

Summary
Autophagy represents a key regulator of aging and metabolism in sensing energy deprivation. We find that fasting in mice activates autophagy in the liver paralleled by activation of hypothalamic AgRP neurons. Optogenetic and chemogenetic activation of AgRP neurons induces autophagy, alters phosphorylation of autophagy regulators, and promotes ketogenesis. AgRP neuron-dependent induction of liver autophagy relies on NPY release in the paraventricular nucleus of the hypothalamus (PVH) via presynaptic inhibition of NPY1R-expressing neurons to activate PVHCRH neurons. Conversely, inhibiting AgRP neurons during energy deprivation abrogates induction of hepatic autophagy and rewiring of metabolism. AgRP neuron activation increases circulating corticosterone concentrations, and reduction of hepatic glucocorticoid receptor expression attenuates AgRP neuron-dependent activation of hepatic autophagy. Collectively, our study reveals a fundamental regulatory principle of liver autophagy in control of metabolic adaptation during nutrient deprivation.
Keywords: AgRP neurons, autophagy, hypothalamus, non-cell autonomous, short-term fasting, liver metabolism, HPA axis, corticosterone, CRH neurons, NPY1R
Graphical abstract
Highlights
-
•
AgRP-mediated activation of hepatic autophagy flux requires NPY expression
-
•
AgRP → PVH neurocircuitry regulates HPA axis and systemic corticosterone levels
-
•
Inhibition of AgRP activity during a short-term fast blunts hepatic autophagy flux
-
•
Aging impairs fasting-induced HPA axis activation and liver autophagosome formation
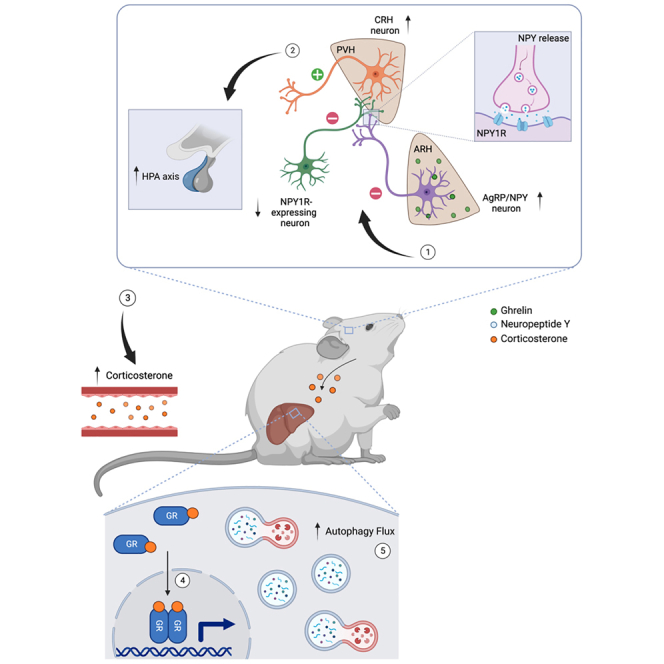
Chen et al. investigate the role of hypothalamic AgRP neurons in the adaptation of liver physiology during energy deprivation. During a short-term fast, AgRP neurons release NPY and engage a downstream NPY1R neurocircuitry to activate PVHCRH neurons. This activates the HPA axis and glucocorticoid release, which ultimately exert top-down control of hepatic autophagy and rewiring of metabolism, providing adaptation to the negative energy state.
Introduction
The hypothalamus represents a key center in the regulation of fundamental homeostatic responses such as feeding, drinking, and temperature regulation. To adapt energy homeostasis according to the energy state and fuel availability of the organism, highly specialized neurons receive hormonal, nutritional, and neuronal inputs, which instruct them about the energy state of the organism.2 Specifically, neurons in the arcuate nucleus of the hypothalamus (ARH) exhibit a privileged localization of an incomplete blood-brain barrier, allowing them to sensitively detect signals originating from circulation. Here, two anatomically neighboring and functionally distinct neuronal populations, namely the agouti-related peptide (AgRP)-expressing and pro-opiomelanocortin (POMC)-expressing neurons reside, which are critical for sensing and integrating metabolic cues to regulate not only food intake and energy expenditure but also systemic glucose homeostasis and proteostasis.3 Selective ablation of AgRP neurons in adult mice reduces fodd intake,4,5 while stimulating AgRP neurons optogenetically and chemogenetically provokes compulsive food seeking and feeding behavior.6,7 In addition to the adaptive feeding responses, the activity of AgRP neurons also coordinates additional autonomic and behavioral responses in concert with the energy availability of the organism. This has been exemplified in the CNS-dependent regulation of glucose homeostasis. Mice with selective inactivation of the insulin receptor in AgRP neurons present with an attenuated ability of insulin to suppress the hepatic glucose production.8 Moreover, acute optogenetic and chemogenetic stimulation of AgRP neurons reduces peripheral insulin sensitivity in part via the inhibition of glucose metabolism in brown adipose tissue (BAT).9 In line with their ability to integrate peripheral signals and adapt their electrical activity according to energy availability, sensory food perception has been implicated in the transient and rapid inhibition of AgRP neurons.10 In contrast, POMC neurons are activated with sensory food perception to regulate liver proteostasis in anticipation of the postprandial state.11 Collectively, these studies have contributed to a holistic view that the melanocortin circuitry contributes to the adaptation of integrative physiology in accordance with actual and even anticipated changes in the metabolic state of the organism.
This coordinated regulation of energy homeostasis and multiple integrated physiological responses through melanocortin neurons is of importance for the survival of the organism, as starvation represents a life-threatening challenge to the organism. Here, AgRP neuron-induced feeding supports systemic survival mechanisms. However, in the absence of food, additional cell-autonomous and systemic mechanisms are activated to compensate for the limited food supply. A major evolutionary conserved pathway in this regulation is autophagy. Traditionally, autophagy is viewed as a cell-autonomous process that targets cytosolic components, such as proteins, lipids, and nucleic acids to lysosomes for the degradation to maintain cellular homeostasis and provide metabolic substrates for energy production during nutrient deprivation.12 Induction of autophagy requires the de novo formation of double-membrane vesicles, termed autophagosomes, that engulf and target cargo for the degradation upon fusing with the lysosome.13 The initiation of autophagy is tightly controlled by the antagonistic kinases Ulk-1 and mammalian target of rapamycin (mTOR). Nutrient starvation induces the activation of 5' adenosine monophosphate-activated protein kinase (AMPK), which in turn phosphorylates and activates Ulk-1 to activate autophagy.14 LC3-II is recognized by autophagy receptors, such as p62, which is linked to targeted cargo for degradation. Elongated autophagosomes undergo further functional maturation to finally fuse with lysosomes to promote cargo degradation. Beyond its energy-regulating role, autophagy plays a critical role in protein quality control, subjecting misfolded proteins and/or damaged organelles to lysosomal degradation. Here, altered autophagy has been closely linked to aging and the onset of multiple aging-associated diseases, including metabolic disorders.12,15
Autophagy thus plays a critical role in the control of multiple metabolic processes. Glycogen accumulation under conditions of impaired autophagy contributes to age-associated diseases. Moreover, in response to starvation, lipid droplets can undergo selective degradation by autophagy, providing an alternative mechanism to lipase-mediated liberation of fatty acids for energy supply.16 In addition to lipophagy, chaperone-mediated autophagy promotes the degradation of lipid droplet-associated proteins Perilipin (Plin)-2 and -3, and an age-dependent decline in liver autophagy has been linked to hepatic steatosis.17 Moreover, autophagy-mediated degradation of the nuclear co-repressor (NCoR1) promotes PPARα activation in the control of hepatic lipid homeostasis, and peroxisome proliferator-activated receptor alpha (PPARα) activation in turn has been shown to promote autophagy.18 Thus, intense bi-directional interactions of autophagy and lipid metabolism control metabolic homeostasis, aging, and the onset of metabolic disorders.
Of note, the induction of autophagy in AgRP and POMC neurons has been linked to their energy state-dependent regulation. Starvation induces autophagy in AgRP neurons to mobilize lipids to generate fatty acids and consequently augment AgRP expression to promote hunger.19 Furthermore, mice with a specific ablation of autophagy gene Atg7 within the AgRP neuron displayed elevated POMC and α-melanocyte-stimulating hormone (α-MSH) levels, which plausibly contribute to their decreased body weight and adipose mass. Conversely, the loss of functional autophagy in POMC neurons impairs axonal growth and projections to downstream target nuclei, resulting in metabolic dysregulation.20 Interestingly, the induction of autophagy in POMC neurons has been linked to cold-induced lipolysis in BAT.21 Together, these studies underscore the critical function of hypothalamic autophagy in the regulation of energy homeostasis, through the modulation of neuronal activity and neuropeptide release.
In this study, we have investigated the potential interaction of fasting-activated AgRP neurons and the adaptation of liver physiology during energy deprivation. We find that the induction of AgRP neuron activity by short-term starvation or optogenetic and chemogenetic activation of AgRP neurons in the fed state promotes hepatic autophagic flux and metabolic remodeling. Interestingly, this phenomenon is abrogated in the absence of neuropeptide (NPY) expression, and selective re-expression of NPY in AgRP neurons restores AgRP neuron-dependent activation of hepatic autophagy. In addition, we show that NPY promotes the presynaptic inhibition of NPY1R-expressing GABAergic neurons subserving CRH neurons in the PVH. Importantly, the chemogenetic inhibition of AgRP neuron during a short-term fast abrogates starvation-induced liver autophagy. Finally, we reveal that the AgRP neuron-dependent induction of liver autophagy is mediated through increased serum corticosterone concentrations and liver glucocorticoid receptor (GR) signaling. Collectively, our study reveals a fundamental regulatory principle of control of liver autophagy in the control of metabolic adaptation during nutrient deprivation.
Results
Short-term fasting and AgRP neuron activation promotes autophagy in the liver
To evaluate the kinetics of fasting-induced activation of hunger-promoting AgRP- and NPY-co-expressing (AgRP) neurons in the ARH, we employed a model of short-term food deprivation, i.e., after 4 and 6 h into the dark cycle (Figure S1A). Limiting access to food during the first 4 and 6 h of the natural feeding cycle reduced blood glucose concentrations and decreased body weight between 1.0 and 1.5 g compared with animals with free access to food (Figures S1B and S1C). Assessing the activation of AgRP neurons via double in situ hybridization for Fos mRNA (magenta) expression in AgRP-expressing neurons (cyan) revealed robust activation of these cells as evidenced by an increase in the proportion of Fos-positive AgRP neurons to 47% in fasted compared with 12% in fed mice after 4 h and to 59% of Fos-positive cells in fasted mice compared with 8% in fed mice after 6 h (Figures S1D and S1F). On the contrary, activation of anorexigenic POMC neurons (yellow) readily decreased in fasted compared with fed mice during the same period (Figures S1D and S1E). These results highlighted the rapid changes in the activities of AgRP and POMC neurons after short-term alterations in nutrient availability.
Given this short-term regulation of AgRP and POMC neurons and their well-documented role in controlling metabolic pathways in peripheral organs such as the liver and adipose tissues,22,23 we next investigated the consequences of activating AgRP neurons via optogenetic stimulation over a time course comparable with what was observed under fasting conditions in control mice. Optical fibers were placed above the ARH of mice expressing the light-activated ion channel Channelrhodopsin-2 (ChR2) specifically in AgRP neurons (Figure 1A). Blue light illumination of AgRP neurons during the light cycle gave rise to an increase in food intake over 4 h, compared with the food intake observed in the absence of laser stimulation as previously described (Figures S2A and S2B).6 Furthermore, we performed the optogenetic activation of AgRP neurons for 1, 2, and 4 h during the light cycle in the absence of food (Figure 1A). We extracted mRNA from the livers of these animals and subjected it to deep mRNA sequencing. Although 1 h of optogenetic AgRP neuron stimulation only resulted in minor changes in the liver transcriptome (data not shown), there was a progressive and overlapping change in gene expression between 2 and 4 h following optogenetic AgRP neuron activation (Figure 1B). In fact, there was a coordinate regulation of 1319 genes in which the genes exhibit a time-dependent increase (n = 707) or decrease (n = 612) after 2 and 4 h of AgRP neuron photostimulation (FDR < 0.05) (Figure 1C). We next subjected these genes to a gene ontology (GO) term enrichment analysis to identify specific pathways in the liver that are regulated by AgRP neuron activation. This analysis revealed that autophagy regulation and catabolic amino acid metabolism pathways were among the top enriched GO terms (Figure 1D).
Figure 1.

Optogenetic stimulation of AgRP neurons induces autophagy in the liver
(A) Experimental design for optogenetic activation of AgRP neurons in the presence and absence of food.
(B) Scatterplot of the Log2-fold changes of 2 h (yellow), 4 h (blue), and overlapping hepatic transcripts (red) between 2 and 4 h stimulations (n = 5 animals/group). Each colored dot represents a significantly regulated gene.
(C) Log2-fold change of hepatic gene expression that increase or decrease after 2 and 4 h of AgRP neuron activation.
(D) Fisher’s exact test results represented in a bar chart showing significantly enriched gene ontology terms and the percentage of significantly regulated genes.
(E) Validation of gene expression related to autophagy, glucose metabolism, and lipid metabolism by quantitative real-time PCR; data are normalized to the respective ChR2WT group and represented as scatter dot plots with individual values relative to Tbp expression (n = 5 animals/group).
(F) Representative western blot analysis of liver homogenates from 1, 2, and 4 h optogenetically stimulated ChR2WT and ChR2AgRP mice, showing autophagic marker proteins LC3-I, LC3-II, and p62 and the corresponding gapdh loading control (see Figure S2C for additional western blots used for quantification).
(G) Densitometric analysis of the autophagic marker proteins in post-nuclear supernatants of liver extracts of 1, 2, and 4 h optogenetic stimulated ChR2WT and ChR2AgRP mice (n = 9–10 animals/group/time point).
(H) Representative TEM images of liver sections of 4 h optogenetic-stimulated ChR2WT and ChR2AgRP mice. (TOP) Autophagic vacuoles are indicated by blue arrows and digital zoom image of a double-membrane autophagosome outlined by the white-dotted box. Scale bars, 1 μm.
(I) Quantification of double-membrane autophagic vacuoles per 100 μm2 following 1, 2, or 4 h of optogenetic stimulation in ChR2WT and ChR2AgRP mice (n = 4 animals/group/time point; ∼20 images/animal/liver).
(J) Experimental design and representative western blot analysis of liver homogenates from leupeptin-based LC3-II flux analysis in vivo following 4 h of optogenetic activation of AgRP neurons. Mice were injected with saline as controls (n = 4 animals/group).
(K) Net LC3-II flux in the liver as described in (J) (n = 4 animals/group).
Data are represented as mean ± SEM. Statistical analyses were performed by two-way ANOVA followed by Šídák post hoc tests (for E and F without repeated-measure [RM]), one-way ANOVA followed by Tukey’s post hoc test (for I) or unpaired two-tailed Student’s t tests (for K). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (see also Figures S1 and S2).
Subsequent validation using quantitative real-time PCR analysis unveiled an increase in hepatic expression of transcripts associated with autophagy (Trp53inp1, Ulk1, Sesn2, and Atp6v0d1). In addition, we detected the coordinate regulation of genes involved in lipid metabolism (Plin5, Lpin1, Lpin2, Lipg, Ppargc1a, and Foxo1) (Figure 1E). AgRP neuron activation also increased expression of key regulators of gluconeogenesis (G6pc, Pck1), which are under transcriptional control of FOXO/Ppargc1a, of which expression of both also increased upon AgRP neuron stimulation (Figure 1E).
Since autophagy in the liver has been linked to both the control of glucose and lipid metabolism, we next assessed whether autophagy is indeed activated after short-term AgRP neuron stimulation. Accordingly, we performed immunoblotting for microtubule-associated protein light chain 3 (LC3, ratio of LC3-II/LC3-I)—an indicator of autophagosome abundance—and p62 (also known as SQSTM1/sequestosome 1)—an autophagy receptor degraded by autophagy—as markers of autophagy induction in the livers of mice with AgRP neuron photostimulation. This analysis revealed a time-dependent increase in LC3 processing 2 and 4 h after the optogenetic stimulation of AgRP neurons, whereas p62 abundance remained unchanged (Figures 1F and 1G).
To further corroborate the notion that AgRP neuron activation is sufficient to promote autophagy in the liver, we employed transmission electron microscopy (TEM) to quantify autophagic vacuoles in liver sections from mice with optogenetic AgRP neuron stimulation. Littermates lacking ChR2 expression in AgRP neurons were photostimulated for 4 h as controls. Here, we detected a progressive increase in the number of autophagic vacuoles amounting to a 3-fold increase in the liver of mice with 4 h optogenetic AgRP neuron stimulation compared with controls (Figures 1H and 1I).
Finally, we assessed LC3-II accumulation following lysosomal inhibition in mice with and without AgRP neuron activation. The extent of LC3-II accumulation in the presence versus absence of a lysosomal inhibitor (leupeptin) reflects the LC3-II flux or the autophagy flux. This method is widely used to study autophagy in vivo.24 Intriguingly, the optogenetic stimulation of AgRP neurons induced an evident increase in the hepatic net LC3-II flux (Figures 1J and 1K). These experiments demonstrate that the optogenetic stimulation of AgRP neurons is sufficient to activate the autophagic flux in the liver.
Central Ghrelin action and short-term fasting promote autophagy in the liver
In light of the notion that the optogenetic stimulation of AgRP neurons activated autophagy in the liver, we aimed to address whether a natural fasting-associated stimulus, well-characterized to act predominantly via the AgRP neurons is capable of promoting hepatic autophagy. Ghrelin is an orexigenic peptide hormone released from the gastrointestinal tract in response to energy deficit and acts through Ghrelin receptors (GhSR) expressed on AgRP neurons to promote feeding.25,26,27 Thus, we asked whether the central administration of Ghrelin is able to promote autophagy in the liver. Ad libitum-fed C57BL/6N mice received two intracerebroventricular (i.c.v.) injections of Ghrelin (1.5 μg) at ZT1 and ZT3 in the light cycle of the day (Figure S3A). As expected, the i.c.v. administration of Ghrelin promoted a significant increase in food intake over a 4-h period (Figures S3B–S3D) and promoted locomotor activity in fasted but not in fed mice (Figures S3E and S3F), similar to what had been described for AgRP neuron activation.9 Repeated Ghrelin injection in the absence of food rapidly increased Fos mRNA expression in AgRP neurons from 9% to 75% compared with saline-injected mice (Figures S3G and S3H). Intriguingly, repeated i.c.v. injections of Ghrelin promoted autophagy as evidenced by a profound 3-fold increase in LC3 lipidation and a ∼30% reduction in p62 protein expression in the liver 4 h after the first application (Figures S4A–S4C). Importantly, i.p. injection of the same dose of Ghrelin failed to induce autophagy in the liver, providing evidence that Ghrelin acts centrally to activate liver autophagy (Figures S4B and S4D). In addition, i.c.v., but not i.p., injection of Ghrelin elicited similar changes, although to weaker extents, in gene expression related to autophagy, glucose metabolism, and lipid metabolism as observed upon optogenetic AgRP neuron activation, and the low dose of peripherally applied Ghrelin induced a comparably mild stimulation of food intake (2-fold for i.p. vs. 5-fold for i.c.v.) (Figures S4E–S4I).
