

Abstract
Tumor development and metastasis are facilitated by the complex interactions between cancer cells and their microenvironment, which comprises stromal cells and extracellular matrix (ECM) components, among other factors. Stromal cells can adopt new phenotypes to promote tumor cell invasion. A deep understanding of the signaling pathways involved in cell‐to‐cell and cell‐to‐ECM interactions is needed to design effective intervention strategies that might interrupt these interactions. In this review, we describe the tumor microenvironment (TME) components and associated therapeutics. We discuss the clinical advances in the prevalent and newly discovered signaling pathways in the TME, the immune checkpoints and immunosuppressive chemokines, and currently used inhibitors targeting these pathways. These include both intrinsic and non‐autonomous tumor cell signaling pathways in the TME: protein kinase C (PKC) signaling, Notch, and transforming growth factor (TGF‐β) signaling, Endoplasmic Reticulum (ER) stress response, lactate signaling, Metabolic reprogramming, cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) and Siglec signaling pathways. We also discuss the recent advances in Programmed Cell Death Protein 1 (PD‐1), Cytotoxic T‐Lymphocyte Associated Protein 4 (CTLA4), T‐cell immunoglobulin mucin‐3 (TIM‐3) and Lymphocyte Activating Gene 3 (LAG3) immune checkpoint inhibitors along with the C‐C chemokine receptor 4 (CCR4)‐ C‐C class chemokines 22 (CCL22)/ and 17 (CCL17), C‐C chemokine receptor type 2 (CCR2)‐ chemokine (C‐C motif) ligand 2 (CCL2), C‐C chemokine receptor type 5 (CCR5)‐ chemokine (C‐C motif) ligand 3 (CCL3) chemokine signaling axis in the TME. In addition, this review provides a holistic understanding of the TME as we discuss the three‐dimensional and microfluidic models of the TME, which are believed to recapitulate the original characteristics of the patient tumor and hence may be used as a platform to study new mechanisms and screen for various anti‐cancer therapies. We further discuss the systemic influences of gut microbiota in TME reprogramming and treatment response. Overall, this review provides a comprehensive analysis of the diverse and most critical signaling pathways in the TME, highlighting the associated newest and critical preclinical and clinical studies along with their underlying biology. We highlight the importance of the most recent technologies of microfluidics and lab‐on‐chip models for TME research and also present an overview of extrinsic factors, such as the inhabitant human microbiome, which have the potential to modulate TME biology and drug responses.
Keywords: 3D‐model, cancer therapy, gut microbiota, immune signaling, metabolism, signaling, tumor microenvironment
Abbreviations
- TME
tumor microenvironment
- ECM
extracellular matrix
- MMP
matrix metalloproteinase
- TAM
tumor‐associated macrophage
- CAF
cancer‐associated fibroblast
- APC
antigen‐presenting cell
- DC
dendritic cell
- Teff
effector T cell
- NK
natural Killer cell
- XCL
X‐C Motif Chemokine Ligand
- CCL
CC motif chemokine ligand
- FLT3LG
FMS Related Receptor Tyrosine Kinase 3 Ligand
- FGF
fibroblast growth factor
- CXCL
CXC motif chemokine ligand
- VEGF
vascular endothelial growth factor
- SDF‐1
stromal cell‐derived factor 1
- TGF‐ß
transforming growth factor ß
- IL
interleukin
- IDO
indoleamine 2,3‐dioxygenase
- TDO2
tryptophan 2,3‐dioxygenase
- SLIT2
slit guidance ligand 2
- CD
cluster of differentiation
- PDGF
platelet‐derived growth factor
- PDGFR
platelet‐derived growth factor receptor
- HGF
hepatocyte growth factor
- MET
mesenchymal‐epithelial transition factor
- EC
Endothelial cell
- CAR‐T Cell
Chimeric antigen receptor‐T cell
- PSMA
prostate‐specific membrane antigen
- TNF α
tumor necrosis factor α
- NICD
notch‐intracellular domain
- ASO
antisense oligonucleotides
- BiP
binding‐immunoglobulin protein
- HIF
hypoxia‐inducible factor
- PD‐1
Programmed cell death protein 1
- PD‐L1
Programmed cell death protein ligand 1
- CCR
C‐C chemokine receptor type
- 3D
Three (3D)‐dimensional
- PDX
patient‐derived tumor xenograft
- WNT
Wingless/Integrated
- PKC
protein kinase C
- ER
Endoplasmic Reticulum
- cGAS
cyclic GMP–AMP synthase
- STING
stimulator of interferon genes
- CTLA4
Cytotoxic T‐Lymphocyte Associated Protein 4
- TIM‐3
T‐cell immunoglobulin mucin‐3
- LAG3
Lymphocyte Activating 3
- PAK4
p21‐Activated kinase 4
- EGFRvIII
Epidermal growth factor receptor variant III
- FGF
Fibroblast growth factor
- PEGPH20
pegvorhyaluronidase alfa
- DON
6‐diazo‐5‐oxo‐l‐norleucine
- GM‐CSF
Granulocyte‐macrophage colony‐stimulating factor
- Th
T helper
- Teff
T effector
- HOX
Homeobox
- DAG
diacylglycerol
- STAT
Signal Transducer And Activator Of Transcription
- NF‐κB
Nuclear factor‐κB
- DLL1
delta like canonical Notch ligand 1
- JAG2
jagged canonical Notch ligand 2
- NICD
intracellular domain of the Notch receptor
- ADC
antibody‐drug conjugates
- IRE1α
inositol‐requiring enzyme 1α
- SLC16A
solute carrier family 16 member
- LDHA
lactate dehydrogenase A
- CHC
cyano‐4‐hydroxycinnamate
- ROS
Reactive Oxygen Species
- IDH
Isocitrate dehydrogenase
- D2HG
D‐2‐Hydroxyglutarate
- GBM
Glioblastoma
- AML
Acute myeloid leukemia
- TCGA
The Cancer Genome Atlas
- TMEM
transmembrane protein 173
- TBK‐1
TANK‐binding kinase 1
- IRF‐3
Interferon regulatory factor 3
- PCDH
protocadherin
- Cx
connexin 43
- IKK
nuclear factor‐κB (IκB) kinase
- IFN‐I
Type I interferons
- MDSC
Myeloid‐derived suppressor cell
- BTLA
B and T lymphocyte attenuator
- scRNA‐seq
single‐cell RNA sequencing
- EVs
extracellular vesicles
- ITSM
immunoreceptor tyrosine‐based switch motif
- NSCLC
Non‐small cell lung cancer
- SHP2
Src‐homology‐region‐2‐containing protein tyrosine phosphatase‐2
- CyTOF
Cytometry by time of flight
- TCR
T cell receptor
- CTLA4
Cytotoxic T‐lymphocyte associated protein 4
- JNK
c‐Jun N‐terminal kinase
- ERK
extracellular signal‐regulated 701 kinases
- TIM‐3
T cell immunoglobulin and mucin‐domain containing‐3
- HLA‐B
human 710 leukocyte antigen B
- LCK
lymphocyte‐specific protein tyrosine kinase
- ITK
inducible T cell kinase
- LAG3
Lymphocyte activation gene 3
- α‐syn
α‐synuclein fibrils
- L‐SECtin
lymph node sinusoidal endothelial cell C‐type lectin
- FGL‐1
fibrinogen‐like protein 1
- TILs
tumor infiltrating lymphocytes
- BTLA
B‐ and T‐lymphocyte attenuator
- HVEM
herpesvirus entry mediator
- HNSCCs
head and neck squamous cell carcinomas
- MDSCs
myeloid‐derived suppressive cells
- PDOs
patient‐derived organoids.
1. BACKGROUND
The concept of tumor microenvironment (TME) research emerged in the 1800s when the relationship between inflammation and cancer was proposed and acknowledged with Paget's theory of “seed and soil” [1]. With the progress made in the following decades, Hanahan and Weinberg enhanced the hallmarks of cancer from six to ten in 2011 with the recognition of the TME [2]. However, with the limited understanding of the TME, the therapies targeted against its components, such as the blood vasculature, were efficient against cancers of all organs regardless of their source [3].
With the advent of single‐cell analysis through sequencing technologies and analytical bioinformatics, the immense complexity of the TME is apparent, and contrary to the earlier notions, the TME is now understood to be either tumor‐supportive or tumor‐suppressive depending on the stage and type of cancer [4, 5]. Currently approved therapeutics of the TME are targeted against immune checkpoints, T cells and blood vasculature. In one study, inhibition of p21‐activated kinase 4 (PAK4), selectively expressed by tumor endothelial cells (ECs) in glioma, re‐sensitizes tumor cells to chimeric antigen receptor (CAR)‐T cell therapy engineered against epidermal growth factor receptor variant III (EGFRvIII) mutation in glioma, allowing engineered T cells to enter the brain and elicit a robust immune response [6]. Another study targeting chemoresistant and desmoplastic colorectal cancer (CRC) cells by targeting vascular endothelial growth factor A (VEGFA) and angiopoietin‐2 (ANGPT2) along with the cluster of differentiation 40 (CD40) agonistic antibodies destroyed tumor fibrosis and induced T cell‐mediated killing [7]. These studies exemplify the successes and current state of the art of TME signaling and mechanisms of unleashing tumor therapeutics.
However, several challenges impede the progression of TME research. One challenge is to create experimental models that can preserve the initial characteristics of the primary tumor to develop personalized medicine tools for drug development and cancer therapy. The recent development of three‐dimensional platforms for cell culture using lab‐on‐chip and microfluidic devices holds enormous potential to better simulate TME processes and bridge the gap between preclinical and clinical translations [8]. Another aspect of personalized medicine that poses a challenge to TME research is to identify responders from non‐responders [4]. To achieve this, several approaches, such as noninvasive liquid biopsies to identify circulating tumor DNA in the blood or the use of extracellular vesicles as diagnostic markers, are underway to improve the prediction of responses in patients [9]. Thus, preclinical studies, especially those involving immune responses, should consider responsiveness of the mechanistic aspects of mutations or molecular subtyping in patients.
Few recent reviews have discussed the advances in TME therapeutics. Baghban et al. explored the molecular interactions between cancer cells and the TME to identify novel cancer therapeutics [10]. Jin et al. classified the chemopathological characteristics of TME, such as the metabolic, immune and acidic niches and advances in drug repurposing in their context [11]. Moreover, Bejarano et al. provided a comprehensive analysis of the current therapies targeting the TME and their clinical evaluation [3]. In this current review, we extensively discuss the advances in the signaling mechanisms, both intrinsic to cancer cells and non‐autonomous signaling prevalent in the TME and the therapeutics targeting those mechanisms from a preclinical and clinical perspective. We further comprehensively assessed the prevalent and newly identified tumor models in TME research and described how the gut microbiome alters the TME and affects treatment response.
2. EXPLOITING MICROENVIRONMENTAL CUES FOR THERAPY
2.1. Components of the TME
Single‐cell‐based technologies have enabled a better dissection of the TME through precise monitoring of cell sub‐populations and spatial architecture, thus revealing the heterogeneous and complex nature of the TME [10, 12]. Other than the cancer cells that form the bulk of the tumor, the other predominant populations include the immune cells constituting the tumor‐associated macrophages (TAMs), natural killer (NK) and dendritic cells (DCs), and T and B lymphocytes. The blood and lymphatic ECs, complex collagen fibers and glycoproteins form the ECM, while the cancer‐associated fibroblasts (CAFs) and mesenchymal stem cells further assist in ECM remodeling and even chemoresistance [13, 14]. The unique signatures of its cellular components, the associated signaling and the diversity of the TME have been targeted in cancer therapy.
The contribution of the immune system in the modulation of cancer has recently gained importance. Other than tumor heterogeneity, the ‘immune system’ forms a crucial aspect of the complex architecture of the TME, and their modulation can be leveraged to overcome the persistent problems of therapy failure and resistance. Cytotoxic CD8+ T cells infiltrate in the TME to get primed by antigen‐presenting cells (APCs), macrophages, B cells and DCs to modulate cytotoxic effector T (Teff) cell response. For instance, DCs secrete the chemoattractants chemokine (C‐X‐C motif) ligand 9 (CXCL9) and 10 (CXCL10) to facilitate the infiltration of CD8+ Teff cells in the TME and for T cell cytotoxic activity [15]. Instead, their engagement in inhibitory crosstalk, such as with the PD‐1/ programmed death‐ligand 1 (PD‐L1) signaling axis, leads to immunosuppression in the TME [16]. Other immune cells facilitating a pro‐tumorigenic response in the TME include the immunomodulatory regulatory T (Treg) cells and the myeloid suppressor (MDSCs) that result in an immunosuppressive TME [17] and therapy failure. However, the presence of NK cells in the TME is believed to be anti‐tumorigenic, resulting from the release of cytokines and chemoattractants such as X‐C motif chemokine ligand 1 (XCL1), chemokine ligand 5 (CCL5) or Fms‐related tyrosine kinase 3 ligand (FLT3LG) and leading to APC accumulation in the TME [18, 19, 20].
CAFs were initially considered a homogeneous population. However, recent studies indicated that CAFs consist of several types of stromal cells that differ in their origin, functions, number, and phenotype [21, 22, 23, 24]. Thus, CAFs can either lead to cancer progression or inhibit cancer growth, depending on their nature. They secrete several growth factors, such as fibroblast growth factor (FGF) and C‐X‐C motif chemokine ligand 12 (CXCL12), to promote angiogenesis via the VEGF, stromal cell‐derived factor 1 (SDF‐1) and TGF‐ß signaling [25, 26]. CAFs recruit and polarize immune cells such as macrophages, neutrophils, T cells and DCs to a pro‐tumorigenic phenotype by secreting several cytokines, chemokines, and other effector molecules such as Interleukin 6 (IL‐6), and 8 (IL‐8), TGF‐β, CXCL12, CCL2, SDF‐1, VEGF, Indoleamine‐pyrrole 2,3‐dioxygenase (IDO) and tryptophan 2,3‐dioxygenase (TDO2) [27]. However, certain CAFs (Slit Guidance Ligand 2 [Slit2]+ and cluster of differentiation 146 [CD146]+ CAFs) were shown to have anti‐tumorigenic effects such as tumor suppression and increased tumor chemosensitivity [28]. Furthermore, downregulating the paracrine signaling of fibroblasts, such as the platelet‐derived growth factor (PDGF)/ PDGF receptor (PDGFR) signaling pathway and hepatocyte growth factor (HGF)/ mesenchymal‐epithelial transition factor (MET) signaling pathway, was shown to promote chemosensitivity in CAFs [29, 30].
The TME modulates ECs to induce an angiogenic response because of the high nutritional demand of the tumor [31]. Hypoxia‐inducible factor 1‐alpha (HIF‐1α) activation in tumor cells promotes the secretion of proangiogenic factors, such as VEGF, FGF‐2, and PDGF, and stimulates angiogenesis [32, 33]. ECs are targeted indirectly for tumor therapy by inhibiting angiogenesis via neutralizing antibodies for VEGF or inhibitors of VEGF activity [34]. In a clinical trial, bevacizumab, a monoclonal antibody against circulating VEGF‐A, combined with chemotherapy was shown to improve the overall and progression‐free survival of colorectal cancer patients compared with chemotherapy alone [35]. However, tumor cells become resistant after long‐term treatment, and these anti‐angiogenic drugs were shown to promote vasoinvasion leading to metastasis and reduced lifespan in mice [36]. Thus, it suggests the need for targeting the multiple oncogenic interactions in the TME rather than individual cell types and molecules.
ECM acts as a scaffold and plays a tumor‐suppressing role in healthy tissues; however, it is modified in tumor tissue to possess a tumor‐promoting role [37]. The ECM components underlying tumor‐promoting activity, such as fibronectin and its splice variants, crosslinked collagen I and tenascin‐C, are induced in the TME [37, 38] and interact with integrins to influence tumor cell migration, proliferation and cellular signaling [39]. Various strategies have been developed to target aberrant ECM components to develop novel treatments, including fresolimumab (to inhibit collagen synthesis), collagenases and matrix metalloproteinases (MMPs) (to promote collagen degradation), 4‐methylumbelliferone (to inhibit hyaluronic acid synthesis), hyaluronidase (to promote hyaluronic acid degradation), Vitaxin and Volociximab (to target integrin and inhibit angiogenesis) [40]. Moreover, Provenzano et al. found that systemic administration of pegvorhyaluronidase alfa (PEGPH20), an enzyme against hyaluronic acid, reduced stromal hyaluronic acid, normalized interstitial fluid pressures, re‐expanded the microvasculature and led to tumor suppression in pancreatic ductal adenocarcinomas murine models [41].
Thus, different components of the TME secrete growth factors, components of the ECM, cytokines and extracellular molecules that are essential for signaling between cells in the TME and systemically. Thus, it would be critical to identify strategies for identifying key vulnerabilities and targeting them to alleviate the immune suppression prevalent in most TMEs.
2.2. TME‐based cancer therapy
In recent times, combining therapies, especially those inducing the engagement of immune cells, have been the primary focus of TME‐based therapeutics. In this section, we review several facets of the TME that have been targeted for therapy. Table 1 shows the clinical trials targeting different components of the TME.
TABLE 1.
Clinical trials conducted to target components of the TME.
| Inhibitor | Functional Mechanism | Cancer Type/Stage | Clinical ID/Phase | References |
|---|---|---|---|---|
| ECM | ||||
| Fresolimumab | mAb inhibits collagen synthesis (targets TGF‐β) | Advanced malignant melanoma or renal cell carcinoma | NCT01401062NCT02581787 | [42] |
| Losartan | Anti‐hypertensive drug: inhibits collagen synthesis | Breast, pancreatic, skin, and ovarian cancer | NCT01821729 | [43, 44] |
| FN‐CH296 | Recombinant fibronectin ‐ stimulates T cells to achieve strong tumor‐inhibitory effects | Advanced cancer | Phase I | [45] |
| Vitaxin | Humanized mAb ‐ targets integrin αvβ3 | Progressive tumors with stage IV disease | Phase I | [46] |
| Cilengitide | Peptide antagonist ‐ targets the binding between integrin αvβ3 and RGD | Head and neck tumor, glioblastoma | Phase II | [42, 47] |
| huBC‐1‐mIL‐12 | Murine mAb ‐ targets extra domain B (EDB) of fibronectin | Malignant melanoma, renal cell carcinoma | NCT00625768 (Phase I) | [48] |
| L19‐IL‐2 | L19, was fused with IL‐2 ‐ targets EDB | Advanced renal cell carcinoma, metastatic melanoma | NCT01058538 (I) NCT01055522 (II) | [49, 50] |
| RO5429083 | CD44 antibody ‐ inhibits the mRNA transcription of CD44 or CD44v | Neoplasms, Myelogenous Leukemia, acute | NCT01358903NCT01641250 | [51, 52] |
| Ronespartat (SST0001) | Heparanase inhibitor | Multiple myeloma | NCT01764880 (Phase I) | [53, 54] |
| Incyclinide (CMT‐3 and COL‐3) | MMP inhibitor | Advanced carcinomas | NCT00004147NCT00003721NCT00001683NCT00020683 | ‐ |
| Immune cells | ||||
| MTP10‐HDL | Immunostimulatory muramyl tripeptide ‐ epigenetic reprogramming of the multipotent progenitor cells in the bone marrow | Mouse melanoma model | preclinical | [55] |
| 6‐diazo‐5‐oxo‐l‐norleucine (DON) | Blocks glutamine metabolism in myeloid precursor cells | 4T1 triple‐negative breast cancer model | preclinical | [56] |
| “designer” T cells (dTc, CAR‐T) against PSMA | PSA declines of 50% and 70% in 2/5 patients | Prostate cancer | phase I trial | [57] |
| CD133‐CAR‐T therapy | CART‐133 transfer for treating patients with CD133‐positive and late‐stage metastasis malignancies. | Advanced metastasis malignancies | phase I trial | [58] |
| HPV16 vaccine (ISA101) | Long immunogenic peptide antigens ‐ induce CD4+T and CD8+T cell cytotoxic activities | HPV16‐induced cancers | Phase I/II study | [59] |
| Pexidartinib (PLX3397) | CSF‐1R 1 inhibitor | Advanced solid tumors | NCT02734433 (1) | [60] |
| Giant cell tumor | NCT02371369 (3) | [61] | ||
| Melanoma | NCT02975700 (1/2) | ‐ | ||
| Pancreatic/colorectal cancer | NCT02777710 (1) | ‐ | ||
| Gastrointestinal stromal cancer | NCT03158103 (1) | [62] | ||
| Advanced solid tumors | NCT01525602 (1) | [63] | ||
| Gastric cancer | NCT03694977 (2) | ‐ | ||
| Canakinumab | Anti‐IL‐1β monoclonal antibody | Lung cancer | NCT01327846 | [64] |
| Sipuleucel‐T | Recombinant fusion protein of prostatic acid phosphatase; PA2024 linked to GM‐CSF | Prostate adenocarcinoma | NCT03686683 | ‐ |
| Cancer‐Associated Fibroblasts | ||||
| Val‐boroPro (talabostat) and Cisplatin | FAP‐targeted inhibitory small‐molecules | Colorectal cancer, melanoma | Phase II | [65, 66] |
| Crenolanib | PDGFR‐targeted inhibitor | Gastro‐intestinal stromal tumor | NCT02847429 (Phase III) | ‐ |
| Endothelial Cells and Pericytes | ||||
| Bevacizumab (Avastin) | Antibody ‐ anti‐angiogenic; targets VEGF | FDA‐approved (in clinics) | [67] | |
| Everolimus (RAD001) | Rapamycin derivative mTOR inhibitor | Renal cell carcinoma | NCT01206764 (Phase 4) | ‐ |
| Pazopanib (Votrient) | Multi‐target tyrosine kinase inhibitor | Advanced renal cell carcinoma and soft tissue sarcoma | [68, 69] | |
Tumors have a high demand for rich vasculature to keep up with the high nutrient and glucose demand for their progression. However, most tumors are poorly vascularized and hypoxic, which advances cancer progression and chemoresistance [70, 71]. Additionally, poor tumor vasculature increases interstitial fluid pressure, protecting the tumor core from cancer therapeutics via the bloodstream [72, 73]. To block neoangiogenesis and mitigate hypoxia, VEGF antagonists have been used as a line of therapy [74]. However, its success depends on other microenvironmental factors. For example, anti‐VEGF treatment is ineffective in obese mice due to increased IL‐6 and FGF‐2 expression by the adipocytes in the TME, while co‐targeting these improve the anti‐VEGF therapy response [75].
