

Abstract
Excitotoxicity is the underlying mechanism for all acute neuronal injury, from cerebral ischemia, status epilepticus, traumatic CNS injury, and hypoglycemia. It causes morphological neuronal necrosis, and it triggers a programmed cell death program. Excessive calcium entry through the NMDA‐receptor‐operated cation channel activates two key enzymes—calpain I and neuronal nitric oxide synthase (nNOS). Calpain I, a cytosolic enzyme, translocates to mitochondrial and lysosomal membranes, causing release of cytochrome c, endonuclease G, and apoptosis‐inducing factor (AIF) from mitochondria and DNase II and cathepsins B and D from lysosomes. These all translocate to neuronal nuclei, creating DNA damage, which activates poly(ADP) ribose polymerase‐1 (PARP‐1) to form excessive amounts of poly(ADP) ribose (PAR) polymers, which translocate to mitochondrial membranes, causing release of truncated AIF (tAIF). The free radicals that are released from mitochondria and peroxynitrite, formed from nitric oxide (NO) from nNOS catalysis of L‐arginine to L‐citrulline, damage mitochondrial and lysosomal membranes and DNA. The end result is the necrotic death of neurons. Another programmed necrotic pathway, necroptosis, occurs through a parallel pathway. As investigators of necroptosis do not recognize the excitotoxic pathway, it is unclear to what extent each contributes to programmed neuronal necrosis. We are studying the extent to which each contributes to acute neuronal necrosis and the extent of cross‐talk between these pathways.
Keywords: excitotoxic neuronal necrosis, necroptosis, programmed cell death, status epilepticus
Key points.
Status epilepticus induces excitotoxicity, which induces programmed neuronal necrosis.
Three key enzymes that are activated to produce neuronal necrosis are calpain I, nNOS and PARP‐1.
Another programmed necrotic pathway, necroptosis, has been proposed as the only pathway producing regulated necrosis.
We are investigating the relative contributions of the two pathways and the extent of crosstalk between them.
1. INTRODUCTION
Prior to the 1980s, based upon extensive research on mechanisms of neuronal damage from hypoxia‐ischemia, hypoglycemia, and status epilepticus (SE), the Swedish investigator Bo Siesjő synthesized the then‐existing knowledge on the potential mechanisms that resulted in “ischemic cell change” in the three conditions. 1 He speculated that an elevation of intracellular Ca2+, which in the case of SE he thought was due to opening of voltage‐sensitive Ca2+ channels, activated phospholipases, with elevation of free fatty acids and in particular arachidonic acid, which generates free radicals that damage intracellular organelles and plasma membranes. Investigations in the 1960s and 1970s into the morphology of ischemic neuronal damage showed electron‐dense, shrunken neurons, which correspond to what we understand as necrotic neurons today. 2 , 3 , 4 , 5 The convergence of the intracellular biochemical findings and morphological findings did not occur until much later, but the seeds of our current understanding had been laid.
In the 1970s, John Olney introduced the phenomenon of excitotoxic neuronal death, which laid the groundwork for an understanding of all acute neuronal injury. 6 , 7 , 8 , 9 In 1983, Steven Rothman changed the paradigm of hypoxic neuronal damage by showing that it is dependent on synaptic connections between neurons, thus pointing to synaptic activity between neurons as the basis of acute neuronal injury. 10
2. THE CONCEPT OF EXCITOTOXICITY
John Olney’s initial description of excitotoxic neuronal death was that produced by monosodium glutamate (MSG) on hypothalamic neurons in infant mice. 6 Electron microscopy showed that the damaged neurons had neuronal nuclei with chromatin clumps and cytoplasm with dilated endoplasmic reticulum and mitochondria, with damaged cristae. 6 Olney found that kainic acid, a glutamate (GLU) analogue, produced electron‐microscopic evidence in the hypothalamus and hippocampus of dark, shrunken neurons 7 (Figure 1) identical with those seen in the brains of adult rats 5 (Figure 2) and macaque monkeys 2 exposed to hypoxia–ischemia. The neurons were shrunken, electron‐dense, with shrunken nuclei containing chromatin clumps and cytoplasm with dilated endoplasmic reticulum and mitochondria, with remnants of cristae. These changes are identical to those we found following status epilepticus (SE) in adult rats. 11 , 12 , 13 In SE, blockade of the N‐methyl‐D‐aspartate (NMDA) subtype of GLU receptors with either competitive 14 or non‐competitive 15 NMDA‐receptor antagonists, given 15 minutes after the onset of SE, provided remarkable neuroprotection to up to 22 of 24 brain regions examined. 15 This suggests that endogenous GLU is responsible for producing SE‐induced neuronal necrosis.