To directly compare the induction of liver autophagy and changes in the liver mRNA expression observed upon optogenetic or Ghrelin-induced activation of AgRP neurons to those observed under short-term nutrient deprivation, we assessed the induction of liver autophagy in mice, which had been fasted for 4 and 6 h. This analysis revealed that 4 h of food restriction into the dark cycle induced a similar increase in LC3 processing compared with optogenetic or Ghrelin-mediated activation of AgRP neurons (Figures S5A and S5B). We then performed a leupeptin-based autophagic flux assessment in C57BL/6N mice, which were fasted for 4 h into the dark cycle. In the presence of leupeptin, fasted mice displayed a marked increase in LC3-II accumulation revealing a clear stimulation of hepatic net LC3-II flux (Figures S5C and S5D). Similarly, the liver mRNA expression profile of the genes associated with autophagy, glucose metabolism, and lipid metabolism was robustly regulated (Figure S5E).
AgRP neuron-dependent activation of liver autophagy requires NPY expression
In addition to AgRP, these neurons also express Neuropeptide Y (NPY) and the inhibitory neurotransmitter GABA, and the release of AgRP, NPY, and GABA from these cells controls different phases during the kinetic activation of food intake.28,29,30 Here, we examined the potential contribution of NPY release from AgRP/NPY co-expressing neurons in the ARH to promote liver autophagy, by activating AgRP neurons in the presence or absence of NPY. To achieve this, we crossed mice expressing ChR2 from the ROSA26 locus in a Cre-dependent manner (ROSA26ChR2) with those heterozygous for a null mutation in the NPY locus (NPYΔ/wt-mice).31 Further intercrosses with AgRP-IRES-Cre mice yielded littermates, which were either controls (ROSA26ChR2fl/wt; AgRPCrewt/wt; NPYwt/wt, i.e., NPYwt/wt), expressed ChR2 in AgRP neurons in the presence of NPY (ROSA26ChR2fl/wt; AgRPCreCre/wt; NPYwt/wt, i.e., ChR2AgRP; NPYwt/wt), lacked NPY expression in the absence of ChR2 expression in AgRP neurons (ROSA26ChR2fl/wt; AgRPCrewt/wt; NPYΔ/Δ, i.e., NPYΔ/Δ), or those expressing ChR2 in AgRP neurons and lacking NPY (ROSA26ChR2fl/wt; AgRPCreCre/wt; NPYΔ/Δ, i.e., ChR2AgRP; NPYΔ/Δ) (Figure 2A). Although optogenetic stimulation of AgRP neurons evoked a clear 3-fold induction of LC3 lipidation in ChR2AgRP; NPYwt/wt mice, this response was abrogated upon optogenetic stimulation of AgRP neurons in NPY knockout mice (Figures 2B, 2C, and S6A). Similarly, the induction of mRNA expression in genes related to autophagy, glucose metabolism, and lipid metabolism, which were clearly induced on AgRP neuron activation in ChR2AgRP; NPYwt/wt mice was similarly reduced upon AgRP neuron activation in the absence of NPY expression (Figure 2D).
Figure 2.

AgRP-mediated control of hepatic autophagy requires expression of NPY
(A) Workflow for virus-mediated re-expression of NPY in the ARH, experimental design for optogenetic stimulation of AgRP neurons in the presence and absence of NPY.
(B) Representative western blots using liver extracts from fed ChR2WT, ChR2AgRP, ChR2 NPYΔ/Δ, and ChR2AgRP-NPYΔ/Δ mice after 4 h of photostimulation (see Figure S6A for additional western blot used for quantification).
(C) Densitometric analysis of the ratio of LC3-II/LC3-I (LC3) and p62/Gapdh as autophagic marker proteins in post-nuclear supernatants of liver homogenates from mice in (B) (n = 9–10 animals/group).
(D) Quantitative real-time PCR analyses of genes related to autophagy, glucose metabolism, and lipid metabolism; data are normalized to ChR2WT littermates and represented as scatter dot plots with individual values relative to Tbp expression (n = 6 animals/group).
(E) Experimental design for DREADD stimulation of AgRP neurons in the presence and absence of NPY.
(F) Representative western blots of liver extracts from fed hM3DgqWT, hM3DgqAgRP, hM3DgqNPYΔ/Δ, and hM3DgqAgRP-NPYΔ/Δ mice sacrificed at ZT7.5 after i.p. injections of CNO (1 mg/kg) at ZT3.5 and ZT5.5 (see also Figure S6F for additional western blots used for quantification).
(G) Densitometric analysis of the ratio of LC3-II/LC3-I (LC3) and p62/Gapdh as autophagic marker proteins in post-nuclear supernatants of liver homogenates from hM3DgqWT (n = 7), hM3DgqAgRP (n = 14), hM3DgqNPYΔ/Δ (n = 9), and hM3DgqAgRP-NPYΔ/Δ (n = 9) mice.
(H) Representative confocal images indicating successful expression of bilaterally injected control virus (AAV-DIO-mCherry) in ChR2AgRP and ChR2AgRP-NPYΔ/Δ mice, and NPY virus (AAV-DIO-NPY) in ChR2AgRP-NPYΔ/Δ mice in the ARH. Note that NPY containing fibers in the ARH and PVH were detected in ChR2AgRP-NPYΔ/Δ mice with re-expression of NPY using immunohistochemistry; this is absent in the corresponding control.
(I) Representative western blots of autophagic marker proteins using liver extracts from mice in (H) after 4 h of optogenetic stimulation (see also Figure S6G for additional replicates used for quantification).
(J) Densitometric analysis of the ratio of LC3-II/LC3-I (LC3) and p62/Gapdh as autophagic marker proteins in post-nuclear supernatants of liver homogenates from mice in (I) (n = 10–17 animals/group).
Data are represented as mean ± SEM. Statistical analyses were performed by one-way ANOVA followed by Tukey’s post hoc test. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (see also Figure S6).
To further confirm the role of NPY-dependent and -independent signaling of AgRP neurons through a complementary approach, we investigated the consequences of chemogenetically activating AgRP neurons in the presence and absence of NPY expression. To this end, we expressed the stimulatory chemogenetic receptor hM3DGq from the ROSA26 locus (ROSA26hM3DGq) in a Cre-dependent manner in AgRP neurons of mice expressing or lacking NPY (Figure 2E). Similar to what was observed upon optogenetic stimulation of AgRP neurons, chemogenetic activation of AgRP neurons also markedly enhanced LC3 lipidation in the liver, and this response was abrogated in hM3DGqAgRP; NPYΔ/Δ mice (Figures 2F, 2G, and S6F).
It is well established that NPY expression is not limited to AgRP neurons in the ARH. In fact, NPY is also widely expressed in both the CNS and peripheral tissues. Here, we aimed to investigate whether the observed alteration in liver autophagy induction upon AgRP neuron activation in NPY-deficient mice was indeed a specific outcome of lacking NPY expression in AgRP neurons. To achieve this, we stereotactically delivered in the ARH either an AAV allowing for Cre-dependent expression of mCherry in ChR2AgRP; NPYwt/wt mice and ChR2AgRP; NPYΔ/Δ mice or an AAV allowing for Cre-dependent expression of NPY in ChR2AgRP; NPYΔ/Δ mice. Immunohistochemical analyses revealed successful mCherry expression in AgRP neurons in the ARH of ChR2AgRP; NPYwt/wt mice and ChR2AgRP; NPYΔ/Δ mice, as well as expression of NPY in the ARH of ChR2AgRP; NPYwt/wt mice injected with the mCherry expressing virus and in ChR2AgRP; NPYΔ/Δ mice that had been injected with the AAV allowing for Cre-dependent expression of NPY (Figure 2H), as previously described.30 Although laser stimulation clearly induced LC3 processing in the liver of ChR2AgRP; NPYwt/wt mice injected with the mCherry-expressing virus, this response was abrogated in ChR2AgRP; NPYΔ/Δ mice with AAV mCherry injections in the ARH but restored in ChR2AgRP; NPYΔ/Δ mice with AgRP neuron-restricted re-expression of NPY (Figures 2I, 2J, and S6G). In addition, the regulation of hepatic genes involved in autophagy, glucose metabolism, and lipid metabolism was also largely recovered upon NPY re-expression in AgRP neurons (Figure S6H).
PVHNPY1R neuron inhibition activates hepatic autophagy
AgRP neurons elicit their food intake and metabolism regulatory functions through a widespread neuronal network in the CNS.9,32 Although the optogenetic stimulation of AgRP projections in the paraventricular nucleus of the hypothalamus (PVH), paraventricular thalamus (PVT), lateral hypothalamus (LHA), and the Bed nucleus of the stria terminalis (BNST) contributes to the food intake-promoting effects of optogenetic AgRP neurons activation,32 projection stimulation in the LHA and BNST contribute to the short-term control of glucose homeostasis.9 To begin unraveling the neurocircuitry downstream of AgRP neurons responsible for the control of liver autophagy, we performed optogenetic neurocircuitry-mapping experiments in ChR2AgRP mice (Figure S8A). Optical fibers were implanted above the PVH, and mice were subjected to subsequent blue light illuminations of 10 and 20 mW (Figures S7A and S7B). Both laser powers evoked a similar increase in food intake as previously described (Figure S7B).32 Subsequent photostimulation of AgRP → PVH projections for 4 h in the absence of food during the day cycle was thus performed using a laser intensity of 10 mW. Photostimulation of AgRP → PVH projections increased hepatic LC3 processing and resulted in coordinated changes in gene expression in the liver related to autophagy, glucose metabolism, and lipid metabolism similar to what was observed on stimulating the AgRP neural somata in the ARH (Figures S8B, S8C, and S9A). Similarly, the stimulation of AgRP → LHA projections also led to a profound increase in food intake (Figures S7C and S7D), and blue light illumination at 10 mW in the absence of food also elevated LC3 processing in the liver, although to lesser extents than photostimulation of AgRP → PVH projections (Figures S8D and S8E). Moreover, gene expression profiling in the liver revealed a profile of mRNA expression changes in genes related to autophagy, glucose metabolism, and lipid metabolism similar to that observed on the AgRP → PVH projections stimulation (Figure S9B). By contrast, despite the robust induction of food intake following the AgRP → BNST projection stimulation (Figures S7E and S7F), LC3 processing in the liver remained unchanged, and expression of genes related to autophagy, glucose metabolism, and lipid metabolism was regulated only to a minor extent (Figures S8F, S8G, and S9C). Despite these data indicating that AgRP neuron projections to PVH and LHA but not BNST may be mainly responsible for mediating control of hepatic autophagy, these findings do not rule not out the possibility that a certain degree of the observed physiological changes may result from backpropagation of action potential to AgRP neuron somata depending on the density of AgRP fibers within the studied projection sites.
To further define the downstream neurocircuitry responsible for brain-mediated liver autophagy via AgRP neuron activation, we interrogated how NPY can potentially mediate this response via the PVH or LHA. The downstream inhibitory effect of NPY on neurons via NPY1R and NPY5R activation and the well-documented role of NPY1R-signaling in mediating the orexigenic effect of AgRP neuron stimulation33 led us to hypothesize that NPY-dependent inhibition of NPY1R-expressing neurons and/or NPY1Rs expressed in the projections within the PVH and/or LHA may contribute to the regulation of hepatic autophagy. Therefore, we bilaterally injected NPY1R-Cre transgenic animals with Cre-dependent adeno-associated viruses (AAV) expressing the inhibitory DREADD receptor hM4Di into the PVH or LHA (Figure 3A). Interestingly, a significantly elevated hepatic LC3 lipidation and a corresponding decrease in the p62 level were observed in the CNO-mediated inhibition of PVHNPY1R neurons (Figures 3B and 3C). In addition, gene transcripts associated with autophagy, glucose metabolism, and lipid metabolism were also induced following the chemogenetic inhibition of PVHNPY1R neurons (Figure 3D). On the contrary, LHANPY1R neuron inhibition affected neither liver autophagy nor the hepatic gene expression profile (Figures 3E–3G).
Figure 3.

NPY1R inhibition in the PVH promotes liver autophagy
(A) Workflow for PVH or LHA-targeted delivery of Cre-dependent AAV-hM4di in NPY1R-Cre mice and experimental design for the CNO-mediated inhibition of NPY1R-expressing neurons.
(B) Representative western blots of 4 h inhibition of PVHNPY1R neurons following saline or CNO (3 mg/kg) treatment.
(C) Densitometric analysis of the ratio of LC3-II/LC3-I (LC3) and p62/Gapdh as autophagic marker proteins in liver homogenates from mice in (B) (n = 10–11 animals/group).
(D) Quantitative real-time PCR analyses of genes related to autophagy, glucose and lipid metabolism; data are normalized to the saline group and represented as scatter dot plots with individual values relative to Tbp expression (n = 7–11 animals/group)
(E) Representative western blots of 4 h inhibition of LHANPY1R neurons following saline or CNO (3 mg/kg) treatment.
(F) Densitometric analysis of the ratio of LC3-II/LC3-I (LC3) and p62/Gapdh as autophagic marker proteins in liver homogenates from mice in (E) (n = 6 animals/group).
(G) Quantitative real-time PCR analyses of genes related to autophagy, glucose metabolism, and lipid metabolism; data are normalized to the saline group and represented as scatter dot plots with individual values relative to Tbp expression (n = 6 animals/group).
Data are represented as mean ± SEM. Statistical analyses were performed by unpaired two-tailed Student’s t tests. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (see also Figure S13).
AgRP neuron activation is required for the induction of hepatic autophagy upon short-term nutrient deprivation
The aforementioned findings have revealed that the activation of AgRP neurons is sufficient to promote liver autophagy. However, a critical question remained whether AgRP neuron activation is required for modulating hepatic autophagy during short-term food restriction. To probe this question, we set out to inhibit AgRP neurons during short-term food restriction to evaluate the induction of hepatic autophagy upon food removal in the presence or absence of AgRP neuron activation.
Specifically, AgRP-IRES-Cre mice received bilateral injections of Cre-dependent AAV in the ARH to express either mCherry (AAV-hSyn-DIO-mCherry; mCherry) or a mutated human glycine receptor (AAV-FLEX-hGlyRa1AG-mCherry; Flex-hGlyRa1AG), which is inert to its endogenous ligand glycine but is controlled by the administration of ivermectin.34 Approximately 50% – 60% of AgRP cells in the ARH successfully expressed either mCherry or Flex-hGlyRa1AG (Figures S10A and S10B). Noteworthy, neurons expressing the mutated hGlyR receptor elicited the long-term inhibition of neuronal activity in response to i.p.-delivered ivermectin.35 In the current study, both groups of mice received an i.p. injection of ivermectin (5 mg/kg) and underwent a subsequent fast for 4 h during the onset of the dark cycle on the same day (Figure 4A). Mice injected with AAV-DIO-mCherry and subjected to ivermectin treatment exhibited the expected proportion (∼40%) of Fos-expressing AgRP neurons in short-term food-deprived mice, whereas mice injected with AAV-Flex hGlyRa1AG-mCherry failed to induce the Fos expression in AgRP neurons during the same period of nutrient deprivation following ivermectin injection (Figures 4B and 4C). This indicates successful suppression of AgRP neuron activity during a short-term fast.
Figure 4.

Hepatic autophagy induction on short-term nutrient deprivation requires AgRP neuron activation
(A) Workflow for virus-mediated expression of either mCherry or hGlyR in AgRP neurons, experimental design for the chemogenetic inhibition of AgRP neuron.
(B) Representative confocal images showing AgRP (Cyan) and cFos (Magenta) mRNA expression in the ARH of 4 h fasted AgRP-IRES-Cre mice with bilateral intra-ARH injection of AAV-DIO-mCherry or AAV-Flex-hGlyR following ivermectin (IVM) injection.
(C) Quantification of cFos-positive AgRP neurons from mice in (B) following i.p. IVM injection and a 4 h, short-term fast (n = 6–7 animals/group).
(D) Representative western blots of liver extracts from mice in (B) after a short-term fasting in the dark cycle (see also Figure S10C for additional replicates used for quantification).
(E) Densitometric analysis of the ratio of LC3-II/LC3-I (LC3) and p62/Gapdh as autophagic marker proteins in liver homogenates from mice in (B) (n = 13–14 animals/group).
(F) Representative western blot analysis of liver homogenates from leupeptin-based LC3-II flux analysis in vivo following the inhibition of AgRP neuron and a 4 h, short-term fast. Mice were injected with saline as controls.
(G) Densitometric analysis of net LC3-II flux from liver homogenates from mice in (F) (n = 5 animals/group).
(H) Quantitative real-time PCR analyses of genes related to autophagy, glucose and lipid metabolism; data are normalized to mice injected with AAV-DIO-mCherry and represented as scatter dot plots with individual values relative to Tbp expression (n = 13–14 animals/group).
(I) Change in body weight between ZT11 and ZT15 on a 4 h fast (n = 13–14 animals/group).
(J) Blood glucose measurements at baseline (ZT11) and after 4 h fast (ZT15); represented as blood glucose levels of individual animal before and after fasting (n = 13–14 animals/group).
Data are represented as mean ± SEM. Statistical analyses were performed by unpaired two-tailed Student’s t tests (for C, E, F, and G) or two-way RM ANOVA followed by Šídák post hoc test (for H). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (see also Figure S10).
Importantly, autophagy flux (Figures 4D–4G and S10C) and expression of mRNA transcripts associated with autophagy, glucose metabolism, and lipid metabolism (Figure 4H) were markedly reduced in the livers of fasted mice injected with AAV-Flex-hGlyR-mCherry compared with the group injected with the control virus. Together, these data establish the necessity of AgRP neuron activation for the induction of liver autophagy during short-term nutrient deprivation. In addition, mice with the suppression of AgRP neuron activation during nutrient deprivation exhibited an attenuated loss of body weight and decline in blood glucose concentrations during food deprivation (Figures 4I and 4J), supporting the importance of the activation of AgRP neurons in mediating the physiological adaptations to fasting.