Tumor‐specific ECM varies among different cancer types. This specificity is exploited for therapies. For example, breast cancer samples have a remarkably high fibrillar collagen content, leading to a poor prognosis [76, 77]. Thus, probes targeted to tumor‐specific collagen help detect tumors and micrometastases [78]. Conjugation or recombinant fusion of therapeutic agents such as cetuximab or lumican to a collagen‐binding domain peptide can increase efficacy and safety [79, 80]. Similar to collagen, TME also displays a unique matrix of fibrin and fibronectin. Specific antibodies (such as L19) against fibronectin to improve tumor response in the clinical trials of patients with glioblastoma by localizing interleukin 2 (IL‐2) or interleukin 12 (IL‐12) in the tumor, leading to increased infiltration of cytotoxic T cells [81, 82, 83].
The abundance of TAMs in TME can be therapeutically harnessed if they can be polarized to their anti‐tumor phenotype. Histone deacetylase inhibitor, TMP195, polarizes TAMs toward an anti‐tumor phenotype and increases the efficacy of carboplatin, paclitaxel, and anti‐PD‐1 therapies [84]. Moreover, the phagocytic role of TAMs can be harnessed to concentrate cytotoxic drugs in the TME [85, 86]. Miller et al. found a reduction in liver metastases due to enhanced delivery of nano‐encapsulated platinum in the TME via TAMs in a breast cancer mouse model [87]. On a similar note, immunosuppressive myeloid cells can be differentiated/polarized for anti‐tumor phenotypes [88, 89]. For this, myeloid progenitor differentiation has been manipulated via a bone marrow homing nanoparticle therapeutically containing the immunostimulatory muramyl tripeptide. This peptide shows anti‐tumor effects by epigenetic reprograming of the multipotent progenitor cells in the bone marrow, which overcomes the immunosuppressive TME [55]. Moreover, a prodrug of 6‐diazo‐5‐oxo‐l‐norleucine (DON) that blocks glutamine metabolism in myeloid precursor cells can differentiate monocytes into anti‐tumor TAMs, leading to tumor regression in mouse models [56].
Stimulating a patient's dominant immune system can have long‐lasting effects against cancer. Targeting the immunosuppressive TGF‐β signaling in T cells by PLGA nanoparticles enhances antitumor immunity in mice. These nanoparticles bind to T cells and release a TGF‐βR1 inhibitor, SD‐208, that stimulates the potency of CD8+ Teff cells while inhibiting the inhibitory Treg cells in the TME [90]. CAR‐T cell therapy by engineering T cells to express synthetic receptors that recognize tumor‐associated antigens in a major histocompatibility complex (MHC)‐independent way has shown huge success in hematological malignancies. Although CAR‐T cell therapy in solid tumors is challenging since CAR‐T cells cannot penetrate solid tumors through vascular endothelium, prostate‐specific membrane antigen (PSMA)‐directed CAR‐T cells have shown success in clinical trials against prostate cancer [57]. CD133‐CAR‐T therapy in colorectal, pancreatic and hepatocellular carcinoma has shown anti‐metastatic potential in a phase I clinical trial [91]. Moreover, therapeutic cancer vaccines have shown promising results in cancer immunotherapy by amplifying tumor‐specific T‐cell responses. They can be categorized as cellular, viral, or molecular vaccines [92]. Viral vaccines using a heterologous prime‐boost strategy to amplify T‐cell responses have been successful in prostate cancer. Here, the delivery of a tumor antigen by a viral vector is boosted by a subsequent delivery of the same antigen by another vector [93]. Also, peptide‐based vaccines that deliver long immunogenic antigens to DCs have been shown to induce CD4+ T and CD8+ T cell cytotoxic activities and improve survival in patients with HPV‐16‐positive cervical cancer and newly diagnosed glioblastoma (NCT02455557) [59].
DCs form a nexus between the adaptive and innate immune responses. DCs present antigen to T cells along with upregulation of co‐stimulatory molecules and cytokine production. The inactivity of DCs in the TME to perform these functions hampers immune response to tumors. DC deficiency in the TME can be caused by several factors. Tumor‐derived exosomes are known to inhibit DC differentiation by releasing immunosuppressive factors such as IL‐6 and TGF‐β [94, 95]. Moreover, high lipid accumulation in DCs facilitated by tumor cells can also decrease the secretion profile and reduce antigen‐presenting capacity, and high amounts of hyaluronic acid in the ECM affect DC maturation [96]. Also, hypoxia in the TME inhibits DC maturation and function by inducing VEGF signaling caused by the binding of hypoxia‐induced VEGF to its receptors on DC membranes [97]. Combinational DC‐based therapy, such as DC‐based vaccine or granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), which stimulates DC differentiation, activation and migration, along with immune‐checkpoint blockade, has shown success in clinics [98]. Immune checkpoint blockers pembrolizumab and nivolumab are among the most frequently used blockers, along with US FDA‐approved PD‐1/PD‐L1 inhibitors [96].
CAFs are unique in expressing their cell surface markers. For example, depleting fibroblast activation protein (FAP)+ CAFs via a FAP vaccine decreases collagen density and improves chemoresistance in mouse models [99]. However, CAF depletion has also been shown to reduce infiltrating immune cells, leading to tumor progression [100]. CAF depletion in the TME leads to a shift from T helper 2 (Th2) to T helper 1 (Th1) to promote inflammation, accompanied by the up‐regulation of IL‐2 and IL‐7 and downregulation of TAMs, Tregs and MDSCs [101]. Moreover, CAF depletion using diphtheria toxin‐based immunotherapy reduced cancer growth by increasing CD8+ Teff‐cell infiltration to the TME [102]. Another approach to target CAFs is to inhibit their tumor‐promoting functions. For example, CAFs are known to activate the transcription of homeobox (HOX) transcript antisense RNA through paracrine TGF‐β1, which leads to epithelial‐mesenchymal transition and thus promotes breast cancer metastasis. Moreover, inhibiting TGF‐β1 was found to significantly inhibit CAF‐induced tumor growth and lung metastasis in an MDA‐MB‐231 orthotopic tumor transplantation nude mouse model [103]. Furthermore, inhibiting the proliferation of CAFs by co‐administering IPI‐926, which inhibits the hedgehog signaling, with the chemotherapeutic drug gemcitabine increasing tumor drug sensitivity in the mouse models of pancreatic ductal adenocarcinoma [104].
Most current therapies directed against the TME target TAMs, tumor vasculature, DCs, ECM, T cells and CAFs. As each of these cell types functions uniquely to modulate the TME, it is important to analyze them and identify critical nodes that could be targeted to inhibit TME support to tumor cells.
3. TARGETING SIGNALING IN THE TME
Tumor cells highjack and modulate various signaling pathways such as the PKC, Notch and TGF‐β signaling pathways, endoplasmic reticulum (ER) stress response, lactate and metabolic signaling, and the most recent cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) and Siglec signaling pathways [105, 106, 107, 108]. These signaling pathways are crucial in maintaining a favorable TME and developing resistance against therapies or multi‐drug resistance [109]. In this section, we review the signaling mechanisms of these pathways in different cancer types and the current status of therapies to target them.
3.1. Protein kinase C (PKC) signaling
The extent to which PKC isoform activation or inactivation affects TME components, including stroma and immune systems, determines their promotion or suppressor functions on tumor growth. PKC is a family of structurally related serine/threonine kinases that functions as the transducer of signals from a variety of molecules ranging from hormones (adrenaline, angiotensin), growth factors (insulin, epidermal growth factor), cytokines (Tumour necrosis factor α (TNF‐α), IL‐1β and IL‐6) and neurotransmitters (i.e., dopamine, endorphins) to regulate cell survival, proliferation, differentiation, apoptosis, adhesion and malignant transformation [105, 110, 111]. The binding of ligands to their receptors can activate phospholipase C, leading to the upregulation of cytosolic concentrations of the activators of PKC signaling, diacylglycerol (DAG) and Ca++ [112, 113]. The activation of PKC can upregulate several molecular pathways, including Akt, signal transducer and activator of transcription 3 (STAT3), nuclear factor‐κB (NF‐κB) and apoptotic pathways, to regulate tumorigenesis and metastasis [112]. PKC alpha, a PKC isoform, showed antitumor activity by inducing the polarization of TAMs within the TME [114]. Additionally, the protein levels of PKC alpha, beta and epsilon were found to be downregulated in cancers such as colon cancer [115, 116]. PKC theta, another isoform of PKC, showed tumor‐suppressive effects by inducing immune suppression within the TME by controlling CTLA4‐mediated regulatory T‐cell function [117, 118, 119]. Conversely, phorbol esters, the naturally occurring activating ligands of PKC, showed tumor‐promoting functions, suggesting that PKC could be an oncogene [111, 120, 121]. Moreover, within the TME, PKC beta, another PKC isoform, is a well‐documented effector of the VEGF signaling that promotes angiogenesis and is required for invasiveness in certain tumors, such as pancreatic tumors [122, 123, 124]. Therefore, it is likely that different isoforms of PKCs or the same isoform within different contexts may act as tumor promotors or tumor suppressors in a context‐dependent manner.
Nonetheless, combining existing therapies with novel molecules to modulate the dysregulated PKC signaling in cancer could be promising. Bryostatins, which are PKC activators, were shown to protect against phorbol ester‐induced tumors [125]. Epoxytiglianes, another class of PKC activators, showed efficacy in preclinical mouse models and clinical mast cell tumors in canine models [126, 127, 128]. Another activator of PKC, tigilanol tiglate, has been approved for use in canine mast cell tumors [129]. Besides activators, CGP 41251, an inhibitor of PKC, has also shown anti‐tumor activity and was found to reverse multidrug resistance when combined with adriamycin [130].
Although preclinical activation has led to the identification of complex PKC functions, their translation in clinical trials is impeded by a lack of mechanistic insights and robust pathological markers. This understanding is required to clinically address the action or inaction of the isoforms and to reveal the gain or loss of function of isoforms required for optimum efficiency of therapeutic interventions.
3.2. Notch signaling
Recent evidence indicates that a distinct population within tumors can express distinct Notch ligands or paralogues, which can both activate and inhibit tumor development in different cancers since the outcome of Notch signaling is highly changeable depending on the context. For instance, the presence of Notch ligand delta‐like canonical Notch ligand 1 (DLL1) on DCs interacts with the NOTCH2 receptor on tumor cells to promote DC immunosuppressive function. However, jagged canonical Notch ligand 2 (JAG2) on DCs plays negative tumor‐promoting roles Moreover, Notch mutations have been suggested to serve as predictive biomarkers for immune checkpoint therapy in various cancers. Thus, although not yet clinically successful, an integrative analysis with newer perspectives holds the potential for clinical developments [131].
Notch signaling plays a key role in determining cell fate and regulating embryonic as well as tumor angiogenesis [132, 133]. Notch receptors are heterodimeric, single‐pass transmembrane receptors that interact with either one of their membrane‐bound ligands (Jagged1, Jagged2, and Delta‐like ligands Dll1, Dll3 and Dll4) to stimulate the expression of target genes [133]. In solid tumors’ TME, the activation of Notch signaling generally promotes oncogenesis [134]. For example, Notch ligand Dll4 is upregulated in tumor samples from clear cell renal cell carcinoma patients [135], and inhibiting Dll4 leads to the disruption of tumor vasculature within the TME [136, 137]. However, Notch signaling functions as a tumor suppressor in the malignancies of myeloid origin, such as acute myelogenous leukemia and chronic myeloid leukemia [138, 139]. Moreover, a chronic blockade of Notch signaling leads to vascular tumors of the liver, skin, ovary, testes and colon in mice [140, 141].
Demcizumab and enoticumab (Dll4 targeting antibodies) alone or in combination with existing antitumor drugs, and Rovalpituzumab (anti‐Dll3 targeting antibody) work by suppressing cancer stem cells and angiogenesis in the TME and have progressed to randomized phase II trials [142, 143, 144]. Brontictuzumab (anti‐Notch1 receptor antibody) and Tarextumab (anti‐Notch2/3 receptor antibody) were tested in relapsed or refractory tumors, small‐cell lung cancer and pancreatic ductal adenocarcinoma, but clinical trials were discontinued owing to a low response rate in patients [145, 146, 147]. MEDI3622 (anti‐TACE antibody) showed promising preclinical activity in human colorectal adenocarcinoma progression [148, 149]. Inhibiting γ‐secretase using small molecules like PF‐03084014 and BMS‐906024 has shown promising results in triple‐negative breast cancer, desmoid tumors and pancreatic ductal adenocarcinoma, and related phase II and III trials are currently underway [150, 151, 152, 153]. Small molecule and peptide inhibitors of the intracellular domain of the Notch receptor (NICD)‐transcriptional complex assembly have been developed, and phase I/IIa clinical trials are being conducted on one such inhibitor, CB‐103 (NCT03422679). Other categories of Notch inhibitors, such as IMR‐1 and PRI‐724, have been found to be effective in inhibiting the Notch‐transcriptional complex in in vitro model of triple‐negative breast cancer [154].
Although extensively studied in the past several decades, Notch signaling therapeutics have failed clinical expectations. The major shortcomings are high cytotoxicity, shown by pan‐NOTCH inhibitors, and low affinity of antibody‐drug conjugates (ADC). To combat these issues, it was suggested to investigate novel isoform‐specific drugs with high ADC affinity. Moreover, combination therapies targeting chemoresistance, endocrine resistance and radio‐resistance also hold promise.
3.3. TGF‐β signaling
TGF‐β therapeutics have recently shown great promise with the use of TGF‐β‐neutralizing antibodies and ligand traps, which inhibit the binding of TGF‐β with its receptors [155]. Moreover, dosing strategies to bypass cellular toxicity or specifically target TGF‐β isoforms that are maximally linked to cancer progression hold promise in clinics to avoid the cytotoxicity of TGF‐β inhibitors [155].
Transforming growth factor‐β (TGF‐β) is a family of cytokines that intricately regulate embryonic development, tissue homeostasis and regeneration [156]. Moreover, they regulate various aspects of cancer cells, such as adhesion, differentiation, cell cycle progression and apoptosis [157, 158]. TGF‐β signaling plays a biphasic role in TME and cancer progression. It acts as a tumor suppressor in the initial stages of malignancies by suppressing cell proliferation and inducing apoptosis [159]. However, cancer cells adapt to the protective TGF‐β signaling and utilize its moonlighting functions to create a conducive TME by activating CAFs, promoting angiogenesis and ECM production, and suppressing anti‐tumor immune responses [160, 161, 162].
Various strategies have been adopted to target the deregulated TGF‐β signaling, including neutralizing antibodies to target either ligands or receptors, ligand traps, small‐molecule inhibitors, and antisense oligonucleotides (ASOs). Fresolimumab, a human monoclonal antibody against TGF‐β in a phase 1 clinical trial (NCT00356460), demonstrated preliminary evidence of anti‐tumor activity in malignant melanoma and renal cell carcinoma [42]. LY3022859, an antibody against TGF‐β receptor 2, showed survival benefits in mouse models [163]. TGF‐β ligand traps are chimeric fusion proteins designed to prevent TGF‐β from binding to its receptors. AVID200, a ligand trap for TGF‐β1 and TGF‐β3, showed anti‐tumor activity in mouse models [164] and feasibility in clinics in patients with advanced solid‐state tumors in a phase 1 clinical trial (NCT03834662) [165]. Moreover, various small molecule inhibitors of TGF‐β, including galunisertib (LY2157299) [166, 167, 168], vactosertib (TEW‐7197) [169, 170, 171] and LY3200882 [172], have shown specific antitumor activity. Lastly, phase 1 clinical studies with trabedersen (AP12009), Lucanix (belagenpumatucel‐L), or with ASOs against TGF‐β2 mRNA, showed better survival in glioblastoma, melanoma, pancreatic cancer or colorectal cancer patients [173, 174].
The pleiotropic effects of TGF‐β in tissue homeostasis have rendered exploiting their pro‐tumorigenic properties difficult, as targeting TGF‐β can systemically affect healthy tissues and tumor cells, thus safety concerns. Thus, a better understanding of the molecular mechanisms of TGF‐β in their regulation of normal and cancerous cells is required. Moreover, stratifying patients based on biomarkers who may benefit from TGF‐β targeting is essential.
3.4. ER stress response pathways
The presence of hypoxia [175, 176], reduced nutrient availability [177, 178], accumulation of reactive oxygen species [179, 180] and a decreased pH within the TME [180, 181] contribute to a chronic upregulation of ER stress in the TME, impacting the fate and survival of cancer cells. Additionally, oncogenic transformation contributes to the constitutive activation of ER stress sensors such as inositol‐requiring enzyme 1α (IRE1α), leading to persistent activation of ER stress by cancer cells [182]. Such chronic activation of ER stress modulates the TME by reducing the surface expression of major histocompatibility complex class 1 and 2 proteins, thereby impeding immune recognition of the cancer cells by NK cells [183, 184]. Therefore, reducing the ER stress load could be utilized to target TME for more favorable treatment outcomes.
Therapies modulating ER stress response pathways by inhibiting IRE1α, PRKR‐like ER kinase (PERK) and molecular chaperone binding‐immunoglobulin protein (BiP) have shown success in various preclinical and clinical studies. IRE1α inhibitors KIRA8 or AMG‐18, STF083010, MKC8866 and MKC3946 reduced tumor growth in various cancers, including multiple myeloma [185, 186, 187], melanoma [188], breast cancer and prostate cancer [189]. Similarly, PERK inhibitors GSK2606414 and GSK2656157 possess antitumor activity and can reactivate T cell function in the TME in mouse embryonic fibroblasts; however, these compounds had adverse effects in mouse models, hindering their progress toward clinical trials [190, 191, 192, 193]. Further, suppression of BiP signaling by KP1339 (also known as IT‐139) or HA15 in glioblastoma, bladder or breast cancer cell lines has been shown to enhance the response to anti‐cancer therapy [194, 195].
3.5. Modulation of the TME by lactate bioavailability
Targeting lactate transporters, such as solute carrier family 16 member 1 (SLC16A1) and member 7 (SLC16A7), to reduce lactate levels in tumors has shown huge preclinical success. Another approach to inhibit the conversion of pyruvate to lactate through targeting lactate dehydrogenase A (LDHA) is useful in inhibiting the oncometabolic lactase functions and revoking T cell‐ and NK cell‐mediated immunosuppression in various cancers [108].
Due to elevated glucose uptake rates, cancer tissues have high levels of metabolic by‐products like lactate that can be partly attributed to the accelerated metabolism of cancer stem cells and other constituents of the TME [2, 196]. Recent reports suggest the role of proton‐coupled lactate efflux in maintaining an acidic phenotype in the TME, thereby promoting angiogenesis [197, 198], cell invasion [198] and metastasis [199, 200, 201, 202]. Additionally, high lactate concentration in the TME inhibits the maturation of monocytes to dendritic cells [203, 204] and reduces cytokine production and cytotoxic activity by T cells and NK cells [205, 206, 207], thereby contributing to suppressed immune recognition of cancer cells.
Suppression of lactate efflux by cancer cells using the small molecule inhibitor AZ3965, α‐cyano‐4‐hydroxycinnamate (CHC), has shown promising results in preclinical models of Burkitt lymphoma, breast cancer, gastric cancer, small cell lung cancer and glioblastoma [208, 209]. Moreover, pharmacological inhibition of lactate dehydrogenase‐A, a key gene involved in lactate synthesis using N‐hydroxyindoles and galloflavin or genetic ablation of LDHA, reduced tumorigenesis in non‐small cell lung cancer[210], pancreatic ductal adenocarcinoma [211, 212] and cervical cancer cells [212, 213].
Despite understanding the pathogenic roles of lactate, how its targeting affects host anti‐tumor immunity and synergizes with other anti‐cancer immune therapies still needs to be explored since lactate also partly modulates the metabolism in the TME as an energy source, signaling molecule, and as a key tumor immunosuppressive factor [202]. Moreover, identifying more potent lactate transporter and LDHA inhibitors warrants improving anti‐cancer therapies.
3.6. Metabolic reprogramming of the TME
Metabolic reprogramming is an essential hallmark of cancer characterized by the ability of cancer cells to reprogram the metabolism of non‐cancerous cells, specifically immune cells, to go against their nature, help in tumor progression, and better adapt to the limited availability of nutrients in the TME [214, 215, 216]. This reprogramming, via regulating metabolic enzymes’ activities, helps enrich the TME with nutrients and plays a causal role in tumor progression [217, 218].
The metabolic niche of the TME is regulated by four key factors: 1) intrinsic metabolism of the tumor cells, 2) tumor‐non‐tumor cell interaction, 3) location and heterogeneity of the tumor, and 4) metabolic homeostasis of the body [219]. Regarding intrinsic metabolism, several studies have demonstrated that tumor cells drive aerobic glycolysis and promote cell proliferation via fueling mitochondrial metabolism [220, 221]. Moreover, apart from glucose and lactate, tumor cells use fatty acids, proteins and amino acids as fuel [222, 223]. For example, glutamate is converted into aspartate in cells with a dysfunctional electron transport chain to promote proliferation, allowing tumor cells to adapt rapidly to the substrates available in their TME niche [224, 225].
T cells provide a natural defense against cancer cells as they can specifically kill tumor cells by recognizing tumor‐specific antigens. However, glycolytic tumors have very low T cell infiltration and proliferation as activated T cells and tumor cells compete for glucose in the TME [226]. Limiting glucose levels leads to cellular competition; this impairs T‐cell function via decreased mTOR signaling. Reduction in mTOR activity diminishes IFNγ transcripts in T cells, mitigating Th1 CD4+ T cell differentiation [227, 228, 229]. Furthermore, tumor cells compete with T cells for amino acids [230]. For example, glutamine is required for T‐cell function and differentiation, and tumor cells use glutamine to activate STAT3 to promote cell proliferation [231].
