

Abstract
Antifreeze proteins (AFPs), found in many cold-adapted organisms, can protect them from cold and freezing damages and have thus been considered as additional protectants in current cold tissue preservation solutions that generally include electrolytes, osmotic agents, colloids and antioxidants, to reduce the loss of tissue viability associated with cold-preservation. Due to the lack of toxicity profile studies on AFPs, their inclusion in cold preservation solutions has been a trial-and-error process limiting the development of AFPs’ application in cold preservation. To assess the feasibility of translating the technology of AFPs for mammalian cell cold or cryopreservation, we determined the toxicity profile of two highly active beetle AFPs, DAFP1 and TmAFP, from Dendroides canadensis and Tenebrio molitor in this study. Toxicity was examined on a panel of representative mammalian cell lines including testicular spermatogonial stem cells and Leydig cells, macrophages, and hepatocytes. Treatments with DAFP1 and TmAFP at up to 500 μg/mL for 48 and 72 hours were safe in three of the cell lines, except for a 20% decrease in spermatogonia treated with TmAFP. However, both AFPs at 500 μg/mL or below reduced hepatocyte viability by 20 to 40 % at 48 and 72h. At 1000 μg/mL, DAFP1 and TmAFP reduced viability in most cell lines. While spermatogonia and Leydig cell functions were not affected by 1000 μg/mL DAFP1, this treatment induced inflammatory responses in macrophages. Adding 1000 μg/ml DAFP1 to rat kidneys stored at 4°C for 48 hours protected the tissues from cold-related damage, based on tissue morphology and gene and protein expression of two markers of kidney function. However, DAFP1 and TmAFP did not prevent the adverse effects of cold on kidneys over 72 hours. Overall, DAFP1 is less toxic at high dose than TmAFP, and has potential for use in tissue preservation at doses up to 500 μg/mL. However, careful consideration must be taken due to the proinflammatory potential of DAFP1 on macrophages at higher doses and the heighten susceptibility of hepatocytes to both AFPs.
Keywords: Antifreeze proteins, DAFP1, toxicity, cell lines, cold protection, kidney
1. INTRODUCTION
Antifreeze proteins (AFPs) were first discovered in some Antarctic fishes (as glycosylated proteins) more than four decades ago (DeVries and Wohlschlag, 1969) and have since been found in many cold-adapted organisms including fishes, insects, plants, and bacteria (DeVries, 1971; Duman, 2001; Griffith et al., 1992). AFPs adopt a wide range of different structures, but they all bind to specific surfaces of ice crystals whereby lowering the freezing point without affecting the melting point of the solution and inhibiting ice growth and recrystallization (Raymond and DeVries, 1977). The antifreeze activities of insect AFPs are generally much higher than that of their fish counterparts and are thus also called hyperactive AFPs. Furthermore, it has been reported recently that insects AFPs can also effectively inhibit the crystallization of certain carbohydrate cryoprotectants (Wang et al., 2014; Wen et al., 2016; Wen et al., 2022). Due to the unique properties of AFPs, they have been investigated as natural protectants for low temperature preservation of biological species (Griffith et al., 2005; Halwani et al., 2014; Kratochvílová et al., 2017; Mangiagalli et al., 2017; Rodriguez et al., 2019; Weng et al., 2019). Most of the studies of preservation using AFPs have been focused on fish AFPs (Hirano et al., 2008a; Huelsz-Prince et al., 2019; Itskovitzeldor et al., 1993; Kamijima et al., 2013; Nguyen et al., 2018; Rubinsky et al., 1990), therefore the safety of fish AFPs as “novel” food ingredients have been thoroughly investigated (Crevel et al., 2002). However, there are few toxicity data available on hyperactive insect AFPs, which limits their potential applications in preservation.
AFPs in different organisms can exhibit different properties such as freeze tolerance, ice restructuring, and ice adhesion (Davies, 2014). AFPs also have another fundamental ability to protect cells and their membranes from hypothermic damage (Rubinsky et al., 1991). DAFP, a family of 30 AFP isoforms derived from the hemolymph of Dendroides canadensis beetles, has been shown to prevent the freezing of larvae through its interaction with trehalose, the sugar source of D. canadensis, which is subjected to precipitation in the cold. The ability of DAFP to inhibit trehalose crystallization may contribute to the cold survival strategy of D. canadensis, allowing them to survive during winter in which temperatures can fluctuate between −18 to −28°C (Wen et al., 2016). More recently, it has been reported that DAFP1 can completely inhibit the nucleation of D-mannitol from its supersaturated solution (Wen et al., 2022). Both trehalose and D-mannitol are effective osmolyte cryoprotectants but associated with notorious spontaneous crystallization concerns. Thus, we aimed to take advantage of these properties and develop D. canadensis and similar insect derived AFPs for future use in human tissue and organ preservation by studying their protective abilities in mammalian cell culture.
In this study, we examine the toxicity of two hyperactive beetle AFPs, TmAFP from Tenebrio molitor and DAFP1 from Dendroides canadensis, on a panel of representative mammalian cell lines including macrophages (innate immunity/inflammatory/antigen-presenting cell), Leydig cell (steroidogenic/endocrine cell), spermatogonial stem cells (germline stem cell), and hepatocytes (capable of xenobiotics metabolism). Cell viability in the presence and absence of a range of AFP concentrations was assayed, as well as expression of functional markers on the cell lines. Moreover, protein localization experiments were performed with FITC-labeled AFPs and the internalization potential of the AFPs was assessed. Lastly, we evaluated the ability of DAFP1 to be tissue protective in cold conditions, using rat kidney as model, to evaluate its potential value in organ preservation. Our results provide the first toxicity assessment of exposure to hyperactive AFPs and thus will facilitate subsequent investigations of their uses as protectants to prevent cells and tissues damage during low-temperature preservation.
2. METHODS
2.1. Materials
All chemicals were purchased from either Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA) at ACS grade or better (unless described elsewhere) and were used as received. Milli-Q water produced from a Synergy water system was used for the preparation of all solutions.
Rat pups were maintained and euthanized in accordance with protocols approved by the IACUC of the University of Southern California.
2.2. Cell line culture
C18-4 Cell Line.
The LTAg-immortalized mouse type A spermatogonia cell line was a gift from Marie-Claude Hofmann (The University of Texas MD Anderson Cancer Center, Houston, Texas, USA). Cells were cultured in Gibco™ DMEM containing 4.5 g/L d-Glucose, L-Glutamine and 110 mg/L of sodium pyruvate (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat inactivated FBS (Sigma-Aldrich, St. Louis, MO, USA) and 1% Penicillin-Streptomycin Solution 100X (Corning™) at 35 °C, 7% CO2. The cells were grown in 75 cm2 Corning™ cell culture-treated flasks to sub-confluence and seeded in culture wells at different densities depending on the experiments.
MA-10 Cell Line.
The mouse MA-10 Leydig cell line derived from culture-derived tumors was a gift from Dr. Mario Ascoli (University of Iowa, Iowa city, IA, USA). Cells were cultured in DMEM-F12+ Glutamax (Invitrogen, Waltham, MA) supplemented with 2.5% horse serum (Invitrogen, Waltham, MA), 10% heat inactivated FBS (Sigma-Aldrich, St. Louis, MO, USA), and 1% Penicillin-Streptomycin Solution 100X (Corning™) at 37 °C, 3.5% CO2. The cells were grown in 25 cm2 Corning™ cell culture-treated flasks, passaged at sub-confluence and seeded in culture wells as needed by the experiments.
RAW 264.7 Cell Line.
The mouse RAW 264.7 macrophage cell line established from a tumor induced by the Abelson murine leukemia virus was purchased from ATCC (Manassas, VA, USA). Cells were cultured in Gibco™ DMEM containing 4.5 g/L d-Glucose, L-Glutamine and 110 mg/L of sodium pyruvate (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat inactivated FBS (Sigma-Aldrich, St. Louis, MO, USA) and 1% Penicillin-Streptomycin Solution 100X (Corning™) at 37 °C, 5% CO2. The cells were grown in 25 cm2 Corning™ cell culture-treated flasks, passaged at sub-confluence and seeded in culture wells as needed by the experiments.
HUH7 Cell Line.
The human HUH7 liver/hepatocyte cell line was established from a liver tumor from a 57-year-old Japanese male and purchased from JCRB Cell Bank. Cells were cultured in Gibco™ DMEM containing 4.5 g/L d-Glucose, L-Glutamine and 110 mg/L of sodium pyruvate (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat inactivated FBS (Sigma-Aldrich, St. Louis, MO, USA) and 1% Penicillin-Streptomycin Solution 100X (Corning™) at 37 °C, 5% CO2. The cells were grown in 25 cm2 Corning™ cell culture-treated flasks to sub-confluence and seeded at various densities in wells in function of the experiments.
2.3. AFP Protein preparation
The expression and purification of the AFPs, an isoform from Dendroides canandensis, and an isoform from Tenebrio molitor followed previously published procedures (Wang et al., 2009; Zamalloa, 2012). Briefly, the AFPs were expressed as fusion proteins in Escherichia coli Origami B cells. The cells were harvested by centrifugation at 4 °C. After the cells were disrupted, the crude protein was purified using immobilized metal ion affinity chromatography (IMAC) (Ni-NTA agarose, Qiagen). The tags of the AFP were cleaved off with enterokinase (New England Biolabs) and then the resulting protein was further purified by using IMAC and ion exchange chromatography. The purified wild-type AFP was characterized using SDS-PAGE gel electrophoresis and high-performance liquid chromatography (HPLC) as previously described (Rodriguez et al., 2019; Wang et al., 2009).
2.4. Preparation of FITC-labeled proteins
DAFP1 and BSA were conjugated with fluorescein isothiocyanate (FITC) following established procedures with minor modifications (Goding, 1976). Briefly, an FITC solution in DMSO was added into the protein solutions in 0.10 M carbonate buffer, pH 9.0, resulting in the molar ratios of FITC:AFP and FITC:BSA of 0.5:1 and 4:1 respectively in the reaction mixtures, which were then incubated in the dark at 4°C for 8 hours and an additional 2 hours after adding ammonium chloride to a final concentration of 50 mM. The FITC-protein conjugates in the reaction mixtures were purified using Sephadex G25 columns (GE Healthcare, Chicago, IL). FITC-protein conjugates were characterized using UV-Vis spectroscopy (Agilent Cary 60, Santa Clara, CA) and florescence spectroscopy (Agilent Cary Eclipse, Santa Clara, CA).
2.5. MTT viability Assay
Cells were plated at 8–10,000 cells/well in 96 well Corning™ culture-treated microplates and treated with PBS diluted in culture media (vehicle control) or AFP proteins diluted in culture media for 48–72 hours. MTT viability assay was conducted following the recommendations of the manufacturer (Roche Cell proliferation kit I MTT, Sigma-Aldrich, St. Louis, MO, USA) and absorbance values were measured with the VICTOR™ X5 Multilabel Plate Reader (PerkinElmer, Inc., Waltham, MA, USA). Results represent the means and SEM of 3 independent experiments conducted each in triplicates.
2.6. EdU Proliferation Assay
Cells were plated in 96 well Corning™ culture-treated black bottom microplates at a density of 8–10,000 cells/well and treated with PBS diluted in culture media (vehicle control) or AFP proteins diluted in culture media for 42 hours. For the last 6 hours of treatment, cells were incubated with 10 μM EdU (5-ethynyl-2’-deoxyuridine) as recommended by the manufacturer (Click-IT® EdU HCS Assay, Invitrogen, Waltham, MA, USA). Cells were washed with PBS and fixed with 4% paraformaldehyde followed by permeabilization with 0.1% Triton X-100. The Click-iT reaction cocktail was then added to each well and incubated in the dark for 30 minutes. Cells were washed and 100 μL 1:2000 HCS NuclearMask was added to stain DNA for 30 minutes in the dark. The plates were imaged and quantified with the Cytation 5 Cell Imaging Multi-Mode Reader (Biotek, Winooksi, VT, USA). Results represent the means and SEM of 3 independent experiments conducted each in triplicates.
2.7. RNA Extraction, cDNA synthesis, and Quantitative real time PCR (qPCR) analysis
C18-4 and Raw 264.7 cells were plated at 100–200,000 cells/well and treated with medium (vehicle control) or AFP proteins diluted in culture medium for 40–48 hours. Cells were collected after treatment, solubilized in RNA lysis buffer and total RNA was extracted based on manufacturer recommendations using the RNAqueous™-Micro Total RNA Isolation Kit (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized from purified RNA using the PrimeScript™ RT Master Mix (Takara Bio, Mountain View, CA, USA) according to manufacturer’s instructions. The results represent the means and SEM of 3 independent experiments conducted each in triplicates. For gene expression analysis of rat kidney tissues, total RNA extraction of kidney fragments was performed using a Nucleospin RNA XS kit (Takara Bio, Mountain View, CA, USA). qPCR analysis was performed as previously described (Walker et al., 2020) using SYBER Green Supermix (Thermo Fisher Scientific, Waltham, MA). A minimum of two pups for each dose were assessed in triplicate. The comparative Ct method was used to calculate relative gene expression. Primers were designed using Primer-BLAST from the NCBI-NIH gene database and are listed in Table 2. QPCR cycling conditions were consistent with previously described conditions.
