, Zheng D, et al. FGF/FGFR signaling: from lung development to respiratory diseases. Cytokine Growth Factor Rev. 2021;62:94‐104. doi: 10.1016/j.cytogfr.2021.09.002 [DOI] [PubMed] [Google Scholar]
- 13. Rajendran R, Bottiger G, Stadelmann C, Karnati S, Berghoff M. FGF/FGFR pathways in multiple sclerosis and in its disease models. Cells. 2021;10:884. doi: 10.3390/cells10040884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rajendran R, Rajendran V, Giraldo‐Velasquez M, et al. Oligodendrocyte‐specific deletion of FGFR1 reduces cerebellar inflammation and neurodegeneration in MOG35‐55‐induced EAE. Int J Mol Sci. 2021;22(17):9495. doi: 10.3390/ijms22179495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mierzwa AJ, Zhou YX, Hibbits N, Vana AC, Armstrong RC. FGF2 and FGFR1 signaling regulate functional recovery following cuprizone demyelination. Neurosci Lett. 2013;548:280‐285. doi: 10.1016/j.neulet.2013.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamali S, Rajendran R, Stadelmann C, et al. Oligodendrocyte‐specific deletion of FGFR2 ameliorates MOG35‐55‐induced EAE through ERK and Akt signalling. Brain Pathol. 2021;31(2):297‐311. doi: 10.1111/bpa.12916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rottlaender A, Villwock H, Addicks K, Kuerten S. Neuroprotective role of fibroblast growth factor‐2 in experimental autoimmune encephalomyelitis. Immunology. 2011;133(3):370‐378. doi: 10.1111/j.1365-2567.2011.03450.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lindner M, Thummler K, Arthur A, et al. Fibroblast growth factor signalling in multiple sclerosis: inhibition of myelination and induction of pro‐inflammatory environment by FGF9. Brain. 2015;138(Pt 7):1875‐1893. doi: 10.1093/brain/awv102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ellidag HY, Yilmaz N, Kurtulus F, et al. The three sisters of fate in multiple sclerosis: Klotho (Clotho), fibroblast growth Factor‐23 (Lachesis), and vitamin D (Atropos). Ann Neurosci. 2016;23(3):155‐161. doi: 10.1159/000449181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183‐193. doi: 10.1016/S1474-4422(14)70256-X [DOI] [PubMed] [Google Scholar]
- 21. Thompson AJ, Kermode AG, MacManus DG, et al. Pathogenesis of progressive multiple sclerosis. Lancet. 1989;1(8650):1322‐1323. doi: 10.1016/s0140-6736(89)92710-4 [DOI] [PubMed] [Google Scholar]
- 22. Dutta R, Trapp BD. Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol. 2014;27(3):271‐278. doi: 10.1097/WCO.0000000000000094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith JA, Nicaise AM, Ionescu RB, Hamel R, Peruzzotti‐Jametti L, Pluchino S. Stem cell therapies for progressive multiple sclerosis. Front Cell Dev Biol. 2021;9:696434. doi: 10.3389/fcell.2021.696434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, Nataf S. Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol. 2010;68(4):465‐476. doi: 10.1002/ana.22054 [DOI] [PubMed] [Google Scholar]
- 25. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol. 2018;9:3116. doi: 10.3389/fimmu.2018.03116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haider L, Zrzavy T, Hametner S, et al. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain. 2016;139(Pt 3):807‐815. doi: 10.1093/brain/awv398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Absinta M, Sati P, Masuzzo F, et al. Association of chronic active multiple sclerosis lesions with disability in vivo. JAMA Neurol. 2019;76(12):1474‐1483. doi: 10.1001/jamaneurol.2019.2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8(11):647‐656. doi: 10.1038/nrneurol.2012.168 [DOI] [PubMed] [Google Scholar]
- 29. McGinley MP, Goldschmidt CH, Rae‐Grant AD. Diagnosis and treatment of multiple sclerosis: a review. JAMA. 2021;325(8):765‐779. doi: 10.1001/jama.2020.26858 [DOI] [PubMed] [Google Scholar]