The polarization of TAMs towards either the M1 (antitumoral) or M2 (protumoral) phenotype is dictated by cellular metabolism and thus regulates its response to tumor cells. Increased glycolysis by tumor cells leads to the formation of TAMs with low glycolytic potential, which promotes metastasis [232]. Moreover, tumor cells produce lactate which induces the M2 phenotype of TAMs via stabilizing HIF‐1α and activating G‐protein‐coupled receptor 132 [210, 233, 234]. Glutamine metabolism in TAMs also promotes an M2 phenotype via the production of α‐ketoglutarate, which aids in fatty acid oxidation and epigenetic activation of M2 genes [235].
Tumor location plays a key role in TME modulation because different organs and tissues have different proteomic and metabolic signatures. These differences determine the metabolite dependencies of tumor cells. Moreover, the perfusion level, tissue function and cell‐type composition within the same organ also contribute to metabolic heterogeneity. For example, blood vessel proximity distinctly defines metabolic niches. Additionally, a significant correlation has been found between glycolysis, mitochondrial metabolism and local oxygen concentrations in a study conducted on human melanoma and head and neck cancers [236]. Glucose tracing studies stipulate that tumor cells in highly perfused locations rely mainly on glucose, whereas those in less perfused regions depend on other carbon sources [237]. Furthermore, solid tumors, being metabolically heterogeneous, show glutamine‐depleted cores, which promote histone hypermethylation, resulting in the reduced expression of differentiation‐related genes and cancer cell dedifferentiation [238, 239].
Although cellular metabolism and its role in the TME are well explored, the role of systemic nutrient levels in characterizing the metabolic environment of the TME is still elusive. Recent studies indicate that dietary restrictions and hormonal modulation affect local metabolism [240]. For example, dietary restriction of serine‐glycine is beneficial in tumors lacking p53 because of their inability to counteract reactive oxygen species (ROS)‐associated oxidative stress [241, 242]. Moreover, the gut microbiome also produces specific microbial metabolites that can be altered by dietary restrictions, thus affecting tumor cell metabolism [243].
Metabolic enzymes were previously considered to catalyze their specific reactions, and their roles were strictly limited to regulating metabolic pathways. However, research in the past decades indicated that these enzymes have a moonlighting function in phosphorylating various proteins that regulate many pathways ranging from cell‐cycle progression and proliferation to apoptosis, autophagy and T‐cell activation. Some of these enzymes are pyruvate kinase M2, phosphoenolpyruvate carboxykinase 1, acylglycerol kinase, hexokinase and phosphoglycerate kinase 1 [244, 245, 246, 247]. Moreover, the metabolic products of these enzymes play a crucial role in regulating gene expression [245, 248]. Interestingly, these enzymes perform their non‐canonical functions via protein‐protein interactions and regulate many central signaling pathways and functions of several organelles, such as the nucleus, ER and mitochondria [249]. Thus, unraveling the moonlighting functions of metabolic enzymes that help in tumor progression helps us to better understand the TME dynamics and can be exploited to develop better therapeutic interventions [250].
Several challenges hinder the targeting of the pro‐tumorigenic metabolic profile of the TME. Targeting the tumor cells’ proliferative profile also affects normal cell metabolism at the systemic level. Combining these drugs with other cancer hallmarks, such as immunity, could increase the therapeutic window for targeting oncometabolites. Another prevalent strategy is to inhibit enzymes mutated in cancer, such as the isocitrate dehydrogenase 1 (IDH) 1/2 mutation‐induced oncometabolite D‐2‐Hydroxyglutarate (D2HG) in glioblastoma (GBM) and acute myeloid leukemia (AML). IDH inhibitors have shown clinical success in AML and are under investigation with combination therapies in GBM. Thus, targeting tumor‐specific metabolites in the optimum therapeutic window and exploiting cancer‐specific vulnerabilities hold great potential in metabolomics [251].
3.7. cGAS‐STING signaling in the TME
Research on the cGAS‐STING signaling promoting tumor progression is emerging in the field of cancer. The Cancer Genome Atlas (TCGA) database categorizes 18 different malignant tumor types. Researchers have observed differences in the expression of essential genes in the cGAS‐STING signaling mechanism between normal and malignant tissues, along with MB21D1‐encoding cGAS, transmembrane protein 173 (TMEM‐173)‐encoding STING, TANK‐binding kinase 1 (TBK‐1) and interferon regulatory factor 3 (IRF‐3). Comprehensive research and recent evidence proved that these four genes were significantly elevated in almost all cancer models, suggesting that cGAS‐STING signal transduction might be stimulated in all cancer types [252, 253]. In some cancer models, extremely invasive tumors can ambiguously depend on and utilize the cGAS‐STING pathway to modulate tumorigenesis with significant implications for cancer therapy [254]. NF‐κB regulates cell proliferation, apoptosis and survival of normal cells, thus acting as a crucial stimulator of the inflammatory response. Additionally, NF‐κB promotes the development of inflammation, tumors and immune dysfunction [255]. Chromosomal instability induces chronic inflammatory signals by constantly activating the cGAS‐STING signaling mechanism, which downstream NF‐κB function and consecutively increases metastatic cancer cell progression [256] (Figure 1).
FIGURE 1.
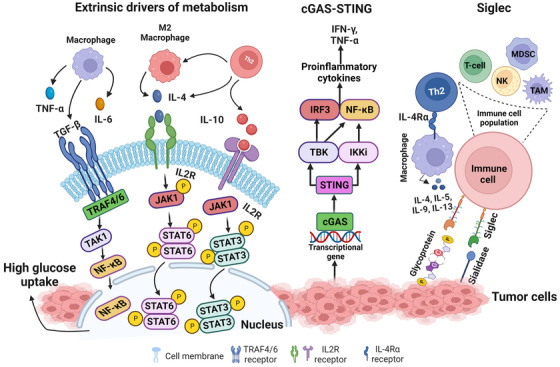
Hormones, metabolites and cytokines released by the microenvironment regulate gene expression to increase glucose uptake and glycolysis in the tumor cell. cGAS‐STING cellular signaling pathway is activated upon recognition of double‐stranded DNA in the cytosol. cGAS in turn activates the STING protein on the ER to initiate downstream signaling, primarily through TBK‐1 and IKK. STING activation typically leads to the activation of transcription factors, IRF3 and NF‐κB1, which is known to partially inhibit the activity of NF‐κB1. STING signaling results in the production of IFN‐I and TNF‐α proinflammatory cytokines. Siglec‐sialic (sialidase) axes signaling to represent siglecs on the surface of immune cells and binding with sialic on tumor cell leads to the deactivation of immune response by all the immune cell population as siglec express on most of the immune cell (e.g., T‐cell, TAM, MDSC, NK and neutrophils). Created with BioRender.com Abbreviations: cGAS: cyclic GMP–AMP synthase; STING: stimulator of interferon genes; ER: Endoplasmic Reticulum; TBK‐1: TANK‐binding kinase 1; IKK: nuclear factor‐κB (IκB) kinase; IRF3: Interferon regulatory factor 3; NF‐κB1: nuclear factor κB1; IFN‐I: Type I interferons; TNF‐α: Tumour necrosis factor α; TAM: Tumor‐Associated Macrophage; MDSC: Myeloid‐derived suppressor cell; NK: Natural Killer, TRAF: Tumor‐necrosis factor Receptor‐Associated Factor, TAK: TGF‐β‐activated kinase.
Moreover, TCGA dataset analysis revealed that the STING expression level in cancer is negatively correlated with infiltrating immune cells in various tumor models, demonstrating that significant upregulation of the cGAS‐STING signaling mechanism predicts a poor prognosis in cancer patients [252]. It has been shown that various tumor cells can specifically advance the accumulation of astrocyte‐gap junctions to enhance brain metastasis by expressing protocadherin 7 (PCDH7), composed of connexin 43 (Cx43)[257]. These junction carriers pass to the cGAMP, from cancer cells to adjacent astrocytes, to stimulate STING by triggering IRF‐3 and TBK‐1 to generate TNF and IFN‐α. Similarly, as paracrine signals, these factors further activate the NF‐κB and STAT‐1 pathway in metastatic brain cells, thus promoting brain metastasis and resistance in lung and breast cancer therapy [257].
3.8. Siglec signaling in the TME
Tumor cells express an abnormal quantity of sialic acids on their cell surface [258]. Most sialic acids belong to negatively charged disaccharides masking the chains of glycan on glycoproteins and glycolipids [258, 259] and are known as sialoglycans. On tumor cells, sialoglycans are involved in tumor cell‐to‐cell interactions within the TME, and it was also suggested that sialoglycans with negatively charged moieties could form an invisible cover by protecting tumor cells from immune recognition [260]. A vaccination trial with autologous sialidase treatment in human breast and melanoma cancer revealed that the elimination of sialic acid elicited a robust antitumor and an immune response [261]. Studies over the past decades have suggested a clear function of sialoglycans in tumors via immune evasion, not only by shielding of antigens but also by sialic acids show robust immunomodulatory properties as sugar moieties [260, 262, 263]. Sialoglycans form different ligands for sialic acid binding proteins, i.e., Factor H (FH), to evade complemented activation and are proposed as a mediator of the selectin‐independent adhesion of lymphocyte trafficking, plays an essential role in metastasis [258, 264, 265]. Sialic acid‐binding immunoglobulin‐like lectins (Siglecs) have specific interactions with the immunomodulatory properties of sialoglycans. In mammals, siglecs are divided into two groups: the structurally preserved siglecs‐ (1, 2, 4 to 15) and the CD33‐related siglecs (3, 5 to 11, 14 to 16) [266, 267]. Tumor cells with abnormal expression of sialic acid upregulate siglec family expression to infiltrate immune cells in the TME. The function of inhibitory siglecs closely resembles the PD‐1 immune checkpoint function (Figure 1) [266, 267]. TME also promotes abnormal sialylation in tumor cells and modulates the expression of siglec on infiltrating immune cells [268]. Future studies could explore the approach to target the dysregulation of the sialoglycan‐siglec axis in cancer, which could contribute to shaping the immunosuppressive TME, composing an obstacle to overcome towards effective immunotherapy in cancers.
4. IMMUNOTHERAPY AND IMMUNOSUPPRESSIVE SIGNALING IN TME
4.1. Checkpoint signaling mechanisms
Several inhibitory immune receptors have been identified and studied intensively in the TME immune population, such as PD‐1, CTLA4, TIM‐3, LAG3 and B and T lymphocyte attenuator (BTLA), and are termed “immune checkpoints”, which essentially indicate a molecule that acts as a guard of immune responses against tumors or pathogens. The immuno‐suppressive functions of the immune checkpoints usually rely upon ligand‐receptor interaction. Recent studies using advanced technologies, like mass cytometry (CyTOF) and single‐cell RNA‐sequencing (scRNA‐seq), followed by functional studies, have shown a dynamic and diverse immune landscape that facilitates the understanding of tumor heterogeneity in various cancers, including various tumor stages and genetic backgrounds. In the TME, exhausted T cells exhibit reduced effector function and increased expression of immune checkpoints (e.g., PD‐1, CTLA4, TIM‐3, LAG3, BTLA) [269]. In this section, we summarize several well‐researched immune checkpoint receptor signaling mechanisms.
4.1.1. Programmed cell death protein 1 (PD‐1) signaling
Some tumors express PD‐L1, which contributes to immune evasion by inhibiting cytotoxic responses. Thus, anti‐PD‐1/PD‐L1 antibodies can promote T cell activation and enhance anti‐tumor immunity by blocking the interaction between PD‐1 and PD‐L1/PD‐L2 [270, 271]. Typically, PD‐L1 or PD‐L2 are expressed on the surface of cancer cells or antigen‐presenting cells and transduce a signal for cooperation with PD‐1 expression on the cell surface of T lymphocytes to stimulate restraint signaling [272, 273]. Tumor cells secrete extracellular vesicles (EVs) having membrane‐bound PD‐L1, mostly to upregulate the PD‐1 mechanism, thus decreasing T lymphocytes [274, 275]. Additionally, PD‐L1 can interact in cis with Cluster of differentiation 80 (CD80) to PD‐1 [276, 277, 278], which may disturb the PD‐L1 and PD‐1 interaction and the CTLA4 and CD80 interaction and also maintain the flexibility of CD80 to trigger the signaling of CD28 [277, 279]. Consequently, the cis PD‐L1 and CD80 interaction influences antitumoral immunity by abolishing PD‐1 and CTLA4 functions. In contrast, ligand involvement with PD‐1 causes a mutation in immunoreceptor tyrosine‐based switch motif (ITSM) in cytotoxic T cells, which significantly abolishes tumor growth in the Non‐small cell lung cancer (NSCLC) model [280, 281]. Phosphorylated immunoreceptor tyrosine‐based switch motif (p‐ITSM) primarily recruits Src‐homology‐region‐2‐containing protein tyrosine phosphatase‐2 (SHP2) to dephosphorylate key signaling molecules to downmodulate PD‐1 activity levels [282, 283]. Even though SHP2 is important for inhibitory signaling of PD‐1 in most cases, T lymphocyte SHP2 deficiency can still be detrimental by reacting to the treatment of anti‐PD‐1 antibodies in vitro, thus revealing an alternate signaling pathway [284, 285]. Reports also showed that phosphorylated PD‐1 ITSM could recruit SHP‐1 to play a role in the T‐cell inhibitory mechanism [286]. Previous studies showed quantitatively applied mass spectrometry of PD‐1 signalosome assembly in primary effector T cells and confirmed that PD‐1 mainly recruited SHP2 [282, 287, 288]. Deep immune profiling of immune checkpoint inhibitor (ICI)‐refractory and responsive in mouse models using Cytometry by the time of flight (CyTOF) showed that ICI‐refractory in glioma tumors was associated with accumulating PD‐L1+ TAMs and lack of MHC‐II+ antigen‐presenting cells [289]. It is important to note that multiple TAM subpopulations likely drive the immune evasion of GBM. In addition to PD‐L1+ TAMs, CyTOF and scRNA‐seq analyses revealed that CD73high macrophages are immunosuppressive cells and have a signature distinct from microglia that persist after anti‐PD‐1 treatment [269, 290, 291]. A transcriptional study of the PD‐1‐modulated activation on individual populations of T cells confirmed that PD‐1 signaling largely inhibits transcriptional genes generated by the powerful T cell receptor (TCR) signaling pathway [292, 293]. Emerging evidence leveraged translationally to improve anti‐PD‐L1 therapy, and future studies can minimize it by avoiding the resistance and using combination therapy, for example, through companion biomarkers and/or identifying novel targets that could be modulated to overcome resistance.
4.1.2. Cytotoxic T‐lymphocyte associated protein 4 (CTLA4) signaling
New studies suggest that CTLA4 blockade within the TME could decrease the activation threshold of T cells while selectively depleting immunosuppressive regulatory T cells, which can increase the number of tumor‐specific CD8+ T cells [294]. CTLA4 binds to CD80 or CD86 with higher binding affinity and impedes CD28 co‐stimulation [295, 296]. T cells expressing CTLA4 on the surface can decrease CD80 and CD86 expressions on APCs by undergoing trans‐endocytosis and inhibiting CD28 signaling [297]. It is commonly known that the CTLA4 cytoplasmic domain recruits SHP2 and contains YVKM motifs, which are considered to recruit SHP2 [298]. Some studies reported that there might be phosphotyrosine‐independent cooperation among SHP2 and CTLA4 as alternated tyrosine CTLA4 could interact with SHP2 and inhibit TCRζ signaling pathways [299, 300]. Evidence also confirms no immediate interaction between CTLA4 and SHP2. However, it is perhaps arbitrated through the PI3K protein [301]. Further studies have suggested a disturbing action of CTLA4 on ZAP70. Micro‐clusters are established when the ligands bind to TCR, and ZAP‐70 reacts and arbitrates on the downstream pathway. CTLA4 disrupted the ZAP70 cluster formation in most T cells, although 5%‐10% of T cells demonstrated cluster forming followed by anti‐CD3 treatment [302, 303, 304]. A study reported almost no effect on ZAP70. Instead, the activation of T cells was associated with impaired c‐Jun N‐terminal kinase (JNK) and extracellular signal‐regulated kinases (ERK) signaling activation [305, 306]. Consequently, the inhibition of CTLA4 could be achieved without the degradation of CTLA4 and adverse events caused by toxicity. Exploring CTLA4's inhibition in combination with other checkpoint inhibitors, such as anti‐PD‐1 and anti‐PD‐L1, could improve the therapeutic efficacy compared to their single inhibition.
4.1.3. T cell immunoglobulin and mucin‐domain containing‐3 (TIM‐3) signaling
The interest in studying the ICI TIM‐3 comes amid growing efforts to boost the efficacy of ICI immunotherapy. TIM‐3 expression has a “complex biology” that negatively affects the immune system. Nevertheless, tyrosine kinase FYN and human leukocyte antigen B (HLA‐B)‐associated transcript 3 (BAT3) were suggested to inhibit the cytoplasmic tail of TIM‐3 signaling [307, 308, 309]. It is hypothesized that when TIM‐3 is unbound, BAT3 is confined to bind TIM‐3 cytoplasmic motif and engage the intermediary form of lymphocyte‐specific protein tyrosine kinase (LCK). In this condition, T cell activity was not inhibited. The binding state of TIM‐3 with ligands activates the phosphorylation of Tyr256 and Tyr263 tyrosine residues by IL‐2‐inducible T cell kinase (ITK), followed by ligand binding, releasing BAT3 into the cytoplasm [310, 311]. Release of BAT3 suggests that FYN tyrosine‐protein kinase links with the TIM‐3 cytoplasmic tail to generate inhibitory signaling that causes anergy of T cells by activating the transmembrane protein phosphoprotein membrane anchor with glycosphingolipid microdomains 1 (PAG1). This prompts the recruitment of tyrosine kinase (CSK), leading to phosphorylating LCK and suppressing T cells [308, 312]. Another inhibitory signaling mechanism discussed is the colocalization of TIM‐3 with CD45 and CD148 at the immunological synapse, where T cell function was suppressed [313]. The latest study showed that the binding for a short term of the extracellular motif of TIM‐3 amidst phosphatidylserine (PS) leads to the activation of TCR signaling. Another data revealed that the blockade activity of the TIM‐3 or galectin 9 axis is due to the action of galectin 3 by clustering TIM‐3 to inhibit the binding to PS [314]. Nevertheless, the binding of TIM‐3 to PS in NK cells abrogated the activity and all‐inclusive cytokines production [315]. It is known that TIM‐3 action is based on different ligand interactions, and investigative efforts are focusing on pairing novel agents directed at TIM‐3 activity with PD‐1/PD‐L1 immune ICI therapy [316, 317].
4.1.4. Lymphocyte activation gene 3 (LAG3) signaling
LAG3 interacts with its ligands to regulate the function of T cells. The ligands of LAG3 are not only limited to MHC II as well as other ligands like galectin 3 (Gal‐3), α‐synuclein fibrils (α‐syn), lymph node sinusoidal endothelial cell C‐type lectin (L‐SECtin) and fibrinogen‐like protein 1 (FGL‐1) [318, 319, 320]. In TCR signaling engagement, the LAG3 cytoplasmic tail arbitrates the inhibitory signaling through 3 conserved motifs, glutamate proline di‐peptide multiple repeats (EP), a KIEELE, and a serine phosphorylation (S484) motif. It is known that the inhibiting action of LAG3 is not directly contemplated by eradicating the interaction between MHC‐II and CD4 [321]. Some confirmed data showed that inhibitory signaling is triggered when LAG3 and CD‐3 interlink. When LAG3 physically interplays with the complex of CD3 or TCR, it decreases the stimulative alteration of the complex. In addition, besides this, it also abolishes the activity of calcium influx. The essential role of LAG3 is to reduce the T‐cell counter, but it does not necessarily induce any apoptosis in T cells [322, 323]. It is already illustrated that the specific KIEELE motif is a vital sequence necessary for blocking the signaling pathway. Notably, a single lysine residue (Lys468) exerts an inhibitory effect in CD4+ T lymphocytes at 468 motifs within the KIEELE [324]. However, LAG3 can positively induce Treg cell activation and stimulate its immunosuppressive function [325, 326]. LAG3 may synergize with other inhibitory molecules (PD‐1, CTLA4) to improve the inhibitory activity of Treg cells, leading to APC‐induced immune tolerance [327]. LAG3 can activate the maturation and stimulation of DCs through the regulation of intracellular phosphorylation protein and promote chemokines like tumor necrosis factor α (TNF‐α) [328]. LAG3, highly expressed on the tumor‐infiltrating lymphocytes (TILs), interacts with ligands located on the surface of tumor cells to cause T cell dysfunction or even exhaustion, promoting tumor immune escape, particularly evident in CD8+ T cells [329, 330].
4.1.5. B‐ and T‐lymphocyte attenuator (BTLA) signaling
BTLA is the essential co‐signaling immune checkpoint protein with bidirectional functions [331]. BTLA contains ITIM and ITSM motifs and stimulates the inhibition of Grb2 protein in its cytoplasmic domain. Due to adaptor signaling, Grb2 recruits the p85 protein subunit of the PI3K or Akt signaling mechanism, inducing the proliferation of B and T lymphocytes [332]. ITIM and ITSM, both tyrosine residues, are phosphorylated in BTLA signaling to recruit SHP1 and SHP2, which inhibit lymphocyte functional signaling. BTLA compels the strongest phosphatase SHP1 to abrogate the CD28 and TCR signaling mechanisms. In contrast, PD‐1 signaling recruits the weakened phosphatase SHP2 [282]. In addition, BTLA and herpesvirus entry mediator (HVEM) shares ligand on the same T lymphocytes (CD8+), and the same cell interaction happens, which suppresses T cell function [333]. On the other hand, on APCs, when HVEM is attached to BTLA on T lymphocytes, it invigorates downstream NF‐κB signaling, which leads to APC maturation [334]. The unique interaction between HVEM and BTLA allows for bidirectional signaling, which elucidates an understanding of the opposite or dual roles of HVEM and BTLA and the approach for their specific targeting in treatment.
