

Summary
N6-adenine DNA methylation (6mA), a rediscovered epigenetic mark in eukaryotic organisms, diversifies in abundance, distribution, and function across species, necessitating its study in more taxa. Paramecium bursaria is a typical model organism with endosymbiotic algae of the species Chlorella variabilis. This consortium therefore serves as a valuable system to investigate the functional role of 6mA in endosymbiosis, as well as the evolutionary importance of 6mA among eukaryotes. In this study, we report the first genome-wide, base pair-resolution map of 6mA in P. bursaria and identify its methyltransferase PbAMT1. Functionally, 6mA exhibits a bimodal distribution at the 5′ end of RNA polymerase II-transcribed genes and possibly participates in transcription by facilitating alternative splicing. Evolutionarily, 6mA co-evolves with gene age and likely serves as a reverse mark of endosymbiosis-related genes. Our results offer new insights for the functional diversification of 6mA in eukaryotes as an important epigenetic mark.
Subject areas: Cellular physiology, Epigenetics, Cell biology
Graphical abstract
Highlights
-
•
The first genome-wide, base pair-resolution map of 6mA in Paramecium bursaria
-
•
6mA participates in transcription by facilitating intron retention
-
•
6mA co-evolves with gene age
-
•
6mA likely serves as a reverse mark of endosymbiosis-related genes
Cellular physiology; Epigenetics; Cell biology
Introduction
As a prevalent epigenetic modification, the crucial roles of N6-adenine DNA methylation (6mA) were first implicated in prokaryotic organisms.1,2,3 With improvement in detection sensitivity, 6mA has recently been detected in a broad range of eukaryotes, from unicellular (protists and basal fungi) to multicellular (animals and plants) organisms, illustrating a wide variety in its abundance, distribution, and function.4,5,6,7,8,9,10,11
Ciliates (i.e., members of the phylum Ciliophora) are unicellular eukaryotes that have served as model systems in many fields of research including molecular biology, epigenetics, and evolutionary biology.12,13,14,15,16,17,18 Notably, 6mA ratio in ciliates is several orders of magnitude higher than that in metazoans (e.g., ∼1% in Tetrahymena vs. ∼0.0006–0.007% in mouse embryonic stem cells),8,19,20 although 6mA findings in higher vertebrates are controversial.21,22,23,24,25,26 Furthermore, another DNA methylation, namely 5-methylcytosine (5mC), is not detectable in most ciliates,27,28,29,30,31,32,33 effectively minimizing its confounding effects on 6mA analysis. Along with the intense research efforts for mammalian 6mA, 6mA in ciliates was also reinvestigated.8,11,19,34 6mA in the oligohymenophorean ciliate Tetrahymena thermophila (referred to hereafter as Tetrahymena) and Paramecium tetraurelia and the hypotrich ciliate Oxytricha trifallax (referred to hereafter as Oxytricha) is located in the sequence of 5′-ApT-3′ and specifically accumulates in nucleosomal linker DNA regions downstream of transcription start sites.11,19 Tetrahymena 6mA functions as an integral part of the chromatin landscape, sharing a similar distribution pattern with the chromatin remodeler-controlled histone variant H2A.Z and affecting the stability of nucleosomes.19 Oxytricha 6mA locally repels nucleosome occupancy in vitro but not in vivo, suggesting that the intrinsic interactivity between 6mA and the nucleosome can be modulated by the endogenous chromatin environment.11,35 P. tetraurelia shows a positive correlation between 6mA enrichment and gene expression.34 More importantly, a distinct clade of MT-A70 family methyltransferase—AMT1 reported in Tetrahymena—differs from metazoan METTL4 homologs in its distribution, sequence preference, methylation abundance, and potential correlation with transcription among different organisms.8 These findings demonstrate that ciliates provide an ideal system for studying the evolutionary diversification of eukaryotic 6mA.36,37,38,39,40
The fact that Ciliophora is such a highly diversified lineage necessitates the investigation of 6mA in more ciliate species. P. bursaria harbors hundreds of endosymbiotic Chlorella variabilis cells in its cytoplasm. The P. bursaria/C. variabilis consortium not only helps to reveal the representative 6mA characteristics of the genus Paramecium but also provides a valuable resource for investigating the role of 6mA in endosymbiosis. In this study, we 1) reanalyze the published SMRT (single molecule, real-time) sequencing (SMRT-seq) data for P. bursaria41; 2) provide the first genome-wide map of 6mA in P. bursaria with single base pair resolution; 3) reveal the correlation of 6mA with ApT dinucleotides, Pol II transcription, alternative splicing (AS), horizontal gene transfer, and endosymbiosis-related genes; and 4) identify and functionally confirm a 6mA methyltransferase PbAMT1 in P. bursaria.
Results
6mA occurs in the macronucleus (MAC) of P. bursaria
To demonstrate the existence of 6mA in P. bursaria, first we imaged living and fixed P. bursaria cells (Figure 1A) and then performed immunofluorescence (IF) staining with anti-6mA antibody.19 Cells were pretreated with cold acetone to preclude the autofluorescence of symbiotic algae.42 In vegetative P. bursaria cells, 6mA was observed in the somatic MAC with active transcription but was undetectable in the germline transcriptionally inert micronucleus (MIC) (Figure 1B), resembling the 6mA distribution pattern in Tetrahymena.19
Figure 1.

Global distribution of 6mA in Paramecium bursaria
(A) Photomicrographs of P. bursaria in vivo (a, b) and after silver carbonate staining (c). Bar = 25 μm. Note the green algae inside the P. bursaria cell indicated by arrowheads.
(B) Immunofluorescence staining of 6mA in vegetative P. bursaria cells. Note the absence of 6mA signal in the micronucleus (MIC, dashed circles). Bar = 15 μm.
(C) Representative interpulse duration (IPD) ratios of SMRT sequencing data of vegetative P. bursaria cells. IPD peaks (columns) and detected 6mA signals (balloons) on both the Watson (purple) and Crick (orange) strands are indicated, respectively. The representative sequence is shown, and only 6mA sites with significantly high IPD ratio are indicated.
(D) Normalized 6mA ratio in Paramecium bursaria (green), Paramecium tetraurelia (purple), Tetrahymena thermophila (red), and Oxytricha trifallax (blue) genomes. 6mA ratio was defined as the number of methylated adenine sites divided by the total adenine sites (6mA/A).
To determine the precise amount of 6mA, we reanalyzed the published SMRT-seq data of P. bursaria.41 6mA was detected with high confidence (coverage >30× and Qv > 30 for mean coverage 123× SMRT-seq data) on 266,215 adenines, all with the typical kinetic signature (Figure 1C), corresponding to 1.28% of the total adenines in the P. bursaria genome (Figure 1D). This content is slightly lower than that in its congener Paramecium aurelia (∼2.5%),28 possibly due to species divergence and the different methods used for 6mA measurement, i.e., strict cutoff with SMRT-seq data in P. bursaria vs. no cutoff with high-performance liquid chromatography (HPLC) in P. aurelia. Furthermore, we reanalyzed the 6mA ratio with the comparable cutoff value of 6mA in P. bursaria in the other three ciliates reported previously, P. tetraurelia (coverage >100× for mean coverage 400× data), Tetrahymena (coverage >36× for mean coverage 144× data), and Oxytricha (coverage >70× for mean coverage 278× data) (Table S1), from published data.11,19,34 Only 6mA sites with high confidence were selected for subsequent analysis. The 6mA ratio in P. bursaria was the highest (1.28%) compared with that in P. tetraurelia (1.06%), Tetrahymena (0.54%), and Oxytricha (0.38%) (Figure 1D and Table S1). The discrepancy of 6mA ratio in Oxytricha (0.38% in this study vs. 0.78–1.04% in Beh et al., 2019)11 and P. tetraurelia (1.06% in this study vs. 1.6% in Hardy et al., 2021)34 resulted from different ways of SMRT-seq data processing (see Table S1).
6mA occurs preferentially in the AT motif (5′-ApT-3′) mainly as symmetrically and highly methylated sites
6mA was distributed throughout the whole genome in P. bursaria (Figures 2A and S1A). The majority of 6mA (99.60%) in P. bursaria was located at the consensus sequence of 5′-ApT-3' (Figures 2B and S1B, Tables S1 and S2), similar to that of P. tetraurelia (99.63%),34 Tetrahymena (87.76%),8,19 and Oxytricha (95.98%).11 It was worth mentioning that 6mA in P. bursaria displayed a slight VATB (V = A, C, or G and B = C, G, or T) motif, as previously observed in Oxytricha11 and P. tetraurelia.34 6mA in P. bursaria presented a stronger preference to the 5′-ApT-3′ sequence than that in Tetrahymena (3.48% vs. 1.38% in 6mApT/total ApT) (Figure 2C, Table S2), even though the former contains a less AT-rich genome than the latter (71.27% vs. 77.70%).43 Consistent with the ApT enrichment, 6mA depositions on three other dinucleotides (ApA, ApC, ApG) were quite low (Figures 2C and S1B, Table S2).
Figure 2.

Different genomic features of 6mA in P. bursaria
(A) Circos plots of 6mA distribution across 10 longest contigs. Six rings from outside to inner show distribution of 6mA in different motifs (symmetric, asymmetric, non-ApT), different methylation levels (high 80%–100%, intermediate 20%–80%), or all 6mA sites. See color notes on the right for more details.
(B) Conserved motif for 6mA sites (at position 0) in P. bursaria, P. tetraurelia, T. thermophila, and O. trifallax.
(C) Relative distribution of 6mA in ApN (ApA/ApT/ApC/ApG) dinucleotides in P. bursaria (green), P. tetraurelia (purple), T. thermophila (red), and O. trifallax (blue). The relative distribution of 6mA in ApN was calculated by the number of methylated adenines in a particular ApN dinucleotide divided by the number of the corresponding ApN dinucleotides in the genome, e.g., 6mApT/ApT.
(D) Classification of 6mA sites based on their symmetry in P. bursaria (green), P. tetraurelia (purple), T. thermophila (red), and O. trifallax (blue). The Y axis represents percentage (of a given class of 6mA/all 6mA). See Table S3 for details.
(E) Density plot of 6mA distribution by methylation levels on Watson (x axis) or Crick strands (y axis) in P. bursaria.
(F) Comparison of 6mA methylation levels in P. bursaria (green), P. tetraurelia (purple), T. thermophila (red), and O. trifallax (blue). Methylation levels are ranked from low to high and divided into 10 quantiles.
(G) Area-proportional Venn diagram shows the overlap of highly methylated (80–100%) and symmetrically methylated 6mA in P. bursaria.
(H) Correlation analysis between methylation levels (x axis) and proportion of symmetric 6mA sites (y axis: symmetric 6mA/total 6mA). Note the percentage of symmetric 6mA is calculated only for 6mA sites, not all A sites.
6mA sites within palindromic ApT dinucleotides consist of two possible methylated status: symmetric (methylated on both DNA strands) and asymmetric (methylated on either one of the two strands). 79.23% of 6mA sites were symmetrically methylated, which fell among the proportions in P. tetraurelia (80.86%), Tetrahymena (61.06%), and Oxytricha (81.27%) (Figure 2D and Table S3). 20.37% of sites were methylated asymmetrically (Figure 2D, Table S3) without strand specificity between the Watson and Crick strands (Figures 2E, S1C, and S1D), like that in P. tetraurelia, Tetrahymena, and Oxytricha.8,11,19,34 Thus, 6mA in P. bursaria occurred mainly as symmetric methylation.