FIGURE 1.
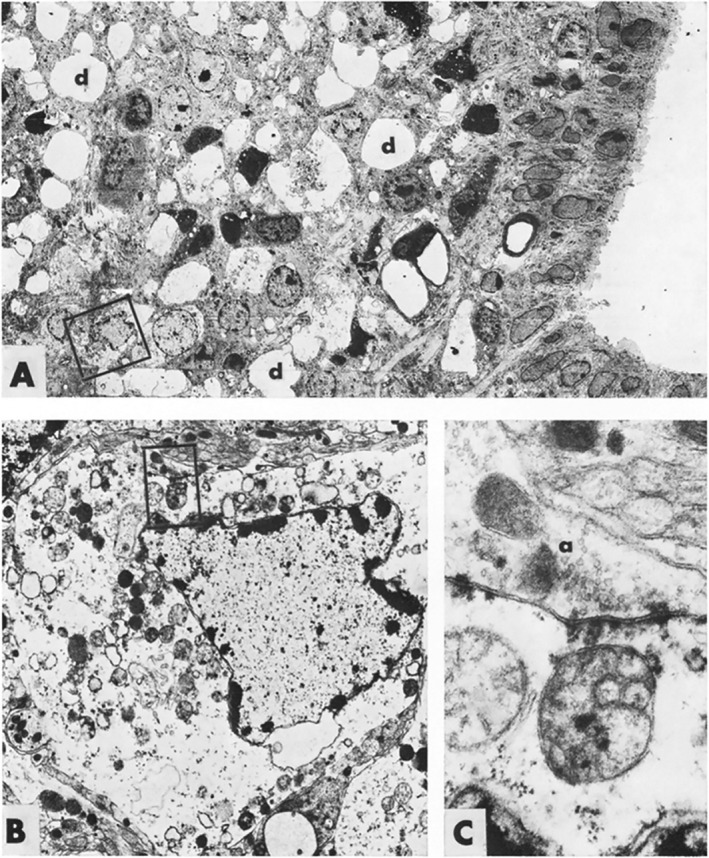
Electron microscopic views of degenerating neurons in the arcuate region of the hypothalamus 110 minutes after receiving kainic acid 50 mg/kg. In A, some neurons have a “dark cell” appearance, whereas others, such as the neuron in the box, shown at higher magnification in B, show a pyknotic nucleus with chromatin clumps at the nuclear membrane and edematous cytoplasm with dilated endoplasmic reticulum and mitochondria. The boxed mitochondrion, shown in C, is irreversibly damaged, swollen, and without visible cristae. From Olney et al. (1974), with permission from Elsevier
FIGURE 2.

Electron microscopic photomicrograph showing a rat pyramidal neuron with “ischemic cell change” following ipsilateral common carotid occlusion and 0% oxygen for 40 minutes with up to 2 h of survival. The nucleus is pyknotic, with scattered chromatin clumps. The cytoplasm shows dilatation of endoplasmic reticulae (arrow and “g”) and mitochondria (“m”) with “bleb‐like” swelling of cristae (hard to see). From Brown and Brierley (1972), with permission from Elsevier
2.1. Is neuronal apoptosis responsible for acute neuronal injury?
Apoptosis is a form of cell death with a specific morphology as well as evidence that it is “programmed” by caspase‐dependent mechanisms. Caspases are enzymes that produce, by intrinsic (mitochondrial) or extrinsic (cell surface), mechanisms apoptotic cell death. 16 In neurons, apoptosis is characterized morphologically by nuclei with large round or crescentic chromatin clumps, with progressively damaged nuclear membranes. 17 , 18 A key point is that apoptotic neuronal death may occur naturally, in the course of brain development, or be induced by exogenous factors, but that it only occurs in the neonatal rodent brain. Another key point is that apoptotic and excitotoxic (necrotic) neuronal death are morphologically distinct.
However, when the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) stain was used to show double‐stranded DNA cleavage, and DNA agarose gel electrophoresis showed double‐stranded DNA cleavage at 180 base‐pair intervals, creating a “laddering” pattern, it was assumed that this was only seen in apoptotic neurons. We showed that both occurred in necrotic neurons as well 12 , 13 (Figures 3, 4). In addition, first reports were that acute neuronal injury was the result of caspase‐dependent mechanisms, and that using an inhibitor of caspase‐3 provided neuroprotection. 19 However, later reports showed that SE‐induced neuronal death in adult rodents did not involve caspase activation, 11 , 20 , 21 which complemented the recognition that apoptosis and caspase activation only occurred in immature brain. Evidence that calpain‐1, 22 , 23 poly(ADP) ribose polymerase‐1 (PARP‐1), 24 , 25 , 26 and neuronal nitric oxide synthase (nNOS) 27 , 28 , 29 activation were involved produced recognition that acute neuronal necrosis is caspase‐independent and is indeed a programmed cell death.