4.2. Immunotherapies targeting immune checkpoint signaling
In recent years, the TME has appeared as a promising approach for targeting several cancers. Here we discuss recent research targeting immune checkpoint signaling by blockade antibodies, ICIs, or various tumor‐suppressive blocking peptides, nanoparticles, and CAR‐T cell therapy to target the receptor‐ligand intercommunication and attenuate T cell function to kill tumor cell progression. Many studies or clinical trials have conquered and are authorized for clinical translation [335]. Nonetheless, the overall survival response for these immunotherapies is not enough and needs a more comprehensive study [335]. Blocking antibodies PD‐1 or PD‐L1 is the most widely used in various cancer models as immunotherapy. T cell‐targeted immune modulators are used as monotherapy or combination treatment with chemotherapies for almost all cancer types [336, 337]. Active clinical trials on different immune checkpoint proteins in several cancer models are shown in Figure 2 and Table 2. Recently small molecules have been studied and shown to have the potential to emerge into immune checkpoint inhibitors with nanomolar binding affinity to PD‐L1. Conceivable data revealed abolished PD‐1 and PD‐L1 interactions, improved T‐cell activity, and enhanced antitumor immunity in colon, breast, pancreatic, and kidney cancer models [338, 339, 340, 341]. Recently, glioblastoma mouse models revealed the inhibition of intratumoral immune‐suppressive microglia or macrophages into the TME and increased CD8+ T cell infiltration, activation, and cytotoxicity into the tumor tissue and synergizing with anti‐PD‐1 combination therapy [342, 343]. In contrast, to intensify the T cells’ function and minimize the adverse effects of using CTLA4 immunotherapies, ICI treatment using combinations of anti‐PD‐1 (nivolumab) and anti‐CTLA (ipilimumab) have shown to boost CD8+ T cell activation, proliferation, enhance the Teff cell memory (CD8+) and produce interferon‐γ and granzyme‐B in malignant pleural mesothelioma (MPM) patients [344]. Recently, the US FDA‐approved PD‐L1 inhibitors atezolizumab and durvalumab were combined with carboplatin plus etoposide (CP/ET) to target and kill aggressively growing tumors in advanced SCLC patients and reported an overall increase in survival [345, 346]. Recent data from several clinical trials are encouraging combination therapy with a TIM‐3 directed antibody (TSR‐022) and PD‐1 together with chemotherapy for patients with NSCLC [347]. The researcher explained that the combination regimen was well tolerated across multiple dosing levels and that the responses showed the strategy is worth pursuing. Anti‐LAG3 monoclonal antibody relatlimab was also evaluated in several advanced solid tumors such as head and neck squamous cell carcinomas (HNSCCs), melanoma, NSCLC, renal cell carcinoma and bladder cancer, and the trial evaluated the efficacy of relatlimab, mono‐immunotherapy or in a combination regimen with an anti‐PD‐1 antibody nivolumab [348]. LAG3 may synergize with other inhibitory molecules (PD‐1, CTLA4) to improve the inhibitory activity of Treg cells, leading to APC‐induced immune tolerance [327]. Several ongoing clinical trials investigating the blockade of TIM‐3 inhibitor (sabatolimab) with or without PD‐1 inhibitors in advanced solid tumors concluded that the doses for combination therapy of sabatolimab and spartalizumab were under tolerance and showed anti‐cancer activity with survival benefits [349]. Moreover, CAR‐T cell therapy is emerging to be a progressive new pillar in immune cell therapy for cancer, which has yielded remarkable clinical responses in patients with B‐cell leukemia. However, many challenges remain to be addressed to overcome its ineffectiveness in treating solid tumors and hematological malignancies [350]. The great potential of CAR‐T cell therapy at the beginning or earlier during the treatment course was unraveled, and the strategy revealed higher success rates and reduced toxicity associated with anticancer treatments [351]. Early administration of the therapy may also provide access to a higher proportion of naïve unexposed T‐cell population, which is beneficial to facilitate the production of CAR‐T cells. Finally, immunotherapeutic development of targeting immune checkpoint signaling molecules has promptly increased over the decade. The development of new biomarkers or identification of new targeting mechanisms with combinational strategies and novel nano‐drug delivery strategies could significantly boost immunotherapy benefits in an effort to improve the quality of life for cancer patients by enhancing overall survival and eventually eliminate cancer.
FIGURE 2.
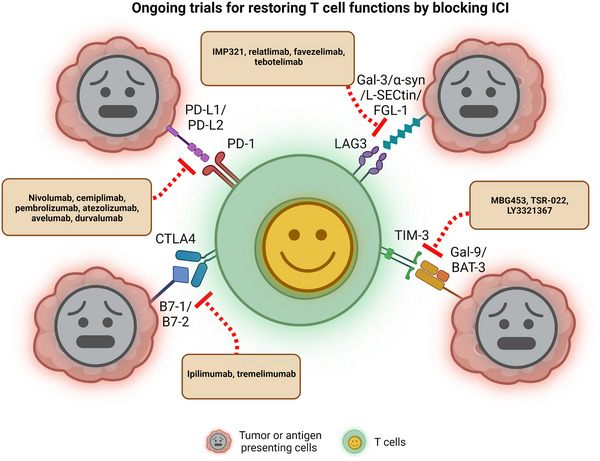
Blockade of ICI restores T cell function. Inhibition of PD‐1, CTLA4, TIM‐3, and LAG3 with interaction by antibodies, drugs, and peptides. The cytotoxic signal is released when the TCR recognizes an antigen on the membrane of tumor cells or (APCs). However, PD‐L1/2, B7‐1/2, Gal‐9/BAT‐3 and Gal‐3/α‐syn/L‐selectin/FGL‐1 are up‐regulated in tumor cells (APCs) when T cells are activated, resulting in inhibitory signals and dampening T cell activation by stimulatory signals. Clinical trials of blocking or inhibiting these interactions using antibodies or a newly designed combination, which restores T cell function and anti‐tumor immunity, are ongoing. Created with BioRender.com.Abbreviations: ICI: Immune checkpoint inhibitor; PD‐1: Programmed cell death protein 1; CTLA4: Cytotoxic T‐lymphocyte associated protein 4; TIM‐3: T‐cell immunoglobulin and mucin domain 3; LAG3: Lymphocyte Activating 3; TCR: T cell receptor; APC: Antigen‐presenting cells; PD‐L1/2: ligands of PD‐1 (PD‐L1 and PD‐L2); Gal‐9: galectin‐9; BAT‐3: HLA‐B associated transcript 3; Gal‐3: Galectin‐3; α‐syn: Alpha‐synuclein; FGL‐1: Fibrinogen like 1.
TABLE 2.
Existing anticancer signaling and immune checkpoint inhibitory molecules in clinical trials.
| Treatment | Drugs/regimen | Condition or disease | Target | Clinical Trial No. | Phase |
|---|---|---|---|---|---|
| Chemotherapy | Leflunomide | smoldering multiple myeloma | Dihydroorotate dehydrogenase | NCT05014646 | Phase 2 |
| Pemetrexed | Non‐Small Cell Lung Cancer | Glycinamide ribonucleotide formyltransferase | NCT00102804 | Phase 3 | |
| Metformin Hydrochloride | HER2‐positive breast cancer | MAP kinase, Akt, mTOR | NCT03238495 | Phase 2 | |
| AZD3965 | Burkitt Lymphoma, Diffuse Large B Cell Lymphoma, Adult Solid Tumor | Monocarboxylate transporter 1 | NCT01791595 | Phase 1 | |
| L‐asparaginase | Pancreatic Adenocarcinoma Metastatic | Asparagine synthetase | NCT02195180 | Phase 2 | |
| Methotrexate | primary CNS lymphoma | Dihydrofolate reductase | NCT04609046 | Phase 1 | |
| 5‐Fluorouracil | Metastatic Pancreatic Cancer | Thymidylate synthase | NCT02620800 | Phase 1 | |
| Hydroxyurea | Leukemia | Ribonucleotide reductase | NCT05005182 | Phase 2 | |
| 6‐Mercaptopurine | Acute Lymphoblastic Leukemia | Phosphoribosyl pyrophosphate amidotransferase | NCT01503632 | Phase 3 | |
| Enasidenib/AG‐221 | Hematologic Neoplasms | Isocitrate dehydrogenase 2 | NCT01915498 | Phase 1/2 | |
| CB‐839 | Non‐Small Cell Lung Cancer | Glutaminase | NCT04250545 | Phase 1 | |
| Immunotherapy | Pembrolizumab | Colorectal cancer | PD‐1 | NCT02563002 | Phase 3 |
| Nivolumab | Melanoma | PD‐1/PD‐L1 | NCT01721772 | Phase 3 | |
| Cemiplimab | NSCLC | PD‐1/PD‐L1 | NCT03088540 | Phase 3 | |
| Nivolumab, Ipilimumab combined with chemotherapy | NSCLC | PD‐L1 | NCT03215706 | Phase 3 | |
| TSR‐022+Nivolumab combined with chemotherapy | Advanced solid tumors | TIM‐3/PD‐1 | NCT02817633 | Phase 1 | |
| Atezolizumab + Carboplatin + Etoposide | SCLC | PD‐L1 | NCT02763579 | Phase 3 | |
| Varlilumab and nivolumab | Recurrent glioblastoma | Anti‐CD27 and anti‐PD‐1 | NCT02335918 | Phase 1/2 | |
| Atezolizumab combination Nab‐Paclitaxel | Breast cancer | PD‐L1 | NCT02425891 | Phase 3 | |
| Pembrolizumab and vorinostat combined with temozolomide | New diagnosis GBM | PD‐L1 | NCT03426891 | Phase 1 | |
| Pembrolizumab and Bevacizumab | Recurrent glioblastoma | PD‐1 and VEGF | NCT02337491 | Phase 2 | |
| Tremelimumab and durvalumab in combination or alone | Malignant Glioma, Recurrent Glioblastoma | CTLA4 and PDL1 | NCT02794883 | Phase 2 | |
| Nivolumab and ipilimumab | Mesothelioma | PD‐L1 | NCT02899299 | Phase 3 | |
| Atezolizumab + Bevacizumab | Hepatocellular Carcinoma | PD‐L1 | NCT03434379 | Phase 3 | |
| ICT‐121 DC vaccine | Recurrent gliomas | Dendritic cell Vaccine | NCT02049489 | Phase 1 | |
| MBG453 | Hematologic malignancy | TIM‐3 | NCT03066648 | Phase 1 | |
| LY3321367 | Advanced solid tumors | TIM‐3 | NCT03099109 | Phase 1 | |
| TTF(Optune), Nivolumab Plus/Minus Ipilimumab | Recurrent glioblastoma | PD‐1/PD‐L1 | NCT03430791 | Phase 2 | |
| Atezolizumab combination with Cobimetinib + Vemurafenib | Melanoma | PD‐L1/BRAF kinase | NCT02908672 | Phase 3 | |
| Nivolumab | Recurrent or progressive IDH‐mutant gliomas | PD‐1/PD‐L1 | NCT03557359 | Phase 2 | |
| IMP321 (anti‐LAG‐3) + Pembrolizumab | Melanoma | LAG‐3/PD‐1 | NCT02676869 | Phase 1 | |
| IMP321 (anti‐LAG‐3) + Paclitaxel | Breast cancer | LAG‐3/PD‐1 | NCT02614833 | Phase 2 | |
| BMS‐986016 + Nivolumab | Melanoma | LAG‐3/PD‐1 | NCT05002569 | Phase 3 | |
| Favezelimab/Pembrolizumab | Colorectal cancer | LAG‐3/PD‐1 | NCT05064059 | Phase 3 |
4.3. Immunosuppressive chemokine cell signaling into the TME
The chemokine gradient determines the composition of the TME. Pro‐tumor chemokine gradient attracts immune suppressive immune cells and excludes effector immune cells. On the other hand, the antitumor chemokine gradient favors the migration of effector immune cells. Here, we discuss key chemokine signaling axes in tumors and immune crosstalk therapeutically targeted to make the TME less immune suppressive.
4.3.1. CCR4‐CCL22/17 signaling axis in the migration of regulatory T cells into the TME
Regulatory T cells suppress immune responses against tumors and their normal function to maintain immune tolerance in physiological conditions [352]. The CCR4‐CCL17 signaling axis induces chemotaxis of CCR4+ T cells, mainly Th2 and Tregs, generating an immunosuppressive TME [353]. CCL17 acts as a ligand for CCR4 and is produced by DCs or endothelial cells [354, 355]. These observations were made in the context of gastric cancer and esophageal squamous cell carcinoma. Similarly, CCL22 is also another ligand of CCR4. In breast cancer, the CCR4‐CCL22 signaling axis was reported to induce increased migration of regulatory T cells in TME, which in turn leads to the exclusion of effector T cells in the TME [356, 357].
4.3.2. CCR2‐CCL2 axis and CCR5‐CCL5 signaling axis in the migration of TAMs into the TME
TAMs are bone marrow‐derived myeloid cells that are part of the tumor‐associated stroma and are usually reprogrammed to perform immune suppressive and pro‐tumor functions [358]. The CCR2‐CCL2 axis is implicated in the recruitment of TAMs and myeloid‐derived suppressive cells (MDSCs) in the tumor bed. These observations were made in different cancers like glioma, renal tumors, lung cancer, prostate cancer, and melanoma [359]. The tumor cells and tumor stroma are known to express CCL2, and the receptor CCR2 is exclusively expressed by myeloid cells [360, 361]. Similarly, CCL5, which is produced by epithelial cells, fibroblasts, monocytes, NK cells, DCs, endothelial cells, macrophages and lymphocytes, acts as a ligand for CCR5, preferentially expressed by Tregs, and the CCL5‐CCR5 signaling axis can induce the migration of TAMs, myeloid‐derived suppressive cells and that of regulatory T cells. Moreover, disrupting the CCL5‐CCR5 axis may inhibit their migration and reduce tumor growth [362, 363].
4.3.3. CCR5‐CCL5 and CCR6‐CCL20 signaling axis in the migration of dendritic cells into the TME
DCs are APCs that help in priming effector T cells. DCs play an important role in regulating the migration of CD8+ T cells into the TME [363]. CCL3, CCL44 and CCL55 are the ligands for CCR5 and are produced by various cell types like epithelial cells, fibroblasts, monocytes, NK cells, DCs, endothelial cells, macrophages and lymphocytes [364]. CCL3, CCL4 and CCL5 form a cytokine gradient that can recruit DCs into the TME. The recruited DCs produced CXCR3 ligands are important for migrating effector CD8 T cells to the tumor [365, 366]. Along with CCR5, the CCR6‐CCL20 axis is known to promote the migration of DCs into sites of inflammation [367]. A preclinical murine melanoma model showed increased migration of DCs in CCL20‐positive tumor cells, indicating the role of CCL20 in attracting DCs to the tumor site [368].
4.4. Therapeutics targeting chemokine signaling in the TME
4.4.1. Abrogation of the CCR4‐CL22/17 axis
The preclinical model of murine Hodgkin tumor with T cells coexpressing CCR4 and a chimeric antigen receptor targeting CD30 demonstrated an improved homing and antitumor activity [369]. Mogamulizumab (KW‐0761), a humanized, glycoengineered IgG1κ monoclonal antibody, is known to bind to CCR4‐expressing cells to induce antibody‐dependent, cell‐mediated cytotoxicity (ADCC) [370]. This antibody is in clinical trials for lung and esophageal cancer patients [371]. Small molecule inhibitors targeting the CCR4‐CCL22/CCL17 axis are also being developed [359].
4.4.2. Abrogation of the CCR2‐CCL2 axis
In preclinical models of breast cancer, anti‐CCL2 blocking antibodies inhibited infiltrating monocytes and subsequent recruitment of metastasis‐associated macrophages, thereby inhibiting metastasis and prolonging the lifespan of tumor‐bearing mice [360]. CCR2 inhibitor (PF‐04136309) demonstrated a reduction in the migration of TAMs toward tumors in the pancreatic ductal adenocarcinoma model [360]. Along the same lines, decreased inflammatory monocytes were found in the peripheral blood of pancreatic ductal adenocarcinoma patients treated with PF‐04136309 in combination with nab‐paclitaxel/gemcitabine [372].
4.4.3. Activation of the CCR5‐CCL3 signaling axis
In the preclinical study on murine melanoma models, adenoviral delivery of CCL3 along with the adoptive transfer of T cells led to significant migration of effector T cells to the tumor bed [373]. Virus‐mediated expression of CXCL10, CXCL11, or recombinant CXCL10 injection in tumors was able to increase the infiltration of anticancer T cells in mice [364, 374, 375]. This approach suggests that pairing adoptive T‐cell therapy with cytokine therapy could be beneficial for the long‐term persistence and function of effector T cells.
Cytokine signaling is important for tumor development and the recognition of tumors by immune cells. Understanding the dynamics of cytokine signaling could allow us to use cytokine signaling to effectively curb tumor growth and elicit anti‐tumor memory to sustain tumor regression.
5. 3D MODELS OF THE TME
A complex TME is driven by physical and biochemical interactions between cancer cells and the tumor stroma, constituting the rest of the TME. To understand these interactions, it is important to mimic these integrated interactions using 3D models of the TME, which closely mimic the patient‐derived heterogeneity and help understand the cellular interactions in the TME to a much deeper extent as compared to the oversimplified 2‐dimensional (2D) models.
5.1. Spheroids
Spheroids are the most widely used 3D tumor models that precisely model in vitro conditions of a TME, including cellular heterogeneity, cell‐cell interactions, cell‐ECM interactions, signaling pathways, and gene expression profiles [376, 377]. Spheroids are created by a forced aggregation of cells, and those larger than 500 μm mimic the TME of micrometastases (microscopic, aggregated cancer cells that have escaped the original tumor) and avascular tumors owing to the presence of a gradient of oxygen and nutrients [378]. Moreover, this gradient leads to the formation of concentric zones in the spheroids with a center containing an anoxic core with necrotic cells, a middle zone containing hypoxic cells, and an outer zone of highly proliferative cells, recapitulating the properties of an in vitro tumor [379].
Spheroids can be associated with microfluidic or scaffold‐based procedures to generate physiologically more relevant tumor models [377]. They are also advantageous over 2D cell culture models in studying the role of ECM stiffness on tumor growth when combined with biomaterials like hydrogel by mimicking cell‐cell/cell‐ECM interactions in the TME [380]. Spheroids can also be fine‐tuned by varying the composition of tumor cells, CAFs, and immune cells to obtain an immunosuppressive TME to study the roles of various immune cells like monocytes, NK cells, or T cells in tumors [381]. The spheroid model of human breast cancer played a key role in discovering the dependence of angiogenesis (sprout formation) on VEGF and FGF growth factors [382]. Spheroid 3D models, therefore, represent a powerful model system for studying tumor biology and drug testing. Nonetheless, there are certain limitations, including 1) poor uniformity in spheroid size or morphology, 2) low throughput, and 3) difficulties in retrieving cells for analyses, etc., in the 3D spheroid models that limit reproducibility while using these models [377].
5.2. Organoids
Organoids are small 3D self‐organized tissue cultures derived from progenitor stem cells that maintain natural cancer cell heterogeneity in vitro, including genetic and phenotypic features of the tissue from which the culture is derived [383, 384]. In contrast to the spheroids, where a forced aggregation of cancer cells forms a 3D structure, organoids follow a natural course of 3D tumor development based on the genetic programming of progenitor cells [377]. Therefore, organoids more closely resemble the in vitro development of tumors and can be cultured from patient‐derived cells from biopsy samples. Since patient‐derived organoids (PDOs) consist of only epithelial cells, to develop a TME in organoids, either of the two approaches for organoid culture can be used: reconstituted the TME (submerged Matrigel culture) model or native TME (air‐liquid interface culture) model.
5.2.1. Reconstituted the TME (submerged matrigel culture) model
In this method, dissociated tumor cells are cultured to produce PDOs in a Matrigel submerged beneath the nutrient media containing supplements depending on the tumor tissue type [383, 385, 386]. The PDOs are subsequently supplemented exogenously with stromal cells (i.e., CAFs), immune cells (i.e., cytotoxic T lymphocytes) or patient‐matched peripheral blood lymphocytes to develop an in vitro TME [387, 388]. The reconstituted TME model recapitulates not only the genetic and phenotypic features of a tumor but also models the functional response of a patient to therapeutic agents [389, 390].
5.2.2. Native TME (air‐liquid interface culture) model
Herein, to develop PDOs, the minced tumor tissue fragments are embedded in a collagen gel where the nutrient media diffuses from the bottom of the gel, and the top layer of the collagen is exposed to air for oxygen diffusion to take place [391, 392]. In contrast to the reconstituted TME model, cancer cells grow alongside the endogenous stromal and immune cells without needing reconstitution [393]. In addition to preserving the genetic and phenotypic constitution of the tumor, native TME models preserve the complex composition of the TME, including parenchyma and stroma [394].
Moreover, organoids are more advantageous due to the ease of culturing and closer resemblance to the TME, thus being more pathophysiologically relevant as compared to the patient‐derived tumor xenograft (PDX) model and other tumor‐bearing animal models, which are more tedious and expensive to maintain [384, 395]. Organoids represent early passage material retaining the genetic makeup of cancer cells compared to long‐passaged cancer cell lines that might have lost the genetic features of their original tumors owing to genomic instability [395, 396]. Moreover, the organoids derived from 2D monolayer cultures or mouse xenograft cultures differ from a primary organoid culture that retains cellular heterogeneity, tumor architecture, cell‐cell interactions, and stemness [377]. These models have been extensively used to assess a patient's response to preclinical treatments. However, the organoid model system has some limitations, including high variability and difficulty in achieving in vitro‐like maturity, lack of vasculature, an immune‐competent TME, and stroma [395]. Nevertheless, as discussed earlier, some of the limitations have been overcome by co‐culturing with other cell types like stromal cells, CAFs, or T cells [175].