In the polyploid MAC of P. bursaria, different 6mA sites possessed various methylation levels. We classified 6mA sites into ten quantiles according to their methylation levels (Figure 2F). Most 6mA sites (97.22%) were methylated with 50–100% methylation level, the majority (71.98% of 6mA) being in the range 70–100% (Figure 2F and Table S4). This is similar to Tetrahymena wherein 64.51% of 6mA sites were enriched with a 60–90% methylation level (Figure 2F)8,19 but slightly different from Oxytricha wherein 6mA has a more even distribution (50–90%) and a slight enrichment (22.53% of 6mA) of 90–100% (Figure 2F)11 and from P. tetraurelia with an enrichment of 40–80% methylation level (75.55% of 6mA).34
Interestingly, symmetric 6mA of P. bursaria was in direct proportion to high-methylated 6mA (80–100%). Symmetric 6mA in P. bursaria was more enriched on 80–100% methylation level compared to asymmetric 6mA and non-ApT 6mA (Figures S2A–S2C). Symmetric 6mA shared a significant overlap with high-methylated 6mA (44.0% of total 6mA; representation factor: 1.1) (Figure 2G). In addition, methylation levels showed an obvious positive association with symmetric 6mA (r = 0.78) (Figure 2H). These findings suggest that 6mA in P. bursaria mainly exists as symmetrically and highly methylated sites.
6mA is exclusively present on Pol II-transcribed genes
Composite analysis showed that 6mA was accumulated toward the 5′ end of the gene body (8,220 well-annotated long genes, >1 kb) in P. bursaria, like in P. tetraurelia, Tetrahymena, and Oxytricha (Figure 3A).8,11,19,34 This distribution was attributable more to symmetric 6mA than asymmetric 6mA (Figure 3B). Interestingly, 6mA in P. bursaria exhibited a bimodal distribution around transcription start sites (TSSs) (Figures 3A and 3C). This pattern is similar to that in Chlamydomonas reinhardtii4 and P. tetraurelia but is different from that in Tetrahymena and Oxytricha wherein 6mA accumulates downstream, but not upstream, of TSS.8,11,34 Fourier analysis showed that 6mA exhibited a periodic profile with a frequency of ∼150 bp (Figures S3A and S3B). This was further confirmed by the genome-wide distribution analysis between 6mA and nucleosomes, revealing two damped oscillations with the same periodicity but with opposite phases (Figure 3C). 6mA, especially high-methylated 6mA, was specifically excluded from nucleosomal DNA (Figure 3D), similar to P. tetraurelia, Tetrahymena, and Oxytricha.8,11,34 In addition, genes bearing 6mA presented more prominent nucleosome arrays than genes without 6mA (Figure S3C).
Figure 3.

6mA in P. bursaria is correlated with Pol II genes transcription
(A) Composite analysis of 6mA distribution on the gene body of P. bursaria (green), P. tetraurelia (purple), T. thermophila (red), and O. trifallax (blue). The gene body length is scaled to one unit and extended to each side for locus statistical analysis (bin size = 0.05). Distribution frequency is calculated as “6mA amount at a certain position/total 6mA amount”.
(B) Composite analysis of symmetric, asymmetric, and ensemble 6mA distribution on the gene body of P. bursaria. Genes are scaled to unit length and extended to each side by unit length as above. Note that symmetric 6mA is a predominant attributor to the trend.
(C) Distribution profiles of 6mA (green) and nucleosome (blue) around transcription start sites (TSS) in P. bursaria. Note the bimodal distribution around TSS (arrowheads).
(D) 6mA distribution relative to the called nucleosome dyad in P. bursaria cells grouped by methylation level. The violin plots indicate the 6mA ratio between adjacent nucleosome dyads, except the low-methylated 6mA (0–30%).
(E) Validating the methylation status of nine GATC sites by DpnI/DpnII digestion coupled with qPCR analysis. The sites were selected for their location on genes transcribed by different RNA polymerases (Pol I, II, and III), and their methylation levels calculated from SMRT sequencing data (H1-H3: high methylation; I1-I2: intermediate methylation; N1-N2: no methylation; all these are transcribed by Pol II. N3: no methylation transcribed by Pol I; N4: no methylation transcribed by Pol III.). See STAR Methods for details. Data are represented as mean ± SD.
(F) The distribution plot of two groups of genes with high (FPKM ≥1) and low (FPKM <1) expression levels around TSS shows that the 6mA methylation is correlated with active genes.
(G) Statistical analysis of expression levels (log2 transformation of FPKM) in genes with (6mA+) or without 6mA (6mA-). Student’s t test was performed. Data are represented as mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns: not significant, p > 0.05.
(H) 6mA amount (number of 6mA sites in the first 1 kb of gene body) of genes with different expression levels downstream the TSS. Genes are ranked from low to high by their expression levels (log2 transformation of FPKM) and divided into ten quantiles.
The aforementioned enrichment of 6mA stimulated us to investigate the relationship between 6mA and three eukaryotic RNA polymerases (Pol I, II, and III) -transcribed genes. We first analyzed 6mA distribution on Pol I-transcribed rRNA and Pol III-transcribed non-coding RNA genes (Table S5). 6mA was not detected on either category. Considering that 6mA was previously reported in the human mitochondrial DNA9,44 transcribed by the mitochondrial RNA polymerase (POLRMT),45 we also examined the presence of mitochondrial 6mA in P. bursaria. 6mA was detected with high confidence (coverage >25×, Qv > 30 for 100×, coverage = 2,684×) on only three sites in the mitochondrial genome, which were likely to be false positives as they had lower interpulse duration (IPD) ratios and methylation levels than Pol II-transcribed genes and presented no 6mA kinetic signature (Figures S4A–S4C). Together, we conclude that 6mA in P. bursaria is exclusively related with Pol II-transcribed genes (Figure 1C).
To validate the SMRT-seq results, we performed restriction-enzyme digestion coupled with qPCR analysis, based on the fact that DpnI cleaves methylated GATC sites with a strong preference for symmetric rather than asymmetric methylation whereas DpnII cuts unmethylated GATC sites.1,46 The results showed that the five methylated sites in Pol II-transcribed genes (three sites at 80–100% high levels, two sites at 20–80% intermediate levels) were all sensitive to DpnI digestion, while unmethylated sites (two for Pol II, one for Pol I, and one for Pol III) were sensitive to DpnII digestion (Figure 3E), further supporting the assertion that 6mA is exclusively present on Pol II-transcribed genes.
The relationship between 6mA and Pol II-transcribed genes inspired us to further investigate whether 6mA is linked to transcription. In one approach, we classified genes by gene expression level (high expression: 80% of all genes; low expression: 20%) and examined their normalized 6mA distribution around TSS. Lowly expressed genes tended to have lower 6mA occupancies around TSS and vice versa (Figure 3F). In another approach, we classified genes by the presence or absence of 6mA. Genes with 6mA were expressed at significantly higher levels than genes without 6mA (p < 0.0001) (Figure 3G). We then compared 6mA amount (number of 6mA sites in the first 1 kb of the gene body) in ten gene quantiles according to their expression levels. 6mA amount was significantly lower in quantile 1 and progressively increased from quantile 2 to quantile 10 (Figure 3H). Consistent with these findings, both of 6mA amount and methylation level were correlated, although weakly, with expression levels of associated genes (r = 0.24, p < 0.001; r = 0.24, p < 0.001). These results indicate that 6mA in P. bursaria was related with transcription of Pol II-transcribed genes, as reported previously in Tetrahymena and Oxytricha.8,11,19
6mA is more enriched in retained introns than that in spliced introns
We next investigated the genomic features of 6mA in P. bursaria and found that 6mA was preferentially localized in genic regions (92.2%) (Figures 4A and S5A and Table S6),47,48 resembling the pattern in P. tetraurelia, Tetrahymena, and Oxytricha.8,11,19,34 Specifically, the 6mA ratio (total 6mA sites number/total A number of corresponding sequence) of exonic, intronic, and intergenic region in P. bursaria is all higher than that in Tetrahymena and Oxytricha (Figure S5A, right panel). In P. bursaria, despite the fact that the average AT content of introns (74.46%) is higher than that of exons (69.75%), the 6mA ratio is significantly lower in intronic regions (1.58% vs. 1.92%, p < 0.0001) (Figure S5B). This was corroborated by observed/simulated ratio analysis, which showed an enrichment of 6mA in exons and a depletion in introns (Figures 4A and S5A).
Figure 4.

6mA is related to alternative splicing
(A) Comparison of observed versus simulated distributions of 6mA in P. bursaria genome suggests that 6mA is enriched in exons rather than introns.
(B) The ratio of different alternative splicing events (IR: intron retention, AE: alternative 5' or 3′ splice-site selection, ES: exon skipping) in P. bursaria.
(C) Statistical analysis of 6mA ratio (left panel) and expression level (right panel) indicated by log2 transformation of FPKM (fragments per kilobase per million) in P. bursaria (n1 = 360; n2 = 17,442), P. tetraurelia (n1 = 3369; n2 = 30,048), and T. thermophila (n1 = 1,066; n2 = 25,151) shows that AS genes contain obviously higher expression levels and amounts of 6mA compared with other genes. Student’s t test was performed, and n represents the number of corresponding genes per group. Data are represented as mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns: not significant, p > 0.05. 6mA ratio was defined as the number of methylated adenine sites divided by the total adenine sites (6mA/A) in the corresponding genomic sequence.
(D) Statistical analysis of methylation level (left panel) and 6mA ratio (right panel) in P. bursaria demonstrates that the level of 6mA in retained introns of AS (AS-RI) genes (n = 150) is higher than that in normal introns of AS (AS-NI) genes (n = 423) and other introns of non-AS (other introns) genes (n = 24,818). Student’s t test was performed, and n represents the number of corresponding introns per group. Data are represented as mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns: not significant (p > 0.05). Methylation level here was calculated as average methylation level of per intron.
Particular exons and introns of a gene are likely to be skipped or retained during the formation of mature messenger RNA (mRNA), which are examples of AS.47,48,49 By analyzing RNA sequencing (RNA-seq) data, it was found that 360 genes (2.02% of all genes) (referred to as AS genes below) in P. bursaria generated AS isoforms, which is consistent with previous findings that AS events in unicellular eukaryotes occur in less than 5% of genes.50 In total, 825 AS events were detected. All three basic types of AS events were found (Figure 4B), i.e., cassette-exon inclusion or skipping (ES, 8 events), alternative 5' or 3′ splice-site selection (AE, 65 events), and intron retention (IR) (752 events, the majority type), indicating that splice-site recognition in P. bursaria is executed by intron definition (Jaillon et al., 2008; Saudemont et al., 2017) as observed in Tetrahymena50 instead of by exon definition in humans.51
It has been suggested that AS events in multicellular eukaryotes are associated with intragenic 5mC.52,53,54 In P. bursaria, the absence of 5 mC and the preferential enrichment of 6mA in exons stimulated us to examine the correlation of 6mA and AS events. On the one hand, compared to genes without 6mA (5,894), methylated genes (11,908) were enriched for splice variants in P. bursaria (89.72% vs. 67.00%), P. tetraurelia (87.80% vs. 85.89%), and Tetrahymena (96.39% vs. 79.88%). On the other hand, the 6mA ratio (Figure 4C, left panel) in AS genes and the expression level of these genes (Figure 4C, right panel) were notably higher than those in other genes, among P. bursaria (p < 0.001), P. tetraurelia (p < 0.001), and Tetrahymena (p < 0.001). Given that the major type of AS event in P. bursaria is IR, we focused on the retained introns. Intriguingly, the retained introns of AS genes in P. bursaria carried higher methylation levels (average methylation level of per intron) (p < 0.01) (Figure 4D, left panel) and 6mA ratios (p < 0.05) (Figure 4D, right panel) than other introns of AS genes or introns of non-AS genes. Therefore, in the category of introns, 6mA preferentially distributes on the retained introns of AS genes.