FIGURE 3.

Electron microscopic photomicrographs show that brain regions with acidophilic neurons 24 h and 72 h after 3‐h lithium‐pilocarpine‐induced SE are necrotic morphologically, and internucleosomal DNA cleavage (DNA laddering) occurs in these brain regions. (A, B, D, and E) The photomicrographs are of ventral CA1 neurons from control and SE rats with 24‐h (A and B, respectively) and 72‐h survival periods (D and E, respectively). A and D show normal neurons with normal nuclei and cytoplasm and normal mitochondria and endoplasmic reticulum. B and E show necrotic neurons with dark, shrunken cell bodies, pyknotic nuclei with small, scattered chromatin clumps, and cytoplasmic vacuolar degeneration. Many of the vacuoles are swollen mitochondria with disrupted cristae. Scale bar = 3 µm. A, B, D, and E are photomicrographs of brain regions used in the data analysis by Fujikawa et al. 12 C and F show agarose gel electrophoresis of DNA extracted from brain regions examined by electron microscopy in control rats and those with SE and a 24‐h (C) or a 72‐h recovery period (F). In both C and F, the first lane (L) is a 100 base‐pair DNA standard ladder, lanes 1 and 2 are negative and positive apoptotic tissue controls, and subsequent lanes are control and SE dorsal hippocampus (3 and 4), ventral hippocampus (5 and 6), neocortex (7 and 8), amygdala and piriform cortex (9 and 10), and entorhinal cortex (11 and 12). Internucleosomal DNA cleavage at approximately 200 base‐pair intervals (DNA laddering) is present in the positive tissue control and in all five brain regions from SE rats, showing that DNA laddering can occur in necrotic as well as apoptotic cells. (from Fujikawa et al. 48 with permission from Blackwell Science Ltd., now Wiley)
FIGURE 4.

Three‐hour status epilepticus (SE) produces acidophilic neurons with pyknotic nuclei which are TUNEL negative 24 h after SE, but which may become TUNEL positive 72 h after SE, and which are electron‐dense, shrunken necrotic neurons by ultrastructural examination. These H&E‐stained (A–D) and adjacent TUNEL‐stained sections with Methyl Green counterstain (E–H) are from layer 2 of the piriform cortex of control rats with 24 h (A and E) and 72‐h (C and G) recovery periods, and rats with 3‐h LPCSE and 24‐h (B and F) and 72‐h (D and H) recovery periods. Normal neurons are present in A, C, E, and G. Necrotic, acidophilic neurons with condensed, pyknotic nuclei, and eosinophilic cytoplasm are evident by H&E stain 24 and 72 h after SE (B and D). The acidophilic neurons 24 h after SE (arrows point to three in B) are TUNEL negative (arrows point to three in F). In D, some of the acidophilic neurons 72 h after SE (arrows point to three) remain TUNEL negative in H (arrows point to three), but many others are positive by TUNEL stain in H (arrowheads point to three). Glial nuclei in A–G are indicated by asterisks. I–P show neurons from layer 2 of the piriform cortex of the opposite hemispheres of the same rats shown in A–H. I and K show 4500× and M and O show 30 000× magnifications of normal neurons from control rats with 24‐h (I and M) and 72‐h (K and O) recovery periods. Normal nuclei and cytoplasm with normal mitochondria (arrows point to three in M and two in O), rough endoplasmic reticulum and free ribosomes are seen. J and L show 4500× and N and P show 30 000× magnifications of typical necrotic neurons from rats with 3‐h SE and 24‐h (J and N) and 72‐h (L and P) recovery periods. The cell bodies are dark and shrunken, the nuclei are dark and pyknotic, with small, scattered, irregular chromatin clumps, and there is cytoplasmic degeneration, with numerous vacuoles (J and L). Many of the vacuoles at higher magnification (N and P) are greatly swollen, spherically shaped mitochondria with disrupted cristae and flocculent matrix densities, indicative of irreversible mitochondrial damage (arrowheads point to one in N and two in P). Scale bars = 20 µm (A–H), 4 µm (I–L), and 1 µm (M–P). From Fujikawa et al. (2000), with permission from Pergamon Press (now Elsevier)