AgRP neuron activation promotes hepatic phosphoproteom changes in pathways controlling autophagy and lipid mobilization
To investigate the regulation of known signaling pathways in the control of autophagy on AgRP neuron stimulation, we assessed the phosphorylation of the ribosomal proteins S6, AKT, Atg14, and AMPK and protein expression of transcription factor EB (TFEB) across the time course of optogenetic AgRP neuron activation. This revealed transiently elevated S6-phosphorylation 1 and 2 h following neuronal activation and mildly activated AKT phosphorylation after 2 h of stimulation, whereas AMPK phosphorylation remained unaltered (Figures S11A–S11C). On the other hand, the phosphorylation of Atg14 and TFEB expression increased on prolonged AgRP activation (Figures S11D and S11E). To further define pathways affected in the liver upon prolonged optogenetic AgRP neuron activation, we investigated phosphoproteome changes in the liver upon optogenetic AgRP neuron activation. Given the clear dependency of driving liver autophagy on NPY expression in AgRP neurons, we performed an unbiased phosphoproteomic analysis comparing mice that were unstimulated and mice with AgRP neurons activated for 4 h in the presence and absence of NPY expression (Figure 5A). Specifically, we performed a one-way ANOVA analysis and identified a total of 92 differently regulated phosphopeptides, of which 62 were increased on 4 h AgRP neuron activation in the presence of NPY, but not in the absence of NPY (Figure 5B). Interestingly, these phosphopeptides were enriched in GO terms related to pre-autophagosomal structure, autophagic vacuole, and macroautophagy, including the well-characterized regulators of autophagy Ulk1 (S450), Sqstm1 (T269, T272), Optn (S345), and Zfyve1 (also known as Dfcp1) (S129) (Figures 5C and 5D). Notably, the protein Zfyve1 is known to play an important role in lipid-droplet formation, and we observed a 1.95-fold upregulated (q value < 0.02) after AgRP neuron activation, which was attenuated on NPY deletion. Moreover, we detected multiple phosphorylation sites in Lipin1 and Lipin2, which showed a similar regulation. Together, concordant with the detected gene expression changes, these experiments provide evidence for a coordinate NPY-dependent regulation of autophagy and lipid mobilization in the liver in response to AgRP neuron activation.
Figure 5.

AgRP neuron activation promotes hepatic phosphoproteom changes in pathways associated with autophagy and lipid mobilization
(A) Schematic representation of the liver phosphoproteomics workflow.
(B) Number of identified phosphorylated peptides. Significantly altered phosphopeptides were identified using a one-way ANOVA and a permutation-based false discovery rate (FDR) cutoff of 0.10.
(C) Hierarchical clustering of significantly altered hepatic phosphopeptides in individual ChR2WT, ChR2AgRP, ChR2 NPYΔ/Δ and ChR2AgRP-NPYΔ/Δ mice after 4 h photostimulation (n = 6 animals/group). The row dendrogram was calculated using Euclidean distance and complete methods.
(D) Scatterplot showing the significant enriched GO terms of differentially regulated phosphopeptides clusters. p values were corrected for multiple testing by the Benjamini-Hochberg (BH) procedure.
(E) Total diglycerides (DAGs), triacylglycerides (TAGs), and acyl carnitines (AcCa) concentrations in the liver of 4 h photostimulated ChR2WT and ChR2AgRP mice (n = 9–14 animals/group).
(F) Liver hydroxybutyric acid concentrations in 4 h photostimulated ChR2WT and ChR2AgRP mice (n = 11–14 animals/group).
(G) Total diglycerides (DAGs), triacylglycerides (TAGs) and acyl carnitines (AcCa) concentrations in the liver following a short-term 4 h fast and the simultaneous chemogenetic inhibition of AgRP neurons (n = 13–14 animals/group).
(H) Liver hydroxybutyric acid concentrations following a short-term 4 h fast and simultaneous chemogenetic inhibition of AgRP neurons (n = 13–14 animals/group).
Data are represented as mean ± SEM. Statistical analyses were performed by unpaired two-tailed Student’s t test (for E, F, G, and H). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (see also Figures S11 and S12).
To investigate the metabolic consequences of the responses elicited via AgRP neuron activation, we performed metabolomic analyses of the livers from two independent experiments; ChR2WT and ChR2AgRP mice in the presence of blue light illumination in the ARH for 4 h and the chemogenetic inhibition of AgRP neuron during a short-term fast. Although the overall abundance of amino acids did not exhibit major changes in the optogenetic activation of AgRP neurons or during the inhibition of AgRP neurons during fasting, there was a reduction in alanine, glutamine, and histidine concentrations on AgRP neuron stimulation and an increase in AgRP neuron inhibition (Figures S12A and S12D). By contrast, concentrations of the branched-chain amino acids specifically isoleucine and valine were increased on AgRP neuron stimulation as previously described,36 and decreased on AgRP neuron inhibition during fasting (Figures S12A and S12D). Although AgRP neuron stimulation did not alter triacylglyceride (TAG) content in the liver, it resulted in a clear 1.5-fold increase in diglyceride (DAG), a 2.5-fold increase in acylcarnitine, and a 2-fold increase in hydroxybutyrate concentrations (Figures 5E, 5F, S12B, and S12C). Conversely, AgRP neuron inhibition during fasting significantly reduced liver hydroxybutyrate concentrations without major alterations in DAG, TAG, and acylcarnitine concentrations (Figures 5G, 5H, S12E, and S12F). Collectively, these data indicated that AgRP neuron activation may promote ß-oxidation and ketogenesis.
AgRP neuron activation increases circulating corticosterone concentrations
Several putative peripheral effector pathways might mediate AgRP neuron-dependent activation of autophagy in the liver. Apart from the well-documented role of AgRP neurons in controlling glucose metabolism via the rapid regulation of autonomic innervation of the liver and BAT, it is plausible that prolonged changes in humoral, circulating signals might relay the signal from the CNS to the liver in control of autophagy. Thus, we first screened whether well-characterized regulators of liver metabolism change in the plasma of mice upon prolonged AgRP neuron activation. This analysis revealed a robust ∼2- to 3-fold increase in circulating corticosterone concentrations following 2 and 4 h after AgRP neuron stimulation, although there was only a trend toward an increase in circulating glucagon concentrations after 4 h of AgRP neuron activation (Figures 6A and S12G). No significant changes were observed in circulating insulin concentrations (Figure S12H). Moreover, optogenetic AgRP neuron activation transiently increased circulating ACTH concentrations after 2 h of photostimulation, followed by a profound suppression of circulating ACTH concentrations after 4 h of optogenetic stimulation (Figure S12I), which may point toward the corticosterone-mediated feedback inhibition of the hypothalamic-pituitary adrenal (HPA) axis following a prolonged activation of AgRP neurons.
Figure 6.

AgRP neuron activation increases circulating corticosterone concentrations to promote hepatic autophagy
(A) Serum corticosterone levels after 2 and 4 h optogenetic stimulation of AgRP neurons (n = 9–11 animals/group).
(B) Serum corticosterone levels after 4 h optogenetic stimulation of AgRP neurons in the presence or absence of NPY (n = 9–10 animals/group).
(C) Serum corticosterone levels after 4 h of optogenetic AgRP → PVH, AgRP → LHA, or AgRP → BNST projection stimulation (n = 7–9 animals/group/projection).
(D) Serum corticosterone levels after 4 h of chemogenetic PVHNPY1R or LHANPY1R neuron inhibition (n = 6–11 animals/group/projection).
(E) Experimental design for i.p. dexamethasone treatment alone and in combination with lysosomal inhibitor (leupeptin) to measure autophagy flux in vivo.
(F) Representative western blots of liver homogenates from C57BL/6N mice which received either a single i.p. injection of saline or dexamethasone. Gapdh was used as loading control.
(G) Densitometric analysis of the ratio of LC3-II/LC3-I (LC3) and p62/Gapdh as autophagic marker proteins in liver homogenates from mice in (F) (n = 7 animals/group).
(H) Representative western blots of liver homogenates from C57BL/6N mice which received either an i.p. injection of saline/leupeptin, 1 h after saline/dexamethasone treatment. Gapdh was used as loading control.
(I) Net LC3-II flux in the liver as described in (H) (n = 5 animals/group).
(J) Experimental design for AAV-mediated expression of either Scrmb- or Nr3c1-shRNA in the liver of ChR2AgRP mice 3 weeks prior to 4 h optogenetic stimulation in the absence of food.
(K) Validation of the knock down of Nr3c1 in the liver by qPCR (n = 7–8 animals/group).
(L) Representative western blots of hepatic autophagic marker proteins from mice in (J).
(M) Densitometric analysis of the ratio of LC3-II/LC3-I (LC3) and p62/Gapdh as autophagic marker proteins in liver homogenates from mice in (L) (n = 7–8 animals/group).
Data are represented as mean ± SEM. Statistical analyses were performed by two-way ANOVA followed by Šídák post hoc tests (for A), one-way ANOVA followed by Tukey’s post hoc tests (for B) or unpaired two-tailed Student’s t test (for C, D, G, I, K, and M). ns, not significant, ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (see also Figure S14).
Interestingly, the ability of AgRP neuron activation to elicit an increase in circulating corticosterone concentrations was abolished in mice lacking NPY; similar to what was observed in the induction of liver autophagy in these animals (Figure 6B). Of note, photostimulation of AgRP → PVH and AgRP → LHA projections similarly increased serum corticosterone concentrations but photostimulation of AgRP → BNST projections failed to raise circulating corticosterone concentrations (Figure 6C). In addition, the chemogenetic inhibition of PVHNPY1R but not LHANPY1R neurons elicited a similar increase in circulating corticosterone concentrations (Figure 6D). Collectively, these experiments revealed a parallel regulation of liver autophagy and circulating corticosterone concentrations under different complementary experimental conditions.
To dissect the nature of AgRP → PVHNPY1R-neurocircuitry in relation to the corticotropin-releasing hormone (CRH)-dependent modulation of the HPA axis, we quantified the expression of NPY1R in CRH neurons in the PVH and LHA. We detected only 14% of PVHCRH neurons and 30% of LHACRH neurons expressing NPY1R, respectively (Figures S13A and S13B). Since CRH neuron activation represents a critical step in the activation of the HPA axis,37 we next investigated Fos expression in PVHCRH neurons using in situ hybridization following the chemogenetic inhibition of NPY1R-neurons, in which AAV-hM4Di was stereotaxically delivered into the PVH of NPY1R-Cre mice. We observed higher Fos-positive PVHCRH neurons after i.p. administration of CNO, showing that the chemogenetic inhibition of targeted NPY1R-neurons activates PVHCRH neurons (Figures S13D and S13E). Thus, it is likely that inhibiting NPY1R-neurons disinhibits PVHCRH neurons in an NPY-dependent manner. This is consistent with the described effect of NPY in the PVH to activate CRH neurons.38 This model would require that NPY1R neurons innervating CRH neurons are likely GABAergic. Since PVH is largely composed of glutamatergic neurons, we next assessed the expression of Slc32Aa1, the vesicular GABA transporter (VGAT), in NPY1R-neurons in and closely surrounding the PVH. This analysis revealed that of the NPY1R-expressing cells in the PVH, 92.1% expressed Slc17a6, the vesicular glutamate transporter VGLUT2, whereas only 7.9% expressed VGAT (Figure S13C). Thus, it is unlikely that direct AgRP projections to PVHNPY1R-neurons inhibit PVHCRH cells. Interestingly, 35.1% of the NPY1R-expressing cells in close proximity to the PVH-expressed VGAT (Figure S13C), and these cells have previously been demonstrated to provide inhibitory inputs to PVHCRH neurons, and in addition, PVHCRH neurons receive long-range GABAergic inputs from the BNST, lateral septum, and raphe magnus nucleus.39,40 To investigate whether our stereotactic injection of AAV-hM4Di may also have targeted GABAergic peri-PVH NPY1R-expressing cells, we analyzed mCherry expression in NPY1R-Cre mice with PVH-targeted AAV-hM4Di injection. Indeed, this analysis revealed mCherry co-expression in VGAT-positive NPY1R neurons immediately surrounding the PVH (Figure S13F). Therefore, it is plausible that NPY released from AgRP neurons inhibits pre-synaptic NPY1R-expressing projections from GABAergic peri-PVH NPY1R-neurons projecting into the PVH. This in turn disinhibits PVHCRH neurons to promote the increase in circulating corticosterone and activate liver autophagy.
To address whether the observed parallel regulation of circulating corticosterone concentrations and liver autophagy induction in the different mouse models could be causally linked, we performed an acute dexamethasone treatment in C57BL/6N mice. To this end, C57BL/6N mice were i.p. injected with a single dose of dexamethasone (5 mg/kg body weight [BW]) at ZT3.5, and liver tissues were analyzed for the induction of autophagy and changes in gene expression related to autophagy, glucose metabolism, and lipid metabolism (Figure 6E). 4 h following dexamethasone delivery, we observed a ∼2.5-fold increase in LC3 processing coupled with a marked decrease in p62 levels in dexamethasone compared with saline-treated mice (Figures 6F and 6G). Importantly, we observed that acute dexamethasone treatment in the presence of the lysosomal inhibitor leupeptin promotes hepatic LC3-II accumulation in vivo (Figures 6H and 6I). Moreover, expression of genes in autophagy, glucose metabolism, and lipid metabolism pathways were altered in a similar direction and magnitude as observed in AgRP neuron activation (Figure S14A). We also tested whether dexamethasone can promote autophagy in cultured hepatocytes. Indeed, dexamethasone treatment in Hepa1-6 cells induced LC3-processing in a time-dependent manner and augments autophagic flux in these cells (Figures S14B and S14C).
Having identified that peripheral dexamethasone application can elicit similar changes in liver autophagy as observed on AgRP neuron stimulation, we next addressed whether hepatic corticosterone action is required for AgRP neuron-dependent regulation of liver autophagy. To this end, ChR2AgRP mice were intravenously (i.v.) injected with either an AAV allowing for liver-specific expression of a scrambled shRNA (control) or an shRNA targeting the GR transcript (Nr3c1). Three weeks following the delivery of the AAVs, AgRP neurons were photostimulated for 4 h in both groups of mice (Figure 6J). mRNA expression in the liver revealed successful reduction of GR expression by 50% in mice, which had received the AAV-Nr3c1-shRNA compared with those injected with AAV-Srmb-shRNA (Figure 6K). Moreover, the reduction of hepatic GR expression reduced the ability of AgRP neuron photostimulation to induce autophagy as evidenced by a significant reduction in LC3 lipidation (Figures 6L and 6M) and to regulate the gene expression network related to autophagy, glucose metabolism, and lipid metabolism (Figure S14D).
Aging attenuates Ghrelin- and fasting-induced activation of the HPA-axis and hepatic autophagy
Autophagy represents a critical regulator of longevity across a broad evolutionary spectrum.41 Accordingly, we aimed to investigate whether the hereto-described HPA axis-dependent, regulation of liver autophagy undergoes age-dependent changes. We applied i.c.v. Ghrelin in C57BL/6J mice at the ages of 15 and 78 weeks to test the ability of central Ghrelin to regulate serum corticosterone concentrations and liver autophagy. First, we validated the action of central Ghrelin to induce feeding in mice of different ages. Interestingly, we observed an attenuated response to Ghrelin-induced feeding in 78-week-old mice compared with 15-week-old mice (Figure 7A). This is consistent with a clear reduction in hepatic LC3 processing after the central administration of Ghrelin in aged mice compared with young mice (Figures 7B, 7C, and S14E). Finally, we assessed the magnitude of Ghrelin-stimulated serum corticosterone concentration, which was markedly blunted in aged compared with young mice (Figure 7D).
Figure 7.

Aging impairs ghrelin and fasting-mediated activation of the HPA-axis and hepatic autophagy
(A) Cumulative food intake over a period of 4 h following either i.c.v. saline (S) or Ghrelin (Ghr) injection in 15 and 78 weeks old C57BL/6J mice as indicated by the black arrows. (Right) Depiction of total food intake after 4 h treatment (n = 10–15 animals/group/treatment; crossover experiment).
(B) Representative Western blots of liver homogenates from 15 and 78 weeks old C57BL/6J mice after i.c.v. treatment of saline or Ghrelin in the absence of food. Gapdh was used as loading control. Bold line indicates dissection.
(C) Densitometric analysis of autophagic marker proteins in liver homogenates from mice in (B) (n = 5–8 animals/group/treatment).
(D) Plasma corticosterone concentrations of 15 and 78 weeks old C57BL/6J mice 4 h after either central saline or ghrelin delivery (n = 5–8 animals/group/treatment).
(E) Representative western blots of liver homogenates from C57BL/6J mice of 10 and 81 weeks old which were either ad libitum fed or fasted for 4 h into the dark cycle. Gapdh was used as the loading control. Bold line indicates dissection.
(F) Densitometric analysis of autophagic marker proteins in liver homogenates from mice in (E) (n = 6–7 animals/group/treatment).
(G) Plasma corticosterone concentrations of mice in (E) (n = 6–7 animals/group/treatment).
(H) Change in body weight after 4 h of fasting into the dark cycle (n = 6–7 animals/group/treatment).
(I) Blood glucose concentrations of ad libitum fed and fasted C57BL/6J mice at ZT11 and ZT15 respectively (n = 6–7 animals/group/time point).
Data are represented as mean ± SEM. Statistical analyses were performed by two-way ANOVA followed by Šídák post hoc tests (for A, C, D, F, and G; without RM, for H and I; RM for treatment). ns, not significant, ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (see also Figure S14 for full western blots).
Next, we exposed C57BL/6J mice aged 10 and 81 weeks to the short-term 4-h fasting paradigm to assess hepatic autophagy. Here, we observed that fasting-induced LC3 lipidation was significantly reduced in aged compared with young mice, and this was accompanied by a marked reduction in circulating corticosterone concentrations during fasting in 81-week-old mice compared with 10-week-old mice (Figures 7E–7G and S14F). Although fasting-induced weight loss was comparable among the two groups of mice, 81-week-old mice presented with a blunted fasting-induced reduction of blood glucose concentrations similar to what was observed in fasted mice with AgRP neuron inhibition (Figures 4J, 7H, and 7I). Collectively, our experiments indicate that the regulation of liver autophagy is impaired in aged animals.