IR events decrease with reduced 6mA ratios
Based on the 6mA comparison in retained introns and spliced introns above, we hypothesized that if 6mA ratio in P. bursaria was reduced, IR events would be decreased. To perturb the 6mA ratio, we first tried to identify 6mA-specific methyltransferase(s) in P. bursaria. By homology search, five proteins from the MT-A70 family were predicted as the putative DNA 6mA methyltransferases of P. bursaria, named here as P. bursaria adenine MTases (PbAMTs) (Figures 5A and 5B and Tables S7 and S8). PbAMT1 was split into two peptides (PbAMT1a and PbAMT1b) according to the initial annotation. We then refined the annotation on the PbAMT1 gene model by sequencing the genomic DNA and cDNA (Figures S6A and S6B, Table S7). The PbAMT1-coding DNA sequence was 1,075 bp in size, and the gene contained five introns and six exons encoding a protein of 309 amino acids (Figure S6A). PbAMT1 contained the conserved MT-A70 domain, which harbors the DPPW signature motif required for substrate recognition and catalytic activity of MTases55,56 at the N terminus and the ciliate-specific GNEL motif at the C terminus, respectively (Figure S6A). PbAMT1 belongs to a distinct eukaryotic subclade (Figure 5B), resembling the major 6mA MTase AMT1 in Tetrahymena,8 with orthologs distributed in lower eukaryotes (protists and basal fungi) but not in higher eukaryotes (animals, plants, and true fungi). PbAMT6 is grouped in the AMT6-AMT7 subclade, members of which are AMT1-associated proteins required for its MTase activity.8,19 PbAMT2 belongs to the protist-specific subclade which is homologous to the AMT2/5 clade that contains ZZ-type zinc fingers.56 PbAMT4 is the METTL3 ortholog, and PbAMT3 is the METTL14 ortholog; both are associated with RNA N6-adenosine methylation (m6A).57 As expected, no PbAMTs clustered with METTL4 homologs, the reported 6mA MTases in metazoans and plants (Figure 5B).58,59
Figure 5.

Phylogenetic analysis of PbAMTs 1–6
(A) Domain structure of PbAMTs 1–6. MT-A70 domains of PbAMTs 1–4 are predicted by CD-Search, while that of PbAMT6 is inferred from sequence alignment with PbAMTs 1–4. The structure of AMTs 1–7 of T. thermophila (AMT2 is also referred to as TAMT-1) is also shown for comparison. MT-A70 domain, DPPW, and GNEL motif are highlighted by purple, red, and green, respectively.
(B) Phylogenetic analysis of MT-A70 proteins. Putative DNA 6mA (subclades AMT1, AMT2/5, AMT6/7, and METTL4/DAMT1) and RNA m6A (subclades METTL3 and METTL14) methyltransferase candidates are separated by a dashed curve. The eukaryotic tree (top left panel) is shown with different colors according to their phylogenetic position. PbAMTs 1–6 of P. bursaria are shown in bold plus cyan in different clades. The scale bar is 0.5. Species full name and NCBI GI number are shown in Table S13.
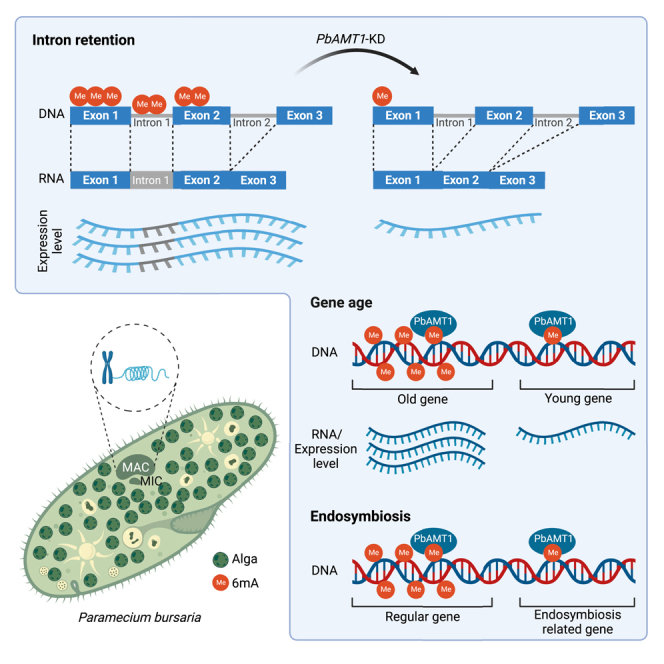
To determine whether PbAMT1 regulates 6mA in P. bursaria, we generated PbAMT1 knockdown cells (PbAMT1-KD) by RNA interference (Figure S6C). The expression of PbAMT1 was downregulated after three days of induction and was more significantly reduced after six days of induction, as demonstrated by the qRT-PCR analysis (Figure 6A). Accordingly, 6mA ratio in PbAMT1-KD cells was significantly lower than that in control cells (fed with empty vector) after 3 days and 6 days of induction, as shown by IF staining (Figures 6B and 6C). Interestingly, PbAMT1-KD cells grew more slowly, and about 22.49% of them contained an abnormally large contractile vacuole (Figure 6D), similar to the phenotype of Tetrahymena cells wherein the 6mA MTase AMT1 was deleted.8 Together, these results indicate that PbAMT1 is a 6mA methyltransferase in P. bursaria, regulating 6mA ratio and being required for normal cell growth.
Figure 6.

Intron retention events decrease with reduced 6mA ratios
(A) Expression profile of PbAMT1 in control and PbAMT1-KD cells. Expression levels are represented by normalized quantitative RT-PCR data. Data are represented as mean ± SD. Student’s t test was performed. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns: not significant, p > 0.05.
(B) Immunofluorescence (IF) staining of DNA 6mA in vegetative control and PbAMT1-KD cells. The absence of 6mA signal in the micronucleus is indicated.
(C) Statistical analysis of 6mA IF signal intensity in B. Cell images were randomly selected (control: n = 110, PbAMT1-KD: n = 119) and processed by ImageJ. Data are shown as boxplots. Student’s t test was performed, and n represents the number of corresponding cells per group. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns: not significant, p > 0.05. The DAPI signal was used to confirm the range of nucleus, and then the 6mA signal of nucleus was circled and the signal intensity was quantified.
(D) The cell proliferation profile of control and PbAMT1-KD cells within six days, feeding with E. coli HT115. The original cell density is 400 cells/mL. Significant differences are labeled by ∗ (Student’s t test was performed). ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns: not significant, p > 0.05. Note that abnormal contractile vacuoles (CVs) were observed in a large portion (22.49%) of PbAMT1-KD cells but not in control cells.
(E) The 6mA profiles on gene body between control and PbAMT1-KD cells. 6mA is significantly decreased in PbAMT1-KD cells, and the 6mA peaks are more enriched in gene body.
(F) Heatmap of intron retention density (characterized by normalized reads of retained introns) in control and PbAMT1-KD cells. IR is significantly reduced in PbAMT1-KD cells.
(G) Alternative splicing events and 6mA distribution on a representative gene locus (partial gene g716, contig GWHAAFB00000006) in control and PbAMT1-KD cells, respectively. The region of intron retention is highlighted by the dashed green box and the yellow asterisk.
To confirm the correlation between 6mA and AS, we performed 6mA immunoprecipitation sequencing (6ma-IP-seq) to track 6mA level. Meanwhile, we explored Oxford Nanopore Technologies (ONT) sequencing (ONT-seq) and Illumina-based RNA-seq to detect the variants of retained introns. Compared to the control cells, global 6mA level in PbAMT1-KD cells showed a significant reduction (Figure 6E), consistent with the IF results (Figures 6B and 6C). Correspondingly, the number of IR events in PbAMT1-KD cells was reduced (PbAMT1-KD vs. control: 609 vs. 689 for ONT, 472 vs. 648 and 412 vs. 540, respectively, for two RNA-seq replicates) (Figure 6F), along with the reduced 6mA ratio (Figures S7A–S7D, Table S16). The two RNA-seq replicates shared a small overlap of the IR events (∼30% in wild type [WT], 18% in PbAMT1-KD) despite the comparable total event number (Figure S7C), possibly attributable to the dynamic nature of AS. In control cells, representative IR regions harbored high levels of 6mA, which were eliminated in PbAMT1-KD cells (Figure 6G). Meanwhile, several IR isoforms with considerable read counts were detected in control cells by ONT-seq and RNA-seq, which were nearly undetectable in PbAMT1-KD cells (Figures 6G, S7D, and S7E). We therefore conclude that 6mA possibly participates in AS especially for IR in P. bursaria.
Potential roles of 6mA in gene age and endosymbiosis
The P. bursaria/C. variabilis consortium serves as a model to investigate endosymbiosis,41 a phenomenon that a symbiont dwells within the body of its symbiotic partner. Given that 6mA ratio of C. variabilis NC64A was reported to be ∼0.6%,60 we supposed that the higher 6mA ratio of P. bursaria might be attributable to horizontal gene transfer (HGT) genes from C. variabilis. In total, we identified 159 putative genes possibly involved in HGT, referred to as HGT-like genes. Indeed, HGT-like genes showed higher 6mA ratios, as well as higher expression levels, than other genes (Figures 7A, 7B, and S8A), partially explaining the higher 6mA ratio in P. bursaria. More interestingly, we found the HGT-like genes with high gene methylation level tend to be much older genes (phylostratigraphic levels 1–2 vs. 1–10) (Figures S8B and S8C), which impelled us to investigate the relationship between 6mA and gene age. We further calculated gene age of all annotated genes in P. bursaria and found that gene methylation level of older genes was significantly higher than that of younger genes (Figure 7C, left panel; p value of Tukey’s multiple comparisons test in Table S9), suggesting that 6mA in P. bursaria co-evolved with gene age. However, the detailed mechanism between 6mA and gene age requires further research. A similar trend was observed with gene expression levels (Figure 7C, right panel; Table S10). Together, these results suggested that the 6mA in P. bursaria co-evolved with gene age.
Figure 7.

Potential roles of 6mA in gene age and endosymbiosis
(A) Composite analysis of 6mA distribution in HGT-like genes (159 genes) and other genes (17,643 genes) on the gene body of P. bursaria. Note that 6mA of HGT-like genes is obviously more than 6mA of other genes.
(B) Statistical analysis of expression levels (log2 transformation of FPKM) of HGT-like genes (159 genes) and other genes (17,643 genes). Student’s t test was performed. Data are represented as mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns: not significant, p > 0.05.
(C) Comparative analysis of gene methylation level and expression level of all genes divided into different gene age groups in P. bursaria. The gene methylation level was calculated by the sum of methylation levels at each site per gene. The expression level was calculated by the TPM methods. Data are represented as mean ± SEM. The detailed information of Tukey’s multiple comparisons test among the gene age groups is shown in Tables S9 and S10. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns: not significant, p > 0.05.
(D) Gene ontology enrichment analysis of unmethylated genes. The R package ggplot2 was performed. The size of dot represents the gene number, and the color bar shows the p value.
(E) The GSEA analysis of 6mA-depleted genes enriched in the ligand-gated ion channel activity pathways.
