

Abbreviations
- ER+
estrogen receptor‐positive
- m6A
N⁶‐methyladenosine
- MeRIPseq
m6A‐specific methylated RNA immunoprecipitation
- RNA‐seq
RNA‐sequencing
- KRAS
Kirsten rat sarcoma virus
- NF‐κB
nuclear factor‐κB
- EMT
epithelial‐mesenchymal transition
- CDS
coding region
- GO
Gene Ontology
- mTORC
mammalian target of rapamycin complex
- FTO
FTO alpha‐ketoglutarate dependent dioxygenase
- ALKBH5
alkB‐homolog‐5‐RNA demethylase
- METTL3
methyltransferase‐3
- METTL14
methyltransferase‐14
- FTO+
FTO positive staining
- OS
overall survival
- PFS
progression‐free survival
- TNBC
triple‐negative breast cancer
- MA2
meclofenamic acid (ethyl‐ester form)
Dear Editor,
The molecular determinants that drive breast cancer progression to metastasis are complex and partly controlled by nucleic acid epi‐modifications [1]. Although DNA‐methyl modifications in breast cancer metastasis have been well described, there is limited understanding of the role of RNA methylation in advanced disease [2]. This study aimed to provide an understanding on the role of the epi‐transcriptome in estrogen receptor‐positive (ER+) breast cancer progression to metastasis.
Numerous RNA modifications have been described to date, of which N⁶‐methyladenosine (m6A) modifications are the most common [3]. Here, we mapped dynamic global m6A‐specific methylated RNA immunoprecipitation sequencing (MeRIPseq), with corresponding RNA‐sequencing (RNA‐seq) and mass spectrometry, in cell models of disease progression to metastasis: endocrine‐sensitive (MCF7/luminal A), endocrine‐resistant (LY2/luminal B) and endocrine‐resistant brain metastatic (T347/patient‐derived luminal B metastatic) (Figure 1A). The relevance of this model system was verified using comparative analysis of patient‐matched brain metastatic samples [4] with RNA‐seq data from our cells (LY2 vs. MCF7 and T347 vs. MCF7) [4] (Supplementary Table S1). Consistent differential gene expression in key oncogenic pathways such as Kirsten rat sarcoma virus (KRAS), nuclear factor‐κB (NF‐κB) and epithelial‐mesenchymal transition (EMT) were observed across patient samples and our cell models (Supplementary Figure S1A).
FIGURE 1.
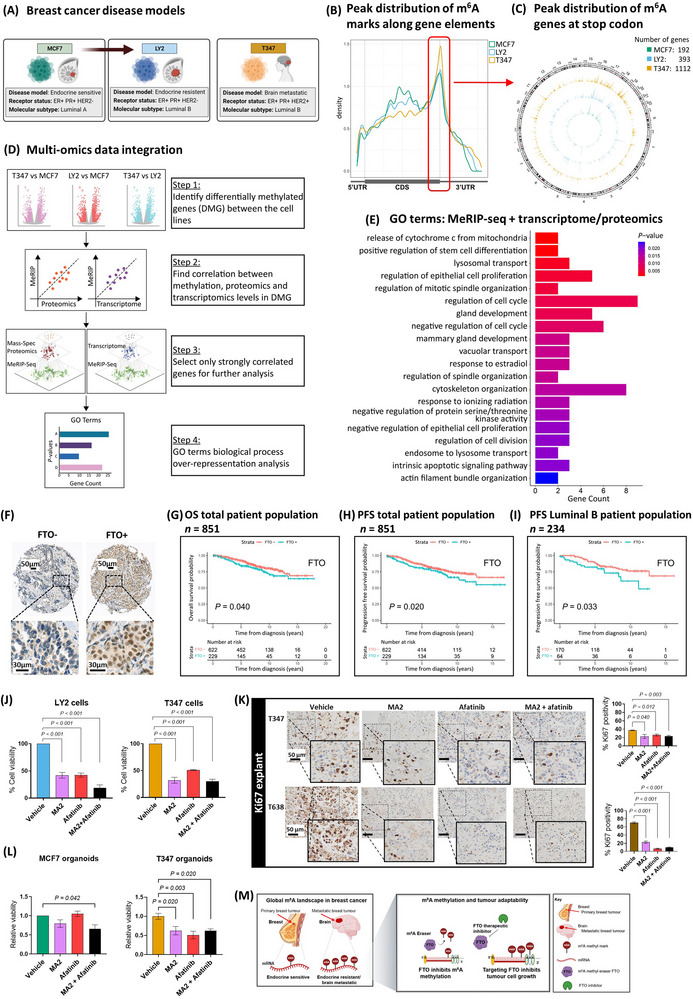
RNA methylation landscape in breast cancer. (A) Schematic representation of breast cancer cells, MCF7 endocrine‐sensitive, LY2 endocrine‐resistant and T347, a model derived from an ER+ treatment‐resistant brain metastatic patient tumor. (B) MeRIPseq density plot of m6A binding along gene elements, 5’UTR, CDS and 3’UTR. The distribution indicates a preference for binding around the stop codon in all three cell models, MCF7, LY2 and T347. (C) Circos plot compares genes with m6A modification at the stop codon for each cell line along chromosomes. The number of genes increases progressively, MCF7 (192), LY2 (393) and T347 (1112). MCF7 (green), LY2 (blue) and T347 (yellow). (D) Data integration multi‐omics factor analysis. Schematic representation of the integration