Discussion
Liver autophagy represents a central integrator of metabolic health. Although autophagy-dependent coordinated protein catabolism is important for liver homeostasis,42,43 this role has expanded to the degradation of lipids (lipophagy) and glycogen in the maintenance of metabolic health.16,44 Here, the role of autophagy in the control of glucose homeostasis is multifaceted. Although some studies indicate a role of autophagy in the control of glucose supply and gluconeogenesis during starvation,45 others have demonstrated the normal maintenance of glucose concentrations during starvation upon compromised liver autophagy.46 In addition, liver autophagy is also critical for ketogenesis during starvation.46 We demonstrate that the inhibition of AgRP neuron activation reduces ketone body production in the liver, whereas optogenetic activation of AgRP neurons induces increases in liver DAG, acylcarnitine, and hydroxybutyrate concentrations, indicative of an increased ß-oxidative flux. Thus, bi-directional regulation of autophagy on AgRP neuron manipulation is paralleled by bi-directional control of fasting-associated changes in the liver lipid metabolism. By contrast, blocking AgRP neuron-dependent activation of autophagy upon fasting prevents the fasting-induced reduction of blood glucose concentrations, despite inhibited expression of the gluconeogenic enzymes Pck1 and G6P, whereas AgRP neuron activation promotes the expression of key genes in gluconeogenesis, consistent with previous findings that insulin-induced inhibition of AgRP neurons suppress gluconeogenesis.8 Thus, AgRP neuron-dependent control of glucose metabolism appears to be more complex and likely involves the regulation of multiple tissues beyond the liver such as BAT.9
On a molecular level, AgRP neuron activation induces expression of the nuclear hormone co-activator PGC1a in the liver, and inhibition of AgRP neurons during starvation reduces hepatic PGC1a expression. Although acting as a co-activator of FOXO-dependent expression of key gluconeogenesis genes,47 PGC1a also functions as a co-activator of PPARa in the control of hepatic lipid metabolism48 and regulates ketogenesis in the liver.49 In addition, we detect a robust upregulation of Plin-5 expression on AgRP neuron activation. Plin5 can traffic lipid droplet-derived monounsaturated fatty acids to allosterically activate SIRT1 to regulate PGC1-dependent PPARa co-activation.50 Moreover, the upregulation of Lipin1 on AgRP neuron activation can act as another amplifier of the hepatic PGC1a/PPARa pathway.51 Finally, PPARa activation is critical for fasting-induced activation of autophagy and lipophagy in the liver.52 Collectively, our experiments indicate that bi-directional AgRP neuron-dependent regulation of PGC1/PPARa affects multiple pathways that coordinate ketogenesis and autophagy during fasting. Future studies are warranted to investigate the specific autophagic cargo subjected to degradation on AgRP neuron activation.
In light of these fundamental regulatory functions in molecular and organellar quality control and metabolic homeostasis, autophagy has emerged as central for the longevity-inducing effect of dietary restriction, impaired insulin signaling, germline ablation, and reduced mitochondrial function in C. elegans.53 Autophagy-dependent regulation of longevity in turn has been linked to its role in the control of the lipid metabolism.54 Therefore, the flexible and dynamic regulation of AgRP-neuron-dependent autophagy induction may potentially be harnessed to improve health span in multicellular organisms.
This may appear counter-intuitive at the first sight in light of the food intake promoting effects of AgRP neuron activation. Strategies to inhibit AgRP neuron activation have been viewed as promising for the development of new therapeutic approaches for obesity.3 However, although the hunger-promoting effect of AgRP neuron activation is rapidly observed during the first hours and days of chemogenetic or optogenetic AgRP neuron activation,6,7 prolonged AgRP neuron activation no longer increases food intake and fails to induce obesity.55 Moreover, as AgRP neurons inhibit anorexigenic POMC neurons via the release of GABA,56 it is hypothesized that increased GABAergic synaptic input onto POMC neurons in obesity is caused by prolonged AgRP neuron activation. Nevertheless, chemogenetic inhibition of AgRP neurons fails to reduce diet-induced inhibitory postsynaptic currents (IPSCs) on POMC neurons.57 We have recently identified prepronociceptin (PNOC)-expressing neurons as a novel class of GABAergic ARH neurons, which are activated by high-fat diet (HFD) feeding to inhibit POMC neurons, thus defining alternative GABAergic neurons in the ARH as diet-induced inhibitors of POMC neuron activity in obesity.58 Thus, the role of AgRP neuron activation in obesity development and the concept to inhibit them as an obesity treatment remains to be further defined.
On the other hand, the fact that the dynamic regulation of AgRP neurons is required for fuel usage adaptation indicates their necessity for their fine-tuned dynamic regulation in metabolic adaptation. This notion is further supported by studies that the ablation of AgRP neurons in newborn mice alters autonomic innervation of multiple organs and leads to altered fuel usage and obesity in normal chow diet (NCD)-fed mice.22 Similarly, chemogenetic activation of AgRP neurons controls fuel usage in part via control of lipolysis in white adipose tissue (WAT).23 Although these studies revealed important roles of AgRP neuron activation and metabolic adaptations in multiple organs, our findings identify a role for their dynamic regulation in the control of autophagy, lipid metabolism, and ketogenesis in the liver.
Given the importance of autophagy in the control of organismal health and survival, the regulatory steps of autophagy regulation have been extensively studied and unraveled over the past decade (for reviews see Kaushik et al.12 and Klionsky et al.15). These studies have largely centered on the processes governing the control of cell autonomous sensing of energy depletion. Here, the coordinated bi-directional control of the key nutrient-sensing pathways TOR and AMPK represent key regulators of autophagy.14 Nevertheless, it is important to consider that under survival-threatening conditions, multiple cell types have to adapt simultaneously for the survival of a multicellular organism. Therefore, the principle of non-cell autonomous control has been revealed for multiple stress-response signaling pathways, including ER-stress and mitochondrial stress across a broad evolutionary spectrum of species.59,60,61,62,63 In mice, specialized energy-sensing melanocortin neurons in the ARH are engaged in a similar regulatory principle. AgRP and POMC neurons integrate multiple signals, such as the nutrient-sensing hormones leptin, insulin, Ghrelin, and GLP-1 and nutrient components such as glucose, amino acids, and fatty acids directly.3 Moreover, through neurocircuits integrating sensory information, they adapt their activity already during anticipated changes of nutrient availability.10 Here, sensory food detection-dependent activation of POMC neurons promotes ER-stress signaling pathways in the liver to adapt to postprandial changes in proteostasis.11 Congruent with POMC neuron-dependent control of hepatic ER homeostasis, Atg7 expression in POMC neurons are critical for cold-induced activation of lipohagy and lipolysis in the liver and BAT.21 Interestingly, this pathway depends on sympathetic innervation of the BAT. Although acute cold stress engages autophagy-dependent activation of POMC neurons to rapidly activate liver autophagy within 1 h, presumably via the activation of SNA, starvation-induced, AgRP-dependent regulation of liver autophagy requires a longer activation, and this pathway depends on increasing circulating corticosterone concentrations. Although it was shown that the autophagy induction in POMC neurons is required for some of the beneficial metabolic effects of intermeal fasting,64 future studies should focus on investigating the potential contribution of AgRP versus POMC neuron-dependent control of liver autophagy to achieve the beneficial healthspan-expanding effects of caloric restriction and intermittent fasting.
Regarding the neurocircuitry involved in AgRP neuron-dependent control of liver autophagy and metabolic adaptation, we demonstrate that the stimulation of AgRP neuron projections in the PVH and LHA but not in the BNST is sufficient to elicit autophagy induction in the liver, paralleled by increased circulating corticosterone concentrations in an NPY-dependent manner. This points to specific effector sites in the AgRP neuron-dependent regulation of peripheral tissue functions. We have previously shown that at least in part AgRP → BNST projections regulate BAT function,9 whereas others have shown that the food intake-promoting effect of AgRP neuron activation is mediated through a broader network of AgRP neuron targets.32 Moreover, our data are consistent with the previous findings showing that the central administration of NPY increases feeding in part through PVH and LHA,65 acute feeding regulatory effects of AgRP neurons in the PVH engage NPY signaling,33 and central NPY action is linked to the control of liver glucose metabolism.66 Our study on re-expression of NPY in AgRP neurons further highlights the necessity for NPY released from AgRP neurons in the control of liver autophagy. Interestingly, regarding autophagy, a few studies have revealed that NPY promotes autophagy within the CNS.67 NPY has been shown to stimulate autophagy in hypothalamic neurons through the activation of NPY Y1 and Y5 receptors.68 Importantly, we have now also unraveled that the inhibition of NPY1R-expressing neurons targeted by PVH-injection of AAV-hM4Di activates hepatic autophagy and activation of CRHPVH neurons to promote HPA-axis activation likely via the pre-synaptic inhibition of GABAergic neurons, which suppress CRHPVH neurons.
Finally, we provide several lines of independent evidence that AgRP neuron/NPY-dependent induction of liver autophagy is mediated via increases in circulating corticosterone concentrations. The role of glucocorticoids in the control of hepatic gluconeogenesis has been extensively studied. Importantly, PGC1a, which is upregulated on AgRP neuron activation, acts as a coactivator for GR-dependent expression of Pck1 and G6P in an HNF4a-dependent manner, whereas the PGC1a-dependent induction of ß-oxidation and ketogenesis is HNF4a-independent.49 Interestingly, Lipin1 expression in the liver, which is upregulated in response to AgRP neuron activation, are a direct target for GR-dependent transcription in both adipocytes and hepatocytes.69,70 In addition to its role in controlling the expression of gluconeogenic enzymes, GR has recently been linked to the expression of Gck in hepatocytes in the control of liver glycogen synthesis. In addition, multiple studies have revealed a role for GR signaling in the control of autophagy across multiple tissues. Although GR activation inhibits chaperone-mediated autophagy and microautophagy,71 dexamethasone treatment induces autophagy in neurons and BAT72,73 and skeletal muscle.74 Thus, further defining the complex targets of GR signaling in the liver may broaden our understanding of its role in the complex metabolic adaptation of the liver to energy deprivation.
Limitations of the study
We acknowledge some limitations of the present study. The induction of liver autophagy was observed in both sexes of mice in response to the activation of AgRP neurons. Although each experimental group was sex-matched, it limits our ability to pinpoint potential sex differences in the degree of liver autophagy induction. Although our study indicates that AgRP neurons play an important role in controlling hepatic autophagy during nutrient deprivation, it will be important to assess the cargo(es) within the autophagosomes to determine whether AgRP neurons potentially regulate selective autophagy such as mitophagy or lipophagy. Interestingly, protein expression of p62 as a marker of autophagy did not always inversely correlate with autophagy activity in our study. However, p62 regulation during autophagy is complex and may be influenced by transcriptional regulation. Therefore, it will be an important aspect to study AgRP neuron-dependent regulation of p62-independent autophagy pathways.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal Gapdh | Cell Signaling Technology | Cat# 2118; RRID:AB_561053 |
| Mouse monoclonal SQSTM1/p62 | Abcam | Cat# ab56416; RRID:AB_945626 |
| Rabbit Polyclonal LC3B | Cell Signaling Technology | Cat# 2775; RRID:AB_915950 |
| Rabbit monoclonal LC3B (D11) XP® | Cell Signaling Technology | Cat# 3868S; RRID:AB_2137707 |
| Rabbit Monoclonal NPY | Cell Signaling Technology | Cat# 11976; RRID:AB_2716286 |
| mCherry Monoclonal Antibody (16D7) | Invitrogen™ | Cat# M11217; RRID:AB_2536611 |
| Donkey Polyclonal anti-Rabbit IgG (H+L), Alexa Fluor 647 | Invitrogen™ | Cat# A-31573; RRID:AB_2536183 |
| Donkey Polyclonal anti-Rat IgG (H+L), Alexa Fluor 594 | Invitrogen™ | Cat# A-21209; RRID:AB_2535795 |
| Goat polyclonal anti-mouse IgG-peroxidase | Sigma Aldrich | Cat# A4416; RRID:AB_258167 |
| Goat polyclonal anti-rabbit IgG-peroxidase | Sigma Aldrich | Cat# A6154; RRID:AB_258284 |
| Rabbit polyclonal TFEB | Cell Signaling Technology | Cat# 4240S; RRID:AB_11220225 |
| Rabbit monoclonal Phospho-Atg14 (Ser29) | Cell Signaling Technology | Cat# 92340S; RRID:AB_2800182 |
| Rabbit monoclonal Atg14 | Cell Signaling Technology | Cat# 96752S; RRID:AB_2737056 |
| Rabbit monoclonal S6 (5G10) | Cell Signaling Technology | Cat# 2217S; RRID:AB_331355 |
| Rabbit polyclonal Phospho-S6 (Ser235/236) | Cell Signaling Technology | Cat# 2211S; RRID:AB_331679 |
| Rabbit polyclonal AMPK | Cell Signaling Technology | Cat# 2532L; RRID:AB_330331 |
| Rabbit monoclonal Phospho-AMPK (Thr172, 40H9) | Cell Signaling Technology | Cat# 2535S; RRID:AB_331250 |
| Rabbit monoclonal AKT (pan, 11E7) | Cell Signaling Technology | Cat# 4685S; RRID:AB_2225340 |
| Rabbit monoclonal Phospho-AKT (Ser473, D9E) | Cell Signaling Technology | Cat# 4060L; RRID:AB_2315049 |
| Chicken polyclonal GFP | Abcam | Cat# ab13970; RRID:AB_300798 |
| Rabbit polyclonal AgRP (83-132) amide (Human) | Phoenix Pharmaceutical | Cat# H-003-53; RRID:AB_2313908 |
| Bacterial and virus strains | ||
| AAV9-hSyn-DIO-mCherry | Addgene | Cat# 50459-AAV9; RRID:Addgene_50459 |
| AAV9-hSyn-DIO-hM4D(Gi)-mCherry | Addgene | Cat# 44362-AAV9; RRID:Addgene_44362 |
| AAV8-EF1a-DIO-mNPY-WPRE-1 | Assoc. Prof. Zachary A. Knight | Chen et al.29 |
| AAV2/10-Flex-hGlyR-mCherry | Dr. Henning Fenselau | Lynagh and Lynch34 |
| AAV8-GFP-U6-Scrmb-shRNA | Vector Biolabs | N/A |
| AAV8-GFP-U6-mNR3C1-shRNA | Vector Biolabs | Cat# shAAV-266053 RefSeq# NM_008173 |
| Chemicals, peptides, and recombinant proteins | ||
| Tris Base | Sigma Aldrich | Cas# 77-86-1 |
| EDTA | Sigma Aldrich | Cat# E6758 |
| PMSF | AppliChem | Cat# A0999 |
| NAF | Sigma Aldrich | Cat# S7920 |
| cOmplete™, Mini Protease Inhibitor Cocktail | Roche | Cat# 11836153001 |
| PhosSTOP™, phosphatase inhibitor | Roche | Cat# 4906837001 |
| Western Blocking Reagent, Solution | Roche | Cat# 11921681001 |
| SignalStain® Antibody Diluent | Cell Signaling Technology | #8112L |
| Sucrose | Sigma Aldrich | Cas# 57-50-1 |
| Paraformaldehyde | Sigma Aldrich | Cas# 30525-89-4 |
| 4x Laemmli sample buffer | Bio-Rad | Cat# 161-0747 |
| Tween® 20 (Polysorbate) | VWR France | Cas# 9005-64-5 |
| β-Mercaptoethanol | Sigma Aldrich | Cat# M3148 |
| DMEM, high glucose, GlutaMAX™ | Gibco™ | Cat# 61965-026 |
| Fetal Calf Serum (FCS; Premium) | PAN Biotech | Cat# P30-3302 |
| Penicillin-Streptomycin | Gibco™ | Cat# 15140-122 |
| Clozapine N-oxide, Metabolite of Clozapine | Abcam | Cat# ab141704 |
| Ghrelin (Rat, Mouse) | Phoenix Pharmaceutical | Cat# 031-31 |
| Dexamethasone (in vitro) | Sigma Aldrich | Cat# D4902-100MG |
| Dexamethasone 21-phosphate disodium salt (in vivo) | Sigma Aldrich | Cat# D1159 |
| Leupeptin | Sigma Aldrich | Cat# L8511-100MG |
| Ammonium Chloride | Sigma Aldrich | Cat# A9434-500G |
| Donkey Serum | Sigma Aldrich | Cat# S30-100ML |
| Buprenorphine | Bayer | PZN 01498870 |
| Meloxicam | Boerhinger Ingelheim | PZN 07578423 |
| Ivermectin | Sigma Aldrich | Cat# I8898 |
| Glycerol formal | Sigma Aldrich | Cat# 49920 |
| 1,2-Propanediol | Sigma Aldrich | Cat# 398039 |
