

Abstract
Normal lung development critically depends on HH (Hedgehog) and PDGF (platelet-derived growth factor) signaling, which coordinate mesenchymal differentiation and proliferation. PDGF signaling is required for postnatal alveolar septum formation by myofibroblasts. Recently, we demonstrated a requirement for HH in postnatal lung development involving alveolar myofibroblast differentiation. Given shared features of HH signaling and PDGF signaling and their impact on this key cell type, we sought to clarify their relationship during murine postnatal lung development. Timed experiments revealed that HH inhibition phenocopies the key lung myofibroblast phenotypes of Pdgfa (platelet-derived growth factor subunit A) and Pdgfra (platelet-derived growth factor receptor alpha) knockouts during secondary alveolar septation. Using a dual signaling reporter, Gli1lZ;PdgfraEGFP, we show that HH and PDGF pathway intermediates are concurrently expressed during alveolar septal myofibroblast accumulation, suggesting pathway convergence in the generation of lung myofibroblasts. Consistent with this hypothesis, HH inhibition reduces Pdgfra expression and diminishes the number of Pdgfra-positive and Pdgfra-lineage cells in postnatal lungs. Bulk RNA sequencing data of Pdgfra-expressing cells from Postnatal Day 8 (P8) lungs show that HH inhibition alters the expression not only of well-established HH targets but also of several putative PDGF target genes. This, together with the presence of Gli-binding sites in PDGF target genes, suggests HH input into PDGF signaling. We identified these HH/PDGF targets in several postnatal lung mesenchymal cell populations, including myofibroblasts, using single-cell transcriptomic analysis. Collectively, our data indicate that HH signaling and PDGF signaling intersect to support myofibroblast/fibroblast function during secondary alveolar septum formation. Moreover, they provide a molecular foundation relevant to perinatal lung diseases associated with impaired alveolarization.
Keywords: hedgehog signaling, platelet-derived growth factor signaling, myofibroblast, matrix fibroblast, postnatal lung
Alveologenesis during the final stage of lung development occurs in two phases and expands surface area for gas exchange with the vascular bed to supply the adult organism with sufficient oxygen (1). During the first phase, termed alveolarization, a scaffold of secondary septa is generated along the saccular walls containing abundant mesenchyme and the developing capillaries. During the second phase, termed maturation, stromal cell numbers decrease, yielding a thin vascular network covered by alveolar epithelium. Alveolarization and maturation are impaired in bronchopulmonary dysplasia (BPD), the most common lung disease of premature infants. BPD lungs exhibit simplified alveolar sacs with disorganized matrix and abnormal vascularity indicative of arrested postnatal development. BPD is associated with significant perinatal morbidity and mortality, and those surviving to adulthood show altered lung structure, decreased functional capacity, and increased lung disease (2, 3). Experimental BPD models, such as hyperoxia, mechanical ventilation, and genetic knockouts (KOs), have highlighted the involvement of HH (Hedgehog) and PDGF (platelet-derived growth factor) signaling (4–6). These pathways share biologic features (ligand dependence, epithelial-to-mesenchymal signal direction, complex paracrine mechanism, and pathway activity involving the primary cilium) (reviewed in References 7 and 8). Both control proliferation and differentiation of proximal and distal mesenchyme during lung development, including myofibroblasts (MyoFBs), lipofibroblasts, and matrix fibroblasts (MFBs) (9). However, it remains unclear whether HH signaling and PDGF signaling act in parallel or converge on shared cell targets.
HH signaling involves HH ligands (SHH [Sonic Hedgehog], IHH [Indian Hedgehog], and DHH [Desert Hedgehog]); repressive receptor and activating transmembrane proteins, Ptch1 (patched 1) and Smo (smoothened) respectively; the adaptor molecule SuFu (suppressor of fused); and the glioma-associated zinc-finger transcription factors Gli1, Gli2, and Gli3, which transduce the signal to the nucleus. Several HH pathway molecules are direct transcriptional targets and faithfully report pathway activity: Gli1 is augmented by pathway activity, whereas Ptch1 and Hhip (hedgehog inhibitory protein) are reduced. PDGF signaling involves PDGF ligands (PDGF-aa, -bb, -cc, -dd, and -ab), which stimulate autophosphorylation of PDGFRa (platelet-derived growth factor receptor alpha) and PDGFRb receptor subunits and promote intracellular signaling via MAPK (mitogen-activated protein kinase) and PIP3 (phosphatidyl inositol 3,4,5 triphosphate). Negative feedback primarily involves post-translational modification rather than increased expression of negative feedback molecules, as with HH signaling. Several direct transcriptional targets of PDGF signaling have been identified (10) that are linked to lung development and fibrosis (11–14).
HH signaling and PDGF signaling are both essential to lung development but differ in the aspect and stage they influence. HH ligands are expressed in embryonic lung epithelium and activate signaling within the mesenchyme to expand mesenchymal lineages (15–18). Mutants in which the HH pathway has been abrogated have hypoplastic lungs (16, 17, 19) that lack myofibroblasts (MyoFBs) (19, 20). Conversely, mutants with augmented HH signaling display an overabundance of alveolar mesenchyme (15, 20). Thus, HH signaling is vital to embryonic lung mesenchyme. Our recent studies revealed that HH activity remains important in postnatal lung (21). Postnatal HH activity peaks during alveolarization, and loss at this stage alters lung structure by reducing septum formation, impairing MyoFB differentiation, decreasing mesenchymal proliferation, and altering extracellular matrix expression. In contrast, although PDGF pathway components are also expressed in embryonic lungs, they are required only in postnatal mesenchyme. PDGFa and PDGFRa are essential for alveolarization. Mice lacking Pdgfa or Pdgfra undergo normal branching morphogenesis but have saccular lungs with decreased elastin and lack of MyoFBs (22, 23). Conversely, Pdgfa-overexpressing lungs display mesenchymal excess (24). In postnatal lung, Pdgfra+ cells aligned along alveolar entry rings at the secondary septal tips express MyoFB markers (SMA [smooth muscle actin]), whereas PDGFRa+ cells located at the base express lipofibroblast markers (ADRP [adipose differentiation-related protein]) (25, 26). Embryonically labeled Pdgfra lineage gives rise to SMA+ MyoFBs (27). Genetic ablation of Pdgfra-expressing cells at Postnatal Day 1 (P1) (27) and inhibition of PDGFRa signaling by imatinib from P1 to P7 impairs alveolarization and normal lung morphology (28). Whereas embryonically labeled Gli1 lineage gives rise to Pdgfra+ MyoFBs (18), it is not clear whether these cells remain receptive to HH signals at the postnatal stage of lung development.
Given the similarity in lung phenotypes produced by disrupted PDGF or HH signaling and previous studies implicating pathway convergence on key lung cells, the alveolar MyoFBs, we sought to better delineate the intersection of HH and PDGF signaling in murine postnatal lung development. Here we used compound HH–PDGF reporters to determine the temporospatial relationship of coordinated pathway action. We combined this information with bulk and single-cell RNA sequencing (scRNA-seq) to identify the host cell type and HH-responsive transcriptional targets of PDGF signaling and to determine single or coreceiver status for HH and PDGF input.
Methods
Complementary details, including detailed references, are available in the data supplement.
Animal Studies
All studies were approved by the New York University School of Medicine Institutional Animal Care and Use Committee in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International guidelines. Heterozygotes of the following genotypes were used: Gli1lZ, Gli1creERT2, PdgfraEGFP, and R26RtdTom; for lineage-tracing experiments, Gli1creERT2; R26RtdTom; and for coexpression studies of HH and PDGF pathway molecules, Gli1lZ;PdgfraEGFP dual reporters. For conditional Pdgfra deletion, we used Gli1creERT2/+;PdgfraFx/Fx mice. C57BL/6J mice were from The Jackson Laboratory. For HH pathway inhibition, we used a single dose of the anti–HH antibody 5E1 or daily cyclopamine (CPM). Conditional cre alleles were activated with a single tamoxifen dose.
Histology and Immunohistochemistry
Lung tissue harvest, processing, immunohistochemistry/immunofluorescence, and immunofluorescence quantification were performed as described (see data supplement). Images, taken with a Nikon Ni epifluorescence microscope and NIS-Elements software and an Akoya Vectra Polaris multispectral imaging system (Akoya Biosciences), were quantified using Visiopharm software.
Morphometric Analysis
Quantification of tissue:alveolar (T:A) airspace ratio, mean linear intercept (MLI), and septal tip:alveolar airspace ratio (secondary alveolar septum formation) was performed as described (21) with minor modifications.
Lung Single-Cell Suspensions
Lung digests were performed as described (21). For the scRNA-seq experiment, lungs of three mice per group were pooled. After digestion, cell count and viability were assessed.
Flow-assisted Cell Sorting
Single-cell suspensions of PgfraEGFP/+ lungs were sorted on the basis of EGFP (enhanced GFP) fluorescence. Cells from Pdgfra+/+ lungs served as controls. Cell viability was assessed using DAPI staining. EGFP expression and viability were examined using flow cytometric analysis with a BD Biosciences LSR II analyzer and FlowJo software (version 9). For cell sorting, 250,000 EGFP+ cells, including high and dim cell fractions, were collected using a BD Biosciences FACSAria II sorter and processed for mRNA extraction followed by either RNA sequencing (RNA-seq) or qRT-PCR.
RNA-Seq
RNA-seq and transcriptomic analysis of total RNA extracted from flow-sorted cells were performed as described by Spina et al., Nat Comm 2022 (see data supplement). Functional gene enrichment and pathway associations were identified using Ingenuity Pathway Analysis (Qiagen). MotifMap (https://motifmap.ics.uci.edu) (29) was used to identify putative Gli-binding sites in the proximity of genes altered by HH inhibition.
scRNA-Seq
Single cells (20,000/sample) were loaded onto a GEM code instrument (10× Genomics). GEMs were created according to the manufacturer’s protocol for library preparation and sequencing using a HiSeq 2500 instrument (Illumina). Reads were aligned using CellRanger 3 (10x Genomics) and downstream analysis done using iCellR as described by Spina et al., Nat Comm 2022 (see data supplement). Ligand–receptor interactions were assessed using CellChat (http://www.cellchat.org/) (30).
qRT-PCR
Total RNA extraction and qRT-PCR were performed as described (21). Relative gene expression was calculated using the 2−Δ(ΔCT) method. Genes were considered differentially expressed if Student’s t test indicated P < 0.05 in both Gapdh- and Hprt (hypoxanthine guanine phosphoribosyl transferase)–normalized samples.