process: DMG discovery (step 1), correlation analysis (steps 2 and 3) and over‐representation analysis (step 4). (E) GO‐term biological process pathways of integration analysis. Pathway analysis of strongly correlated features between MeRIP expression levels and either transcriptomic or proteomic expression, as identified by BioInfoMiner (P < 0.05). Displayed are the top 20 identified pathways. (F) TMA inserts display representative patient tumors with negative (FTO‐) and positive (FTO+) protein expression. Kaplan‐Meier estimates for FTO (G) OS (n = 851, P = 0.040) and (H) PFS (n = 851, P = 0.020) in the total patient population, and (I) PFS in the luminal B molecular subtype (n = 234, P = 0.033). Blue line indicates FTO+, and red line indicates FTO‐ expression. (J) Cell viability assays. LY2 and T347 cells were treated with vehicle (DMSO), MA2 (8 × 10−2 mmol/L), and afatinib (2.5 × 10−5 mmol/L) alone and in combination for 72 hours. Significant reduction in cell viability with each treatment was observed (P < 0.001). Assays were performed in triplicate (mean ± SEM, n = 3). (K) IHC analysis for Ki67 protein expression in T347 and T638 PDX explant tissue. Explants were treated for 72 hours with vehicle (DMSO), MA2 (8 × 10−2 mmol/L), and afatinib (2.5 × 10−5 mmol/L) alone and in combination. Expression of Ki67 was assessed and quantified using Aperio imaging software. In T347 and T638 explants, MA2 and afatinib alone and in combination significantly reduced Ki67 expression compared to vehicle (P < 0.050, T347 and P < 0.001, T638, n = 6 images/group). (L) Three‐dimensional organoid culture assay. MCF7 and T347 cells were assayed for organoid viability following treatment with vehicle (DMSO), MA2 (8 × 10−2 mmol/L), and afatinib (2.5 × 10−5 mmol/L) alone and in combination for 72 hours. MCF7 organoid viability was significantly reduced with combination treatment (P = 0.042) but not with MA2 or afatinib alone. T347 organoid viability was significantly reduced with each treatment (MA2, P = 0.020; Afatinib, P = 0.003; Combination, P = 0.020) (mean ± SEM, n = 3). (M) Schematic of RNA methylation in breast cancer brain metastasis. At a global level, there is a gain in m6A methylated genes with progression to brain metastasis. Pharmacological targeting of the RNA methyl‐eraser FTO inhibits tumor cell growth. Abbreviations: ER+: estrogen receptor‐positive; MeRIPseq: m6A‐specific methylated RNA immunoprecipitation; m6A: N⁶‐methyladenosine; 5’UTR: 5‐prime untranslated region; CDS: coding region; 3’UTR: 3‐prime untranslated region; DMG: differentially methylated genes; GO: Gene Ontology; TMA: tissue microarray; FTO: FTO alpha‐ketoglutarate dependent dioxygenase; FTO+: positive staining; FTO‐: negative staining; OS: overall survival; PFS: progression‐free survival; DMSO: dimethyl‐sulfoxide; MA2: meclofenamic acid (ethyl‐ester form); SEM: standard error of the mean; IHC: immunohistochemistry; Ki67: marker of proliferation; PDX: patient‐derived xenograft.