2.8. Progesterone ELISA Assay
MA-10 cells were plated in 10,000 cells/well in 96 well Corning™ culture-treated microplates and treated with AFP proteins diluted in culture media or PBS diluted in culture media (vehicle control) for 48 hours. After treatment duration, samples were washed with PBS and were either unstimulated (kept in serum free culture media) or stimulated with 50 ng/ml hCG (human chorionic gonadotropin) in serum free culture media for 2 hours. Supernatant was collected for use immediately for progesterone measurement or stored in the −80c freezer for future analysis. Cell layers were solubilized using 0.1 N NaOH for protein quantification and data normalization, as previously described (Boisvert et al., 2016). Progesterone was measured using the Progesterone ELISA Kit (Cayman Chemical, Ann Arbor MI). Basal (unstimulated) samples were diluted at 1:20 and stimulated samples were diluted at 1:1000 in ELISA buffer provided. The VICTOR™ X5 Multilabel Plate Reader (PerkinElmer, Inc., Waltham, MA, USA) was used to detect OD measurement of Ellman’s reagent at wavelength 405 nm. Data was analyzed using the manufacturer’s provided spreadsheet and %B/B0 from Standards 1–8 vs. progesterone concentration was plotted in Prism version 7.0 (GraphPad Software, San Diego, CA) software using a 4-parameter logistic fit. Progesterone concentrations were normalized to total protein/well quantified with Bradford reagent according to the manufacturer’s protocol (VWR Life Science, Solon, OH, USA). Results represent the means and SEM of 3 independent experiments conducted each in triplicates.
2.9. TNFa ELISA Assay
For TNFa quantification, RAW 264.7 cells were plated at 100,000 cells/well in 12 well Corning™ culture-treated plates and treated with AFP proteins diluted in culture media or PBS diluted in culture media (vehicle control). After treatments, supernatant was collected and saved for cytokine analysis and RNA lysis buffer was added to the cell layer for qPCR. Samples were stored in −80c until use. TNFa levels were measured using the Quantikine™ ELISA kit (R&D Systems, Minneapolis, MN) according to manufacturer’s protocol. Undiluted samples were evaluated against standard curve diluted in cell culture media. The VICTOR™ X5 Multilabel Plate Reader (PerkinElmer, Inc., Waltham, MA, USA) was used to detect OD measurement of Ellman’s reagent at wavelength 450 nm. TNFa concentration was plotted in Prism version 7.0 (GraphPad Software, San Diego, CA) software using a 4-parameter logistic fit. TNFa concentrations were normalized to RNA concentration/well. RNA was extracted from cells using the RNAqueous™-Micro Total RNA Isolation Kit (Invitrogen, Carlsbad, CA, USA) as described above. The results represent the means and SEM of 3 independent experiments conducted each in triplicates.
2.10. Internalization of FITC-labelled proteins
Cells were plated in 96 well Corning™ culture-treated black bottom microplates at 8–10,000 cells/well and allowed to adhere overnight. The next day, FITC-labeled DAFP1 or BSA at 1000 μg/ml, prepared as described above, were diluted at 1000 μg/mL concentrations in cell culture media. Cells were either incubated with medium alone, FITC-BSA or FITC-DAFP1 for 6 hours. Cells were then washed three times with sterile PBS (Corning Inc., Corning, NY, USA) and visualized with Revolve Fluorescence Microscope (Echo, San Diego, CA) or the Cytation 5 Cell Imaging Multi-Mode Reader (Biotek, Winooksi, VT, USA). Afterwards, cells were trypsinized with 0.25% trypsin (Gibco Thermo-Fisher Scientific, Waltham, MA), neutralized with cell culture media, and lysed with RIPA lysis buffer (Chemcruz Santa Cruz Biotech, Dallas, TX). Spent treatment media, washes, and lysates were collected and fluorescence was measured with the VICTOR™ X5 Multilabel Plate Reader (PerkinElmer, Inc., Waltham, MA, USA) at 485 nm. The amounts of FITC-conjugated protein internalized in cells were calculated from the cell lysate data after subtracting the background fluorescence of control wells where cells were cultured with medium alone and no FITC.
2.11. Rat Kidney Cold Treatment
Kidneys were isolated from postnatal day (PND) 6 rats and rinsed with sterile PBS. For each rat pup, both kidneys were transferred into microcentrifuge tubes containing 500 μl of vehicle control medium or DAFP1 at 1000 μg/ml, diluted in sterile RPMI 1640 medium (Gibco Thermo-Fisher Scientific, Waltham, MA) and samples were kept at 4°C for 48 hours (3 rats per condition) or 72 hours (2–3 rats per condition). At the end of the cold treatment, one kidney per animal was frozen in liquid nitrogen and stored at −80°C until further processing for qPCR analysis, while the other kidney was fixed in 4% PFA in PBS (Santa Cruz Biotech, Dallas, TX) overnight for future paraffin embedding and immunofluorescence analysis. In both cases, the effects of cold treatments were compared to the same endpoints measured in kidneys that had been either frozen in liquid nitrogen or fixed in PFA at the time of dissection (Time 0).
2.12. Tissue processing, paraffin embedding and immunofluorescence analysis
Tissue processing, paraffin embedding:
Rat kidney tissues were fixed with 10% formalin and kept in 70% EtOH at 4°C until processing. They were automatically processed by Spin Tissue Processor Microm STP-120 (Thermo Fisher Scientific, Waltham, MA) with a program to process tissues using the following reagents (90min/step): dehydrant (70%, 80%, 95%, 100%) (Richard-Allan Scientific), 2 cycles of xylene (Cancer Diagnostics, Durham, NC) and 2 rounds of paraffin (Type9, Thermo Fisher Scientific, Waltham, MA). After processing, tissues were embedded using Tissue Embedding Center EC-350 (Thermo Fisher Scientific, Waltham, MA) at 60°C in tissue block molds filled with hot paraffin. Tissue blocks were cooled down at room temperature for 30 min and stored at 4°C until sectioning. Tissues were trimmed and rehydrated on ice water for 30 min. After rehydration, tissues were sectioned at 5 μm using the Rotary Microtome HM-310 (Thermo Fisher Scientific, Waltham, MA), transferred to the water bath (14792V, VWR Life Science, Solon, OH) at 37°C, and picked up by VistaVision™ HistonBond® slides (16004406, VWR Life Science, Solon, OH). Sections were dried overnight at room temperature.
Immunofluorescence (IF) analysis:
IF was performed as previously described (Manku et al., 2016). Briefly, slides were first dewaxed and rehydrated using Citrisol and Trilogy solution (Cell Marque, Rocklin, CA). Following treatment with DAKO Target Antigen Retrieval Solution (Agilent Technologies Inc., Santa Clara, CA), the sections were incubated with PBS containing 10% BSA and 10% donkey serum for one hour to block non-specific protein interactions. The sections were then incubated with anti-Aquaporin 1 (AQP1) antibody (ab189291, Abcam, Cambridge, UK) or anti-POD1 (Tcf21) (bs-8688r, Bioss Antibodies, Woburn, MA) antibody diluted in PBS containing 10% BSA, 0.1% Triton-X 100, and donkey serum overnight at 4°C. Once the overnight incubation was complete, slides were incubated with a secondary goat anti-rabbit Alexa Flour 488 antibody (Invitrogen, Waltham, MA) diluted in PBS containing 1% BSA for one hour at room temperature. Nuclear staining was performed using DAPI staining (Vector Labs, Burlingame, CA) for 5 minutes. The slides were then mounted with one drop of room temperature Fluoromount-G (Electron Microscopy Sciences) and cover-slipped. Slides were imaged on the Cytation 5 Cell Imaging Multi-Mode Reader (Biotek, Winooksi, VT, USA).
2.13. Statistical analysis
Statistical analysis was conducted by Student’s T-test or one-way ANOVA with post-hoc multiple comparison analysis using the Graphpad Prism Software (San Diego, CA, USA). Statistical significance was considered at p ≤ 0.05. All experiments were performed in duplicates or triplicates using a minimum of three independent experiments. Results are shown as mean ± standard error of the mean (SEM). Values of * p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001 indicate significance compared to vehicle controls.
3. RESULTS
3.1. Effects of DAFP1 48 hours treatment on cell viability and proliferation in C18-4, MA-10, RAW 264.7, and HUH7 cell lines.
We did not observe any loss of viability measured with the MTT assay at up to 500 μg/mL treatment dose over 72 hours in several representative cell lines. The C18-4 mouse spermatogonial cell line withstood up to 2000 μg/mL treatment of DAFP1 with no loss of cell viability over 48 hours, while 500 μg/mL DAFP1 induced a 25% increase in cell viability (Fig. 1A). After 72 hours, there was a small (10 to 15%) viability loss at the 250 and 2000 μg/mL (Fig. 1B). TmAFP had more impact on C18-4 cells, decreasing viability by 20% at 250 and 500 μg/mL after 48 hours (Fig. 1C) and 72 hours (Fig. 1D).
Figure 1. Effect on AFPs on the viability of testicular C18-4 spermatogonia and MA-10 Leydig cells.
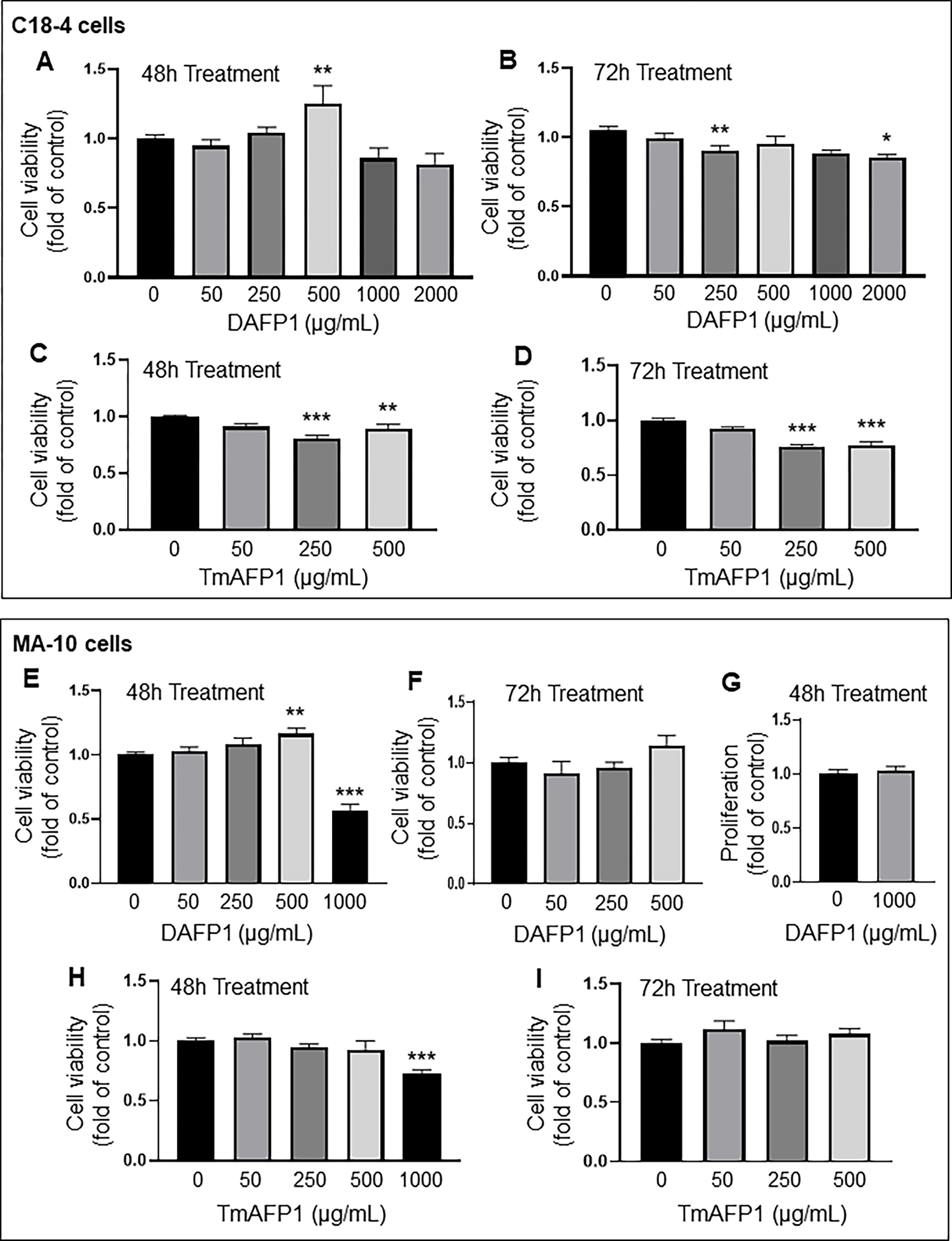
(A-B) Effect of 0 to 2000 μg/mL DAFP1 on C18-4 cell viability measured by MTT assay over (A) 48h and (B) 72h treatments. (C-D) Changes in viability with TmAFP treatments at 0 to 500 μg/mL over (C) 48h and (D) 72h. (E-G) Effect of 0 to 1000 μg/mL DAFP1 on MA-10 cell viability measured by MTT assay over (E) 48h and (F) 72h treatments. (G) MA-10 cell proliferation measured by EDU incorporation assay in control cells (0) and cells treated with 1000 μg/mL DAFP1 for 48h. (H-I) Changes in MA-10 cell viability with TmAFP treatments at 0 to 1000 μg/mL over (H) 48h and (I) 72h. Results are presented as fold change of vehicle control. N=3 independent experiments conducted in triplicates. Significant difference relative to vehicle control with One way ANOVA test and multiple comparisons: * (p≤0.05), ** (p<0.01), *** (p<0.001).