5.3. Microfluidic models
Microfluidic models consist of a network of microfluidic channels where tumor spheroids can be continuously perfused and grown in the presence of fine‐tuned concentrations of growth factors or drugs [397, 398]. These models allow fine control over the mechanical forces, the orientation of the tissue interfaces, and chemical gradients, and they can be modified for high throughput screening [399, 400]. Moreover, microfluidic devices used in these models use microscale volumes, which can drastically reduce their operating costs compared to 3D TME models [401]. They have been extensively used for studying tumor‐stroma interactions and the effects of growth factors or drugs in a biomimetic TME for various cancer types, including breast cancer, colorectal cancer, melanoma, and Merkel cell carcinoma [376, 402]. Specific cell‐cell interaction can also be studied using microfluidic models, for example, in tumor cell and stromal interaction studies. Culturing organoids in the microfluidic device have helped us understand tumor‐stroma interactions and their systemic effects [403, 404]. They have also been used to mimic microvascular functions and TME modeling [175]. Limitations of the microfluidic models include the requirement of specialized skills to perform the experiments, edge effects and high shear pressure in the device can compromise the reproducibility of the experimental outcomes, and the most used material, polydimethylsiloxane (PDMS), to fabricate microfluidic devices indiscriminately absorbs small molecules [397, 398, 405, 406].
6. ROLE OF GUT MICROBIOTA IN SHAPING THE TME
The role of gut microbiota in tumorigenesis has been disputed for centuries, and its importance in tumor progression has only been recognized in the past few decades. Although only 11 microbial species are known to directly cause carcinogenesis [407], a category of microbes and their functions promote tumorigenesis but are insufficient to cause tumorigenesis on their own. These microbiotas utilize their immunomodulatory functions and metabolites to upregulate tumor progression. For example, p53 mutations in tumor cells are carcinogenic only in the presence of microbially produced gallic acid in the gut; otherwise, mutant p53 was found to be more protective than wild‐type p53 in tumor regression in mouse models of WNT‐driven intestinal cancer [408]. Moreover, Kras mutation and loss of p53 failed to promote lung tumorigenesis in antibiotic‐treated/germ‐free mice because lung microbiota could promote tumorigenesis via IL‐23 and IL‐17 production, leading to tumor‐promoting inflammation [409]. Furthermore, mounting evidence suggests the presence of intratumoral microbiota [410]. These microbes regulate tumors in several ways. They affect gastrointestinal and urinary tract mutagenesis by secreting genotoxins [411, 412, 413], promote cytotoxin‐associated gene A‐ or T cell‐mediated inflammation as shown in stomach and lung cancers, respectively [409, 414], regulate chemoresistance via direct drug metabolism by microbes [415, 416], promote tumor progression via fungal activation of the host's C3 complement system [417] and even advance metastasis by upregulating tumor MMPs or by reducing infiltrating immune cells [418, 419].
Recent studies indicate that information on gut microbiota can be channelized for novel therapeutic approaches. However, depending on various factors, the gut microbiome can lead to either tumorigenesis or tumor regression [420, 421]. For example, chemotherapy may lead to gut microbiome changes that increase drug response efficiency. The chemotherapeutic agent, cyclophosphamide, was shown to alter intestinal microbiota in mice and promote “pathogenic” Th17 cell production [422]; however, removing gut microbiota via antibiotic treatment led to drug resistance [423, 424]. Additionally, if chemotherapy was degraded by microbiota, it led to tumor progression [415]. Geller et al. showed that intratumor Gammaproteobacteria in a tumor could metabolize gemcitabine into its inactive form leading to chemoresistance in a murine model of colon cancer [415]. Furthermore, certain bacteria stimulate tumorigenesis by blocking immune effectors that normally inhibit tumorigenesis. For example, Fusobacterium nucleatum inhibits the host's NK cells from recruiting myeloid suppressor cells at the site of the infection, thus indirectly helping in cancer genesis. This is mediated by Fap2, a bacterial virulence factor, which can bind and block the NK inhibitory receptor T cell immunoreceptor with Ig and ITIM domains (TGIT), thus arresting the NK cell‐mediated tumor cell attack [425].
Furthermore, targeting microbiota for tumor regression is easy as it can be easily modified by many approaches, such as probiotics, antibiotics, and fecal microbiota transplantation (FMT) [426]. Probiotics increase anticancer immunity by decreasing Treg levels and enhancing CD4+ T cell differentiation, CD8+ T cell activation, and intratumoral infiltration of NK cells [427, 428]. Moreover, probiotics, such as L. acidophilus NCFM, increase butyrate‐producing species in the gut, leading to improved immunotherapy response [429, 430]. FMT has also been exploited for tumor suppression. For example, mice transplanted with liquefied and filtered stool from patients with bacteroides‐rich microbiota population respond better to CTLA4 inhibitor treatment [431]. Antibiotics have also been shown to have anti‐cancer properties. For example, adriamycin is used in many childhood cancer patients. Bleomycin effectively treats germ cell cancers, lymphomas, and squamous cell carcinomas [432].
Lastly, certain microbial species may interfere with the host's hormonal metabolism. When gut microbiota depletion occurs along with an increase in the β‐glucuronidase‐secreting bacteria, such as Clostridium leptum and Clostridium coccoides, due to gut dysbiosis, the enzyme deconjugates liver‐catabolized and plant‐derived estrogens and enables them to activate host's estrogen receptors [430, 433, 434]. Estrogen receptor activation promotes cell proliferation in estrogen‐responsive tissues (breast and endometrium) [435]. Thus, this sudden rise in estrogen increases the risk of developing breast cancer [436]. It further confirms that gut dysbiosis may be a risk factor for breast cancer. Thus, diet, chemotherapy, antibiotics, probiotics, and hormones all play a role in modulating the TME microbiota [437, 438].
7. CONCLUSIONS
Accumulating evidence demonstrates that stromal cells and other TME components undergo reprogramming to support tumor growth and metastasis. Transformed cancer cells invade normal, healthy cells in their vicinities, such as immune cells and fibroblasts, to counter stress, nutrient deprivation, and hypoxia. As previously described, CAFs and immune cells such as TAMs, both of which are major parts of the heterogenous tumor ecosystem, further support tumor progression by upregulating signaling pathways associated with inflammation and angiogenesis. Tumor‐derived exosomes prevent DC differentiation by the secretion of immunosuppressive molecules such as IL‐6. Furthermore, gut microbiota plays a crucial role in tumorigenesis via their immunomodulatory abilities.
Preclinical research efforts are focused on developing therapeutic strategies that disrupt the crosstalk between tumor cells and the TME, leading to better patient outcomes. 3D cultures such as spheroids and organoids have been extensively used to model tumor heterogeneity and cellular behavior. Several clinical trials are underway to assess candidates that have shown efficacy at preclinical levels, including monoclonal antibodies against proangiogenic or prosurvival proteins.
Due to the involvement of many signaling pathways and dynamic interactions between cancer cells and ECM, combinational approaches may likely show better efficacy than implementing a single strategy. Firstly, an improved understanding of metabolic and signaling pathways associated with the TME could largely help with better drug design. Secondly, exosomes may serve as powerful tools due to their role in early diagnosis and their unique signatures. At the same time, therapeutic targeting of tumor‐derived exosomes and cell‐free DNA is urgently needed. In addition, modulation of gut microbiota could be a potential strategy for tumor suppression and warrants further investigation. Taken together, the TME can be modulated by targeting these complex interactions and signaling pathways to improve the effectiveness of currently existing therapeutic strategies in treating cancers.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Anshika Goenka, Prakash Gangadaran and Byeong‐Cheol Ahn contributed to the conception, writing, and discussion of this manuscript. Anshika Goenka, Fatima Khan, Bhupender Verma, and Priyanka Sinha equally contributed and wrote the initial draft of the manuscript. Crismita C. Dmello and Manasi P. Jogalekar contributed to the initial draft of the manuscript. All authors have approved the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable
CONSENT FOR PUBLICATION
Not applicable
ACKNOWLEDGMENTS
The authors have nothing to report.
Goenka A, Khan F, Verma B, Sinha P, Dmello CC, Jogalekar MP, et al. Tumor microenvironment signaling and therapeutics in cancer progression. Cancer Commun. 2023;43:525–561. 10.1002/cac2.12416
Anshika Goenka, Fatima Khan, Bhupender Verma and Priyanka Sinha contributed equally to this work
Contributor Information
Anshika Goenka, Email: anshika.goenka@northwestern.edu.
Prakash Gangadaran, Email: prakashg@knu.ac.kr.
Byeong‐Cheol Ahn, Email: abc2000@knu.ac.kr.
DATA AVAILABILITY STATEMENT
Not applicable
REFERENCES
- 1. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. 2018;18(6):359–76. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 3. Bejarano L, Jordāo MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021;11(4):933–59. [DOI] [PubMed] [Google Scholar]
- 4. Gohil SH, Iorgulescu JB, Braun DA, Keskin DB, Livak KJ. Applying high‐dimensional single‐cell technologies to the analysis of cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(4):244–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer. 2019;19(4):215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma W, Wang Y, Zhang R, Yang F, Zhang D, Huang M, et al. Targeting PAK4 to reprogram the vascular microenvironment and improve CAR‐T immunotherapy for glioblastoma. Nature Cancer. 2021;2(1):83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ragusa S, Prat‐Luri B, González‐Loyola A, Nassiri S, Squadrito ML, Guichard A, et al. Antiangiogenic immunotherapy suppresses desmoplastic and chemoresistant intestinal tumors in mice. J Clin Invest. 2020;130(3):1199–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ayoubi‐Joshaghani MH, Dianat‐Moghadam H, Seidi K, Jahanban‐Esfahalan A, Zare P, Jahanban‐Esfahlan R. Cell‐free protein synthesis: The transition from batch reactions to minimal cells and microfluidic devices. Biotechnol Bioeng. 2020;117(4):1204–29. [DOI] [PubMed] [Google Scholar]
- 9. Nabet BY, Esfahani MS, Moding EJ, Hamilton EG, Chabon JJ, Rizvi H, et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell. 2020;183(2):363–76.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baghban R, Roshangar L, Jahanban‐Esfahlan R, Seidi K, Ebrahimi‐Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell commun signal. 2020;18(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin M‐Z, Jin W‐L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther. 2020;5(1):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu Y, Su G‐H, Ma D, Xiao Y, Shao Z‐M, Jiang Y‐Z. Technological advances in cancer immunity: from immunogenomics to single‐cell analysis and artificial intelligence. Signal Transduct Target Ther. 2021;6(1):312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Metcalf KJ, Alazzeh A, Werb Z, Weaver VM. Leveraging microenvironmental synthetic lethalities to treat cancer. J Clin Invest. 2021;131(6):e143765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang T, Song X, Xu D, Tiek D, Goenka A, Wu B, et al. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics. 2020;10(19):8721–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fuertes MB, Kacha AK, Kline J, Woo S‐R, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caronni N, Montaldo E, Mezzanzanica L, Cilenti F, Genua M, Ostuni R. Determinants, mechanisms, and functional outcomes of myeloid cell diversity in cancer. Immunol Rev. 2021;300(1):220–36. [DOI] [PubMed] [Google Scholar]
- 17. Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20(11):662–80. [DOI] [PubMed] [Google Scholar]
- 18. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza‐Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018;172(5):1022–37.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature. 2019;574(7776):45–56. [DOI] [PubMed] [Google Scholar]
- 20. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer‐dendritic cell axis defines checkpoint therapy‐responsive tumor microenvironments. Nat Med. 2018;24(8):1178–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer‐Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers. 2015;7(4):2443–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer‐associated fibroblasts: an emerging target of anti‐cancer immunotherapy. J Hematol Oncol. 2019;12(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu X, Zheng L, Yuan Q, Zhen G, Crane JL, Zhou X, et al. Transforming growth factor‐β in stem cells and tissue homeostasis. Bone Res. 2018;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte‐derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73(18):5657–68. [DOI] [PubMed] [Google Scholar]
- 25. Tang D, Gao J, Wang S, Ye N, Chong Y, Huang Y, et al. Cancer‐associated fibroblasts promote angiogenesis in gastric cancer through galectin‐1 expression. Tumour Biol. 2016;37(2):1889–99. [DOI] [PubMed] [Google Scholar]
- 26. Wang F‐T, Sun W, Zhang J‐T, Fan Y‐Z. Cancer‐associated fibroblast regulation of tumor neo‐angiogenesis as a therapeutic target in cancer. Oncol Lett. 2019;17(3):3055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer‐associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mhaidly R, Mechta‐Grigoriou F. Role of cancer‐associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev. 2021;302(1):259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Z, Yang Q, Tan Y, Tang Y, Ye J, Yuan B, et al. Cancer‐Associated Fibroblasts Suppress Cancer Development: The Other Side of the Coin. Front Cell Dev Biol. 2021;9:613534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol Lett. 2017;14(3):2611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sobierajska K, Ciszewski WM, Sacewicz‐Hofman I, Niewiarowska J. Endothelial Cells in the Tumor Microenvironment. Adv Exp Med Biol. 2020;1234:71–86. [DOI] [PubMed] [Google Scholar]
- 32. Masoud GN, Li W. HIF‐1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015;5(5):378–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia‐inducible factor‐1alpha promote growth factor‐induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ. 2006;290(6):H2528–34. [DOI] [PubMed] [Google Scholar]
- 34. Medinger M, Mross K. Clinical trials with anti‐angiogenic agents in hematological malignancies. J angiogenes res. 2010;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42. [DOI] [PubMed] [Google Scholar]
- 36. Ebos JML, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robertson C. The extracellular matrix in breast cancer predicts prognosis through composition, splicing, and crosslinking. Exp Cell Res. 2016;343(1):73–81. [DOI] [PubMed] [Google Scholar]
- 38. Insua‐Rodríguez J, Oskarsson T. The extracellular matrix in breast cancer. Adv Drug Deliv Rev. 2016;97:41–55. [DOI] [PubMed] [Google Scholar]
- 39. Singh C, Shyanti RK, Singh V, Kale RK, Mishra JPN, Singh RP. Integrin expression and glycosylation patterns regulate cell‐matrix adhesion and alter with breast cancer progression. Biochem Biophys Res Commun. 2018;499(2):374–80. [DOI] [PubMed] [Google Scholar]
- 40. Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I study of GC1008 (fresolimumab): a human anti‐transforming growth factor‐beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9(3):e90353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Diop‐Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Nat Acad Sci USA. 2011;108(7):2909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao Y, Cao J, Melamed A, Worley M, Gockley A, Jones D, et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc Nat Acad Sci USA. 2019;116(6):2210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishikawa T, Kokura S, Enoki T, Sakamoto N, Okayama T, Ideno M, et al. Phase I clinical trial of fibronectin CH296‐stimulated T cell therapy in patients with advanced cancer. PLoS One. 2014;9(1):e83786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gutheil JC, Campbell TN, Pierce PR, Watkins JD, Huse WD, Bodkin DJ, et al. Targeted antiangiogenic therapy for cancer using Vitaxin: a humanized monoclonal antibody to the integrin alphavbeta3. Clin Cancer Res. 2000;6(8):3056–61. [PubMed] [Google Scholar]
- 47. Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open‐label, controlled, randomized phase II CORE study. Neuro‐oncol. 2015;17(5):708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rudman SM, Jameson MB, McKeage MJ, Savage P, Jodrell DI, Harries M, et al. A phase 1 study of AS1409, a novel antibody‐cytokine fusion protein, in patients with malignant melanoma or renal cell carcinoma. Clin Cancer Res. 2011;17(7):1998–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johannsen M, Spitaleri G, Curigliano G, Roigas J, Weikert S, Kempkensteffen C, et al. The tumour‐targeting human L19‐IL2 immunocytokine: preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur J Cancer (Oxford, England: 1990). 2010;46(16):2926–35. [DOI] [PubMed] [Google Scholar]
- 50. Eigentler TK, Weide B, de Braud F, Spitaleri G, Romanini A, Pflugfelder A, et al. A dose‐escalation and signal‐generating study of the immunocytokine L19‐IL2 in combination with dacarbazine for the therapy of patients with metastatic melanoma. Clin Cancer Res. 2011;17(24):7732–42. [DOI] [PubMed] [Google Scholar]
- 51. Birzele F, Voss E, Nopora A, Honold K, Heil F, Lohmann S, et al. CD44 Isoform Status Predicts Response to Treatment with Anti‐CD44 Antibody in Cancer Patients. Clin Cancer Res. 2015;21(12):2753–62. [DOI] [PubMed] [Google Scholar]
- 52. Vey N, Delaunay J, Martinelli G, Fiedler W, Raffoux E, Prebet T, et al. Phase I clinical study of RG7356, an anti‐CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget. 2016;7(22):32532–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cassinelli G, Lanzi C, Tortoreto M, Cominetti D, Petrangolini G, Favini E, et al. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem Pharmacol. 2013;85(10):1424–32. [DOI] [PubMed] [Google Scholar]
- 54. Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan‐1 axis. Clin Cancer Res. 2011;17(6):1382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Priem B, van Leent MMT, Teunissen AJP, Sofias AM, Mourits VP, Willemsen L, et al. Trained Immunity‐Promoting Nanobiologic Therapy Suppresses Tumor Growth and Potentiates Checkpoint Inhibition. Cell. 2020;183(3):786–801.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oh M‐H, Sun I‐H, Zhao L, Leone RD, Sun I‐M, Xu W, et al. Targeting glutamine metabolism enhances tumor‐specific immunity by modulating suppressive myeloid cells. J Clin Invest. 2020;130(7):3865–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo ASY, et al. Phase I Trial of Anti‐PSMA Designer CAR‐T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2‐T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate. 2016;76(14):1257–70. [DOI] [PubMed] [Google Scholar]
- 58. Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133‐directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology. 2018;7(7):e1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Melief CJM, Welters MJP, Vergote I, Kroep JR, Kenter GG, Ottevanger PB, et al. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci Transl Med. 2020;12(535):eaaz8235. [DOI] [PubMed] [Google Scholar]
- 60. Lee J‐H, Chen TW‐W, Hsu C‐H, Yen Y‐H, Yang JC‐H, Cheng A‐L, et al. A phase I study of pexidartinib, a colony‐stimulating factor 1 receptor inhibitor, in Asian patients with advanced solid tumors. Invest New Drugs. 2020;38(1):99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay J‐Y, et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised phase 3 trial. Lancet (London, England). 2019;394(10197):478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rosenbaum E, Kelly C, D'Angelo SP, Dickson MA, Gounder M, Keohan ML, et al. A Phase I Study of Binimetinib (MEK162) Combined with Pexidartinib (PLX3397) in Patients with Advanced Gastrointestinal Stromal Tumor. Oncologist. 2019;24(10):1309–e983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wesolowski R, Sharma N, Reebel L, Rodal MB, Peck A, West BL, et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF‐1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol. 2019;11:1758835919854238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin‐1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double‐blind, placebo‐controlled trial. Lancet (London, England). 2017;390(10105):1833–42. [DOI] [PubMed] [Google Scholar]
- 65. Narra K, Mullins SR, Lee H‐O, Strzemkowski‐Brun B, Magalong K, Christiansen VJ, et al. Phase II trial of single agent Val‐boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol Ther. 2007;6(11):1691–9. [DOI] [PubMed] [Google Scholar]
- 66. Eager RM, Cunningham CC, Senzer NN, Stephenson J, Anthony SP, O'Day SJ, et al. Phase II assessment of talabostat and cisplatin in second‐line stage IV melanoma. BMC Cancer. 2009;9:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fukumura D, Jain RK. Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. J Cell Biochem. 2007;101(4):937–49. [DOI] [PubMed] [Google Scholar]
- 68. Nakano K, Funauchi Y, Hayakawa K, Tanizawa T, Ae K, Matsumoto S, et al. Relative Dose Intensity of Induction‐Phase Pazopanib Treatment of Soft Tissue Sarcoma: Its Relationship with Prognoses of Pazopanib Responders. Journal of Clinical Medicine. 2019;8(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Noda S, Yoshida T, Hira D, Murai R, Tomita K, Tsuru T, et al. Exploratory Investigation of Target Pazopanib Concentration Range for Patients With Renal Cell Carcinoma. Clinical Genitourinary Cancer. 2019;17(2):e306–e13. [DOI] [PubMed] [Google Scholar]
