

Abstract
Background
Several clinical studies have uncovered a negative correlation between baseline tumor burden and the efficacy of immune checkpoint inhibitor (ICI) treatment. This study aimed to uncover the specific mechanisms underlying the difference in sensitivity to ICI treatment between tumors with high (HTB) and low (LTB) tumor burden.
Methods
For in vivo studies, several mouse models of subcutaneous tumors were established, and transcriptome sequencing, immunohistochemistry, and flow cytometry assays were used to detect the immune status in these subcutaneous tumors. For in vitro experiments, co‐culture models, cytokine antibody arrays, western blotting, flow cytometry, and enzyme‐linked immunosorbent assays were used to explore the underlying molecular mechanisms
Results
We found that MC38 or B16 subcutaneous tumors from the HTB group did not show any response to anti‐programmed cell death protein‐1 (PD‐1) therapy. Through flow cytometry assays, we found that the infiltration with CD8+ T cells was significantly decreased whereas M2‐like macrophages were enriched in subcutaneous tumors of HTB groups compared with those of LTB group. These changes were not affected by the initial number of injected tumor cells or tumor age, nor could they be reversed by surgical tumor reduction. Intraperitoneal colony‐stimulating factor 1 receptor (CSF‐1R) inhibitor PLX3397 injection at different time points of tumor growth only had an effect when administered in the early tumor stage to maintain the “heat” of the tumor microenvironment during the process of tumor growth, thereby achieving a response to ICI treatment when the tumor grew to a large size. Mechanistically, we found that insulin‐like growth factor binding protein 2 (IGFBP2) expression levels were significantly elevated in HTB tumor tissues. IGFBP2 promoted the programmed death‐ligand 1 (PD‐L1) expression in M2‐like macrophages by activating signal transducer and activator of transcription 3 (STAT3), and PD‐L1+ M2‐like macrophages exerted an immunosuppressive effect by inhibiting the proliferation and activation of CD8+ T cells in a PD‐L1‐dependent fashion.
Conclusions
This study suggested that the low efficacy of ICI treatment in HTB tumors is mainly attributed to the intratumoral accumulation of PD‐L1+ M2‐like macrophages via the IGFBP2‐STAT3‐PD‐L1 signaling pathway and their substantial inhibitory effects on T cell proliferation and activation.
Keywords: CD8+ T cell, IGFBP2, immune checkpoint inhibitor, macrophage, PD‐L1, STAT3, tumor burden, tumor immune microenvironment
List of abbreviations
- bFGF
basic fibroblast growth factor
- CAR
Chimeric Antigen Receptor
- CCL2
chemokine ligand 2
- CSF‐1R
macrophage colony‐stimulating factor‐1 receptor
- CTL
cytotoxic T lymphocyte
- DMEM
Dulbecco's modified Eagle medium
- ELISA
enzyme‐linked immunosorbent assay
- GO
gene‐ontology
- HIF‐1α
hypoxia‐inducible factor 1α
- HTB
high‐baseline tumor burden
- HTCS
HTB tissue culture supernatant
- ICI
immune checkpoint inhibitor
- IgG
immunoglobulin G
- IGFBP2
insulin‐like growth factor binding protein 2
- IHC
immunohistochemistry
- IOD
integrated optical density
- LTB
low‐baseline tumor burden
- LTCS
LTB tissue culture supernatant
- Mac‐HTB
macrophages from SS2W‐H subcutaneous tumor
- Mac‐LTB
macrophages from SS2W‐L subcutaneous tumor
- Mac‐P
peritoneal macrophage
- MCSF
macrophage colony‐stimulating factor
- MMP‐2
matrix metalloprotein‐2
- MMP‐9
pro‐matrix metalloprotein‐9
- NC
control culture medium
- NSCLC
non‐small cell lung cancer
- PBS
phosphate‐buffered saline
- PBS
phosphate buffer saline
- PD‐1
programmed cell death protein‐1
- PD‐L1
programmed death‐ligand 1
- PF4
platelet factor 4
- RNA‐seq
RNA sequencing
- RPMI
Roswell Park Memorial Institute
- SS2W‐L
single side, two weeks, LTB
- SS2W‐H
single side, two weeks, HTB
- SS3W‐H
single side, three weeks, HTB
- STAT3
signal transducer and activator of transcription 3
- TAM
tumor‐associated macrophage
- TCGA
The Cancer Genome Atlas
- TCS
tissue culture supernatant
- TGF‐β
transforming growth factor β
- TIMP‐2
tissue inhibitor of metalloproteinases 2
- TIME
tumor immune microenvironment
- Tregs
regulatory T cells
- TS3W‐H
two sides, three weeks, HTB2
1. BACKGROUND
In recent decades, cancer immunotherapy, especially immune checkpoint inhibitors (ICIs), has opened a new chapter in cancer treatment [1]. Compared to traditional cancer therapies, ICIs enable cytotoxic T cells to overcome immunosuppression and activate their cytotoxic effects on tumor cells by disrupting the interactions between the ligand‐receptor pairings of immune checkpoint molecules, such as programmed cell death protein 1 (PD‐1) and its receptor programmed death‐ligand 1 (PD‐L1) [2]. Although profound and durable therapeutic responses to ICIs have been reported in various malignancies, a substantial fraction of tumors, especially “cold tumors”, which are characterized by insufficient T‐cell infiltration and low immunogenicity, remain poorly responsive to ICI therapy [3]. Therefore, better screenings for patients with such low immunotherapy responses and a better understanding of the mechanisms underlying ICI resistance in such patients would help to develop effective immunotherapy sensitization strategies and expand the population suitable for immunotherapy.
To date, baseline tumor burden has been shown in several malignancies to be a predictive and prognostic biomarker for patients receiving ICI treatment. A smaller tumor burden usually indicates a better therapeutic response to ICI treatment and longer overall or progression‐free survival [4, 5, 6, 7]. However, the specific mechanisms underlying such a negative correlation between tumor size and ICI sensitivity are still unclear. A few studies have indicated that differences in tumor microenvironment (TME) characteristics exist between tumors with high tumor burden (HTB) and those with low tumor burden (LTB), but the findings are inconclusive. For example, in patients with diffuse large B lymphoma, HTB was associated with decreased infiltration of Ki‐67+ T cells in either the solid tumor tissue or circulating blood [8]. By contrast, in tissue samples of ductal breast cancer, the infiltration of cytotoxic T lymphocytes was significantly increased in tumor tissue with large size [9]. Similarly, at the preclinical level, the intratumoral abundance of CD8+ T cells was significantly diminished in a murine model of renal adenocarcinoma as the tumors progressed in size, but was found to be increased in large vs. small lesions in CT26 tumors [10]. Therefore, the correlation between tumor size and immunophenotype may vary among different tumor types and requires more studies to elucidate this connection.
In this study, we aimed to explore whether differences in TME characteristics are responsible for different sensitivity of HTB and LTB tumors to ICI treatment in colon cancer and melanoma. Additionally, we intended to explore the specific mechanisms for the formation of a tumor burden‐related immunophenotype.
2. MATERIALS AND METHODS
2.1. Clinical specimens and datasets
This study was approved by the Nanfang Hospital Ethics Review Board (Guangzhou, Guangdong, China). The informed consent was signed by every patient before we got the tissue specimen. The tissue sample cohort in this study consists of two types: (1) Radical resection cohort: The cohort included colorectal cancer patients with indications for radical resection at different T stages. A total of 51 paraffin‐embedded samples from patients with colon cancer who underwent radical tumor resection were collected between January 2012 and January 2018. According to the medium tumor size, patients were divided into an LTB (n = 26; average TB = 13.00 cm3) and HTB (n = 25; average TB = 88.11 cm3) group. (2) Colorectal liver metastases cohort: This cohort included patients who had been histologically diagnosed with clorectal liver metastases at Nanfang Hospital, Southern Medical University (Guangzhou, Guangdong, China). And these patients must have liver metastases of sufficient size to biopsy. In 20 patients with colon cancer, needle biopsy samples of liver metastases were collected.
We also analyzed the differences in immune cell infiltration in the TME of patients with HTB and LTB in the colon cancer (COAD) and melanoma (SKCM) cohorts from The Cancer Genome Atlas (TCGA) databases (https://portal.gdc.cancer.gov/). The estim ated proportion of immune cells in the TME of each sample of the TCGA‐COAD and TCGA‐SKCM cohorts were retrieved from the study by Thorsson et al. [11]. The information on TNM staging, a classification method proposed by the International Anti‐cancer Association (UICC) based on the scope of the tumor lesions, of eligible TCGA‐COAD and TCGA‐SKCM samples was downloaded from the UCSC Xena website (https://xenabrowser.net/) [12]. Patients were grouped into HTB and LTB groups respectively according to the T category of the TNM staging system. Patients with T stage of T0 or T1 stage were assigned to the LTB group, whereas those with T4 stage were allocated to the HTB group.
2.2. Cell lines and cell culture
The colon cancer cell line MC38, melanoma cell line B16, and mouse macrophage cell line RAW264.7 were purchased from Jennio Biotech Co. Ltd. (Guangzhou, Guangdong, China). MC38 colon cancer cell line was cultured in 1640 Roswell Park Memorial Institute (RPMI; Gibco, NY, Grand Island, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, Thermo Scientific, Logan, UT, USA). B16 melanoma cell line was cultured in Dulbecco's modified Eagle medium (DMEM; Hyclone) supplemented with 10% FBS. All cells were incubated in a humidified atmosphere with 5%CO2 and 37°C. All cell lines were authenticated by the short tandem repeat profiling at January 27, 2015.
2.3. Mouse model construction
All animal procedures were approved by the Nanfang Hospital Animal Ethics Committee (Guangzhou, Guangdong, China; Application No.: NFYY‐2017‐0926). C57BL/6J mice were purchased from Guangdong Medical Laboratory Animal Center (SCXK 2013‐0002, Guangzhou, Guangdong, China). They were housed at 25°C, and humidity was 50%‐60%. The light cycle consisted of light for 12 h and dark for 12 h. In this study humane endpoints were used including: moribund were humanely euthanized. Based on the animal use protocol, we used two criteria to identify the moribund animals: (1) mice that showed signs of desease like difficulty breathing, eating, or drinking; (2) a mouse loses ≥15% body weight in 4 days. The mice were euthanized after deep anesthesia. The total number of mice used in this study is 156: anti‐PD‐1 treatment mouse model (5 mice per groups), mouse used for RNA‐sequencing (5 mice per groups), various tumor burden mouse model (3 mice per groups), surgery mouse model (12 mice per groups), PLX3397 combined with anti‐PD‐1 treatment mouse model (3 mice per groups), IGFBP2‐neutralizing treatment (3 mice per groups), mouse used for cytokine antibody array (1 mice per groups), mouse used for cell extraction (20 mice).