In mouse MA-10 Leydig cells, DAFP1 did not change cell viability at concentrations below 500 μg/mL after 48h and 72h treatments, while 500 μg/mL DAFP1 resulted in a slight (16%) increase in viability (Fig. 1E–F). However, 1000 μg/mL DAFP1 induced a significant 44% decrease in MA-10 cell viability (Fig. 1E). MA-10 cell proliferation was further assessed for DAFP1 at 1000 μg/mL using the EDU incorporation assay, showing no effect on cell proliferation, which confirmed that this treatment induced loss of cell viability and not a cytostatic effect (Fig. 1G). A 25 % decrease in cell viability was found in MA-10 cells treated with 1000 μg/mL TmAFP for 48 hours, while lower concentrations of TmAFP had no effect (Fig. 1H–I).
DAFP1 did not have an adverse effect on cell viability in RAW 264.7 mouse macrophages at up to 500 μg/mL at 48h and 72h (Fig. 2A–B). However, a limited loss of cell viability (21%) was observed with 1000 μg/mL DAFP1 at 48h (Fig. 2A). TmAFP did not affect RAW 264.7 cell viability at both times (Fig. 2 C–D).
Figure 2. Effect on AFPs on RAW 264.7 macrophage viability.
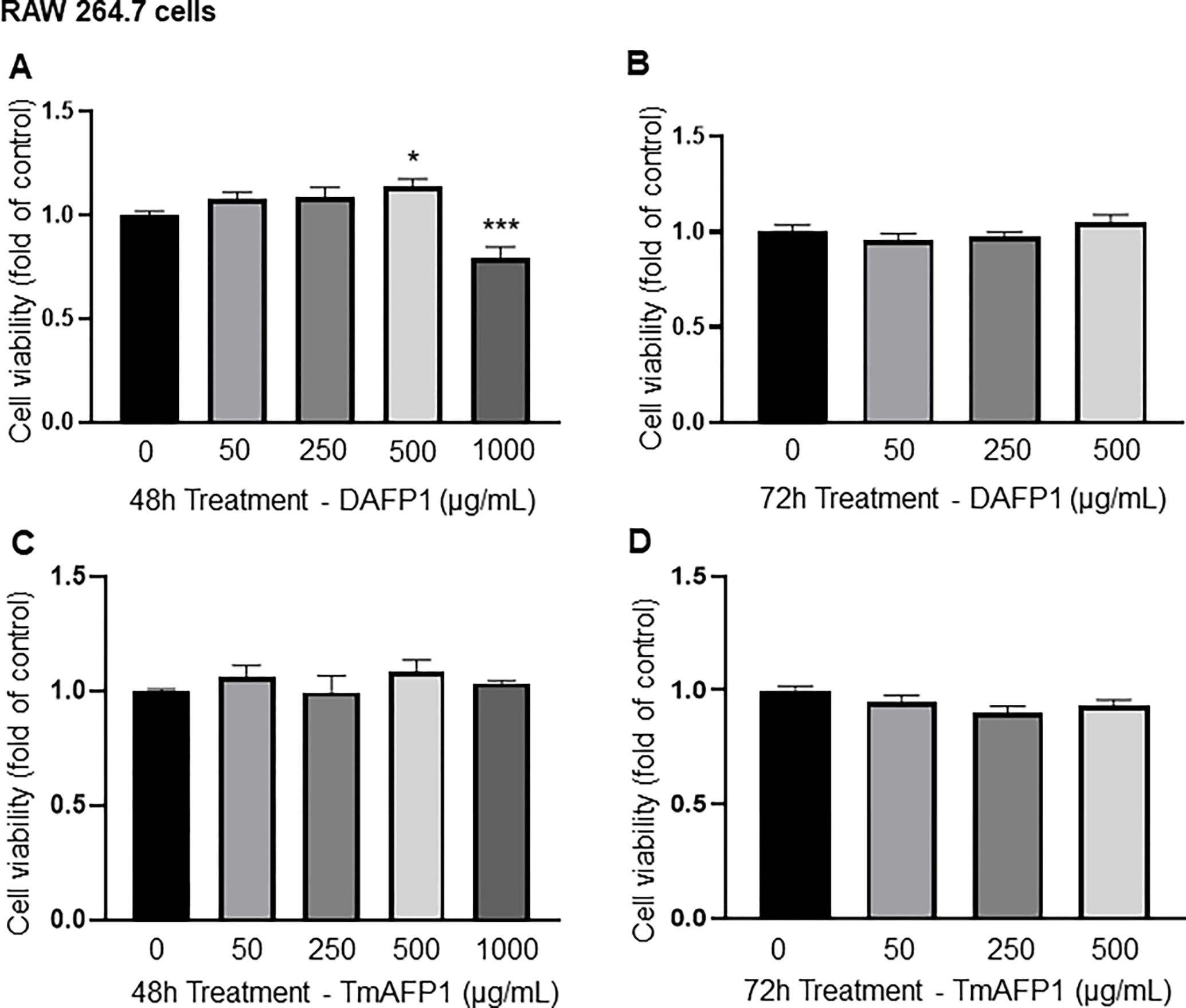
(A-B) Effect of vehicle (0) and up to 1000 μg/mL DAFP1 on RAW 264.7 macrophage viability measured by MTT assay over (A) 48h and (B) 72h treatments. (C-D) Effect of vehicle (0) and TmAFP at up to 1000 μg/mL on RAW 264.7 cell viability measured by MTT assay over (C) 48h and (D) 72h treatments. Results are presented as fold change of vehicle control. N=3 independent experiments conducted in triplicates. Significant difference relative to vehicle control with One way ANOVA test and multiple comparisons: * p≤0.05, ** p≤0.01, *** p≤0.001.
The HUH7 human hepatocyte cell line was the only one to show decreases in cell viability, ranging from 25% to 35%, with DAFP1 at 500 and 1000 μg/mL after 48h treatment, and with 50, 500 and 1000 DAFP1 at 72h (Fig. 3A, B). However, 1000 μg/mL DAFP1 did not affect cell proliferation measured by EDU-incorporation assay, confirming that this treatment decreased cell viability (Fig. 3C, D). In HUH7 hepatocytes, TmAFP at 500 and 1000 μg/mL decreased viability by 37% and 43%, respectively at 48h, and by around 25% for 50 to 1000 μg/mL at 72h (Fig. 3E, F). Overall, these data indicated a higher sensitivity of the HUH7 human hepatocyte cell line to the AFP proteins, compared to the other cell lines.
Figure 3. Effect of AFPs on HUH7 hepatocyte Viability and proliferation.
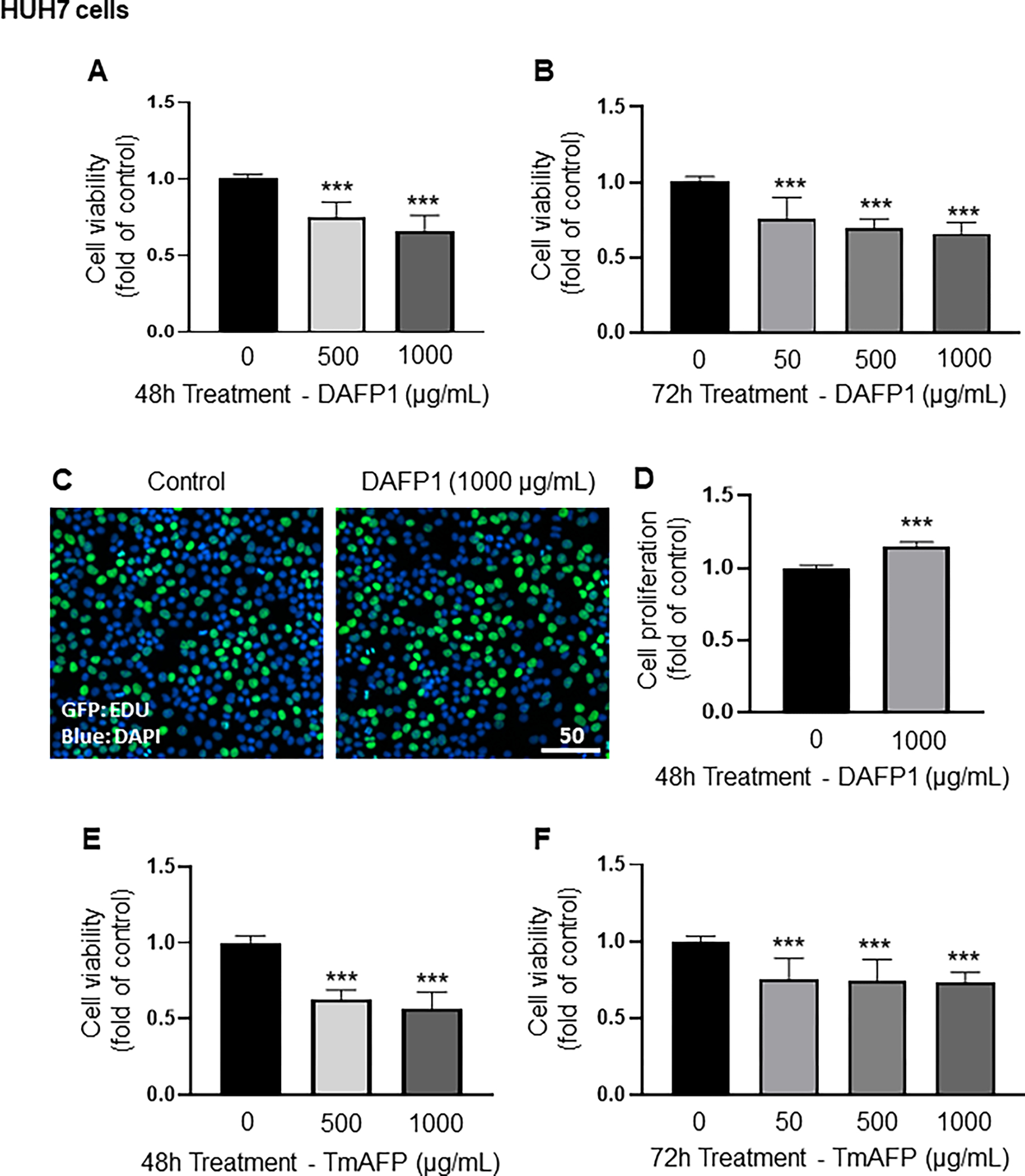
Cell viability was determined in HUH7 cells treated with vehicle (0) or up to 1000 μg/mL of DAFP1 (A-D) or TmAFP (E-F) for 48h or 72h by MTT assay. Proliferation was measured by EDU incorporation assay, with representative images of EDU incorporation shown in C (scale in μm.), and quantification shown in D. Results represent the means ± SEM of 3 independent experiments conducted each in triplicates and are presented as fold change of vehicle control (0 μg/mL). Significant differences relative to vehicle control were calculated with One way ANOVA test and multiple comparisons or t-test: * p≤0.05, ** p≤0.01, *** p≤0.001.
While DAFP1 did not affect cell viability after 48 hours treatment at up to 500 μg/mL in 3 mammalian cell lines, a lower concentration of 50 μg/mL was sufficient to decrease cell viability in the hepatic cell line. Moreover, at the concentration of 1000 μg/mL, DAFP1 decreased cell viability by 20 to 40% in 3 out of 4 cell types. TmAFP decreased viability even at the concentration of 500 μg/mL in 3 out of 4 cell lines, as well as at 1000 μg/mL, and at 50 μg/mL in one cell line. Thus, TmAFP exerted more adverse effects on cell viability than DAFP1. Thus, subsequent functional experiments were performed with DAFP1 only, while cold-preservation cold preservation experiments were performed with both AFP proteins.