| Benzoylchloride | Sigma Aldrich | Cat# 259950-5ML |
| Ammonium acetate | Sigma Aldrich | Cat# 73594 |
| Ammonium formate | Sigma Aldrich | Cat# 70221 |
| 13C10 ATP internal standard | Sigma Aldrich | Cat# 710695 |
| Formic acid | Signa Aldrich | Cat# 27001 |
| Glacial Acetic acid | Sigma Aldrich | Cat# 338826 |
| Equisplash Lipido Mix internal standard | Avanti | Cat# 330731 |
| Metabolomics Amino acid Mix internal standard | Cambridge Isotopes | MSK A2 |
| Optima Acetonitrile LC/MS Grade | Fisher Scientific | Cat# A955-212 |
| Optima Methanol LC/MS Grade | Fisher Scientific | Cat# A456-212 |
| Optima Isopropanol LC/MS Grade | Fisher Scientific | Cat# A461-212 |
| Methyl-tert-butyl-ether | Sigma Aldrich | Cat# 306975 |
| Chromasolv LC-MS Ultra Water | Honeywell | Cat# 14263 |
| Critical commercial assays | ||
| Mouse ACTH ELISA Kit | Abcam | Cat# ab263880 |
| Mouse Glucagon ELISA Kit | Crystal Chem | Cat# 81518 |
| Mouse Corticosterone ELISA Kit | Crystal Chem | Cat# 80556 |
| Ultra-Sensitive Mouse Insulin ELISA Kit | Crystal Chem | Cat# 90080 |
| mirVana™ miRNA Isolation Kit | Invitrogen™ | Cat# AM1561 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| High-Capacity CDNA Reverse Transcription Kit | Applied Biosystems™ | Cat# 4368814 |
| TruSeq® Stranded mRNA Library Prep | Illumina | Cat# 20020595 |
| RNA 6000 Nano Kit | Agilent Technologies | Cat# 5067-1511 |
| RNA 6000 Nano Ladder | Agilent Technologies | Cat# 5067-1529 |
| RNAscope Fluorescent Multiplex Detection Reagents v2 | ACD | Cat# 323100 |
| SuperSignal™ West Dura Extended Duration Substrate | Thermo Fisher Scientific | Cat# 34075 |
| Takyon™ Low ROX Probe 2X MasterMix dTTP blue | Eurogentec | Cat# UF-LPMT-B0701 |
| Probe Mm-cFos-C1 | ACD-RNAscope Probe | Cat# 316921 |
| Probe Mm-cFos-C2 | ACD-RNAscope Probe | Cat# 316921-C2 |
| Probe Mm-AgRP-C2 | ACD-RNAscope Probe | Cat# 400711-C2 |
| Probe Mm-POMC-C3 | ACD-RNAscope Probe | Cat# 426141-C3 |
| Probe Mm-GhSR-C3 | ACD-RNAscope Probe | Cat# 314081-C3 |
| Probe Mm-NPY-C3 | ACD-RNAscope Probe | Cat# 313321-C3 |
| Probe Mm-NPY1R-C1 | ACD-RNAscope Probe | Cat# 427021 |
| Probe Mm-NPY1R-C3 | ACD-RNAscope Probe | Cat# 427021-C3 |
| Probe Mm-CRH-C1 | ACD-RNAscope Probe | Cat# 316091 |
| Probe Mm-Slc17a6-C2 | ACD-RNAscope Probe | Cat# 428871-C2 |
| Probe Mm-Slc17a6-C3 | ACD-RNAscope Probe | Cat# 428871-C3 |
| Probe Mm-Slc32a1-C2 | ACD-RNAscope Probe | Cat# 319191-C2 |
| Probe Mm-Slc32a1-C3 | ACD-RNAscope Probe | Cat# 319191-C3 |
| Opal 520 Fluorophore | Akoya Bioscience | Cat# FP1487001KT |
| Opal 570 Fluorophore | Akoya Bioscience | Cat# FP1488001KT |
| Opal 650 Fluorophore | Akoya Bioscience | Cat# FP1496001KT |
| Opal 690 Fluorophore | Akoya Bioscience | Cat# FP1497001KT |
| High-Select™ TiO2 Phosphopeptide Enrichment Kit | Thermo Fisher Scientific | Cat# A32993 |
| Deposited data | ||
| RNAseq | This paper | NCBI GEO: GSE188472 |
| Phosphoproteomics | This paper | PRIDE: PXD029247 |
| Experimental models: Cell Lines | ||
| Hepa1-6 | ATCC | (ATCC CRL-1830™) passage number 33; RRID: CVCL_0327 |
| Experimental models: Organisms/strains | ||
| C57BL/6N | Charles River Laboratories | Strain# 027 |
| Aged C57BL/6JRj | Janvier labs | Strain# C57BL/6JRj |
| AgRP-IRES-Cre | Tong et al.75 | |
| ChR2fl/fl mice | Madisen et al.76 | |
| NPYΔ/Δ mice | Erickson et al.31 | |
| hM3DGqfl/fl | Steculorum et al.9 | |
| NPY1R-Cre | Padilla et al.77 | |
| Oligonucleotides | ||
| TaqMan-Probe Trp53inp1 | Thermo Fisher Scientific | Mm00458141_m1 |
| TaqMan-Probe ULK1 | Thermo Fisher Scientific | Mm00437238_m1 |
| TaqMan-Probe Bag3 | Thermo Fisher Scientific | Mm00443474_m1 |
| TaqMan-Probe Sesn2 | Thermo Fisher Scientific | Mm00460679_m1 |
| TaqMan-Probe Atp6v0d1 | Thermo Fisher Scientific | Mm00442694_m1 |
| TaqMan-Probe Plin5 | Thermo Fisher Scientific | Mm00508852_m1 |
| TaqMan-Probe Lpin1 | Thermo Fisher Scientific | Mm00550511_m1 |
| TaqMan-Probe Lpin2 | Thermo Fisher Scientific | Mm00522390_m1 |
| TaqMan-Probe Lipg | Thermo Fisher Scientific | Mm00495368_m1 |
| TaqMan-Probe Pck1 | Thermo Fisher Scientific | Mm01247058_m1 |
| TaqMan-Probe G6pc | Thermo Fisher Scientific | Mm00839363_m1 |
| TaqMan-Probe Ppara | Thermo Fisher Scientific | Mm00440939_m1 |
| TaqMan-Probe Ppargc1a | Thermo Fisher Scientific | Mm01208835_m1 |
| TaqMan-Probe Cd36 | Thermo Fisher Scientific | Mm00432403_m1 |
| TaqMan-Probe Foxo1 | Thermo Fisher Scientific | Mm00490671_m1 |
| TaqMan-Probe Nr3c1 | Thermo Fisher Scientific | Mm00433832_m1 |
| TaqMan-Probe Tbp (Dye: VIC-MGB) | Thermo Fisher Scientific | Mm00446973_m1 |
| Software and algorithms | ||
| Fiji/ImageJ software | Schindelin et al.78 | https://imagej.net/software/fiji/ |
| GraphPad Prism (Version 9.1.0) | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| 2100 Expert Software | Agilent Technologies | Part Number:G2953CA |
| Spectronaut 15.5.211111.50606 | Biognosys AG | www.biognosys.com |
| InstantClue 0.11.0 | Nolte et al.79 | www.instantclue.de |
| TraceFinder 4.1 | Thermo Fisher Scientific | OPTON-30626 |
| LIPIDSEARCH 4.2 | Thermo Fisher Scientific | OPTON-30880 |
| Other | ||
| Normal Chow Diet (ssniff R/M-H Phytoestrogenarm) | ssniff Spezialdiäten GmbH | Cat# V1554 |
| 4–15% Criterion™ TGX™ Precast Midi Protein Gel | Bio-Rad | Cat# 5671085 |
| Any kD™ Criterion™ TGX™ Precast Midi Protein Gel | Bio-Rad | Cat# 5671125 |
| Trans-Blot (R)Turbo, Midi Format, 0.2mm PVDF | Bio-Rad | Cat# 1704157 |
| Epredia™ SuperFrost Ultra Plus™ GOLD Adhesion Slides | Thermo Fisher Scientific | Cat# 11976299 |
| Vectashield Antifade Mounting-Medium with DAPI | Vector Laboratories | Cat# H-1200 |
| Contour Blood Glucose Meter | Bayer | N/A |
| Contour Next strips | Bayer | Cat# #84167879 |
| Leica TCS SP-8-X Confocal microscope | Leica Microsystems | https://www.leica-microsystems.com/products/confocal-microscopes/p/leica-tcs-sp8-x/ |
| 2100 Bioanalyzer Instrument | Agilent Technologies | Part Number:G2939BA |
| FUSION Solo | Vilber Lourmat | http://www.vilber.de/produkte/chemilumineszenz/ |
| Laser Diode Fiber Light Source (473 nM) | Doric Lenses | LDFLS_473/070 |
| Mono Fiber-optic Patch Cords (200 μM – NA0.48) | Doric Lenses | MFP_200/230/900-0.48_0.32m_FC-CM3(P) |
| Phenomaster | TSE Systems | https://www.tse-systems.com/service/phenomaster/ |
| Exploris 480 Mass Spectrometer | Thermo Fisher Scientific | Cat# BRE725532 |
| nLC 1200 Liquid Chromatography | Thermo Fisher Scientific | Cat# LC140 |
| Buffer A: 0.1% (v/v) formic acid | Thermo Fisher Scientific | LS118-1 |
| Buffer B: 0.1% (v/v) formic acid in 80% acetonitrile | Thermo Fisher Scientific | LS122 |
| PoroShell 120 EC-C18 2.7-μm beads | Agilent Technologies | 899999-77 |
| ORBITRAP ID-X, | Thermo Fisher Scientific | FSN05-10001 |
| Q-Exactive HF | Thermo Fisher Scientific | 0726055 |
| Q-Exactive Plus | Thermo Fisher Scientific | IQLAAEGAAPFALGMBDK |
| Waters i-Class UPLC | Waters | www.waters.com |
| CQUITY UPLC BEH C8 Column, 130Å, 1.7 μm, 2.1 mm X 100 mm | Waters | 176000885 |
| ACQUITY UPLC HSS T3 Column, 100Å, 1.8 μm, 2.1 mm X 100 mm | Waters | 186003539 |
| Integrion Anion Exchange chromatography system | Thermo Fisher Scientific | 22153-60306 |
| IonPac AS11-HC, 4μm, 2x250mm | Thermo Fisher Scientific | 078035 |
| VANQUISH FLEX QUATERNARY UHPLC chromatograpy system | Thermo Fisher Scientific | 5400.0205 |
| Screw caps for glass and polypropylene HPLC vials | CZT | Cat# 3111L3010 |
| Polyprpylene HPLC vials | CZT | Cat# 451127756 |
| Glass HPLC vials | CZT | Cat# 451100012_E |
| 200 μL-glass inserts for glass HPLC vials | CZT | Cat#501106012 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jens Claus Brüning (bruening@sf.mpg.de)
Materials availability
No unique reagents were generated in this study.
Experimental model and subject details
Animal husbandry
All animal procedures were conducted in compliance with protocols approved by local government authorities (Bezirksregierung Köln) and were in accordance with NIH guidelines. Mice breeding and maintenance was permitted by the Department for Environment and Consumer Protection – Veterinary Section, Köln, North Rhine-Westphalia, Germany. Mice were housed in individually ventilated cages (IVCs) within a controlled environment at 22°C – 24°C and a 12-hr light/12-hr dark cycle. Mice were checked daily to verify animal health and the health status monitoring of the animal facility was performed quarterly every year. Mice were housed in groups of not more than 5 mice per cage after weaning. Animals received ad libitum access standard rodent chow (ssniff V1554, 59494 Soest, Germany) and water. Unless otherwise indicated, all the mouse lines generated in the paper were maintained in house on a C57BL/6N background. Experimental groups were matched for both male and female sexes. Majority of the experimental procedures were performed in adult mice between 10 and 22 wks of age. Aging studies were performed on C57BL/6J male mice between 10 and 81 wks of age.
Mouse lines
C57BL/6N
This mouse line was obtained from Charles River, Germany.
Aged C57BL/6J
This mouse line was obtained from Janvier, France.
AgRP-IRES-Cre
Mice were obtained from breeding in house in the facility of the Max Planck Institute for Metabolism Research, Cologne, NRW, Germany, and their generation has been described.75
NPY1R-Cre
Mice have been previously described,77 and were obtained from the Jackson Laboratories (stock# 030544)
ChR2fl/fl mice
(ROSA26loxSTOPloxChR2(H134R)-EYFP-WPRE) with a conditional allele (Ai32),76 were obtained from the Jackson Laboratories (stock# 012569). These mice were maintained as homozygous animals in the facility of the Max Planck Institute for Metabolism Research, Cologne, NRW, Germany.
NPYΔ/Δ mice
Whole body NPY-deficient mice were on a 129sv background.31
hM3DGqfl/fl
Mice (ROSA26CAGSloxSTOPloxhM3DGq) has been previously described.9 These mice were maintained as homozygous fl/fl animals.
Generation of experimental mouse Lines
ChR2AgRP
Heterozygous AgRP-IRES-Cre mice were bred to homozygous ChR2fl/fl mice to generate ChR2AgRP mice as experimental animals. ChR2WT littermates were used as controls.
ChR2AgRP-NPYΔ/Δ
The resultant offspring from breeding AgRP-IRES-Cre mice and ChR2fl/fl mice were crossed with whole body NPY deficient (NPYΔ/Δ) mice. Further breedings were performed with ChR2fl/fl mice in order to generate homozygosity for the ChR2fl/fl allele. To generate the four experimental groups, ChR2fl/fl; AgRP-IRES-CreCre/wt; NPYΔ/wt mice were bred with heterozygous NPYΔ/wt mice. The resulting four experimental groups were all heterozygous ChR2fl/wt.
hM3DGqAgRP-NPYΔ/Δ
Similar breeding strategy were performed to generate mice for the chemogenetics experiments. AgRP-IRES-CreCre/wt mice with hM3DGqfl/fl mice and subsequently with the NPYΔ/Δ mice. To generate the four experimental groups, hM3DGqfl/fl; AgRP-IRES-CreCre/wt; NPYΔ/wt mice were bred with hM3DGqfl/fl; AgRP-IRES-Crewt/wt; NPYΔ/wt. The resulting four experimental groups were all homozygous hM3DGqfl/fl.
Cell line
Hepa1-6 cells (gender: female) were obtained from ATCC (ATCC® CRL-1830) passage number 33 and were not further authenticated in our laboratory. They were tested negative for ectromelia virus by ATTC and for mycoplasma in our laboratory. Cells were maintained under standard conditions in humidified incubator (Binder International) at 37°C, 5% CO2 in DMEM (1x) + GlutaMAX™ Dulbeccos Modified Eagle Medium; 4.5 g/l D-glucose (GIBCO) supplemented with 10% fetal bovine serum and 5% penicillin/streptomycin. Cells were passaged every 3 days at 80 - 90% confluency.
Method details
Mouse experiments
Short-term fasting in the dark cycle
To circumvent potential disruption of circadian rhythms resulting from light exposure, mice were individually housed at least one week prior to fasting experiments. At ZT11, basal body weight and blood glucose levels were recorded before food was removed. At the same time, mice and their respective nesting were transferred into clean cages to minimise stress to ensure no access to residual food spills at the bottom of the cage. The control group received ad libitum access to food and water throughout the fasting period. After 4 or 6 hours of fasting, body weight and blood glucose levels were measured, mice were anesthetized in the dark and sacrificed at ZT15 and ZT17 respectively (see Figure S1A). Similar paradigm was used to study the effects of aging (10 wks, and 81 wks old C57BL/6J mice) on short-term fasting-induced HPA axis activation and hepatic autophagy.
Intraperitoneal injection in C57BL/6N
Ip injection of Ghrelin. In a separate cohort of C57BL/6N mice, animals received an ip injection of Ghrelin (1.5 μg in 100 μL) or saline at ZT1 and ZT3 in the absence of food. Mice were sacrificed at ZT5 and liver samples were removed for post hoc analysis of autophagic markers using Western blotting. Thereafter, animals were perfused transcardially, fixed with 4% PFA and brains were removed for the staining of ghrelin receptor expressing AgRP neurons using RNAscope.
Ip injection of Dexamethasone. In a separate cohort of C57BL/6N mice, food was removed 1 h (ZT2.5) before they receive a single ip injection of dexamethasone (5 mg/kg) or saline at ZT3.5 in the absence of food. Mice were rapidly decapitated at ZT7.5 and liver and trunk blood samples were collected (see Figure 6E).
AAV mediated knockdown of glucocorticoid receptor (Nr3c1) in the liver. An adeno-associated virus (AAV) serotype 8 encoding an shRNA under the control of the U6 promoter, targeting murine Nr3c1 was used (AAV8-GFP-U6-mNr3c1-shRNA; Vector Biolabs). Briefly, a total volume of 100 μL of AAV8-GFP-U6-mNr3c1-shRNA or a respective control virus (AAV8-GFP-U6-scrambled-shRNA; Vector Biolabs), containing 2.51011 GC in PBS was delivered via the tail vein injection into ChR2AgRP mice. The insertion of an optical fiber cannula above the ARH was performed one week later (see Figure 6J).
Stereotaxic surgical procedures
Mice were anesthetized with isoflurane and placed onto a stereotaxic frame (David Kopf Instruments). To relief postoperative pain, mice received buprenorphine (0.1 mg/kg) and meloxicam (5 mg/kg). Three consecutive days of post-surgical care included tramadol in the drinking water (1 mg/mL), twice daily inspection and once daily body weight measurement to document full recovery of the experimental mice.
Fiber placements. Flat tip optical fibers (fiber core = 200 μm, numerical AP = 0.48; Doric lenses Inc.) were implanted above the targeted region using the corresponding coordinates from Bregma: ARH (antero-posterior (AP): -1.45 mm; medio-lateral (ML): 0.2 mm; dorso-ventral (DV): -5.2 mm), PVH (AP: -0.82 mm, ML: 0.25 mm, DV: -4.2 mm), LHA (AP: -1.6mm, ML: +1.05 mm, DV: -4.55 mm) and BNST (AP: 0.45 mm, ML: 0.5 mm, DV: -3.8 mm). Dental acrylic was applied to affix the cannula securely to the skull. For viral-mediated NPY re-expression and liver-specific shRNA knock-down of Nr3c1 experiments, optical fibers were placed above the ARH (AP: -1.45 mm; ML: 0.0 mm; DV: -5.2 mm).
Virus injections. For chemogenetic inhibition of AgRP neurons, ∼300 nL of adeno-associated virus (AAV) expressing mCherry or hGlyR (AAV9-hSyn-DIO-mCherry or AAV2/10-FLEX-hGlyR-mCherry) was bilaterally injected into the ARH (coordinates from bregma AP: -1.45 mm; ML: ± 0.25 mm; DV: -5.75 mm to -5.93 mm) at 100 nL per min and the glass pipette was withdrawn 5 min after injection. Similar technique was employed for the re-expression of NPY in the ARH, in which ∼300 nL of AAV8-EF1a-DIO-NPY virus (kindly provided by Dr. Z. Knight) or control virus AAV8-hSyn-DIO-mCherry was bilaterally injected followed by the insertion of an optical fiber cannula. For the inhibition of PVHNPY1R and LHANPY1R neurons, ∼50-75 nL of adeno-associated virus (AAV) expressing hM4Di (AAV8-DIO-hM4Di-mCherry) was bilaterally injected into the PVH (coordinates from bregma AP: -0.70 mm; ML: ± 0.20 mm; DV: -4.60 mm) or LHA (coordinates from bregma AP: -1.55 mm; ML: ± 1.10 mm; DV: -5.00 mm) respectively.
Intracerebroventricular cannulation. Briefly, 26-gauge icv cannulas were stereotaxically placed in the lateral ventricle of ∼10-12 weeks old C57BL/6N mice using coordinates from bregma, AP: -0.2 mm, ML: 1.0 mm, DV: -2.1 mm). Dental acrylic (Super Bond C&B) was used to secure the cannula to the surface of the skull. The cannula was sealed at the external opening with a dummy cannula. Animals were housed individually after surgery and given a week of recovery before experimental manipulations. Similar technique was used for central delivery of Ghrelin in 15 wks and 78 wks old C57BL/6J mice for the aging study.