m6A modifications of long internal exons are thought to be involved in gene splicing, whereas methylation near stop‐codons is associated with translational control [5]. Analysis of MeRIPseq revealed the highest density of m6A modifications for each model at or near the stop codon (Figure 1B). Comparative analysis displayed global increases in genes with m6A modification marks with disease progression to metastasis (circos plot, Supplementary Figure S1B), including both the coding sequence (CDS) and stop‐codon (Supplementary Figure S1B and Figure 1C). Notably, greater total m6A activity was observed in the resistant and metastatic cells compared to MCF7 cells (Supplementary Figure S1C). At the stop‐codon, we observed hypermethylated Gene Ontology (GO) features gained with disease progression, including DNA methylation (LY2 vs. MCF7) and gene expression (T347 vs. MCF7) regulation. Although smaller in number, hypomethylated features were associated with apoptotic signaling (LY2 vs. MCF7) and stem cell differentiation genes (T347 vs. MCF7) (Supplementary Figure S1D, Supplementary Tables S2‐S3). Comparable differentially hyper‐ and hypo‐methylated features were observed at the CDS region, including RNA transcription and gene expression regulation (LY2 vs. MCF7 and T347 vs. MCF7), apoptosis (LY2 vs. MCF7) and stem cell differentiation (T347 vs. MCF7), respectively (Supplementary Figure S1E, Supplementary Tables S2‐S3).
To understand the impact of perturbed RNA stop‐codon methylation on translational control, we analyzed RNA‐seq (GSE1765325) and mass spectrometry data (Supplementary Table S4) and observed differential expression patterns across the progressive disease models (Supplementary Figure S2A‐B). Pathway analysis revealed gains in mammalian target of rapamycin complex (mTORC) signaling at both the gene and protein levels on brain metastasis (Supplementary Figure S2C‐D, Supplementary Tables S1 and S5, respectively). Data integration with differential gene and protein expression has the potential to identify key epi‐transcriptomic pathways of functional consequence. Taking a two‐step multi‐omics factor analysis approach [6], we integrated MeRIPseq with RNA‐seq and protein expression data from MCF7, LY2 and T347 cells (Figure 1D, Supplementary Table S6). Integrated genes and proteins with the highest correlation scores were ranked by gene set enrichment (Supplementary Table S7). Identified pathways were epithelial cell proliferation, migration, response to estradiol and regulation of kinase activity. Consistent with differential hypo‐methylated genes, regulation of stem cell differentiation was a top‐ranked integrated pathway (Figure 1E).
m6A modifications are regulated through the key members of the RNA methylation machinery, including methyl erasers FTO alpha‐ketoglutarate dependent dioxygenase (FTO) and alkB‐homolog‐5‐RNA demethylase (ALKBH5) and methyl writers, methyltransferase‐3 (METTL3) and methyltransferase‐14 (METTL14). The role of these players in cancer progression remains controversial. In breast cancer, METTL3 and METTL14 have been associated with tumor cell proliferation [7], whereas FTO and ALKBH5 have been associated with poor prognosis and metastatic potential [2, 7]. Conversely, FTO silencing has recently been reported to promote breast cancer cell growth and invasion, and has been associated with EMT [8]. The tissue of origin, molecular subtype and site of metastasis could provide a greater understanding of the role of specific methyl modulators.