3.2. Effect of DAFP1 on functional measurements in C18-4 spermatogonia cell line
Next, we examine whether DAFP1 treatment affected cellular function by studying functional responses in spermatogonia, Leydig and macrophage cell lines. Gene expression of spermatogonial markers was evaluated to determine whether 500 and 1000 μg/mL DAFP1 treatment would alter the stemness and differentiation status of C18-4 cells. qCR analysis was conducted to evaluate changes in expression of spermatogonial stem cell and undifferentiated spermatogonial markers: Id4 (Inhibitor of DNA Binding 4, HLH Protein), Foxo1 (Forkhead Box O1) and Mcam (Melanoma Cell Adhesion Molecule), as well as spermatocyte marker Sycp1 (Synaptonemal Complex Protein 1). There was a slight but significant (29%) decrease in Id4 expression at 1000 μg/mL DAFP1, but no effect at 500 μg/mL (Fig. 4A). Moreover, no changes were observed in the other germ cell markers Foxo1, Mcam, and Sycp1 (Fig. 4B–D). Among seven other germ cell markers examined, none showed significant changes, with only Nanos2 presenting a non-significant decreasing trend at 1000 μg/mL (data not shown). Thus, the overall results suggest that DAFP1 at 1000 μg/mL has minimal effect on the functional state of C18-4 spermatogonia.
Figure 4. Effect of DAFP1 on germ cell markers in C18-4 Spermatogonia.
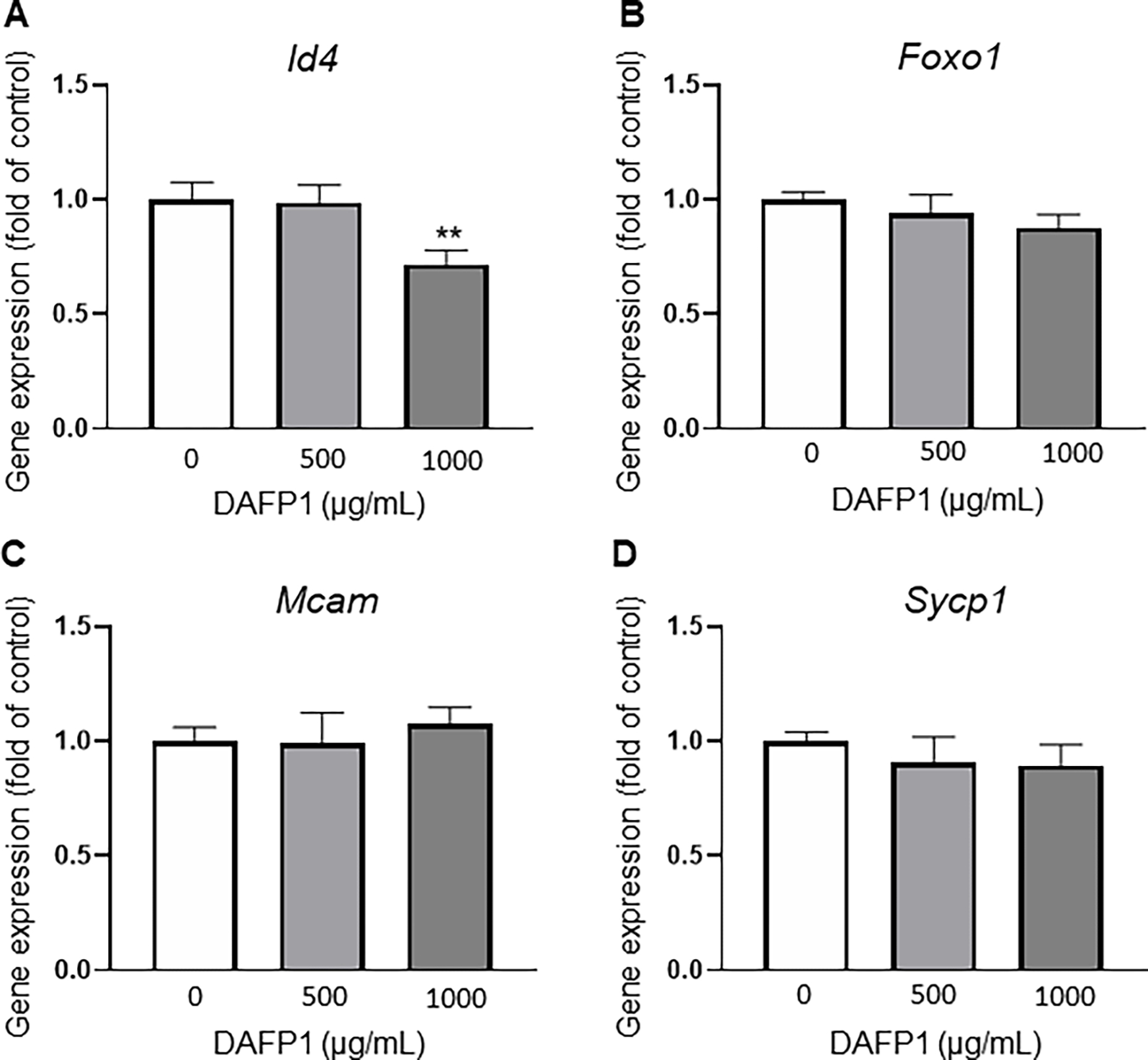
Effect of 48h treatments with vehicle or 1000 μg/mL DAFP1 on select spermatogonial markers (A) Id4 (B) Foxo1 and (C) Mcam, and on spermatocyte marker Sycp1 (D). Results are presented as fold change of vehicle control. qPCR data are normalized to GAPDH. N=3 independent experiments conducted in triplicates. Significant difference relative to vehicle control with t-test: * p≤0.05, ** p≤0.01, *** p≤0.001.
3.3. Effect of DAFP1 on functional measurements in MA-10 Leydig cell line
The main function of Leydig cells is to produce testosterone, as well as other steroid hormones such as progesterone. The MA-10 mouse Leydig cell line is a well-established functional Leydig cell model that is responsive to hormonal stimulation and secretes high amounts progesterone upon stimulation with human chorionic gonadotropin (hCG) hormone (Boisvert et al., 2016). To evaluate whether DAFP1 at 1000 μg/mL altered the functionality of MA-10 cells, progesterone production at both basal state and hormone-induced state was evaluated. The cells were pre-treated with vehicle or 1000 μg/mL DAFP1 for 48 hours, then treated with medium or 50 ng/ml hCG for 2 hours, and progesterone production in cell supernatants was measured. Pretreatment with DAFP1 had no effect on basal progesterone production but reduced by 32% the large increase in production induced by hCG stimulation (Fig. 5). Nonetheless, the hormonal response of the cells treated with this high concentration of DAFP1 remained robust, with a 23-fold increase in progesterone production compared to basal levels, indicating a minimal effect on Leydig cell function.
Figure 5. Effect of DAFP1 on MA-10 Leydig cell Progesterone Synthesis.
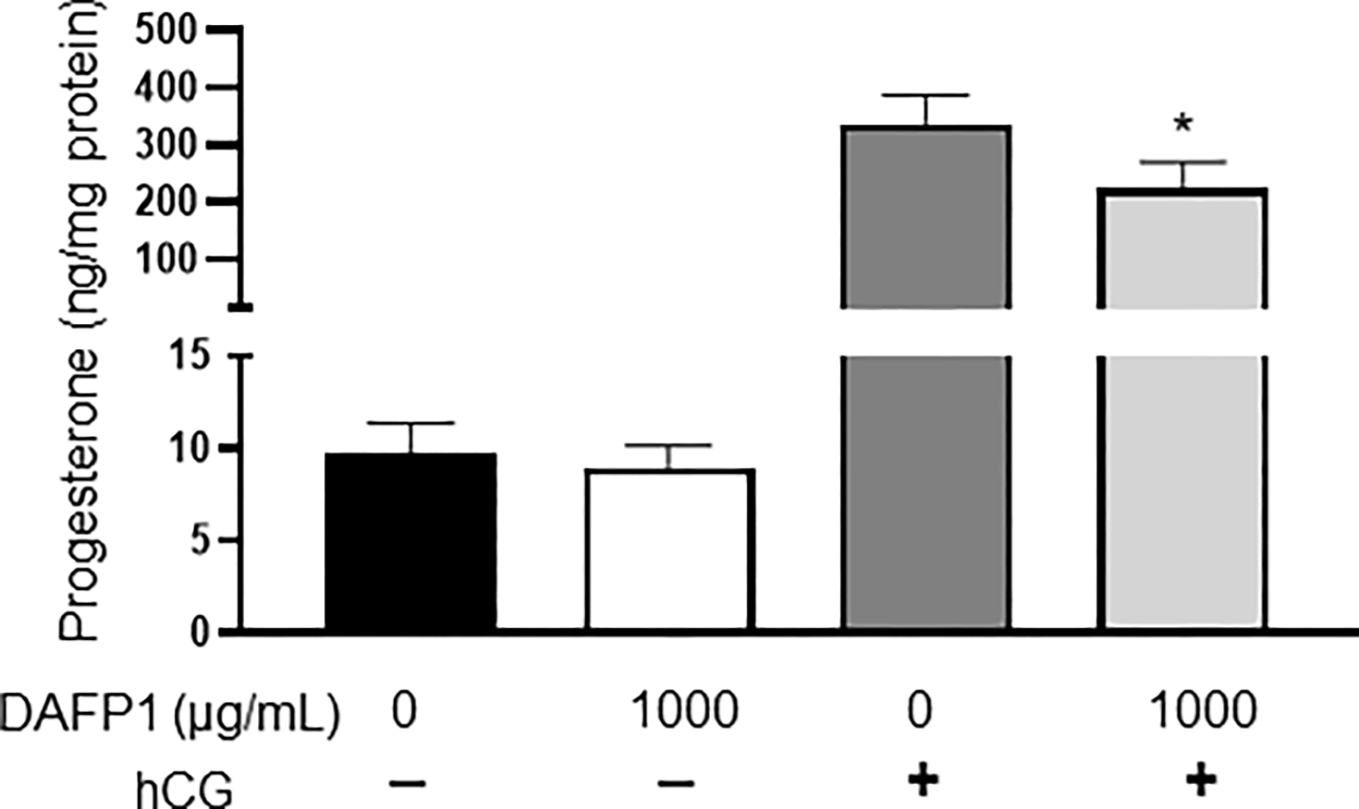
Effect of 48h treatments with vehicle or 1000 μg/mL DAFP1 on MA-10 cells progesterone production in the presence or absence of 50 ng/mL hCG added for 2h at the end of treatment. Results are normalized to total protein. N=3 independent experiments conducted in triplicates. Significant difference relative to vehicle control with t-test: * p≤0.05, ** p≤0.01, *** p≤0.001.
3.4. Effect of DAFP1 on functional measurements in RAW 264.7 macrophage cell line
To better characterize the toxicity profile of DAFP1 on RAW 264.7 cells, we first examined the cytokine expression profile of the cells exposed to 1000 μg/mL DAFP1. DAFP1 significantly upregulated the gene expression of pro-inflammatory genes Il1b, Cxcl2, Cxcl10 and Tnfa by 5303-fold, 1396-fold, 22.6-fold and 14.7-fold respectively upon 40h treatment (Fig. 8A–D). Furthermore, the secretion of the pro-inflammatory cytokine TNFa was increased by 18-fold in cells treated with 1000 μg/mL DAFP1 compared to control cells after 40h treatment (Fig. 8E), in agreement with the 14-fold increase observed in Tnfa gene expression.
Figure 8. DAFP1 internalization in mammalian cell lines.