Considering that 5mC methylation in the gene body is essential for the maintenance of the symbiotic states in the model coral Aiptasia,61 we next investigated whether 6mA is associated with endosymbiosis-correlated genes. Gene Ontology analysis of differentially methylated genes revealed that methylated genes were not enriched in any pathways (p > 0.05) (Figures S8D–S8F). This finding is not surprising as most genes (∼67%) in P. bursaria are methylated with 6mA. In contrast, unmethylated genes (∼33% of all annotated genes) were significantly enriched in pathways such as ion channel activity (p = 3.00E-09), response to mechanism stimulus (p = 1.31E-06), and transmembrane transporter activity (p = 3.64E-06) (Figures 7D and 7E), partial members of which are reported to be involved in endosymbiosis of P. bursaria.62,63 Therefore, we focused on some well-annotated endosymbiosis-related proteins, including seven cation channel family proteins, one globin, and three multidrug and toxin extrusion (MATE) family genes.41 All of these contained little or even no 6mA, but there was no clear trend in their expression level, which was higher in some genes but lower in other genes (Table S11). These results lead us to speculate that 6mA might act as a reverse mark to distinguish the endosymbiosis-related genes.
Discussion
Here we focused on its congener P. bursaria to delineate the features of 6mA and its role in endosymbiosis. Four model ciliates, P. bursaria, P. tetraurelia, Tetrahymena, and Oxytricha, have similar 6mA features including distribution patterns (5′-ApT-3' and 5′ end of gene body around TSS) and correlated factors (Pol II-transcribed genes and gene transcription), but the 6mA ratio in P. bursaria is higher than that in Tetrahymena and Oxytricha.8,11,19 A comparatively high 6mA ratio is likely a common feature of the genus Paramecium, given that the 6mA ratio of its congener P. aurelia has been reported to be ∼2.5% detected by DNA chromatography28 and 1.6% detected by SMRT-seq in P. tetraurelia during late autogamy (T50).34 In the present study, we found that vegetative P. tetraurelia, Paramecium caudatum, and Paramecium multimicronucleatum, each of which was fed with dam-/dcm- Escherichia coli, also had a higher 6mA ratio (2.35%, 1.64%, and 1.66%, respectively) than Tetrahymena (0.54%) and Oxytricha (0.38%) (Figure S9A). P. bursaria has a bimodal distribution of 6mA around TSS, unlike Tetrahymena and Oxytricha both of which have a unimodal distribution downstream of TSS (Figure 3C).8,11,19,34 This bimodal distribution of 6mA was also reported in the green alga Chlamydomonas,4 raising the possibility that the endosymbiotic alga C. variabilis may also exhibit a bimodal distribution of 6mA and in turn affect its host P. bursaria. However, P. tetraurelia without endosymbiotic algae also shows a weak bimodal peak, arguing for the alternative scenario that the bimodal distribution is a feature to the genus Paramecium. Considering that the intergenic region in P. bursaria is considerably shorter than that in Tetrahymena (average 1,184 bp vs. 2,339 bp), i.e., compact genome (Figure S9B), we selected divergently transcribed genes with long intergenic regions (>500 bp and >1,000 bp) to rule out interference from neighboring genes. However, the bimodal distribution of 6mA around TSS still exists (Figure S9C), excluding the possibility that 6mA peaks upstream of TSS are actually the peaks downstream of TSS in adjacent genes. Further studies of more species of Paramecium, especially those without endosymbiotic algae, and of other ciliate species will help to elucidate the phenomenon.
6mA is depleted at the TSS in P. bursaria, P. tetraurelia, Tetrahymena, and Oxytricha,8,11,19,34 which could be attributable to two not mutually exclusive factors. On the one hand, many transcription factors, such as TFIIE and TFIIH of the preinitiation complex (PIC),64 assemble at TSS and might hinder 6mA methyltransferase binding, thus contributing to the absence of 6mA. On the other hand, the intrinsic interactivity between 6mA and the nucleosome may also be an important contributor. In Tetrahymena, as 6mA is preferentially localized in linker DNA regions flanked by well-positioned/H2A.Z-containing nucleosomes,19 the nucleosome-free regions (NFRs) wherein TSS resides are not ideal for the occurrence of 6mA. In Oxytricha, TSS is flanked on only one side by +1 nucleosome as nucleosome upstream TSS is precluded in nanochromosomes, thereby reducing the 6mA at TSS.11 In P. bursaria, TSS was flanked by well-positioned nucleosomes downstream and by a slightly above-background nucleosome upstream; such an imbalance may also result in the absence of 6mA at TSS. Given that 6mA locally affects nucleosome occupancy and stability,8,11,19 we speculate that 6mA may act both as a contributor and as a recipient of the chromatin structure.
AS-generated protein diversity is essential for normal cell development in higher eukaryotes.47,48,65,66,67 It has been reported that RNA m6A plays an important role in mRNA splicing of the Drosophila sex determination gene Sxl68,69 and MyD88 of human dental pulp cells.70 Nuclear m6A reader YTHDC1 has also been implicated in the maintenance of a dynamic interaction network of different families of splicing-related proteins.71 5mC is linked with AS by two different mechanisms. The first is executed by CCCTC-binding factor (CTCF) and methyl-CpG binding protein 2 (MeCP2) to regulate the elongation rate of RNA polymerase II (Pol II); the other maybe involves recruiting splicing factors by the H3K9 trimethylation (H3K9me3) reading protein HP1.66,67 6mA could influence the binding between specific proteins or transcription factors and DNA by steric hindrance, which alters Pol II elongation and downstream splicing. Furthermore, 6mA was reported to be related with H3K4me3 in Tetrahymena,8 the level of the latter being crucial for the recruitment of the early spliceosome.72 Thus, 6mA might regulate splicing by its steric hindrance per se or by the correlation with H3K4me3. In addition, high nucleosome occupancy has been associated with low splicing efficiency in P. tetraurelia.73 The positive correlation between 6mA and nucleosome occupancy in P. bursaria (Figures 2C and 2D) raises the possibility that higher 6mA ratio in intron in company with higher nucleosome occupancy decreases splicing efficiency, thus resulting in IR. Nonetheless, there was no correlation between IR degree (defined as reads corresponding to a retained intron/all reads corresponding to the flanking exons) and either the methylation level or 6mA ratio of introns (r = 0.07; r = 0.07). It suggests that 6mA may contribute to the occurrence of AS, but the retained amount of a particular intron may be co-regulated by other factors.
The previous study showed that not all genes in P. bursaria have the same copy number.74 To confirm whether the gene dosage is related to 6mA levels, we analyzed the copy number of different genes/contigs (Figure S10). Our analysis showed that different genes/contigs had different copy numbers, with a positive correlation between the copy number of two-telomere contigs/chromosomes and their 6mA ratio (r = 0.32) (Figures S10A and S10B). This was also the case for genes longer than 1 kb (r = 0.29; r = 0.29) (Figure S10C). Then we compared the gene copy number between AS genes and non-AS genes. It showed that copy number of AS genes was significantly higher than that in non-AS genes (Figure S10D). Overall, AS genes were related with higher 6mA level and higher copy number. Then, to correct the potential bias, we normalized genes’ 6mA level (both 6mA ratio and 6mA amount) by their own copy number. After normalization, 6mA levels of AS genes and non-AS genes were comparable (Figure S10E, left panel), suggesting that copy number may be a major factor to distinguish these two. However, within AS genes, their retained introns still carried significantly higher 6mA level (p < 0.05) than other introns (Figure S10E, right panel). Therefore, 6mA is indeed related with IR. Similarly, we normalized endosymbiotic-related genes’ 6mA level by copy number. Their normalized 6mA level still was obviously lower than the mean level (Table S11), except for one value (g5940), supporting that 6mA might act as a reverse mark to distinguish endosymbiosis-related genes.
A 6mA methyltransferase in P. bursaria, PbAMT1, belongs to the AMT1 clade that includes 6mA MTase AMT1 in Tetrahymena and MTA1 in Oxytricha8,11 (Figure 5A). Loss of function of these MTases results in severe defects, such as an abnormally large contractile vacuole (CV) both in P. bursaria and Tetrahymena8 and the malfunction of the sexual cycle in Oxytricha.8,11 6mA MTases of multicellular eukaryotes belong to the METTL4 subclade (Figure 5B), including DAMT-1 related with epigenetic inheritance in Caenorhabditis elegans, BmMETTL4 regulating normal proliferation in Bombyx mori, and METTL4 contributing to the attenuation of transcription and copy number in mitochondria.5,44,75 The evolutionary divergence of 6mA MTases may thus be an important force driving the diversification of eukaryotes.
Limitations of the study
The high level of 6mA exists in P. bursaria, but we cannot explain it in this paper. The endosymbiotic alga C. variabilis have a high level of 6mA and a bimodal distribution of 6mA around TSS, raising the possibility that the endosymbiotic alga in turn affects its host P. bursaria. Still, it is also possible that the high level of 6mA and bimodal distribution are the features to the genus Paramecium. Further studies of more species of Paramecium, especially those without endosymbiotic algae, and of other ciliate species will help to elucidate the phenomenon in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-6mA | Synaptic Systems | Cat#202003; RRID:AB_2279214 |
| Goat anti-Rabbit IgG (H+L) | Invitrogen | Cat#A-21428; RRID:AB_2535849 |
| Bacterial and virus strains | ||
| Klebsiella pneumonia | repository | N/A |
| Escherichia coli | repository | HST04 |
| Deposited data | ||
| MNase-seq, RNA-seq, 6mA-IP-seq and Oxford Nanopore Technologies (ONT) data of P. bursaria | This paper | NCBI: PRJNA673334 |
| SMRT-seq and RNA-seq data of P. bursaria | He et al.41 | BIGD: PRJCA001086 |
| RNA-seq dataset of P. bursaria | Kodama and Fujishima76 | NCBI: DRP000940 |
| SMRT-seq of Tetrahymena | Wang et al.57 | NCBI: SRX5993111 |
| SMRT-seq of Oxytricha | Beh et al.11 | NCBI: SRX5944401 |
| SMRT-seq of P. tetraurelia | Hardy et al.34 | ENA: PRJEB40264 |
| RNA-seq of P. tetraurelia | Hardy et al.34 | ENA: ERR1827399 |
| Customized scripts | This paper | https://github.com/Bryan0425/Paramecium-6mA-related |
| Software and algorithms | ||
| RepeatMasker v4.0.9 | Tarailo-Graovac and Chen77 | http://www.repeatmasker.org/RMDownload.html |
| Trimmomatic v0.39 | Bolger et al.78 | http://www.usadellab.org/cms/?page = trimmomatic |
| Augustus v3.3.3 | Stanke et al.79 | https://github.com/Gaius-Augustus/Augustus |
| SMRT link v7.0 | Pacific Biosciences | https://www.pacb.com/support/software-downloads/ |
| R package circlize v0.4.4 | Gu et al.80 | https://jokergoo.github.io/circlize_book/book/ |
| R package ggplot2 | Wickham81 | http://had.co.nz/ggplot2/ |
| WebLogo3 | Crooks et al.82 | https://weblogo.threeplusone.com/ |
| RNAmmer v1.2 | Lagesen et al.83 | https://services.healthtech.dtu.dk/service.php?RNAmmer-1.2 |
| tRNAscan-SE v2.0.5 | Lowe and Eddy84 | http://lowelab.ucsc.edu/tRNAscan-SE/ |
| Rfam v11.0 | Griffiths-Jones et al.85 | ftp://ftp.ebi.ac.uk/pub/databases/Rfam/12.2/Rfam.cm.gz |
| guppy v3.2.10 | Oxford Nanopore Technologies | https://github.com/metagenomics/denbi-nanopore-training/blob/master/docs/basecalling/basecalling.rst |
| Nanofilt v2.5.0 | De Coster et al.86 | https://pypi.org/project/NanoFilt/ |
| Pychopper | Oxford Nanopore Technologies | https://github.com/nanoporetech/pychopper |
| minimap2 v2.16 | Li87 | https://github.com/lh3/minimap2 |
| Pinfish | Oxford Nanopore Technologies | https://github.com/nanoporetech/pinfish |
| Gffcompare | Pertea and Pertea88 | https://github.com/gpertea/gffcompare |
| Tophat2 v2.1.1 | Trapnell et al.89 | http://ccb.jhu.edu/software/tophat/index.shtml |
| cufflinks v2.2.1 | Trapnell et al.89 | https://cole-trapnell-lab.github.io/cufflinks/ |
| ASprofile v1.0.4 | Florea et al.90 | https://ccb.jhu.edu/software/ASprofile/ |
| SQANTI3 v3.0 | Tardaguila et al.91 | https://github.com/ConesaLab/SQANTI3 |
| Cupcake v24.3.0 | Elizabeth Tseng | https://github.com/Magdoll/cDNA_Cupcake |
| FLAIR v1.5 | Tang et al.92 | https://github.com/BrooksLabUCSC/FLAIR |
| eggNOG-mapper | Huerta-Cepas et al.93 | http://eggnog-mapper.embl.de/ |
| GSEA v4.1.0 | Mootha et al.94; Subramanian et al.95 | http://www.gsea-msigdb.org/gsea/index.jsp |
| PSI-BLAST v2.9.0+ | Altschul et al.96 | https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/ |
| CD-HIT | Huang et al.97 | http://cd-hit.org |
| MUSCLE v3.8 | Edgar98 | http://www.drive5.com/muscle/ |
| FastTree v2.1 | Price et al.99 | http://www.microbesonline.org/fasttree/ |
| NCBI Conserved Domain Search | Lu et al.100 | https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml |
| Phylostratigraphic analysis v0.0.5 | Drost et al.101; Domazet-Loso et al.102 | https://github.com/AlexGa/Phylostratigraphy |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yuanyuan Wang (wangyuanyuan@ouc.edu.cn).