- 70. Forster JC, Harriss‐Phillips WM, Douglass MJ, Bezak E. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia (Auckland, NZ). 2017;5:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Goenka A, Tiek D, Song X, Huang T, Hu B, Cheng S‐Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells. 2021;10(3):484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Siemann DW, Horsman MR. Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacol Ther. 2015;153:107–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor‐Vascular Disrupting Agents. Cancer Treat Rev. 2011;37(1):63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Niu G, Chen X. Vascular endothelial growth factor as an anti‐angiogenic target for cancer therapy. Curr Drug Targets. 2010;11(8):1000–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity promotes resistance to anti‐VEGF therapy in breast cancer by up‐regulating IL‐6 and potentially FGF‐2. Sci Transl Med. 2018;10(432):eaag0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bourgot I, Primac I, Louis T, Noël A, Maquoi E. Reciprocal Interplay Between Fibrillar Collagens and Collagen‐Binding Integrins: Implications in Cancer Progression and Metastasis. Front Oncol. 2020;10:1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McConnell JC, O'Connell OV, Brennan K, Weiping L, Howe M, Joseph L, et al. Increased peri‐ductal collagen micro‐organization may contribute to raised mammographic density. Breast Cancer Res. 2016;18(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gao M, Yu F, Lv C, Choo J, Chen L. Fluorescent chemical probes for accurate tumor diagnosis and targeting therapy. Chem Soc Rev. 2017;46(8):2237–71. [DOI] [PubMed] [Google Scholar]
- 79. Liang H, Li X, Chen B, Wang B, Zhao Y, Zhuang Y, et al. A collagen‐binding EGFR single‐chain Fv antibody fragment for the targeted cancer therapy. J Control Release. 2015;209:101–9. [DOI] [PubMed] [Google Scholar]
- 80. Ayalew L, Acuna J, Urfano SF, Morfin C, Sablan A, Oh M, et al. Conjugation of Paclitaxel to Hybrid Peptide Carrier and Biological Evaluation in Jurkat and A549 Cancer Cell Lines. ACS Med Chem Lett. 2017;8(8):814–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Weiss T, Puca E, Silginer M, Hemmerle T, Pazahr S, Bink A, et al. Immunocytokines are a promising immunotherapeutic approach against glioblastoma. Sci Transl Med. 2020;12(564):eabb2311. [DOI] [PubMed] [Google Scholar]
- 82. Zhou Z, Lu Z‐R. Molecular imaging of the tumor microenvironment. Adv Drug Deliv Rev. 2017;113:24–48. [DOI] [PubMed] [Google Scholar]
- 83. Raavé R, van Kuppevelt TH, Daamen WF. Chemotherapeutic drug delivery by tumoral extracellular matrix targeting. J Control Release Society. 2018;274:1–8. [DOI] [PubMed] [Google Scholar]
- 84. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti‐tumour macrophages. Nature. 2017;543(7645):428–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lecoultre M, Dutoit V, Walker PR. Phagocytic function of tumor‐associated macrophages as a key determinant of tumor progression control: a review. J Immunother Cancer. 2020;8(2):e001408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor‐associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18(1):177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Miller MA, Zheng Y‐R, Gadde S, Pfirschke C, Zope H, Engblom C, et al. Tumour‐associated macrophages act as a slow‐release reservoir of nano‐therapeutic Pt(IV) pro‐drug. Nat Commun. 2015;6:8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid‐Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37(3):208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schmid D, Park CG, Hartl CA, Subedi N, Cartwright AN, Puerto RB, et al. T cell‐targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat Commun. 2017;8(1):1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jou J, Harrington KJ, Zocca M‐B, Ehrnrooth E, Cohen EEW. The Changing Landscape of Therapeutic Cancer Vaccines‐Novel Platforms and Neoantigen Identification. Clin Cancer Res. 2021;27(3):689–703. [DOI] [PubMed] [Google Scholar]
- 93. Kantoff PW, Gulley JL, Pico‐Navarro C. Revised Overall Survival Analysis of a Phase II, Randomized, Double‐Blind, Controlled Study of PROSTVAC in Men With Metastatic Castration‐Resistant Prostate Cancer. J Clin Oncol. 2017;35(1):124–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor‐released microvesicles promote the differentiation of myeloid cells with transforming growth factor‐beta‐mediated suppressive activity on T lymphocytes. Cancer Res. 2006;66(18):9290–8. [DOI] [PubMed] [Google Scholar]
- 95. Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol. 2007;178(11):6867–75. [DOI] [PubMed] [Google Scholar]
- 96. Zhu S, Yang N, Wu J, Wang X, Wang W, Liu Y‐J, et al. Tumor microenvironment‐related dendritic cell deficiency: a target to enhance tumor immunotherapy. Pharmacol Res. 2020;159:104980. [DOI] [PubMed] [Google Scholar]
- 97. Han Z, Dong Y, Lu J, Yang F, Zheng Y, Yang H. Role of hypoxia in inhibiting dendritic cells by VEGF signaling in tumor microenvironments: mechanism and application. Am J Cancer Res. 2021;11(8):3777–93. [PMC free article] [PubMed] [Google Scholar]
- 98. Mastelic‐Gavillet B, Balint K, Boudousquie C, Gannon PO, Kandalaft LE. Personalized Dendritic Cell Vaccines‐Recent Breakthroughs and Encouraging Clinical Results. Front Immunol. 2019;10:766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting tumor‐associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116(7):1955–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One. 2009;4(11):e7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP‐expressing carcinoma‐associated fibroblasts synergizes with anti‐PD‐L1 immunotherapy in pancreatic cancer. Proc Nat Acad Sci USA. 2013;110(50):20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ren Y, Jia H‐H, Xu Y‐Q, Zhou X, Zhao X‐H, Wang Y‐F, et al. Paracrine and epigenetic control of CAF‐induced metastasis: the role of HOTAIR stimulated by TGF‐ß1 secretion. Mol Cancer. 2018;17(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein kinase C and cancer: what we know and what we do not. Oncogene. 2014;33(45):5225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell. 2019;24(1):25–40. [DOI] [PubMed] [Google Scholar]
- 107. Oakes SA. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am J Pathol. 2020;190(5):934–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Certo M, Tsai C‐H, Pucino V, Ho P‐C, Mauro C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol. 2021;21(3):151–61. [DOI] [PubMed] [Google Scholar]
- 109. Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist Updat. 2020;53:100715. [DOI] [PubMed] [Google Scholar]
- 110. Mochly‐Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discovery. 2012;11(12):937–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kikkawa U, Takai Y, Tanaka Y, Miyake R, Nishizuka Y. Protein kinase C as a possible receptor protein of tumor‐promoting phorbol esters. J Biol Chem. 1983;258(19):11442–5. [PubMed] [Google Scholar]
- 112. Sadeghi MM, Salama MF, Hannun YA. Protein Kinase C as a Therapeutic Target in Non‐Small Cell Lung Cancer. Int J Mol Sci. 2021;22(11):5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Black AR, Black JD. Protein kinase C signaling and cell cycle regulation. Front Immunol. 2012;3:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cheng Y, Zhu Y, Xu W, Xu J, Yang M, Chen P, et al. PKCα in colon cancer cells promotes M1 macrophage polarization via MKK3/6‐P38 MAPK pathway. Mol Carcinog. 2018;57(8):1017–29. [DOI] [PubMed] [Google Scholar]
- 115. Pongracz J, Clark P, Neoptolemos JP, Lord JM. Expression of protein kinase C isoenzymes in colorectal cancer tissue and their differential activation by different bile acids. Int J Cancer. 1995;61(1):35–9. [DOI] [PubMed] [Google Scholar]
- 116. Suga K, Sugimoto I, Ito H, Hashimoto E. Down‐regulation of protein kinase C‐alpha detected in human colorectal cancer. Biochem Mol Biol Int. 1998;44(3):523–8. [DOI] [PubMed] [Google Scholar]
- 117. Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, et al. Protein kinase C theta affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med. 2003;197(11):1525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kwon M‐J, Ma J, Ding Y, Wang R, Sun Z. Protein kinase C‐θ promotes Th17 differentiation via upregulation of Stat3. J Immunol. 2012;188(12):5887–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. He X, Koenen HJPM, Smeets RL, Keijsers R, van Rijssen E, Koerber A, et al. Targeting PKC in human T cells using sotrastaurin (AEB071) preserves regulatory T cells and prevents IL‐17 production. J Invest Dermatol. 2014;134(4):975–83. [DOI] [PubMed] [Google Scholar]
- 120. Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium‐activated, phospholipid‐dependent protein kinase by tumor‐promoting phorbol esters. J Biol Chem. 1982;257(13):7847–51. [PubMed] [Google Scholar]
- 121. Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7(4):281–94. [DOI] [PubMed] [Google Scholar]
- 122. Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS, et al. Characterization of vascular endothelial growth factor's effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J Clin Invest. 1996;98(9):2018–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Suzuma K, Takahara N, Suzuma I, Isshiki K, Ueki K, Leitges M, et al. Characterization of protein kinase C beta isoform's action on retinoblastoma protein phosphorylation, vascular endothelial growth factor‐induced endothelial cell proliferation, and retinal neovascularization. Proc Nat Acad Sci USA. 2002;99(2):721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Goicoechea SM, García‐Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG, et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer‐associated fibroblasts. Oncogene. 2014;33(10):1265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hennings H, Blumberg PM, Pettit GR, Herald CL, Shores R, Yuspa SH. Bryostatin 1, an activator of protein kinase C, inhibits tumor promotion by phorbol esters in SENCAR mouse skin. Carcinogenesis. 1987;8(9):1343–6. [DOI] [PubMed] [Google Scholar]
- 126. Boyle GM, D'Souza MMA, Pierce CJ, Adams RA, Cantor AS, Johns JP, et al. Intra‐lesional injection of the novel PKC activator EBC‐46 rapidly ablates tumors in mouse models. PLoS One. 2014;9(10):e108887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Miller J, Campbell J, Blum A, Reddell P, Gordon V, Schmidt P, et al. Dose Characterization of the Investigational Anticancer Drug Tigilanol Tiglate (EBC‐46) in the Local Treatment of Canine Mast Cell Tumors. Front vet sci. 2019;6:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. De Ridder TR, Campbell JE, Burke‐Schwarz C, Clegg D, Elliot EL, Geller S, et al. Randomized controlled clinical study evaluating the efficacy and safety of intratumoral treatment of canine mast cell tumors with tigilanol tiglate (EBC‐46). J Vet Intern Med. 2021;35(1):415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Parker PJ, Brown SJ, Calleja V, Chakravarty P, Cobbaut M, Linch M, et al. Equivocal, explicit and emergent actions of PKC isoforms in cancer. Nat Rev Cancer. 2021;21(1):51–63. [DOI] [PubMed] [Google Scholar]
- 130. Utz I, Hofer S, Regenass U, Hilbe W, Thaler J, Grunicke H, et al. The protein kinase C inhibitor CGP 41251, a staurosporine derivative with antitumor activity, reverses multidrug resistance. Int J Cancer. 1994;57(1):104–10. [DOI] [PubMed] [Google Scholar]
- 131. Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu K, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther . 2022;7(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, et al. Haploinsufficiency of delta‐like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Nat Acad Sci USA. 2004;101(45):15949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kofler NM, Shawber CJ, Kangsamaksin T, HO Reed, Galatioto J, Kitajewski J. Notch signaling in developmental and tumor angiogenesis. Genes Cancer. 2011;2(12):1106–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Qiao L, Wong BCY. Role of Notch signaling in colorectal cancer. Carcinogenesis. 2009;30(12):1979–86. [DOI] [PubMed] [Google Scholar]
- 135. Patel NS, Li J‐L, Generali D, Poulsom R, Cranston DW, Harris AL. Up‐regulation of delta‐like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 2005;65(19):8690–7. [DOI] [PubMed] [Google Scholar]
- 136. Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E, et al. Dosage‐sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18(20):2474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang W‐C, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444(7122):1083–7. [DOI] [PubMed] [Google Scholar]
- 138. Kannan S, Sutphin RM, Hall MG, Golfman LS, Fang W, Nolo RM, et al. Notch activation inhibits AML growth and survival: a potential therapeutic approach. J Exp Med. 2013;210(2):321–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Liu N, Zhang J, Ji C. The emerging roles of Notch signaling in leukemia and stem cells. Biomark Res. 2013;1(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Liu Z, Turkoz A, Jackson EN, Corbo JC, Engelbach JA, Garbow JR, et al. Notch1 loss of heterozygosity causes vascular tumors and lethal hemorrhage in mice. J Clin Invest. 2011;121(2):800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, et al. Chronic DLL4 blockade induces vascular neoplasms. Nature. 2010;463(7282):E6–7. [DOI] [PubMed] [Google Scholar]
- 142. McKeage MJ, Kotasek D, Markman B, Hidalgo M, Millward MJ, Jameson MB, et al. Phase IB Trial of the Anti‐Cancer Stem Cell DLL4‐Binding Agent Demcizumab with Pemetrexed and Carboplatin as First‐Line Treatment of Metastatic Non‐Squamous NSCLC. Target Oncol. 2018;13(1):89–98. [DOI] [PubMed] [Google Scholar]
- 143. Mayer RJ, Ohtsu A, Yoshino T, Falcone A, Garcia‐Carbonero R, Tabernero J, et al. TAS‐102 versus placebo plus best supportive care in patients with metastatic colorectal cancer refractory to standard therapies: Final survival results of the phase III RECOURSE trial. J Clin Oncol. 2016;34(4_suppl):634. [Google Scholar]
- 144. Jimeno A, LoRusso P, Strother RM, Diamond JR, Plato L, Younger A, et al. Phase I study of REGN421 (R)/SAR153192, a fully‐human delta‐like ligand 4 (Dll4) monoclonal antibody (mAb), in patients with advanced solid tumors. J Clin Oncol. 2013;31(15_suppl):2502. [Google Scholar]
- 145. Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, Heymach JV, et al. A phase I dose‐escalation and dose‐expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol: Official Journal of the European Society for Medical Oncology. 2018;29(7):1561–8. [DOI] [PubMed] [Google Scholar]
- 146. Casulo C, Ruan J, Dang NH, Gore L, Diefenbach C, Beaven AW, et al. Safety and Preliminary Efficacy Results of a Phase I First‐in‐Human Study of the Novel Notch‐1 Targeting Antibody Brontictuzumab (OMP‐52M51) Administered Intravenously to Patients with Hematologic Malignancies. Blood. 2016;128(22):5108. [Google Scholar]
- 147. Hu ZI, Bendell JC, Bullock A, LoConte NK, Hatoum H, Ritch P, et al. A randomized phase II trial of nab‐paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019;8(11):5148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Peng L, Cook K, Xu L, Cheng L, Damschroder M, Gao C, et al. Molecular basis for the mechanism of action of an anti‐TACE antibody. mAbs. 2016;8(8):1598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Dosch J, Ziemke E, Wan S, Luker K, Welling T, Hardiman K, et al. Targeting ADAM17 inhibits human colorectal adenocarcinoma progression and tumor‐initiating cell frequency. Oncotarget. 2017;8(39):65090–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, et al. A Phase I, dose‐finding study in patients with advanced solid malignancies of the oral γ‐secretase inhibitor PF‐03084014. Clin Cancer Res. 2015;21(1):60–7. [DOI] [PubMed] [Google Scholar]
- 151. Kummar S, O'Sullivan Coyne G, Do KT, Turkbey B, Meltzer PS, Polley E, et al. Clinical Activity of the γ‐Secretase Inhibitor PF‐03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis). J Clin Oncol. 2017;35(14):1561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Papayannidis C, DeAngelo DJ, Stock W, Huang B, Shaik MN, Cesari R, et al. A Phase 1 study of the novel gamma‐secretase inhibitor PF‐03084014 in patients with T‐cell acute lymphoblastic leukemia and T‐cell lymphoblastic lymphoma. Blood Cancer J. 2015;5(9):e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zweidler‐McKay PA, DeAngelo DJ, Douer D, Dombret H, Ottmann OG, Vey N, et al. The Safety and Activity of BMS‐906024, a Gamma Secretase Inhibitor (GSI) with Anti‐Notch Activity, in Patients with Relapsed T‐Cell Acute Lymphoblastic Leukemia (T‐ALL): Initial Results of a Phase 1 Trial. Blood. 2014;124(21):968. [Google Scholar]
- 154. Nasser F, Moussa N, Helmy MW, Haroun M. Dual targeting of Notch and Wnt/β‐catenin pathways: Potential approach in triple‐negative breast cancer treatment. Naunyn‐Schmiedeb Arch Pharmacol. 2021;394(3):481–90. [DOI] [PubMed] [Google Scholar]
- 155. Liu S, Ren J, Ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chaudhury A, Howe PH. The tale of transforming growth factor‐beta (TGFbeta) signaling: a soigné enigma. IUBMB Life. 2009;61(10):929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Syed V. TGF‐β Signaling in Cancer. J Cell Biochem. 2016;117(6):1279–87. [DOI] [PubMed] [Google Scholar]
- 158. Katz LH, Li Y, Chen J‐S, Muñoz NM, Majumdar A, Chen J, et al. Targeting TGF‐β signaling in cancer. Expert Opin Ther Targets. 2013;17(7):743–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Yang L, Moses HL. Transforming growth factor beta: tumor suppressor or promoter? Are host immune cells the answer? Cancer Res. 2008;68(22):9107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Batlle E, Massagué J. Transforming Growth Factor‐β Signaling in Immunity and Cancer. Immunity. 2019;50(4):924–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Derynck R, Turley SJ, Akhurst RJ. TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18(1):9–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Xie F, Ling L, van Dam H, Zhou F, Zhang L. TGF‐β signaling in cancer metastasis. Acta Biochim Biophy Sin. 2018;50(1):121–32. [DOI] [PubMed] [Google Scholar]
- 163. Zhong Z, Carroll KD, Policarpio D, Osborn C, Gregory M, Bassi R, et al. Anti‐transforming growth factor beta receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin Cancer Res. 2010;16(4):1191–205. [DOI] [PubMed] [Google Scholar]
- 164. Kim B‐G, Malek E, Choi SH, Ignatz‐Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the TGF‐β pathway. J Hematol Oncol. 2021;14(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Yap TA, Lakhani NJ, Araujo DV, Rodon Ahnert J, Chandana SR, Sharma M, et al. AVID200, first‐in‐class TGF‐beta 1 and 3 selective and potent inhibitor: Safety and biomarker results of a phase I monotherapy dose‐escalation study in patients with advanced solid tumors. J Clin Oncol. 2020;38(15_suppl):3587.32776807 [Google Scholar]
- 166. Ikeda M, Morimoto M, Tajimi M, Inoue K, Benhadji KA, Lahn MMF, et al. A phase 1b study of transforming growth factor‐beta receptor I inhibitor galunisertib in combination with sorafenib in Japanese patients with unresectable hepatocellular carcinoma. Invest New Drugs. 2019;37(1):118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Yingling JM, McMillen WT, Yan L, Huang H, Sawyer JS, Graff J, et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first‐in‐class transforming growth factor‐β receptor type I inhibitor. Oncotarget. 2018;9(6):6659–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ikeda M, Takahashi H, Kondo S, Lahn MMF, Ogasawara K, Benhadji KA, et al. Phase 1b study of galunisertib in combination with gemcitabine in Japanese patients with metastatic or locally advanced pancreatic cancer. Cancer Chemother Pharmacol. 2017;79(6):1169–77. [DOI] [PubMed] [Google Scholar]
- 169. Keedy VL, Bauer TM, Clarke JM, Hurwitz H, Baek I, Ha I, et al. Association of TGF‐β responsive signature with anti‐tumor effect of vactosertib, a potent, oral TGF‐β receptor type I (TGFBRI) inhibitor in patients with advanced solid tumors. J Clin Oncol. 2018;36(15_suppl):3031.30199311 [Google Scholar]
- 170. Kim B‐G, Choi SH, Luo G, Sergeeva O, Lee Z, Driscoll J, et al. Vactosertib, a TGF‐ß Receptor I Kinase/ALK5 Inhibitor, Diminishes Tumor Progression and Bone Disease in a Mouse Model of Multiple Myeloma and Overcomes Resistance to Proteasome Inhibitors. Blood. 2018;132(Supplement 1):1918. [Google Scholar]
- 171. Kim HS, Ahn J‐H, Kim JE, Hong JY, Lee J, Kim SH, et al. A phase I study of TGF‐β inhibitor, vactosertib in combination with imatinib in patients with advanced desmoid tumor (aggressive fibromatosis). J Clin Oncol. 2020;38(15_suppl):11557‐. [Google Scholar]
- 172. Pei H, Parthasarathy S, Joseph S, McMillen W, Xu X, Castaneda S, et al. Abstract 955: LY3200882, a novel, highly selective TGFβRI small molecule inhibitor. Cancer Res. 2017;77(13_Supplement):955. [Google Scholar]
- 173. Schlingensiepen K‐H, Schlingensiepen R, Steinbrecher A, Hau P, Bogdahn U, Fischer‐Blass B, et al. Targeted tumor therapy with the TGF‐beta 2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006;17(1‐2):129–39. [DOI] [PubMed] [Google Scholar]
- 174. Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, et al. Targeted therapy for high‐grade glioma with the TGF‐β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro‐oncol. 2011;13(1):132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Koritzinsky M, Levitin F, van den Beucken T, Rumantir RA, Harding NJ, Chu KC, et al. Two phases of disulfide bond formation have differing requirements for oxygen. J Cell Biol. 2013;203(4):615–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. May D, Itin A, Gal O, Kalinski H, Feinstein E, Keshet E. Ero1‐L alpha plays a key role in a HIF‐1‐mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene. 2005;24(6):1011–20. [DOI] [PubMed] [Google Scholar]
- 177. Moore CE, Omikorede O, Gomez E, Willars GB, Herbert TP. PERK activation at low glucose concentration is mediated by SERCA pump inhibition and confers preemptive cytoprotection to pancreatic β‐cells. Mol Endocrinol (Baltimore, Md). 2011;25(2):315–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. [DOI] [PubMed] [Google Scholar]
- 179. Shimizu Y, Hendershot LM. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid Redox Signaling. 2009;11(9):2317–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36(10):1302–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Dong L, Krewson EA, Yang LV. Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells. Int J Mol Sci. 2017;18(2):278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Chen X, Cubillos‐Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21(2):71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. de Almeida SF, Fleming JV, Azevedo JE, Carmo‐Fonseca M, de Sousa M. Stimulation of an unfolded protein response impairs MHC class I expression. J Immunol (Baltimore, Md: 1950). 2007;178(6):3612–9. [DOI] [PubMed] [Google Scholar]
- 184. Obiedat A, Seidel E, Mahameed M, Berhani O, Tsukerman P, Voutetakis K, et al. Transcription of the NKG2D ligand MICA is suppressed by the IRE1/XBP1 pathway of the unfolded protein response through the regulation of E2F1. FASEB J. 2019;33(3):3481–95. [DOI] [PubMed] [Google Scholar]
- 185. Harnoss JM, Le Thomas A, Shemorry A, Marsters SA, Lawrence DA, Lu M, et al. Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc Nat Acad Sci USA. 2019;116(33):16420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Vincenz L, Jäger R, O'Dwyer M, Samali A. Endoplasmic reticulum stress and the unfolded protein response: targeting the Achilles heel of multiple myeloma. Mol Cancer Ther. 2013;12(6):831–43. [DOI] [PubMed] [Google Scholar]
- 187. Mimura N, Fulciniti M, Gorgun G, Tai Y‐T, Cirstea D, Santo L, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood. 2012;119(24):5772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Tay KH, Luan Q, Croft A, Jiang CC, Jin L, Zhang XD, et al. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell Signalling. 2014;26(2):287–94. [DOI] [PubMed] [Google Scholar]