Statistical Analysis
Statistical significance was determined using GraphPad Prism. Data were analyzed using the unpaired Student’s t test. Time-course data were tested using one-way ANOVA. Data are expressed as mean ± SD, with P values <0.05 considered to indicate statistical significance.
Results
HH Inhibition Induces Lung Phenotypes Resembling Those of PDGF Pathway KOs
We have shown that postnatal HH pathway activity peaks when secondary septum formation is maximal (P6–P10) and that HH inhibition by the pan–HH antibody 5E1 at P3 causes abnormal lung structure, impaired MyoFB differentiation, and decreased proliferation of Gli1+ cells and their progeny (21). The observation that HH inhibition before P3 also impaired secondary septum formation suggested inhibition of a pathway essential to alveolarization, such as PDGF signaling. Therefore, we confirmed our previous observation and tested whether earlier perinatal inhibition (Embryonic Day 18.5 [E18.5]) would further exacerbate the lung phenotype (Figure 1). These HH-inhibited lungs, examined at P8, exhibit only rare secondary alveolar septa and a saccular appearance, similar to Pdgfa−/− and Pdgfra−/− lungs (22) (Figure 1A). Consistent with this finding, morphometric analysis shows further increase in MLI, as well as further decrease of the T:A airspace ratio and the septal tip:alveolar airspace ratio compared with lungs inhibited at later stages (P1 and P3) (Figure 1B). To test whether HH pathway inhibition at a different level reproduces the 5E1-induced lung phenotype, we used the Smo inhibitor CPM. Treatment with CPM from P3 to P7, but not its control tomatidine, resulted in a similar lung phenotype with enlarged airspaces, increased MLI, and decreased T:A airspace ratio, accompanied by decreased expression of HH transcriptional targets (Gli1, Ptch1, Hhip) and the MyoFB marker Acta2 (actin alpha 2, smooth muscle) (see Figures E1A and E1B in the data supplement). The similar impairment in septal MyoFB differentiation produced by loss of either HH or PDGF signaling suggests these pathways may affect each other.
Figure 1.

HH (Hedgehog) inhibition induces lung phenotypes resembling those of PDGF pathway knockout mice. Late embryonic HH inhibition induces a saccular lung phenotype, similar to PDGFa−/− and PDGFRa−/− lungs (22, 23). Lungs of C57BL6/J mice, treated with 5E1 or IgG control at Postnatal Day 3 (P3), P1, and Embryonic Day 18.5 (E18.5), were harvested at P8 for morphology and morphometry (n = 3–8/group and condition). (A) Hematoxylin and eosin–stained lung sections demonstrate enlargement and simplification of air spaces after HH inhibition, which is most pronounced with treatment at E18.5. (B) Lung morphometric assessment shows time point–dependent increase in MLI, as well as decrease in T:A ratio and of secondary alveolar septa number per airspace with HH inhibition. Data are mean ± SD. P values <0.05 indicate statistical significance. Scale bars, 50 μm. MLI = mean linear intercept; PDGF = platelet-derived growth factor; PDGFa = platelet-derived growth factor subunit A; PDGFRa = platelet-derived growth factor receptor alpha; T:A = tissue area per alveolar area.
Joint Expression of HH and PDGF Signaling Intermediates Coincides with MyoFB Accumulation during Postnatal Alveolar Septation
Given the observed similarities in HH and PDGF phenotypes, we explored whether these pathways may affect each other first by testing for coexpression of their pathway molecules over the time course of postnatal lung development using Gli1lacZ/+; PdgfraEGFP/+ dual reporter mice. We used lacZ expression from the Gli1 locus as a faithful indicator of HH pathway activity and Pdgfra-EGFP expression to identify cells capable of receiving PDGF signals, as no unambiguous direct target for PDGF pathway activity has been established. We assessed late embryonic (E18.5), postnatal (P0, P2, P5, P8, P11, and P14), and adult (P33) mice, on the basis of the observations that MyoFBs appear in the saccular alveolar walls at the beginning of alveolarization (31) and that ∼50% of Gli1+;SMA+ MyoFBs are Pdgfra positive at P4 and P6 (21). At the different time points, alveolar Gli1+;Pdgfra− (potential single HH receiver), Gli1−;Pdgfra+ (potential single PDGF receiver), and Gli1+;Pdgfra+ (potential HH–PDGF coreceiver) cells were localized and quantified in postnatal lungs by immunofluorescence-based staining for nuclear b-galactosidase (Gli1 reporter) and nuclear EGFP (Pdgfra reporter) (Figures 2A and 2B; see Figure E2).
Figure 2.
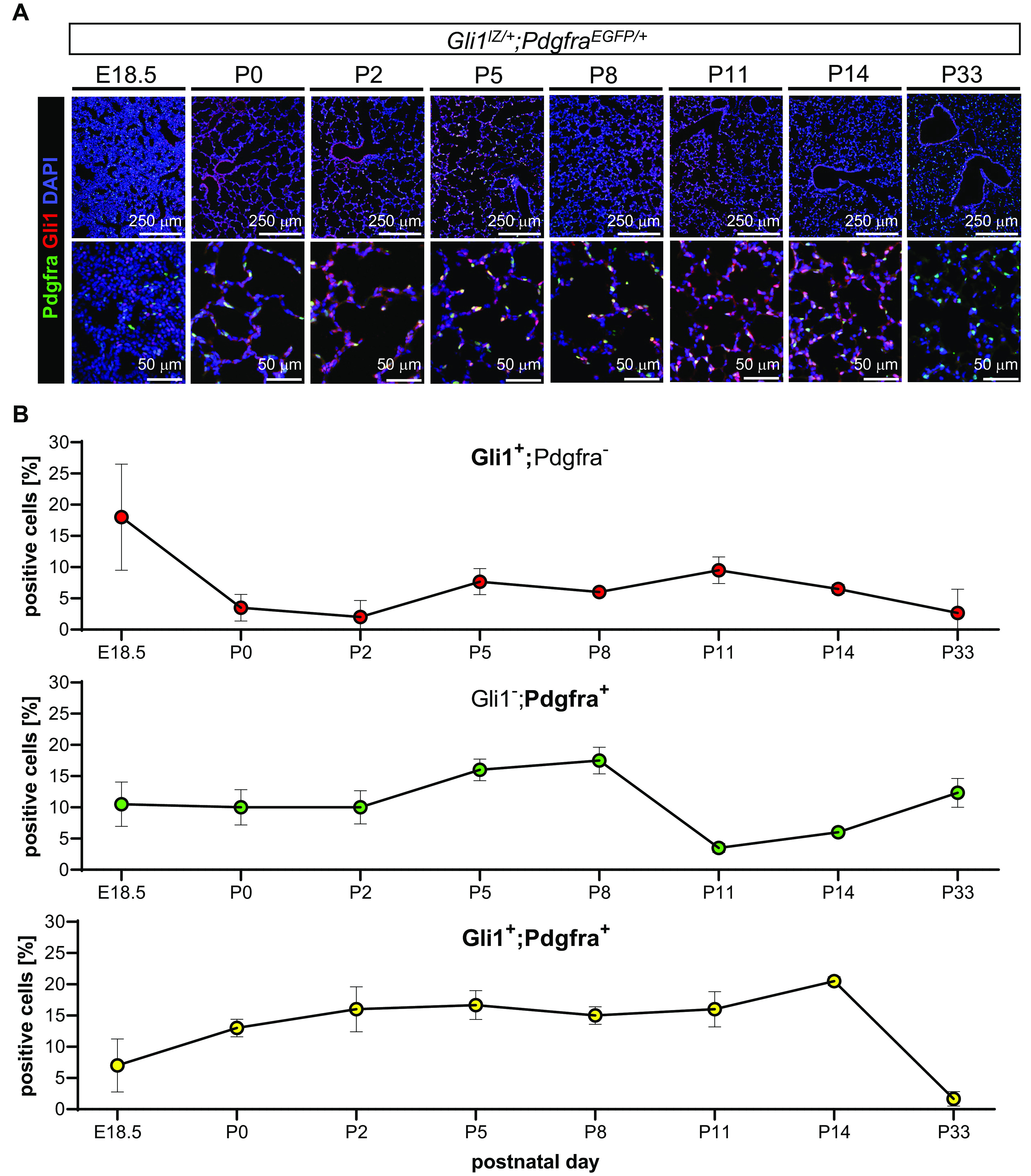
Joint expression of HH and PDGF signaling intermediates peaks during alveolarization in postnatal lung. (A) Alveolar Gli1+ (GLI family zinc finger 1) cells (red), Pdgfra+ cells (green), and Gli1+;Pdgfra+ cells (yellow) were detected using coimmunofluorescence in embryonic (E18.5) to adult lung (P33) tissue of Gli1lZ/+;PdgfraEGFP/+ reporter mice (n = 3 or 4/time point). (B) Quantification of cell percentages of Gli1+ cells (red), Pdgfra+ cells (green), and Gli1+;Pdgfra+ cells, expressed as ratio of single- or double-positive cells over all nucleated cells, shows significant representation of double-positive cells throughout alveolarization, with a steady increase in cell number from E18.5 to P14 (lower panel) and strong alignment with the typical location of alveolar myofibroblast at the growing alveolar septal tips. In contrast, Gli1+;Pdgfra+ cells are almost absent in adult lung. B-galactosidase (lZ) expression detects Gli1+ cells (red). EGFP expression detects Pdgfra+ cells. Nuclei are detected by DAPI (blue). Data are mean ± SD. Scale bars, 250 μm (upper panel) and 50 μm (lower panel). P = 0.01 (Gli1+;Pdgfra−), P < 0.001 (Gli1−;Pdgfra+), and P < 0.001 (Gli1+;Pdgfra+) (one-way ANOVA). EGFP = enhanced GFP.