In vivo optogenetic photostimulation
Following bilateral virus injection and/or optical fiber implantation, mice were allowed a post-surgical recovery period of 1-3 weeks. Subsequently, mice were acclimated in experimental cages (TSE system), for optimal habituation to food hoppers and water dispensers. One week prior to experimental day, mice were habituated to a fiber-optic patch cord (core diameter 200 μm, numerical AP 0.48; Doric lenses) connected to a rotary joint (Doric lenses). On the day of experiment, food spills were removed from the food hoppers, mice were moved to clean cages and new fiber-optic patch cord was attached at ZT1. At ZT3.5 (4 h stimulation) and ZT5.5 (2 h stimulation), blue laser (473 nm) light stimulation consisting of pulse trains (10 ms pulses of 20 Hz; 1 s on, 3 s off) was delivered. For ARH photostimulation, a laser power of ∼20 mW was employed. For PVH, LHA and BNST photostimulation, two respective laser powers of ∼20 mW and ∼10 mW were used. The irradiance in the targeted region was calculated using the online tool at https://web.stanford.edu/group/dlab/cgi-bin/graph/chart.php, hence above the threshold for activation of ChR2 (∼1 mW/mm2). All photostimulated mice were decapitated at ZT7.5 and liver and blood samples are rapidly retrieved. Brains were post fixed in 4% PFA and the placements of the optical fibers were verified post hoc.
Intracerebroventricular injection of Ghrelin in C57BL/6N mice
Mice had ad libitum access to food and water. On the experimental day, food intake and locomotor activity were measured after 6 h, which consists of 1 h (Pre-Ghrelin injection between ZT0 and ZT1), 4 h (Ghrelin stimulated between ZT1 and ZT5) and 1h (Post-ghrelin injection between ZT5 and ZT6) measurements. Given the short half-life of Ghrelin, mice received an icv injection of either ghrelin (1.5 μg) or saline in a final volume of 1.5 μL at ZT1 and ZT3 to mimic 4 h activation of AgRP neurons. Subsequent food intake and locomotion measurement were conducted in a crossover fashion with one week wash-out period between the experiments. On the day of sacrifice, food was removed and mice were transferred to clean cages at ZT0 to ensure no food access. Similarly, a single icv Ghrelin injection was performed at ZT1 and ZT3 respectively. At ZT5, mice were deeply anesthetized, liver samples were rapidly removed and snap frozen in liquid nitrogen. Then, animals were perfused transcardially, fixed with 4% PFA and brains were removed. Finally, the activation of ghrelin receptor expressing AgRP neurons was verified using RNAscope (see Figure S3G).
Food intake and locomotor activity measurements
An automated recording system (PhenoMaster, TSE Systems) was employed for simultaneous measurement of food, water and locomotor activity. Mice were acclimatized to food hoppers and water dispensers placed in training cages one week prior to the experiment. For consistency, mouse bedding was changed at the beginning of the light cycle (between ZT0 and ZT2) on experimental day to remove food spills. For optogenetic experiments, old mouse nesting was returned to each individual mouse to facilitate acclimatization following 1 h of laser off basal recordings (pre). After which, mice received a total of 4 h optogenetic laser stimulation, followed by 1 h of post-stimulation recordings (laser off). For ip or icv ghrelin experiments, see above.
Chemogenetic activation and inhibition of AgRP neurons
Ip injection of CNO. Clozapine-N-Oxide (CNO) powder was dissolved in DMSO (10 mg/mL) and diluted 1:100 in 0.9% NaCl. Mice were individually housed at least one week prior to CNO injections. hM3dGqWT, hM3dGqAgRP, hM3dGqNPY Δ/Δ and hM3dGqAgRP-NPY Δ/Δ mice were fed ad libitum and food was removed 1 h (ZT2.5) prior to intraperitoneal injections of CNO (1 mg/kg body weight) at ZT3.5 and ZT5.5 respectively. Mice were sacrificed at ZT7.5 on the experimental day (see Figure 2E). For the inhibition of PVHNPY1R and LHANPY1R neurons, mice received either saline or CNO (3 mg/kg body weight).
Ip injection of Ivermectin. Singly housed AgRP-IRES-CreCre/wt mice injected with AAV expressing mCherry or hGlyR received ip injections of Ivermectin (5 mg/kg in propylene glycol) at ZT3,35 and mice received ad libitum access to food and water. Food was removed at ZT11 and mice were sacrificed at ZT15 (see Figure 4A).
Autophagy flux assay
Ip injection of Leupeptin. Autophagic flux is inferred on the basis of LC3-II turnover assessed by Western blotting in the presence and absence of lysosomal inhibitor; leupeptin is a protease inhibitor which blocks the autophagosome-lysosome fusion step.24 For autophagy flux assessment in vivo, leupeptin (40 mg/kg body weight) was delivered ip 1 hour after a short-term fast into the night feeding cycle (see Figure S5), optogenetic stimulation of AgRP neuron (see Figure 1J) or ip Dexamethasone treatment (see Figure 6E). Livers were then extracted and immunoblotted for LC3B after each respective experimental paradigm. Rate of ‘destined’ autophagolysosome fusion was determined by dividing each LC3-II densitometric value by the average LC3-II of Leu+ treated+ value. Net LC3-II flux termed autophagy flux was calculated by subtracting the densitometric value of Leu-untreated LC3-II from corresponding Leu-treated LC3-II value.
Analysis of gene expression
Total murine liver RNA was isolated using the mirVana Isolation Kit (Ambion) in accordance to the manufacturer’s instructions. The isolated RNA was reverse-transcribed with High-Capacity cDNA RT Kit and amplified with Takyon LowRox MasterMix dTTP Blue (Eurogentec). Quantitative PCR was performed using the QuantStudio 7 Flex Real-Time PCR System using the QuantStudio Real-Time PCR Software v1.7.1 (Life Technologies). Relative expression of samples was adjusted for total RNA content and normalized to the mRNA expression levels of the TATA-binding protein (Tbp). Data was analyzed using the 2-ΔΔCt method (see key resources table for list of primers).
Transcriptomics
RNA sequencing. Murine liver RNA concentration was measured by Nanodrop ND-100 (Peqlab Biotechnology, France), and diluted to a final concentration of 200 – 250 ng/mL. RNA integrity number (RIN) was analyzed with the Agilent RNA 6000 Nano Kit (5067-1511, Agilent Technologies) and the Agilent 2100 Bioanalyzer. All samples displayed a RIN of >8. RNA libraries were prepared with 1 μg of total RNA using the Illumina® TruSeq® mRNA stranded sample preparation Kit. After poly-A selection (using poly-T oligo-attached magnetic beads), mRNA was purified and fragmented using divalent cations under elevated temperatures. The RNA fragments underwent reverse transcription using random primers. This is followed by second-strand cDNA synthesis with DNA Polymerase I and RNase H. After end repair and A-tailing, indexing adapters were ligated. The products were then purified and amplified (14 PCR cycles) to create the final cDNA libraries. After library validation and quantification (Agilent tape station), equimolar amounts of the library were pooled. The pool was quantified by using the Peqlab KAPA Library Quantification Kit and the Applied Biosystems 7900HT Sequence Detection System. The pool was sequenced for 37.5 million reads using an Illumina NovaSeq6000 instrument and a PE100 sequencing protocol.
Transcriptomics analysis. We applied the community-curated nfcore rnaseq analysis pipeline80 version 3.0.81 The gene-level quantification was carried out using Salmon 1.4.0 using the reference genome GRCm38.82 The differential gene expression analysis was done using the DESeq2 1.32.0 R package based on a model using a negative binomial distribution.83 P-values were FDR adjusted using Wald test. Gene-ontology term analysis was carried out using the clusterProfiler R package implementing an over-representation test using hypergeometric distribution modelling.84 P-values were FDR adjusted using Benjamini–Hochberg correction.
Phosphoproteomics
Protein Digestion. Liver lysates were generated using the Tris 50 mM, NaCl 130 mM, EDTA 5 mM, NP-40 1%, PMSF 100 mM, NaF 100 mM, cOmplete mini protease inhibitor cocktail (1 tablet/10 mL) and PhosSTOP™ protease inhibitor (1 tablet/10 mL) were freshly added. Proteins were reduced using TCEP and alkylated by CAA for 45 min at 45°C by adding both chemicals simultaneously to a final concentration of 10 mM and 20 mM respectively. Protein concentration was determined and 4000 μg protein were subjected for acetone precipitation (4x volume, -20°C overnight). The pellet was washed twice using ice-cold 80% acetone. Trypsin (Sigma Aldrich)/LysC (Wako) mixture in 100 mM HEPES pH=8.5 was added on the pellet in a 1:100 enzyme:substrate ratio and incubated overnight at 37°C on a ThermoMixer (shaking: 850rpm). Generated peptides were desalted using the SepPak (Waters, 186000308, 50 mg sorbent). Peptides were eluted using 80% acetonitrile and 0.1% formic acid and 30 μg peptides were removed for whole proteome measurements. Samples were dried to complete dryness in a SpeedVac and stored at -20°C.
Phosphopeptide Enrichment. Phosphorylated peptides were enriched using the HighSelect TiO2 Phosphopeptide enrichment kit (Thermo Fisher Scientific, A32993) following the manufacture’s protocol, using peptide equivalent to a total of 1500 μg protein input. The eluate containing enriched p-sites was dried in a SpeedVac and resuspended in 8 μL 2% formic acid and 2.5% acetonitrile.
Liquid Chromatography and Mass Spectrometry. The mass spectrometry instrumentation consisted out of a nanoLC 1200 (Thermo Fisher) coupled via a nano-electrospray ionization source to an Exploris480. The mass spectrometer operated in a data-independent mode. MS1 spectra were acquired at a resolution of 60.000. The covered mass range by data-independent acquisition (DIA) was 320-1150 m/z using 48 windows. The MS2 resolution at 200 m/z was set to 15,000, the maximal injection time was defined as 23ms, the AGC target was 3e6 and the collision energy was set to 26,28,30 (stepped mode). MS2 Spectra were acquired in centroid mode.
Data Analysis. The mass spectrometry data were correlated against the mouse reference proteome (Mus Musculus Uniprot) using the Spectronaut directDIA approach. Phosphorylation, acetylation the protein N-term as well as methionine oxidation were defined as a variable modification. PTM localization was enabled and the probability cutoff was set to 0.75. The maximum of variable modification was set to 4. Caramidmethylation at cysteine residues were set as a fixed modification. The PRIDE upload (see below) contains parameter files for more details about the Spectronaut settings. Peptide grouping was done on the modified sequence and MS2 peptide intensity was used for statistical testing. The normalization strategy was set to ‘Global Normalization’ however the data were quantile normalized prior to statistical assessment. Statistical analysis as well as visualizations were performed using the InstantClue software.79
Metabolomics
2-phase metabolite extraction of polar and lipophilic metabolites. For the extraction of total lipids 9-22 mg of snap-frozen murine liver tissue were collected in 2 mL round bottom Eppendorf tubes. For the extraction of the snap frozen material, the cells were homogenized to a fine powder using a ball mill-type grinder (Tissue Lyser2, qiagen) equipped with a 10 samples holder. For the homogenization of the cell pellets one liquid nitrogen cooled 5 mm stainless steel metal balls was added to each Eppendorf tube and the frozen material was disintegrated for 1 min at 25 Hz.
Lipids were extracted by adding 1 mL of pre-cooled (-20°C) extraction buffer (methyl tert-butyl ether (MTBE): methanol: UPLC-grade water 5:3:2 [v:v:v]), containing a 2 μL of EquiSplash Lipidomix (avantilipids) as an internal standard. The tubes were immediately vortexed until the sample was well re-suspended in the extraction buffer. The homogenized samples were then incubated on a cooled (4°C) orbital mixer at 1500 rpm for 30 min. After this step, the metal ball was removed using a magnet and the samples were centrifuged for 10 min at 21.100 x g in a cooled table top centrifuge (4°C). The supernatant was transferred to a fresh 2 mL Eppendorf tube and 250 μL of MTBE and 150 μL of UPLC-grade water were added to each sample. The tubes were immediately vortexed before incubating them for an additional 10 min on a cooled (15°C) orbital mixer at 1500 rpm, before centrifuging them for 10 min at 15°C and 16.000 x g. After the centrifugation, the tubes contained two distinct phases. The upper, MTBE phase, contains the lipids, while the lower, methanol-water, phase contains the polar and semi-polar metabolites.
For the lipidomic analysis 600 μL of the upper lipid phase were collected into fresh 1.5 mL Eppendorf tubes, which were stored at -80°C, until mass spectrometric analysis. The remaining polar phase (∼800 μL) were immediately dried in a SpeedVac concentrator and stored dry at -80°C until mass spectrometric analysis.
Liquid Chromatography-High Resolution Mass Spectrometry-based (LC-HRMS) analysis of lipids. The stored (-80°C) lipid extracts were immediately before analysis dried in a SpeedVac concentrator and lipid pellets were re-suspended in 200 μL of a UPLC-grade acetonitrile: isopropanol (70:30 [v:v]) mixture, followed by vortexing and 10 min incubation on a thermomixer at 4°C. Following 5 min centrifugation at 10.000 x g, the cleared supernatant was transferred to 2 ml glass vials with 200 μL glass inserts (Chromatography Zubehör Trott, Germany), which were placed in an Acquity iClass UPLC (Waters) sample manager at 6°C. The UPLC was connected to a Tribrid Orbitrap HRMS, equipped with a heated ESI (HESI) source (ID-X, Thermo Fischer Scientific).
Of each lipid sample 1 μL were injected onto a 100 x 2.1 mm BEH C8 UPLC column, packed with 1.7 μm particles (Waters). The flow rate of the UPLC was set to 400 μl/min and the buffer system consisted of buffer A (10 mM ammonium acetate, 0.1% acetic acid in UPLC-grade water) and buffer B (10 mM ammonium acetate, 0.1% acetic acid in UPLC-grade acetonitrile/isopropanol 7:3 [v/v]). The UPLC gradient was as follows: 0-1 min 45% A, 1-4 min 45-25% A, 4-12 min 25-11% A, 12-15 min 11-1% A, 15-20 min 1% A, 20-20.1 min 1-45% A and 20.1-24 min re-equilibrating at 45% A. This leads to a total runtime of 24 min per sample.
The ID-X mass spectrometer was operating either for the first injection in positive ionization mode or for the second injection in negative ionization mode. In both cases, the analyzed mass range was between m/z 150-1500. The resolution was set to 120.000, leading to approximately 4 scans per second. The RF lens was set to 60%, while the AGC target was set to 250%. The maximal ion time was set to 100 ms and the HESI source was operating with a spray voltage of 3.5 kV in positive ionization mode, while 3.2 kV were applied in negative ionization mode. The ion tube transfer capillary temperature was 300°C, the sheath gas flow 60 arbitrary units (AU), the auxiliary gas flow 20 AU and the sweep gas flow was set to 1 AU at 340°C.
All samples were analyzed in a randomized run-order.
Targeted data analysis was performed using the quan module of the TraceFinder 4.1 software (Thermo Fischer Scientific) in combination with a sample-specific in-house generated compound database.
Semi-targeted liquid chromatography-high-resolution mass spectrometry-based (LC-HRS-MS) analysis of amine-containing metabolites. The LC-HRMS analysis of amine-containing compounds was performed using an adapted benzoylchlorid-based derivatisation method.85
In brief: The polar fraction of the metabolite extract was re-suspended in 200 μL of LC-MS-grade water (Optima-Grade, Thermo Fisher Scientific) and incubated at 4°C for 15 min on a thermomixer. The re-suspended extract was centrifuged for 5 min at 21,100 x g at 4°C and 50 μL of the cleared supernatant were mixed in an auto-sampler vial with a 200 μL glass insert (Chromatography Accessories Trott, Germany). The aqueous extract was mixed with 25 μL of 100 mM sodium carbonate (Sigma), followed by the addition of 25 μL 2% [v/v] benzoylchloride (Sigma) in acetonitrile (Optima-Grade, Thermo Fisher Scientific). Samples were vortexed and kept at 20°C until analysis.
For the LC-HRMS analysis, 1 μL of the derivatized sample was injected onto a 100 x 2.1 mm HSS T3 UPLC column (Waters). The flow rate was set to 400 μL/min using a binary buffer system consisting of buffer A (10 mM ammonium formate (Sigma), 0.15% [v/v] formic acid (Sigma) in LC-MS-grade water (Optima-Grade, Thermo Fisher Scientific). Buffer B consisted solely of acetonitrile (Optima-grade, Thermo Fisher-Scientific). The column temperature was set to 40°C, while the LC gradient was: 0% B at 0 - 4.1min; 0-15% B 4.1 – 4.5 min; 15-17% B 4.5-11 min; 17-55% B 11 – 11.5 min, 55-70% B 11.5 - 13 min; 70-100% B 13 - 14 min; 100% B 14 -14.1 min; 100-0% B 14.1-19 min; 0% B. The mass spectrometer (Orbitrap ID-X, Thermo Fisher Scientific) was operating in positive ionization mode recording the mass range m/z 100-1000. The heated ESI source settings of the mass spectrometer were: Spray voltage 3.5 kV, capillary temperature 300°C, sheath gas flow 60 AU, aux gas flow 20 AU at a temperature of 340°C and the sweep gas to 2 AU. The RF-lens was set to a value of 60%. Semi-targeted data analysis for the samples was performed using the TraceFinder software (Version 4.1, Thermo Fisher Scientific). The identity of each compound was validated by authentic reference compounds, which were run before and after every sequence. Peak areas of [M + nBz + H]+ ions were extracted using a mass accuracy (<5 ppm) and a retention time tolerance of <0.05 min. Areas of the cellular pool sizes were normalized to the internal standards (U-15N;U-13C amino acid mix (MSK-A2-1.2), Cambridge Isotope Laboratories), which were added to the extraction buffer, followed by a normalization to the fresh weight of the analyzed sample.
Anion-Exchange Chromatography Mass Spectrometry (AEX-MS) for the analysis of anionic metabolites. Extracted metabolites were re-suspended in 200 μL of Optima UPLC/MS grade water (Thermo Fisher Scientific). After 15 min incubation a thermomixer at 4°C and a 5 min centrifugation at 21,100 x g at 4°C, 100 μL of the cleared supernatant were transferred to polypropylene autosampler vials (Chromatography Accessories Trott, Germany).
The samples were analysed using a Dionex ionchromatography system (Integrion, Thermo Fisher Scientific) as described previously.86 In brief, 5 μL of polar metabolite extract were injected in push partial mode using an overfill factor of 1, onto a Dionex IonPac AS11-HC column (2 mm × 250 mm, 4 μm particle size, Thermo Fisher Scientific) equipped with a Dionex IonPac AG11-HC guard column (2 mm × 50 mm, 4 μm, Thermo Fisher Scientific). The column temperature was held at 30°C, while the auto sampler was set to 6°C. A potassium hydroxide gradient was generated using a potassium hydroxide cartridge (Eluent Generator, Thermo Scientific), which was supplied with deionized water. The metabolite separation was carried at a flow rate of 380 μL/min, applying the following gradient conditions: 0-3 min, 10 mM KOH; 3-12 min, 10−50 mM KOH; 12-19 min, 50-100 mM KOH, 19-21 min, 100 mM KOH, 21-22 min, 100-10 mM KOH. The column was re-equilibrated at 10 mM for 8 min.