MC38 cells or B16 cells were inoculated subcutaneously into 4‐week‐old female mice to generate the mouse model. MC38 cells or B16 cells were subcutaneously injected on the left (2.0 × 106 cells) and right (5 × 105 cells) sides (hind limb) of the same mouse. The tumor dimensions were measured with vernier calipers.Tumor volumes were calculated using the equation Volume = (length × width2)/2. In order to clarify the impact of surgical tumor reduction on the tumor microenvironment, we surgically removed a portion of the tumor on the left side on the 14th day after subcutaneous tumor implantation to ensure that the size of the tumors on both sides was consistent. The tumors were partially resected through a longitudinal skin incision along the major axis of the tumor. On days 14, 16, 18, and 21, CD3+ CD8+ T cells of subcutaneous tumors were detected by flow cytometry.
For drug treatment assays, mice were randomly assigned to (without blinding) different treatment groups and received intraperitoneally different drugs or antibodies according to the specific experimental purpose: (1) for anti‐PD‐1 or IgG treatment group, the anti‐PD‐1 (10 mg/kg) or IgG (10 mg/kg) was intraperitoneal injected into mice on day 14, 17, 20, 23; (2) for PLX3397 (d7) combined anti‐PD‐1 treatment group, the PLX3397 (40 mg/kg) was administered orally to mice on day 7 and day 9, anti‐PD‐1 (10 mg/kg) was intraperitoneal injected into mice on day 21, 23, 25; (3) for PLX3397 (d13) combined anti‐PD‐1 treatment group, the PLX3397 (40 mg/kg) was administered orally to mice on day 13 and day 15, anti‐PD‐1 (10 mg/kg) was intraperitoneal injected into mice on day 17, 19, 21; (4) for IGFBP2‐neutralizing treatment group, anti‐IGFBP2 (10 mg/kg) was intraperitoneal injected into mice on day 7; (5) for saline + IgG treatment group, saline (100 μL) was administered orally to mice on day 7 and day 9, IgG (10 mg/kg) was intraperitoneal injected into mice on day 17, 19, 21; (6) for saline + anti‐PD‐1 treatment group, saline (100 μL) was administered orally to mice on day 7 and day 9, anti‐PD‐1 (10 mg/kg) was intraperitoneal injected into mice on day 17, 19, 21.The tumor volume was measured with vernier caliper every 3 days. At the end of the experiment, mice were euthanized, and the tumor xenografts were immediately dissected to detect CD3+ CD8+ T cells or macrophages of subcutaneous tumors.
2.4. Reagents
Anti‐PD‐1 antibody (BE0146, 10 mg/kg) and anti‐IGFBP2 antibody (AF797‐SP, 10 mg/kg) were purchased from BioXCell (Beijing, China) for intraperitoneal injection. FLLL32 (1226895‐15‐3, 5 μmol/L) and HJC0152 (S8561, 5 μmol/L) were purchased from Selleck Chemicals (Shanghai, China) for inhibiting the phosphorylation of STAT3. IGFBP2 enzyme‐linked immunosorbent assay (ELISA) (F5217‐A) was purchased from Jiangsu Yutong (Changzhou, Jiangsu, China) to detect the IGFBP2 content between tumor tissue culture supernatants. Annexin V‐FITC/PI apoptosis detection Kit (CA1020) was purchased from Solarbio (Beijing, China) to test the proportion of apoptotic RAW264.7 after exposure to tissue culture supernatants. Mouse cytokine antibody array C1 (AAM‐CYT‐1000‐2) was purchased from RayBiotech (Atlanta, Georgia, USA) for detecting the cytokines content between mouse LTB and HTB MC38 subcutaneous tumors environments. Detailed information on other reagents and antibodies in this work are described in Supplementary Table S1 and Supplementary Table S2.
2.5. Immunohistochemistry (IHC) staining
IHC was routinely performed, as previously described [13], on 4‐mm sections of formalin‐fixed paraffin‐embedded (FFPE) tissues. Paraffin sections were heated at 65°C for 4 h. Tumor tissue sections were deparaffinized and hydrated through an alcohol gradient to water. The sections were subjected to microwave antigen retrieval in 0.01 μmol/L citrate buffer (pH = 6.0; Zeye Biotech, Shanghai, China) for 10 min. Then, endogenous peroxidase activity was blocked with 3% hydrogen peroxide (Zeye Biotech, Shanghai, China) for 10 min. After incubating with 5% bovine serum albumin (20 μL, Macklin, Shanghai, China) for 1 h at room temperature, sections were incubated with anti‐mouse CD8 (1:50, 66868‐1, Proteintech, Rosemont, IL, USA) overnight at 4°C. Subsequently, sections were incubated with rabbit anti‐mouse CD8 antibody (1:200, ab217344, Abcam, Cambridge, MA, USA) for 1 h at 37°C. Sections were afterward incubated following the instructions of a DAB substrate kit (G1212, Servicebio, Wuhan, Hubei, China). Then, the sections were counterstained with hematoxylin (20 μL, aladdin, Shanghai, China), dehydrated (70%, 80%, 90%, 95%, 100%, 100% ethanol, Macklin, Shanghai, China), and mounted. Integrated optical density values for CD8 protein were calculated using Image‐Pro Plus 6.0 (MEDIA CYBERNETICS, Rockville, Maryland, USA).
2.6. Extraction of tissue lymphocytes and macrophages
For the extraction of tumor‐infiltrating lymphocytes, subcutaneous tumors were digested with collagenase I (1 mg/mL, A004194, Sangon Biotech, Shanghai, China). Cells were isolated using a lymphocyte separation medium (MLSM1092, MultiSciences, Hangzhou, Zhejiang, China). Cells were washed twice and suspended in phosphate‐buffered saline (PBS). Purified tumor‐infiltrating lymphocytes were used for flow cytometry.
For the extraction of spleen CD8+ T cells, spleens were digested with collagenase I (1 mg/mL) and isolated using a lymphocyte separation medium (1:1, Multisciences, Hangzhou, Zhejiang Province, China). Cells were washed twice and suspended in PBS. Single‐cell suspensions of the harvested lymphocytes were treated with the Mouse CD8+ T Cell Enrichment Kit (8804‐6822, Miltenyi, Bergisch Gladbach, Rheinisch‐Bergischer Kreis, Germany). Purified spleen CD8+ T cells were used for co‐culture experiments.
For the extraction of tumor‐infiltrating macrophages, subcutaneous tumor tissues were digested with collagenase I (1 mg/mL), and cells were isolated using a monocyte separation medium (P3970, Solarbio). Cells were cultured in DMEM supplemented with 10% FBS at 5% CO2, 37 °C for 3 h. Nonadherent cells were removed, and the adherent cells were cultured continuously or used for flow cytometry.
Peritoneal macrophages (mac‐P) were harvested from 4‐week‐old female C57BL/6J mice. Mice were euthanized, the abdomen was sterilized, and 3 mL PBS was intraperitoneally injected. The abdomen was gently massaged for 5 min. Ascites cells were collected by centrifugation (3000 rpm, 10min) and cultured in DMEM supplemented with 10% FBS at 5% CO2, 37°C for 3 h. Nonadherent cells were removed, and the adherent cells were cultured continuously.
2.7. Preparation of conditioned medium and supernatant‐stimulated macrophages
LTB and HTB subcutaneous tumor tissues were cut into small pieces of equal volume. Tumor pieces were placed into 1 mL DMEM at 4°C for 24 h. The supernatant was harvested and centrifuged (1000 rpm, 5min). Then, the conditioned medium was prepared in the following proportion: control medium (50% complete medium + 50% serum‐free medium), LTB subcutaneous tumor tissue culture supernatants (LTCS, 50% complete medium + 50% MC38 or B16 LTB subcutaneous tumor tissue culture supernatants), and HTB subcutaneous tumor tissue culture supernatants (HTCS, 50% complete medium + 50% MC38 or B16 HTB subcutaneous tumor tissue culture supernatants). To generate supernatant‐stimulated macrophages, peritoneal macrophages (mac‐Ps) were cultured with control medium, LTCS, or HTCS for 24 h, then used for flow cytometry or co‐culture experiments.
2.8. Co‐culture system
Purified spleen CD8+ T cells (1 × 105 cells/well in 96‐well plates) and supernatant‐stimulated macrophages (0.5 × 105 cells/well in 96‐well plates) were co‐cultured in 1640 RPMI supplemented with 10% FBS, recombinant mouse interleukin‐2 (rmIL‐2, 2 ng/mL, P00198, Solarbio), and CD3/CD28 antibody (2 μL per 8 × 104 cells, ThermoFisher, Wilmington, Massachusetts, USA) with or without anti‐PD‐L1 antibody (5 μg/mL, BE0101, BioXCell, Beijing, China) for 5 days. Then, the CD8+ T cells were washed twice and suspended in PBS. The cells were analyzed by flow cytometry.
2.9. Flow cytometry
For analyses of tumor‐infiltrating CD8+ T cells, single‐cell suspensions of the harvested mouse lymphocytes were prepared and stained with a cocktail of antibodies against the following surface markers: CD45 (1:100; AM04505‐100, MultiSciences, Hangzhou, Zhejiang, China), CD3 (1:100; 69‐0032‐82, eBioscience, San Diego, California, USA), and CD8a (1:100; 56‐0081‐82, eBioscience). For analyses of M2 macrophages, single‐cell suspensions of the harvested macrophages were incubated with CD11b (1:100; AH011B01‐100, MultiSciences), CD11c (1:100; AM011C05‐100, MultiSciences), CD206 (1:100; 25‐2061‐80, MultiSciences), F4/80 (1:100; AM048004‐100, MultiSciences), and PD‐L1 (1:100; AM27407‐20, MultiSciences). For analyses of co‐cultured system CD8+ T cells, cells were incubated with CD45 (1:100, AM04505‐100, MultiSciences), CD3 (1:100), and CD8a (1:100) and subsequently with Ki‐67 (1:100; 12‐5698‐80, MultiSciences) after treatment with the Fixation/Permeabilization Kit (GAS003/2, MultiSciences). After incubation with antibodies at 4°C for 30 min, 1 × 106 cells per sample were washed twice and suspended in PBS. The cells were subsequently analyzed by flow cytometry (BD Bioscience, Lake Franklin New Jersey, USA).