2.2. Is acute status epilepticus‐induced neuronal necrosis programmed?
It has been established that SE‐induced neuronal death is morphologically necrotic, yet induced 180 base‐pair double‐stranded DNA cleavage 11 , 12 , 13 (Figure 3), reinforced by immunohistochemical evidence of double‐stranded DNA breaks 11 , 12 , 13 , 20 (Figure 4), with no evidence of neuronal apoptosis or activation of the intrinsic or extrinsic caspase programmed pathways. 11 , 12 , 13 , 20 , 30 Double‐stranded DNA cleavage is dependent on activation of an endogenous endonuclease or endonucleases. In this regard, lysosomal DNase II and mitochondrial endonuclease G are released and translocate to neuronal nuclei within the first 60 minutes of lithium‐pilocarpine‐induced SE 31 (Figure 5). In addition, mitochondrial cytochrome c and apoptosis‐inducing factor (AIF) and lysosomal cathepsins B and D are also released and translocate to neuronal nuclei within the first 60 minutes of SE as well 31 (Figure 5). Translocation of all of these mitochondrial and lysosomal enzymes and proteins shows that SE‐induced neuronal necrosis is a programmed process.
FIGURE 5.

Two mitochondrial proteins (cytochrome c and apoptosis‐inducing factor) and endonuclease G and two lysosomal proteases (cathepsins B and D) and DNase II translocate to neuronal nuclei within the first 60 minutes of status epilepticus (SE). The first row for each show control neurons in rat piriform cortex layer 3, showing cytoplasmic staining in a, DAPI nuclear staining in b, and merged images in c. The second row shows piriform cortex 60 minutes after the onset of SE. The arrows in each panel point to neurons showing translocation to nuclei in d, with merged images in f confirming the translocation with purple to purplish‐white nuclei. Arrowheads point to neurons in d that are already pyknotic (shrunken) with nuclear translocation that also show purple to purplish‐white nuclei in f. The scale bar in a = 10 µm. Unpublished photomicrographs from Zhao et al. (2010)
3. ANOTHER PROGRAMMED MECHANISM, CELLULAR NECROPTOSIS, OCCURS IN ACUTE CELLULAR INJURY
In cell culture, when human or murine T cells were exposed to Fas‐induced cell death with inhibition of the caspase pathways with the non‐specific caspase inhibitor zVAD.fmk, instead of an apoptotic death they died a cell death with necrotic features. 32 They also found that tumor necrosis factor (TNF) did not induce cell death in T cells if they were deficient in receptor‐interacting protein (RIP) kinase. 32 Eight years later, investigators used a RIP1 kinase (RIPK1) inhibitor, 7‐Cl‐O‐necrostatin‐1 (Nec‐1), delivered intracerebroventricularly (i.c.v.), that showed neuroprotection in a mouse model of 2‐hours middle cerebral artery (MCA) occlusion. 33 The untreated group showed neuronal necrosis, so the investigators called this type of neuronal death “necroptosis,” a combination of necrosis and apoptosis.
Investigators today think that necroptosis is the only form of programmed necrosis (aside from the variants ferroptosis, pyroptosis and a combination of pyroptosis, apoptosis, and necroptosis [PANoptosis 34 ], which are beyond the scope of this review). None of these investigators refer to excitotoxic neuronal necrosis from NMDA‐receptor activation, 35 , 36 , 37 which we have documented. 8 , 9
3.1. Necroptosis in cerebral ischemia
A study that investigated the role of necroptosis in oxygen‐glucose deprivation (OGD) of hippocampal neurons for 2 hours in cell culture and 15 minutes of transient global cerebral ischemia (TGCI) found that receptor‐interacting protein kinases 1 (RIPK1) and 3 (RIPK3) mRNA and protein were upregulated in hippocampal neurons in OGD. 38 RIPK3 down‐regulation was neuroprotective and up‐regulation exacerbated neuronal damage. 38
3.2. Necroptosis in status epilepticus
A study that investigated the role of necroptosis in SE showed that hydrogen‐rich saline (HRS), injected i.p. in Sprague‐Dawley rats subjected to lithium‐pilocarpine‐induced SE (LPCSE) showed down‐regulation of phosphorylated mixed lineage kinase domain‐like protein (MLKL) and reduction of cognitive impairment on the Morris Water Maze. 39 They also claimed to show down‐regulation of activated caspase‐3, which points to the involvement of neuronal apoptosis, but the electron photomicrographs (Figures 4D and E) show necrotic neurons. 12 , 13 The method by which cells were quantified was not described in the article.