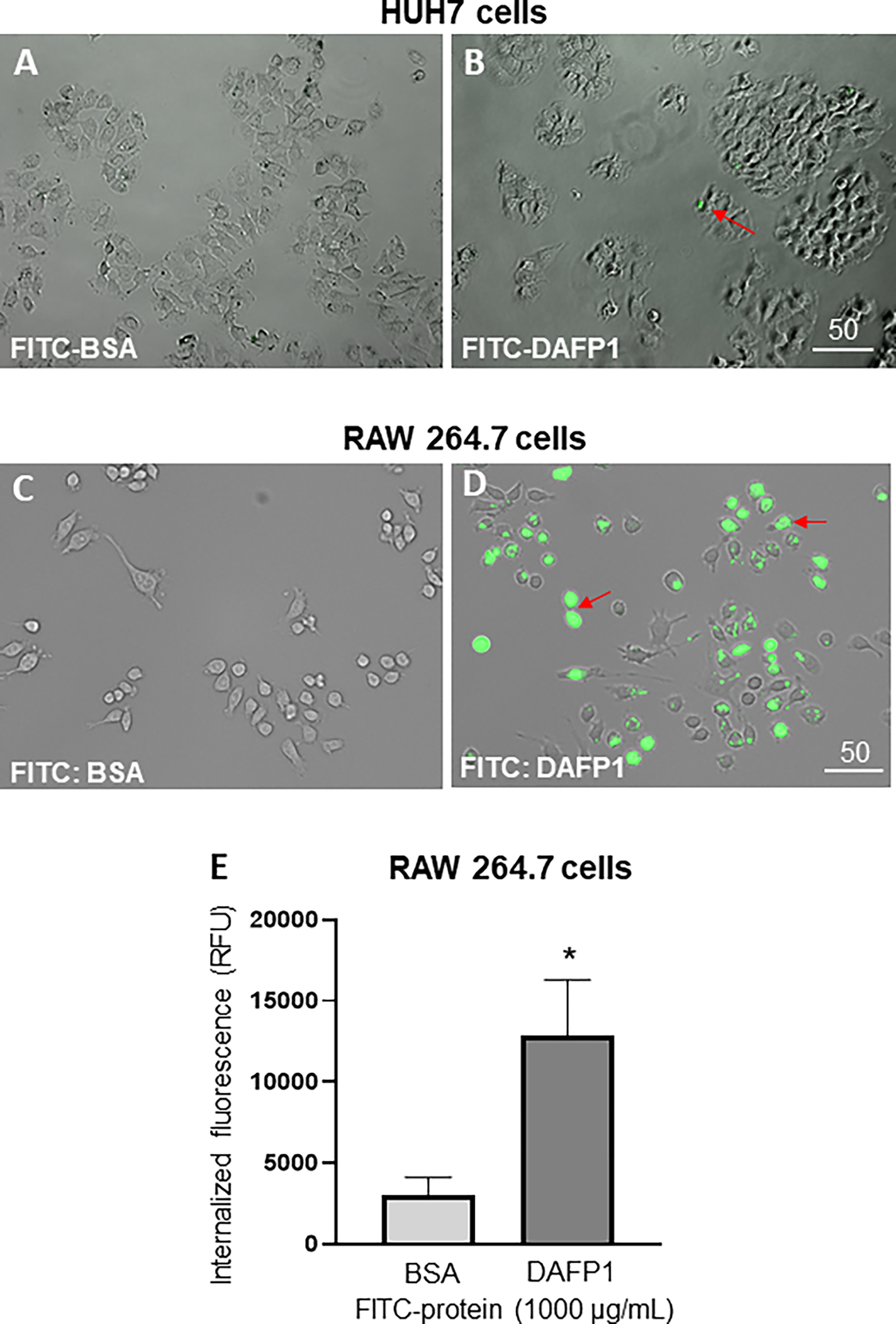
Representative merge images of bright field and FITC signal in cells illustrating the internalization of FITC-BSA and FITC-DAFP1 into HUH7 cells (A-B) and RAW 264.7 cells (C-D) after 6h of treatment with either medium, 1000 μg/mL of FITC-BSA or FITC-DAFP1. Scales in μm. (E) Quantification of relative fluorescent units (RFU) measured in RAW 264.7 cell lysates. N=2 independent experiments conducted in triplicates. Significant difference relative to FITC-BSA fluorescence using One way ANOVA test and multiple comparisons. * p≤0.05.
Because 1000 μg/mL DAFP1 significantly reduced RAW 264.7 macrophage viability when measured with MTT assay, we further evaluated if this effect was due to an increase in apoptosis or decreased proliferation, by determining the expression levels of two proapoptotic genes, Bad and Bim, the antiapoptotic gene Bcl2, and the proliferation marker gene Pcna, respectively. The effect of DAFP1 in RAW 264.7 cells revealed a complex response with both the pro-apoptotic gene Bad and anti-apoptotic gene Bcl2 being significantly increased by DAFP1, and no effect on Bim expression, indicating a dual effect that might explain the limited decrease (21%) in viability (Fig. 7A–C). No changes were observed in the expression of Pcna, a proliferative marker, upon DAFP1 treatment (Fig. 7D). Overall, this suggests that DAFP1 has only a limited effect on RAW 264.7 cell viability, but that it exerts a strong inflammatory response in these cells.
Figure 7. Effect of DAFP1 on the expression of apoptosis-related genes and proliferation marker PCNA in RAW 264.7 macrophages.
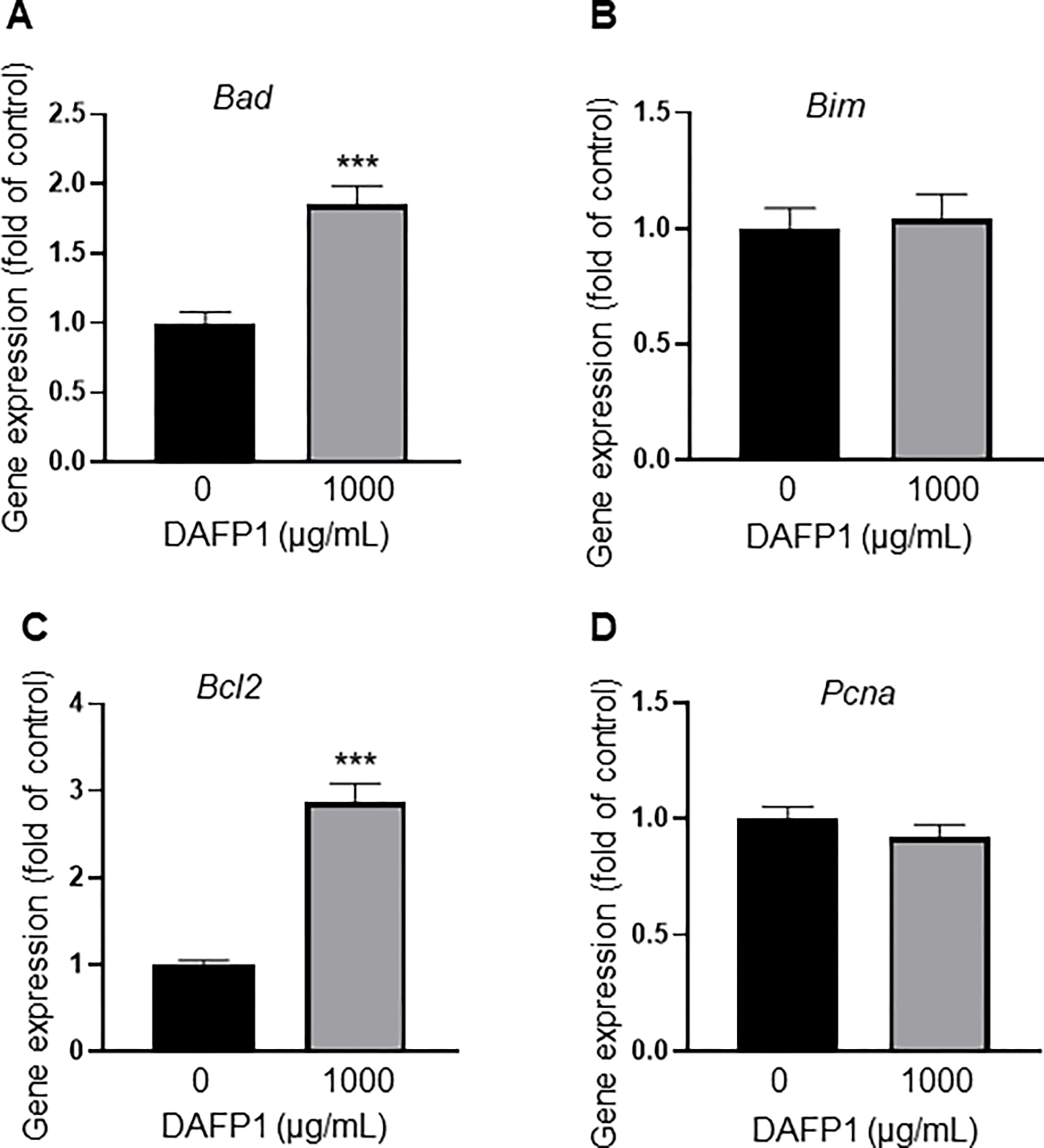
Effect of 40h treatment with vehicle or 1000 μg/mL DAFP1 on (A-B) pro-apoptotic genes Bad and Bim, and (C) anti-apoptotic gene Bcl2; and (D) proliferation marker Pcna. qPCR data are normalized to HPRT. N=3 independent experiments conducted in triplicates. Significant difference relative to vehicle control with Student t-test: *** p≤0.001
3.5. Analysis of DAFP1 internalization in MA-10, HUH7, and RAW 264.7 cells
To further characterize the potential toxicity of DAFP1 on the cell lines, we next evaluated the ability of three cell lines to internalize DAFP1. MA-10, HUH7, and RAW 264.7 cells were incubated with either media containing FITC alone, 1000 μg/mL of FITC-tagged DAFP1 or 1000 μg/mL FITC-tagged BSA, for 6 hours. Pictures were taken and the media, washings and cell layers were collected to determine the percent recovery of FITC in each fraction. In all instances, small amounts of DAFP1 were recovered with only 0.04%, 0.35%, and 0.44% recovery in the MA-10, HUH7, and RAW 264.7 cell lysates respectively. Representative images of HUH7 cells clearly showed that DAFP1 was not internalized into these cells and the rare fluorescence observed in the treated condition was background signal (Fig. 8A–B). A similar absence of internalization was observed with MA-10 cells (data not shown). This contrasted with a clear FITC-DAFP1 internalization in RAW 264.7 cells, where a strong signal of FITC-conjugated DAFP1 protein was observed in many cells (Fig. 8D), compared with no visible internalization of FITC-conjugated BSA (Figure 8C). Quantification of relative fluorescent units (RFU) in cell lysates showed a significant induction of DAFP1 internalization upon 6h treatment with 1000 μg/mL FITC-DAFP1 that was 4-fold higher than the levels found in cells incubated with FITC-BSA (Fig. 8E). These data suggest that only RAW 264.7 macrophages had the ability to incorporate DAFP1.
3.6. Effects of DAFP1 and TmAFP on rat kidneys after 48 and 72 hours of hypothermic conditions
To assess the potential use of DAFP1 and TmAFP in low temperature organ preservation, we evaluated the effect of 1000 μg/mL AFP treatment on the expression of two kidneys markers, Tcf21/pod1, a gene critical for glomerulogenesis and kidney function (Cui et al., 2003), and Aquaporin 1 (Aqp1), a water channel expressed in the proximal tubule and Henle’s loop (Knepper et al., 1996), and assessment of tissue morphology in freshly collected postnatal day 6 rat kidneys kept at 4°C for 48 and 72 hours. While there was a significant decrease in Tcf21/Pod1 expression in cold-treated samples in medium alone for 48 hours compared to Time zero, in the presence of DAFP1, Tcf21/Pod1 expression returned to levels similar to those in samples frozen at Time 0 (Fig. 9A). Aqp1 was significantly induced after 48 hours with 1000 μg/mL DAFP1 treatment compared to time 0 controls (Fig. 9B). However, when the kidneys were kept for 72 hours at 4°C, DAFP1 was no longer able to rescue Tcf21/Pod1 and Aqp1 gene expression, inducing significant decreases, further lowering gene expression below 50 % of the decreases induced by cold exposure (Fig. 9C, D). While 1000 μg/mL TmAFP was not sufficient to recover gene expression to levels found at Time zero, its presence did not aggravate the effects of cold on the two genes (Fig. 9C, D). The differential effects of DAFP1 and TmAFP during 3-days prolonged exposure suggest that there might be some benefit in using TmAFP over DAFP1, depending on the conditions. Upon evaluation of immunofluorescent tissue section images, the morphological appearance of the tissues after 48 hours was disrupted in kidneys kept at 4°C in medium alone, as shown by multiple areas of degeneration and loss of organization of the fluorescent signals both proteins (Fig. 9F) compared to samples fixed immediately after dissection (Fig. 9E). In contrast, the sections from kidney kept in cold in the presence of DAFP1 presented a more normal morphology and fluorescent signals for Tcf21 and Aqp1 comparable to those seen in Time zero samples (Fig. 9G). Thus, DAFP1 appeared to prevent the loss of structural integrity of kidney tubules and glomeruli and the expression of kidney gene markers that were apparent in the absence of DAFP1 after 48 hours in cold condition and maintained kidney structure closer to that observed at Time 0 condition.
Figure 9. Effect of DAFP1 and TmAFP on the morphology and expression of functional markers in rat kidneys kept under hypothermic conditions.