- 30. Absinta M, Lassmann H, Trapp BD. Mechanisms underlying progression in multiple sclerosis. Curr Opin Neurol. 2020;33(3):277‐285. doi: 10.1097/WCO.0000000000000818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yeung MSY, Djelloul M, Steiner E, et al. Dynamics of oligodendrocyte generation in multiple sclerosis. Nature. 2019;566(7745):538‐542. doi: 10.1038/s41586-018-0842-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lubetzki C, Zalc B, Williams A, Stadelmann C, Stankoff B. Remyelination in multiple sclerosis: from basic science to clinical translation. Lancet Neurol. 2020;19(8):678‐688. doi: 10.1016/S1474-4422(20)30140-X [DOI] [PubMed] [Google Scholar]
- 33. Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. 2006;26(1):328‐332. doi: 10.1523/JNEUROSCI.2615-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gruchot J, Weyers V, Gottle P, et al. The molecular basis for remyelination failure in multiple sclerosis. Cells. 2019;8(8):825. doi: 10.3390/cells8080825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lampron A, Larochelle A, Laflamme N, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. 2015;212(4):481‐495. doi: 10.1084/jem.20141656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ornitz DM, Itoh N. New developments in the biology of fibroblast growth factors. WIREs Mech Dis. 2022;14:e1549. doi: 10.1002/wsbm.1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y, Liu D, Zhang T, Xia L. FGF/FGFR signaling in hepatocellular carcinoma: from carcinogenesis to recent therapeutic intervention. Cancers. 2021;13(6):1360. doi: 10.3390/cancers13061360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Belov AA, Mohammadi M. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol. 2013;5(6):a015958. doi: 10.1101/cshperspect.a015958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prudovsky I. Cellular mechanisms of FGF‐stimulated tissue repair. Cells. 2021;10(7):1830. doi: 10.3390/cells10071830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Francavilla C, O'Brien CS. Fibroblast growth factor receptor signalling dysregulation and targeting in breast cancer. Open Biol. 2022;12(2):210373. doi: 10.1098/rsob.210373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dolegowska K, Marchelek‐Mysliwiec M, Nowosiad‐Magda M, Slawinski M, Dolegowska B. FGF19 subfamily members: FGF19 and FGF21. J Physiol Biochem. 2019;75(2):229‐240. doi: 10.1007/s13105-019-00675-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16(2):233‐247. doi: 10.1016/j.cytogfr.2005.01.007 [DOI] [PubMed] [Google Scholar]
- 43. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116‐129. doi: 10.1038/nrc2780 [DOI] [PubMed] [Google Scholar]
- 44. Brewer JR, Mazot P, Soriano P. Genetic insights into the mechanisms of Fgf signaling. Genes Dev. 2016;30(7):751‐771. doi: 10.1101/gad.277137.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perez Pinero C, Giulianelli S, Lamb CA, Lanari C. New insights in the interaction of FGF/FGFR and steroid receptor signaling in breast cancer. Endocrinology. 2022;163(2):bqab265. doi: 10.1210/endocr/bqab265 [DOI] [PubMed] [Google Scholar]
- 46. Wiedemann M, Trueb B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics. 2000;69(2):275‐279. doi: 10.1006/geno.2000.6332 [DOI] [PubMed] [Google Scholar]
- 47. Chiu IM, Touhalisky K, Baran C. Multiple controlling mechanisms of FGF1 gene expression through multiple tissue‐specific promoters. Prog Nucleic Acid Res Mol Biol. 2001;70:155‐174. doi: 10.1016/s0079-6603(01)70016-5 [DOI] [PubMed] [Google Scholar]
- 48. Mason I. Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci. 2007;8(8):583‐596. doi: 10.1038/nrn2189 [DOI] [PubMed] [Google Scholar]