As predicted from our prior studies, the percentage of alveolar Gli1+;Pdgfra− cells decreases from E18.5 to P0 but peaks during alveolarization and declines to low values in adult lung. The fraction of Gli1−;Pdgfra+ cells remains stable from E18.5 to P2, increases around P5–P8, falls around P11–P14, and stabilizes at about 12% of all cells in adult lung. The fraction of Gli1+;Pdgfra+ cells increases from 7% at E18.5 to 15% at P2 and constitutes up to 22% of all alveolar cells between P2 and P14. This suggests expansion of a cell population capable of responding to HH and PDGF during initiation of postnatal alveolar septation. High-magnification images show that the predominant sites of Gli1+;Pdgfra+ cells are the growing secondary septa, the location of the alveolar MyoFBs (Figure 2A). In contrast, in adult lung (P33), the double-positive cell percentage declines to ∼2%, whereas single PDGFRa+ cells remain prominent in the alveoli. We examined body weight and lung morphometry in Gli1lZ/+;PdgfraEGFP/+ and Gli1lZ/+;Pdgfra+/+ mice to test whether concomitant loss of one Gli1 and one Pdgfra allele alters overall growth and lung structure. Body weight was not different. T:A ratio and MLI were indistinguishable from wild type during the time window of our study, although the T:A ratio was slightly increased and MLI was lower in Gli1lZ/+;PdgfraEGFP/+ lungs around P11 but normalized in adulthood (P33) (see Figure E3).
HH Inhibition Reduces Pdgfra Expression and Numbers of Pdgfra+ Cells in Postnatal Lungs
Given the overlap of Gli1+ and Pdgfra+ cells during alveolarization, and that Pdgfra+;SMA+ MyoFBs are implicated as HH targets (25), we tested the hypothesis that HH signaling affects PDGF signaling–mediated events that control alveolar septation. We examined lungs after HH inhibition at P1 and E18.5. Expression analysis of HH and PDGF pathway genes 48 hours after HH inhibition shows that in P1-inhibited lung, Gli1 and Ptch1 are decreased, whereas expression of Pdgfa, Pdgfra, and Pdgfrb is unchanged (Figure 3A). In contrast, E18.5-inhibited lungs had significantly lower Pdgfra expression than control lungs, in addition to decreased Gli1 and Ptch1 expression (Figure 3B). These data indicate developmental stage–specific effects on Pdgfra expression. Next, we assessed cellular expression of Pdgfra and markers of two key cell types belonging to the Pdgfra lineage (26), SMA (MyoFBs) and ADRP (lipofibroblasts), in P8 lungs that were blocked with 5E1 at either P1 or E18.5 (Figures 3C and 3D). In P1-inhibited lungs, Pdgfra+ septal cell percentage is decreased. Many of the residual Pdgfra+ cells are SMA negative, as predicted from our prior work (21). In E18.5-inhibited lungs, the fraction of septal Pdgfra+ cells is further decreased, consistent with our gene expression data, and they are mostly SMA negative. Whereas ADRP expression was similar in P1-inhibited lungs, it was decreased in E18.5-inhibited lungs. We also examined proliferation in Pdgfra+ cells and found that the fractions of both Ki67+ and Pdgfra+ cells were decreased (Figure 4). However, the percentage of Ki67+;Pdgfra+ cells of all Pdgfra+ cells remained similar. Given the finding that conditional Pdgfra knockout (KO) at P2 in the Gli1 cell lineage decreases alveolar type 2 (AT2) epithelial cell number (32), we assessed the effect of HH inhibition on AT2 cells. HH inhibition at neither P1 nor E18.5 alters the fraction of AT2 cells at P8 (Figures 3C and 3D).
Figure 3.
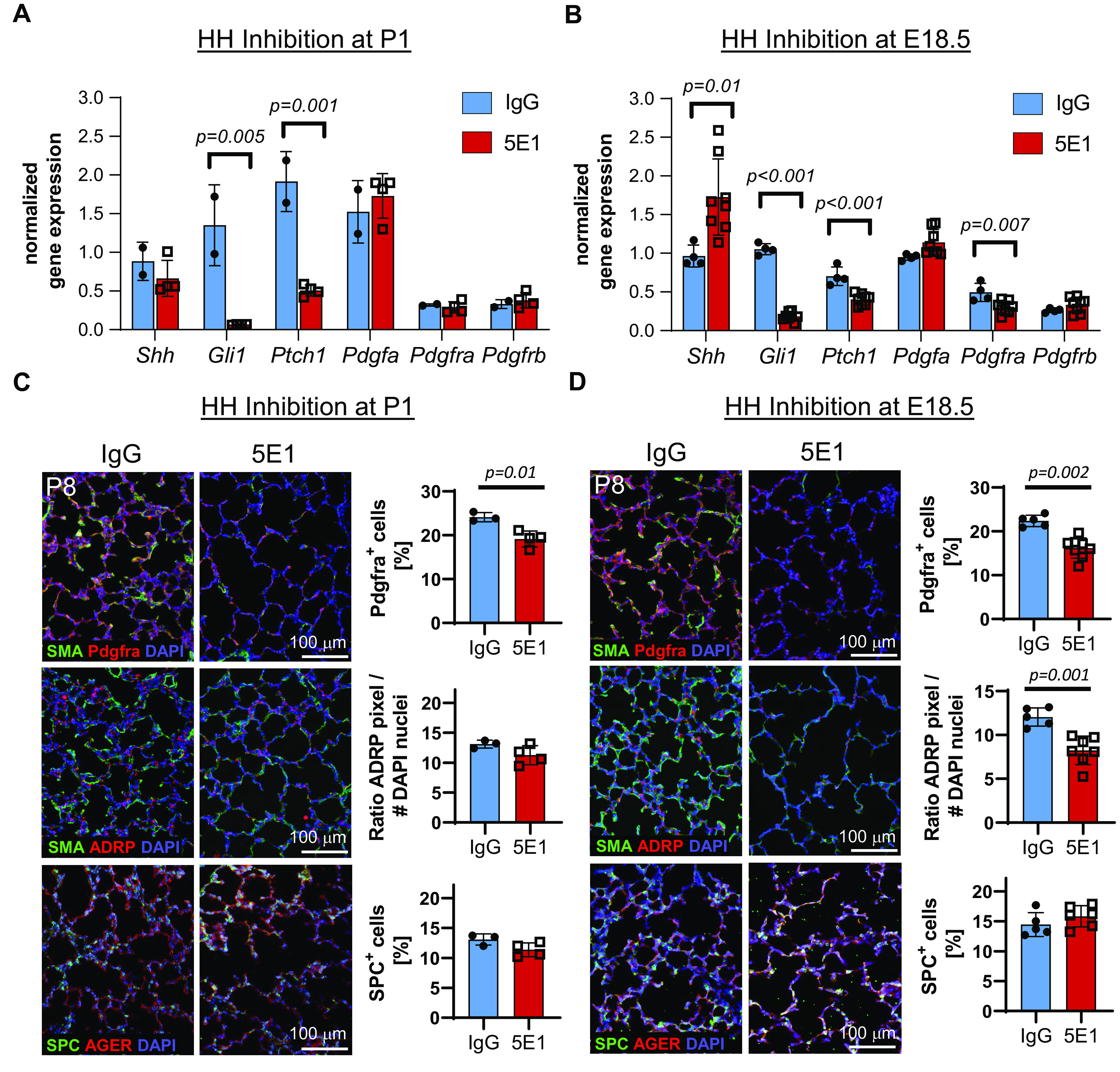
HH inhibition reduces Pdgfra expression and numbers of Pdgfra+ and Pdgfra-lineage cells in postnatal lungs. (A–D) To measure the effect of HH inhibition timing on gene expression of HH and PDGF pathway molecules, HH was inhibited at either P1 (A and C) or E.18.5 (B and D). Gene expression was assessed 48 hours after inhibition using qRT-PCR on total lung RNA at either P3 (A) or P1 (B). Tissue expression of selected cell markers (SMA [myofibroblasts], ADRP [lipofibroblasts], Pdgfra, SPC [alveolar type 2 cells], and AGER [alveolar type 1 cells]) was assessed using immunohistochemistry/immunofluorescence in the lungs at P8. Nuclei are stained with DAPI (blue). Quantitated image data and gene expression data are normalized mean ± SD. P values <0.05 (t test) indicate statistical significance; scale bars, 100 μm. ADRP = adipose differentiation-related protein; AGER = advanced glycosylation end-product specific receptor; Ptch1 = patched 1; Shh = Sonic Hedgehog; SMA = smooth muscle actin; SPC = surfactant protein C.
Figure 4.