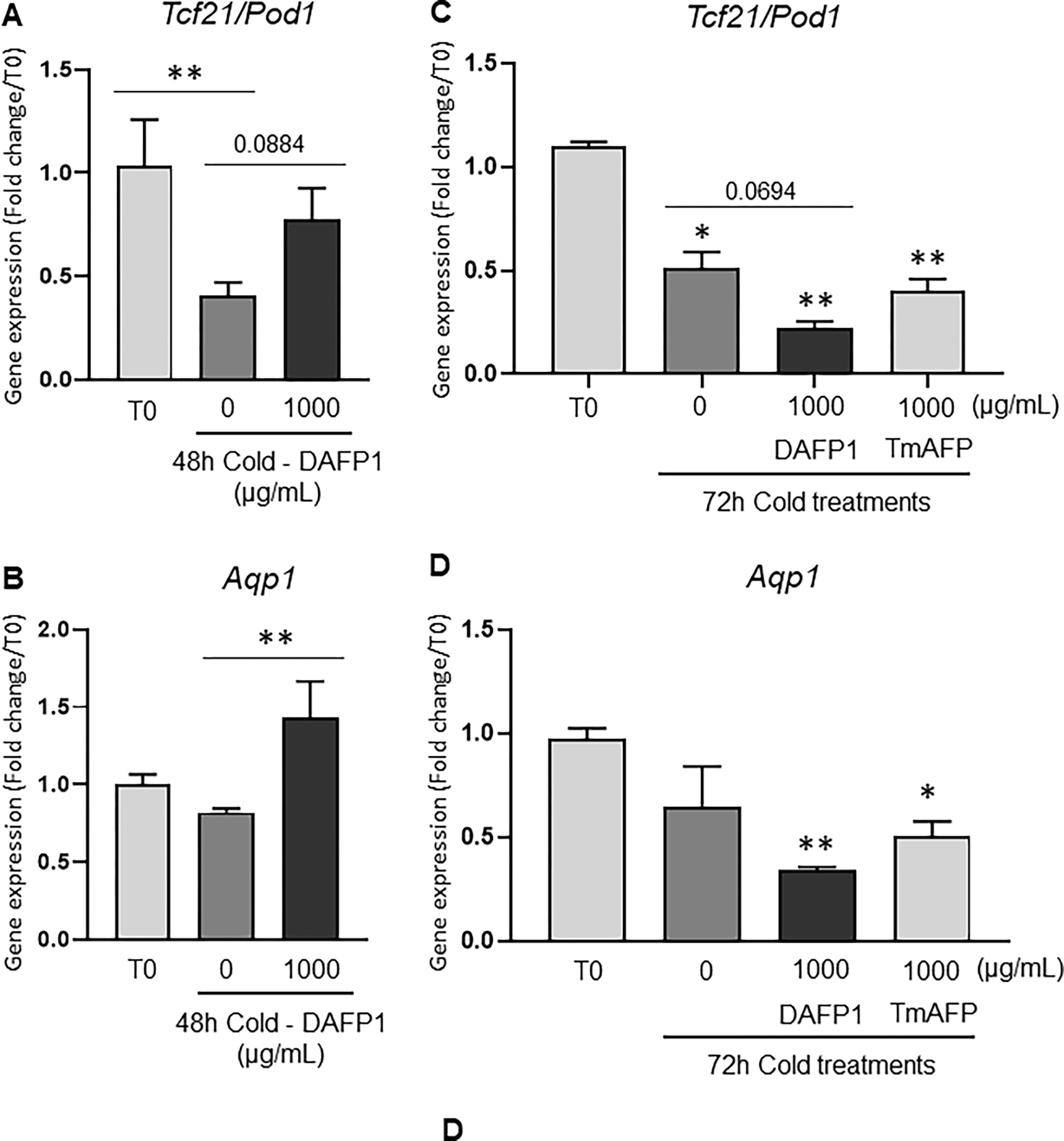
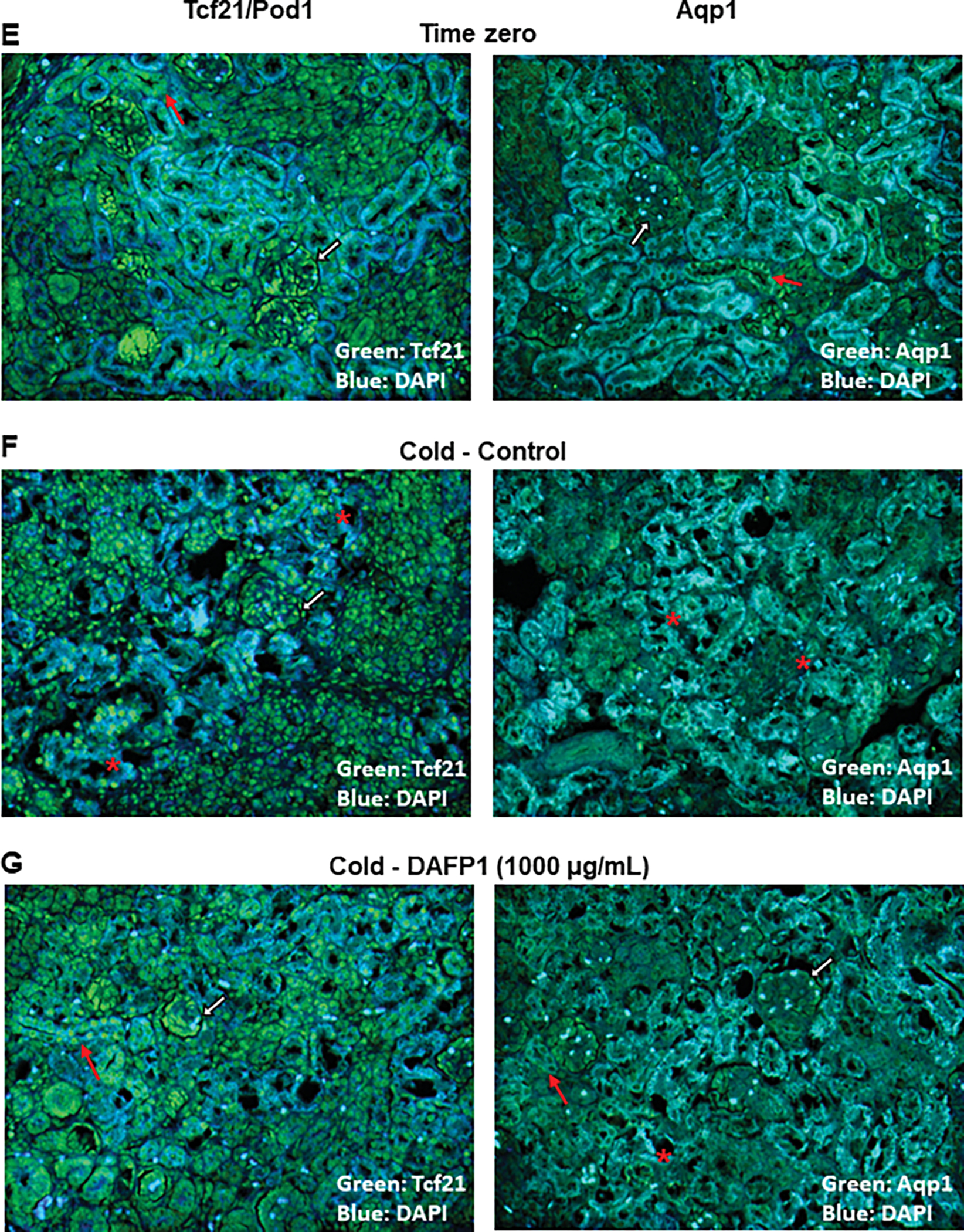
The expression of gene and protein levels of Aqp1 (Aquaporin water channel 1) and Tcf21 (Pod1) were examined in PND6 rat kidneys kept at 4°C for 48h (A, B, E-G) or 72h (C, D) in the presence of medium (Control/0) or 1000 μg/mL DAFP1 or TmAFP, in comparison to kidneys frozen at the time of dissection (Time zero; T0). The kidneys of 3 rats were used per condition for 48h cold exposure and 2 to 3 rats per condition for 72h cold exposure. After 48h cold treatment, for each rat, one kidney was frozen for mRNA analysis and the other kidney was fixed in paraformaldehyde for immunofluorescence analysis. After 72h cold treatment, all kidneys were processed for qPCR analysis. The mRNA levels of Tcf21 (A, C) and Aqp1 (B, D) were determined by qPCR analysis and normalized to GAPDH. Gene expression in control and DAFP1- or TmAFP- treated tissues kept in cold condition is expressed as fold change of the levels measured in samples frozen after dissection (T0). Statistical significance: * p≤0.05, ** p≤0.01. (E-G) Representative pictures showing the general morphology and immunofluorescence signals of Tcf21 and Aqp1 in kidneys at time zero (E) vs tissues kept at 4°C for 48h with medium (Control) (F) or 1000 μg/mL DAFP1 (G). White arrows point at glomeruli, red arrows point at tubules, and red stars indicate areas of tissue degeneration.
4. DISCUSSION
To our knowledge, little is known about the toxicity associated with insect derived AFPs. Studies have been primarily conducted on the more well-studied AFPs derived from fish species, such as the Notch-fin eelput, with no effects on cellular viability using doses as high as 20 mg/ml (Hirano et al., 2008b). A literature review examining the effects of consuming AFPs, intrinsic to fish and many edible plants, also report no adverse effects associated with protein toxicity at consumption levels reaching 10 mg/day in the USA and up to 500 mg in parts of Europe (Crevel et al., 2002). But it may be difficult to draw any specific conclusion due to the lack of any obvious adverse toxicological health outcomes. Moreover, little is known about the effect of insect derived AFPs on mammalian cells and their toxicity profiles have never been established. The published results of preserving cells and tissues using AFPs are controversial, largely due to this lack of knowledge. Thus, this study aimed to establish the toxicity profiles of two highly active AFPs on a panel of mammalian cells to better assess the possibility of using them as additives in cold preservation solutions. The results of this study will guide the use of AFPs in the preservation of mammalian cells, tissues, and organs, by contrasting the lack of adverse effects on viability and biological functions in some of the cell lines with increased toxicity or undesirable effects in other cell lines.
Consistent with the literature, we report no observed toxic effect on cell viability in DAFP1, derived from D. canadensis, at doses up to 500 μg/mL for up to 48 hrs of treatment in the MA-10 Leydig cells and RAW264.7 macrophage cell lines. However, the spermatogonial C18-4 cell line presented a non-monotonic viability dose-response, with 250 μg/mL of DAFP1 or TmAFP inducing small but significant 10 to 20 % decrease of control levels after 72 hours treatment. Non-monotonic and hormetic dose-responses are proposed to reflect adaptive responses of cells or tissues, and have been observed with chemicals such as hormones and endocrine disruptor chemicals (Calabrese and Mattson, 2017; Varret et al., 2018). Thus, it is possible that a similar effect takes place in this cell line. The other cell line with decreased cell viability in response to DAFP1 was the human hepatocyte HUH7 cell line, in which adverse effects were found at 50 and 500 μg/mL, suggesting an increased susceptibility of hepatocytes to this AFP. At the higher concentration of 1000 μg/mL, variable effects on cellular viability were observed depending on the AFP isoform, cell type, and treatment duration. Proliferation was not affected in MA-10, HUH7 and RAW 264.7 cells, eliminating the possibility of a cytostatic effect on these cell types, and suggesting that the changes in MTT readings reflected cell death.
Although the AFP derived from Tenebrio molito, TmAFP, had no effect on cell viability at up to 500 μg/mL for up to 72 hours on RAW 264.7 and MA-10 cells, it exerted more adverse effects than DAFP1 on cell viability, by decreasing viability in C18-4 spermatogonia and HUH7 hepatocyte cell lines at 500 μg/mL and lower concentrations. Indeed, HUH7 cells showed heightened susceptibility to both DAFP1 and TmAFP compared to the other mammalian cell types. Since we did not examine cell viability in hepatocytes from other species, we cannot conclude at present if this lower tolerance to AFPs is inherent to hepatocytes or if it is related to the human origin of the cell line. These data imply that the same amounts of AFPs may have cytotoxic effects on specific cell types while being innocuous for others, calling for caution and designing toxicity studies including several tissues and species.
Since our goal is to repurpose AFP proteins for human use for organ cold preservation, and the data indicated a lower cytotoxicity for DAFP1 in representative cell lines, we performed subsequent functional assays on cell lines using 1000 μg/mL DAFP1, to determine whether DAFP1 could disrupt cell function at high concentration and better assess its safety margin. Moreover, in a previous study on rat pancreatic INS-1 β-cells, we found this concentration to be optimal for maintaining cell viability for up to 5 days at 4°C for both AFPs (Gonzalez, 2019). The further characterization of DAFP1 potential toxicity was conducted by evaluating its effects on the specific functions on three of the cell lines. The mouse C18-4 cell line is representative of the spermatogonia stem cell (SSC), a pool of cells that support lifelong spermatogenesis in the adult male. SSCs rely on molecular signals and secreted factors to either self-renew to maintain the SSC pool or differentiate through spermatogenesis to form spermatozoa. Premature differentiation can reduce the number of SSCs available for maturation into sperm and thus contribute to fertility issues (Culty, 2013; Manku and Culty, 2015). Despite a slight but significant decrease in the SSC marker Id4 after 48-hour treatment with 1000 μg/mL DAFP1, the lack of significant changes in the gene expression of 10 gene markers representing undifferentiated and progenitor spermatogonia, as well as differentiated spermatogonia and spermatocyte, indicates that DAFP1 did not alter the differentiation or self-renewing properties of the spermatogonial cell line at up to 500 μg/mL over 48 hours, and had only a minimal effect at 1000 μg/mL.