For apoptosis analyses, mac‐Ps were cultured with control medium, LTCS, or HTCS for 24 h and harvested. Cells were washed twice with PBS and stained using the Annexin V‐FITC Apoptosis Detection Kit (CA1020, Solarbio). A total of 1 × 106 cells per sample were washed twice and suspended in PBS. The cells were analyzed by flow cytometry. Raw data were analyzed through the FlowJo_V10 software (BD Bioscience, Lake Franklin, New Jersey, USA).
2.10. RNA‐sequencing (RNA‐seq) analysis of MC38 subcutaneous tumor cells
Total RNA in the tissue samples was extracted using Trizol (Invitrogen, Carlsbad, CA, USA). The mRNA was isolated and purified from the total RNA using Oligo (dT) magnetic beads (Beyotime, Shanghai, China). First‐strand Illumina‐barcoded libraries were generated using the NEB RNA Ultra Directional kit (NEB, Ipswich, Massachusetts, USA) according to the manufacturer's instructions. Sequencing was performed using BGIseq500 platform (BGI, Shenzhen, Guangdong, China). Data were aligned to mouse reference genome NCBI_GCF_000001635.26_GRCm38.p6 (https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/635/GCF_000001635.26_GRCm38.p6/) using Bowtie2 (https://github.com/BenLangmead/bowtie2).
2.11. RNA‐seq data processing and analysis
We used the R package limma to transform the count data of transcriptome sequencing using the voom algorithm and obtained the differentially expressed genes between HTB and LTB tumor tissues based on the voom‐transformed transcriptome data. Gene set enrichment analysis was used to estimate the pathway activation level based on the gene set files of the Gene Ontology (GO) database with the online enrichment tool “KOBAS” (http://kobas.cbi.pku.edu.cn/genelist/) [14]. Brifely, we input high‐expression and low‐expression gene symbol of the HTB group into the corresponding input box on the web page (http://kobas.cbi.pku.edu.cn/genelist/), selected “species” as “human” and “enriched database” as the “Gene Ontology” to download the output result.
2.12. Mouse cytokine antibody array
Cytokines from LTB and HTB MC38 or B16 subcutaneous tumors were detected using the AAM‐CYT‐1000‐2 mouse cytokine antibody array (RayBiotech, Norcross, Georgia, USA). Briefly, samples were added to the incubation well and incubate overnight at 4°C with shaking. Membrane was washed with shaking at room temperature, then sealing solution was added and incubated at room temperature for 2 hours. 1 mL detection solution C and 1mL detection solution D were mixed. 500 μL C/D mixture was added onto each membrane. Membranes were shaken slowly for 2 minutes and removed the mixture. The original data obtained from the scans were background‐corrected and normalized between chips using Raybiotech software (Ray Biotech, Norcross, Georgia,, USA). Differential proteins were screened using fold change (expression difference multiple) under the following selection conditions: (1) fold change ≤ 0.83 or fold change ≥ 1.2; (2) mean signal value per group > 150.
2.13. ELISA
The IGFBP2 levels in subcutaneous tumors were detected using the IGFBP2 ELISA assay kit (F5217‐A, Jiangsu Yutong) according to the manufacturer's instructions. Briefly, the subcutaneous tumor lysis solution was added to 96‐well plates and incubated with the antibody from the kit. After washing nonadherent antibodies away, reaction reagents were added, and the absorbance was measured at 450 nm by spectrophotometry (SPECTRO, Kleve, Germany).
2.14. Western blotting
Total proteins from peritoneal macrophages, MC38 or B16 cells were extracted using RIPA lysis buffer (Macklin, Shanghai, China) combined with protease and phosphatase inhibitors (Beyotime, Shanghai, China). Then, Sodium Dodecyl Sulfate‐polyacrylamide gel electrophoresis was applied to estimate and equalize protein quantities. Polyvinylidene fluoride membranes (Merck millipore, Darmstadt, Germany) with the transferred proteins were incubated with primary antibodies at 4°C overnight and fluorophore‐conjugated goat anti‐rabbit (FDR007, Fdbio science, Shanghai, China) or anti‐mouse secondary antibodies (FDM007, Fdbio science) for 1 h, then scanned using an Odyssey Infrared Imaging System (LI‐COR; Lincoln, NE, USA).The following primary antibodies were used for western blotting: rabbit anti‐signal transducer and activator of transcription 3 (STAT3) (1:1000, 4904T, Cell Signaling Technology, Danvers, MA, USA), anti‐phosphorylated‐STAT3 (Y705) (1:1000, 9145T, Cell Signaling Technology), and mouse anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (1:5000, 5174, Cell Signaling Technology).
2.15. Statistical analyses
All statistical analyses were performed using R software (version 4.0.5), GraphPad Prism software (version 8.0.1; GraphPad Software, San Diego, California, USA), and Statistical Package for the Social Science (SPSS) software (version 26.0; IBM SPSS, Chicago, IL, USA). Quantitative data are shown as the mean ± standard deviation. The statistical significance of comparisons between two groups was estimated using Student's t‐tests and Mann‐Whitney U tests for normally distributed variables and nonnormally distributed variables, respectively. For comparisons of more than two groups, Kruskal‐Wallis (nonparametric) and one‐way ANOVA (parametric) tests were used when appropriate. Fisher's exact test was used to compare the difference in categorical data. P < 0.05 were considered significant.
3. RESULTS
3.1. High baseline tumor burden is correlated with an “immune‐desert” phenotype in clinical tumor samples
First, we compared the immune infiltration of tumor tissues between HTB and LTB groups in the TCGA‐COAD and TCGA‐SKCM cohorts. Patients were grouped into HTB and LTB groups respectively according the strategy we mentioned in the Methods part. As shown in Figure 1A‐B, we found that in both patients with colon cancer and those with melanoma, the relative expression levels of signatures of tumor‐infiltrating lymphocytes regional fraction and lymphocytes in the HTB group were significantly lower than those in the LTB group, indicating that the TME characteristics of HTB tumors were closely linked to an immune‐desert phenotype. To further verify these results, we collected postoperative tissues from 51 patients with colon cancer and needle biopsy samples of liver metastasis from 20 patients with colon cancer for IHC staining. Using the medium tumor size as the threshold, the 51 patients with colon cancer were divided into 26 with LTB and 25 with HTB, with an average tumor volume of 13.00 cm3 in the LTB group and 88.11 cm3 in the HTB group. IHC staining showed that the number of CD8+ T cells in the HTB group was significantly lower than that in the LTB group (Figure 1C). Similar results were observed in the needle biopsy samples of liver metastases from patients with colon cancer, the population of CD8+ T cells in a larger metastatic tumor was lower than that in a smaller metastatic tumor (Figure 1D). These results suggested that the TME of HTB tumors features as an “immune‐desert” phenotype, which was characterized by obviously reduced intratumoral infiltration of CD8+ T cells. However, whether this affects the efficacy of ICIs through a transformation of the TME, as well as the specific mechanisms remain to be explored.
FIGURE 1.
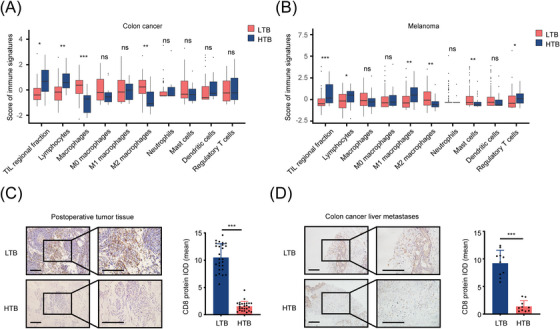
HTB is associated with the decreased amount of CD8+ T cells in TME, (A‐B) Box plots show the scores of immune signatures in the TCGA‐COAD (A) and TCGA‐SKCM (B) cohorts. Boxes represent 25%‐75% of values, lines in boxes represent median values, whiskers represent 1.5 interquartile ranges, and black dots represent outliers, (C) IHC staining showed the amount of CD8+ T cells in colon cancer postoperative tissues (LTB, n = 26; HTB, n = 25; scale bar = 100 μm), (D) IHC staining revealed the population of CD8+ T cells in the needle biopsy samples of liver metastases from patients with colon cancer (LTB, n = 10; HTB, n = 10; scale bar = 100 μm). ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Abbreviations: HTB, high tumor burden; TME, tumor microenvironment; LTB, low tumor burden; TCGA, The Cancer Genome Atlas; IHC, immunohistochemistry; IOD, integrated option density.
3.2. High baseline tumor burden is correlated with the “immune‐desert” phenotype and low efficacy of anti‐PD‐1 treatment in mouse subcutaneous tumor models
To further confirm the relationship between tumor burden, immunophenotype, and therapeutic efficacy of ICIs, we established HTB and LTB subcutaneous tumor mouse models using 2 × 106 or 5 × 105 tumor cells (MC38 cells or B16 cells), respectively, and randomly divided these mice into an anti‐PD‐1 treatment group and an Immunoglobulin G (IgG) control group (Figure 2A, Supplementary Figure S1A‐B). Consistent with the results of clinical studies, in both MC38 (Figure 2B‐D) and B16 (Figure 2E‐G) tumor‐bearing mice, the tumor growth in the LTB group was significantly inhibited by anti‐PD‐1 treatment, whereas tumors in the HTB group did not show any response to anti‐PD‐1 treatment. To investigate the specific mechanism by which HTB weakens the efficacy of ICIs, we obtained the MC38 subcutaneous tumors from the HTB and the LTB groups for RNA‐seq. A total of 108 significantly upregulated genes and 667 significantly downregulated genes in HTB tumors were identified (Figure 2H). The GO enrichment analysis showed that the genes upregulated in HTB tumors were mainly enriched in items related to tumor proliferation and metastasis‐related pathways, such as extracellular matrix regulation and angiogenesis; whereas genes downregulated in HTB tumors were enriched in immune‐related pathways (Figure 2I), including innate and adaptive immune responses. Consistent with the findings of the GO enrichment analysis, a decreased number of CD8+ T cells was also observed in HTB tumors of both MC38 and B16 models (Figure 2J‐K). Taken together, these results suggest that the HTB was correlated with the “immune‐desert” phenotype and poor ICI efficacy in mouse subcutaneous tumor models.
FIGURE 2.