For the analysis of metabolic pool sizes, the eluting compounds were detected in negative ion mode using full scan measurements in the mass range m/z 50 – 750 on a Q-Exactive HF high resolution MS (Thermo Fisher Scientific). The heated electrospray ionization (ESI) source settings of the mass spectrometer were: Spray voltage 3.2 kV, capillary temperature was set to 300°C, sheath gas flow 60 AU, aux gas flow 20 AU at a temperature of 300°C and a sweep gas glow of 2 AU. The S-lens was set to a value of 60.
Hepa1-6 cell culture
48 hours before the experiment, the cells were trypsinized for 3 mins, counted by using an automated cell counter (EVE automatic Cell counter NanoEnTek) and plated at a density of 2.0 × 105 cells/well in 12-well plates.
Longitudinal Dexamethasone treatment. Dexamethasone (D4902) was dissolved in 100% ethanol. Cells in each well were added with 1 mL of medium containing a final concentration of 5 μM of dexamethasone and then transferred to a 37°C incubator at 5% CO2 for 15’, 30’, 60’, 120’ and 240’. For 0’, cells were left untreated.
Autophagic flux assay. Autophagic flux determines LC3-II accumulation in lysosomes when cells are treated with a cocktail of lysosomal inhibitor, consisting of leupeptin (100 μM) and ammonium chloride (20 mM). Specifically, hepa1-6 cells were treated with medium with and without lysosomal inhibitor 60’ prior to Dexamethasone treatment. At 0’, medium was replaced with medium containing 5 μM of Dexamethasone or PBS in the presence and absence of lysosomal inhibitor. Both experiments were stopped by rapid removal of the cell medium at 240’, washed 1x with cold PBS, snapped frozen on dry ice and kept at -80°C until protein extraction. For technical replicates the mean of three wells were used. At least three independent experiments were performed.
Serum and plasma preparation and analyses
Blood glucose measurement. Blood glucose was measured by a hand-held glucometer ContourXT with Contour Next strips.
Serum and plasma analysis using enzyme-linked immunosorbent assay (ELISA). Experiments related to optogenetic activation of AgRP neurons in the presence or absence of NPY and projection sites stimulation (PVH, LHA, BNST), serum corticosterone, glucagon, or insulin concentrations were determined in duplicates using specific amount of serum according to the manufacturer’s kit instructions. To generate serum, whole blood was allowed to clot at RT for 30 minutes followed by a 30 minutes centrifugation at 4°C, 4000 x g and stored at -80°C until use.
For mice with chemogenetic inhibition of AgRP neurons, PVH-targeted NPY1R inhibition or aging cohorts, plasma was used to determine the concentrations of corticosterone at basal and after a short-term 4 h fast in the dark cycle. ∼100 - 500 μL of intracardial blood was collected using EDTA-coated tubes and centrifuged for 30 minutes at 4°C, 4000 x g. A similar plasma collection strategy was used to investigate the consequence of aging on circulating corticosterone concentrations after central ghrelin delivery or a short-term fast.
For longitudinal plasma ACTH measurement during optogenetic stimulation of AgRP neuron, ∼30 – 50 μL of blood was collected from the tail before the start of the laser (0’) and 120’ after laser stimulation. After 240’ of optogenetic stimulation, mice were decapitated and trunk blood was collected using EDTA-coated tubes and centrifuged for 30 minutes at 4°C, 4000 x g. 10 μL of plasma was used to determine circulating ACTH concentrations according to the manufacturer’s kit instructions.
Protein isolation from murine liver
Ceramic (zirconium oxide) beads were added into 2 mL round-bottom tubes containing 50 – 100 mg of murine liver for total RNA isolation. Livers were homogenized for 100 s at RT in 500 μL of the following buffer: Tris 50 mM, NaCl 130 mM, EDTA 5 mM, NP-40 1%, PMSF 100mM, NaF 100mM, cOmplete mini protease inhibitor cocktail (1 tablet/10 mL) and PhosSTOP™ protease inhibitor (1 tablet/10 mL) were freshly added. The samples were left on ice for 5 min, visually inspected for complete homogenization. If samples were not completely homogenized then the procedure was repeated. Samples were then centrifuged for 30 min at 17,000 x g, the supernatant was taken for determination of protein concentration using the BCA assay (Pierce) relative to a BSA standard. Protein was mixed with Laemmli buffer (Bio-Rad) (1:4) containing b-Mercaptoethanol (1:10) and H2O was added to a final concentration of 2 mg protein per mL.
Western Blotting
Western Blots were performed using 26 well pre-cast 4%-15% gradient gels from BioRad. The voltage was set to 50 V for 5 min, followed by 120 V for 65 min. Tris/glycine solution containing 10% SDS was used as running buffer. Transferring of proteins into the PVDF membranes was performed using the recommended 7 min protocol on the Trans-Blot Turbo system (BioRad). Following the transfer, membranes were blocked with 1:10 western blotting blocking reagent (Roche 11829200) in TBST pH 7.4 for 60 min and incubated at 4°C in primary antibody overnight. Membranes were washed 3 times for 10 min in TBST pH 7.4 the following day. Next, membranes were incubated for 60 min in secondary antibody conjugated to horseradish peroxidase (HRP) at RT, followed by 4 times of 10 min washing in TBST pH 7.4. Membranes were dabbed on paper towels to remove residual washing buffer before incubating it with SuperSignal ECL Western Blotting Substrate (Thermo Scientific) for ∼1-2 min. Luminescence was detected either with a Fusion Solo imaging system and FusionCapt Advance software (Vilber, France). Densitometric analyses of protein bands were performed using imageJ (Fiji)-software package (National Institutes of Health, Bethesda, United States). As a loading control, all membranes were probed with Gapdh antibody. Primary antibodies were diluted as follows: anti-LC3B (1:1000); anti-p62 (1:1000); anti-GAPDH (1:4000) in TBS-T pH 7.4, 5% western blotting blocking reagent. Secondary antibodies were diluted 1:4000 in TBS-T pH 7.4, 5% western blotting blocking reagent. LC3 lipidation was calculated as LC3-II/LC3-I ratio and p62 was normalized to GAPDH.
Perfusion for RNA scope and immunohistochemistry, confocal imaging, and quantification
Mice were deeply anesthetized and transcardially perfused with phosphate-buffered saline (PBS, pH 7,4) followed by 4% paraformaldehyde (PFA). Brains were extracted and post-fixed in 4% PFA at 4°C overnight, then moved to 20% sucrose solution in 0.1 M phosphate buffered saline (PBS, pH 7.4) at 4°C until cutting. 20 μm thick sections were generated with a cryostat and transferred to SuperFrost Plus Gold slides (ThermoFisher).
Immunohistochemistry
Brain slides were washed (3x 10 min) with 0,1M PBS containing 0.1% Triton X-100 (PBST), followed by blocking for 60 min at room temperature with blocking solution (PBST, 3% donkey serum). After blocking slides were incubated in primary antibody solution (Rabbit-anti-NPY, 1:1000, Cell-Signaling Technology, and mCherry-monoclonal, 1:1000, Invitrogen in SignalStain® Antibody Diluent, Cell-Signaling Technology) overnight at 4°C. Slides were then washed (3x 10 min) with PBST at room temperature and incubated in secondary antibody solution (Alexa 647 donkey anti-rabbit, 1:500, Invitrogen, and Alexa 594 donkey anti-rat, 1:500, Invitrogen in SignalStain® Antibody Diluent, Cell-Signaling Technology) for 60 min followed by washing (3x 10 min) with PBST at room temperature. Sections were then mounted with DAPI using Vectashield Antifade Mounting Medium (Vector Laboratories), covered using a coverslip and stored at 4°C in the dark.
Immunofluorescent staining for ChR2-EYFP validation. Adult ChR2AgRP mice were anesthetized and perfused transcardially with 4% paraformaldehyde. Perfused brains were frozen, sectioned at 30 μm for ARH, PVH, LHA and BNST, and processed for immunofluorescence as described.9 Briefly, after an overnight blocking step (2% serum, 0,3% triton) sections (4 sections per well) were incubated for 72 h in primary antibody (chicken anti-GFP 1:1000; rabbit anti-AgRP: 1:2000, Phoenix Pharmaceuticals #H-003-53). The primary antibodies were localized with corresponding affinity- purified IgGs conjugated with Alexa-fluorophores (Life Technologies, 1:500). Images were acquired using a confocal Leica TCS SP-8-X microscope equipped with a 10x or 20x objective.
In situ hybridization
Anaesthetized mice were perfused with PBS (pH 7.4) followed by 4% PFA dissolved in PBS (pH 7.4). Brains were removed from the skull, post-fixed in 4% PFA at 4°C overnight, then moved to 20% sucrose solution (in 1X PBS) at 4°C until cutting. 20 μm thick sections were generated with a cryostat. Detection of AgRP, POMC, cFos, GhsR, CRH, NPY1R, SLC17A6 (VGLUT2) or SLC32A1 (VGAT) was performed using a fluorescent in situ hybridization technique (RNAscope, Advanced Cell Diagnostics) according to the manufacturer’s instructions. 3-plex negative and 3-plex positive control probes were processed in parallel with the target probes. Sections were mounted on SuperFrost Plus Gold slides (ThermoFisher), dried at RT, briefly rinsed in autoclaved Millipore water, air dried and baked at 60°C overnight. Afterward, slides were submerged in Target Retrieval (Cat No. 322000) at 95.0-99.0°C for 9 min, followed by a brief rinse in autoclaved Millipore water and dehydration in 100% ethanol. A hydrophobic barrier was then created around the sections using an ImmEdge hydrophobic barrier pen (Cat No. 310018). All incubation steps were performed at 40°C using the ACD HybEz hybridization system (Cat No. 321462). Sections were incubated with Protease III (Cat No. 322340) for 30 min. The subsequent hybridization, amplification and detection steps were performed according to the manufacturer’s instructions (Multiplex Fluorescent Detection kit v2, Cat No. 323110). For simultaneous detection of genes within each experiment, the respective tyramide-based probes used and diluted as follows:
| Experiment | Gene-Channel | Fluorophore | Figure |
|---|---|---|---|
| AgRP and cFos mRNA expression in the ARH of 4 h fasted AgRP-IRES-Cre mice with bilateral intra-ARH injection of AAVDIO-mCherry or AAV-Flex-hGlyR following ivermectin (IVM) injection | cFOS-C1 AgRP-C2 |
Opal-520 (1:500) Opal-690 (1:750) |
Figure 5B |
| AgRP, POMC and cFos mRNA expression in the ARH of 4 h and 6 h ad libitum fed or fasted mice | cFOS-C1 AgRP-C2 POMC-C3 |
Opal-570 (1:750) Opal-650 (1:1000) Opal-520 (1:1000) |
Figure S1D |
| AgRP, GhSR and cFos mRNA expression in the ARH of C57BL/6N mice icv injected with either saline or Ghrelin | cFOS-C1 AgRP-C2 GhSR-C3 |
Opal-570 (1:750) Opal-650 (1:1000) Opal-520 (1:1000) |
Figure S3G |
| CRH and NPY1R mRNA expression in the PVH and LHA of C57BL/6N mice | CRH-C1 NPY1R-C3 |
Opal-520 (1:750) Opal-690 (1:1000) |
Figure S13A Figure S13B |
| CRH and cFos mRNA expression in the PVH of saline or CNO-injected miceNPY1R | CRH-C1 cFOS-C2 |
Opal-520 (1:750) Opal-690 (1:1000) |
Figure S13D |
| NPY1R, SLC17A6 (VGLUT2) or SLC32A1 (VGAT) mRNA expression in the PVH of C57BL/6N mice | NPY1R-C1 Slc17a6-C2 Slc32a1-C3 |
Opal-520 (1:750) Opal-620 (1:750) Opal-690 (1:1000) |
Figure S13C Figure S13F |
Sections were then mounted with DAPI using Vectashield Antifade Mounting Medium (Vector Laboratories), covered using a coverslip and stored at 4°C in the dark.
Imaging and quantification
Images were acquired with a confocal Leica TCS microscope, using the 20x/0.75 or 40x/1.30 immersion objectives. Laser intensities for the probe channels were kept constant throughout the imaging process. Images of the ARH, PVH and LHA were captured from rostral to caudal, rendering approximately 5-6 sections per animal. Images were imported into Fiji ImageJ (NIH), where the DAPI and the probe channels were adjusted regarding brightness and contrast. The mouse brain atlas published by Paxinos and Franklin was used as a reference to identify the ROIs. Cells stained for the respective target were manually quantified. Neuronal activation was determined by manually counting each individual cFos positive cell; an active cell contained three or more cFos dots, see Figures S1D and S13D.
For CRH and NPY1R co-localization in the PVH and LHA. Cells were counted using the DAPI staining. Cells with more than 50% of CRH fluorescent signal were considered positive CRH cells. Of those, cells with NPY1R signal, i.e., three or more NPY1R dots, were considered NPY1R positive. The same principle was adopted for the quantification of CRH and NPY1R-signals in LHA (see Figure S13B).
For NPY1R/VGLUT2/VGAT or NPY1R/mCherry/VGLUT2/VGAT co-localization in and surrounding the PVH. Individual cells within and surrounding the vicinity of PVH were defined and outlined using the signal. In order to determine whether cells were targeted, mCherry-positive cells were counted. Cells were considered targeted when they showed both NPY1R and mCherry signal. Of those, cells with a VGAT or VGLUT2 signal, i.e., more than 50% of the cell, were counted as VGAT or VGLUT2 positive, respectively. Cells displaying both VGAT and VGLUT2 signals were not considered for the quantification (see Figures S13C and S13F).
Assessment of autophagic vacuoles via Transmission Electron-Microscopy (TEM)
After 1, 2 or 4 hours photostimulation, ChR2AgRP mice were deeply anesthetized and intracardially flushed with 0,5% (w/v) procainhydrochlorid and 0,2% (v/v) heparin in 0.1 M cacodylate buffer and perfused with 2% PFA, 2.5% glutaraldehyde and 0.2% (v/v) saturated picric acid in 0.15 M cacodylate buffer. Control littermates were photostimulated for 4 hours. From the large liver lobe, 1x1x1 mm tissue cubes were prepared with a thin sharp razor blade and liver samples were post-fixed in 2.5% glutaraldehyde/ 2% formaldehyde in 0.15 M cacodylate buffer (pH 7.2) overnight at 4°C. Afterwards, samples were rinsed in 0.1 M cacodylate buffer (pH 7.2) and incubated with 1% OsO4 and 1% potassium ferrocyanid in 0.1 M cacodylate buffer (pH 7.2) for 3 h at 4°C. Subsequently, tissues were dehydrated using ascending ethanol series for 15 min each at 4°C. Tissues were dehydrated using ascending ethanol series, transferred to propylene oxide and finally embedded in epoxyresin for 72 hours at 62°C. Ultrathin sections (70 nm) were cut with a diamond knife (Diatome, Biel,Switzerland) on an ultramicrotome (EM-UC6, Leica Microsystems) and placed on copper grids. Ultrathin sections were contrasted with 1.5% uranylacetate and lead citrate (Reynolds solution). Images were acquired with a transmission electron microscope (JEOL JEM 2100Plus), camera OneView 4K 16bit (Gatan), and software DigitalMicrograph (Gatan) at 80 kV at room temperature. Images were analyzed using the ImageJ (Fiji)-software package. Autophagic vacuoles of at least 20 images/liver at a 10,000x magnification were taken at random.
Quantification and statistical analysis
Group sizes commonly applied in murine studies were used. Sample size is the number of animal subjects per group and is annotated within the figure legends (Figures 1, 2, 3, 4, 5, 6, 7, and S1–S14). Unpaired two-tailed Student’s t test was performed to compare two independent groups. Datasets consisting of more than two groups were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc multiple comparison test. For statistical analyses of groups comparing two variables (e.g., genotype and treatment), a two-way ANOVA was conducted followed by Šídák post hoc multiple comparison test. Longitudinal data were analyzed using two-way repeated-measures (RM) ANOVA and Šídák post hoc post hoc multiple comparison test. Three-way RM ANOVA followed by Šídák post hoc multiple comparison test was performed in the case of comparing groups with three variables (e.g. genotype, laser power and time). Statistical Analysis was performed using GraphPad Prism 9 Software. No specific methods were used to determine whether the data met assumptions of the statistical approaches.
Acknowledgments
We thank Jens Alber, Nadine Evers, Nadine Spenrath, and Pia Scholl for their technical assistance. We thank Peter Wienand and Jakob Nissen for advice relating to cell cultures. We thank Dr. Astrid Schauss and Janine Heise from the CECAD Imaging Facility for their support in assessing autophagosomes via TEM. We thank Dr. Henning Fenselau for providing the AAV-Flex-hGlyRa1AG virus for the chemogenetic inhibition of AgRP neurons. All the schematic diagrams in this manuscript were created with www.biorender.com.
W.C. was supported by the Humboldt research fellowship program of the Alexander Von Humboldt Foundation and received a senior postdoctoral grant from CECAD. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no 742106), funding by BMBF through the German Center for Diabetes Research (DZD), and support through the EASD-Novo Nordisk Foundation Diabetes Prize for Excellence of the European Diabetes Association (EASD) to J.C.B.
Author contributions
W.C. and J.C.B. conceived the present study, designed the experiments, and wrote the original draft of the manuscript. All co-authors reviewed and edited the manuscript. All authors agreed on the final version of the manuscript. W.C. performed majority of the in vivo surgeries/injections, executed majority of the presented experiments, and analyzed the majority of the results. O.M. performed RT-PCR, in situ hybridization, and histological quantifications in this study. A.S. performed and analyzed results relating to RT-PCR and western blotting. P.K. and L.S. analyzed the RNA-seq data. S.H. performed the phosphopeptide enrichment, and H.N. performed and analyzed the phosphoproteomics of livers. T.S.-H. was involved in performing optogenetic fiber canulations in the LHA and BNST projection sites. E.K. provided help for immunohistochemical staining and valuable opinions on autophagy flux. N.L.K. provided help, valuable discussions, and guidance with the optimization and identification of autophagosomes. P.G. performed the metabolomics experiments and analyzed the data.
Declaration of interests
The authors declare no competing interests.
Published: April 18, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2023.03.019.
Supplemental information
Data and code availability
-
•
The mass spectrometry proteomics data have been deposited into the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier: PXD029247.1 RNA sequencing data have been deposited into NCBI Gene Expression Omnibus with an identifier: GSE188472.