- 49. Mocchetti I, Wrathall JR. Neurotrophic factors in central nervous system trauma. J Neurotrauma. 1995;12(5):853‐870. doi: 10.1089/neu.1995.12.853 [DOI] [PubMed] [Google Scholar]
- 50. Lee DC, Hsu YC, Chung YF, et al. Isolation of neural stem/progenitor cells by using EGF/FGF1 and FGF1B promoter‐driven green fluorescence from embryonic and adult mouse brains. Mol Cell Neurosci. 2009;41(3):348‐363. doi: 10.1016/j.mcn.2009.04.010 [DOI] [PubMed] [Google Scholar]
- 51. Hsu YC, Liao WC, Kao CY, Chiu IM. Regulation of FGF1 gene promoter through transcription factor RFX1. J Biol Chem. 2010;285(18):13885‐13895. doi: 10.1074/jbc.M109.081463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gritti A, Parati EA, Cova L, et al. Multipotential stem cells from the adult mouse brain proliferate and self‐renew in response to basic fibroblast growth factor. J Neurosci. 1996;16(3):1091‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tao Y, Black IB, DiCicco‐Bloom E. Neurogenesis in neonatal rat brain is regulated by peripheral injection of basic fibroblast growth factor (bFGF). J Comp Neurol. 1996;376(4):653‐663. doi: [DOI] [PubMed] [Google Scholar]
- 54. Rai KS, Hattiangady B, Shetty AK. Enhanced production and dendritic growth of new dentate granule cells in the middle‐aged hippocampus following intracerebroventricular FGF‐2 infusions. Eur J Neurosci. 2007;26(7):1765‐1779. doi: 10.1111/j.1460-9568.2007.05820.x [DOI] [PubMed] [Google Scholar]
- 55. Tao Y, Black IB, DiCicco‐Bloom E. In vivo neurogenesis is inhibited by neutralizing antibodies to basic fibroblast growth factor. J Neurobiol. 1997;33(3):289‐296. [PubMed] [Google Scholar]
- 56. Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF‐2‐deficient mice. EMBO J. 1998;17(15):4213‐4225. doi: 10.1093/emboj/17.15.4213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Werner S, Unsicker K, von Bohlen und Halbach O. Fibroblast growth factor‐2 deficiency causes defects in adult hippocampal neurogenesis, which are not rescued by exogenous fibroblast growth factor‐2. J Neurosci Res. 2011;89(10):1605‐1617. doi: 10.1002/jnr.22680 [DOI] [PubMed] [Google Scholar]
- 58. Song Y, Lee S, Jho EH. Enhancement of neuronal differentiation by using small molecules modulating nodal/Smad, Wnt/beta‐catenin, and FGF signaling. Biochem Biophys Res Commun. 2018;503(1):352‐358. doi: 10.1016/j.bbrc.2018.06.033 [DOI] [PubMed] [Google Scholar]
- 59. Yellapragada V, Eskici N, Wang Y, et al. FGF8‐FGFR1 signaling regulates human GnRH neuron differentiation in a time‐ and dose‐dependent manner. Dis Model Mech. 2022;15(8):dmm049436. doi: 10.1242/dmm.049436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gao XZ, Ma RH, Zhang ZX. miR‐339 promotes hypoxia‐induced neuronal apoptosis and impairs cell viability by targeting FGF9/CACNG2 and mediating MAPK pathway in ischemic stroke. Front Neurol. 2020;11:436. doi: 10.3389/fneur.2020.00436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen J, Wang Z, Zheng Z, et al. Neuron and microglia/macrophage‐derived FGF10 activate neuronal FGFR2/PI3K/Akt signaling and inhibit microglia/macrophages TLR4/NF‐kappaB‐dependent neuroinflammation to improve functional recovery after spinal cord injury. Cell Death Dis. 2017;8(10):e3090. doi: 10.1038/cddis.2017.490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kang K, Xu P, Wang M, et al. FGF21 attenuates neurodegeneration through modulating neuroinflammation and oxidant‐stress. Biomed Pharmacother. 2020;129:110439. doi: 10.1016/j.biopha.2020.110439 [DOI] [PubMed] [Google Scholar]
- 63. Warrington AE, Barbarese E, Pfeiffer SE. Differential myelinogenic capacity of specific developmental stages of the oligodendrocyte lineage upon transplantation into hypomyelinating hosts. J Neurosci Res. 1993;34(1):1‐13. doi: 10.1002/jnr.490340102 [DOI] [PubMed] [Google Scholar]