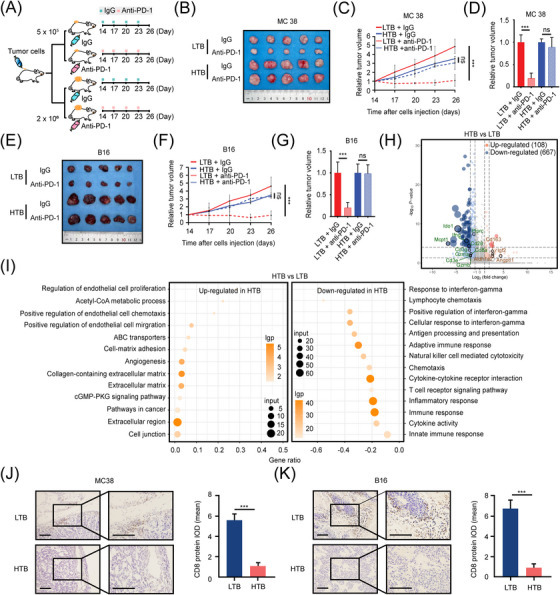
HTB attenuates the efficacy of ICIs and may transform TME. (A) Schematic diagram of LTB and HTB animal model construction for anti‐PD‐1 treatment. Mice were injected with IgG (n = 5) or anti‐PD‐1 antibody (10 mg/kg, intraperitoneal injection, n = 5) every three days from day 14, (B) Subcutaneous tumor of MC38 group mice on day 26, (C) Relative tumor growth curve of MC38 group mice (n = 5), (D) Relative subcutaneous tumor volume of MC38 group mice on day 26, (E) Subcutaneous tumor of B16 group mice on day 26 (n = 5), (F) Relative tumor growth curve of B16 group mice, (G) Relative subcutaneous tumor volume of B16 group mice on day 26, (H) There are 775 genes significantly changed in HTB colon cancer compared to LTB colon cancer. The results were determined by RNA‐seq and shown by the volcano plot. 108 genes were highly expressed in HTB group, and 667 genes were low expressed, (I) GO Enrichment analysis showed that the immune relevant biological process is strongly correlated with tumor burden, (J) The population of CD8+ T cells in MC38 subcutaneous tumor is less in HTB group than in LTB group (scale bar = 100 μm), (K) The amount of CD8+ T cells in B16 subcutaneous tumor is less in HTB group than in LTB group (scale bar = 100 μm). ns, not significant; ***, P < 0.001. Abbreviations: HTB, high tumor burden; ICI, immune checkpoint inhibitor; lgp, ‐log10(p‐value); TME, tumor microenvironment; LTB, low tumor burden; PD‐1, programmed cell death protein 1; RNA‐seq, RNA sequencing; GO, gene ontology.
3.3. The high baseline tumor burden‐related “immune‐desert” phenotype is neither caused by tumor age nor by the morphological factor of tumor size
Next, we tried to determine whether the tumor age or the morphological factor of tumor size contributed to the development of the “immune‐desert” phenotype in HTB tumors. A total of 30 mice were randomly divided into 5 MC38 groups and 5 B16 groups to generate different types of HTB and LTB subcutaneous tumor models (Figure 3A, Supplementary Figure S2A‐B). In group 1, mice received subcutaneous injections of 5 × 105 cells on the left side for two weeks, which was marked as “single side, two weeks, LTB (SS2W‐L)”; in group 2, mice received subcutaneous injections of 2.0 × 106 cells on the left side for two weeks which was marked as “single side, two weeks, HTB (SS2W‐H)”; in group 3, mice received subcutaneous injections of 5 × 105 cells on the left side for three weeks which was marked as “single side, three weeks, HTB (SS3W‐H)”; in group 4, mice received subcutaneous injections of 5 × 105 cells on both left and right sides simultaneously, which was marked as “two sides, two weeks, LTB1 (TS2W‐L1)” and “two sides, two weeks, LTB2 (TS2W‐L2)” respectively; in group 5, mice received subcutaneous injections of 5 × 105 cells on the left side for two weeks and on the right side for three weeks simultaneously, which was marked as “two sides, two weeks, LTB3 (TS2W‐L3)” and “two sides, three weeks, HTB2 (TS3W‐H)”, respectively. As shown in Figure 3B‐C, in both MC38 and B16 models, compared to the SS2W‐L group, the infiltration abundance of CD8+ T cells in the SS2W‐H and SS3W‐H groups was significantly decreased, whereas there was no significant difference in the CD8+ T‐cell infiltration level between the SS2W‐H group and the SS3W‐H group, suggesting that the formation of an HTB‐related immunosuppressive TME was independent of the initial tumor cell number and the time of tumor growth. In the group of mice receiving bilateral subcutaneous tumor implantation, although the total tumor burden increased, the proportion of CD8+ T cells in TS2W‐L1 and TS2W‐L2 tissues was not significantly different from that in the SS2W‐L group, suggesting that the TME characteristic of each individual tumor tissue was determined by its own size rather than the total tumor burden the mice bore. Furthermore, the comparison between TS2W‐L3 and TS3W‐H showed that the proportion of CD8+ T cells in TS3W‐H tissue was also significantly lower than that in TS2W‐L3 tissue in the same mouse, indicating that the decrease in CD8+ T‐cell infiltration was dynamically realized along with the progression in tumor size. Finally, to assess the role of the morphological factor of tumor size on the TME, we generated a debulking surgery model using MC38 colon cancer cells; mice initially received subcutaneous injections of 2.0 × 106 cells on the left side and 5 × 105 cells on the right side (Figure 3A and D). After 14 days, we surgically reduced the mass on the left side of the mouse (HTB tumor) to a size similar to the mass on the right side (LTB tumor) and then detected the intratumoral quantity of CD8+ T cells on days 14 (before surgery), 16, 18, and 21 (Figure 3E‐H, Supplementary Figure S2C). Interestingly, for left‐side tumors, we found that the levels of intratumoral CD8+ T cells did not significantly vary among the four time points before and after surgical tumor reduction, whereas the CD8+ T‐cell infiltration in the TME of right‐side tumors gradually decreased from the day 14 to a level similar to that of the surgical intervention group on the day 21 (Figure 3E‐H, Supplementary Figure S2D). Overall, the stable infiltration level of intratumoral CD8+ T cells at different postoperative time points indicated that the immunosuppressive microenvironment of HTB tumors is not caused by the morphological factor of tumor size.
FIGURE 3.
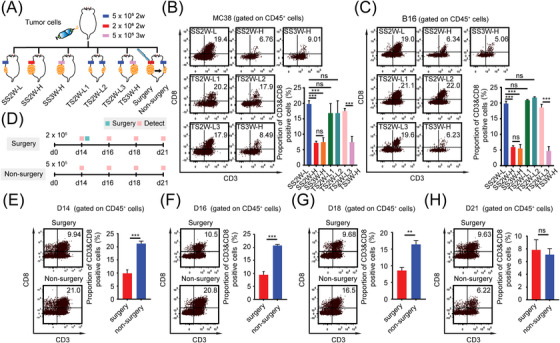
The HTB‐related “immune‐desert” phenotype is neither caused by tumor age nor by the morphological factor of tumor size, (A) Schematic diagram of various tumor burden animal model construction. MC38 cells or B16 cells were used to construct SS2W‐L (5 × 105 cells, subcutaneous growth for 2 weeks), SS2W‐H (2.0 × 106 cells, subcutaneous growth for 2 weeks), SS3W‐H (5 × 105 cells, subcutaneous growth for 3 weeks), TS2W‐L1 & TS2W‐L2 (TS2W‐L1: 5 × 105 cells, subcutaneous growth for 2 weeks on left side. TS2W‐L2: 5 × 105 cells, subcutaneous growth for 2 weeks on right side), TS2W‐L3 & TS3W‐H (TS2W‐L3: 5 × 105 cells, subcutaneous growth for 2 weeks on left side. TS3W‐H: 5 × 105 cells, subcutaneous growth for 3 weeks on right side), Surgery & Non‐surgery (Surgery: 2.0 × 106 cells, subcutaneous growth for 2 weeks, tumor reduction surgery was performed on day 14. Non‐surgery: 2.0 × 106 cells, subcutaneous growth for 2 weeks, without tumor reduction surgery on day 14.) mice models, (B) Proportion of CD3+ CD8+ T cell in indicated MC38 subcutaneous tumors group detected by flow cytometry (n = 3), (C) Proportion of CD3+ CD8+ T cell in indicated B16 subcutaneous tumors group detected by flow cytometry (n = 3), (D) Schematic diagram of surgery mice model construction. MC38 cells were injected subcutaneously on the left (2.0 × 106 cells) and right (5 × 105 cells) sides of the same mouse. In order to make the tumor volume on both sides equal, tumor reduction surgery was performed on the left subcutaneous tumor on day 14. The intratumoral quantity of CD8+ T cells on days 14 (before surgery), 16, 18, and 21 were detected by flow cytometry (n = 3), (E) Proportion of CD3+ CD8+ T cell in subcutaneous tumor on day 14 detected by flow cytometry (n = 3), (F) Proportion of CD3+ CD8+ T cell in subcutaneous tumor on day 16 detected by flow cytometry (n = 3), (G) Proportion of CD3+ CD8+ T cell in subcutaneous tumor on day 18 detected by flow cytometry (n = 3), (H) Proportion of CD3+ CD8+ T cell in subcutaneous tumor on day 21 detected by flow cytometry (n = 3). ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Abbreviations: HTB, high tumor burden; LTB, low tumor burden; SS, single side were injected subcutaneously; TS, two side were injected subcutaneously.