Materials availability
-
•
Plasmids generated in this study have been deposited to our lab.
-
•
This study did not generate new unique reagents.
Experimental model and subject details
Cell culture
The Paramecium bursaria 110224 strain used in the present study was initially collected from Zhongshan Park, Qingdao, China (120.20°E, 36.03°N) in 2011 and maintained in the Laboratory of Protozoology at Ocean University of China. It was cultivated at 25 °C in an incubator with constant illumination in wheat grass powder (WGP, Pines International) bacterized with a non-virulent strain of Klebsiella pneumonia. β-Sitosterol (1 mg/L) was added to the bacterized WGP medium before feeding P. bursaria.103 The other three species of genus Paramecium from the Laboratory of Protozoology at Ocean University of China, i.e., P. tetraurelia, P. caudatum and P. multimicronucleatum, which were also used in the present study, were fed with dam-/dcm- Escherichia coli.
Method details
Photomicrographs and silver carbonate staining
Live cells were observed and photographed using differential interference contrast microscopy at 400× to 1,000× magnification. The silver carbonate method104 was used to reveal the infraciliature.
Immunofluorescence staining
Cells were fixed and permeabilized in 5 mL chilled acetone for 5 min before spreading onto polylysine-coated coverslips.42 The 6mA staining experiment followed previously described procedures.19 Antibody information: the primary antibody (α-6mA, Synaptic Systems, 202003, 1:2000); the secondary antibody (Goat anti-Rabbit IgG (H+L), Invitrogen, A-21428, 1:4000).
Gene model and annotation update
SMRT sequencing and RNA-seq data of P. bursaria were downloaded from the public database BIGD.41 Another published RNA-seq dataset of P. bursaria with or without Chlorella variabilis symbionts76 was also used to improve the accuracy of gene model prediction in this study.
To identify protein-coding genes, repeat regions of genomes were recognized and masked by RepeatMasker v4.0.977 with default parameters. All RNA-seq reads were trimmed by Trimmomatic v0.3978 to remove adapters and filter low-quality reads. Subsequently, transcripts were generated by mapping to the masked P. bursaria genome using BLAT (-noHead -stepSize = =5 -minIdentity=93). All RNA-seq data were incorporated into Augustus v3.3.3 scripts by generating the hints file, including intron and exonpart hints and wiggle track. Augustus79 (--species=tetrahymena) was employed for gene model and protein prediction. In all, 17,825 protein-coding genes were predicted with high confidence, 8,220 of which were longer than 1 kb, representing a significant improvement over the published versions (17,825 vs. 17,266; 8,220 vs. 7,654) (Table S12).41
SMRT sequencing data analysis
The latest published MAC genome of P. bursaria downloaded from ParameciumDB (https://paramecium.i2bc.paris-saclay.fr/)105 was used as a reference for subreads mapping (123× available SMRT-seq data). 6mA was called by the SMRT link v7.0 (Pacific Biosciences, Base Modification and Motif Analysis protocol with default parameters). Strict cut-offs (Qv > 30 and coverage > 25× for normalizing to 100×) were used to filter out unauthentic modifications.8
As defined previously,8 6mA was divided into different groups according to their methylation level (0-100%) or motifs (symmetric/ asymmetric/ non-ApT). All 6mA calculations of sites and percentages were implemented by customized Perl scripts and diagrams were plotted by GraphPad Prism 7.106 Methylation level was defined as the ratio between reads counts of methylated adenines (6mA) and total adenines (A) at a specific position (6mA/A). Each 6mA site has its own methylation level. For circle diagrams generated by R package circlize v0.4.4,80 the 6mA ratio was defined as the ratio between the total number of methylated A sites (6mA) and the total number of all A sites in the genome or in a particular genomic sequence (∑6mA/∑A). 6mA amount is the number of 6mA sites in the genome or in a particular genomic sequence (∑6mA), not taking into account the effect of the methylation level of each 6mA site. Neither 6mA amount nor methylation level is related to the ApT frequency of the genome. The density plot of methylation levels on Watson and/or Crick strands was generated by R package ggplot2.81 To analyze the distribution of 6mA, we scaled the gene body length to one unit and extended it to each side for locus statistical analysis by customized Perl scripts (bin size = 0.05). The numbers of 6mA sites were accumulated from 1000-nt upstream to 2000-nt downstream transcription start sites to explore patterns of 6mA distribution.
Conserved motifs, which are sequences from 5-nt upstream to 5-nt downstream of the methylated adenines, were selected and identified by WebLogo3.82
RNA polymerase I (Pol I) and III (Pol III) -transcribed genes were detected in the P. bursaria MAC genome41 using RNAmmer v1.2,83 tRNAscan-SE v2.0.584 and Rfam v11.0,85 as described previously.19
Oxford Nanopore Technologies (ONT) data generation and analysis
Oxford PromethION 2D amplicon libraries were generated according to the Nanopore community protocol using library preparation kit SQK-LSK109 and sequenced on R9 flowcells to generate fast5 files. All generated fast5 reads were then basecalled in guppy v3.2.10 (https://github.com/metagenomics/denbi-nanopore-training/blob/master/docs/basecalling/base calling.rst) with the default options to yield fastq files. The fastq reads for each sample were filtered using Nanofilt v2.5.086 with options -l 100 -q 7. The full-length reads were detected using Pychopper (https://github.com/nanoporetech/pychopper) with options -b primer.fa -i raw.fq -t 200 -s 98, and were aligned to the genome with minimap2 v2.16.87 Pinfish was run with default parameters, yielding polished consensus reads. Structure annotations were performed using Gffcompare88 to identify known and novel transcripts.
Isoform identification and alternative splicing analysis
The alternative splicing (AS) events were predicted using both RNA-seq and the ONT full-length transcript sequencing data of P. bursaria according to the coverage of transcripts to distinguish multiple RNA isoforms from same gene. For illumina sequencing, the raw reads were trimmed and filtered as above. After mapping back to genome using Tophat2 v2.1.1 (--library-type fr-unstranded, --no-discordant),89 the RNA isoforms were reconstructed by cufflinks v2.2.1 (-I 100 --min-intron-length 10 -u --library-type fr-unstranded)89 and all the type of alternative splicing were identified by ASprofile v1.0.4 (-r tmap.file).90 The AS gene list of Tetrahymena was provided by Dr. Jie Xiong (Chinese Academy of Sciences, Wuhan, China).50 Custom scripts uploaded on Github (https://github.com/Bryan0425) were employed to extract and calculate the information. For ONT full-length transcript sequencing, we employed SQANTI3 v3.091 and Cupcake v24.3.0 (https://github.com/Magdoll/cDNA_Cupcake) with default parameters to identify isoforms and classify AS events. FLAIR v1.592 (the process of align, correct, collapse, quantity and diffSplice was run followed its handbook) was employed to analyze the differential isoforms of the AS events between two conditions (control and PbAMT1-KD cells).
To calculate the intron retention degree, the ASprofile as above was used to identify the alternative splicing events which contained the intron retention in control RNA-seq data, the intron retention degree (IRD) was calculated by the formula (IRD = reads corresponding to a retained intron/all reads corresponding to the flanking exons). As for the heatmap of intron retention density, we calculated the normalized FPKM of retained intron to show the variances in both control and PbAMT1-KD cell. The higher intron retention density represented more retained introns.
Gene ontology annotation and gene set enrichment analysis
Gene Ontology (GO) enrichment analysis of all predicted genes was implemented by the eggNOG-mapper with default parameters93 and 8,992 proteins were annotated into 4,686 GO pathways. The gene set enrichment analysis was performed by GSEA v4.1.0.94,95 By calculating the gene methylation level defined as the sum of the methylation levels of each 6mA site on the gene body of a particular gene (∑ (6mA/total A), we transformed the gene methylation level into Gaussian distribution by log2 and the mean (μ) of the distribution was determined. Then we constructed the gct file by the observed value and the mean methylation level per gene. The results of GSEA analysis were narrowed down by P-value < 0.05 and FDR < 25%.
MNase-seq sample preparation and analysis
Approximately 5×105 P. bursaria cells were collected, incubated on ice for 5 min with lysis buffer (0.25 M sucrose, 10 mM MgCl2, 10 mM Tris-HCl pH 7.4, 1× Protease inhibitor, 1 mM dithiothreitol and 0.2% NP-40), and then homogenized with douncer. The lysate was digested by 120 U/mL Micrococcal Nuclease (MNase, NEB, M0247S) at 25 ◦C for 15 min. The mono-nucleosome sized DNA was selected by agarose electrophoresis and purified with Zymoclean gel extraction kit (D4008). After reads mapping,41 only the mono-nucleosome sized fragments (120-260 bp) were analyzed. Nucleosomes dyads were defined as previously reported to calculate nucleosome distribution around TSS.8
Phylogenetic analysis
The putative N6-adenine methyltransferases sequences of Tetrahymena8 were queried against the P. bursaria proteins to identify the putative methyltransferases by PSI-BLAST v2.9.0+ (maximum E-value = 1e-4).26,96 The resulting sequences were added to perform the alignments of MT-A70 candidates from Wang et al, 2019.8 Retrieved hits were collapsed to remove redundant sequences using CD-HIT (-c 0.97)97 and sequences were aligned by the MUSCLE v3.8 program.98 A phylogenetic tree was acquired by FastTree v2.1 program99 with the approximate maximum-likelihood method. All the protein sequences used for phylogenetic analysis is listed in Table S13. NCBI Conserved Domain Search (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml)100 was used to predict the conserved domain of proteins. IBS v1.0107 was used to plot the sketch.