- 189. Jin Y, Saatcioglu F. Targeting the Unfolded Protein Response in Hormone‐Regulated Cancers. Trends Cancer. 2020;6(2):160–71. [DOI] [PubMed] [Google Scholar]
- 190. Hurst KE, Lawrence KA, Essman MT, Walton ZJ, Leddy LR, Thaxton JE. Endoplasmic Reticulum Stress Contributes to Mitochondrial Exhaustion of CD8+ T Cells. Cancer Immunol Res. 2019;7(3):476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Atkins C, Liu Q, Minthorn E, Zhang S‐Y, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013;73(6):1993–2002. [DOI] [PubMed] [Google Scholar]
- 192. Rojas‐Rivera D, Delvaeye T, Roelandt R, Nerinckx W, Augustyns K, Vandenabeele P, et al. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017;24(6):1100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Nie Z, Chen M, Wen X, Gao Y, Huang D, Cao H, et al. Endoplasmic Reticulum Stress and Tumor Microenvironment in Bladder Cancer: The Missing Link. Front Cell Dev Biol. 2021;9:683940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Fu Y, Wey S, Wang M, Ye R, Liao C‐P, Roy‐Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Nat Acad Sci USA. 2008;105(49):19444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase‐7 activation. J Biol Chem. 2003;278(23):20915–24. [DOI] [PubMed] [Google Scholar]
- 196. Racker E. Bioenergetics and the problem of tumor growth. Am Sci. 1972;60(1):56–63. [PubMed] [Google Scholar]
- 197. Constant JS, Feng JJ, Zabel DD, Yuan H, Suh DY, Scheuenstuhl H, et al. Lactate elicits vascular endothelial growth factor from macrophages: a possible alternative to hypoxia. Wound Repair and Regeneration: Official Publication of the Wound Healing Society [and] the European Tissue Repair Society. 2000;8(5):353–60. [DOI] [PubMed] [Google Scholar]
- 198. Sun K, Tang S, Hou Y, Xi L, Chen Y, Yin J, et al. Oxidized ATM‐mediated glycolysis enhancement in breast cancer‐associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine. 2019;41:370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Walenta S, Salameh A, Lyng H, Evensen JF, Mitze M, Rofstad EK, et al. Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am J Pathol. 1997;150(2):409–15. [PMC free article] [PubMed] [Google Scholar]
- 200. Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, et al. Gpr132 sensing of lactate mediates tumor‐macrophage interplay to promote breast cancer metastasis. Proc Nat Acad Sci USA. 2017;114(3):580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, et al. Lactate‐Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver‐Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol Res. 2019;7(2):335–46. [DOI] [PubMed] [Google Scholar]
- 202. Ippolito L, Morandi A, Giannoni E, Chiarugi P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem Sci. 2019;44(2):153–66. [DOI] [PubMed] [Google Scholar]
- 203. Gottfried E, Kunz‐Schughart LA, Ebner S, Mueller‐Klieser W, Hoves S, Andreesen R, et al. Tumor‐derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107(5):2013–21. [DOI] [PubMed] [Google Scholar]
- 204. Puig‐Kröger A, Pello OM, Muñiz‐Pello O, Selgas R, Criado G, Bajo M‐A, et al. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte‐derived dendritic cells: effect of lactate and glucose‐degradation products. J Leukocyte Biol. 2003;73(4):482–92. [DOI] [PubMed] [Google Scholar]
- 205. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell‐derived lactic acid on human T cells. Blood. 2007;109(9):3812–9. [DOI] [PubMed] [Google Scholar]
- 206. Xia H, Wang W, Crespo J, Kryczek I, Li W, Wei S, et al. Suppression of FIP200 and autophagy by tumor‐derived lactate promotes naïve T cell apoptosis and affects tumor immunity. Sci Immunol. 2017;2(17):eaan4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Husain Z, Seth P, Sukhatme VP. Tumor‐derived lactate and myeloid‐derived suppressor cells: Linking metabolism to cancer immunology. Oncoimmunology. 2013;2(11):e26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Polański R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res. 2014;20(4):926–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Mathupala SP, Parajuli P, Sloan AE. Silencing of monocarboxylate transporters via small interfering ribonucleic acid inhibits glycolysis and induces cell death in malignant glioma: an in vitro study. Neurosurgery. 2004;55(6):1410–9; discussion 9. [DOI] [PubMed] [Google Scholar]
- 210. Xie H, Hanai J‐I, Ren J‐G, Kats L, Burgess K, Bhargava P, et al. Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor‐initiating cells. Cell Metab. 2014;19(5):795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Maftouh M, Avan A, Sciarrillo R, Granchi C, Leon LG, Rani R, et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br J Cancer. 2014;110(1):172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, et al. Discovery of N‐hydroxyindole‐based inhibitors of human lactate dehydrogenase isoform A (LDH‐A) as starvation agents against cancer cells. J Med Chem. 2011;54(6):1599–612. [DOI] [PubMed] [Google Scholar]
- 213. Manerba M, Vettraino M, Fiume L, Di Stefano G, Sartini A, Giacomini E, et al. Galloflavin (CAS 568‐80‐9): a novel inhibitor of lactate dehydrogenase. ChemMedChem. 2012;7(2):311–7. [DOI] [PubMed] [Google Scholar]
- 214. Phan LM, Yeung S‐CJ, Lee M‐H. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti‐cancer therapies. Cancer Biol Med. 2014;11(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Lyssiotis CA, Kimmelman AC. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017;27(11):863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Allison KE, Coomber BL, Bridle BW. Metabolic reprogramming in the tumour microenvironment: a hallmark shared by cancer cells and T lymphocytes. Immunology. 2017;152(2):175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Lane AN, Higashi RM, Fan TW‐M. Metabolic reprogramming in tumors: Contributions of the tumor microenvironment. GENES DIS. 2020;7(2):185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Elia I, Haigis MC. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab. 2021;3(1):21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Martinez‐Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles' heel of tumor cells. Cell Cycle (Georgetown, Tex). 2011;10(24):4208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. [DOI] [PubMed] [Google Scholar]
- 222. Keenan MM, Chi J‐T. Alternative fuels for cancer cells. Cancer j (Sudbury Mass). 2015;21(2):49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Clement E, Lazar I, Attané C, Carrié L, Dauvillier S, Ducoux‐Petit M, et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020;39(3):e102525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Birsoy K, Wang T, Chen WW, Freinkman E, Abu‐Remaileh M, Sabatini DM. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162(3):540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Garcia‐Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. 2018;20(7):775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Yin Z, Bai L, Li W, Zeng T, Tian H, Cui J. Targeting T cell metabolism in the tumor microenvironment: an anti‐cancer therapeutic strategy. J Exp Clin Cancer Res. 2019;38(1):403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38(9):2438–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T‐cell differentiation and function. Immunol Rev. 2012;249(1):43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Cham CM, Gajewski TF. Glucose availability regulates IFN‐gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol (Baltimore, Md: 1950). 2005;174(8):4670–7. [DOI] [PubMed] [Google Scholar]
- 230. Kedia‐Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun. 2019;10(1):2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Cacace A, Sboarina M, Vazeille T, Sonveaux P. Glutamine activates STAT3 to control cancer cell proliferation independently of glutamine metabolism. Oncogene. 2017;36(15):2074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Wenes M, Shang M, Di Matteo M, Goveia J, Martín‐Pérez R, Serneels J, et al. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016;24(5):701–15. [DOI] [PubMed] [Google Scholar]
- 233. Colegio OR, Chu N‐Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour‐associated macrophages by tumour‐derived lactic acid. Nature. 2014;513(7519):559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Liu P‐S, Wang H, Li X, Chao T, Teav T, Christen S, et al. α‐ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18(9):985–94. [DOI] [PubMed] [Google Scholar]
- 236. Xiao Z, Dai Z, Locasale JW. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat Commun. 2019;10(1):3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Hensley CT, Faubert B, Yuan Q, Lev‐Cohain N, Jin E, Kim J, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164(4):681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18(10):1090–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer. 2017;3(3):169–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Lien EC, Vander Heiden MG. A framework for examining how diet impacts tumour metabolism. Nat Rev Cancer. 2019;19(11):651–61. [DOI] [PubMed] [Google Scholar]
- 241. Humpton TJ, Hock AK, Maddocks ODK, Vousden KH. p53‐mediated adaptation to serine starvation is retained by a common tumour‐derived mutant. Cancer Metab. 2018;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Flöter J, Kaymak I, Schulze A. Regulation of Metabolic Activity by p53. Metabolites. 2017;7(2):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Cogdill AP, Gaudreau PO, Arora R, Gopalakrishnan V, Wargo JA. The Impact of Intratumoral and Gastrointestinal Microbiota on Systemic Cancer Therapy. Trends Immunol. 2018;39(11):900–20. [DOI] [PubMed] [Google Scholar]
- 244. Hu Z, Qu G, Yu X, Jiang H, Teng X‐L, Ding L, et al. Acylglycerol Kinase Maintains Metabolic State and Immune Responses of CD8+ T Cells. Cell Metab. 2019;30(2):290–302.e5. [DOI] [PubMed] [Google Scholar]
- 245. Li X, Qian X, Jiang H, Xia Y, Zheng Y, Li J, et al. Nuclear PGK1 Alleviates ADP‐Dependent Inhibition of CDC7 to Promote DNA Replication. Mol Cell. 2018;72(4):650–60e8. [DOI] [PubMed] [Google Scholar]
- 246. Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y, et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580(7804):530–5. [DOI] [PubMed] [Google Scholar]
- 247. Tuo L, Xiang J, Pan X, Hu J, Tang H, Liang L, et al. PCK1 negatively regulates cell cycle progression and hepatoma cell proliferation via the AMPK/p27Kip1 axis. J Exp Clin Cancer Res. 2019;38(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Yamaguchi N, Weinberg EM, Nguyen A, Liberti MV, Goodarzi H, Janjigian YY, et al. PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. eLife. 2019;8:e52135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, et al. Mammalian target of rapamycin up‐regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Nat Acad Sci USA. 2011;108(10):4129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Pan C, Li B, Simon MC. Moonlighting functions of metabolic enzymes and metabolites in cancer. Mol Cell. 2021;81(18):3760–74. [DOI] [PubMed] [Google Scholar]
- 251. Dey P, Kimmelman AC, DePinho RA. Metabolic Codependencies in the Tumor Microenvironment. Cancer Discov. 2021;11(5):1067–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. An X, Zhu Y, Zheng T, Wang G, Zhang M, Li J, et al. An Analysis of the Expression and Association with Immune Cell Infiltration of the cGAS/STING Pathway in Pan‐Cancer. Mol Ther Nucleic Acids. 2019;14:80–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Bakhoum SF, Ngo B, Laughney AM, Cavallo J‐A, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553(7689):467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Woo S‐R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING‐dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS‐STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21(9):548–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Li J, Duran MA, Dhanota N, Chatila WK, Bettigole SE, Kwon J, et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021;11(5):1212–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez‐Soto A, et al. Carcinoma‐astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533(7604):493–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Varki A, Schnaar RL, Schauer R. Sialic Acids and Other Nonulosonic Acids. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al., editors. Essentials of Glycobiology. 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [PubMed] [Google Scholar]
- 259. Mereiter S, Balmaña M, Campos D, Gomes J, Reis CA. Glycosylation in the Era of Cancer‐Targeted Therapy: Where Are We Heading? Cancer Cell. 2019;36(1):6–16. [DOI] [PubMed] [Google Scholar]
- 260. Büll C, den Brok MH, Adema GJ. Sweet escape: sialic acids in tumor immune evasion. Biochim Biophys Acta. 2014;1846(1):238–46. [DOI] [PubMed] [Google Scholar]
- 261. Rosato FE. Active specific immunotherapy of human solid tumors. Ann NY Acad Sci. 1976;277(00):332–8. [DOI] [PubMed] [Google Scholar]
- 262. RodrÍguez E, Schetters STT, van Kooyk Y. The tumour glyco‐code as a novel immune checkpoint for immunotherapy. Nat Rev Immunol. 2018;18(3):204–11. [DOI] [PubMed] [Google Scholar]
- 263. Pearce OMT, Läubli H. Sialic acids in cancer biology and immunity. Glycobiology. 2016;26(2):111–28. [DOI] [PubMed] [Google Scholar]
- 264. Büll C, Stoel MA, den Brok MH, Adema GJ. Sialic acids sweeten a tumor's life. Cancer Res. 2014;74(12):3199–204. [DOI] [PubMed] [Google Scholar]
- 265. Moore SR, Menon SS, Cortes C, Ferreira VP. Hijacking Factor H for Complement Immune Evasion. Front Immunol. 2021;12:602277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Büll C, Heise T, Adema GJ, Boltje TJ. Sialic Acid Mimetics to Target the Sialic Acid‐Siglec Axis. Trends Biochem Sci. 2016;41(6):519–31. [DOI] [PubMed] [Google Scholar]
- 267. Duan S, Paulson JC. Siglecs as Immune Cell Checkpoints in Disease. Annu Rev Immunol. 2020;38:365–95. [DOI] [PubMed] [Google Scholar]
- 268. Jiang K‐Y, Qi L‐L, Kang F‐B, Wang L. The intriguing roles of Siglec family members in the tumor microenvironment. Biomark Res. 2022;10(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Pang L, Khan F, Dunterman M, Chen P. Pharmacological targeting of the tumor‐immune symbiosis in glioblastoma. Trends Pharmacol Sci. 2022;43(8):686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Takehara T, Wakamatsu E, Machiyama H, Nishi W, Emoto K, Azuma M, et al. PD‐L2 suppresses T cell signaling via coinhibitory microcluster formation and SHP2 phosphatase recruitment. Commun Biol. 2021;4(1):581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Pawelczyk K, Piotrowska A, Ciesielska U, Jablonska K, Gletzel‐Plucinska N, Grzegrzolka J, et al. Role of PD‐L1 Expression in Non‐Small Cell Lung Cancer and Their Prognostic Significance according to Clinicopathological Factors and Diagnostic Markers. Int J Mol Sci. 2019;20(4):824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Hudson K, Cross N, Jordan‐Mahy N, Leyland R. The Extrinsic and Intrinsic Roles of PD‐L1 and Its Receptor PD‐1: Implications for Immunotherapy Treatment. Front Immunol. 2020;11:568931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Zhou K, Guo S, Li F, Sun Q, Liang G. Exosomal PD‐L1: New Insights Into Tumor Immune Escape Mechanisms and Therapeutic Strategies. Front Cell Dev Biol. 2020;8:569219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Liu J, Peng X, Yang S, Li X, Huang M, Wei S, et al. Extracellular vesicle PD‐L1 in reshaping tumor immune microenvironment: biological function and potential therapy strategies. Cell Commun Signal: CCS. 2022;20(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, et al. PD‐L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun. 2020;11(1):4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Zhao Y, Lee CK, Lin C‐H, Gassen RB, Xu X, Huang Z, et al. PD‐L1:CD80 Cis‐Heterodimer Triggers the Co‐stimulatory Receptor CD28 While Repressing the Inhibitory PD‐1 and CTLA‐4 Pathways. Immunity. 2019;51(6):1059–73.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Sugiura D, Maruhashi T, Okazaki I‐M, Shimizu K, Maeda TK, Takemoto T, et al. Restriction of PD‐1 function by cis‐PD‐L1/CD80 interactions is required for optimal T cell responses. Science (New York, NY). 2019;364(6440):558–66. [DOI] [PubMed] [Google Scholar]
- 279. Nishimura CD, Pulanco MC, Cui W, Lu L, Zang X. PD‐L1 and B7‐1 Cis‐Interaction: New Mechanisms in Immune Checkpoints and Immunotherapies. Trends Mol Med. 2021;27(3):207–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Qi T, Fu J, Zhang W, Cui W, Xu X, Yue J, et al. Mutation of PD‐1 immune receptor tyrosine‐based switch motif (ITSM) enhances the antitumor activity of cytotoxic T cells. Transl Cancer Res. 2020;9(11):6811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Sahillioglu AC, Schumacher TN. Multimodular Optimization of Chemically Regulated T Cell Switches Demonstrates Flexible and Interchangeable Nature of Immune Cell Signaling Domains. Hum Gene Ther. 2021;32(19‐20):1029–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Celis‐Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Grégoire C, et al. Quantitative Interactomics in Primary T Cells Provides a Rationale for Concomitant PD‐1 and BTLA Coinhibitor Blockade in Cancer Immunotherapy. Cell Rep. 2019;27(11):3315–30.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD‐1 stimulation. Sci Adv. 2020;6(5):eaay4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Fan Z, Tian Y, Chen Z, Liu L, Zhou Q, He J, et al. Blocking interaction between SHP2 and PD‐1 denotes a novel opportunity for developing PD‐1 inhibitors. EMBO Mol Med. 2020;12(6):e11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp‐2 Is Dispensable for Establishing T Cell Exhaustion and for PD‐1 Signaling In Vivo. Cell Rep. 2018;23(1):39–49. [DOI] [PubMed] [Google Scholar]
- 286. Patsoukis N, Duke‐Cohan JS, Chaudhri A, Aksoylar H‐I, Wang Q, Council A, et al. Interaction of SHP‐2 SH2 domains with PD‐1 ITSM induces PD‐1 dimerization and SHP‐2 activation. Commun Biol. 2020;3(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Sheppard K‐A, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD‐1 inhibits T‐cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574(1‐3):37–41. [DOI] [PubMed] [Google Scholar]
- 288. Wang Q, Bardhan K, Boussiotis VA, Patsoukis N. The PD‐1 Interactome. Adv Biol. 2021;5(9):e2100758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Simonds EF, Lu ED, Badillo O, Karimi S, Liu EV, Tamaki W, et al. Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor‐refractory glioblastoma. J Immunother Cancer. 2021;9(6):e002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26(1):39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Pang L, Khan F, Heimberger AB, Chen P. Mechanism and therapeutic potential of tumor‐immune symbiosis in glioblastoma. Trends Cancer. 2022;8(10):839–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Lerrer S, Tocheva AS, Bukhari S, Adam K, Mor A. PD‐1‐stimulated T cell subsets are transcriptionally and functionally distinct. iScience. 2021;24(9):103020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Shimizu K, Sugiura D, Okazaki I‐M, Maruhashi T, Takegami Y, Cheng C, et al. PD‐1 Imposes Qualitative Control of Cellular Transcriptomes in Response to T Cell Activation. Mol Cell. 2020;77(5):937–50.e6. [DOI] [PubMed] [Google Scholar]
- 294. Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human Treg and effector T cells in tumor immunity by Fc‐engineered anti‐CTLA‐4 antibody. Proc Nat Acad Sci USA. 2019;116(2):609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7‐1) binds both CD28 and CTLA‐4 with a low affinity and very fast kinetics. J Exp Med. 1997;185(3):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Halliday N, Williams C, Kennedy A, Waters E, Pesenacker AM, Soskic B, et al. CD86 Is a Selective CD28 Ligand Supporting FoxP3+ Regulatory T Cell Homeostasis in the Presence of High Levels of CTLA‐4. Front Immunol. 2020;11:600000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg‐expressed CTLA‐4 depletes CD80/CD86 by trogocytosis, releasing free PD‐L1 on antigen‐presenting cells. Proc Nat Acad Sci USA. 2021;118(30):e2023739118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Chikuma S, Kanamori M, Mise‐Omata S, Yoshimura A. Suppressors of cytokine signaling: Potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci. 2017;108(4):574–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Bagchi S, Yuan R, Engleman EG. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annual Review of Pathology. 2021;16:223–49. [DOI] [PubMed] [Google Scholar]
- 300. Miyatake S, Nakaseko C, Umemori H, Yamamoto T, Saito T. Src family tyrosine kinases associate with and phosphorylate CTLA‐4 (CD152). Biochem Biophys Res Commun. 1998;249(2):444–8. [DOI] [PubMed] [Google Scholar]
- 301. Schneider H, Rudd CE. Tyrosine phosphatase SHP‐2 binding to CTLA‐4: absence of direct YVKM/YFIP motif recognition. Biochem Biophys Res Commun. 2000;269(1):279–83. [DOI] [PubMed] [Google Scholar]
- 302. Schneider H, Smith X, Liu H, Bismuth G, Rudd CE. CTLA‐4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur J Immunol. 2008;38(1):40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Li K, Yuan Z, Lyu J, Ahn E, Davis SJ, Ahmed R, et al. PD‐1 suppresses TCR‐CD8 cooperativity during T‐cell antigen recognition. Nat Commun. 2021;12(1):2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Liu H, Purbhoo MA, Davis DM, Rudd CE. SH2 domain containing leukocyte phosphoprotein of 76‐kDa (SLP‐76) feedback regulation of ZAP‐70 microclustering. Proc Nat Acad Sci USA. 2010;107(22):10166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Calvo CR, Amsen D, Kruisbeek AM. Cytotoxic T lymphocyte antigen 4 (CTLA‐4) interferes with extracellular signal‐regulated kinase (ERK) and Jun NH2‐terminal kinase (JNK) activation, but does not affect phosphorylation of T cell receptor zeta and ZAP70. J Exp Med. 1997;186(10):1645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Ohtsuka S, Ogawa S, Wakamatsu E, Abe R. Cell cycle arrest caused by MEK/ERK signaling is a mechanism for suppressing growth of antigen‐hyperstimulated effector T cells. Int Immunol. 2016;28(11):547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20(3):173–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim‐3–mediated cell death and exhaustion. Nat Med. 2012;18(9):1394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine‐dependent coupling of Tim‐3 to T‐cell receptor signaling pathways. Mol Cell Biol. 2011;31(19):3963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim‐3 is phosphorylated upon stimulation by its ligand galectin‐9. Biochem Biophys Res Commun. 2006;351(2):571–6. [DOI] [PubMed] [Google Scholar]
- 311. Das M, Zhu C, Kuchroo VK. Tim‐3 and its role in regulating anti‐tumor immunity. Immunol Rev. 2017;276(1):97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Zhu C, Dixon KO, Newcomer K, Gu G, Xiao S, Zaghouani S, et al. Tim‐3 adaptor protein Bat3 is a molecular checkpoint of T cell terminal differentiation and exhaustion. Sci Adv. 2021;7(18):eabd2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Clayton KL, Haaland MS, Douglas‐Vail MB, Mujib S, Chew GM, Ndhlovu LC, et al. T cell Ig and mucin domain‐containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol (Baltimore, Md: 1950). 2014;192(2):782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Smith CM, Li A, Krishnamurthy N, Lemmon MA. Phosphatidylserine binding directly regulates TIM‐3 function. Biochem J. 2021;478(17):3331–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Tan S, Xu Y, Wang Z, Wang T, Du X, Song X, et al. Tim‐3 Hampers Tumor Surveillance of Liver‐Resident and Conventional NK Cells by Disrupting PI3K Signaling. Cancer Res. 2020;80(5):1130–42. [DOI] [PubMed] [Google Scholar]