The mouse MA-10 cell line is representative of Leydig cells, the interstitial cells in the testis whose function is to synthesize and secrete androgens. Androgen production is essential for the development of male genitalia in utero and after birth, regulating the growth and function of the testis and secondary male sex characteristics, as well as spermatogenesis and non-reproductive biological functions (Walker et al., 2021). MA-10 cell functionality was assessed by measuring progesterone secretion in basal and hormone-induced conditions and showed that testicular steroidogenesis was not affected by DAFP1 in basal conditions, and only minimally when stimulated by hormone, since the cells showed a strong increase in steroid production in response to hCG stimulation, even after 48-hour treatment with 1000 μg/mL DAFP1.
Since significant decreases in viability were observed in MA-10, HUH7 and RAW 264.7 cell lines with the high concentration of DAFP1 (1000 μg/mL), we next examined if DAFP1 could be internalized to exert its effects on intracellular targets in these cell types. In the case of Leydig cells and hepatocytes, there was minimal FITC recovered in the cell lysates and upon further inspection, there was insignificant localization of DAFP1 in the cells. This suggests that the adverse effects observed on the viability of Leydig cells and hepatocytes were due to an interaction of DAFP1 at the surface of the cells. It is possible that DAFP1 at high dose could interact directly or indirectly with plasma membrane receptors mediating apoptosis in these cells. On the other hand, FITC-DAFP1 internalization was observed in RAW 264.7 macrophages. These data suggest that only RAW 264.7 macrophages had the ability to incorporate DAFP1, which may be related to the induction of the inflammatory response in these cells. Moreover, the absence of incorporation of FITC-BSA, a large protein usually not cell permeable, by RAW 264.7 cells suggests that the internalization of DAFP1 might be related to the nature of this protein triggering an endocytic response, whereas BSA did not induce such an effect in macrophages.
It is apparent that DAFP1 treatment induces an inflammatory response in the macrophage cell line by markedly increasing TNFa production and the gene expression of all inflammatory genes evaluated in the study. Phagocytes play a key role during inflammation as the first line of defense against pathogens, initiating inflammatory responses by releasing cytokines in attempts to clear debris (Oishi and Manabe, 2018). Taken together, the small decrease in cell viability with 1000 μg/mL DAFP1, likely due to increased apoptosis, and the inflammatory response this treatment induces in RAW 264.7 cells, suggest that this concentration of DAFP1 has negative effects on macrophages, and perhaps is stimulating the cells to phagocytize the protein as a defense mechanism.
Lastly, the potential of DAFP1 and TmAFP to exert protective effect on tissues kept in cold conditions was assessed. Improvements in transplantation techniques may be achieved by improving the preservation of cell and tissue for extended periods. However, cold temperatures may have detrimental effects on cells, as increasingly cooler temperatures can cause cell membrane to become leaky, allowing ions to uncontrollably enter the cell. This can lead to cell swelling and ultimately induce cell bursting (Rubinsky, 2003). AFPs may be a simple and cost-effective solution for use in the hypothermic preservation of cells and tissues. Tatsuro et al. found that fish AFP was able to preserve rat insulinoma cells RIN-5F over 5 days at 4°C at a survival rate of 78% while still preserving its ability to secrete insulin (Kamijima et al., 2013). NfeAFPs extracted from notched-fin eelputs was shown to protect cold sensitive human liver cells HepG2 exposed at 4°C conditions for 24–72 hours. NfeAFPs decreased hypothermia induced cytotoxicity, preserved intracellular ATP levels, and improved viability (Hirano et al., 2008b). Additionally, NfeAFPs were able to preserve bovine embryos for up to 7 days at 4°C and when the surviving embryos were transferred to recipient heifers, it resulted in a 50% pregnancy rate (Ideta et al., 2014). Therefore, to assess the transferability of DAFP1 and TmAFP for cold preservation applications, we evaluated the ability of these proteins to preserve isolated kidneys from PND6 rats in cold storage conditions (4°C) for 48 and 72 hours. We found that keeping the kidneys in a medium containing DAFP1 for 48 hours prevented the cold-driven reduction in the mRNA expression of two kidney markers, Tcf21, a transcription factor expressed in kidney epithelial cells (Gooskens et al., 2018), and Aqp1, encoding a water channel protein involved in regulating water transport across kidney tubules (Knepper et al., 1996). While DAFP1 induced a return of Tcf21 transcripts to levels comparable to that of freshly dissected kidneys, it increased the mRNA expression of Aqp1 beyond the original levels. The increase in Aqp1 might correspond to a protective response of the cells against cold, as suggested by a study performed on rat kidney ductal cells which showed that increasing aquaporin 2 expression protected the cells from cold-induced damages (Wang and Ben, 2004). Moreover, the levels of both proteins in paraffin sections of kidneys stored with DAFP1 appeared similar to those observed in kidneys fixed after dissection. Furthermore, kidneys kept in cold conditions without DAFP1 presented exacerbated disruption of tubule structure and disorganized pattern of Tcf21 fluorescent signal, as well as lower signal for Aqp1 protein, which was not observed in the Time 0 condition. The alterations that were observed in the vehicle-treated group appeared to be prevented in the kidneys stored in the presence of DAFP1 treatment, suggesting that DAFP1 exhibited protective effects for 48 hours. However, the protective effects of DAFP were lost after 72 hours in cold, at which point DAFP1 exacerbated the effect of cold on gene expression, suggesting a long-term toxicity of this AFP not observed with TmAFP.
Although DAFP1 and TmAFP at 1000 μg/mL were not able to protect kidneys against the insults of cold over 72 hours in culture media, it might not be critical considering that kidneys destined for organ transplantation are not kept for such prolonged periods of time, since they were found to remain viable only for up to 36 hours in cold preservation solutions. To mimic whole organ preservation at 4°C, prolonging tissue integrity for up to 2 days at 4°C, as observed in the presence of DAFP1, may be sufficient to gain more flexibility in organ transfer from donor to recipient. Moreover, this series of experiments was limited in the high dose of AFPs that affected viability in some cell lines and induced macrophage activation. It is possible that treating a whole tissue limits the penetration of DAFP1 and exposure of sensitive cell types within the tissue. Thus, the effects of lower doses of the AFPs should be tested more extensively in future studies and it would not be surprising that optimal protective effects may be observed at some of the lower doses. Future studies will aim to further characterize this protective effect through assessing multiple doses and in multiple organ systems. Although this might not be a concern with antifreeze proteins added in a fluid surrounding an intact organ kept in cold conditions, as tested here with kidneys, the results on a macrophage cell line suggesting its activation by an insect AFP stress the need to design experiments verifying that AFPs do not affect immune cells in cold-preserved organs.
A potential mechanism behind the protective effects of DAFP1 may be due to the ability of D. canadensis AFPs to reduce hypothermia-induced cellular damage. Prior studies have proposed that AFPs can slow down cold swelling due to their ability to preserve intracellular ATP levels, which is required for cellular ion homeostasis and cell size regulation. Cold stress utilizes more ATP, which can be attributed to characteristic membrane damage and cell swelling that can ultimately cause cells to rupture (Hirano et al., 2008a). We observed a significant increase in expression of the gene encoding aquaporin, whose main role in the kidney is to transfer water across the membrane to preserve the concentration gradient of solutes within the cell. Perhaps in response to cold stress, DAFP1 is inducing the synthesis of aquaporins to maintain integrity of the tissue. However, future studies will have to be conducted to further explore this hypothesis.
In conclusion, by exhibiting an overall safe toxicity profile at doses up to 500 μg/mL, minimal effects on the functions of two cell types at high dose, and unique hypothermic-preservation properties for 2-days, DAFP1 has potential for a variety of cold preservation applications at doses up to 500 μg/mL. However, since cell viability was reduced with a DAFP1 dose of 1000 μg/mL, which also induced macrophage activation, and at lower doses for two of the cell lines examined, careful consideration must be taken to minimize potentially toxic effects in case high doses of AFPs are used. Additionally, the study of the mechanisms behind the differential effects of the two AFP proteins on cells and tissues could be instrumental in understanding key properties associated with beneficial effects as compared to adverse effects. Large-scale adaptation of these compounds could aid in various applications, such as in tissue storage for restoring fertility in cancer patients and providing additional pre-clinical models for drug development studies.
Figure 6. Effect of DAFP1 on the expression of inflammatory genes and TNFa cytokine in RAW 264.7 macrophages.
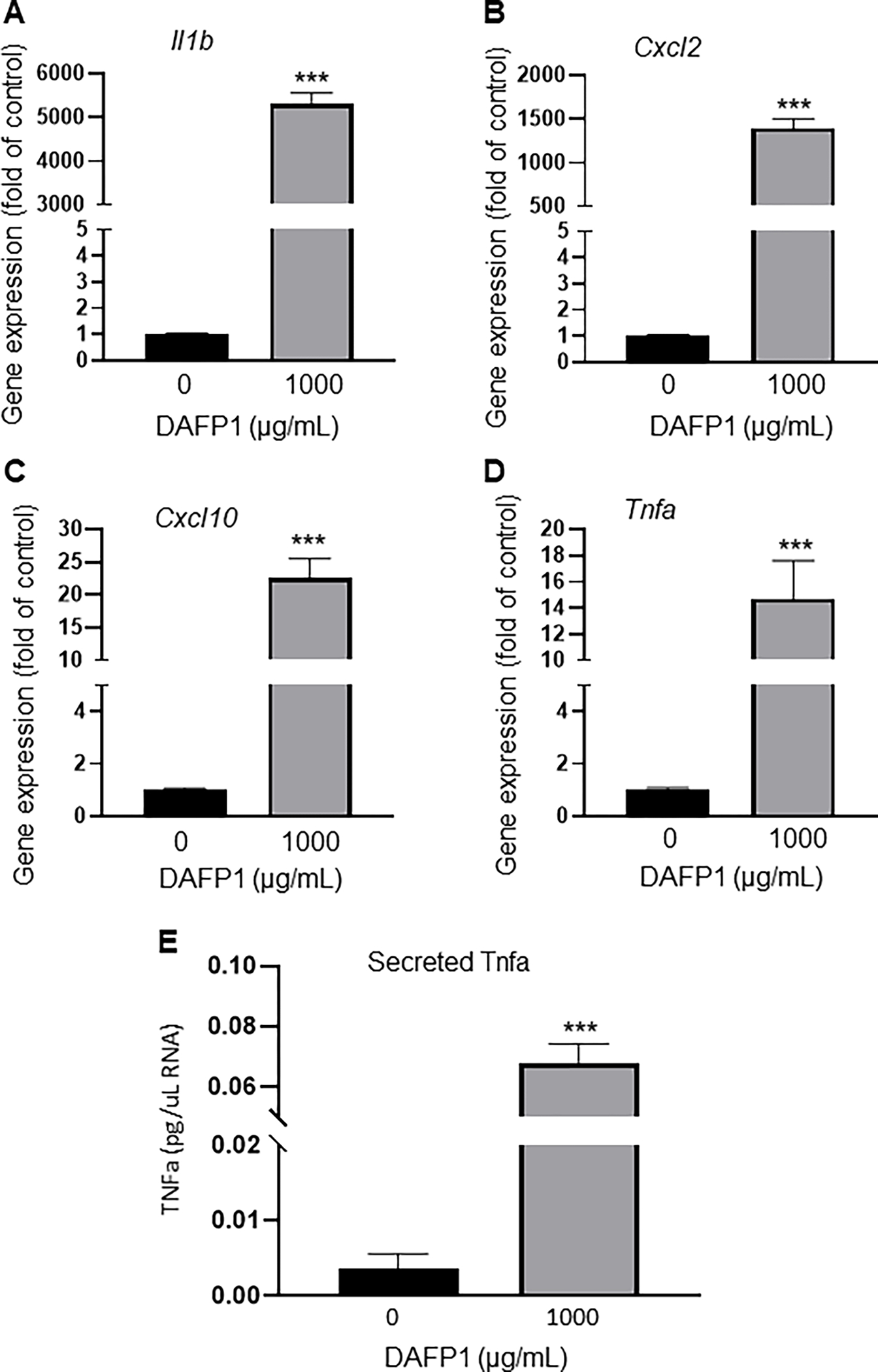
Effect of 40h treatment with vehicle or 1000 μg/mL DAFP1 on (A-D) gene expression of the inflammatory genes Il1b, Cxcl2, Tnfa, and Cxcl10 measured by qPCR analysis, normalized to HPRT. (E) Effect of 40h treatment with vehicle or 1000 μg/mL DAFP1 on TNFa secreted protein measured by ELISA assay. Results are normalized to total RNA content in cells. N=3 independent experiments conducted in triplicates. Significant difference relative to vehicle control with Student t-test: *** p≤0.001.
Table 1-.