RNAi knockdown
Sequences used for silencing the PbAMT1 gene were amplified from the cDNA of P. bursaria, using primers PbAMT1_f1437_PstI (CTGCAG GTAAATGATGATGCTATACC) and PbAMT1_r1667_KpnI (GGTACC GCATTTTACTTATCATATCG). The amplified fragment was 231 bp in size, complementary to the partial second and third exons of PbAMT1. This fragment was ligated into the pEASY-Blunt cloning vector (TransGen Biotech, M20514) and transformed into Trans1-T1 competent cells (TransGen Biotech, CD501-03). Positive clones were selected for plasmid extraction. The fragment released by PstI (NEB, R3140V) and KpnI (NEB, R3142S) digestion was ligated into the L4440 plasmid by T4 DNA Ligase (NEB, M0202V), which was then transformed into HT115 competent cells (Shanghai Weidi Biotechnology, EC2010). Positive clones were cultured and induced by 0.4 mmol/L isopropyl β-D-thiogalactoside (IPTG) to produce double-stranded RNA (dsRNA) when they reached the logarithmic growth period. The HT115 cells were harvested when grown to an OD600 value of 1. P. bursaria RNAi knockdown cells were fed on the transformed E. coli HT115 with the L4440 plasmid containing the target gene fragment, while control cells were fed on the transformed E. coil HT115 with the L4440 empty vector. Fresh E. coil HT115 was cultured and added to the experimental system every single day. P. bursaria cells were collected for subsequent experiments after three and six days of feeding, respectively.
RNA extraction and cDNA preparation
Total RNA was extracted with the RNeasy Plus Mini kit (Qiagen, 74134) and cDNA was synthesized using a synthetic oligo-dT primer and M-MLV Reverse Transcriptase (Invitrogen, 28025013) after DNase treatment (Invitrogen, AM1907).
Reverse transcription polymerase chain reaction (RT-PCR) and quantitative reverse transcription PCR (qRT-PCR)
RT-PCR was performed using Premix Taq (TaKaRa, RR901A) to confirm the PbAMT1 gene model. For the qRT-PCR analysis of PbAMT1 expression in control and PbAMT1-KD cells, the housekeeping gene GAPDH was used for loading control and normalization (Holding stage: 50 °C, 2 min; 95 °C, 10 min. Cycling stage: 95 °C, 15 s; 50 °C, 2 min; 60 °C, 1 min. Melt curve stage: 95 °C, 15 s; 60 °C, 1 min; 95 °C, 30 s; 60 °C, 15 s.). All PCR primers are listed in Table S14.
DpnI/DpnII digestion and quantitative PCR (qPCR) analysis
Approximately 2 μg purified genomic DNA was treated with 40 U DpnI (NEB, R0176v) overnight or with 40 U DpnII (NEB, R0543L) overnight at 37 °C. The samples were heat-inactivated for 20 min at 80 °C (DpnI) or 65 °C (DpnII) after digestion. A total of 4 ng digested and non-digested DNA was subjected to quantitative PCR (qPCR) analysis using EvaGreen Express 2× qPCR Master Mix-Low ROX (Abm, MasterMix-LR). Primers flanking selected GATC sites were used to validate the methylation status at a specific position (Table S15). Primers matched to the coding DNA sequence (CDS) of GAPDH were used as internal controls. Methylation status are reflected by normalized Ct difference (ΔCt) between digested and undigested samples.
Identification of horizontal gene transfer
P. bursaria horizontal gene transfer (HGT) genes were identified by two steps, similar to the strategy widely used.108,109,110 Firstly, the potential genes were screened by using RBH BLASTP (Reciprocal best hit) against the Chlorella variabilis genes with a high cut-off, i.e., 1e-10 e-value and 30% identity. This step acquired 871 candidates. Secondly, the candidates were BLASTP searched (1e-10) against both genus Paramecium and genus Chlorella in the protein database (NCBI Reference Sequence Database, RefSeq). The retrieved homologs and candidates were aligned by MUSCLE98 and phylogenetic trees were constructed by FastTree.99 Under manual scrutiny, a gene clustered in the Chlorella clade which had a Paramecium outgroup was accepted as an HGT-like gene (159 such genes were recovered).
Gene age estimation and gene methylation level calculation
Gene ages were estimated using the phylostratigraphic approach as previously described by Domazet-Lošo and Tautz.102 Phylostratigraphic analysis v0.0.5101 was employed to trace the evolutionary origin of protein coding genes.102 All genes in P. bursaria were divided into different phylostratigraphic levels (psl), i.e., gene age groups psl1 to psl10 (no genes were divided into psl4, psl7 and psl8 groups) corresponding old-to-young groups. The gene methylation level (the sum of the methylation levels of each 6mA site on the gene body of a particular gene, ∑ (6mA/total A)) and expression level of different gene ages were calculated by customized Perl scripts.
UHPLC-QQQ-MS/MS analysis
The experiment followed previously described procedures.8 DNA was denatured and digested into mononucleotides using a mixture of enzymes, including DNase I (1 U, NEB, M0303L), calf intestinal phosphatase (1 U, NEB, M0290L), and snake venom phosphodiesterase I (0.005 U, Sigma, P4506). Digested DNA was diluted, purified, and analyzed by ultra-high-performance liquid chromatography tandem mass spectrometry (UHPLC-QQQ-MS/MS) on an Acquity BEH C18 column (75 mm × 2.1 mm, 1.7 μm, Waters, MA, USA) using a Xevo TQ-S triple quadrupole mass spectrometer (Waters, Milford, MA, USA).10
6mA immunoprecipitation (6mA-IP)
6mA IP experiment followed previously described procedures.4,111 gDNA was isolated using phenol-chloroform-isoamyl alcohol extraction and digested by RNase A (sigma, R4642). Then gDNA was sonicated to 200-400 bp using Diagenode Bioruptor UCD 300. DNA was denatured at 95 °C and chilled for 10 minutes on ice. A portion of 10 μL DNA was saved as input, i.e., an internal control. The rest of DNA was incubated with α-6mA antibody (Synaptic Systems, 1:500) in 500 mL of 1× IP buffer (50mM Tris-HCl pH7.4, 750mM NaCl, 0.5% Triton X-100 and 20mM EDTA) at 4 °C overnight. Protein A magnetic beads were pre-washed three times using 1 mL of 1× IP buffer, before mixing with DNA-6mA antibody complex at 4 °C. 2h rotation was needed for the sufficient ligation between the complex and beads. After incubation, the beads were washed three times with 1 mL of 1× IP buffer. Then the methylated DNA was eluted twice by 100 μL elution buffer at 4°C for 1 h.4,111 The input and IP DNA were sequenced respectively.
Quantification and statistical analysis
All bioinformatical data are expressed as mean ± SEM, and n represents the number of independent replicates per group, as detailed in each figure legend. qRT-PCR data are shown as mean ± SD. For comparisons between two groups only, Student’s t-test was used. ∗∗∗∗ p < 0.0001; ∗∗∗ p < 0.001; ∗∗ p < 0.01; ∗ p < 0.05; ns: not significant, p > 0.05. For comparisons among multi-groups (gene age part; Tables S9 and S10), Tukey’s multiple comparisons test was used. Correlation calculation was performed using two-tailed Pearson correlation analysis. Statistical testing was performed in GraphPad Prism 8.106
Acknowledgments
The authors would like to thank the following people for assistance with this study: Dr. Jie Xiong (Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China) for sharing the AS gene list of Tetrahymena; Dr. Chundi Wang (Marine College, Shandong University, Weihai, China) for guidance of RNAi experiments; Mr. Ming He (Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China) for sharing PacBio and RNA-seq data; Dr. Jun Wang (Institute of Microbiology, Chinese Academy of Sciences, Beijing, China) for suggestions on ONT data analysis; and Ms. Jinghan Diao (OUC) for English editing. Our special thanks are given to Dr. Weibo Song (OUC) for his kind suggestions during preparation of the manuscript. We also appreciate both the computing resources provided on IEMB-1, a high-performance computing cluster operated by the Institute of Evolution and Marine Biodiversity, and the Center for High Performance Computing and System Simulation, Laoshan Laboratory (Qingdao).
This work was supported by the National Natural Science Foundation of China (32125006, 32070437, 32200399), Natural Science Foundation of Shandong Province of China (ZR2021QC046), and Laoshan Laboratory (LSKJ202203203).
Author contributions
B.P., Y.W., and S.G. conceived the study; B.P., F.Y., T.L., and F.W. performed the experiments; B.P. and F.Y. performed the bioinformatic analysis; B.P., Y.W., and S.G. wrote the paper. A.W. edited the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. We avoided “helicopter science” practices by including the participating local contributors from the region where we conducted the research as authors on the paper.
Published: April 14, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106676.