- 64. Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Prog Neurobiol. 2002;67(6):451‐467. doi: 10.1016/s0301-0082(02)00058-8 [DOI] [PubMed] [Google Scholar]
- 65. Bansal R. Fibroblast growth factors and their receptors in oligodendrocyte development: implications for demyelination and remyelination. Dev Neurosci. 2002;24(1):35‐46. doi: 10.1159/000064944 [DOI] [PubMed] [Google Scholar]
- 66. Klimaschewski L, Claus P. Fibroblast growth factor signalling in the diseased nervous system. Mol Neurobiol. 2021;58(8):3884‐3902. doi: 10.1007/s12035-021-02367-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li JS, Yao ZX. Modulation of FGF receptor signaling as an intervention and potential therapy for myelin breakdown in Alzheimer's disease. Med Hypotheses. 2013;80(4):341‐344. doi: 10.1016/j.mehy.2012.12.008 [DOI] [PubMed] [Google Scholar]
- 68. Furusho M, Ishii A, Bansal R. Signaling by FGF receptor 2, Not FGF receptor 1, regulates myelin thickness through activation of ERK1/2‐MAPK, which promotes mTORC1 activity in an Akt‐independent manner. J Neurosci. 2017;37(11):2931‐2946. doi: 10.1523/JNEUROSCI.3316-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Furusho M, Dupree JL, Nave KA, Bansal R. Fibroblast growth factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. J Neurosci. 2012;32(19):6631‐6641. doi: 10.1523/JNEUROSCI.6005-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25(32):7470‐7479. doi: 10.1523/JNEUROSCI.2120-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cohen RI, Chandross KJ. Fibroblast growth factor‐9 modulates the expression of myelin related proteins and multiple fibroblast growth factor receptors in developing oligodendrocytes. J Neurosci Res. 2000;61(3):273‐287. doi: [DOI] [PubMed] [Google Scholar]
- 72. Tan BK, Hallschmid M, Adya R, Kern W, Lehnert H, Randeva HS. Fibroblast growth factor 21 (FGF21) in human cerebrospinal fluid: relationship with plasma FGF21 and body adiposity. Diabetes. 2011;60(11):2758‐2762. doi: 10.2337/db11-0672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Forouzanfar F, Sadeghnia HR, Hoseini SJ, et al. Fibroblast growth factor 1 gene‐transfected adipose‐derived mesenchymal stem cells modulate apoptosis and inflammation In the chronic constriction injury model of neuropathic pain. Iran J Pharm Res. 2020;19(4):151‐159. doi: 10.22037/ijpr.2020.113223.14176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tang MM, Lin WJ, Zhang JT, Zhao YW, Li YC. Exogenous FGF2 reverses depressive‐like behaviors and restores the suppressed FGF2‐ERK1/2 signaling and the impaired hippocampal neurogenesis induced by neuroinflammation. Brain Behav Immun. 2017;66:322‐331. doi: 10.1016/j.bbi.2017.05.013 [DOI] [PubMed] [Google Scholar]
- 75. Tang MM, Lin WJ, Pan YQ, Li YC. Fibroblast growth factor 2 modulates hippocampal microglia activation in a neuroinflammation induced model of depression. Front Cell Neurosci. 2018;12:255. doi: 10.3389/fncel.2018.00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bovolenta R, Zucchini S, Paradiso B, et al. Hippocampal FGF‐2 and BDNF overexpression attenuates epileptogenesis‐associated neuroinflammation and reduces spontaneous recurrent seizures. J Neuroinflammation. 2010;7:81. doi: 10.1186/1742-2094-7-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Deng B, Lv W, Duan W, et al. FGF9 modulates Schwann cell myelination in developing nerves and induces a pro‐inflammatory environment during injury. J Cell Biochem. 2018;119(10):8643‐8658. doi: 10.1002/jcb.27105 [DOI] [PubMed] [Google Scholar]