List of qPCR primers
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Mouse primers | ||
| Foxo1 | CTTCAAGGATAAGGGCGACA | GACAGATTGTGGCGAATTGA |
| Id4 | CAGGGTGACAGCATTCTCTG | CCGGTGGCTTGTTTCTCTTA |
| Mcam | CAAACTGGTGTGCGTCTTCTT | CTTTTCCTCTCCTGGCACAC |
| Cxcl10 | AAAAAGGTCTAAAAGGGCTC | AATTAGGACTAGCCATCCAC |
| Il1b | GGATGATGATGATAACCTGC | CATGGAGAATATCACTTGTTGG |
| Cxcl2 | GGGTTGACTTCAAGAACATC | CCTTGCCTTTGTTCATCAGTATC |
| TNFa | CTATGTCTCAGCCTCTTCTC | CATTTGGGAACTTCTCATCC |
| Sycp1 | CATGCTCGAACAGGTTGCTA | GTCTGCTCATTGGCTCTGAA |
| Bad | CTCGAGATCGGGCTTGGG | ATAGCCCCTGCGCCTCC |
| Bcl2 | TCTTTGAGTTCGGTGGGGTC | TAGTTCCACAAAGGCATCCCAG |
| Bim | GTGCAGGGGTCGTTCGAT | TGCCGGGCTCCTGTCTTG |
| Ki67 | ACCATCATTGACCGCTCCTT | TTGACCTTCCCCATCAGGGT |
| Pcna | CTAGCCATGGGCGTGAAC | GAATACTAGTGCTAAGGTGTCTGCAT |
| Nanos2 | AGTCTCTCTACCGACGCAGT | AAACGTTGAGATCGGGGGAC |
| Vasa | GAAGTGGGTTTCCTTCTGGA | TTCCAAAGCCACCAGTTGTA |
| Dazl | GCTGATATTTTGCCCAATGAA | TCTGTATGCTTCGGTCCACA |
| Oct4 | CCTGCAGAAGGAGCTAGAACAGT | TGTTCTTAAGGCTGAGCTGCAA |
| Thy1 | GGAGTCCAGAATCCAAGTCGG | TATTCTCATGGCGGCAGTCC |
| Gfra1 | GCGTGTGAAGCACTGAAGTC | GGTTCAGTTCCGACCCAAC |
| Rat primers | ||
| Gapdh | CGGAGCAAAAGGGTCATCATCTCC | TGGTTCACACCCATCACAAACAT |
| Aqp1 | CACCTGCTGGCCATTGACTA | ACTGGTCCACACCTTCATGC |
| Tcf21 | CAGTCAACCTGACTTGGCCC | TAAGTGTTCTTGCGGGGTGG |
Acknowledgements
We thank Drs. Marie Claude Hofmann and Mario Ascoli for the donation of C18-4 and MA-10 cell lines, respectively. We thank Drs. John Duman at University of Notre Dame and Peter Davies at Queen’s University for donating the cDNAs of DAFP1 and TmAFP.
Funding
This work was supported by a National Science Foundation PFI-TT grant (grant number 1827782) and the National Institutes of Health (SC3GM086249) to XW and by research funds from the School of Pharmacy of University of Southern California to MC.
REFERENCES
- Boisvert A, Jones S, Issop L, Erythropel HC, Papadopoulos V, Culty M, 2016. In vitro functional screening as a means to identify new plasticizers devoid of reproductive toxicity. Environ Res 150, 496–512. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Mattson MP, 2017. How does hormesis impact biology, toxicology, and medicine? NPJ Aging Mech Dis 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevel R, Fedyk J, Spurgeon M, 2002. Antifreeze proteins: characteristics, occurrence and human exposure. Food and Chemical Toxicology 40, 899–903. [DOI] [PubMed] [Google Scholar]
- Cui S, Schwartz L, Quaggin SE, 2003. Pod1 is required in stromal cells for glomerulogenesis. Dev Dyn 226, 512–522. [DOI] [PubMed] [Google Scholar]
- Culty M, 2013. Gonocytes, from the fifties to the present: is there a reason to change the name? Biology of reproduction 89, 46, 41–46. [DOI] [PubMed] [Google Scholar]
- Davies PL, 2014. Ice-binding proteins: a remarkable diversity of structures for stopping and starting ice growth. Trends in biochemical sciences 39, 548–555. [DOI] [PubMed] [Google Scholar]
- DeVries AL, 1971. Glycoproteins as biological antifreeze agents in antarctic fishes. Science 172, 1152–1155. [DOI] [PubMed] [Google Scholar]
- DeVries AL, Wohlschlag DE, 1969. Freezing resistance in some Antarctic fishes. Science 163, 1073–1075. [DOI] [PubMed] [Google Scholar]
- Duman JG, 2001. Antifreeze and ice nucleator proteins in terrestrial arthropods. Annu Rev Physiol 63, 327–357. [DOI] [PubMed] [Google Scholar]
- Goding JW, 1976. Conjugation of antibodies with fluorochromes: Modifications to the standard methods. Journal of Immunological Methods 13, 215–226. [DOI] [PubMed] [Google Scholar]
- Gonzalez I, Omori K, Perez RI, Al-Abdullah I, Wen X, 2019. Hypothermic preservation of pancreatic insulin producing B-cells using antifreeze proteins. Cryobiology 91, 189. [Google Scholar]
- Gooskens SL, Klasson TD, Gremmels H, Logister I, Pieters R, Perlman EJ, Giles RH, van den Heuvel-Eibrink MM, 2018. TCF21 hypermethylation regulates renal tumor cell clonogenic proliferation and migration. Mol Oncol 12, 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, Ala P, Yang DS, Hon WC, Moffatt BA, 1992. Antifreeze protein produced endogenously in winter rye leaves. Plant Physiol 100, 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, Lumb C, Wiseman SB, Wisniewski M, Johnson RW, Marangoni AG, 2005. Antifreeze proteins modify the freezing process in planta. Plant Physiology 138, 330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halwani DO, Brockbank KG, Duman JG, Campbell LH, 2014. Recombinant Dendroides canadensis antifreeze proteins as potential ingredients in cryopreservation solutions. Cryobiology 68, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Nishimiya Y, Kowata K, Mizutani F, Tsuda S, Komatsu Y, 2008a. Construction of time-lapse scanning electrochemical microscopy with temperature control and its application to evaluate the preservation effects of antifreeze proteins on living cells. Analytical chemistry 80, 9349–9354. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Nishimiya Y, Matsumoto S, Matsushita M, Todo S, Miura A, Komatsu Y, Tsuda S, 2008b. Hypothermic preservation effect on mammalian cells of type III antifreeze proteins from notched-fin eelpout. Cryobiology 57, 46–51. [DOI] [PubMed] [Google Scholar]
- Huelsz-Prince G, DeVries AL, Bakker HJ, Van Zon JS, Meister K, 2019. Effect of antifreeze glycoproteins on organoid survival during and after hypothermic storage. Biomolecules 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideta A, Aoyagi Y, Tsuchiya K, Nakamura Y, Hayama K, Shirasawa A, Sakaguchi K, Tominaga N, Nishimiya Y, Tsuda S, 2014. Prolonging hypothermic storage (4 C) of bovine embryos with fish antifreeze protein. Journal of Reproduction and Development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itskovitzeldor J, Levron J, Arav A, BARAMI S, Stein D, Fletcher G, Rubinsky B, 1993. Hypothermic preservation of human oocytes with antifreeze proteins from subpolar fish. Cryo-Letters 14, 235–242. [Google Scholar]
- Kamijima T, Sakashita M, Miura A, Nishimiya Y, Tsuda S, 2013. Antifreeze protein prolongs the life-time of insulinoma cells during hypothermic preservation. PloS one 8, e73643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper MA, Wade JB, Terris J, Ecelbarger CA, Marples D, Mandon B, Chou CL, Kishore BK, Nielsen S, 1996. Renal aquaporins. Kidney Int 49, 1712–1717. [DOI] [PubMed] [Google Scholar]
- Kratochvílová I, Golan M, Pomeisl K, Richter J, Sedláková S, Šebera J, Mičová J, Falk M, Falková I, Řeha D, 2017. Theoretical and experimental study of the antifreeze protein AFP752, trehalose and dimethyl sulfoxide cryoprotection mechanism: correlation with cryopreserved cell viability. RSC advances 7, 352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiagalli M, Bar-Dolev M, Tedesco P, Natalello A, Kaleda A, Brocca S, de Pascale D, Pucciarelli S, Miceli C, Braslavsky I, 2017. Cryo-protective effect of an ice-binding protein derived from Antarctic bacteria. The FEBS journal 284, 163–177. [DOI] [PubMed] [Google Scholar]
- Manku G, Culty M, 2015. Mammalian gonocyte and spermatogonia differentiation: recent advances and remaining challenges. Reproduction 149, R139–R157. [DOI] [PubMed] [Google Scholar]
- Manku G, Hueso A, Brimo F, Chan P, Gonzalez-Peramato P, Jabado N, Gayden T, Bourgey M, Riazalhosseini Y, Culty M, 2016. Changes in the expression profiles of claudins during gonocyte differentiation and in seminomas. Andrology 4, 95–110. [DOI] [PubMed] [Google Scholar]
- Nguyen T-V, Tanihara F, Hirata M, Hirano T, Nishio K, Do LTK, Van Nguyen T, Nii M, Otoi T, 2018. Effects of antifreeze protein supplementation on the development of porcine morulae stored at hypothermic temperatures. CryoLetters 39, 131–136. [PubMed] [Google Scholar]
- Oishi Y, Manabe I, 2018. Macrophages in inflammation, repair and regeneration. International immunology 30, 511–528. [DOI] [PubMed] [Google Scholar]
- Raymond JA, DeVries AL, 1977. Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proceedings of the National Academy of Sciences 74, 2589–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez C, Sajjadi S, Abrol R, Wen X, 2019. A beetle antifreeze protein protects lactate dehydrogenase under freeze-thawing. International journal of biological macromolecules 136, 1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsky B, 2003. Principles of low temperature cell preservation. Heart failure reviews 8, 277–284. [DOI] [PubMed] [Google Scholar]
- Rubinsky B, Arav A, Fletcher G, 1991. Hypothermic protection—a fundamental property of “antifreeze” proteins. Biochemical and biophysical research communications 180, 566–571. [DOI] [PubMed] [Google Scholar]
- Rubinsky B, Arav A, Mattioli M, Devries AL, 1990. The effect of antifreeze glycopeptides on membrane potential changes at hypothermic temperatures. Biochemical and biophysical research communications 173, 1369–1374. [DOI] [PubMed] [Google Scholar]
- Varret C, Beronius A, Bodin L, Bokkers BGH, Boon PE, Burger M, De Wit-Bos L, Fischer A, Hanberg A, Litens-Karlsson S, Slob W, Wolterink G, Zilliacus J, Beausoleil C, Rousselle C, 2018. Evaluating the evidence for non-monotonic dose-response relationships: A systematic literature review and (re-)analysis of in vivo toxicity data in the area of food safety. Toxicol Appl Pharmacol 339, 10–23. [DOI] [PubMed] [Google Scholar]
- Walker C, Garza S, Papadopoulos V, Culty M, 2021. Impact of endocrine-disrupting chemicals on steroidogenesis and consequences on testicular function. Mol Cell Endocrinol 527, 111215. [DOI] [PubMed] [Google Scholar]
- Walker C, Ghazisaeidi S, Collet B, Boisvert A, Culty M, 2020. In utero exposure to low doses of genistein and di-(2-ethylhexyl) phthalate (DEHP) alters innate immune cells in neonatal and adult rat testes. Andrology 8, 943–964. [DOI] [PubMed] [Google Scholar]
- Wang S, Amornwittawat N, Juwita V, Kao Y, Duman JG, Pascal TA, Goddard III WA, Wen X, 2009. Arginine, a key residue for the enhancing ability of an antifreeze protein of the beetle Dendroides canadensis. Biochemistry 48, 9696–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wen X, DeVries AL, Bagdagulyan Y, Morita A, Golen JA, Duman JG, Rheingold AL, 2014. Molecular recognition of methyl α-D-mannopyranoside by antifreeze (glyco) proteins. Journal of the American Chemical Society 136, 8973–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ben RN, 2004. Upregulation and protein trafficking of aquaporin-2 attenuate cold-induced osmotic damage during cryopreservation. In Vitro Cell Dev Biol Anim 40, 67–70. [DOI] [PubMed] [Google Scholar]
- Wen X, Wang S, Duman JG, Arifin JF, Juwita V, Goddard WA, Rios A, Liu F, Kim S-K, Abrol R, 2016. Antifreeze proteins govern the precipitation of trehalose in a freezing-avoiding insect at low temperature. Proceedings of the National Academy of Sciences 113, 6683–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Wang S, Ramji R, Butler LO, Bagdagulyan Y, Kishishita A, Golen JA, Rheingold AL, Kim SK, Goddard WA 3rd, Pascal TA, 2022. Complete inhibition of a polyol nucleation by a micromolar biopolymer additive. Cell reports Physical science 3, 100723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng L, Stott SL, Toner M, 2019. Exploring dynamics and structure of biomolecules, cryoprotectants, and water using molecular dynamics simulations: implications for biostabilization and biopreservation. Annual review of biomedical engineering 21, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamalloa JA, 2012. Role of conserved threonine residues in an antifreeze protein from Dendroides canadensis. MAI, Los Angeles, CA, pp. 85. [Google Scholar]