3.4. M2‐like macrophages accumulation fosters an “immune‐desert” phenotype and impedes the anti‐PD‐1 response in high‐burden tumors
Since both tumor age and tumor size were not the cause of the immunosuppressive TME in HTB tumors, we speculated that the HTB‐associated immunophenotype might be regulated by biological factors in tumor tissues. We noticed in the analysis of clinical specimens of the TCGA database that in addition to lymphocytes, only M2 macrophage abundance was significantly upregulated in the HTB group of patients with colon cancer or melanoma (Figure 1A‐B). Therefore, we hypothesized that compared to macrophages in LTB tumors, more macrophages infiltrating in HTB tumors would display an M2‐like phenotype, and these M2‐like HTB‐associated macrophages might act as the culprit in inhibiting the infiltration of CD8+ T cells and preventing the response to PD‐1 blockade. To test this hypothesis, we first used flow cytometry to detect the proportion of CD206+ macrophages (M2‐like macrophages [15]) in subcutaneous tumors of different groups of mice, as shown in Figure 3A. Consistent with the analysis results of the clinical TCGA samples, the amount of CD206+ macrophages in the TME among different groups showed a completely opposite trend to that of CD8+ T cells; CD206+ macrophage infiltration was significantly elevated in the SS2W‐H, SS3W‐H, and TS3W‐H groups as compared to SS2W‐L, TS2W‐L1, TS2W‐L2, and TS2W‐L3 tumors (Figure 4A‐B). To further clarify the role of macrophages, we treated mice with CSF‐1R inhibitor PLX3397, an Food and Drug Administration‐approved small‐molecule drug that shows remarkable selectivity to block colony‐stimulating factor 1 receptor (CSF1Rs) on tumor‐infiltrating macrophages, to explore the alteration of CD8+ T‐cell infiltration in HTB subcutaneous tumors after inhibiting polarization, cellular proliferation, and chemotaxis of intratumoral macrophages. Mice initially received subcutaneous injections of 2 × 106 B16 melanoma cells or MC38 colon cancer cells and were then divided into four treatment groups (Figure 4C): saline + IgG, PLX (d7) + anti‐PD‐1, PLX (d13) + anti‐PD‐1, and saline + anti‐PD‐1. PLX (d7) and PLX (d13) were defined as an injection of PLX3397 on days 7 and 13, respectively, after subcutaneous tumor implantation. In the saline + IgG, PLX(d13) + anti‐PD‐1, and saline + anti‐PD‐1 groups, mice received intraperitoneal injection of the anti‐PD‐1 or IgG on day 17 after subcutaneous tumorigenesis. However, as PLX (d7) treatment slowed the tumor growth, we set the time point of intraperitoneal anti‐PD‐1 injection in the PLX(d7) + anti‐PD‐1 group to day 21 after subcutaneous tumor inoculation to ensure that the baseline tumor burden of subcutaneous tumors in the PLX (d7) + anti‐PD‐1 group was similar to that in other groups before PD‐1 treatment.
FIGURE 4.
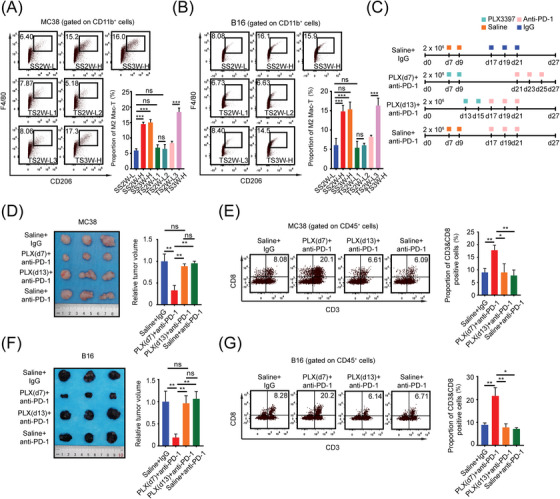
M2‐like macrophages accumulation fosters the “immune‐desert” phenotype and impedes the anti‐PD‐1 response in high‐burden tumors, (A) Proportion of M2‐like macrophages in each MC38 subcutaneous tumors group of Figure 3A detected by flow cytometry (n = 3), (B) Proportion of M2‐like macrophages in each B16 subcutaneous tumors group of Figure 3A detected by flow cytometry (n = 3), (C) Schematic diagram of mice treated with PLX3397 (40 mg/kg) and anti‐PD‐1 antibody (10 mg/kg), (D) Subcutaneous tumor and relative tumor volume of each MC38 group on day 27 (n = 3), (E) Before ICI treatment began, proportion of CD3+ CD8+ T cell in MC38 subcutaneous tumors of 4 groups were detected by flow cytometry, (F) Subcutaneous tumor and relative tumor volume of each B16 group on day 27 (n = 3), (G) Before ICI treatment began, proportion of CD3+ CD8+ T cell in B16 subcutaneous tumors of 4 groups were detected by flow cytometry. ns, not significant; *, P < 0.05;, **, P < 0.01; ***, P < 0.001. Abbreviations: HTB, high tumor burden; TME, tumor microenvironment; LTB, low tumor burden; PLX, PLX3397; PD‐1, programmed cell death protein 1.
The tumor volume and growth curves of all treatment groups are shown in Figure 4D and Supplementary Figure S3A, and the infiltration levels of intratumoral CD8+ T cells are presented in Figure 4E. The results showed that, compared with other groups, the proportion of CD8+ T cells in the TME of the PLX (d7) + anti‐PD‐1 group on the day when the anti‐PD‐1 treatment began was significantly higher, while there was no significant difference in intratumoral CD8+ T‐cell infiltration levels among the saline + IgG, PLX (d13) + anti‐PD‐1, and saline + anti‐PD‐1 groups. In terms of macrophages, F4/80+ cells were dramatically decreased in the subcutaneous tumor tissues obtained from both PLX(d7) + anti‐PD‐1 and PLX(d13) + anti‐PD‐1 groups (Supplementary Figure S3B). Moreover, we found that anti‐PD‐1 significantly retarded tumor growth in the PLX(d7) + anti‐PD‐1 group but not in the PLX(d13) + anti‐PD‐1 group (Supplementary Figure S3A). Similar results were observed in B16 models (Figure 4F‐G, Supplementary Figure S3C‐D). Taken together, these results indicated that the inhibitory effect of M2‐like macrophages on CD8+ T‐cell accumulation is incessant while the tumor progresses in size. Only when PLX3397 had been administered in the early stage, it could play a role in maintaining the “heat” of the TME during the process of tumor growth and thus achieve a response to ICI treatment when the tumor grew to a large size.
3.5. PD‐L1+ M2‐like macrophages induced by soluble factors dampen T cell function in the microenvironment of high‐burden tumors
Next, we intended to explore the mechanism underlying M2‐like macrophage accumulation and mutual exclusion between M2‐like macrophages and CD8+ T cells in HTB tumors. To simulate the complex conditions under which macrophages grow in TMEs with different tumor burdens, we prepared tissue supernatants of SS2W‐H and SS2W‐L tumors (HTCS and LTCS) as described in the “Materials and Methods” section. We mixed 50% complete medium and 50% serum‐free medium to get control medium, then we configured LTCS and HTCS by mixing 50% complete medium whith 50% SS2W‐L or SS2W‐H subcutaneous tumor tissue culture supernatants, next exposed mac‐Ps to LTCS and HTCS, respectively. Using flow cytometry, we found that compared to the control and LTCS treatment groups, macrophage apoptosis was significantly inhibited by HTCS treatment (Figure 5A‐B) while the proportion of CD206+ macrophages was significantly increased (Figure 5C‐D). We also noted that HTCS significantly increased by approximately 3 times the proportion of PD‐L1+ cell subsets among CD206+ macrophages (Figure 5E‐F). These results indicated that soluble factors in the TME of HTB tumors were conducive to macrophage survival and their transformation into an M2‐like phenotype.
FIGURE 5.
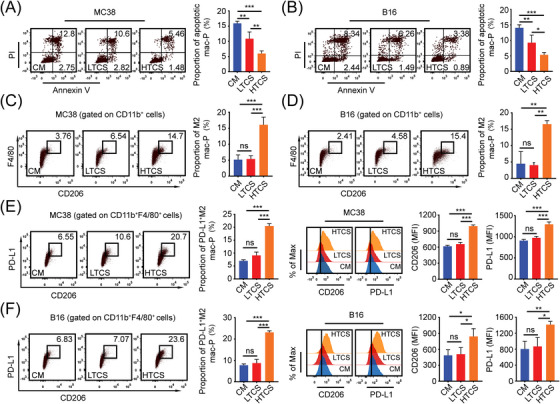
HTB promotes the formation of PD‐L1+ M2 macrophages, (A) Flow cytometry revealed the proportion of apoptotic Mac‐Ps after exposure to control medium (CM, 50% complete medium + 50% serum free medium) or LTB MC38 subcutaneous tumor tissue culture supernatants (LTCS) or HTB MC38 subcutaneous tumor tissue culture supernatants (HTCS) for 24 hours (n = 3), (B) Flow cytometry revealed the proportion of apoptotic Mac‐Ps after exposure to control medium (CM) or LTB B16 subcutaneous tumor tissue culture supernatants (LTCS) or HTB B16 subcutaneous tumor tissue culture supernatants (HTCS) for 24 h (n = 3), (C) Flow cytometry revealed the proportion of M2 Mac‐Ps after exposure to CM or MC38 subcutaneous tumor tissue culture supernatants for 24 h (n = 3), (D) Flow cytometry revealed the proportion of M2 Mac‐Ps after exposure to CM or B16 subcutaneous tumor tissue culture supernatants for 24 h (n = 3), (E) Flow cytometry revealed the proportion of PD‐L1+ M2 Mac‐Ps after exposure to CM or MC38 subcutaneous tumor tissue culture supernatants for 24 h (n = 3), (F) Flow cytometry revealed the proportion of PD‐L1+ M2 Mac‐Ps after exposure to CM or B16 subcutaneous tumor tissue culture supernatants for 24 h (n = 3). ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Abbreviations: LTB, low tumor burden; HTB, high tumor burden; PD‐L1, programmed death‐ligand 1; mac‐P, peritoneal macrophage; CM, control medium; LTCS, low tumor burden subcutaneous tumor tissue culture supernatants (50% complete medium + 50% MC38 or B16 low tumor burden subcutaneous tumor tissue culture supernatants); HTCS, high tumor burden subcutaneous tumor tissue culture supernatants (50% complete medium + 50% MC38 or B16 high tumor burden subcutaneous tumor tissue culture supernatants).
M2‐like macrophages can exert inhibitory effects on T cells in various ways, including secretion of inhibitory factors, metabolic competition, or expression of inhibitory effector molecules [16, 17, 18]. In view of the high PD‐L1 expression on HTB‐associated macrophages, we speculated that the mutual exclusion between M2‐like macrophages and CD8+ T cells in HTB tumors might be mediated by the PD‐1/PD‐L1 axis. Two macrophage‐peripheral T cell co‐culture models were developed (Figure 6A). In the first model, we co‐cultured CD8+ T cells with mac‐Ps, mac‐LTBs (macrophages from SS2W‐L subcutaneous tumors), and mac‐HTBs (macrophages from SS2W‐H subcutaneous tumors), respectively. In the second model, the mac‐Ps would first be exposed to the control medium, LTCS, or HTCS for 24 h and then co‐cultured with CD8+ T cells as described for the first model. Anti‐PD‐L1 was added to supernatants of mac‐HTB & CD8+ T cell co‐culture system or HTCS‐exposed mac‐P & CD8+ T cell co‐culture system to explore the role of the PD‐1/PD‐L1 axis in the reduction of CD8+ T cell numbers induced by HTB‐associated PD‐L1+ M2‐like macrophages.
FIGURE 6.
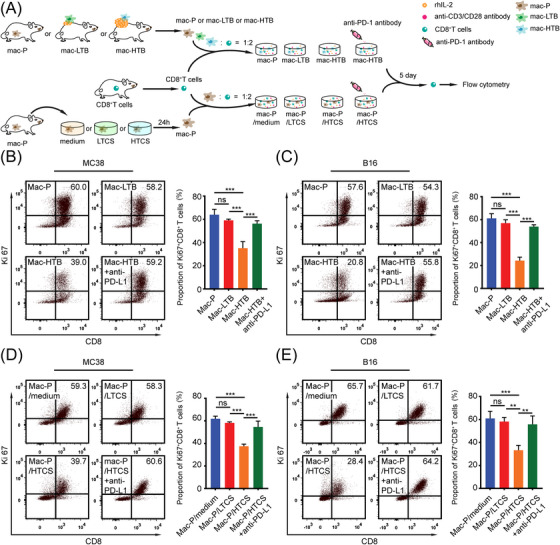
HTB‐associated PD‐L1+ M2‐like macrophages inhibited CD8+ T cells through PD‐1/PD‐L1, (A) Schematic diagram of co‐cultured system of macrophages and T cells. In the first model, we co‐cultured CD8+ T cells with control macrophages (mac‐Ps, from tumor free mice), LTB MC38 subcutaneous tumor‐infiltrating macrophages (mac‐LTB), and HTB MC38 subcutaneous tumor‐infiltrating macrophages (mac‐HTB), respectively. In the second model, the mac‐Ps would first be exposed to the control medium, LTCS, or HTCS for 24 h and then co‐cultured with CD8+ T cells as described for the first model. Anti‐PD‐L1 was added to supernatants of mac‐HTB & CD8+ T cell and HTCS‐exposed mac‐Ps & CD8+ T cell co‐culture systems, (B) Purified peripheral CD8+ T cells were co‐cultured with control macrophages (mac‐Ps) or mac‐LTB or mac‐HTB 2:1 for 5 days with or without anti‐PD‐L1 antibody (5 μg/mL). Ki‐67+ CD8+ T cells were detected by flow cytometry (n = 3), (C) Purified peripheral CD8+ T cells were co‐cultured with mac‐Ps or mac‐LTB or mac‐HTB from B16 subcutaneous tumor 2:1 for 5 days with or without anti‐PD‐L1 antibody (5 μg/mL). Ki‐67+ CD8+ T cells were detected by flow cytometry (n = 3), (D) Purified peripheral CD8+ T cells were co‐cultured with mac‐Ps which stimulated with medium (50% complete medium + 50% serum free medium) or MC38 subcutaneous tumor tissue culture supernatants 2:1 for 5 days with or without anti‐PD‐L1 antibody (5 μg/mL). Ki‐67+ CD8+ T cells were detected by flow cytometry (n = 3), (E) Purified peripheral CD8+ T cells were co‐cultured with mac‐Ps which stimulated with medium or B16 subcutaneous tumor tissue culture supernatants 2:1 for 5 days with or without anti‐PD‐L1 antibody (5 μg/mL). Ki‐67+ CD8+ T cells were detected by flow cytometry (n = 3). ns, not significant; **, P < 0.01; ***, P < 0.001. Abbreviations: LTB, low tumor burden; HTB, high tumor burden; PD‐1, programmed cell death protein 1; PD‐L1, programmed death‐ligand 1; mac‐P, peritoneal macrophage; mac‐T, tumor‐infiltrating macrophage; mac‐LTB, macrophage from low tumor burden subcutaneous tumor; mac‐HTB, macrophage from high tumor burden subcutaneous tumor; LTCS, low tumor burden subcutaneous tumor tissue culture supernatants (50% complete medium + 50% MC38 or B16 low tumor burden subcutaneous tumor tissue culture supernatants); HTCS, high tumor burden subcutaneous tumor tissue culture supernatants (50% complete medium + 50% MC38 or B16 high tumor burden subcutaneous tumor tissue culture supernatants); rhIL‐2, recombinant human interleukin‐2.
Among different co‐culture groups in the first model, we found that compared to mac‐Ps and mac‐LTBs, mac‐HTBs significantly inhibited the proliferation and activation of CD8+ T cells as represented by the proportions of Ki‐67+ CD8+ T cells and IFN‐γ+ CD8+ T cells, respectively, and these inhibitory effects of mac‐HTBs could be reversed by anti‐PD‐L1 treatment (Figure 6B‐C, Supplementary Figure S4A‐B). Similarly, the HTCS‐exposed macrophages showed the same inhibitory effect on the proliferation and activation of CD8+ T cells as mac‐HTBs, and blocking PD‐L1 was also able to restore the proliferation and IFN‐γ expression of CD8+ T cells (Figure 6D‐E). Overall, these findings indicate that HTB‐associated macrophages induced the “immune‐desert” phenotype through the PD‐1/PD‐L1 axis.
Interestingly, in addition to T cells, we found that the expression of PD‐L1 in MC38 and B16 cells was also significantly upregulated in co‐cultures with mac‐HTBs rather than mac‐LTBs (Supplementary Figure S5A), suggesting that mac‐HTB could also indirectly induce immunosuppressor microenvironment by regulating tumor cells.
3.6. The IGFBP2‐STAT3‐PD‐L1 axis induces PD‐L1+ M2‐like macrophages in high‐burden tumors
To detect which cytokine activates and induces PD‐L1 expression on macrophages, we first screened cytokines in both SS2W‐L and SS2W‐H B16 tumor models using a commercial mouse cytokine antibody array (Supplementary Figure S6A‐B). Based on high‐throughput detection, we identified a series of cytokines upregulated in the microenvironment of HTB tumors, including pro‐MMP‐9, bFGF, MMP‐2, IGFBP‐2, TIMP‐2, MCSF, and PF4. Among the top 6 differentially expressed cytokines (pro‐MMP‐9, MMP‐2, IGFBP‐2, TIMP‐2, PF4, and MCSF), we found that only IGFBP2 was able to remarkably induce mac‐Ps into PD‐L1+ CD206+ macrophages (Figure 7A, Supplementary Figure S6C) and was also upregulated in HTCS from both MC38 and B16 models (Figure 7B). Moreover, IGFBP2‐neutralizing antibody (anti‐IGFBP2) blocked M2‐like polarization of intratumoral macrophages in SS2W‐H subcutaneous tumors in vivo (Figure 7C‐D) and in mac‐Ps cultured in HTCS in vitro (Supplementary Figure S6D). Next, we examined the effects of IGFBP2‐activated macrophages on CD8+ T cells. For the in vitro model, mac‐Ps were preactivated by IGFBP2 for 24 h and co‐cultured with CD8+ T cells. IGFBP2‐induced macrophages effectively suppressed CD8+ T cells proliferation, and blockade of PD‐L1 efficiently prevented T cell suppression mediated by such macrophages (Figure 7E). In accordance with the results of the in vitro studies, anti‐IGFBP2 injection at an early tumor age promoted the infiltration of intratumoral CD8+ T cells when MC38 and B16 subcutaneous tumors grew into large size as compared to tumor‐bearing mice injected with IgG control (Figure 7F‐G). Based on this result, we concluded that HTB mainly promotes the formation of the “immune‐desert” phenotype through PD‐L1+ M2‐like macrophages induced by IGFBP2. Previous studies on the effects of IGFBP2 on macrophages are limited. Only one study reported that IGFBP2 can activate STAT3 leading to an immunosuppressive TME infiltrated with M2‐type macrophages in pancreatic duct cancer [19]. Consistent with this report, we also found the phosphorylation degrees of STAT3 in IGFBP2 and HTCS‐stimulated mac‐Ps were significantly stronger than those in the corresponding control groups (Supplementary Figure S6E‐F). The HTCS‐related STAT3 phosphorylation could be further blocked by anti‐IGFBP2, whereas the IGFBP2‐stimulated STAT3 phosphorylation could be further blocked by STAT3 inhibitors (FLLL32 or HJC0152) (Supplementary Figure S6F‐G). Moreover, abolishing the phosphorylation of STAT3 with small ‐molecule inhibitors effectively suppressed the formation of PD‐L1+ M2‐like macrophages induced by IGFBP2 or HTCS (Figure 7H, Supplementary Figure S6D). Taken together, these results suggest that the accumulation of PD‐L1+ M2‐like macrophages in HTB tumors occurred mainly through the IGFBP2‐STAT3 pathway (Figure 8).
FIGURE 7.