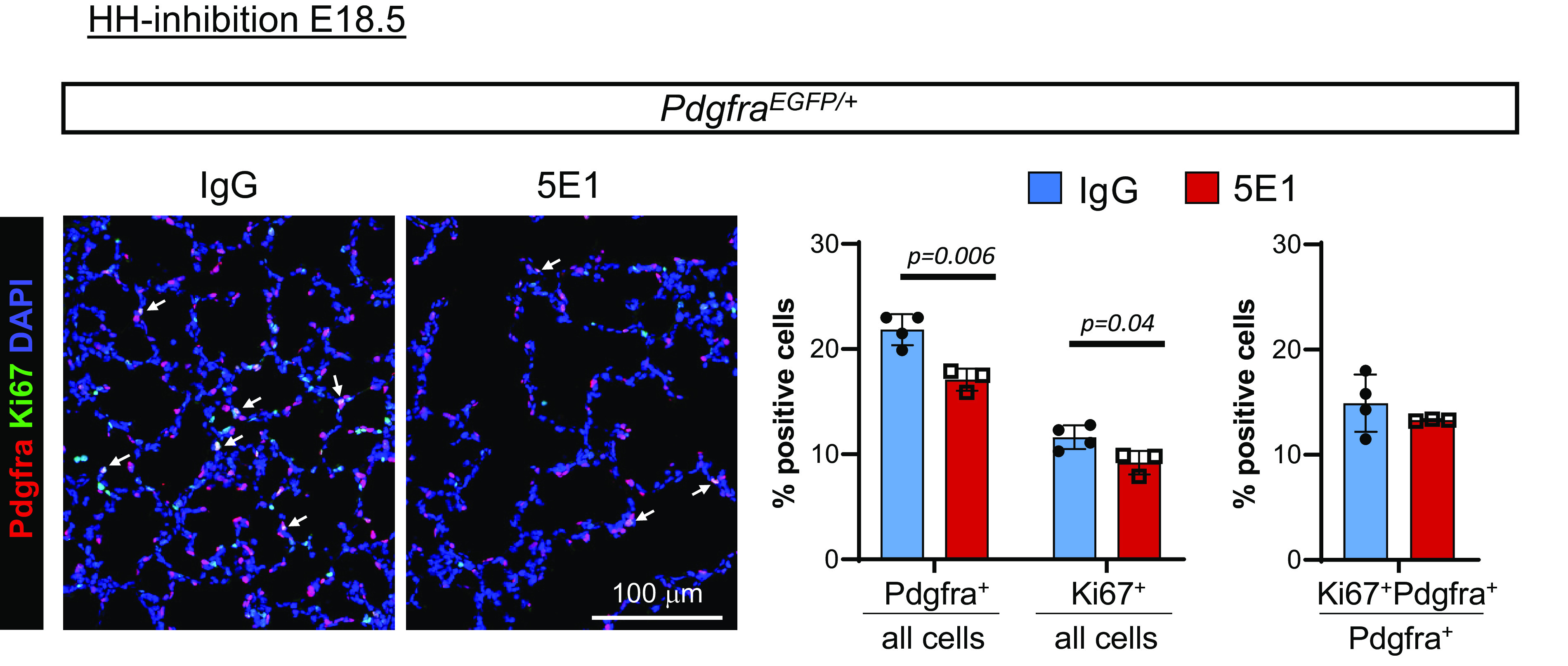
HH inhibition does not alter the proliferative index of Pdgfra-expressing cells. PdgfraEGFP/+ lungs from mice treated with 5E1 or IgG control at E18.5 were harvested at P8 and stained for GFP (red) and Ki67 (green). Nuclei are stained with DAPI (blue). Data are normalized mean ± SD. P values <0.05 (t test) indicate statistical significance; scale bar, 100 μm.
HH Inhibition Alters Transcriptional Target Genes of PDGF Signaling
To test if HH influences PDGF signaling through intracellular cross-talk, we evaluated the effect of HH inhibition on the transcriptomic profile of Pdgfra-expressing cells, looking for signs of reduced PDGF signaling. We inhibited HH signaling with 5E1 in PdgfraEGFP/+ mice at E18.5 and enriched for EGFP+ (Pdgfra-expressing) cells using FACS in lung single-cell suspensions at P8 (Figure 5). Flow cytometric evaluation shows that, as expected, HH inhibition decreases the fraction of total Pdgfra+ and Pdgfrahigh cells (presumably MyoFBs) (Figure 5A). RNA-seq of total RNA from sorted Pdgfra+ cells revealed 1,905 differentially expressed genes in 5E1-treated lungs (two-fold change, adjusted P < 0.05), of which 1,257 were upregulated and 648 were downregulated (Figure 5B). As seen on the volcano plot, Hhip and HH pathway receptor Ptch1 were among the most differentially expressed genes; both are direct transcriptional targets of HH signaling and were downregulated, confirming HH inhibition in Pdgfra-expressing cells (Figure 5C). Pathway enrichment analysis identified several gene sets indicative of altered PDGF signaling and associated with lung diseases (Figure 5D; see Table E1). Further analysis showed alterations in genes relevant to MyoFB (Acta2, Actg2 [actin gamma 2, smooth muscle], Fgf18 [fibroblast growth factor 18], Wnt5a, Igf1 [insulin like growth factor 1], Tgfbi [transforming growth factor beta induced], and Aspn [asporin]) and fibroblast (Vcan [versican] and Col13a1 [collagen type XIII alpha 1 chain]) function (Figure 5E). Expression of Pdgfra, Pdgfrb, and Plin2 (perilipin 2) (lipofibroblast markers) was not significantly different. Two genes that were significantly altered by HH inhibition are Klf9 (KLF transcription factor 9) and Txnip (thioredoxin interacting protein). These genes, together with Klf2 and Cdh11 (cadherin 11), are direct transcriptional targets of PDGF signaling in mouse embryonic fibroblasts (10). Next, we compared our transcriptomic data set from P8 with a published data set of PdgfraEGFP sorted cells from P7 after hyperoxia-induced postnatal disruption of alveolarization starting at P1 (33), a model in which altered PDGF signaling has been implied (27). Comparison of differentially expressed genes shows an overlap of a total of 281 genes. Among them are several key MyoFB markers (Acta2, Actg2, Fgf18, Wnt5a, Igf1, and Tgfbi), the HH target Ptch1, and one of the PDGF targets, Cdh11, which are downregulated with both HH inhibition and hyperoxia (see Figure E4). These data suggest that other perturbations of postnatal lung development affect expression of similar MyoFB-relevant genes as with HH loss in recipient cells of PDGF signaling.
Figure 5.
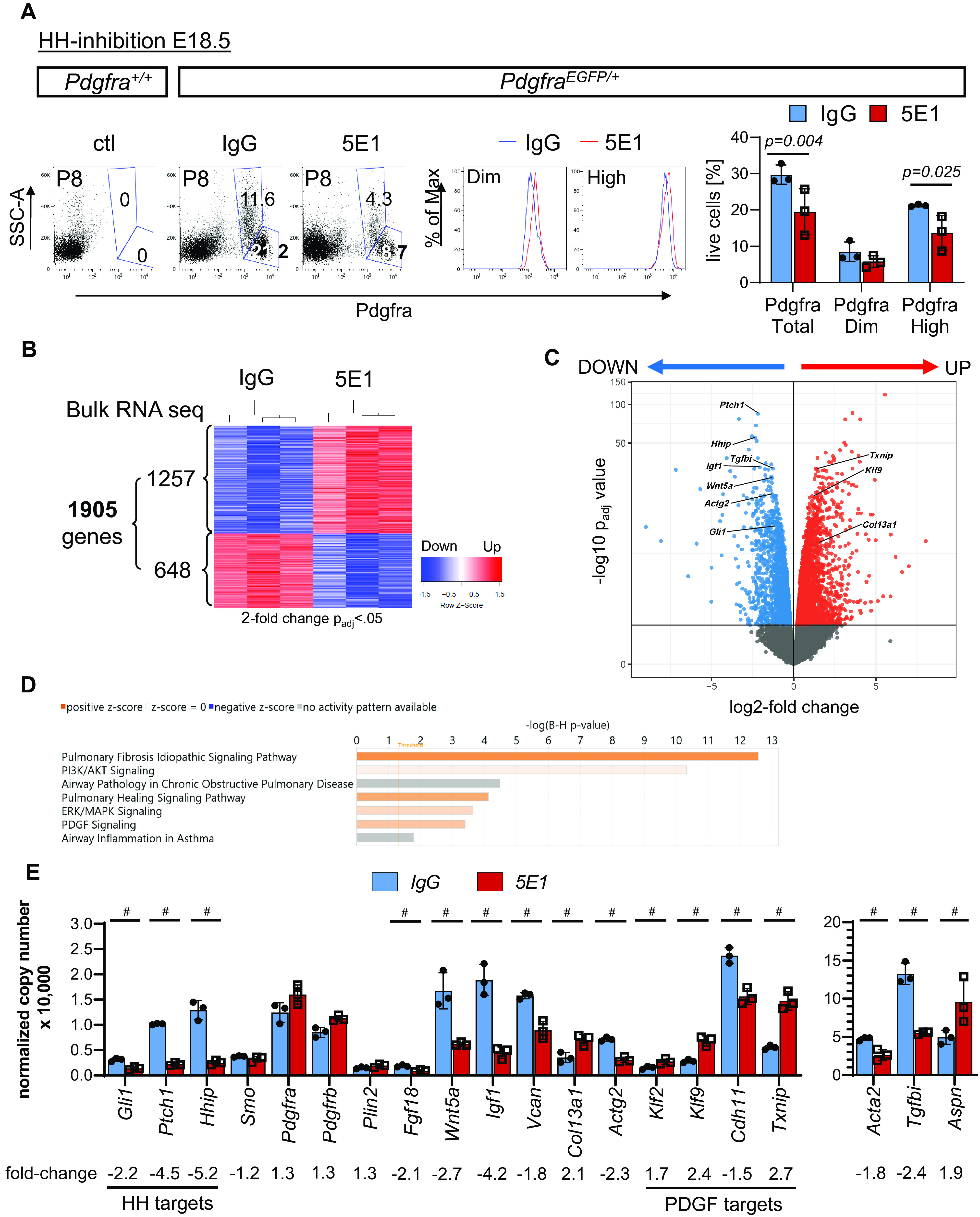
HH inhibition alters key differentiation markers and PDGF pathway targets in Pdgfra-expressing cells. (A) Whole-lung single-cell suspensions of PdgfraEGFP/+ mice, treated with 5E1 or IgG control at E18.5, were sorted for EGFP+ (Pdgfra-expressing) cells. Representative plots of Pdgfra+/+ control lungs (left panel) and PdgfraEGFP/+ lungs (middle panels) show detection of PdgfraDim and PdgfraHigh cell populations. Quantification of PdgfraDim and PdgfraHigh cell populations shows decreased number of total Pdgfra- and PdgfraHigh-expressing cells. (B) RNA-seq analysis of Pdgfra+ sorted cells from P8 PdgfraEGFP/+ lungs, treated with 5E1 or IgG at E18.5, identified 48,216 genes, of which 1,905 were significantly different. (C) Volcano plot of up- and downregulated genes (fold change > 2; adjusted P (Padj) < 0.05). (D) Pathway enrichment analysis using Ingenuity Pathway Analysis shows enrichment in gene sets for PDGF signaling and lung diseases. (E) Effect of HH inhibition on key genes of HH (Gli1, Ptch1, Hhip, and Smo) and PDGF signaling molecules (Pdgfra and Pdgfrb); on markers of Pdgfra-lineage cells: lipofibroblasts (Plin2), myofibroblasts (Acta2, Actg, Wnt5a, Fgf18, and Igf1), and matrix fibroblasts (Col13a1, Vcan, and Fgfra2); and on potential transcriptional targets of PDGF signaling (Klf2, Klf9, Cdh11, and Txnip). Data are normalized mean ± SD. #Adjusted P < 0.001 (t test). Acta2 = actin alpha 2, smooth muscle; Actg = actin gamma 2, smooth muscle; Aspn = asporin; B-H = Benjamini-Hochberg; Cdh11 = cadherin 11; Col13a1 = collagen type XIII alpha 1 chain; ERK = extracellular signal-related kinase; Fgf18 = fibroblast growth factor 18; Fgfra2 = fibroblast growth factor receptor A2; Hhip = hedgehog inhibitory protein; Igf1 = insulin like growth factor 1; Klf = KLF transcription factor; MAPK = mitogen-activated protein kinase; Max = maximum; Plin2 = perilipin 2; RNA-seq = RNA sequencing; Smo = smoothened; SSC-A = side scatter area; Tgfbi = transforming growth factor beta induced; Txnip = thioredoxin interacting protein; Vcan = versican.