3.3. Crosstalk between the excitotoxic and necroptotic pathways
There is evidence that rather than being two independent pathways producing neuronal necrosis that there is crosstalk between them. For example, the non‐competitive NMDA‐receptor antagonist MK‐801 (dizocilpine) and the nNOS inhibitor 7‐nitroindazole both reduced RIPK3 nitrosylation, phosphorylation, and neuronal necrosis in the hippocampal CA1 region following TGCI 40 (Figure 6). In addition, Nec‐1 reduced cathepsin B release from lysosomes, which occurs in excitotoxicity, following TGCI 41 (Figure 6).
FIGURE 6.

A schematic overview of the excitotoxic and necroptotic programmed pathways—are they parallel, independent pathways, or is there interaction between the two, and if the latter, what is the relative importance of each? Significantly more is known about the excitotoxic pathway and its role in acute neuronal injury. 8 , 9 NMDA‐receptor antagonists, such as MK‐801 (dizocilpine), block entry of calcium (Ca2+) through its cation channel, 49 , 50 thereby preventing excessive Ca2+ influx into post‐synaptic neurons. If Ca2+ influx is not blocked, the Ca2+‐activated cytosolic enzymes calpain I and neuronal nitric oxide synthase (nNOS) are activated. Calpain I, in turn, causes release of mitochondrial cytochrome c (cyt c), 51 endonuclease G (endo G) 52 and apoptosis‐inducing factor (AIF) 52 and lysosomal cathepsins B 53 and D 54 and DNase II, 54 all of which translocate to neuronal nuclei within the first 60 minutes of SE. 31 Neuronal NOS forms nitric oxide (NO) from the conversion of L‐arginine to L‐citrulline, and NO combines with superoxide radical (O2 ‐) to form peroxynitrite (OONO‐), a transient yet highly toxic free radical. 55 Peroxynitrite and other free radicals, such as O2 ‐ and the hydroxyl radical (OH‐) damage mitochondrial, lysosomal membranes, and DNA. Poly(ADP)ribose polymerase‐1 (PARP‐1) forms poly(ADP)ribose (PAR) polymers to repair DNA, but with excessive DNA damage, excessive amounts of PAR exit the nucleus and translocate to mitochondrial membranes, causing release of truncated AIF (tAIF), 45 which in turn translocates to neuronal nuclei and with histone 2AX, creates large‐scale (50 kB‐pair) DNA cleavage. 46 With respect to the necroptotic pathway, the in vivo trigger for activation of receptor‐interacting protein kinase 1 (RIPK1) is not known, but its phosphorylation activates phosphorylation of RIPK3, which in turn activates mixed lineage kinase domain‐like protein (MLKL), which translocates to the plasma membrane, damaging it. All three protein kinases translocate to neuronal nuclei prior to necroptosis. 56 Our current research involves investigating the role of each pathway in programmed necrosis from DFP‐induced SE and to determine any cross‐talk between the two pathways. ADP ribose, necrosulfonamide (NSA), poly(ADP) ribose glycohydrolase (PARG), and the TRMP2 and Na+‐Ca2+ exchanger channels, are not discussed in this article. Abbreviations: Excitotoxic pathway: Nec‐1, 7‐Cl‐O‐necrostatin‐1; MK‐801,dizocilpine; Glu, glutamate; Ca2+, calcium; nNOS, neuronal nitric oxide synthase; 7‐NI, 7‐nitroindazole; Lys, lysosome; Mit, mitochondria; O2 ‐, superoxide radical; ONOO ‐ , peroxynitrite; NO, nitric oxide; cath B/D, cathepsins B and D; endo G, endonuclease G, cyt c, cytochrome c; tAIF, truncated apoptosis‐inducing factor; OH ‐ hydroxyl radical; PARP‐1, poly(ADP) ribose polymerase‐1; PAR, poly(ADP) ribose; PARG, poly(ADP) ribose glycohydrolase; TRPM2, transient receptor melastatin 2. Necroptotic pathway: RIPK1, receptor‐interacting protein kinase 1; RIPK3, receptor‐interacting protein kinase 3; MLKL, mixed lineage domain‐like protein kinase; NSA, necrosulfonamide; P, phosphorylated
We are currently investigating the roles of excitotoxic neuronal necrosis and neuronal necroptosis in SE‐induced neuronal necrosis induced by the organophosphate diisopropylfluorophosphate (DFP). The mechanism of neuronal damage is likely to be identical with that of LPCSE, because neuronal damage from organophosphate agents such as soman are reduced by ketamine given up to 2 hours after the onset of soman‐induced SE. 42 We are studying the crosstalk between, and relative contributions of, excitotoxic‐programmed mechanisms and necroptotic mechanisms to DFP‐induced neuronal necrosis (Figure 6). Those currently investigating acute neuronal necrosis focus exclusively on necroptotic mechanisms. 35 , 36 , 37 In fact, excitotoxic neuronal necrosis, a programmed process, in not even mentioned, despite its prominence in studies of programmed necrosis for over 20 years and excitotoxic neuronal necrosis for 50 years.