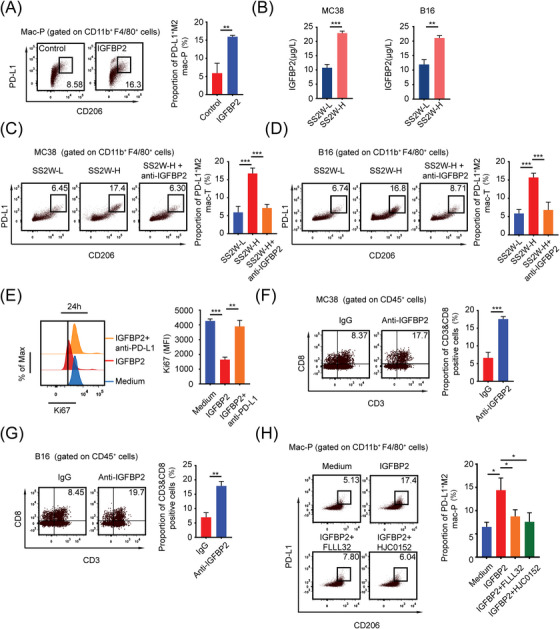
HTB induces PD‐L1+ M2‐like macrophages through IGFBP2‐STAT3 axis, (A) Mac‐Ps were stimulated with or without IGFBP2 (10 ng/mL) for 24 h. PD‐L1+ M2 mac‐Ps were detected by flow cytometry (n = 3), (B) ELISA detected IGFBP2 content between SS2W‐L and SS2W‐H MC38 or B16 subcutaneous tumor conditioned medium (n = 3), (C) Flow cytometry revealed that IGFBP2‐neutralizing antibody (anti‐IGFBP2, 10 mg/kg, intraperitoneal injection on day 7) blocked M2‐like polarization of intratumoral macrophages in HTB MC38 subcutaneous tumors in vivo (n = 3), (D) Flow cytometry revealed that IGFBP2‐neutralizing antibody (10 mg/kg, intraperitoneal injection on day 7) blocked M2‐like polarization of intratumoral macrophages in HTB B16 subcutaneous tumors in vivo (n = 3), (E) Mac‐Ps were preactivated by IGFBP2 for 24 h, co‐cultured with CD8+ T cells with or without anti‐PD‐L1 antibody (5 μg/mL). Flow cytometry revealed that IGFBP2‐induced mac‐Ps inhibit CD8+ T cells through PD‐1/PD‐L1 (n = 3), (F) Flow cytometry revealed that anti‐IGFBP2 injection at an early tumor age (day 7) promoted the infiltration of intratumoral CD8+ T cells when MC38 subcutaneous tumors grew into HTB (n = 3), (G) Flow cytometry revealed that anti‐IGFBP2 injection at an early tumor age (day 7) promoted the infiltration of intratumoral CD8+ T cells when B16 subcutaneous tumors grew into HTB (n = 3), (H) Flow cytometry revealed that IGFBP2 induces PD‐L1+ M2 mac‐Ps through STAT3. Mac‐Ps were stimulated with complete medium or IGFBP2 (10 ng/mL) or IGFBP2 & FLLL32 (5 μmol/L) or IGFBP2 & HJC0152 (5 μmol/L) for 24 hours. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Abbreviations: LTB, low tumor burden; HTB, high tumor burden; mac‐P, peritoneal macrophage; mac‐T, tumor‐infiltrating macrophage; PD‐L1, programmed death‐ligand 1; IGFBP2, insulin‐like growth factor binding protein 2; PD‐1, programmed cell death 1; STAT3, signal transducer and activator of transcription 3.
FIGURE 8.
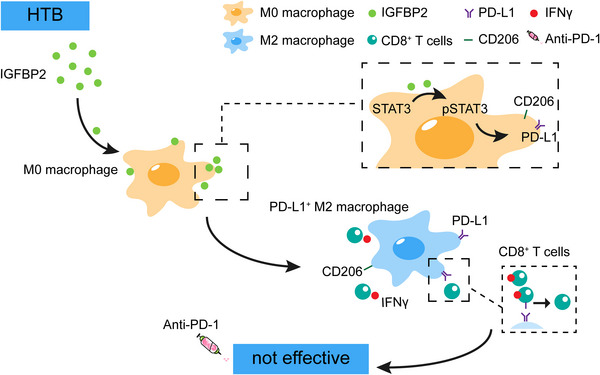
Working model of this study, HTB‐associated macrophages promote an immunosuppressive microenvironment and restrain the efficacy of ICIs through IGFBP2‐STAT3‐PD‐L1 signaling pathway. Abbreviations: LTB, low tumor burden; HTB, high tumor burden; IGFBP2, insulin‐like growth factor binding protein 2; STAT3, signal transducer and activator of transcription 3; PD‐L1, programmed death‐ligand 1; IFNγ, interferon gamma; ICI, immune checkpoint inhibitor.
4. DISCUSSION
Although the correlations between high baseline tumor burden and poorer response to various immunotherapies for diverse malignancies have been borne out in several clinical studies, the underlying mechanisms are still unclear. In this study, by constructing a series of subcutaneous tumor models with different tumor burdens, we not only confirmed that HTB tumors did not respond to ICI treatment and showed an “immune‐desert” phenotype, which was characterized by diminished CD8+ T‐cell infiltration and suppression of immune‐related pathways, but also uncovered for the first time that the formation of an HTB‐associated immunosuppressive TME was determined by the tumor size of each tumor rather than tumor age and total tumor volume. More interestingly, we found that physical shrinkage of subcutaneous tumors in the HTB group did not effectively augment the intratumoral abundance of CD8+ T‐cells, which suggests that the correlation between tumor burden and immune status is independent of the morphological factor of tumor burden and the palliative surgical reduction of tumors with large sizes may not be able to remodel the immunosuppressive TME that already had formed. To date, no studies have directly compared the immune microenvironment of residual tumor tissues before and after palliative surgery. However, some research indicated that residual tumor tissues after incomplete radiofrequency ablation (one of the methods for physical tumor reduction) of large primary or metastatic tumors of the liver also exhibited low T cell infiltration and resistance to PD‐1 blockage [20, 21, 22], which supports our findings.
As one of the important components of the TME, macrophages play crucial roles in regulating anti‐tumor immunity [16, 18, 23]. After recruitment into tumor tissue, macrophages can be polarized into an M2‐like phenotype by tumor cells and are mainly responsible for creating an immunosuppressive TME that is more suitable for tumor cell evasion of immune surveillance, proliferation, and metastasis [16‐18, 23]. In the current study, we also uncovered that the intratumoral accumulation of M2‐like macrophages and the mutual exclusion between M2‐like macrophages and CD8+ T cells were the main reasons contributing to the formation of an immunosuppressive TME and ICI resistance related to HTB tumors. Moreover, by performing co‐culture experiments, we demonstrated that the inhibitory effects of HTB‐associated macrophages on the proliferation and activation of CD8+ T cells occurred directly by activating the PD‐1/PD‐L1 axis. However, besides direct effects, macrophages can also exert their inhibitory effects on T‐cells indirectly by regulating various other cellular components in the TME. For example, regulatory T cells (Tregs) always act as accomplices for macrophages in maintaining a suppressive immune microenvironment, and macrophages can promote Treg function in a bidirectional interaction. Although we did not observe in the TCGA database analysis an increase in Treg infiltration in the clinical samples of the HTB groups in both TCGA‐COAD and TCGA‐SKCM cohorts, whether macrophages and Tregs can also cooperate in HTB tumors still needs to be confirmed by future in vivo and in vitro studies. PD‐L1 expression on the tumor cells quantified using IHC assays (termed as the tumor proportion score, TPS) is currently the basis for screening patients suitable for ICI treatment in most clinical trials [24]. Although a higher TPS is linked with a higher likelihood of responsiveness to anti‐PD‐1/PD‐L1 therapy, a substantial percentage of patients with high PD‐L1 expression do not respond to ICI treatment [24], exposing the fact that PD‐L1 expression may represent more intricate signatures in human cancers. Previously, Wei et al. [25] reported that M2‐like macrophages could induce the expression of PD‐L1 on the surface of tumor cells and endow the latter with capabilities of supporting angiogenesis, metastasis, and resistance to conventional chemotherapy and anti‐PD‐1‐mediated T‐cell cytotoxicity. Based on these findings, we think that PD‐L1+ cancer cells generated by HTB‐related macrophages may be another reason why HTB tumors do not respond to PD‐1 monoclonal antibody therapy.
The clear role of M2‐like macrophages in impeding PD‐1 immunotherapy has led to an interest in developing therapeutic strategies targeting the accumulation of macrophages or inhibiting their M2 polarization for immunotherapy sensitization [26]. Current macrophage‐targeting strategies include CSF‐1R inhibitors [27], Chimeric Antigen Receptor (CAR) T cells [28], and composite nanoparticles [29], among others. However, all these methods have had limited success in clinical trials. In this study, we found that the CSF‐1R inhibitors PLX3397 must be used to clear macrophages when the tumor is small so that the tumor can respond to ICI treatment when it grows into an HTB tumor and the level of intratumoral T cells is comparable to that in the TME of LTB tumors. Based on this finding, we propose that the inhibitory effect of M2‐like macrophages on the intratumoral accumulation of CD8+ T cells exists when the tumor size is still small and such effect is incessant during tumor size progression and is constantly amplified with the increase in M2‐like macrophage infiltration. We believe that this finding may partly explain why therapies targeting macrophages tend to be effective in mouse models (mainly used when tumors are small in most preclinical studies [30]) but not in real clinical practice.
Soluble cytokines are considered to be important mediators regulating cell‐cell interaction and reshaping the immune microenvironment. For example, the recruitment of macrophages by tumor cells can be accomplished by secreting chemokine ligand 2 (CCL2) [31], and the remodeling of the TME by M2‐like macrophages is also inseparable from the secretion of transforming growth factor β (TGF‐β), interleukin 10, and other immunosuppressive cytokines [31]. In this study, we reported for the first time that IGFBP2 is a key cytokine mediating the formation of an HTB‐related immunosuppressive TME. The content of IGFBP2 in HTB tumors was significantly higher than that in LTB tumors, and IGFBP2 promoted the formation of PD‐L1+ M2‐like macrophages in vitro. Moreover, the use of anti‐IGFBP2 reduced the level of PD‐L1+ M2 polarization in vivo. In addition, we confirmed that the IGFBP2–STAT3 axis, which has been validated in several studies[19, 32], also participated in the accumulation of PD‐L1+ M2‐like macrophages in HTB tumors. IGFBP2 is found to be highly expressed in many cancers, and its overexpression is associated with worse prognoses in various malignancies [33]. Although IGFBP2 was identified as a modulator of the IGF system, it exerts several pro‐tumorigenic activities independently of IGF by regulating various signal transductions pathways, including cancer cell migration, invasion, proliferation, vascular formation, and epithelial‐mesenchymal transition [25, 26, 27, 28], and thus may act as a master regulator of a complex oncogenic network. Although our study established an association between IGFBP2 and HTB‐related immunosuppression, the specific mechanism for the upregulation of IGFBP2 in HTB tumors is still unknown. Some studies reported that hypoxia could upregulate IGFBP2 expression [34], and the hypoxia‐responsive element sequences located in the IGFBP2 promoter region can directly bind to the hypoxia‐inducible factor 1α (HIF‐1α) [35]. Since HTB tumors have higher hypoxia levels than LTB tumors [36], we hypothesized that IGFBP2 enrichment in HTB tissues may be attributed to elevated HIF‐1α transcriptional activation, which needs to be further investigated. Moreover, as IGFBP2 can be secreted by various cell types, including cancer‐associated fibroblasts, tumor cells, and mesenchymal stem cells, it would be important to clarify which cells secrete IGFBP2 into the TME of HTB tumors to understand in more detail on IGFBP2‐mediated cell interactions.