- 78. Wang D, Liu F, Zhu L, et al. FGF21 alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J Neuroinflammation. 2020;17(1):257. doi: 10.1186/s12974-020-01921-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mohan H, Friese A, Albrecht S, et al. Transcript profiling of different types of multiple sclerosis lesions yields FGF1 as a promoter of remyelination. Acta Neuropathol Commun. 2014;2:168. doi: 10.1186/s40478-014-0168-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mayer M, Bhakoo K, Noble M. Ciliary neurotrophic factor and leukemia inhibitory factor promote the generation, maturation and survival of oligodendrocytes in vitro. Development. 1994;120(1):143‐153. doi: 10.1242/dev.120.1.143 [DOI] [PubMed] [Google Scholar]
- 81. Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long‐term oligodendrocyte survival. Development. 1993;118(1):283‐295. doi: 10.1242/dev.118.1.283 [DOI] [PubMed] [Google Scholar]
- 82. Butzkueven H, Zhang JG, Soilu‐Hanninen M, et al. LIF receptor signaling limits immune‐mediated demyelination by enhancing oligodendrocyte survival. Nat Med. 2002;8(6):613‐619. doi: 10.1038/nm0602-613 [DOI] [PubMed] [Google Scholar]
- 83. Ishibashi T, Dakin KA, Stevens B, et al. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49(6):823‐832. doi: 10.1016/j.neuron.2006.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kerr BJ, Patterson PH. Leukemia inhibitory factor promotes oligodendrocyte survival after spinal cord injury. Glia. 2005;51(1):73‐79. doi: 10.1002/glia.20177 [DOI] [PubMed] [Google Scholar]
- 85. Laterza C, Merlini A, De Feo D, et al. iPSC‐derived neural precursors exert a neuroprotective role in immune‐mediated demyelination via the secretion of LIF. Nat Commun. 2013;4:2597. doi: 10.1038/ncomms3597 [DOI] [PubMed] [Google Scholar]
- 86. Kelland EE, Gilmore W, Weiner LP, Lund BT. The dual role of CXCL8 in human CNS stem cell function: multipotent neural stem cell death and oligodendrocyte progenitor cell chemotaxis. Glia. 2011;59(12):1864‐1878. doi: 10.1002/glia.21230 [DOI] [PubMed] [Google Scholar]
- 87. Omari KM, John GR, Sealfon SC, Raine CS. CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain. 2005;128(Pt 5):1003‐1015. doi: 10.1093/brain/awh479 [DOI] [PubMed] [Google Scholar]
- 88. Clemente D, Ortega MC, Arenzana FJ, de Castro F. FGF‐2 and Anosmin‐1 are selectively expressed in different types of multiple sclerosis lesions. J Neurosci. 2011;31(42):14899‐14909. doi: 10.1523/JNEUROSCI.1158-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sarchielli P, Di Filippo M, Ercolani MV, et al. Fibroblast growth factor‐2 levels are elevated in the cerebrospinal fluid of multiple sclerosis patients. Neurosci Lett. 2008;435(3):223‐228. doi: 10.1016/j.neulet.2008.02.040 [DOI] [PubMed] [Google Scholar]
- 90. Messersmith DJ, Murtie JC, Le TQ, Frost EE, Armstrong RC. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J Neurosci Res. 2000;62(2):241‐256. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Albrecht PJ, Murtie JC, Ness JK, et al. Astrocytes produce CNTF during the remyelination phase of viral‐induced spinal cord demyelination to stimulate FGF‐2 production. Neurobiol Dis. 2003;13(2):89‐101. doi: 10.1016/s0969-9961(03)00019-6 [DOI] [PubMed] [Google Scholar]
- 92. Frost EE, Nielsen JA, Le TQ, Armstrong RC. PDGF and FGF2 regulate oligodendrocyte progenitor responses to demyelination. J Neurobiol. 2003;54(3):457‐472. doi: 10.1002/neu.10158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ruffini F, Furlan R, Poliani PL, et al. Fibroblast growth factor‐II gene therapy reverts the clinical course and the pathological signs of chronic experimental autoimmune encephalomyelitis in C57BL/6 mice. Gene Ther. 2001;8(16):1207‐1213. doi: 10.1038/sj.gt.3301523 [DOI] [PubMed] [Google Scholar]
- 94. Armstrong RC, Le TQ, Flint NC, Vana AC, Zhou YX. Endogenous cell repair of chronic demyelination. J Neuropathol Exp Neurol. 2006;65(3):245‐256. doi: 10.1097/01.jnen.0000205142.08716.7e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Butt AM, Dinsdale J. Fibroblast growth factor 2 induces loss of adult oligodendrocytes and myelin in vivo. Exp Neurol. 2005;192(1):125‐133. doi: 10.1016/j.expneurol.2004.11.007 [DOI] [PubMed] [Google Scholar]
- 96. Tobin JE, Xie M, Le TQ, Song SK, Armstrong RC. Reduced axonopathy and enhanced remyelination after chronic demyelination in fibroblast growth factor 2 (Fgf2)‐null mice: differential detection with diffusion tensor imaging. J Neuropathol Exp Neurol. 2011;70(2):157‐165. doi: 10.1097/NEN.0b013e31820937e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci. 2002;22(19):8574‐8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hao Y, Tang S, Yuan Y, Liu R, Chen Q. Roles of FGF8 subfamily in embryogenesis and oralmaxillofacial diseases (review). Int J Oncol. 2019;54(3):797‐806. doi: 10.3892/ijo.2019.4677 [DOI] [PubMed] [Google Scholar]
- 99. Cruz‐Martinez P, Martinez‐Ferre A, Jaramillo‐Merchan J, Estirado A, Martinez S, Jones J. FGF8 activates proliferation and migration in mouse post‐natal oligodendrocyte progenitor cells. PLoS One. 2014;9(9):e108241. doi: 10.1371/journal.pone.0108241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Menn B, Garcia‐Verdugo JM, Yaschine C, Gonzalez‐Perez O, Rowitch D, Alvarez‐Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26(30):7907‐7918. doi: 10.1523/JNEUROSCI.1299-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sim FJ, Zhao C, Penderis J, Franklin RJ. The age‐related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 2002;22(7):2451‐2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Malekzadeh A, Leurs C, van Wieringen W, et al. Plasma proteome in multiple sclerosis disease progression. Ann Clin Transl Neurol. 2019;6(9):1582‐1594. doi: 10.1002/acn3.771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lu Y, Li R, Zhu J, et al. Fibroblast growth factor 21 facilitates peripheral nerve regeneration through suppressing oxidative damage and autophagic cell death. J Cell Mol Med. 2019;23(1):497‐511. doi: 10.1111/jcmm.13952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shahror RA, Linares GR, Wang Y, et al. Transplantation of mesenchymal stem cells overexpressing fibroblast growth factor 21 facilitates cognitive recovery and enhances neurogenesis in a mouse model of traumatic brain injury. J Neurotrauma. 2020;37(1):14‐26. doi: 10.1089/neu.2019.6422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wang Z, Leng Y, Wang J, et al. Tubastatin a, an HDAC6 inhibitor, alleviates stroke‐induced brain infarction and functional deficits: potential roles of alpha‐tubulin acetylation and FGF‐21 up‐regulation. Sci Rep. 2016;6:19626. doi: 10.1038/srep19626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF‐23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277(2):494‐498. doi: 10.1006/bbrc.2000.3696 [DOI] [PubMed] [Google Scholar]
- 107. Bhattacharyya N, Chong WH, Gafni RI, Collins MT. Fibroblast growth factor 23: state of the field and future directions. Trends Endocrinol Metab. 2012;23(12):610‐618. doi: 10.1016/j.tem.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Goetz R, Nakada Y, Hu MC, et al. Isolated C‐terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23‐FGFR‐Klotho complex formation. Proc Natl Acad Sci U S A. 2010;107(1):407‐412. doi: 10.1073/pnas.0902006107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bergwitz C, Juppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91‐104. doi: 10.1146/annurev.med.051308.111339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Blau JE, Collins MT. The PTH‐vitamin D‐FGF23 axis. Rev Endocr Metab Disord. 2015;16(2):165‐174. doi: 10.1007/s11154-015-9318-z [DOI] [PubMed] [Google Scholar]
- 111. Stein MS, Ward GJ, Butzkueven H, Kilpatrick TJ, Harrison LC. Dysequilibrium of the PTH‐FGF23‐vitamin D axis in relapsing remitting multiple sclerosis: a longitudinal study. Mol Med. 2018;24(1):27. doi: 10.1186/s10020-018-0028-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Vlot MC, Boekel L, Kragt J, et al. Multiple sclerosis patients show lower bioavailable 25(OH)D and 1,25(OH)2D, but No difference in ratio of 25(OH)D/24,25(OH)2D and FGF23 concentrations. Nutrients. 2019;11(11):2774. doi: 10.3390/nu11112774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Emami Aleagha MS, Siroos B, Allameh A, Shakiba S, Ranji‐Burachaloo S, Harirchian MH. Calcitriol, but not FGF23, increases in CSF and serum of MS patients. J Neuroimmunol. 2019;328:89‐93. doi: 10.1016/j.jneuroim.2018.12.011 [DOI] [PubMed] [Google Scholar]
- 114. Shirazi HA, Rasouli J, Ciric B, Rostami A, Zhang GX. 1,25‐Dihydroxyvitamin D3 enhances neural stem cell proliferation and oligodendrocyte differentiation. Exp Mol Pathol. 2015;98(2):240‐245. doi: 10.1016/j.yexmp.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Masuda Y, Ohta H, Morita Y, et al. Expression of Fgf23 in activated dendritic cells and macrophages in response to immunological stimuli in mice. Biol Pharm Bull. 2015;38(5):687‐693. doi: 10.1248/bpb.b14-00276 [DOI] [PubMed] [Google Scholar]
- 116. Han X, Li L, Yang J, King G, Xiao Z, Quarles LD. Counter‐regulatory paracrine actions of FGF‐23 and 1,25(OH)2 D in macrophages. FEBS Lett. 2016;590(1):53‐67. doi: 10.1002/1873-3468.12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rajendran R, Bottiger G, Dentzien N, et al. Effects of FGFR tyrosine kinase inhibition in OLN‐93 oligodendrocytes. Cells. 2021;10(6):1318. doi: 10.3390/cells10061318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Rajendran R, Giraldo‐Velasquez M, Stadelmann C, Berghoff M. Oligodendroglial fibroblast growth factor receptor 1 gene targeting protects mice from experimental autoimmune encephalomyelitis through ERK/AKT phosphorylation. Brain Pathol. 2018;28(2):212‐224. doi: 10.1111/bpa.12487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Furusho M, Roulois AJ, Franklin RJ, Bansal R. Fibroblast growth factor signaling in oligodendrocyte‐lineage cells facilitates recovery of chronically demyelinated lesions but is redundant in acute lesions. Glia. 2015;63(10):1714‐1728. doi: 10.1002/glia.22838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ziemssen T, Akgun K, Bruck W. Molecular biomarkers in multiple sclerosis. J Neuroinflammation. 2019;16:272. doi: 10.1186/s12974-019-1674-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Paul A, Comabella M, Gandhi R. Biomarkers in multiple sclerosis. Cold Spring Harb Perspect Med. 2019;9(3):a029058. doi: 10.1101/cshperspect.a029058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Huang Y, Dreyfus CF. The role of growth factors as a therapeutic approach to demyelinating disease. Exp Neurol. 2016;283(Pt B):531‐540. doi: 10.1016/j.expneurol.2016.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Grothe C, Meisinger C, Claus P. In vivo expression and localization of the fibroblast growth factor system in the intact and lesioned rat peripheral nerve and spinal ganglia. J Comp Neurol. 2001;434(3):342‐357. doi: 10.1002/cne.1181 [DOI] [PubMed] [Google Scholar]
- 124. Takase N, Koma Y, Urakawa N, et al. NCAM‐ and FGF‐2‐mediated FGFR1 signaling in the tumor microenvironment of esophageal cancer regulates the survival and migration of tumor‐associated macrophages and cancer cells. Cancer Lett. 2016;380(1):47‐58. doi: 10.1016/j.canlet.2016.06.009 [DOI] [PubMed] [Google Scholar]
- 125. Peiris MN, Meyer AN, Nelson KN, Bisom‐Rapp EW, Donoghue DJ. Oncogenic fusion protein BCR‐FGFR1 requires the breakpoint cluster region‐mediated oligomerization and chaperonin Hsp90 for activation. Haematologica. 2020;105(5):1262‐1273. doi: 10.3324/haematol.2019.220871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Hu T, Chong Y, Qin H, et al. The miR‐17/92 cluster is involved in the molecular etiology of the SCLL syndrome driven by the BCR‐FGFR1 chimeric kinase. Oncogene. 2018;37(14):1926‐1938. doi: 10.1038/s41388-017-0091-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhao XM, Byrd VM, McKeehan WL, Reich MB, Miller GG, Thomas JW. Costimulation of human CD4+ T cells by fibroblast growth factor‐1 (acidic fibroblast growth factor). J Immunol. 1995;155(8):3904‐3911. [PubMed] [Google Scholar]
- 128. Byrd V, Zhao XM, McKeehan WL, Miller GG, Thomas JW. Expression and functional expansion of fibroblast growth factor receptor T cells in rheumatoid synovium and peripheral blood of patients with rheumatoid arthritis. Arthritis Rheum. 1996;39(6):914‐922. doi: 10.1002/art.1780390607 [DOI] [PubMed] [Google Scholar]
- 129. Kos FJ, Chin CS. Costimulation of T cell receptor‐triggered IL‐2 production by Jurkat T cells via fibroblast growth factor receptor 1 upon its engagement by CD56. Immunol Cell Biol. 2002;80(4):364‐369. doi: 10.1046/j.1440-1711.2002.01098.x [DOI] [PubMed] [Google Scholar]
- 130. Petri S, Krampfl K, Kuhlemann K, Dengler R, Grothe C. Preserved expression of fibroblast growth factor (FGF)‐2 and FGF receptor 1 in brain and spinal cord of amyotrophic lateral sclerosis patients. Histochem Cell Biol. 2009;131(4):509‐519. doi: 10.1007/s00418-008-0549-x [DOI] [PubMed] [Google Scholar]
- 131. Liu X, Mashour GA, Webster HF, Kurtz A. Basic FGF and FGF receptor 1 are expressed in microglia during experimental autoimmune encephalomyelitis: temporally distinct expression of midkine and pleiotrophin. Glia. 1998;24(4):390‐397. doi: [DOI] [PubMed] [Google Scholar]
- 132. Balaci L, Presta M, Ennas MG, et al. Differential expression of fibroblast growth factor receptors by human neurones, astrocytes and microglia. Neuroreport. 1994;6(1):197‐200. doi: 10.1097/00001756-199412300-00050 [DOI] [PubMed] [Google Scholar]
- 133. Song J, Kang SM, Lee KM, Lee JE. The protective effect of melatonin on neural stem cell against LPS‐induced inflammation. Biomed Res Int. 2015;2015:854359. doi: 10.1155/2015/854359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hu JG, Wu XJ, Feng YF, et al. PDGF‐AA and bFGF mediate B104CM‐induced proliferation of oligodendrocyte precursor cells. Int J Mol Med. 2012;30(5):1113‐1118. doi: 10.3892/ijmm.2012.1110 [DOI] [PubMed] [Google Scholar]
- 135. Zingone A, Cultraro CM, Shin DM, et al. Ectopic expression of wild‐type FGFR3 cooperates with MYC to accelerate development of B‐cell lineage neoplasms. Leukemia. 2010;24(6):1171‐1178. doi: 10.1038/leu.2010.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bansal R, Lakhina V, Remedios R, Tole S. Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with Pdgfralpha, Olig2 and Plp/dm20: implications for oligodendrocyte development. Dev Neurosci. 2003;25(2–4):83‐95. doi: 10.1159/000072258 [DOI] [PubMed] [Google Scholar]
- 137. Pringle NP, Yu WP, Howell M, Colvin JS, Ornitz DM, Richardson WD. Fgfr3 expression by astrocytes and their precursors: evidence that astrocytes and oligodendrocytes originate in distinct neuroepithelial domains. Development. 2003;130(1):93‐102. doi: 10.1242/dev.00184 [DOI] [PubMed] [Google Scholar]
- 138. Kokkinakis DM, Rushing EJ, Shareef MM, et al. Physiology and gene expression characteristics of carcinogen‐initiated and tumor‐transformed glial progenitor cells derived from the CNS of methylnitrosourea (MNU)‐treated Sprague‐Dawley rats. J Neuropathol Exp Neurol. 2004;63(11):1182‐1199. doi: 10.1093/jnen/63.11.1182 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.