4. CONCLUDING REMARKS
All acute neuronal injury in the adult mammalian brain, whether from cerebral ischemia, SE, traumatic CNS damage, or hypoglycemia, is excitotoxic, brought on by excessive activation of NMDA receptors, 8 , 9 or by reversal of astrocytic extracellular glutamate uptake. 43 This opens the receptor‐operated cation channel, permitting excessive amounts of Ca2+ to enter neurons, activating calpain I (µ‐calpain) and nNOS (Figure 6). Calpain I in turn triggers release of mitochondrial cytochrome c, endonuclease G, and AIF and lysosomal cathepsins B and D and DNase II, all of which translocate to neuronal nuclei, promoting DNA damage (Figure 6). The DNA damage causes PARP‐1 to assemble excessive amounts of PAR polymers, which translocate to mitochondrial membranes, causing release of truncated AIF (t‐AIF) 44 , 45 (Figure 6). The t‐AIF translocates to neuronal nuclei, which with histone‐2AX and cyclophilin A causes 150 kb DNA cleavage. 46 This sequence of events defines excitotoxic programmed necrosis.
However, in recent years another programmed necrotic pathway, necroptosis (from “necrosis” and “apoptosis”) has been described 47 and is discussed in a previous section. The trigger for in vivo activation of the necroptotic cascade in cerebral ischemia, TBI and SE is not known, and there are only a few articles devoted to this pathway. In contrast, excitotoxic programmed necrosis is well‐established, but ignored by those investigating acute necroptosis. We are investigating the cross‐talk between the two pathways and to what extent each contributes to acute neuronal necrosis. This is an area of research which future investigations will elucidate.
CONFLICT OF INTEREST
The author declares no conflict of interest.
ETHICAL PUBLICATION
I confirm that I have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
Our current research is funded by VA Merit Review grant 1I01BX004632‐01. The work of Jo Kung‐Chiao Hsieh, Ph.D., Keng‐Tee Chew, M.S., Sunil Kumar, Ph.D., and Seema Rai, Ph.D., Pharm.D. is gratefully acknowledged.
Fujikawa DG. Programmed mechanisms of status epilepticus‐induced neuronal necrosis. Epilepsia Open. 2023;8(Suppl. 1):S25–S34. 10.1002/epi4.12593
It is a pleasure contributing to this special issue of Epilepsia Open, dedicated to Dr. Claude Wasterlain, who has mentored most of the investigators who contributed to this issue. I began as an Epilepsy Fellow in Dr. Wasterlain’s Epilepsy Research Laboratory at Sepulveda VA Medical Center in 1981. Dr. Wasterlain was responsible for my interest in the basic mechanisms of status epilepticus‐induced neuronal damage, which has continued to this day, and for which I am forever grateful.
REFERENCES
- 1. Siesjö BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1:155–85. [DOI] [PubMed] [Google Scholar]
- 2. Brierley JB, Brown AW, Meldrum BS. The nature and time course of the neuronal alterations resulting from oligaemia and hypoglycemia in the brain of Macacca Mulatta. Brain Res. 1971;25:483–99. [DOI] [PubMed] [Google Scholar]
- 3. Brown AW. Structural abnormalities in neurones. J Clin Pathol. 1977;30(Suppl 11):155–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown AW. The nature, distribution and earliest stages of anoxic‐ischaemic nerve cell damage in the rat brain as defined by the optical microsope. Br J Exp Pathol. 1968;49:87–106. [PMC free article] [PubMed] [Google Scholar]
- 5. Brown AW, Brierley JB. Anoxic‐ischaemic cell change in rat brain light microscopic and fine‐structural observations. J Neurol Sci. 1972;16:59–84. [DOI] [PubMed] [Google Scholar]
- 6. Olney JW. Glutamate‐induced neuronal necrosis in the infant mouse hypothalamus. J Neuropathol Exp Neurol. 1971;30:75–90. [DOI] [PubMed] [Google Scholar]
- 7. Olney JW, Rhee V, Ho OL. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;13(77):507–12. [DOI] [PubMed] [Google Scholar]
- 8. Fujikawa DG, editor. Acute Neuronal Injury: The Role of Excitotoxic Programmed Cell Death Mechanisms. New York: Springer; 2010. p. 306. [Google Scholar]
- 9. Fujikawa DG, editor. Acute neuronal injury: the role of excitotoxic programmed cell death mechanisms. 2nd ed. Springer International Publishing AG; 2018. p. 215. [Google Scholar]
- 10. Rothman SM. Synaptic activity mediates death of hypoxic neurons. Science. 1983;220:536–7. [DOI] [PubMed] [Google Scholar]