5. CONCLUSIONS
In this study, we found that high baseline tumor burden was strongly associated with an immunosuppressive TME and low efficacy of ICI treatment. The PD‐L1+ M2‐like macrophages accumulated in HTB tumors through the IGFBP2‐STAT3‐PD‐L1 pathway exhibited clear inhibitory effects on CD8+ T cell proliferation and activation. Based on our results, we propose that baseline tumor burden may play a significant role in realizing individualized immunotherapy that ICI treatment is more suitable for patients with low baseline tumor burden. However, for patients with HTB tumors, effective strategies to improve the efficacy of immunotherapies still warrent further investigation.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Zhaowei Wen, Rui Zhou, Huiying Sun, and Wangjun Liao contributed to the planning of the study and drafted the manuscript. Wangjun Liao contributed to manuscript revision. Zhaowei Wen, Rui Zhou, Huiying Sun, Yannan Zheng and Zhihua Zhang performed all the experiments and prepared all the figures and tables. Siting Zheng, Jianping Bin, Yulin Liao, and Min Shi contributed to interpretation of data, and review of the manuscript. All the authors reviewed and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
FUNDING INFORMATION
This work was supported by the National Natural Science Foundation of China (82073303), National Natural Science Foundation of China (82103335), and National Natural Science Foundation of China (82102731), Science and Technology Planning Project of Guangzhou (202201011560), and Natural Science Foundation of Guangdong Province of China (2022A1515012418).
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Human Research Ethics Committee of Nanfang Hospital (permit number: NFEC‐2017‐206). The tissue samples were obtained with written informed consent from each patient. The animal study was carried out in compliance with the guidance suggestion of Animal Care Committee of Nanfang Hospital (permit number: NFYY‐2017‐0926).
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
We thank the members of Wangjun Liao's laboratory (Nanfang Hospital, Guangzhou, Guangdong, China) for advice and discussion. We would like to thank Editage (www.editage.cn) for English language editing.
Wen Z, Sun H, Zhang Z, Zheng Y, Zheng S, Bin J, et al. High baseline tumor burden‐associated macrophages promote an immunosuppressive microenvironment and reduce the efficacy of immune checkpoint inhibitors through the IGFBP2‐STAT3‐PD‐L1 pathway. Cancer Commun. 2023;43:562–581. 10.1002/cac2.12420
Zhaowei Wen and Huiying Sun contributed equally to this work.
Email addresses for all authors:
Zhaowei Wen, Email: wenzhaowei0769@163.com, Huiying Sun, Email: 1714761643@qq.com;, Zhihua Zhang, Email: 779499326@qq.com, Yannan Zheng, Email: syalonyui@163.com, Siting Zheng, Email: 875841014@qq.com, Jianping Bin, Email: jianpingbin@126.com, Yulin Liao, Email: liao18@msn.com, Min Shi, Email: nfyyshimin@163.com, Rui Zhou, Email: 363799445@qq.com, Wangjun Liao, Email: nfyyliaowj@163.com.
Contributor Information
Rui Zhou, Email: 363799445@qq.com.
Wangjun Liao, Email: nfyyliaowj@163.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. The RNA‐seq data was uploaded to GEO (GSE227270).
REFERENCES
- 1. Wang Y, Wang M, Wu HX, Xu RH. Advancing to the era of cancer immunotherapy. Cancer Commun (Lond). 2021;41(9):803–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang X, Wang B, Cao X. Transcriptional suppression of CD8(+) T cell exhaustion for improving T cell immunotherapy. Cancer Commun (Lond). 2021;41(11):1228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joseph RW, Elassaiss‐Schaap J, Kefford R, Hwu WJ, Wolchok JD, Joshua AM, et al. Baseline Tumor Size Is an Independent Prognostic Factor for Overall Survival in Patients with Melanoma Treated with Pembrolizumab. Clin Cancer Res. 2018;24(20):4960–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hopkins AM, Kichenadasse G, McKinnon RA, Rowland A, Sorich MJ. Baseline tumor size and survival outcomes in lung cancer patients treated with immune checkpoint inhibitors. Semin Oncol. 2019;46(4‐5):380–4. [DOI] [PubMed] [Google Scholar]
- 6. Nishino M, Giobbie‐Hurder A, Ramaiya NH, Hodi FS. Response assessment in metastatic melanoma treated with ipilimumab and bevacizumab: CT tumor size and density as markers for response and outcome. J Immunother Cancer. 2014;2(1):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chardin D, Paquet M, Schiappa R, Darcourt J, Bailleux C, Poudenx M, et al. Baseline metabolic tumor volume as a strong predictive and prognostic biomarker in patients with non‐small cell lung cancer treated with PD1 inhibitors: a prospective study. J Immunother Cancer. 2020;8(2):e000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ji H, Niu X, Yin L, Wang Y, Huang L, Xuan Q, et al. Ratio of Immune Response to Tumor Burden Predicts Survival Via Regulating Functions of Lymphocytes and Monocytes in Diffuse Large B‐Cell Lymphoma. Cell Physiol Biochem. 2018;45(3):951–61. [DOI] [PubMed] [Google Scholar]
- 9. Glajcar A, Szpor J, Hodorowicz‐Zaniewska D, Tyrak KE, Okoń K. The composition of T cell infiltrates varies in primary invasive breast cancer of different molecular subtypes as well as according to tumor size and nodal status. Virchows Arch. 2019;475(1):13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu JW, Bhattacharya S, Yanamandra N, Kilian D, Shi H, Yadavilli S, et al. Tumor‐immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS One. 2018;13(11):e0206223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang TH Ou, et al. The Immune Landscape of Cancer. Immunity. 2018;48(4):812–30.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang L, Wu Y, Lin L, Liu P, Huang H, Liao W, et al. Metastasis‐associated in colon cancer‐1 upregulation predicts a poor prognosis of gastric cancer, and promotes tumor cell proliferation and invasion. Int J Cancer. 2013;133(6):1419–30. [DOI] [PubMed] [Google Scholar]
- 14. Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, et al. KOBAS‐i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021;49(W1):W317–W325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye‐Ogunniyan A, et al. Mannose receptor (CD206) activation in tumor‐associated macrophages enhances adaptive and innate anti‐tumor immune responses. Sci Transl Med. 2020;12(530):eaax6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bronte V, Zanovello P. Regulation of immune responses by L‐arginine metabolism. Nat Rev Immunol. 2005;5(8):641–54. [DOI] [PubMed] [Google Scholar]
- 17. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anti‐cancer therapies. Cancer Cell. 2013;23(3):277–86. [DOI] [PubMed] [Google Scholar]
- 18. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage‐secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18(5):549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun L, Zhang X, Song Q, Liu L, Forbes E, Tian W, et al. IGFBP2 promotes tumor progression by inducing alternative polarization of macrophages in pancreatic ductal adenocarcinoma through the STAT3 pathway. Cancer Lett. 2021;500:132–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi L, Wang J, Ding N, Zhang Y, Zhu Y, Dong S, et al. Inflammation induced by incomplete radiofrequency ablation accelerates tumor progression and hinders PD‐1 immunotherapy. Nat Commun. 2019;10(1):5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li K, Niu Y, Yuan Y, Qiu J, Shi Y, Zhong C, et al. Insufficient ablation induces E3‐ligase Nedd4 to promote hepatocellular carcinoma progression by tuning TGF‐β signaling. Oncogene. 2022;41(23):3197–209. [DOI] [PubMed] [Google Scholar]
- 22. Wu H, Li SS, Zhou M, Jiang AN, He Y, Wang S, et al. Palliative Radiofrequency Ablation Accelerates the Residual Tumor Progression Through Increasing Tumor‐Infiltrating MDSCs and Reducing T‐Cell‐Mediated Anti‐Tumor Immune Responses in Animal Model. Front Oncol. 2020;10:1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7‐H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203(4):871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marletta S, Fusco N, Munari E, Luchini C, Cimadamore A, Brunelli M, et al. Atlas of PD‐L1 for Pathologists: Indications, Scores, Diagnostic Platforms and Reporting Systems. J Pers Med. 2022;12(7):1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wei Y, Zhao Q, Gao Z, Lao XM, Lin WM, Chen DP, et al. The local immune landscape determines tumor PD‐L1 heterogeneity and sensitivity to therapy. J Clin Invest. 2019;129(8):3347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor‐associated macrophages with anti‐CSF‐1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–59. [DOI] [PubMed] [Google Scholar]
- 28. Sánchez‐Paulete AR, Mateus‐Tique J, Mollaoglu G, Nielsen SR, Marks A, Lakshmi A, et al. Targeting Macrophages with CAR T Cells Delays Solid Tumor Progression and Enhances Anti‐tumor Immunity. Cancer Immunol Res. 2022;10(11):1354–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zang X, Cheng M, Zhang X, Chen X. Targeting macrophages using nanoparticles: a potential therapeutic strategy for atherosclerosis. J Mater Chem B. 2021;9(15):3284–94. [DOI] [PubMed] [Google Scholar]
- 30. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF‐1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21(11):799–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li T, Zhang C, Zhao G, Zhang X, Hao M, Hassan S, et al. IGFBP2 regulates PD‐L1 expression by activating the EGFR‐STAT3 signaling pathway in malignant melanoma. Cancer Lett. 2020;477:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei LF, Weng XF, Huang XC, Peng YH, Guo HP, Xu YW. IGFBP2 in cancer: Pathological role and clinical significance (Review). Oncol Rep. 2021;45(2):427–38. [DOI] [PubMed] [Google Scholar]
- 34. Rahman MS, Thomas P. Characterization of three IGFBP mRNAs in Atlantic croaker and their regulation during hypoxic stress: potential mechanisms of their upregulation by hypoxia. Am J Physiol Endocrinol Metab. 2011;301(4):E637–48. [DOI] [PubMed] [Google Scholar]
- 35. Liu Y, Nelson MV, Bailey C, Zhang P, Zheng P, Dome JS, et al. Targeting the HIF‐1α‐IGFBP2 axis therapeutically reduces IGF1‐AKT signaling and blocks the growth and metastasis of relapsed anaplastic Wilms tumor. Oncogene. 2021;40(29):4809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hildingsson S, Gebre‐Medhin M, Zschaeck S, Adrian G. Hypoxia in relationship to tumor volume using hypoxia PET‐imaging in head & neck cancer ‐ A scoping review. Clin Transl Radiat Oncol. 2022;36:40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. The RNA‐seq data was uploaded to GEO (GSE227270).