Supplemental information
Data and code availability
-
•
All sequencing data, including MNase-seq, 6mA-IP-seq, RNA-seq and ONT (Oxford Nanopore Technologies) data, generated in this study have been deposited to NCBI: PRJNA673334. SMRT-seq and RNA-seq data of Paramecium bursaria were downloaded from the public database BIGD (http://bigd.big.ac.cn, BioProject accession: PRJCA001086).41 Another published RNA-seq dataset of P. bursaria with or without Chlorella variabilis symbionts (NCBI BioProject accession: DRP000940)76 was used to improve the prediction accuracy. SMRT-seq of Tetrahymena and Oxytricha were downloaded from the public NCBI dataset SRX5993111 and SRX5944401.8,11 SMRT-seq and RNA-seq data of Paramecium tetraurelia were downloaded from the ENA dataset PRJEB40264 and ERR1827399.34
-
•
All original code has been deposited at Github and is publicly available as of the date of publication (https://github.com/Bryan0425/Paramecium-6mA-related).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Luo G.-Z., Blanco M.A., Greer E.L., He C., Shi Y. DNA N6-methyladenine: a new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol. 2015;16:705–710. doi: 10.1038/nrm4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasu K., Nagaraja V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 2013;77:53–72. doi: 10.1128/mmbr.00044-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wion D., Casadesús J. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 2006;4:183–192. doi: 10.1038/nrmicro1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu Y., Luo G.-Z., Chen K., Deng X., Yu M., Han D., Hao Z., Liu J., Lu X., Doré L.C., et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell. 2015;161:879–892. doi: 10.1016/j.cell.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greer E.L., Blanco M.A., Gu L., Sendinc E., Liu J., Aristizábal-Corrales D., Hsu C.-H., Aravind L., He C., Shi Y. DNA methylation on N6-adenine in C. elegans. Cell. 2015;161:868–878. doi: 10.1016/j.cell.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang Z., Shen L., Cui X., Bao S., Geng Y., Yu G., Liang F., Xie S., Lu T., Gu X., Yu H. DNA N6-adenine methylation in Arabidopsis thaliana. Dev. Cell. 2018;45:406–416.e3. doi: 10.1016/j.devcel.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 7.Mondo S.J., Dannebaum R.O., Kuo R.C., Louie K.B., Bewick A.J., LaButti K., Haridas S., Kuo A., Salamov A., Ahrendt S.R., et al. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 2017;49:964–968. doi: 10.1038/ng.3859. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y., Sheng Y., Liu Y., Zhang W., Cheng T., Duan L., Pan B., Qiao Y., Liu Y., Gao S. A distinct class of eukaryotic MT-A70 methyltransferases maintain symmetric DNA N6-adenine methylation at the ApT dinucleotides as an epigenetic mark associated with transcription. Nucleic Acids Res. 2019;47:11771–11789. doi: 10.1093/nar/gkz1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao C.-L., Zhu S., He M., Chen D., Zhang Q., Chen Y., Yu G., Liu J., Xie S.-Q., Luo F., et al. N6-methyladenine DNA modification in the human genome. Mol. Cell. 2018;71:306–318.e7. doi: 10.1016/j.molcel.2018.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Zhang G., Huang H., Liu D., Cheng Y., Liu X., Zhang W., Yin R., Zhang D., Zhang P., Liu J., et al. N6-methyladenine DNA modification in Drosophila. Cell. 2015;161:893–906. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 11.Beh L.Y., Debelouchina G.T., Clay D.M., Thompson R.E., Lindblad K.A., Hutton E.R., Bracht J.R., Sebra R.P., Muir T.W., Landweber L.F. Identification of a DNA N6-adenine methyltransferase complex and its impact on chromatin organization. Cell. 2019;177:1781–1796.e25. doi: 10.1016/j.cell.2019.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Z., Zha L., Ma X., Xu J., Huang D., Wu W., Chen L., Yang F., Liao W., Wang W. Group-specific functional patterns of mitochondrion-related organelles shed light on their multiple transitions from mitochondria in ciliated protists. Mar. Life Sci. Technol. 2022;200:609–617. doi: 10.1007/s42995-022-00147-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao F., Warren A., Zhang Q., Gong J., Miao M., Sun P., Xu D., Huang J., Yi Z., Song W. The all-data-based evolutionary hypothesis of ciliated protists with a revised classification of the Phylum Ciliophora (Eukaryota, Alveolata) Sci. Rep. 2016;6:24874. doi: 10.1038/srep24874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan B., Chen X., Hou L., Zhang Q., Qu Z., Warren A., Miao M. Comparative genomics analysis of ciliates provides insights on the evolutionary history within "Nassophorea-Synhymenia-Phyllopharyngea" assemblage. Front. Microbiol. 2019;10:2819. doi: 10.3389/fmicb.2019.02819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian M., Cai X., Liu Y., Liucong M., Howard-Till R. A practical reference for studying meiosis in the model ciliate Tetrahymena thermophila. Mar. Life Sci. Technol. 2022;4:595–608. doi: 10.1007/s42995-022-00149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C., Solberg T., Maurer-Alcalá X.X., Swart E.C., Gao F., Nowacki M. A small RNA-guided PRC2 complex eliminates DNA as an extreme form of transposon silencing. Cell Rep. 2022;40:111263. doi: 10.1016/j.celrep.2022.111263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei F., Pan B., Diao J., Wang Y., Sheng Y., Gao S. The micronuclear histone H3 clipping in the unicellular eukaryote Tetrahymena thermophila. Mar. Life Sci. Technol. 2022;4:584–594. doi: 10.1007/s42995-022-00151-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao L., Gao F., Gao S., Liang Y., Long H., Lv Z., Su Y., Ye N., Zhang L., Zhao C., et al. Biodiversity-based development and evolution: the emerging research systems in model and non-model organisms. Sci. China Life Sci. 2021;64:1236–1280. doi: 10.1007/s11427-020-1915-y. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y., Chen X., Sheng Y., Liu Y., Gao S. N6-adenine DNA methylation is associated with the linker DNA of H2A.Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena. Nucleic Acids Res. 2017;45:11594–11606. doi: 10.1093/nar/gkx883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu T.P., Wang T., Seetin M.G., Lai Y., Zhu S., Lin K., Liu Y., Byrum S.D., Mackintosh S.G., Zhong M., et al. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature. 2016;532:329–333. doi: 10.1038/nature17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X., Lai W., Li Y., Chen S., Liu B., Zhang N., Mo J., Lyu C., Zheng J., Du Y.-R., et al. N6-methyladenine is incorporated into mammalian genome by DNA polymerase. Cell Res. 2021;31:94–97. doi: 10.1038/s41422-020-0317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musheev M.U., Baumgärtner A., Krebs L., Niehrs C. The origin of genomic N6-methyl-deoxyadenosine in mammalian cells. Nat. Chem. Biol. 2020;16:630–634. doi: 10.1038/s41589-020-0504-2. [DOI] [PubMed] [Google Scholar]
- 23.O’Brown Z.K., Boulias K., Wang J., Wang S.Y., O’Brown N.M., Hao Z., Shibuya H., Fady P.-E., Shi Y., He C., et al. Sources of artifact in measurements of 6mA and 4mC abundance in eukaryotic genomic DNA. BMC Genom. 2019;20:445. doi: 10.1186/s12864-019-5754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Douvlataniotis K., Bensberg M., Lentini A., Gylemo B., Nestor C.E. No evidence for DNA N6-methyladenine in mammals. Sci. Adv. 2020;6:eaay3335. doi: 10.1126/sciadv.aay3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong Y., Cao L., Deikus G., Fan Y., Mead E.A., Lai W., Zhang Y., Yong R., Sebra R., Wang H., et al. Critical assessment of DNA adenine methylation in eukaryotes using quantitative deconvolution. Science. 2022;375:515–522. doi: 10.1126/science.abe7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiffers S., Ebert C., Rahimoff R., Kosmatchev O., Steinbacher J., Bohne A.-V., Spada F., Michalakis S., Nickelsen J., Müller M., Carell T. Quantitative LC-MS provides no evidence for m6da or m4dc in the genome of mouse embryonic stem cells and tissues. Angew. Chem. 2017;56:11268–11271. doi: 10.1002/anie.201700424. [DOI] [PubMed] [Google Scholar]
- 27.Bracht J.R., Perlman D.H., Landweber L.F. Cytosine methylation and hydroxymethylation mark DNA for elimination in Oxytricha trifallax. Genome Biol. 2012;13:R99. doi: 10.1186/gb-2012-13-10-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cummings D.J., Tait A., Goddard J.M. Methylated bases in DNA from Paramecium aurelia. Biochim. Biophys. Acta. 1974;374:1–11. doi: 10.1016/0005-2787(74)90194-4. [DOI] [PubMed] [Google Scholar]
- 29.Gorovsky M.A., Hattman S., Pleger G.L. [6N] methyl adenine in the nuclear DNA of a eucaryote, Tetrahymena pyriformis. J. Cell Biol. 1973;56:697–701. doi: 10.1083/jcb.56.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palacios G., Martin-Gonzalez A., Gutierrez J.C. Macronuclear DNA demethylation is involved in the encystment process of the ciliate Colpoda inflata. Cell Biol. Int. 1994;18:223–228. doi: 10.1006/cbir.1994.1067. [DOI] [PubMed] [Google Scholar]
- 31.Pratt K., Hattman S. Deoxyribonucleic acid methylation and chromatin organization in Tetrahymena thermophila. Mol. Cell Biol. 1981;1:600–608. doi: 10.1128/mcb.1.7.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rae P.M., Steele R.E. Modified bases in the DNAs of unicellular eukaryotes: an examination of distributions and possible roles, with emphasis on hydroxymethyluracil in dinoflagellates. BioSyst. 1978;10:37–53. doi: 10.1016/0303-2647(78)90027-8. [DOI] [PubMed] [Google Scholar]
- 33.Salvini M., Barone E., Ronca S., Nobili R. DNA methylation in vegetative and conjugating cells of a protozoan ciliate: Blepharisma japonicum. Dev. Genet. 1986;7:149–158. doi: 10.1002/dvg.1020070304. [DOI] [Google Scholar]
- 34.Hardy A., Matelot M., Touzeau A., Klopp C., Lopez-Roques C., Duharcourt S., Defrance M. DNAModAnnot: a R toolbox for DNA modification filtering and annotation. Bioinformatics. 2021;37:2738–2740. doi: 10.1093/bioinformatics/btab032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo G.-Z., Hao Z., Luo L., Shen M., Sparvoli D., Zheng Y., Zhang Z., Weng X., Chen K., Cui Q., et al. N6-methyldeoxyadenosine directs nucleosome positioning in Tetrahymena DNA. Genome Biol. 2018;19:200. doi: 10.1186/s13059-018-1573-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen X., Wang C., Pan B., Lu B., Li C., Shen Z., Warren A., Li L. Single-cell genomic sequencing of three Peritrichs (Protista, Ciliophora) reveals less biased stop codon usage and more prevalent programmed ribosomal frameshifting than in other ciliates. Front. Mar. Sci. 2020;7 doi: 10.3389/fmars.2020.602323. [DOI] [Google Scholar]
- 37.Zhao X., Li Y., Duan L., Chen X., Mao F., Juma M., Liu Y., Song W., Gao S. Functional analysis of the methyltransferase SMYD in the single-cell model organism Tetrahymena thermophila. Mar. Life Sci. Technol. 2020;2:109–122. doi: 10.1007/s42995-019-00025-y. [DOI] [Google Scholar]
- 38.Zheng W., Wang C., Lynch M., Gao S. The compact macronuclear genome of the ciliate Halteria grandinella: a transcriptome-Like genome with 23,000 nanochromosomes. mBio. 2021;12:e01964. doi: 10.1128/mBio.01964-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu J., Chi Y., Lu X., Gao F., Al-Farraj S.A., Petroni G., Jiang J. Doublets of the unicellular organism Euplotes vannus (Alveolata, Ciliophora, Euplotida): the morphogenetic patterns of the ciliary and nuclear apparatuses associated with cell division. Mar. Life Sci. Technol. 2022;4:527–535. doi: 10.1007/s42995-022-00150-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma M., Li Y., Maurer-Alcalá X.X., Wang Y., Yan Y. Deciphering phylogenetic relationships in class Karyorelictea (Protista, Ciliophora) based on updated multi-gene information with establishment of a new order Wilbertomorphida n. Mol. Phylogenet. Evol. 2022;169:107406. doi: 10.1016/j.ympev.2022.107406. [DOI] [PubMed] [Google Scholar]
- 41.He M., Wang J., Fan X., Liu X., Shi W., Huang N., Zhao F., Miao M. Genetic basis for the establishment of endosymbiosis in Paramecium. ISME J. 2019;13:1360–1369. doi: 10.1038/s41396-018-0341-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J., Nie Y., Dang X., Liang A., Chai B., Wang W. Characterization of a Rab11 homologue, EoRab11a, in Euplotes octocarinatus. FEMS Microbiol. Lett. 2009;292:222–230. doi: 10.1111/j.1574-6968.2009.01485.x. [DOI] [PubMed] [Google Scholar]
- 43.Sheng Y., Duan L., Cheng T., Qiao Y., Stover N.A., Gao S. The completed macronuclear genome of a model ciliate Tetrahymena thermophila and its application in genome scrambling and copy number analyses. Sci. China Life Sci. 2020;63:1534–1542. doi: 10.1007/s11427-020-1689-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao Z., Wu T., Cui X., Zhu P., Tan C., Dou X., Hsu K.-W., Lin Y.-T., Peng P.-H., Zhang L.-S., et al. N6-deoxyadenosine methylation in mammalian mitochondrial DNA. Mol. Cell. 2020;78:382–395.e8. doi: 10.1016/j.molcel.2020.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fusté J.M., Wanrooij S., Jemt E., Granycome C.E., Cluett T.J., Shi Y., Atanassova N., Holt I.J., Gustafsson C.M., Falkenberg M. Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol. Cell. 2010;37:67–78. doi: 10.1016/j.molcel.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 46.Vovis G.F., Lacks S. Complementary action of restriction enzymes endo R · DpnI and endo R · DpnII on bacteriophage f1 DNA. J. Mol. Biol. 1977;115:525–538. doi: 10.1016/0022-2836(77)90169-3. [DOI] [PubMed] [Google Scholar]