-
•
This paper does not report original code.
-
•
All values used to generate the graphs and uncropped images of representative Western blots in the paper can be found in the file Data S1 - Source data. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
References
- 1.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Timper K., Brüning J.C. Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis. Model. Mech. 2017;10:679–689. doi: 10.1242/dmm.026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jais A., Brüning J.C. Arcuate nucleus-dependent regulation of metabolism – pathways to obesity and diabetes mellitus. Endocr. Rev. 2022;43:314–328. doi: 10.1210/endrev/bnab025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gropp E., Shanabrough M., Borok E., Xu A.W., Janoschek R., Buch T., Plum L., Balthasar N., Hampel B., Waisman A., et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat. Neurosci. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 5.Luquet S., Perez F.A., Hnasko T.S., Palmiter R.D. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 6.Aponte Y., Atasoy D., Sternson S.M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci. 2011;14:351–355. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krashes M.J., Koda S., Ye C., Rogan S.C., Adams A.C., Cusher D.S., Maratos-Flier E., Roth B.L., Lowell B.B. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Könner A.C., Janoschek R., Plum L., Jordan S.D., Rother E., Ma X., Xu C., Enriori P., Hampel B., Barsh G.S., et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Steculorum S.M., Ruud J., Karakasilioti I., Backes H., Engström Ruud L., Timper K., Hess M.E., Tsaousidou E., Mauer J., Vogt M.C., et al. AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell. 2016;165:125–138. doi: 10.1016/j.cell.2016.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y., Lin Y.C., Kuo T.W., Knight Z.A. Sensory detection of food rapidly modulates arcuate feeding circuits. Cell. 2015;160:829–841. doi: 10.1016/j.cell.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandt C., Nolte H., Henschke S., Engström Ruud L., Awazawa M., Morgan D.A., Gabel P., Sprenger H.G., Hess M.E., Günther S., et al. Food perception primes hepatic ER homeostasis via melanocortin-dependent control of mTOR activation. Cell. 2018;175 doi: 10.1016/j.cell.2018.10.015. 1321–1335.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaushik S., Tasset I., Arias E., Pampliega O., Wong E., Martinez-Vicente M., Cuervo A.M. Autophagy and the hallmarks of aging. Ageing Res. Rev. 2021;72:101468. doi: 10.1016/j.arr.2021.101468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He C., Klionsky D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R., et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klionsky D.J., Petroni G., Amaravadi R.K., Baehrecke E.H., Ballabio A., Boya P., Bravo-San Pedro J.M., Cadwell K., Cecconi F., Choi A.M.K., et al. Autophagy in major human diseases. EMBO J. 2021;40:e108863. doi: 10.15252/embj.2021108863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A.M., Czaja M.J. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulze R.J., Drižytė K., Casey C.A., McNiven M.A. Hepatic lipophagy: new insights into autophagic catabolism of lipid droplets in the liver. Hepatol. Commun. 2017;1:359–369. doi: 10.1002/hep4.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saito T., Kuma A., Sugiura Y., Ichimura Y., Obata M., Kitamura H., Okuda S., Lee H.C., Ikeda K., Kanegae Y., et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019;10:1567. doi: 10.1038/s41467-019-08829-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaushik S., Rodriguez-Navarro J.A., Arias E., Kiffin R., Sahu S., Schwartz G.J., Cuervo A.M., Singh R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011;14:173–183. doi: 10.1016/j.cmet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coupé B., Ishii Y., Dietrich M.O., Komatsu M., Horvath T.L., Bouret S.G. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab. 2012;15:247–255. doi: 10.1016/j.cmet.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Lopez N., Garcia-Macia M., Sahu S., Athonvarangkul D., Liebling E., Merlo P., Cecconi F., Schwartz G.J., Singh R. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016;23:113–127. doi: 10.1016/j.cmet.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joly-Amado A., Denis R.G., Castel J., Lacombe A., Cansell C., Rouch C., Kassis N., Dairou J., Cani P.D., Ventura-Clapier R., et al. Hypothalamic AgRP-neurons control peripheral substrate utilization and nutrient partitioning. EMBO J. 2012;31:4276–4288. doi: 10.1038/emboj.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavalcanti-de-Albuquerque J.P., Bober J., Zimmer M.R., Dietrich M.O. Regulation of substrate utilization and adiposity by Agrp neurons. Nat. Commun. 2019;10:311. doi: 10.1038/s41467-018-08239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klionsky D.J., Abdel-Aziz A.K., Abdelfatah S., Abdellatif M., Abdoli A., Abel S., Abeliovich H., Abildgaard M.H., Abudu Y.P., Acevedo-Arozena A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy. 2021;17:1–382. doi: 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kojima M., Hosoda H., Date Y., Nakazato M., Matsuo H., Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 26.Tschöp M., Smiley D.L., Heiman M.L. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 27.Cowley M.A., Smith R.G., Diano S., Tschöp M., Pronchuk N., Grove K.L., Strasburger C.J., Bidlingmaier M., Esterman M., Heiman M.L., et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37:649–661. doi: 10.1016/s0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- 28.Krashes M.J., Shah B.P., Koda S., Lowell B.B. Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab. 2013;18:588–595. doi: 10.1016/j.cmet.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y., Essner R.A., Kosar S., Miller O.H., Lin Y.C., Mesgarzadeh S., Knight Z.A. Sustained NPY signaling enables AgRP neurons to drive feeding. eLife. 2019;8 doi: 10.7554/eLife.46348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engström Ruud L., Pereira M.M.A., de Solis A.J., Fenselau H., Brüning J.C. NPY mediates the rapid feeding and glucose metabolism regulatory functions of AgRP neurons. Nat. Commun. 2020;11:442. doi: 10.1038/s41467-020-14291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erickson J.C., Clegg K.E., Palmiter R.D. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 32.Betley J.N., Cao Z.F., Ritola K.D., Sternson S.M. Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell. 2013;155:1337–1350. doi: 10.1016/j.cell.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atasoy D., Betley J.N., Su H.H., Sternson S.M. Deconstruction of a neural circuit for hunger. Nature. 2012;488:172–177. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynagh T., Lynch J.W. An improved ivermectin-activated chloride channel receptor for inhibiting electrical activity in defined neuronal populations. J. Biol. Chem. 2010;285:14890–14897. doi: 10.1074/jbc.M110.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Todd W.D., Fenselau H., Wang J.L., Zhang R., Machado N.L., Venner A., Broadhurst R.Y., Kaur S., Lynagh T., Olson D.P., et al. A hypothalamic circuit for the circadian control of aggression. Nat. Neurosci. 2018;21:717–724. doi: 10.1038/s41593-018-0126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gannaban R.B., NamKoong C., Ruiz H.H., Choi H.J., Shin A.C. Central regulation of branched-chain amino acids is mediated by AgRP neurons. Diabetes. 2021;70:62–75. doi: 10.2337/db20-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith S.M., Vale W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006;8:383–395. doi: 10.31887/DCNS.2006.8.4/ssmith. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dimitrov E.L., DeJoseph M.R., Brownfield M.S., Urban J.H. Involvement of neuropeptide Y Y1 receptors in the regulation of neuroendocrine corticotropin-releasing hormone neuronal activity. Endocrinology. 2007;148:3666–3673. doi: 10.1210/en.2006-1730. [DOI] [PubMed] [Google Scholar]
- 39.Herman J.P., Mueller N.K., Figueiredo H. Role of GABA and glutamate circuitry in hypothalamo-pituitary-adrenocortical stress integration. Ann. NY Acad. Sci. 2004;1018:35–45. doi: 10.1196/annals.1296.004. [DOI] [PubMed] [Google Scholar]
- 40.Yuan Y., Wu W., Chen M., Cai F., Fan C., Shen W., Sun W., Hu J. Reward inhibits paraventricular CRH neurons to relieve stress. Curr. Biol. 2019;29 doi: 10.1016/j.cub.2019.02.048. 1243e4–1251.e4. [DOI] [PubMed] [Google Scholar]
- 41.Hansen M., Rubinsztein D.C., Walker D.W. Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018;19:579–593. doi: 10.1038/s41580-018-0033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mortimore G.E., Lardeux B.R., Adams C.E. Regulation of microautophagy and basal protein turnover in rat liver. Effects of short-term starvation. J. Biol. Chem. 1988;263:2506–2512. [PubMed] [Google Scholar]
- 44.Kotoulas O.B., Kalamidas S.A., Kondomerkos D.J. Glycogen autophagy in glucose homeostasis. Pathol. Res. Pract. 2006;202:631–638. doi: 10.1016/j.prp.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 45.Ezaki J., Matsumoto N., Takeda-Ezaki M., Komatsu M., Takahashi K., Hiraoka Y., Taka H., Fujimura T., Takehana K., Yoshida M., et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7:727–736. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takagi A., Kume S., Kondo M., Nakazawa J., Chin-Kanasaki M., Araki H., Araki S., Koya D., Haneda M., Chano T., et al. Mammalian autophagy is essential for hepatic and renal ketogenesis during starvation. Sci. Rep. 2016;6:18944. doi: 10.1038/srep18944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoon J.C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C.R., Granner D.K., et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 48.Vega R.B., Huss J.M., Kelly D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000;20:1868–1876. doi: 10.1128/MCB.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rhee J., Inoue Y., Yoon J.C., Puigserver P., Fan M., Gonzalez F.J., Spiegelman B.M. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc. Natl. Acad. Sci. USA. 2003;100:4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Najt C.P., Khan S.A., Heden T.D., Witthuhn B.A., Perez M., Heier J.L., Mead L.E., Franklin M.P., Karanja K.K., Graham M.J., et al. Lipid droplet-derived monounsaturated fatty acids traffic via PLIN5 to allosterically activate SIRT1. Mol. Cell. 2020;77 doi: 10.1016/j.molcel.2019.12.003. 810–824.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finck B.N., Gropler M.C., Chen Z., Leone T.C., Croce M.A., Harris T.E., Lawrence J.C., Jr., Kelly D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Lee J.M., Wagner M., Xiao R., Kim K.H., Feng D., Lazar M.A., Moore D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nieto-Torres J.L., Hansen M. Macroautophagy and aging: the impact of cellular recycling on health and longevity. Mol. Aspects Med. 2021;82:101020. doi: 10.1016/j.mam.2021.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lapierre L.R., Meléndez A., Hansen M. Autophagy links lipid metabolism to longevity in C. elegans. Autophagy. 2012;8:144–146. doi: 10.4161/auto.8.1.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ewbank S.N., Campos C.A., Chen J.Y., Bowen A.J., Padilla S.L., Dempsey J.L., Cui J.Y., Palmiter R.D. Chronic Gq signaling in AgRP neurons does not cause obesity. Proc. Natl. Acad. Sci. USA. 2020;117:20874–20880. doi: 10.1073/pnas.2004941117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cowley M.A., Smart J.L., Rubinstein M., Cerdán M.G., Diano S., Horvath T.L., Cone R.D., Low M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 57.Rau A.R., Hentges S.T. The relevance of AgRP neuron-derived GABA inputs to POMC neurons differs for spontaneous and evoked release. J. Neurosci. 2017;37:7362–7372. doi: 10.1523/JNEUROSCI.0647-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jais A., Paeger L., Sotelo-Hitschfeld T., Bremser S., Prinzensteiner M., Klemm P., Mykytiuk V., Widdershooven P.J.M., Vesting A.J., Grzelka K., et al. PNOC(ARC) neurons promote hyperphagia and obesity upon high-fat-diet feeding. Neuron. 2020;106 doi: 10.1016/j.neuron.2020.03.022. 1009–1025.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Durieux J., Wolff S., Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor R.C., Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153:1435–1447. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Imanikia S., Özbey N.P., Krueger C., Casanueva M.O., Taylor R.C. Neuronal XBP-1 activates intestinal lysosomes to improve proteostasis in C. elegans. Curr. Biol. 2019;29 doi: 10.1016/j.cub.2019.06.031. 2322–2338.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imanikia S., Sheng M., Castro C., Griffin J.L., Taylor R.C. XBP-1 remodels lipid metabolism to extend longevity. Cell Rep. 2019;28 doi: 10.1016/j.celrep.2019.06.057. 581–589.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor R.C., Hetz C. Mastering organismal aging through the endoplasmic reticulum proteostasis network. Aging Cell. 2020;19:e13265. doi: 10.1111/acel.13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinez-Lopez N., Tarabra E., Toledo M., Garcia-Macia M., Sahu S., Coletto L., Batista-Gonzalez A., Barzilai N., Pessin J.E., Schwartz G.J., et al. System-wide benefits of intermeal fasting by autophagy. Cell Metab. 2017;26 doi: 10.1016/j.cmet.2017.09.020. 856–871.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tiesjema B., Adan R.A., Luijendijk M.C., Kalsbeek A., la Fleur S.E. Differential effects of recombinant adeno-associated virus-mediated neuropeptide Y overexpression in the hypothalamic paraventricular nucleus and lateral hypothalamus on feeding behavior. J. Neurosci. 2007;27:14139–14146. doi: 10.1523/JNEUROSCI.3280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van den Hoek A.M., van Heijningen C., Schröder-van der Elst J.P., Ouwens D.M., Havekes L.M., Romijn J.A., Kalsbeek A., Pijl H. Intracerebroventricular administration of neuropeptide Y induces hepatic insulin resistance via sympathetic innervation. Diabetes. 2008;57:2304–2310. doi: 10.2337/db07-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferreira-Marques M., Aveleira C.A., Carmo-Silva S., Botelho M., Pereira de Almeida L., Cavadas C. Caloric restriction stimulates autophagy in rat cortical neurons through neuropeptide Y and ghrelin receptors activation. Aging (Albany, NY) 2016;8:1470–1484. doi: 10.18632/aging.100996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aveleira C.A., Botelho M., Carmo-Silva S., Pascoal J.F., Ferreira-Marques M., Nóbrega C., Cortes L., Valero J., Sousa-Ferreira L., Álvaro A.R., et al. Neuropeptide Y stimulates autophagy in hypothalamic neurons. Proc. Natl. Acad. Sci. USA. 2015;112:E1642–E1651. doi: 10.1073/pnas.1416609112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manmontri B., Sariahmetoglu M., Donkor J., Bou Khalil M., Sundaram M., Yao Z., Reue K., Lehner R., Brindley D.N. Glucocorticoids and cyclic AMP selectively increase hepatic lipin-1 expression, and insulin acts antagonistically. J. Lipid Res. 2008;49:1056–1067. doi: 10.1194/jlr.M800013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang P., O'Loughlin L., Brindley D.N., Reue K. Regulation of lipin-1 gene expression by glucocorticoids during adipogenesis. J. Lipid Res. 2008;49:1519–1528. doi: 10.1194/jlr.M800061-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sato M., Ueda E., Konno A., Hirai H., Kurauchi Y., Hisatsune A., Katsuki H., Seki T. Glucocorticoids negatively regulates chaperone mediated autophagy and microautophagy. Biochem. Biophys. Res. Commun. 2020;528:199–205. doi: 10.1016/j.bbrc.2020.04.132. [DOI] [PubMed] [Google Scholar]
- 72.Deng J., Guo Y., Yuan F., Chen S., Yin H., Jiang X., Jiao F., Wang F., Ji H., Hu G., et al. Autophagy inhibition prevents glucocorticoid-increased adiposity via suppressing BAT whitening. Autophagy. 2020;16:451–465. doi: 10.1080/15548627.2019.1628537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang Y., Botchway B.O.A., Hu Z., Fang M. Overexpression of SIRT1 inhibits corticosterone-induced autophagy. Neuroscience. 2019;411:11–22. doi: 10.1016/j.neuroscience.2019.05.035. [DOI] [PubMed] [Google Scholar]
- 74.Troncoso R., Paredes F., Parra V., Gatica D., Vásquez-Trincado C., Quiroga C., Bravo-Sagua R., López-Crisosto C., Rodriguez A.E., Oyarzún A.P., et al. Dexamethasone-induced autophagy mediates muscle atrophy through mitochondrial clearance. Cell Cycle. 2014;13:2281–2295. doi: 10.4161/cc.29272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tong Q., Ye C.P., Jones J.E., Elmquist J.K., Lowell B.B. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Madisen L., Mao T., Koch H., Zhuo J.M., Berenyi A., Fujisawa S., Hsu Y.W., Garcia A.J., 3rd, Gu X., Zanella S., et al. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 2012;15:793–802. doi: 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Padilla S.L., Qiu J., Soden M.E., Sanz E., Nestor C.C., Barker F.D., Quintana A., Zweifel L.S., Rønnekleiv O.K., Kelly M.J., Palmiter R.D. Agouti-related peptide neural circuits mediate adaptive behaviors in the starved state. Nat. Neurosci. 2016;19:734–741. doi: 10.1038/nn.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schindelin J., Rueden C.T., Hiner M.C., Eliceiri K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015;82:518–529. doi: 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nolte H., MacVicar T.D., Tellkamp F., Krüger M. Instant clue: A software suite for interactive data visualization and analysis. Sci. Rep. 2018;8:12648. doi: 10.1038/s41598-018-31154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patel H., Ewels P., Peltzer A., Hammarén R., Botvinnik O., Sturm G., Moreno D., Vemuri P., silviamorins. Garcia L.P.F.M., et al. 2020. nf-core/rnaseq: nf-core/rnaseq v3.0 - Silver Shark. Zenodo. [DOI] [Google Scholar]
- 81.Ewels P.A., Peltzer A., Fillinger S., Patel H., Alneberg J., Wilm A., Garcia M.U., Di Tommaso P., Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020;38:276–278. doi: 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 82.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wong J.M., Malec P.A., Mabrouk O.S., Ro J., Dus M., Kennedy R.T. Benzoyl chloride derivatization with liquid chromatography-mass spectrometry for targeted metabolomics of neurochemicals in biological samples. J. Chromatogr. A. 2016;1446:78–90. doi: 10.1016/j.chroma.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schwaiger M., Rampler E., Hermann G., Miklos W., Berger W., Koellensperger G. Anion-exchange chromatography coupled to high-resolution mass spectrometry: a powerful tool for merging targeted and non-targeted metabolomics. Anal. Chem. 2017;89:7667–7674. doi: 10.1021/acs.analchem.7b01624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The mass spectrometry proteomics data have been deposited into the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier: PXD029247.1 RNA sequencing data have been deposited into NCBI Gene Expression Omnibus with an identifier: GSE188472.
-
•
This paper does not report original code.
-
•
All values used to generate the graphs and uncropped images of representative Western blots in the paper can be found in the file Data S1 - Source data. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.