Next, we established the expression time course for Klf2, Klf9, Cdh11, and Txnip throughout postnatal lung development (see Figure E5A). Cdh11 expression peaks at P8, when alveolarization is maximal, and declines thereafter. Klf2 and Klf9 nadir around P4-P6, followed by a peak around P16–P18 during maturation. Txnip expression decreases after birth and remains low thereafter. Given the temporal changes seen for the four genes during alveolarization, we next assessed whether timing of HH inhibition affects their expression. We compared their expression in total RNA 48 hours after HH inhibition at E18.5 or P3 and found that expression of Klf9, Cdh11, and Txnip significantly changed but showed patterns of opposite directionality at the two time points (indicated by arrows in Figure E5B). We validated their response pattern to HH inhibition by treatment with the Smo inhibitor CPM starting at P3 and found that the expression pattern of Klf2, Klf9, Cdh11, and Txnip mirrors the findings of 5E1 treatment at P3 (see Figure E1B). To ensure that allelic loss of Pdgfra did not affect expression of HH and PDGF pathway molecules and targets, we compared their gene expression in PdgfraEGFP/+ and Pdgfra+/+ lungs using qRT-PCR and found no significant differences except the expected decrease in Pdgfra expression (see Figure E6). We also performed a search for putative Gli-binding sites in these genes using MotifMap (29) and identified sites in most of them (see Table E2).
To test the effect of reduced PDGF signaling in the potential HH–PDGF coreceiver cells, we conditionally ablated Pdgfra in Gli1-expressing cells in Gli1creERT2/+;PdgfraFx/Fx mice at P2 and assessed lung phenotype and gene expression at P8 (see Figure E7). Conditional Pdgfra deletion histologically phenocopied HH-inhibited lungs resulting in enlarged airspaces, increased MLI, and decreased T:A ratio. However, expression of the HH targets Ptch1 and Hhip remained unaltered, and similarly Gli1 was reduced only because of heterozygous presence of Gli1creERT2 transgene. Decreased Pdgfra expression indicated successful conditional gene knockdown. As expected, the MyoFB marker Acta2 was significantly reduced. Of the potential PDGF pathway targets, Klf9 was upregulated and Cdh11 downregulated, a pattern similar to that produced by HH loss at P3.
Collectively, these data suggest that HH input alters both transcriptional targets of PDGF signaling and genes important to MyoFB and fibroblast function in postnatal lung.
To uncover the HH effect on different cell populations, including Gli1- and Pdgfra-expressing cells in postnatal lung, we labeled Gli1 cell lineage in Gli1creERT2;R26tdTom lungs at P1 before HH inhibition at P3 and performed scRNA-seq of whole-lung single-cell suspensions at P6. After alignments of reads and quality control measures, nonhierarchical clustering and KNetL-based dimensionality reduction were performed and revealed 23 cell clusters, for which origin (epithelial, mesenchymal, or immune cell) and individual cell types could be identified from the most differentially expressed genes using cell annotations from postnatal lung (LungGENS) (https://research.cchmc.org/pbge/lunggens/) (34) and known marker genes (Figure 6A; see Figures E8 and E9). On closer examination, expression of tdTom (Gli1 lineage) is almost completely restricted to mesenchymal clusters and most prominent in MyoFBs, MFBs, and mesothelium (Figure 6B; see Figure E10A), consistent with other results (18, 21). As expected, the ligands Pdgfa and Shh are expressed in epithelial cells (alveolar type 1 [AT1] > AT2 and ciliated/club), while downstream PDGF pathway (Pdgfra) and HH pathway (Ptch1 and Hhip) components are expressed in mesenchymal cells (Figure 6B). Acta2 expression is restricted to smooth muscle cells and MyoFBs (MyoFB1/2 and MyoFBprog), the latter cells also expressing Pdgfra, Ptch1, and the Gli1 lineage marker tdTom. Pdgfra is also expressed in MFBs (MFB2/3), some of which also express the lipofibroblast marker Plin2, even though a distinguishable cluster for this cell type could not be established in our analysis. However, we were able to distinguish two MyoFB populations, for which distinct alveolar locations have been described (35): “ductal” MyoFBs (MyoFB2) expressing Hhip and tdTom but mostly negative for Pdgfra expression, which likely correspond to the 50% of Gli1+;SMA+ alveolar MyoFBs that are Pdgfra negative (21), and “alveolar” MyoFBs (MyoFB1 and MyoFBprog) that express Fgf18 and Wnt5a, for which expression was decreased only in the Pdgfra-expressing cells. One marker specific to ductal MyoFBs is Aspn, a mesenchymal differentiation marker, implicated in lung fibrosis (36). Col13a1, on the other hand, localizes mostly to MFBs (MFB2/3), which also express Tcf21 (transcription factor 21), similar to postnatal and bleomycin-injured lungs (34, 37). We also localized expression of the PDGF targets (Klf2, Klf9, Cdh11, and Txnip), which we had identified in the transcriptome of Pdgfra-expressing cells. Although the majority of Klf9- and Cdh11-expressing cells are in MyoFB and MFB clusters, Klf2 and Txnip expression were greater in other cell types, including endothelial cells and immune cells. Next, we examined the effect of HH inhibition on each identified cluster (see Figures E10B and E10C). MyoFB (MyoFB1/MyoFB2 and MyoFBprog) and MFB (MFB2) clusters are among the clusters with the most transcriptomic alterations and change in cell numbers after HH inhibition. HH inhibition decreased numbers of MyoFBs (MyoFBprog), which express Pdgfra and Acta2, and increased numbers of MFBs (MFB2), which contain cells expressing the lipofibroblast marker Plin2, hinting at a potential shift from Pdgfra+ MyoFBs to lipofibroblasts, as observed with hyperoxia (33).
Figure 6.
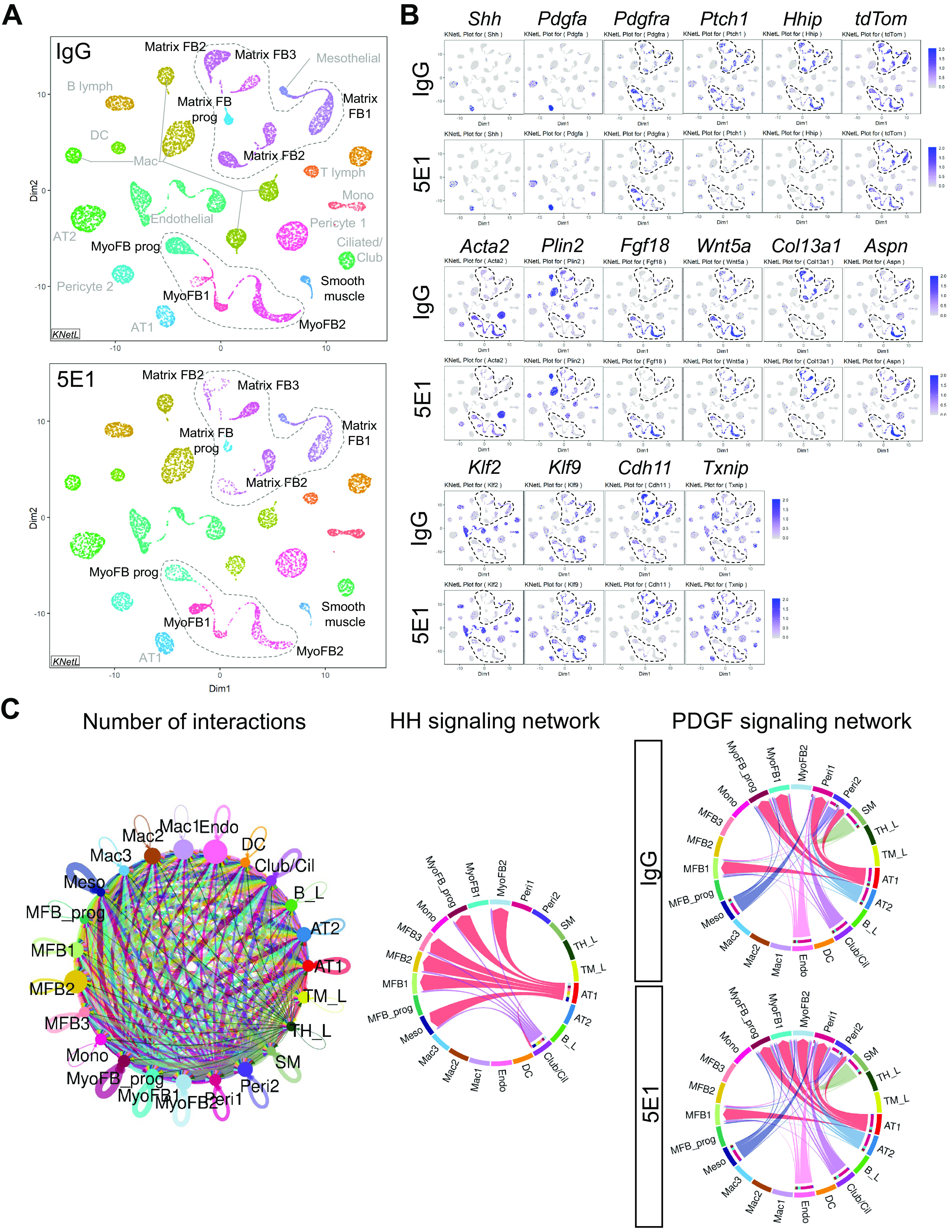
Single-cell transcriptomic analysis identifies HH–PDGF targets in several postnatal lung mesenchymal cell populations. Whole-lung single-cell suspensions of Gli1creERT2;R26tdTom lungs, lineage labeled at P1 and 5E1 or IgG treated at P3 (n = 3/group), were pooled per condition and loaded onto the 10x Genomics Chromium platform for construction of sequencing libraries. Data analysis was performed using the iCellR pipeline. (A) All major lung cell types were identified by hierarchical clustering, dimensionality reduction, and determination of cluster cell types on the basis of key cell identification markers following the annotations from the LungGENS study (34). Shown are KNetL plots of the 23 clusters, obtained through hierarchical clustering comparing IgG control-treated (top panel) and 5E1-treated (bottom panel) lungs. (B) Selected KNetL plots of 5E1- and IgG-treated lungs illustrate overlap of key pathway genes of HH and PDGF signaling in mesenchymal cell clusters (dotted circles), such as myofibroblasts (MyoFBs) (Acta2) and lipofibroblast (Plin2) and matrix fibroblast (MFB) clusters, and effects of HH inhibition in these clusters. tdTom expression reveals tamoxifen-labeled Gli1 cell lineage. Pdgfra-expressing clusters show changes in PDGF signaling targets (Klf2, Klf9, Txnip, and Cdh11). Gene combinations identify specific clusters; for example, Aspn and Hhip present, Pdgfra absent marks cluster MyoFB2 (potential HH single-receiver cells). (C) Sets of differentially expressed genes for each cluster and condition (5E1 and IgG) were analyzed for ligand receptor interactions using CellChat analysis (see Methods). Chord plots depict the total number of projected interactions between different cells types (left), the cell-specific interactions for the HH signaling network (middle), and the PDGF signaling network with and without HH inhibition (right) with significant communication probability (P < 0.05). Identified are mesenchymal cell types that receive single input of either HH ligand (MyoFB2, MFB2, MFB3, and Meso) or PDGFa ligand (MyoFB1) or combined input from HH and PDGFa (MyoFBprog and MFB1). Of note, cluster MyoFB2 gains PDGFa input with HH inhibition. B_L =B-Lymphocyte; Cil = ciliated; DC = dendritic cell; Endo = endothelial; FB = fibroblast; Mac = macrophage; Meso = mesothelial; Mono = monocyte; Peri = pericyte; SM = smooth muscle; TH_L = T helper lymphocyte; TM_L = T IgM-Fc lymphocyte.