- 47.Jaillon O., Bouhouche K., Gout J.F., Aury J.M., Noel B., Saudemont B., Nowacki M., Serrano V., Porcel B.M., Ségurens B., et al. Translational control of intron splicing in eukaryotes. Nature. 2008;451:359–362. doi: 10.1038/nature06495. [DOI] [PubMed] [Google Scholar]
- 48.Saudemont B., Popa A., Parmley J.L., Rocher V., Blugeon C., Necsulea A., Meyer E., Duret L. The fitness cost of mis-splicing is the main determinant of alternative splicing patterns. Genome Biol. 2017;18:208. doi: 10.1186/s13059-017-1344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Black D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 50.Xiong J., Lu X., Zhou Z., Chang Y., Yuan D., Tian M., Zhou Z., Wang L., Fu C., Orias E., Miao W. Transcriptome analysis of the model protozoan, Tetrahymena thermophila, using deep RNA sequencing. PLoS One. 2012;7:e30630. doi: 10.1371/journal.pone.0030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berget S.M. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- 52.Elango N., Yi S.V. DNA methylation and structural and functional bimodality of vertebrate promoters. Mol. Biol. Evol. 2008;25:1602–1608. doi: 10.1093/molbev/msn110. [DOI] [PubMed] [Google Scholar]
- 53.Flores K., Wolschin F., Corneveaux J.J., Allen A.N., Huentelman M.J., Amdam G.V. Genome-wide association between DNA methylation and alternative splicing in an invertebrate. BMC Genom. 2012;13:480. doi: 10.1186/1471-2164-13-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lyko F., Foret S., Kucharski R., Wolf S., Falckenhayn C., Maleszka R. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 2010;8:e1000506. doi: 10.1371/journal.pbio.1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iyer L.M., Abhiman S., Aravind L. In: Progress in Molecular Biology and Translational Science. Cheng X., Blumenthal R.M., editors. Academic Press; 2011. Chapter 2 - Natural history of eukaryotic DNA methylation systems; pp. 25–104. [DOI] [PubMed] [Google Scholar]
- 56.Iyer L.M., Zhang D., Aravind L. Adenine methylation in eukaryotes: apprehending the complex evolutionary history and functional potential of an epigenetic modification. Bioessays. 2016;38:27–40. doi: 10.1002/bies.201500104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang P., Doxtader K.A., Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell. 2016;63:306–317. doi: 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Richards E.J., Elgin S.C.R. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002;108:489–500. doi: 10.1016/S0092-8674(02)00644-X. [DOI] [PubMed] [Google Scholar]
- 59.Van Driel R., Fransz P. Nuclear architecture and genome functioning in plants and animals: what can we learn from both? Exp. Cell Res. 2004;296:86–90. doi: 10.1016/j.yexcr.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 60.Van Etten J.L., Burbank D.E., Schuster A.M., Meints R.H. Lytic viruses infecting a chlorella-like alga. Virology. 1985;140:135–143. doi: 10.1016/0042-6822(85)90452-0. [DOI] [PubMed] [Google Scholar]
- 61.Li Y., Liew Y.J., Cui G., Cziesielski M.J., Zahran N., Michell C.T., Voolstra C.R., Aranda M. DNA methylation regulates transcriptional homeostasis of algal endosymbiosis in the coral model Aiptasia. Sci. Adv. 2018;4:eaat2142. doi: 10.1126/sciadv.aat2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farhat N., Rabhi M., Krol M., Barhoumi Z., Ivanov A.G., McCarthy A., Abdelly C., Smaoui A., Hüner N.P.A. Starch and sugar accumulation in Sulla carnosa leaves upon Mg2+ starvation. Acta Physiol. Plant. 2014;36:2157–2165. doi: 10.1007/s11738-014-1592-y. [DOI] [Google Scholar]
- 63.Lowe C.D., Minter E.J., Cameron D.D., Brockhurst M.A. Shining a light on exploitative host control in a photosynthetic endosymbiosis. Curr. Biol. 2016;26:207–211. doi: 10.1016/j.cub.2015.11.052. [DOI] [PubMed] [Google Scholar]
- 64.Chen H.T., Warfield L., Hahn S. The positions of TFIIF and TFIIE in the RNA polymerase II transcription preinitiation complex. Nat. Struct. Mol. Biol. 2007;14:696–703. doi: 10.1038/nsmb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iñiguez L.P., Ramírez M., Barbazuk W.B., Hernández G. Identification and analysis of alternative splicing events in Phaseolus vulgaris and Glycine max. BMC Genom. 2017;18:650. doi: 10.1186/s12864-017-4054-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lev Maor G., Yearim A., Ast G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015;31:274–280. doi: 10.1016/j.tig.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 67.Luco R.F., Allo M., Schor I.E., Kornblihtt A.R., Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haussmann I.U., Bodi Z., Sanchez-Moran E., Mongan N.P., Archer N., Fray R.G., Soller M. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–304. doi: 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- 69.Lence T., Akhtar J., Bayer M., Schmid K., Spindler L., Ho C.H., Kreim N., Andrade-Navarro M.A., Poeck B., Helm M., Roignant J.-Y. m6A modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540:242–247. doi: 10.1038/nature20568. [DOI] [PubMed] [Google Scholar]
- 70.Feng Z., Li Q., Meng R., Yi B., Xu Q. METTL3 regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J. Cell Mol. Med. 2018;22:2558–2568. doi: 10.1111/jcmm.13491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiao W., Adhikari S., Dahal U., Chen Y.S., Hao Y.J., Sun B.F., Sun H.Y., Li A., Ping X.L., Lai W.Y., et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 72.Sims R.J., Millhouse S., Chen C.-F., Lewis B.A., Erdjument-Bromage H., Tempst P., Manley J.L., Reinberg D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell. 2007;28:665–676. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gnan S., Matelot M., Weiman M., Arnaiz O., Guérin F., Sperling L., Bétermier M., Thermes C., Chen C.L., Duharcourt S. GC content, but not nucleosome positioning, directly contributes to intron splicing efficiency in Paramecium. Genome Res. 2022;32:699–709. doi: 10.1101/gr.276125.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheng Y.H., Liu C.F.J., Yu Y.H., Jhou Y.T., Fujishima M., Tsai I.J., Leu J.Y. Genome plasticity in Paramecium bursaria revealed by population genomics. BMC Biol. 2020;18:180. doi: 10.1186/s12915-020-00912-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li B., Wang X., Li Z., Lu C., Zhang Q., Chang L., Li W., Cheng T., Xia Q., Zhao P. Transcriptome-wide analysis of N6-methyladenosine uncovers its regulatory role in gene expression in the lepidopteran Bombyx mori. Insect Mol. Biol. 2019;28:703–715. doi: 10.1111/imb.12584. [DOI] [PubMed] [Google Scholar]
- 76.Kodama Y., Fujishima M. Synchronous induction of detachment and reattachment of symbiotic Chlorella spp. from the cell cortex of the host Paramecium bursaria. Protist. 2013;164:660–672. doi: 10.1016/j.protis.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 77.Tarailo-Graovac M., Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. 2009;4:4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 78.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gu Z., Gu L., Eils R., Schlesner M., Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 81.Wickham H. ggplot2. WIREs. Comp. Stat. 2011;3:180–185. doi: 10.1002/wics.147. [DOI] [Google Scholar]
- 82.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lagesen K., Hallin P., Rødland E.A., Stærfeldt H.-H., Rognes T., Ussery D.W. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Griffiths-Jones S., Moxon S., Marshall M., Khanna A., Eddy S.R., Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.De Coster W., D’Hert S., Schultz D.T., Cruts M., Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pertea G., Pertea M. GFF utilities: GffRead and GffCompare. F1000Res. 2020;9 doi: 10.12688/f1000research.23297.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Florea L., Song L., Salzberg S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res. 2013;2:188. doi: 10.12688/f1000research.2-188.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tardaguila M., de la Fuente L., Marti C., Pereira C., Pardo-Palacios F.J., Del Risco H., Ferrell M., Mellado M., Macchietto M., Verheggen K., et al. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 2018;28:396–411. doi: 10.1101/gr.222976.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tang A.D., Soulette C.M., van Baren M.J., Hart K., Hrabeta-Robinson E., Wu C.J., Brooks A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020;11:1438. doi: 10.1038/s41467-020-15171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huerta-Cepas J., Forslund K., Coelho L.P., Szklarczyk D., Jensen L.J., von Mering C., Bork P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017;34:2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mootha V.K., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 95.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang Y., Niu B., Gao Y., Fu L., Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Price M.N., Dehal P.S., Arkin A.P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lu S., Wang J., Chitsaz F., Derbyshire M.K., Geer R.C., Gonzales N.R., Gwadz M., Hurwitz D.I., Marchler G.H., Song J.S., et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48:D265–D268. doi: 10.1093/nar/gkz991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Drost H.-G., Gabel A., Grosse I., Quint M. Evidence for active maintenance of phylotranscriptomic hourglass patterns in animal and plant embryogenesis. Mol. Biol. Evol. 2015;32:1221–1231. doi: 10.1093/molbev/msv012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Domazet-Loso T., Brajković J., Tautz D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007;23:533–539. doi: 10.1016/j.tig.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 103.Beisson J., Bétermier M., Bré M.H., Cohen J., Duharcourt S., Duret L., Kung C., Malinsky S., Meyer E., Preer J.R., Jr., Sperling L. Mass culture of Paramecium tetraurelia. Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.prot5362. pdb.prot5362. [DOI] [PubMed] [Google Scholar]
- 104.Ma H., Choi J.K., Song W. An improved silver carbonate impregnation for marine ciliated protozoa. Acta Protozool. 2003;42:161–164. [Google Scholar]
- 105.Arnaiz O., Meyer E., Sperling L. ParameciumDB 2019: integrating genomic data across the genus for functional and evolutionary biology. Nucleic Acids Res. 2020;48:D599–D605. doi: 10.1093/nar/gkz948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Swift M.L. GraphPad prism, data analysis, and scientific graphing. J. Chem. Inf. Comput. Sci. 1997;37:411–412. doi: 10.1021/ci960402j. [DOI] [Google Scholar]
- 107.Liu W., Xie Y., Ma J., Luo X., Nie P., Zuo Z., Lahrmann U., Zhao Q., Zheng Y., Zhao Y., et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31:3359–3361. doi: 10.1093/bioinformatics/btv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li Z.-W., Shen Y.-H., Xiang Z.-H., Zhang Z. Pathogen-origin horizontally transferred genes contribute to the evolution of Lepidopteran insects. BMC Evol. Biol. 2011;11:356. doi: 10.1186/1471-2148-11-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ricard G., McEwan N.R., Dutilh B.E., Jouany J.-P., Macheboeuf D., Mitsumori M., McIntosh F.M., Michalowski T., Nagamine T., Nelson N., et al. Horizontal gene transfer from Bacteria to rumen Ciliates indicates adaptation to their anaerobic, carbohydrates-rich environment. BMC Genom. 2006;7:22. doi: 10.1186/1471-2164-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xiong J., Wang G., Cheng J., Tian M., Pan X., Warren A., Jiang C., Yuan D., Miao W. Genome of the facultative scuticociliatosis pathogen Pseudocohnilembus persalinus provides insight into its virulence through horizontal gene transfer. Sci. Rep. 2015;5:15470. doi: 10.1038/srep15470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dominissini D., Moshitch-Moshkovitz S., Salmon-Divon M., Amariglio N., Rechavi G. Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013;8:176–189. doi: 10.1038/nprot.2012.148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All sequencing data, including MNase-seq, 6mA-IP-seq, RNA-seq and ONT (Oxford Nanopore Technologies) data, generated in this study have been deposited to NCBI: PRJNA673334. SMRT-seq and RNA-seq data of Paramecium bursaria were downloaded from the public database BIGD (http://bigd.big.ac.cn, BioProject accession: PRJCA001086).41 Another published RNA-seq dataset of P. bursaria with or without Chlorella variabilis symbionts (NCBI BioProject accession: DRP000940)76 was used to improve the prediction accuracy. SMRT-seq of Tetrahymena and Oxytricha were downloaded from the public NCBI dataset SRX5993111 and SRX5944401.8,11 SMRT-seq and RNA-seq data of Paramecium tetraurelia were downloaded from the ENA dataset PRJEB40264 and ERR1827399.34
-
•
All original code has been deposited at Github and is publicly available as of the date of publication (https://github.com/Bryan0425/Paramecium-6mA-related).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.