We analyzed the expression of key RNA methyl‐modulators in patients’ tumor with regard to molecular subtypes. Firstly, we conducted transcriptomic and proteomic analyses of ER+ patient primary tumors with subsequent good response (no recurrence) versus poor response (disease recurrence). Gains in FTO and corresponding loss of METTL3 were observed in poor responders, though these differences were not significant (Supplementary Figure S3A, Supplementary Tables S8‐S9). Of note, elevations in FTO protein were confirmed in the resistant (LY2) and metastatic (T347) cells compared to MCF7 by immuno‐blotting, with no difference observed in ALKBH5, METTL3 or METTL14 (Supplementary Figure S3B). Next, we evaluated the protein expression of the RNA methyl‐modulators in primary breast cancer tumors from 870 patients, 20.7% of whom had disease recurrence (Supplementary Table S10). Positive FTO expression (FTO+ staining, Figure 1F) was significantly associated with poor overall survival (OS) (P = 0.040) (Figure 1G) and progression‐free survival (PFS) (P = 0.020) (Figure 1H) in the FTO stained patient population (n = 852, one patient lost to follow‐up). Of note, FTO was also associated with PFS in the luminal B subtype (P = 0.033) (Figure 1I) and to a lesser extent in triple‐negative breast cancer (TNBC) though this did not reach significance (P = 0.190) (Supplementary Figure S3C). Neither eraser ALKBH5 nor the methyl‐writers METTL3 and METTL14 were significantly associated with PFS (Supplementary Figure S3D). Consistent with these observations, FTO expression was positively associated with high histological grade (P < 0.001), whereas METTL3 was inversely associated with high histological grade tumors (P = 0.030) (Supplementary Table S10). Taken together, these data support an association between FTO and aggressive breast cancer.
Next, we determined the efficacy of targeting FTO in advanced breast cancer. Given enhanced kinase activity associated with brain metastasis [9], the efficacy of meclofenamic acid 2 (ethyl‐ester form) (MA2), a selective FTO inhibitor [10], was evaluated alone and in combination with the tyrosine kinase inhibitor afatinib in cells, organoids and explant tumor models. In resistant and metastatic cells, treatment with MA2 and/or afatinib reduced cell viability (LY2 and T347, P < 0.005) (Figure 1J). In two ER+/FTO+ patient brain metastatic xenograft explant models (Supplementary Figure S4), MA2 and afatinib treatment reduced cancer cell proliferation, as indicated by a significant reduction in Ki67 expression (Figure 1K). Furthermore, in 3‐dimensional organoid cultures of T347 cells, treatment with MA2 and/or afatinib significantly reduced organoid viability, whereas in MCF7 cells, only combination treatment affected organoid viability (P = 0.042) (Figure 1L).
Here, we chart the dynamic transcriptome‐wide m6A‐methylome with progression to brain metastasis in ER+ breast cancer. Our models provide evidence of overall global methyl gains, with specific aberrant gains and losses in key oncogenic pathways, including stem cell differentiation. In primary breast cancer patients, FTO expression was significantly associated with OS and PFS (Figure 1G‐I). Further, we report that FTO inhibition in patient models of breast cancer brain metastasis was associated with reduced tumor growth (Figure 1M).
In conclusion, this study showed that aberrant RNA m6A methyl‐gains were associated with breast cancer disease progression to metastasis and identified stem cell differentiation as a key regulatory output of the altered epi‐transcriptome. At a clinical level, targeting methyl modulators could reverse cellular adaptability, offering new therapeutic strategies for metastatic disease.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Study concept and design (Sara Charmsaz, Sinéad Cocchiglia, Leonie Young); Acquisition, analysis, or interpretation of data (Stephen Keelan, Sara Charmsaz, Sinéad Cocchiglia, Daniela Ottaviani, Seán Hickey, Siobhan Purcell, Fiona Bane, Ben Doherty, Benjamin Roux, Damir Varešlija, Leonie Young), Bioinformatic analyses (Mihaela Ola, Nicola Cosgrove); Provision of administrative, technical, or material support (Sinéad Cocchiglia, Fiona Bane, Katherine Sheehan, Muriel Lane, Geoffrey Greene, Arnold Konrad Hill); Drafting of the manuscript (Stephen Keelan, Mihaela Ola, Sara Charmsaz, Sinéad Cocchiglia, Daniela Ottaviani, Leonie Young), critical revision of the manuscript (all authors); Study supervision (Leonie Young).