To uncover potential pathway interactions between distinct cell types, we analyzed our scRNA-seq data set for ligand–receptor interactions for all annotated 23 cell clusters with and without HH inhibition (5E1 vs. IgG) using CellChat (30) (Figure 6C; see Figure E11 and Table E3). CellChat detected a total of 384 significant ligand–receptor interactions between the 23 cell clusters, which were further classified into 75 signaling pathways in the IgG group and reduced to 74 pathways in the 5E1 group because of loss of HH pathway interactions. We further examined interactions for HH and PDGF signaling to determine the signal input among different target cell populations. MyoFB2, MFB2, MFB3, and mesothelial cells receive single input of HH ligand; MyoFB1 receives single input of PDGFa ligand; and MyoFBprog and MFB1 clusters received combined input from HH and PDGF. Of note, cluster MyoFB2 gained PDGF input with HH inhibition. These CellChat findings further support the presence of both single and dual recipients of HH and PDGF signals.
Our scRNA-seq data confirm the cellular overlap of HH and PDGF pathway expression we observed in postnatal Gli1lZ;PdgfraEGFP reporter lungs during alveolarization and show significant transcriptomic changes in response to HH input in distinct MyoFB and MFB clusters of functional relevance (e.g., Pdgfra/Acta2 MyoFBs).
Discussion
Formation of alveoli during postnatal lung development relies on the well-orchestrated activity of patterning pathways that signal to the cells of Pdgfra lineage, such as MyoFBs, lipofibroblasts, and MFBs. Several individual pathways, namely, HH, PDGF, FGF (fibroblast growth factor), HGF (hepatocyte growth factor), and WNT5A, have been causally linked to normal postnatal lung development (21–23, 38, 39). An increasing diversity of signaling among multiple cell types during lung development has been uncovered by scRNA-seq and cell lineage tracing (9, 34, 35). Studies such as ours that combine the power of unbiased high-throughput single and bulk RNA-seq, advanced bioinformatics with reporters for single and combined pathway input, and means of conditional pathway KO help establish a descriptive framework, from which we can begin to understand how pathways coordinate to regulate cell function. This may serve to untangle complexity and provide insights for ameliorating impacts of aberrant signal coordination during perinatal lung injury, as seen with BPD, mechanical ventilation, and hyperoxia.
The intersection of HH and PDGF signaling on alveolar MyoFBs, lipofibroblasts, and MFBs has hitherto remained obscure. Here we reveal joint expression of HH and PDGF pathway intermediates and coordinate activity in alveolar cells signifying their potential to receive concomitant HH and PDGF signal input during alveolarization, a period critical to postnatal lung development. Our results not only demonstrate the striking similarity between the lung phenotypes detected with functional loss HH or PDGF signals but provide substantial evidence of the profound effect of altering HH signaling on the transcriptome and differentiation of Pdgfra-expressing MyoFBs and lipofibroblasts. We show that HH signaling regulates PDGF transcriptional targets with roles in developing or injured lung. Collectively, our data support the central hypothesis that HH signaling and PDGF signaling cooperatively regulate key mesenchymal cells during postnatal lung development.
Several prior observations support this concept. Lungs deficient in the HH pathway molecule SuFu exhibit decreased Pdgfra expression and lack MyoFBs at sites of secondary septum formation (20). Gli1-binding sites are present in the promoters of Pdgfra and Acta2 (20, 40). Embryonically labeled Gli1 cell lineages generate Pdgfra+ MyoFBs in alveolar septa and selective ablation of Pdgfra in the Gli1 lineage impairs postnatal alveolarization (18, 32). Notably, expression of Vcan, a provisional matrix proteoglycan, present in alveolar mesenchyme during saccular and alveolar stages of lung development (41) is similarly decreased, by Pdgfra KO in the Gli1 lineage or by HH inhibition in Pdgfra+ (present findings) or Gli1+ cells (21).
Our transcriptomic data indicate that HH input affects multiple Pdgfra lineages and is most pronounced within MyoFB populations. There, it increases differentiation genes (e.g., Acta2, Actg2) and genes indicative of alveolar MyoFB cell fate, such as Fgf18 (42). Fgf18-expressing MyoFBs are part of the Gli1 lineage, and their disappearance from the alveolar walls during maturation is an important step in the generation of adult lung. Wnt5a is another gene required for MyoFB differentiation during postnatal lung development (39). Our scRNA-seq data indicate that Wnt5a is expressed in MyoFBs that also express Fgf18 and the mesenchymal differentiation marker Aspn (36). In contrast to most of the MyoFB markers, expression of MFB markers (e.g., Col13a1) is increased after HH inhibition, suggesting a shift from MyoFB to MFB differentiation in the HH-inhibited lung.
Our data support the concept that HH signaling coregulates PDGF transcriptional targets. In HH-inhibited lungs, we identified the transcription factors Klf2, Klf9, and Txnip, and the cell–cell junction molecule Cdh11, as potential transcriptional targets of HH signaling. Importantly, previous studies have linked each of these genes to PDGF regulation and/or lung development and disease processes. First, they are all direct transcriptional targets of PDGF signaling (10) and linked to development and disease phenotypes. Klf2−/− lungs exhibit developmental arrest in the late canalicular stage (11). Klf9 (Bteb1) KO decreases bleomycin-induced lung injury (12). CDH11 expression is increased in idiopathic pulmonary fibrosis and bleomycin-injured lungs (43), and abrogating Cdh11 was protective in models of the latter (13). Second, Klf9, Cdh11, and Txnip show significant changes in Pdgfra+ lung cells between E16 and P28 (26), mirroring our expression time-course data. Third, our postnatal scRNA-seq data set localizes Klf9 and Cdh11 expression to MyoFB and fibroblast clusters that are known to be regulated by PDGF signaling. Fourth, conditional Pdgfra loss in Gli1-expressing cells also alters these targets. Collectively, these data indicate that Klf2, Klf9, Cdh11, and Txnip are targets for HH refining PDGF signaling, supporting the concept of intracellular cross-talk as one way the pathways interact. The presence of Gli1-binding sites around these HH-responsive genes (MotifMap) and the time-dependent response pattern of these genes to HH loss further suggest convergence on the same promoters. These four PDGF targets might also serve as a new way to assess PDGF pathway activity.
In the present study, we observed that the joint expression of HH and PDGF pathway intermediates coincides with MyoFB accumulation during postnatal alveolar septation and that the conditional loss of Pdgfra in these cells impairs alveolarization and alters expression of the MyoFB marker Acta2 and the potential PDGF targets. This suggests that these pathways converge within, and possibly generate, a shared cell lineage of HH- and PDGF-coresponsive cells during a defined time window in postnatal lung development (see Figure E12). This idea is further supported by our CellChat-based ligand–receptor analysis showing that lung mesenchymal cells can be divided into three different groups on the basis of receiving either single HH or PDGF ligand input (potential single receivers) or combined input (potential HH–PDGF coreceivers). Pathway integration can result in unique target gene expression, as has been shown for HH and EGF (epidermal growth factor) signaling in cancer, in which the combined HH–EGF signature was distinct from individual HH and EGF signatures (44). Similarly, PDGFRa signaling has been shown to activate ERK (extracellular signal-regulated kinase)/JUN (Jun proto-oncogene, AP-1 transcription factor subunit) and synergize with HH signaling in gastrointestinal stroma tumors (45). One report has suggested that during alveolarization, PDGF and SHH gradients direct lung fibroblasts along the elongating alveolar septa (46).