- 11. Fujikawa DG, Ke X, Trinidad RB, Shinmei SS, Wu A. Caspase‐3 is not activated in seizure‐induced neuronal necrosis with internucleosomal DNA cleavage. J Neurochem. 2002;83:229–40. [DOI] [PubMed] [Google Scholar]
- 12. Fujikawa DG, Shinmei SS, Cai B. Lithium‐pilocarpine‐induced status epilepticus produces necrotic neurons with internucleosomal DNA fragmentation in adult rats. Eur J Neurosci. 1999;11:1605–14. [DOI] [PubMed] [Google Scholar]
- 13. Fujikawa DG, Shinmei SS, Cai B. Kainic acid‐induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98:41–53. [DOI] [PubMed] [Google Scholar]
- 14. Fujikawa DG, Daniels AH, Kim JS. The competitive NMDA receptor antagonist CGP 40116 protects against status epilepticus‐induced neuronal damage. Epilepsy Res. 1994;17:207–19. [DOI] [PubMed] [Google Scholar]
- 15. Fujikawa DG. Neuroprotective effect of ketamine administered after status epilepticus onset. Epilepsia. 1995;36:186–95. [DOI] [PubMed] [Google Scholar]
- 16. Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. [DOI] [PubMed] [Google Scholar]
- 17. Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler V, Dikranian K, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. [DOI] [PubMed] [Google Scholar]
- 18. Ishimaru MJ, Ikonomidou C, Tenkova TI, Der TC, Dikranian K, Sesma MA, et al. Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. J Comp Neurol. 1999;408:461–76. [PubMed] [Google Scholar]
- 19. Henshall DC, Chen J, Simon RP. Involvement of caspase‐3‐like protease in the mechanism of cell death following focally evoked limbic seizures. J Neurochem. 2000;74:1215–23. [DOI] [PubMed] [Google Scholar]
- 20. Fujikawa DG, Shinmei SS, Zhao S, Aviles ER Jr. Caspase‐dependent programmed cell death pathways are not activated in generalized seizure‐induced neuronal death. Brain Res. 2007;1135:206–18. [DOI] [PubMed] [Google Scholar]
- 21. Narkilahti S, Pirtillä TJ, Lukasiuk K, Tuunanen J, Pitkänen A. Expression and activation of caspase 3 following status epilepticus. Eur J Neurosci. 2003;18:1486–96. [DOI] [PubMed] [Google Scholar]
- 22. Bi X, Chang V, Siman R, Tocco G, Baudry M. Regional distribution and time‐course of calpain activation following kainate‐induced seizure activity in adult rat brain. Brain Res. 1996;8(726):98–108. [DOI] [PubMed] [Google Scholar]
- 23. Siman R, Noszek JC, Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci. 1989;9:1579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eliasson MJL, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, et al. Poly (ADP‐ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nature Med. 1997;3:1089–95. [DOI] [PubMed] [Google Scholar]
- 25. Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A, et al. NMDA but not non‐NMDA excitotoxicity is mediated by poly(ADP‐ribose) polymerase. J Neurosci. 2000;20:8005–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, et al. Poly(ADP‐ribose) (PAR) binding to apoptosis‐inducing factor is critical for PAR polymerase‐1‐dependent cell death (parthanatos). Sci Signal. 2011;4:ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dawson VL, Kizushi VM, Huang PL, Snyder SH, Dawson TM. Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase‐deficient mice. J Neurosci. 1996;15(16):2479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lallement G, Shih TM, Pernot‐Marino I, Baubichon D, Foquin A, McDonough JH. The role of nitric oxide in soman‐induced seizures, neuropathology, and lethality. Pharmacol Biochem Behav. 1996;54:731–7. [DOI] [PubMed] [Google Scholar]
- 29. Park EM, Cho S, Frys K, Racchumi G, Zhou P, Anrather J, et al. Interaction between inducible nitric oxide synthase and Poly(ADP‐ribose) polymerase in focal ischemic brain injury. Stroke. 2004;35(12):2896–901. [DOI] [PubMed] [Google Scholar]
- 30. Fujikawa DG, Zhao S, Ke X, Shinmei SS, Allen SG. Mild as well as severe insults produce necrotic, not apoptotic, cells: Evidence from 60‐min seizures. Neurosci Lett. 2010;469(3):333–7. [DOI] [PubMed] [Google Scholar]
- 31. Zhao S, Aviles ER Jr, Fujikawa DG. Nuclear translocation of mitochondrial cytochrome c, lysosomal cathepsins B and D, and three other death‐promoting proteins within the first 60 minutes of generalized seizures. J Neurosci Res. 2010;88:1727. [DOI] [PubMed] [Google Scholar]
- 32. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase‐8‐independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95. [DOI] [PubMed] [Google Scholar]
- 33. Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng M, Kanneganti TD. The regulation of the ZBP1‐NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev. 2020;297:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–43. [DOI] [PubMed] [Google Scholar]
- 36. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vanlangenakker N, Vanden Berghe T, Krysko D, Festjens N, Vandenabeele P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med. 2008;8:207–20. [DOI] [PubMed] [Google Scholar]
- 38. Vieira M, Fernandes J, Carreto L, Anuncibay‐Soto B, Santos M, Han J, et al. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up‐regulation of endogenous RIP3. Neurobiol Dis. 2014;68:26–36. [DOI] [PubMed] [Google Scholar]
- 39. Ruihua J, Jia J, Yang F, Liu Z, Rui L, Jiang Y, et al. Hydrogen alleviates necroptosis and cognitive deficits in lithium–pilocarpine model of status epilepticus. Cell Mol Neurobiol. 2019;39:857–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miao W, Qu Z, Shi K, Zhang D, Zong Y, Zhang G, et al. RIP3 S‐nitrosylation contributes to cerebral ischemic neuronal injury. Brain Res. 2015;19(1627):165–76. [DOI] [PubMed] [Google Scholar]
- 41. Yin B, Xu Y, Wei RL, He F, Luo BY, Wang JY. Inhibition of receptor‐interacting protein 3 upregulation and nuclear translocation involved in Necrostatin‐1 protection against hippocampal neuronal programmed necrosis induced by ischemia/reperfusion injury. Brain Res. 2015;1609:63–71. [DOI] [PubMed] [Google Scholar]
- 42. Dorandeu F, Carpentier P, Baubichon D, Four E, Bernabe D, Burckhart MF, et al. Efficacy of the ketamine–atropine combination in the delayed treatment of soman‐induced status epilepticus. Brain Res. 2005;27(1051):164–75. [DOI] [PubMed] [Google Scholar]
- 43. Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells. 2019;8(2):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andrabi SA, Kim S‐W, Wang H, Koh DW, Sasaki M, Klaus JA, et al. Poly(ADP‐ribose) (PAR) polymer is a death signal. Proc Natl Aca Sci. 2006;103(48):18308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu S‐W, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, et al. Apoptosis‐inducing factor mediates poly(ADP‐ribose) (PAR) polymer‐induced cell death. Proc Natl Aca Sci. 2006;103(48):18314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Artus C, Boujrad H, Bouharrour A, Brunelle MN, Hoos S, Yuste VJ, et al. AIF promotes chromatinolysis and caspase‐independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;5(29):1585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. [DOI] [PubMed] [Google Scholar]
- 48. Fujikawa DG. In: Wasterlain CG, Treiman DM, editors. Neuroprotective strategies in status epilepticus. Cambridge, MA: MIT Press; 2006. p. 463–480. [Google Scholar]
- 49. Huettner JE, Bean BP. Block of N‐methyl‐D‐aspartate‐activated current by the anticonvulsant MK‐801: selective binding to open channels. Proc Natl Acad Sci USA. 1988;85:1307–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA‐receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321(6069):519–22. [DOI] [PubMed] [Google Scholar]
- 51. Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, et al. Critical role of calpain I in mitochondrial release of apoptosis‐inducing factor in ischemic neuronal injury. J Neurosci. 2007;29(27):9278–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Takano J, Tomioka M, Tsubuki S, Higuchi M, Nobuhisa Iwata N, Itohara S, et al. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem. 2005;280:16175–84. [DOI] [PubMed] [Google Scholar]
- 53. Yamashima T, Kohda Y, Tsuchiya K, Ueno T, Yamashita J, Yoshioka T, et al. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA‐074: a novel strategy for neuroprotection based on 'calpain‐cathepsin hypothesis'. Eur J Neurosci. 1998;10:1723–33. [DOI] [PubMed] [Google Scholar]
- 54. Villalpando Rodriguez GE, Torriglia A. Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim Biophys Acta. 2013;1833:2244‐53. [DOI] [PubMed] [Google Scholar]
- 55. Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, et al. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;15(19):5910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]