CONFLICTS OF INTEREST
The authors declare there is no conflict of interest.
CONSENT FOR PUBLICATION
Informed and written consent to publish results from this study was obtained before patient participation. This article does not contain any individual person's data.
ETHICS AND CONSENT TO PARTICIPATE
Ethical approval for this study was obtained by Beaumont Hospital Medical Research Ethics Committee (Dublin, Ireland, REC13/09). Patient clinical material was collected under the regulation of clinical trial NCT01840293 (https://clinicaltrials.gov). Informed and written consent was obtained from each patient prior to patient participation in the clinical trial.
Supporting information
Supplementary Materials
Supplementary Materials
ACKNOWLEDGMENTS
We are thankful to the patients who generously participated in our Cancer Trials Ireland observational clinical trial (C‐TRIAL 09/07/NCT01840293 [https://clinicaltrials.gov]) and to the surgical, pathology and tissue bank colleagues for their substantial assistance and support. Figures 1A and 1M were created with BioRender.com. We kindly acknowledge the funding support from Breast Cancer Ireland (GR 14‐0883) Science Foundation Ireland Investigator Award (12/IA/1294), Science Foundation Ireland Frontiers for the Future Award (19/FFP/6443), Science Foundation Ireland Strategic Partnership Programme, Precision Oncology Ireland (18/SPP/3522) (Leonie S. Young) and Breast Cancer NOW grant, Project (2018JulPR1094) (Leonie S. Young, Damir Varešlija), Breast Cancer NOW Fellowship Award (2019AugSF1310) (Damir Varešlija) and Irish Research Council Postgraduate Award (GOIPG/2020/361) (Stephen Keelan).
Stephen Keelan, Mihaela Ola, Sara Charmsaz and Sinéad Cocchiglia contributed equally to this work
DATA AVAILABILITY STATEMENT
The RNA‐sequencing and MeRIP sequencing data generated from this study have been deposited in the Gene Expression Omnibus database under accession number GSE176535. Source data for figures in the paper are available from the corresponding authors upon request.
REFERENCES
- 1. Wilting RH, Dannenberg JH. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat. 2012;15(1‐2):21‐38. [DOI] [PubMed] [Google Scholar]
- 2. Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, et al. RNA N6‐methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhao BS, Roundtree IA, He C. Post‐transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cosgrove N, Varešlija D, Keelan S, Elangovan A, Atkinson JM, Cocchiglia S, et al. Mapping molecular subtype specific alterations in breast cancer brain metastases identifies clinically relevant vulnerabilities. Nat Commun. 2022;13(1):514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sibbritt T, Patel HR, Preiss T. Mapping and significance of the mRNA methylome. Wiley Interdiscip Rev RNA. 2013;4(4):397‐422. [DOI] [PubMed] [Google Scholar]
- 6. Li Z, Zhuang X, Zeng J, Tzeng CM. Integrated Analysis of DNA Methylation and mRNA Expression Profiles to Identify Key Genes in Severe Oligozoospermia. Front Physiol. 2017;8:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shen H, Lan Y, Zhao Y, Shi Y, Jin J, Xie W. The emerging roles of N6‐methyladenosine RNA methylation in human cancers. Biomark Res. 2020;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jeschke J, Collignon E, Al Wardi C, Krayem M, Bizet M, Jia Y, et al. Downregulation of the FTO m(6)A RNA demethylase promotes EMT‐mediated progression of epithelial tumors and sensitivity to Wnt inhibitors. Nat Cancer. 2021;2(6):611‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Varešlija D, Priedigkeit N, Fagan A, Purcell S, Cosgrove N, O'Halloran PJ, et al. Transcriptome Characterization of Matched Primary Breast and Brain Metastatic Tumors to Detect Novel Actionable Targets. J Natl Cancer Inst. 2019;111(4):388‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43(1):373‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials
Supplementary Materials
Data Availability Statement
The RNA‐sequencing and MeRIP sequencing data generated from this study have been deposited in the Gene Expression Omnibus database under accession number GSE176535. Source data for figures in the paper are available from the corresponding authors upon request.