In this work, we aimed to better define the principles underlying intersection of HH and PDGF signaling, including degree of interdependence, dynamics of pathway dominance, and synergy on cellular targets. Here we show that only very early postnatal HH inhibition phenocopies the saccular phenotype of Pdgfa−/− and Pdgfra−/− lungs, whereas later inhibition (P1 and P3) results in less severe phenotypes and less impaired alveolar septation. One possible explanation for this phenomenon is that early PDGF signaling is dependent on HH signaling and not able to compensate for the defects caused by HH loss. In support, the PDGF target response we observed 48 hours after HH loss at E18.5 was different from the response after loss at P3. Furthermore, loss of HH input resulted in PDGF signal gain in MyoFB cluster MyoFB2, but no compensatory PDGF input into MFB clusters MFB2 and MFB3, suggesting an equally important contribution of HH-induced fibroblast differentiation to alveolar septum formation apart from MyoFB differentiation. Another possibility is that HH and PDGF act independently to achieve similar outputs, but because Pdgfa expression is low in immature AT1 cells, HH is the dominant of the two factors at these early time points (47). Three observations suggest HH dominance over PDGF signaling: HH pathway intermediates are expressed in more mesenchymal cell types than are PDGF pathway intermediates. HH targets were not altered by decreased PDGF signaling. And early genetic studies showed that the requirement for PDGF signaling is limited to postnatal lung development (22, 23), whereas HH signaling is required for both embryonic and postnatal lung development (16, 17, 19). Loss of HH or PDGF alone results in the most severe postnatal lung phenotype, saccular lung with absent secondary septa, suggesting against pathway synergy, which would require concomitant loss of both to induce this phenotype. Of note, two important aspects of our work demonstrate that the nature of their intersection in postnatal lung is likely more complex, making delineation of dominance and additive/synergistic effects challenging. First, the presence of several mesenchymal cell populations, in particular MyoFBs and fibroblasts, which receive either single or combined input of HH and PDGF, points to the impact of signal coordination in the regulation of their functions during alveolarization. Second, both pathways undergo substantial changes over a short time period in terms of expression and activity, rendering effects on recipient cells transient in nature. This could be explained by the timely constellation of pathway activity providing a specific signal to induce differentiation in a particular cell population, such as the proposed HH–PDGF coreceiver cells (see Figure E12). In support of this concept, our CellChat data show at P6 input of PDGF but not HH into MyoFB cluster MyoFB1, while successful Gli1 lineage labeling and HH target gene depression by HH inhibition at P1 indicate active HH signaling at that time.
One limitation of our study is that the data are largely correlative in nature. Further studies are needed to identify the intracellular mechanism(s) involved in the pathway cross-talk. One potential mechanism is by gene coregulation at the transcriptional level. The in vitro use of reporter constructs to establish the dependence of PDGF targets on Gli transcription factor binding, and site-directed mutagenesis of HH-responsive or PDGF-responsive promoter elements to determine synergistic or antagonistic convergence of both pathways and their intracellular hierarchy, are ways to test this hypothesis. Determination of intercellular pathway hierarchy will likely require in vivo conditional KO in single or coreceiver cells and lung phenotype rescue by constitutive pathway activation in the presence of inhibition of the other one. Another limitation is that we cannot completely exclude the possibility of late embryonic effects of HH inhibition at E18.5. However, the facts that HH pathway expression and activity significantly decrease before birth (15) and that E18.5 HH-inhibited postnatal lung shows normal AT1 and AT2 epithelial differentiation suggest that inhibition affected predominantly postnatal lung development.
The pathology of BPD lungs indicates perturbed mesenchymal signaling (4, 48) and could be explained largely by loss of either HH signaling or PDGF signaling. BPD lungs and models support the idea that loss of PDGF signaling is relevant (6, 26, 27, 33, 46). Our findings show that HH inhibition also causes a BPD-like saccular lung phenotype and thus reveals that HH signaling might be relevant in BPD. In support of this concept, Shh and Ptch1 are increased in an animal model of BPD (5). HHIP, an inhibitor of HH signaling, is increased in BPD lungs, and certain genetic variants of HHIP are protective against BPD (49). Our transcriptomic data from Pdgfra-expressing cells and the comparison with a data set from the hyperoxia model show that HH inhibition affects expression of several genes (Wnt5a, Tgfbi, Igf1, Acta2, and Actg2) that are also altered by hyperoxic postnatal lung injury in Pdgfra-expressing cells with a “MyoFB signature” (33). The directionality of these changes raises the possibility that HH inhibition may mimic the hyperoxic effects on Pdgfra cell lineage. Another example is the MFB marker Col13a1, for which increased numbers of expressing cells have been reported with hyperbaric postnatal lung injury (50).
Conclusions
Our data provide evidence for the critical importance of HH signaling for MyoFB/fibroblast function during the PDGF signaling–mediated events that lead to secondary alveolar septum formation. On the basis of our present and prior findings, we propose a model in which time-dependent single and combined HH and PDGF pathway input controls alveolar MyoFB differentiation from progenitors during secondary alveolar septum initiation and elongation in early postnatal lung (see Figure E12). Finally, our findings highlight a potential role for HH signaling in perinatal lung diseases associated with impaired alveolarization.
Acknowledgments
Acknowledgment
The authors thank Adriana Heguy and the Genome Technology Center for expert library preparation and sequencing, the Applied Bioinformatics Laboratories for providing bioinformatics support and helping with data analysis and interpretation, and the New York University School of Medicine High Performance Computing Facility for computing resources. The authors thank Drs. Alexandra Joyner (Memorial Sloan-Kettering Cancer Center) and Sumru Bayin (Gurdon Institute, University of Cambridge, United Kingdom) for invaluable experimental support and Drs. Pamela Cowin and Leopoldo Segal for critical review of the manuscript. M.C.K. thanks the Will Rogers Foundation and the Stony Wold-Herbert Fund for their generous support.
Footnotes
Supported by National Heart, Lung, and Blood Institute grants K08-HL128842 and R56-HL151700 (M.C.K. and V.M.), National Cancer Institute Cancer Center Support Grant P30-CA016087 (C.A.L., V.M., and M.A.), National Eye Institute grants R01-EY031383 and R01-EY033495 (J.N.), a Will Rogers Foundation Research Fellowship (M.C.K.), and a Stony Wold-Herbert Fund Grant-in-Aid (M.C.K.). The New York University microscope core Genome Technology Center and the Applied Bioinformatics Laboratories are partially supported by National Cancer Institute New York University Cancer Center Support Grant P30CA016087 (M.C. and A.K.).
Author Contributions: M.C.K. conceived the study and designed the experiments with input from C.A.L. and J.S.M. T.-A.Y., C.A.L., J.N., M.C., M.A., J.A.N., and M.C.K. performed the experiments. T.-A.Y., C.A.L., J.N., A.K.-J., Z.L., C.B., V.M., J.A.N., and M.C.K. analyzed the data. M.C.K. wrote the manuscript.
Data accessibility: All the raw sequencing reads and processed files for bulk RNA sequencing and single-cell RNA sequencing are deposited in Gene Expression Omnibus (accession number GSE207062).
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0269OC on January 24, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Burri PH. Structural aspects of postnatal lung development—alveolar formation and growth. Biol Neonate . 2006;89:313–322. doi: 10.1159/000092868. [DOI] [PubMed] [Google Scholar]
- 2. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med . 2001;163:1723–1729. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 3. Wong PM, Lees AN, Louw J, Lee FY, French N, Gain K, et al. Emphysema in young adult survivors of moderate-to-severe bronchopulmonary dysplasia. Eur Respir J . 2008;32:321–328. doi: 10.1183/09031936.00127107. [DOI] [PubMed] [Google Scholar]
- 4. Ahlfeld SK, Conway SJ. Aberrant signaling pathways of the lung mesenchyme and their contributions to the pathogenesis of bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol . 2012;94:3–15. doi: 10.1002/bdra.22869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dang H, Wang S, Yang L, Fang F, Xu F. Upregulation of Shh and Ptc1 in hyperoxia-induced acute lung injury in neonatal rats. Mol Med Rep . 2012;6:297–302. doi: 10.3892/mmr.2012.929. [DOI] [PubMed] [Google Scholar]
- 6. Oak P, Pritzke T, Thiel I, Koschlig M, Mous DS, Windhorst A, et al. Attenuated PDGF signaling drives alveolar and microvascular defects in neonatal chronic lung disease. EMBO Mol Med . 2017;9:1504–1520. doi: 10.15252/emmm.201607308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kugler MC, Joyner AL, Loomis CA, Munger JS. Sonic hedgehog signaling in the lung—from development to disease. Am J Respir Cell Mol Biol . 2015;52:1–13. doi: 10.1165/rcmb.2014-0132TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Noskovičová N, Petřek M, Eickelberg O, Heinzelmann K. Platelet-derived growth factor signaling in the lung. From lung development and disease to clinical studies. Am J Respir Cell Mol Biol . 2015;52:263–284. doi: 10.1165/rcmb.2014-0294TR. [DOI] [PubMed] [Google Scholar]
- 9. Riccetti M, Gokey JJ, Aronow B, Perl AT. The elephant in the lung: integrating lineage-tracing, molecular markers, and single cell sequencing data to identify distinct fibroblast populations during lung development and regeneration. Matrix Biol . 2020;91–92:51–74. doi: 10.1016/j.matbio.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen WV, Delrow J, Corrin PD, Frazier JP, Soriano P. Identification and validation of PDGF transcriptional targets by microarray-coupled gene-trap mutagenesis. Nat Genet . 2004;36:304–312. doi: 10.1038/ng1306. [DOI] [PubMed] [Google Scholar]
- 11. Wani MA, Wert SE, Lingrel JB. Lung Kruppel-like factor, a zinc finger transcription factor, is essential for normal lung development. J Biol Chem . 1999;274:21180–21185. doi: 10.1074/jbc.274.30.21180. [DOI] [PubMed] [Google Scholar]
- 12. Gu Y, Wu YB, Wang LH, Yin JN. Involvement of Kruppel-like factor 9 in bleomycin-induced pulmonary toxicity. Mol Med Rep . 2015;12:5262–5266. doi: 10.3892/mmr.2015.4015. [DOI] [PubMed] [Google Scholar]
- 13. Schneider DJ, Wu M, Le TT, Cho SH, Brenner MB, Blackburn MR, et al. Cadherin-11 contributes to pulmonary fibrosis: potential role in TGF-β production and epithelial to mesenchymal transition. FASEB J . 2012;26:503–512. doi: 10.1096/fj.11-186098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tipple TE, Welty SE, Nelin LD, Hansen JM, Rogers LK. Alterations of the thioredoxin system by hyperoxia: implications for alveolar development. Am J Respir Cell Mol Biol . 2009;41:612–619. doi: 10.1165/rcmb.2008-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development . 1997;124:53–63. doi: 10.1242/dev.124.1.53. [DOI] [PubMed] [Google Scholar]
- 16. Motoyama J, Liu J, Mo R, Ding Q, Post M, Hui CC. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat Genet . 1998;20:54–57. doi: 10.1038/1711. [DOI] [PubMed] [Google Scholar]
- 17. Chuang PT, Kawcak T, McMahon AP. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev . 2003;17:342–347. doi: 10.1101/gad.1026303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li C, Li M, Li S, Xing Y, Yang CY, Li A, et al. Progenitors of secondary crest myofibroblasts are developmentally committed in early lung mesoderm. Stem Cells . 2015;33:999–1012. doi: 10.1002/stem.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller LA, Wert SE, Clark JC, Xu Y, Perl AK, Whitsett JA. Role of Sonic hedgehog in patterning of tracheal-bronchial cartilage and the peripheral lung. Dev Dyn . 2004;231:57–71. doi: 10.1002/dvdy.20105. [DOI] [PubMed] [Google Scholar]
- 20. Lin C, Chen MH, Yao E, Song H, Gacayan R, Hui CC, et al. Differential regulation of Gli proteins by Sufu in the lung affects PDGF signaling and myofibroblast development. Dev Biol . 2014;392:324–333. doi: 10.1016/j.ydbio.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kugler MC, Loomis CA, Zhao Z, Cushman JC, Liu L, Munger JS. Sonic hedgehog signaling regulates myofibroblast function during alveolar septum formation in murine postnatal lung. Am J Respir Cell Mol Biol . 2017;57:280–293. doi: 10.1165/rcmb.2016-0268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boström H, Gritli-Linde A, Betsholtz C. PDGF-A/PDGF alpha-receptor signaling is required for lung growth and the formation of alveoli but not for early lung branching morphogenesis. Dev Dyn . 2002;223:155–162. doi: 10.1002/dvdy.1225. [DOI] [PubMed] [Google Scholar]
- 23. Gouveia L, Kraut S, Hadzic S, Vazquéz-Liébanas E, Kojonazarov B, Wu CY, et al. Lung developmental arrest caused by PDGF-A deletion: consequences for the adult mouse lung. Am J Physiol Lung Cell Mol Physiol . 2020;318:L831–L843. doi: 10.1152/ajplung.00295.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J, Hoyle GW. Overexpression of PDGF-A in the lung epithelium of transgenic mice produces a lethal phenotype associated with hyperplasia of mesenchymal cells. Dev Biol . 2001;239:338–349. doi: 10.1006/dbio.2001.0441. [DOI] [PubMed] [Google Scholar]
- 25. Kimani PW, Holmes AJ, Grossmann RE, McGowan SE. PDGF-Ralpha gene expression predicts proliferation, but PDGF-A suppresses transdifferentiation of neonatal mouse lung myofibroblasts. Respir Res . 2009;10:119. doi: 10.1186/1465-9921-10-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Endale M, Ahlfeld S, Bao E, Chen X, Green J, Bess Z, et al. Temporal, spatial, and phenotypical changes of PDGFRα expressing fibroblasts during late lung development. Dev Biol . 2017;425:161–175. doi: 10.1016/j.ydbio.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li R, Bernau K, Sandbo N, Gu J, Preissl S, Sun X. Pdgfra marks a cellular lineage with distinct contributions to myofibroblasts in lung maturation and injury response. eLife . 2018;7:e36865. doi: 10.7554/eLife.36865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lau M, Masood A, Yi M, Belcastro R, Li J, Tanswell AK. Long-term failure of alveologenesis after an early short-term exposure to a PDGF-receptor antagonist. Am J Physiol Lung Cell Mol Physiol . 2011;300:L534–L547. doi: 10.1152/ajplung.00262.2010. [DOI] [PubMed] [Google Scholar]
- 29. Daily K, Patel VR, Rigor P, Xie X, Baldi P. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics . 2011;12:495. doi: 10.1186/1471-2105-12-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun . 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamada M, Kurihara H, Kinoshita K, Sakai T. Temporal expression of alpha-smooth muscle actin and drebrin in septal interstitial cells during alveolar maturation. J Histochem Cytochem . 2005;53:735–744. doi: 10.1369/jhc.4A6483.2005. [DOI] [PubMed] [Google Scholar]
- 32. Li C, Lee MK, Gao F, Webster S, Di H, Duan J, et al. Secondary crest myofibroblast PDGFRα controls the elastogenesis pathway via a secondary tier of signaling networks during alveologenesis. Development . 2019;146:dev176354. doi: 10.1242/dev.176354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riccetti MR, Ushakumary MG, Waltamath M, Green J, Snowball J, Dautel SE, et al. Maladaptive functional changes in alveolar fibroblasts due to perinatal hyperoxia impair epithelial differentiation. JCI Insight . 2022;7:e152404. doi: 10.1172/jci.insight.152404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo M, Du Y, Gokey JJ, Ray S, Bell SM, Adam M, et al. Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat Commun . 2019;10:37. doi: 10.1038/s41467-018-07770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Narvaez Del Pilar O, Gacha Garay MJ, Chen J. Three-axis classification of mouse lung mesenchymal cells reveals two populations of myofibroblasts. Development . 2022;149:dev200081. doi: 10.1242/dev.200081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang S, Lai X, Yang L, Ye F, Huang C, Qiu Y, et al. Asporin promotes TGF-β-induced lung myofibroblast differentiation by facilitating rab11-dependent recycling of TβRI. Am J Respir Cell Mol Biol . 2022;66:158–170. doi: 10.1165/rcmb.2021-0257OC. [DOI] [PubMed] [Google Scholar]
- 37. Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, et al. Single-cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep . 2018;22:3625–3640. doi: 10.1016/j.celrep.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development . 1998;125:3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- 39. Li C, Smith SM, Peinado N, Gao F, Li W, Lee MK, et al. Wnt5a-ror signaling is essential for alveologenesis. Cells . 2020;9:384. doi: 10.3390/cells9020384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu B, Liu J, Wu Z, Liu T, Ullenbruch MR, Ding L, et al. Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am J Respir Cell Mol Biol . 2015;52:418–428. doi: 10.1165/rcmb.2014-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Faggian J, Fosang AJ, Zieba M, Wallace MJ, Hooper SB. Changes in versican and chondroitin sulfate proteoglycans during structural development of the lung. Am J Physiol Regul Integr Comp Physiol . 2007;293:R784–R792. doi: 10.1152/ajpregu.00801.2006. [DOI] [PubMed] [Google Scholar]
- 42. Hagan AS, Zhang B, Ornitz DM. Identification of a FGF18-expressing alveolar myofibroblast that is developmentally cleared during alveologenesis. Development . 2020;147:dev181032. doi: 10.1242/dev.181032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lodyga M, Cambridge E, Karvonen HM, Pakshir P, Wu B, Boo S, et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci Signal . 2019;12:eaao3469. doi: 10.1126/scisignal.aao3469. [DOI] [PubMed] [Google Scholar]
- 44. Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol . 2006;26:6283–6298. doi: 10.1128/MCB.02317-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pelczar P, Zibat A, van Dop WA, Heijmans J, Bleckmann A, Gruber W, et al. Inactivation of Patched1 in mice leads to development of gastrointestinal stromal-like tumors that express Pdgfrα but not kit. Gastroenterology . 2013;144:134–144.e6. doi: 10.1053/j.gastro.2012.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McGowan SE, McCoy DM. Platelet-derived growth factor-A and Sonic hedgehog signaling direct lung fibroblast precursors during alveolar septal formation. Am J Physiol Lung Cell Mol Physiol . 2013;305:L229–L239. doi: 10.1152/ajplung.00011.2013. [DOI] [PubMed] [Google Scholar]
- 47. Gouveia L, Betsholtz C, Andrae J. Expression analysis of platelet-derived growth factor receptor alpha and its ligands in the developing mouse lung. Physiol Rep . 2017;5:e13092. doi: 10.14814/phy2.13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Toti P, Buonocore G, Tanganelli P, Catella AM, Palmeri ML, Vatti R, et al. Bronchopulmonary dysplasia of the premature baby: an immunohistochemical study. Pediatr Pulmonol . 1997;24:22–28. doi: 10.1002/(sici)1099-0496(199707)24:1<22::aid-ppul4>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 49. Amatya S, Rajbhandari S, Pradhan S, Trinh V, Paudel U, Parton LA. Hedgehog signaling pathway gene variant influences bronchopulmonary dysplasia in extremely low birth weight infants. World J Pediatr . 2021;17:298–304. doi: 10.1007/s12519-021-00427-y. [DOI] [PubMed] [Google Scholar]
- 50. Yuan Y, Zhou Y, Li Y, Hill C, Ewing RM, Jones MG, et al. Deconvolution of RNA-seq analysis of hyperbaric oxygen-treated mice lungs reveals mesenchymal cell subtype changes. Int J Mol Sci . 2020;21:1371. doi: 10.3390/ijms21041371. [DOI] [PMC free article] [PubMed] [Google Scholar]