- 316. Acharya N, Sabatos‐Peyton C, Anderson AC. Tim‐3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. 2020;8(1):e000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Friedlaender A, Addeo A, Banna G. New emerging targets in cancer immunotherapy: the role of TIM3. ESMO open. 2019;4(Suppl 3):e000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Huo J‐L, Wang Y‐T, Fu W‐J, Lu N, Liu Z‐S. The promising immune checkpoint LAG‐3 in cancer immunotherapy: from basic research to clinical application. Front Immunol. 2022;13:956090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Burnell SEA, Capitani L, MacLachlan BJ, Mason GH, Gallimore AM, Godkin A. Seven mysteries of LAG‐3: a multi‐faceted immune receptor of increasing complexity. Adv Immunol. 2022;2(1):ltab025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin‐3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG‐3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res. 2015;3(4):412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Maruhashi T, Okazaki I‐M, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG‐3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation‐dependent recognition of pMHCII. Nat Immunol. 2018;19(12):1415–26. [DOI] [PubMed] [Google Scholar]
- 322. Hannier S, Tournier M, Bismuth G, Triebel F. CD3/TCR complex‐associated lymphocyte activation gene‐3 molecules inhibit CD3/TCR signaling. J Immunol (Baltimore, Md: 1950). 1998;161(8):4058–65. [PubMed] [Google Scholar]
- 323. Castro‐Sanchez P, Teagle AR, Prade S, Zamoyska R. Modulation of TCR Signaling by Tyrosine Phosphatases: From Autoimmunity to Immunotherapy. Front Cell Dev Biol. 2020;8:608747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Workman CJ, Dugger KJ, Vignali DAA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene‐3. J Immunol (Baltimore, Md: 1950). 2002;169(10):5392–5. [DOI] [PubMed] [Google Scholar]
- 325. Okamura T, Yamamoto K, Fujio K. Early Growth Response Gene 2‐Expressing CD4+LAG3+ Regulatory T Cells: The Therapeutic Potential for Treating Autoimmune Diseases. Front Immunol. 2018;9:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG‐3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol (Baltimore, Md: 1950). 2010;184(11):6545–51. [DOI] [PubMed] [Google Scholar]
- 327. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene‐3 engagement of MHC class II. J Immunol (Baltimore, Md: 1950). 2008;180(9):5916–26. [DOI] [PubMed] [Google Scholar]
- 328. Gandhi MK, Lambley E, Duraiswamy J, Dua U, Smith C, Elliott S, et al. Expression of LAG‐3 by tumor‐infiltrating lymphocytes is coincident with the suppression of latent membrane antigen‐specific CD8+ T‐cell function in Hodgkin lymphoma patients. Blood. 2006;108(7):2280–9. [DOI] [PubMed] [Google Scholar]
- 329. Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor‐specific CD8⁺ T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Matsuzaki J, Gnjatic S, Mhawech‐Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor‐infiltrating NY‐ESO‐1‐specific CD8+ T cells are negatively regulated by LAG‐3 and PD‐1 in human ovarian cancer. Proc Nat Acad Sci USA. 2010;107(17):7875–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA‐4 and PD‐1. Nat Immunol. 2003;4(7):670–9. [DOI] [PubMed] [Google Scholar]
- 332. Gavrieli M, Murphy KM. Association of Grb‐2 and PI3K p85 with phosphotyrosile peptides derived from BTLA. Biochem Biophys Res Commun. 2006;345(4):1440–5. [DOI] [PubMed] [Google Scholar]
- 333. Steinberg MW, Huang Y, Wang‐Zhu Y, Ware CF, Cheroutre H, Kronenberg M. BTLA interaction with HVEM expressed on CD8(+) T cells promotes survival and memory generation in response to a bacterial infection. PLoS One. 2013;8(10):e77992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Ning Z, Liu K, Xiong H. Roles of BTLA in Immunity and Immune Disorders. Front Immunol. 2021;12:654960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (New York, NY). 2018;359(6382):1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Xin Yu J, Hubbard‐Lucey VM, Tang J. Immuno‐oncology drug development goes global. Nat Rev Drug Discovery. 2019;18(12):899–900. [DOI] [PubMed] [Google Scholar]
- 337. Haanen JBAG, Robert C. Immune Checkpoint Inhibitors. Prog Tumor Res. 2015;42:55–66. [DOI] [PubMed] [Google Scholar]
- 338. Yin H, Zhou X, Huang Y‐H, King GJ, Collins BM, Gao Y, et al. Rational Design of Potent Peptide Inhibitors of the PD‐1:PD‐L1 Interaction for Cancer Immunotherapy. J Am Chem Soc. 2021;143(44):18536–47. [DOI] [PubMed] [Google Scholar]
- 339. Gurung S, Khan F, Gunassekaran GR, Yoo JD, Poongkavithai Vadevoo SM, Permpoon U, et al. Phage display‐identified PD‐L1‐binding peptides reinvigorate T‐cell activity and inhibit tumor progression. Biomaterials. 2020;247:119984. [DOI] [PubMed] [Google Scholar]
- 340. Vadevoo SMP, Gurung S, Khan F, Haque ME, Gunassekaran GR, Chi L, et al. Peptide‐based targeted therapeutics and apoptosis imaging probes for cancer therapy. Arch Pharmacal Res. 2019;42(2):150–8. [DOI] [PubMed] [Google Scholar]
- 341. Haque ME, Khan F, Chi L, Gurung S, Vadevoo SMP, Park R‐W, et al. A Phage Display‐Identified Peptide Selectively Binds to Kidney Injury Molecule‐1 (KIM‐1) and Detects KIM‐1‐Overexpressing Tumors in vivo. Cancer Treat Res Commun. 2019;51(3):861–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Xuan W, Hsu W‐H, Khan F, Dunterman M, Pang L, Wainwright DA, et al. Circadian Regulator CLOCK Drives Immunosuppression in Glioblastoma. Cancer Immunol Res. 2022;10(6):770–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Xuan W, Khan F, James CD, Heimberger AB, Lesniak MS, Chen P. Circadian regulation of cancer cell and tumor microenvironment crosstalk. Trends Cell Biol. 2021;31(11):940–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Mankor JM, Disselhorst MJ, Poncin M, Baas P, Aerts JGJV, Vroman H. Efficacy of nivolumab and ipilimumab in patients with malignant pleural mesothelioma is related to a subtype of effector memory cytotoxic T cells: Translational evidence from two clinical trials. EBioMedicine. 2020;62:103040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Liu SV, Reck M, Mansfield AS, Mok T, Scherpereel A, Reinmuth N, et al. Updated Overall Survival and PD‐L1 Subgroup Analysis of Patients With Extensive‐Stage Small‐Cell Lung Cancer Treated With Atezolizumab, Carboplatin, and Etoposide (IMpower133). J Clin Oncol. 2021;39(6):619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Paz‐Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum‐etoposide versus platinum‐etoposide in first‐line treatment of extensive‐stage small‐cell lung cancer (CASPIAN): a randomised, controlled, open‐label, phase 3 trial. Lancet (London, England). 2019;394(10212):1929–39. [DOI] [PubMed] [Google Scholar]
- 347. 33rd Annual Meeting & Pre‐Conference Programs of the Society for Immunotherapy of Cancer (SITC 2018) : Washington, D.C., USA. 7‐11 November 2018. Journal for Immunotherapy of Cancer. 2018;6(Suppl 1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Yu X, Huang X, Chen X, Liu J, Wu C, Pu Q, et al. Characterization of a novel anti‐human lymphocyte activation gene 3 (LAG‐3) antibody for cancer immunotherapy. mAbs. 2019;11(6):1139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti‐TIM‐3 Antibody, Alone and in Combination with Spartalizumab, an Anti‐PD‐1 Antibody, in Advanced Solid Tumors. Clin Cancer Res. 2021;27(13):3620–9. [DOI] [PubMed] [Google Scholar]
- 350. Jogalekar MP, Rajendran RL, Khan F, Dmello C, Gangadaran P, Ahn B‐C. CAR T‐Cell‐Based gene therapy for cancers: new perspectives, challenges, and clinical developments. Front Immunol. 2022;13:925985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Krishnan A, Bhatia S, Slovak ML, Arber DA, Niland JC, Nademanee A, et al. Predictors of therapy‐related leukemia and myelodysplasia following autologous transplantation for lymphoma: an assessment of risk factors. Blood. 2000;95(5):1588–93. [PubMed] [Google Scholar]
- 352. Khor B. Regulatory T Cells: Central Concepts from Ontogeny to Therapy. Transfus Med Rev. 2017;31(1):36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9. [DOI] [PubMed] [Google Scholar]
- 354. Maruyama T, Kono K, Izawa S, Mizukami Y, Kawaguchi Y, Mimura K, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to infiltration of regulatory T cells in esophageal squamous cell carcinoma. Dis Esophagus. 2010;23(5):422–9. [DOI] [PubMed] [Google Scholar]
- 355. Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. 2008;122(10):2286–93. [DOI] [PubMed] [Google Scholar]
- 356. Gobert M, Treilleux I, Bendriss‐Vermare N, Bachelot T, Goddard‐Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009;69(5):2000–9. [DOI] [PubMed] [Google Scholar]
- 357. Faget J, Biota C, Bachelot T, Gobert M, Treilleux I, Goutagny N, et al. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res. 2011;71(19):6143–52. [DOI] [PubMed] [Google Scholar]
- 358. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen‐presenting functions. Nat Rev Immunol. 2017;17(6):349–62. [DOI] [PubMed] [Google Scholar]
- 359. Kohli K, Pillarisetty VG, Kim TS. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022;29(1):10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Qian B‐Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast‐tumour metastasis. Nature. 2011;475(7355):222–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007;252(1):86–92. [DOI] [PubMed] [Google Scholar]
- 362. Soria G, Yaal‐Hahoshen N, Azenshtein E, Shina S, Leider‐Trejo L, Ryvo L, et al. Concomitant expression of the chemokines RANTES and MCP‐1 in human breast cancer: a basis for tumor‐promoting interactions. Cytokine. 2008;44(1):191–200. [DOI] [PubMed] [Google Scholar]
- 363. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5‐dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Zhang Y, Guan X‐Y, Jiang P. Cytokine and Chemokine Signals of T‐Cell Exclusion in Tumors. Front Immunol. 2020;11:594609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother (Hagerstown, Md: 1997). 2010;33(8):780–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Spranger S, Dai D, Horton B, Gajewski TF. Tumor‐Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31(5):711–23.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med. 1999;190(10):1417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Furumoto K, Soares L, Engleman EG, Merad M. Induction of potent antitumor immunity by in situ targeting of intratumoral DCs. J Clin Invest. 2004;113(5):774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Wilcox RA. Mogamulizumab: 2 birds, 1 stone. Blood. 2015;125(12):1847–8. [DOI] [PubMed] [Google Scholar]
- 371. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase Ia Study of FoxP3+ CD4 Treg Depletion by Infusion of a Humanized Anti‐CCR4 Antibody, KW‐0761, in Cancer Patients. Clin Cancer Res. 2015;21(19):4327–36. [DOI] [PubMed] [Google Scholar]
- 372. Noel M, O'Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, et al. Phase 1b study of a small molecule antagonist of human chemokine (C‐C motif) receptor 2 (PF‐04136309) in combination with nab‐paclitaxel/gemcitabine in first‐line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2020;38(3):800–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Gough M, Crittenden M, Thanarajasingam U, Sanchez‐Perez L, Thompson J, Jevremovic D, et al. Gene therapy to manipulate effector T cell trafficking to tumors for immunotherapy. J Immunol (Baltimore, Md: 1950). 2005;174(9):5766–73. [DOI] [PubMed] [Google Scholar]
- 374. Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. CXCL11‐Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology. 2016;5(3):e1091554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Arenberg DA, White ES, Burdick MD, Strom SR, Strieter RM. Improved survival in tumor‐bearing SCID mice treated with interferon‐gamma‐inducible protein 10 (IP‐10/CXCL10). Cancer Immunol Immunother. 2001;50(10):533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Costa EC, Moreira AF, de Melo‐Diogo D, Gaspar VM, Carvalho MP, Correia IJ. 3D tumor spheroids: an overview on the tools and techniques used for their analysis. Biotechnol Adv. 2016;34(8):1427–41. [DOI] [PubMed] [Google Scholar]
- 377. Rodrigues J, Heinrich MA, Teixeira LM, Prakash J. 3D In Vitro Model (R)evolution: Unveiling Tumor‐Stroma Interactions. Trends Cancer. 2021;7(3):249–64. [DOI] [PubMed] [Google Scholar]
- 378. Däster S, Amatruda N, Calabrese D, Ivanek R, Turrini E, Droeser RA, et al. Induction of hypoxia and necrosis in multicellular tumor spheroids is associated with resistance to chemotherapy treatment. Oncotarget. 2017;8(1):1725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Nath S, Devi GR. Three‐dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol Ther. 2016;163:94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Yue X, Nguyen TD, Zellmer V, Zhang S, Zorlutuna P. Stromal cell‐laden 3D hydrogel microwell arrays as tumor microenvironment model for studying stiffness dependent stromal cell‐cancer interactions. Biomaterials. 2018;170:37–48. [DOI] [PubMed] [Google Scholar]
- 381. Rebelo SP, Pinto C, Martins TR, Harrer N, Estrada MF, Loza‐Alvarez P, et al. 3D‐3‐culture: A tool to unveil macrophage plasticity in the tumour microenvironment. Biomaterials. 2018;163:185–97. [DOI] [PubMed] [Google Scholar]
- 382. Correa de Sampaio P, Auslaender D, Krubasik D, Failla AV, Skepper JN, Murphy G, et al. A heterogeneous in vitro three dimensional model of tumour‐stroma interactions regulating sprouting angiogenesis. PLoS One. 2012;7(2):e30753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer. 2018;18(7):407–18. [DOI] [PubMed] [Google Scholar]
- 384. Fan H, Demirci U, Chen P. Emerging organoid models: leaping forward in cancer research. J Hematol Oncol. 2019;12(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell. 2016;18(6):827–38. [DOI] [PubMed] [Google Scholar]
- 386. Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M, et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell. 2018;22(3):454–67.e6. [DOI] [PubMed] [Google Scholar]
- 387. Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q, et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer. 2018;18(1):335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161(4):933–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Yan HHN, Siu HC, Law S, Ho SL, Yue SSK, Tsui WY, et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell. 2018;23(6):882–97.e11. [DOI] [PubMed] [Google Scholar]
- 391. Ootani A, Li X, Sangiorgi E, Ho QT, Ueno H, Toda S, et al. Sustained in vitro intestinal epithelial culture within a Wnt‐dependent stem cell niche. Nat Med. 2009;15(6):701–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Li X, Nadauld L, Ootani A, Corney DC, Pai RK, Gevaert O, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med. 2014;20(7):769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Yuki K, Cheng N, Nakano M, Kuo CJ. Organoid Models of Tumor Immunology. Trends Immunol. 2020;41(8):652–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell. 2018;175(7):1972–88.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Dutta D, Heo I, Clevers H. Disease Modeling in Stem Cell‐Derived 3D Organoid Systems. Trends Mol Med. 2017;23(5):393–410. [DOI] [PubMed] [Google Scholar]
- 396. Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, et al. Tumor Evolution and Drug Response in Patient‐Derived Organoid Models of Bladder Cancer. Cell. 2018;173(2):515–28.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Bhatia SN, Ingber DE. Microfluidic organs‐on‐chips. Nat Biotechnol. 2014;32(8):760–72. [DOI] [PubMed] [Google Scholar]
- 398. Sontheimer‐Phelps A, Hassell BA, Ingber DE. Modelling cancer in microfluidic human organs‐on‐chips. Nat Rev Cancer. 2019;19(2):65–81. [DOI] [PubMed] [Google Scholar]
- 399. Langhans SA. Three‐Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front Pharmacol. 2018;9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Sung KE, Beebe DJ. Microfluidic 3D models of cancer. Adv Drug Deliv Rev. 2014;79‐80:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Avnet S, Lemma S, Cortini M, Di Pompo G, Perut F, Baldini N. Pre‐clinical Models for Studying the Interaction Between Mesenchymal Stromal Cells and Cancer Cells and the Induction of Stemness. Front Oncol. 2019;9:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex Vivo Profiling of PD‐1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018;8(2):196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Shang M, Soon RH, Lim CT, Khoo BL, Han J. Microfluidic modelling of the tumor microenvironment for anti‐cancer drug development. Lab Chip. 2019;19(3):369–86. [DOI] [PubMed] [Google Scholar]
- 404. Rothbauer M, Zirath H, Ertl P. Recent advances in microfluidic technologies for cell‐to‐cell interaction studies. Lab Chip. 2018;18(2):249–70. [DOI] [PubMed] [Google Scholar]
- 405. Shemesh J, Jalilian I, Shi A, Heng Yeoh G, Knothe Tate ML, Ebrahimi Warkiani M. Flow‐induced stress on adherent cells in microfluidic devices. Lab Chip. 2015;15(21):4114–27. [DOI] [PubMed] [Google Scholar]
- 406. Halldorsson S, Lucumi E, Gómez‐Sjöberg R, Fleming RMT. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens Bioelectron. 2015;63:218–31. [DOI] [PubMed] [Google Scholar]
- 407. IWGotEoCRt H. Biological agents. IARC monographs on the evaluation of carcinogenic risks to humans. 2012;100(Pt B):1–441. [PMC free article] [PubMed] [Google Scholar]
- 408. Kadosh E, Snir‐Alkalay I, Venkatachalam A, May S, Lasry A, Elyada E, et al. The gut microbiome switches mutant p53 from tumour‐suppressive to oncogenic. Nature. 2020;586(7827):133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell. 2019;176(5):998–1013.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type‐specific intracellular bacteria. Science (New York, NY). 2020;368(6494):973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Pleguezuelos‐Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature. 2020;580(7802):269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science (New York, NY). 2018;359(6375):592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Boot A, Ng AWT, Chong FT, Ho S‐C, Yu W, Tan DSW, et al. Characterization of colibactin‐associated mutational signature in an Asian oral squamous cell carcinoma and in other mucosal tumor types. Genome Res. 2020;30(6):803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, et al. Helicobacter pylori CagA phosphorylation‐independent function in epithelial proliferation and inflammation. Cell Host & Microbe. 2009;5(1):23–34. [DOI] [PubMed] [Google Scholar]
- 415. Geller LT, Barzily‐Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (New York, NY). 2017;357(6356):1156–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell. 2017;170(3):548–63.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574(7777):264–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Parhi L, Alon‐Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod‐Levi T, et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun. 2020;11(1):3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Le Noci V, Guglielmetti S, Arioli S, Camisaschi C, Bianchi F, Sommariva M, et al. Modulation of Pulmonary Microbiota by Antibiotic or Probiotic Aerosol Therapy: A Strategy to Promote Immunosurveillance against Lung Metastases. Cell Rep. 2018;24(13):3528–38. [DOI] [PubMed] [Google Scholar]
- 420. Vivarelli S, Salemi R, Candido S, Falzone L, Santagati M, Stefani S, et al. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers. 2019;11(1):E38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Shui L, Yang X, Li J, Yi C, Sun Q, Zhu H. Gut Microbiome as a Potential Factor for Modulating Resistance to Cancer Immunotherapy. Front Immunol. 2019;10:2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Cheng WY, Wu C‐Y, Yu J. The role of gut microbiota in cancer treatment: friend or foe? Gut. 2020;69(10):1867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science (New York, NY). 2013;342(6161):971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Viaud S, Flament C, Zoubir M, Pautier P, LeCesne A, Ribrag V, et al. Cyclophosphamide induces differentiation of Th17 cells in cancer patients. Cancer Res. 2011;71(3):661–5. [DOI] [PubMed] [Google Scholar]
- 425. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42(2):344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Ting NL‐N, Lau HC‐H, Yu J. Cancer pharmacomicrobiomics: targeting microbiota to optimise cancer therapy outcomes. Gut. 2022;71(7):1412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Jacouton E, Chain F, Sokol H, Langella P, Bermúdez‐Humarán LG. Probiotic Strain Lactobacillus casei BL23 Prevents Colitis‐Associated Colorectal Cancer. Front Immunol. 2017;8:1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Hu J, Wang C, Ye L, Yang W, Huang H, Meng F, et al. Anti‐tumour immune effect of oral administration of Lactobacillus plantarum to CT26 tumour‐bearing mice. J Biosci. 2015;40(2):269–79. [DOI] [PubMed] [Google Scholar]
- 429. Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, et al. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia (New York, NY). 2017;19(10):848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol: Official Journal of the European Society for Medical Oncology. 2017;28(6):1368–79. [DOI] [PubMed] [Google Scholar]
- 431. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science (New York, NY). 2015;350(6264):1079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Barlow JJ, Piver MS, Chuang JT, Cortes EP, Onuma T, Holland JF. Adriamycin and bleomycin, alone and in combination, in gynecologic cancers. Cancer. 1973;32(4):735–43. [DOI] [PubMed] [Google Scholar]
- 433. Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host & Microbe. 2011;10(4):324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Dabek M, McCrae SI, Stevens VJ, Duncan SH, Louis P. Distribution of beta‐glucosidase and beta‐glucuronidase activity and of beta‐glucuronidase gene gus in human colonic bacteria. FEMS Microbiol Ecol. 2008;66(3):487–95. [DOI] [PubMed] [Google Scholar]
- 435. Doisneau‐Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer. 2003;10(2):179–86. [DOI] [PubMed] [Google Scholar]
- 436. Fernández MF, Reina‐Pérez I, Astorga JM, Rodríguez‐Carrillo A, Plaza‐Díaz J, Fontana L. Breast Cancer and Its Relationship with the Microbiota. Int J Environ Res Public Health. 2018;15(8):1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Shida K, Nomoto K. Probiotics as efficient immunopotentiators: translational role in cancer prevention. Indian J Med Res. 2013;138(5):808–14. [PMC free article] [PubMed] [Google Scholar]
- 438. Eslami‐S Z, Majidzadeh‐A K, Halvaei S, Babapirali F, Esmaeili R. Microbiome and Breast Cancer: New Role for an Ancient Population. Front Oncol. 2020;10:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable