

Abstract
The low-density lipoprotein receptor (LDLR) controls cellular delivery of cholesterol and clears LDL from the bloodstream, protecting against atherosclerotic heart disease, the leading cause of death in the United States. We therefore sought to identify regulators of the LDLR beyond the targets of current therapies and known causes of familial hypercholesterolemia. We found that cold shock domain-containing protein E1 (CSDE1) enhanced hepatic LDLR mRNA decay via its 3’ untranslated region and regulated atherogenic lipoproteins in vivo. Using parallel phenotypic genome-wide CRISPR interference screens in a tissue culture model, we identified 40 specific regulators of the LDLR that were not previously identified by observational human genetics studies. Among these, we demonstrated that, in HepG2 cells, CSDE1 regulated the LDLR at least as strongly as statins and proprotein convertase subtilisin/kexin type 9 PCSK9 inhibitors. In addition, we showed that hepatic gene silencing of Csde1 treated diet-induced dyslipidemia in mice to a similar degree as Pcsk9 silencing. These results suggest the therapeutic potential of targeting CSDE1 to manipulate the post-transcriptional regulation of the LDLR mRNA for the prevention of cardiovascular disease. Our approach of modeling a clinically relevant phenotype in a forward genetic screen, followed by mechanistic pharmacologic dissection and in vivo validation, may serve as a generalizable template for the identification of therapeutic targets in other human disease states.
One Sentence Summary:
A genome-wide CRISPRi screen identifies CSDE1 as a regulator of hepatic LDLR mRNA decay, suggesting it as a target for the treatment of heart disease.
Graphical Abstract

Introduction
The low-density lipoprotein receptor (LDLR) delivers cholesterol from low-density lipoprotein (LDL) to cells to maintain membrane homeostasis (1). By clearing atherogenic LDL particles from the bloodstream, the hepatic LDLR protects against atherosclerotic heart disease (2). Despite successful therapies that upregulate the hepatic LDLR and reduce heart attacks, such as 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) inhibitors (statins), Neimann-Pick C1-like (NPC1L1) inhibitors, or proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors (3), cardiovascular disease remains the leading cause of death in Western countries (4). Lowering LDL beyond that achieved by statins improves clinical outcomes without adverse effects (5). Although there is a theoretical concentration at which LDL concentrations could get too low (6), this has yet to be discovered in large randomized trials (7). If other LDLR regulatory mechanisms could be leveraged to further treat heart disease remains unknown.
The genetics of familial hypercholesterolemia (FH), which manifests as an isolated elevation in serum LDL, underlies the clinical success of LDLR upregulation by statins and PCSK9 inhibitors. Estimates suggest that 20–40% of FH phenotypes remain unexplained outside of the four major causes: LDLR, apolipoprotein B (APOB), PCSK9, and low density lipoprotein receptor adaptor protein 1 (LDLRAP1) (8). Although polygenic causes drive some unexplained phenotypes (9–11), additional regulators of the LDLR may still exist. Advances in forward genetics using clustered regularly interspaced short palindromic repeats (CRISPR)-based technologies (12–14) can now enable searches for tissue and disease-specific effects across the entire genome that may elude the sporadic natural variants found in observational studies, which themselves require compatibility throughout the entire lifespan and in all cell types. Moreover, hepatic delivery of gene silencing agents has been shown to be effective in the clinic (15), providing a therapeutic modality against hits whose phenotypes are driven by expression in the liver. We therefore employed a genome-wide CRISPR interference (CRISPRi) screen for factors involved in hepatic LDLR regulation, both to understand the biology of this important receptor and to uncover potential therapeutic targets in cardiovascular disease.
Results
A Genome-Wide CRISPR interference screen for LDL receptor regulation
We engineered the HepG2 cell line, which models the regulation of the LDLR (16–20), to constitutively express an inactive Cas9 protein fused to the Krüppel associated box transcriptional repressor (dCas9-KRAB), enabling the knockdown of any given gene with an appropriate single guide RNA (sgRNA, Fig. 1A) (12, 13). Because statins (21) and PCSK9 inhibitors (22–24) increase cell surface LDLR, we scored surface LDLR abundance. To focus on factors that preferentially affect LDLR abundance over other receptors, we performed a parallel screen for regulators of the transferrin receptor (TFR). This critical player in iron metabolism shares a clathrin-mediated intake mechanism, but is otherwise orthogonally regulated from the LDLR (25, 26). Before our screen, we confirmed dCas9-KRAB activity (fig. S1A) and an appropriate dynamic range for both LDLR and TFR regulation by transduction with sgRNAs expected to alter receptor abundance in either direction. For LDLR, we targeted LDLR and myosin regulatory light chain interacting protein (MYLIP), which encodes an E3 ligase that ubiquitinates the LDLR and causes lysosomal degradation (fig. S1B) (27). For TFR, we targeted TFRC and zinc finger CCCH-type containing 12A (ZC3H12A), which encodes an endoribonuclease that degrades transferrin receptor (TFRC) mRNA (fig. S1C) (28).
Figure 1: Results from the genome-wide CRISPR interference screen.
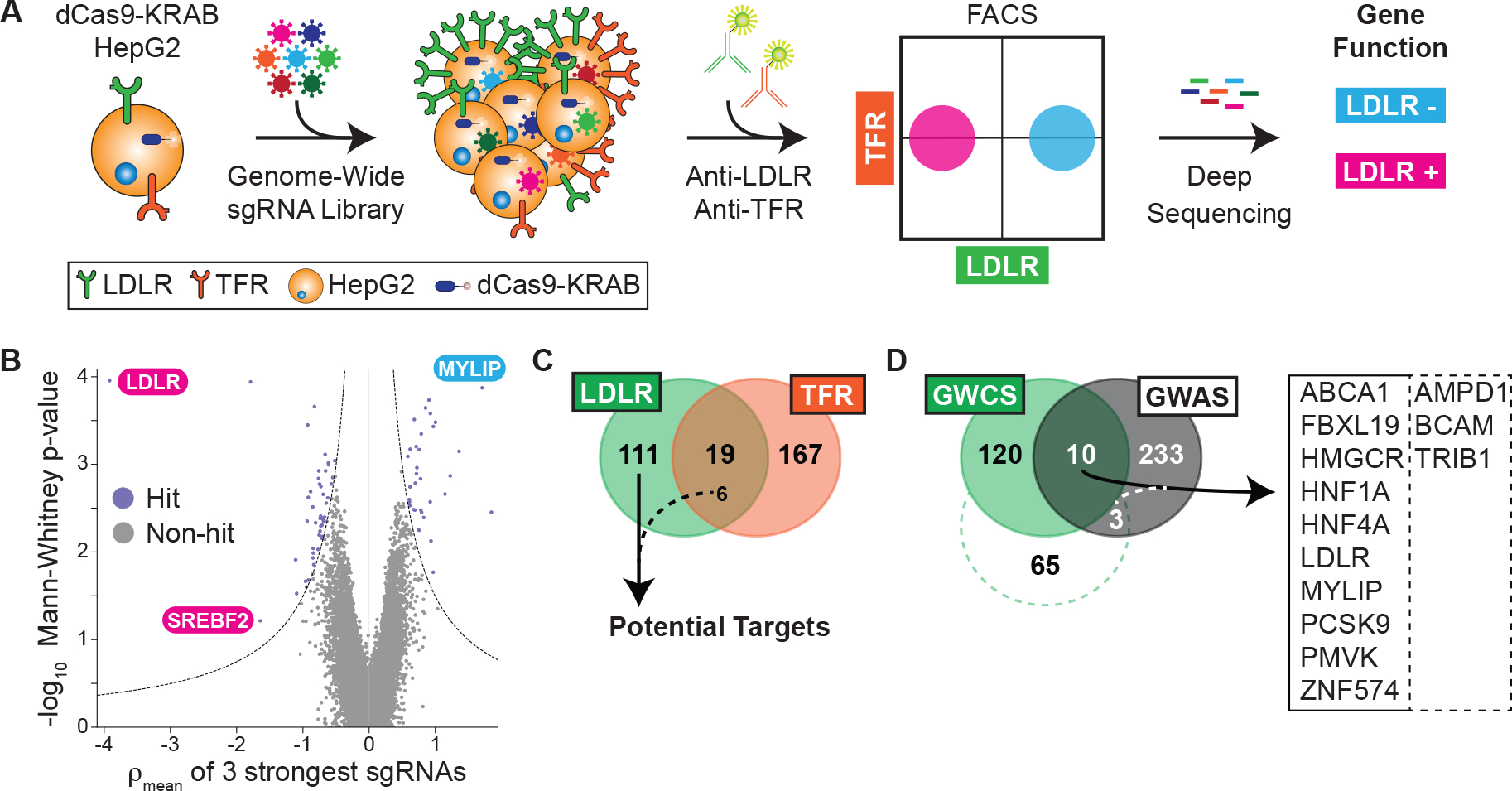
A) Overall schematic of phenotypic selection. CRISPRi-ready cells are transduced with a genome-wide library of sgRNAs, surface labeled with antibody, sorted by flow cytometry, and deep sequenced to deconvolute putative gene functions. See text for details. B) Volcano plot showing the statistical significance (Mann-Whitney test) of the guides recovered for each gene against the mean ρ phenotype of the 3 guides with the strongest effect. ρ is defined as the log2-fold enrichment for sgRNAs recovered from cells with high LDLR abundance cells to those recovered from cells with low LDLR abundance. Guides targeting known regulators of the LDLR are noted. C) Venn diagram showing the overlap between parallel LDLR and TFR screens. Six guides common to both had opposing abundance phenotypes in the respective screens and were included as specific hits. D) Venn diagram of hits between the LDLR genome-wide CRISPRi screen (GWCS) and putative genes correlated with serum LDL-C from GWAS. The dotted line indicates a relaxed threshold for hit selection from LDLR screen, with only an additional 3 genes in the overlap. Overlap genes shown at right.
We next performed our pooled screens in parallel by transducing a library encoding sgRNAs with 5-fold coverage of the entire protein-coding human genome (13). We selected the cells at the upper and lower third of receptor abundance by fluorescence-activated cell sorting (FACS) and quantified the sgRNAs for each population via deep sequencing (fig. S2, A to F, Tables S1 to S4). We compared the degree of enrichment of LDLR or TFR surface amounts in the high abundance to the low abundance cells (defined as ρ, Fig. 1B). We also compared the cells with high and low receptor abundance to the unsorted population (defined as τ or γ, respectively) and included these results in our final hit count. This resulted in 130 total hits for the LDLR and 186 hits for the TFR (Tables S5 and S6). We hypothesized that hits with shared phenotypes would likely have global effects on surface receptors, leaving us with 117 hits specific for LDLR regulation (Fig. 1C, Table S5). Gene ontology (GO) analysis (29) revealed a 15-fold enrichment for cholesterol metabolism as a biologic process (11 total hits, P = 5.7 × 10−10), providing confidence that we recapitulated our target biology. The hits also included 48 members of potentially druggable protein classes, including 29 with proposed enzymatic activity, and 22 hits were unclassified in GO databases (fig. S3A).
Cross-referencing human genetic datasets identifies LDLR regulators in vivo
We next compared genes associated with serum LDL cholesterol (LDL-C) from published genome-wide association studies (GWAS) (30–32) to our list of hits. However, only 13 of these genes overlapped with our results (Fig. 1D), even when we relaxed our threshold for hit selection. To improve power for multiple hypothesis testing across the entire genome, we analyzed 390,375 UK Biobank participants with genome-wide genotypes and known plasma lipids (Table S7) to search for variants associated with LDL-C amongst only our hits (33). We filtered to nonsynonymous protein coding variants in these hits by a threshold minor allele frequency (>0.001) and minimum statistical significance (P = 0.000427, Table 2). For basal cell adhesion molecule (BCAM), we found both an association between higher LDL-C and a nonsense variant, along with bidirectional associations between LDL-C and missense variants, suggesting that this pathway may be tunable. We also found associations between elevated LDL-C and variants in methylsterol monooxygenase 1 (MSMO1), chromosome 6 open reading frame 132 (C6orf132), hepatocyte nuclear factor 4 alpha (HNF4A), and timeless circadian regulator (TIMELESS), suggesting that these hits may be functional in the human and warrant further evaluation. The results also suggested that the accessible “genomic space” of the CRISPRi and GWAS strategies was only partially overlapping.
Regulators of surface LDL receptor abundance affect functional uptake of LDL
To validate our screen results, we generated CRISPRi HepG2 cells harboring either of the two top-scoring sgRNAs for 77 of our hits as well as established controls. We preferentially tested hits with an increase in surface LDLR upon inhibition, as well as those with potentially druggable functions or lacking associated GO terms. Because surface receptor abundance might not necessarily correlate to increased function, we evaluated both LDLR and TFR surface phenotypes alongside LDL uptake (34). This functional assay involved a pulse treatment of exogenous, fluorophore-labeled LDL followed by a similar flow cytometric readout. Lastly, because knockdowns could also cause growth phenotypes, we assayed the number of cells surviving to FACS analysis as a proxy for viability.
We recapitulated the phenotypes for receptor abundance for at least one of the guides in most of the hits (55 genes, 71% of those tested, Table S8). Moreover, for 40 of these genes, both sgRNAs independently validated, suggesting against an off-target effect. We visualized these hits based on their effects, at single cell resolution, on LDLR and TFR abundance, the LDLR/TFR ratio, functional LDL uptake and number of cells surviving to analysis (Fig. 2, fig. S3B). Notably, in this tissue culture model, most knockdowns had independently validated effects on LDLR abundance and LDL uptake of similar or greater magnitude than the HMGCR or PCSK9 controls.
Figure 2: Validation of LDLR CRISPRi hits.

Heatmap showing receptor abundance (LDLR, TFR, and LDLR/TFR ratio) and function (LDL uptake) for dCas9-KRAB HepG2 cells transduced with sgRNA targeting the indicated gene, analyzed by flow cytometry. Hits are grouped according to directional effect on LDLR abundance, and then within groups, by effect on LDL uptake (with uptake from FOXL3-OT1, CIT, and DHX15 sgRNAs not significantly different, at P > 0.05, from negative control sgRNA). CSDE1 is highlighted in blue. Control sgRNAs are shown at right. Readouts show log2 fold change compared to transduction with negative control sgRNA and represent the weighted average of the effects from both sgRNAs targeting each gene. Viability indicates the relative number of cells surviving to flow cytometry in the experiments. Functional classification of genes is shown in fig. S3. Note that LDLR/TFR is a separately ascertained value from individual cells, and not a derived parameter from aggregate data. Only the hits for which two separate sgRNAs independently validated for receptor expression are shown, defined as P < 0.05 via Holm-Sidak corrected t-test. Data represent summary information from 3 to 4 independent experiments.
Knockdown of hits expected to alter cellular cholesterol balance or transcriptionally regulate the LDLR showed directionally consistent effects between LDLR abundance and function (Fig. 2). For genes in the enzymatic pathway of cholesterol metabolism (35) (3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1) and MSMO1), this was consistent with activation of sterol regulatory element-binding protein 2 (SREBP2)-mediated LDLR transcription. For genes encoding certain transcription factors [hepatocyte nuclear factor 1 homeobox A (HNF1A) (36), HNF4A (37), one cut homeobox 1 (ONECUT1) (38), and zinc finger E-box binding homeobox 1 (ZEB1) (39)], this was consistent with an effect on LDLR transcription itself. Knockdowns of solute carrier family 25 member 27 (SLC25A27), which encodes a mitochondrial uncoupling protein (40), and ATP binding cassette subfamily A member 4 (ABCA4), encoding a known lipid transporter (41), both exhibited reductions in LDLR abundance and function (Fig. 2). These genes could plausibly induce a negative lipid balance, increasing LDL uptake via both LDLR-dependent and independent mechanisms.
Targeting of hits that either affected multiple transcriptional pathways or regulated endocytosis showed opposite effects on LDLR abundance and function. Knockdown of tribbles pseudokinase 1 (TRIB1), a GWAS hit (30) encoding a pseudokinase that regulates the constitutive photomorphogenic (COP1) E3 ligase (42) and affects multiple transcription factors (43), showed this phenotype. In the mouse, TRIB1 overexpression lowers serum cholesterol, whereas the knockout has the opposite effect (44, 45), consistent with our results. Knockdown of adaptor related protein complex 2 subunit mu 1 (AP2M1), a TFR screen hit that encodes an adaptor protein required for endocytosis (46), was similar, consistent with an accumulation of non-functional receptors at the cell surface. This phenotype, although specific to the LDLR, was also seen with knockdown of BCAM, which encodes a membrane cell adhesion molecule (47) identified by GWAS (32), and transmembrane protein 217 (TMEM217), which encodes an uncharacterized transmembrane protein (Fig. 2, fig. S4). This suggested that these proteins could have a similar endocytosis adaptor function specific for the LDLR, akin to LDLRAP1 (48), in which mutations cause a recessive form of FH.
Pharmacologic inhibition of clinically relevant pathways provides mechanistic insight into putative LDLR regulators
We next used pharmacologic approaches to perturb specific pathways of LDLR regulation. We hypothesized that hits might alter cholesterol metabolism, LDLR recycling, or a yet unspecified pathway. By combining CRISPRi knockdown with either a statin to inhibit endogenous cholesterol biosynthesis (21), or a PCSK9 inhibitor to arrest LDLR lysosomal degradation (24), and assessing the combined effect, we inferred mechanistic information about the target gene. Furthermore, we hypothesized that either additive or potentiating effects between a clinically validated therapy and a hit gene might suggest potential therapeutic targets.
We evaluated the receptor abundance and function phenotypes for 29 of our validated hits in the presence or absence of simvastatin (49) or PF-846, a selective inhibitor of PCSK9 translation (50) (Fig. 3, Table S9). We calculated a synergy score by subtracting the differential effects of CRISPRi knockdown, compared to the control, in the presence of compound from that with the dimethyl sulfoxide (DMSO) vehicle. A more positive value indicated synergy and a more negative value indicated antagonism.
Figure 3: Synergy of CRISPRi knockdowns with simvastatin or PF-846.
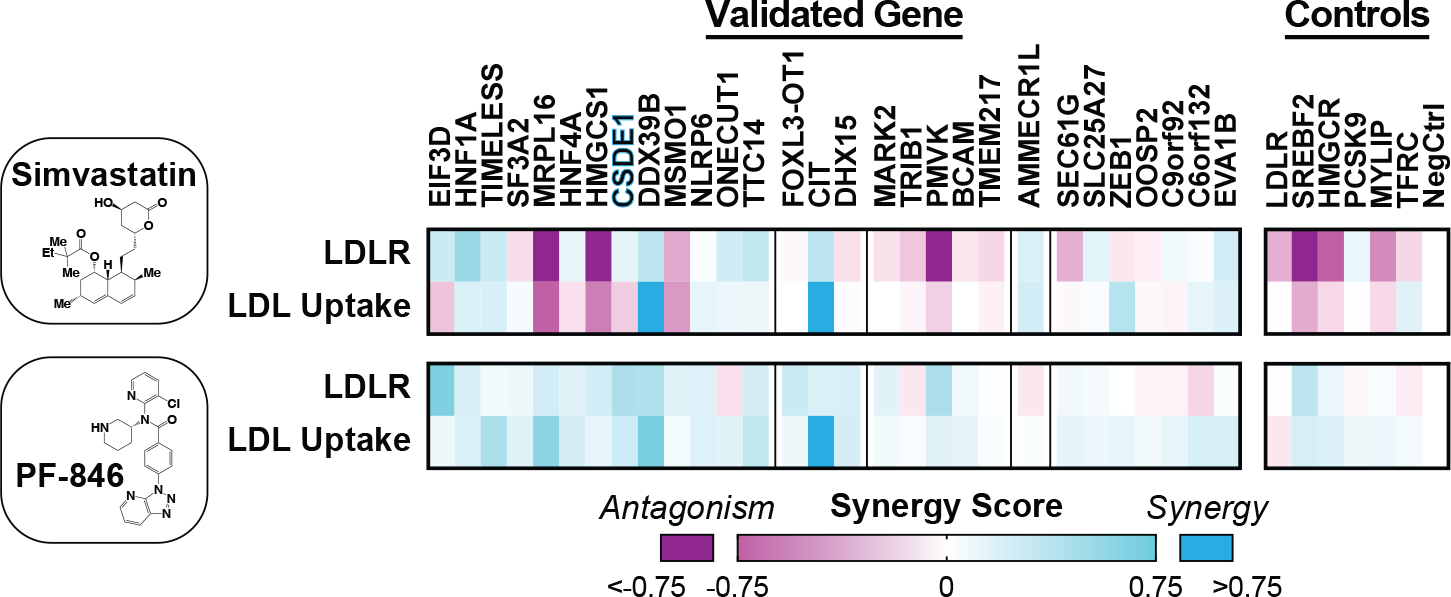
Heatmap showing synergy score for knockdowns of indicated genes combined with simvastatin (top) or PF-846 (bottom). Separate LDLR abundance and function (LDL uptake) experiments are shown. Hits are grouped first according to overall effect on LDLR abundance, and secondarily by effect on LDL uptake, as in Fig. 2. CSDE1 is highlighted in blue. Data represent summary information from 4 independent experiments.
After knockdown, regulators of cholesterol biosynthesis [sterol regulatory element binding transcription factor 2 (SREBF2), HMGCR, HMGCS1, MSMO1, and phosphomevalonate kinase (PMVK)] showed antagonism with the statin, but mild synergy with PCSK9 inhibition (Fig. 3). The antagonism with statins was expected, given that SREBP2 drives LDLR transcription. Because SREBP2 also induces PCSK9 expression, knockdown of these genes raises total PCSK9, explaining both the synergy with PCSK9 inhibition we observed here and that observed with statins in the clinic (51). The synergy phenotypes for knockdown of mitochondrial ribosomal protein L16 (MRPL16), which encodes a structural component of the mitochondrial ribosome (52), mirrored these cholesterol biosynthetic genes (Fig. 3), suggesting that MRP-L16 might play a role in the mitochondrial generation of metabolic precursors to sterol biogenesis. In contrast, C6orf132 knockdown showed the opposite phenotype: mild synergy with a statin, and mild antagonism with PF-846 (Fig. 3). C6orf132 localizes to the Golgi (53), suggesting that it may function by facilitating LDLR delivery to the cell surface, prior to any interaction with extracellular PCSK9. For some transcription factors, the synergy phenotypes pointed to their downstream targets. For example, synergy of HNF1A knockdown with a statin (Fig. 3) is consistent with disruption of hepatocyte nuclear factor 1-alpha (HNF1-α) mediated PCSK9 transcription (54).
CSDE1 regulates the stability of LDLR mRNA
One of the strongest hits, cold shock domain containing E1 (CSDE1), also known as upstream of N-ras (UNR), encodes an RNA binding protein with varied regulatory functions (55–57), including mRNA decay (58). Because the LDLR 3’ untranslated region (UTR) consists of adenylate-uridylate (AU)-rich elements (AREs) implicated in mRNA stability (59), we hypothesized that CSDE1 could mediate the degradation of the LDLR transcript, thereby explaining its observed receptor abundance, function, and synergy phenotypes.
Upon CSDE1 knockdown in HepG2 cells, we observed increased LDLR abundance in both sterol-replete and sterol-depleted conditions (Fig. 4A). Moreover, we observed progressively higher LDLR amounts with sterol-depletion and the addition of a statin (Fig. 4A, fig. S5, A to C), suggesting that the mechanism of CSDE1 disruption is at least additive with SREBP2-mediated LDLR transcription and statin therapy. We reproduced our flow cytometry results in CSDE1-depleted cells (fig. S6A) with immunoblots against both total and surface LDLR (fig. S6B), capturing the latter via a cell-surface biotinylation assay. HepG2 cells transfected with CSDE1-targeting small interfering RNA (siRNA) exhibited similarly increased LDLR abundance (fig. S7A) and LDL uptake (fig. S7B) to the CRISPRi results. siRNA against CSDE1 increased both LDLR transcripts in Huh7 cells (fig. S8) and LDL uptake in primary mouse hepatocytes (1.6-fold increase, P = 0.0372, fig. S9A). Similar results were observed in primary mouse hepatocytes using adeno-associated virus serotype 8 (AAV8)-delivered short hairpin RNA (shRNA) against Csde1, though these outcomes were not statistically significant (P = 0.373, fig. S9B). Together, these observations confirm that the effects of CSDE1 are not limited to a single cell line.
Figure 4: CSDE1 mediates LDLR mRNA decay.
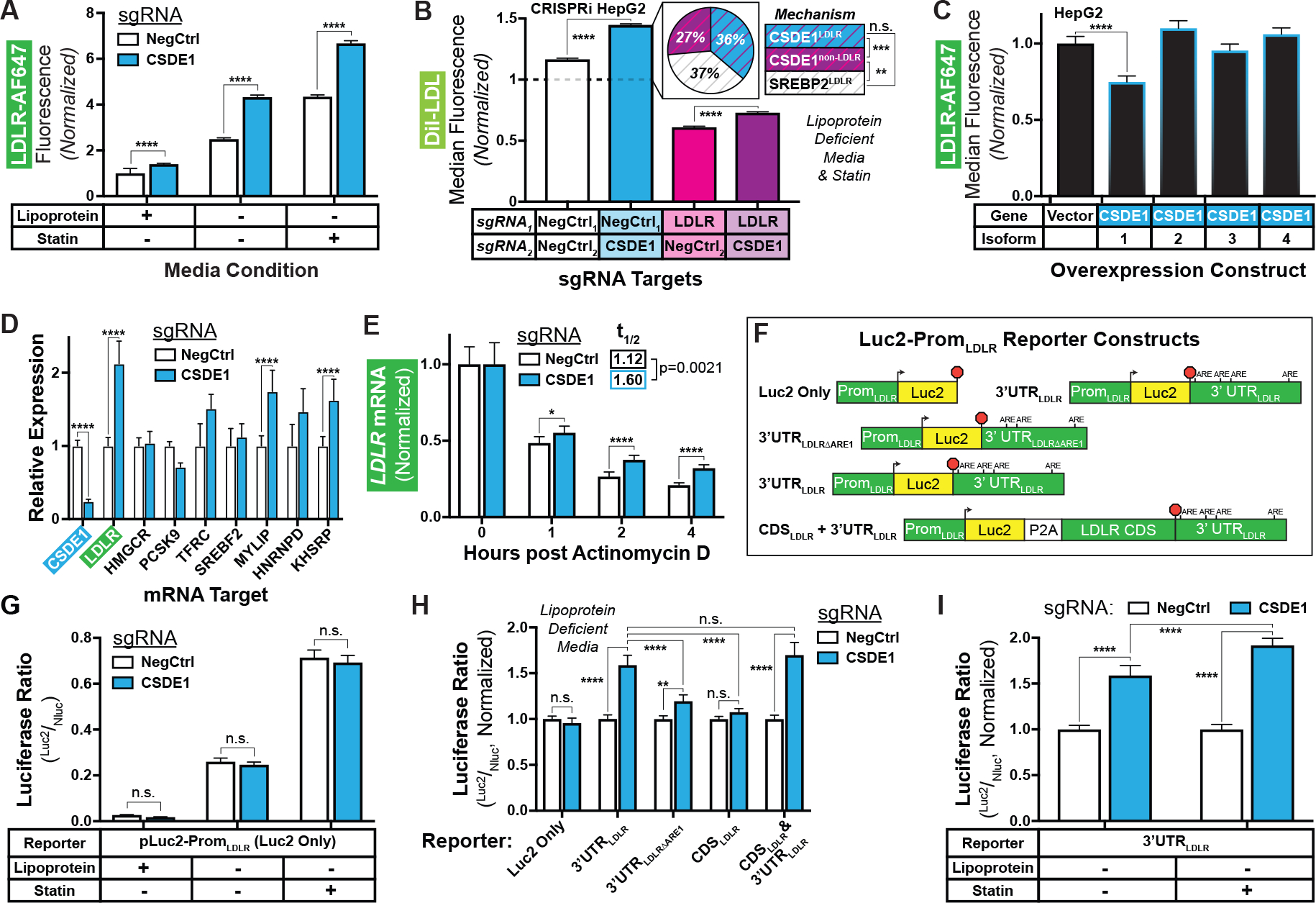
A) Relative LDLR abundance in CRISPRi HepG2 cells transduced with indicated sgRNAs and grown in the indicated media. B) Relative LDL uptake in dual-sgRNA CRISPRi HepG2 cells. The pie chart shows the relative contribution of LDLR-dependent (CSDE1LDLR, blue with purple stripes) and LDLR-independent (CSDE1non-LDLR, purple with magenta stripes) CSDE1-mediated mechanisms, as well as SREBP2-mediated mechanisms (SREBP2LDLR, white with grey stripes) to LDL uptake. Data is normalized to the control cells in standard media (dashed line, data shown in fig. S10C). C) Relative LDLR abundance in HepG2 cells overexpressing indicated CSDE1 isoforms. D) Relative expression of indicated mRNA targets in CRISPRi cells under sterol-depleted conditions. E) Relative expression of LDLR mRNA in CRISPRi cells after arrest of transcription with actinomycin D. Data normalized at T=0 within the sgRNA evaluated to illustrate the change in time, and thus no comparison can be made at T=0. t1/2 indicates data fit to a one-stage exponential decay equation and analyzed by extra sum-of-squares F test. E) Schematics (not to scale) of Luc2-PromLDLR reporter constructs, illustrating LDLR promoter, start site (arrowhead), P2A ribosomal skipping sequence, AREs in 3’ UTR, stop codon (red octagon), and indicated regions of the LDLR gene. G to I) Ratiometric luciferase outputs of CRISPRi cells transfected with indicated reporters. “Luc2 Only” used in G, all constructs used in H, and “3’UTRLDLR” used in I. Outputs normalized to negative control in H and I. All panels) Data represent summary information from 3 to 4 independent experiments. n.s. = non-significant (P ≥ 0.05), * = P < 0.05, ** = P < 0.01,*** = P < 0.001, and **** = P < 0.0001.
Returning to our CRISPRi HepG2 system, we generated a combined CSDE1/LDLR knockdown cell line harboring sgRNAs against each target (60). As expected, the double knockdown had no additional effect on surface LDLR as compared with the LDLR knockdown alone (fig. S10, A and B). However, the double CSDE1/LDLR knockdown exhibited a small but significant increase in LDL uptake (P < 0.0001) compared with the single LDLR knockdown (CSDE1nonLDLR, Fig. 4B, fig. S10, C and D). This LDLR-independent effect constituted about 40% of the total increase of LDL uptake driven by CSDE1 knockdown in the LDLR sufficient background (compare CSDE1nonLDLR to total CSDE1 in pie charts, Fig. 4B, fig. S10C). Both LDLR-dependent (CSDE1LDLR) and LDLR-independent components of CSDE1’s effect on LDL uptake were additive with and unaffected by sterol-depletion and statin therapy (SREBP2LDLR, Fig. 4B, fig. S10, C and D). Further, there was no significant difference in the magnitude of LDL uptake between that driven solely from the LDLR-dependent mechanism of CSDE1 and the sterol-depletion and statin (P = 0.9002, see pie chart, Fig. 4B). Yet in contrast to its equivalent effect on LDL uptake, CSDE1 knockdown was less effective at upregulating surface LDLR than SREBP2 activation (fig. S10, A and B), raising the possibility that CSDE1 knockdown may also result in a more functional LDLR. Taken together, these data suggest that the main effect of CSDE1 on LDL uptake is LDLR-dependent, with an additional but smaller LDLR-independent effect also present.
We next observed that overexpression of isoform 1 of CSDE1, but not isoforms 2 through 4, reduced surface LDLR in HepG2 cells (Fig. 4C, fig. S11, A to D). Overexpression of all four isoforms of CSDE1 downregulated LDLR abundance in the CSDE1 CRISPRi knockdown cells, though isoform 1 showed the strongest effect (fig. S11, E to I). The opposing directional effects of CSDE1 knockdown and overexpression suggested that, under physiologic expression conditions, isoform 1 of CSDE1 is a rate-limiting regulator of the LDLR.
Consistent with our mechanistic hypothesis, we noted over a 2-fold increase in steady-state LDLR mRNA (Fig. 4D), as well as depleted CSDE1 (Fig. 4D, fig. S6A), in the CSDE1 knockdown cells (Fig. 4D). Among control mRNA targets, we also observed significant increases in MYLIP and KH-type splicing regulatory protein (KHSRP) mRNA (P < 0.0001), but not in SREBF2, PCSK9, HMGCR, or TFRC mRNA (Fig. 4D). The gene products of MYLIP and KHSRP downregulate the LDLR (27, 61), which is the opposite of our observed phenotype, suggesting that the direct effect of CSDE1 knockdown on the LDLR mRNA predominates in our tissue culture model. To specifically evaluate transcriptional decay, we treated cells with actinomycin D and measured LDLR transcripts over time. We observed significantly higher LDLR mRNA in the CSDE1 knockdown cells at all subsequent timepoints (P < 0.05, Fig. 4E). The mRNA half-life, modeled by a single-phase decay equation, was nearly 1.5-fold longer in the CSDE1 knockdown cells compared to controls (P = 0.0021, Fig. 4E). CSDE1 knockdown had no significant effect on HMGCR, SREBF2, or TFRC mRNA over time (P > 0.05, fig. S12, A to C). CSDE1 knockdown cells exhibited reductions in PCSK9 and KHSRP mRNA at several timepoints, but the mRNA abundance over time did not fit a decay equation, suggesting against a CSDE1-mediated effect on transcript stability (fig. S12, D and E). By contrast, CSDE1 knockdown showed a similar extension of transcript half-life on MYLIP mRNA as LDLR mRNA (fig. S12F), suggesting that MYLIP may also be negatively regulated by CSDE1. Because the increase in MYLIP transcripts seen with CSDE1 knockdown will downregulate the LDL receptor, the targeting of MYLIP or its encoded protein, inducible degrader of the LDL receptor (IDOL), could act synergistically with the targeting of CSDE1 in lowering LDL cholesterol.
To probe the relationship of CSDE1 to the LDLR 3’ UTR, we transiently expressed luciferase constructs (Fig. 4F) under control of the native LDLR promoter in the CSDE1 knockdown cells. The luciferase-only constructs showed appropriate physiologic upregulation by sterols, regardless of CSDE1 knockdown (Fig. 4G). Constructs fused to the LDLR 3’ UTR, but not those fused to the LDLR coding sequence alone, exhibited increased reporter activity with CSDE1 knockdown (Fig. 4H). This increase in activity was attenuated by removing the first of four AREs (59, 62) from the 3’ UTR (Fig. 4H). Activity of the 3’ UTR-fused construct increased further with statin coadministration (Fig. 4I), suggesting that CSDE1 knockdown may be synergistic with statins, consistent with our prior results (Fig. 3). Taken together, these findings suggest that under physiologic conditions, CSDE1 mediates decay of the LDLR mRNA through its 3’ UTR, with the first ARE of the UTR required for its full effect.
Disruption of CSDE1 upregulates Ldlr mRNA expression and protects from cholesterol loading in zebrafish and mice
We then turned to an in vivo model in zebrafish, as the 3’ UTR of its ortholog ldlra (XM_005163870.4) is AU-rich and contains at least two canonical ARE sequences for mRNA regulation (63). The ldlra knockout in zebrafish results in hyperlipidemia and vascular lipid accumulation, and challenging larvae with a high cholesterol diet is sufficient to increase their overall cholesterol content (64). We employed yolk microinjection of a Cas9-ribonucleoprotein (RNP) complex containing redundant guides to achieve near-saturation gene disruption (65), followed with dietary cholesterol supplementation, and evaluated total cholesterol in the larvae (64). Targeting of csde1 protected against total cholesterol accumulation, with a modest (12%) but significant reduction (P = 0.0017) in total cholesterol in 8-day post fertilization (dpf) zebrafish (fig. S13, A to C), without any obvious phenotypic abnormalities (fig. S13, D to F). By contrast, targeting of ldlra showed the expected 1.4-fold increase in total larval cholesterol (fig. S13A), consistent with prior studies (64).
We then probed the effect of Csde1 gene silencing in the mouse as a therapeutic proof-of-principle, given even greater homology between the 3’ UTRs of the murine and human LDLR orthologs (66). Using wild-type C57BL/6 mice, we delivered shRNA against Csde1 (57) or a scramble control via an AAV8 vector. One week after delivery of moderate dose AAV8 (3 × 1011 genomes/mouse), we harvested blood and tissue samples from the mice. We found that, compared to scramble controls, the Csde1-targeted mice exhibited about 30% reduction in hepatic Csde1 mRNA and about 1.8-fold increase in hepatic Ldlr mRNA (Fig. 5A), no gross histologic abnormalities in the liver (fig. S14, A to D), and robust expression of the enhanced green fluorescent protein (eGFP) reporter encoded by the AAV8 vector (fig. S14, E to L). With Csde1 knockdown, we also observed a corresponding reduction in hepatic CSDE1 protein (Fig. 5B). However, steady-state hepatic LDLR protein amounts were not significantly different (P = 0.826, Fig. 5B). We also saw no differences in the hepatic lipoprotein receptors LDLR-related protein 1 (LRP1) or scavenger receptor class B type 1 (SR-B1, Fig. 5B), hepatic PCSK9 protein (Fig. 5B, fig. S14, E‘ to L‘), the appearance or behavior of the mice, plasma alanine or aspartate aminotransferase activity (fig. S15A), or total hepatic bile acids (fig. S15B). These observations suggested a lack of toxicity of either the Csde1 shRNA or the gene knockdown.
Figure 5: CSDE1 disruption upregulates Ldlr mRNA and LDLR function in mice.
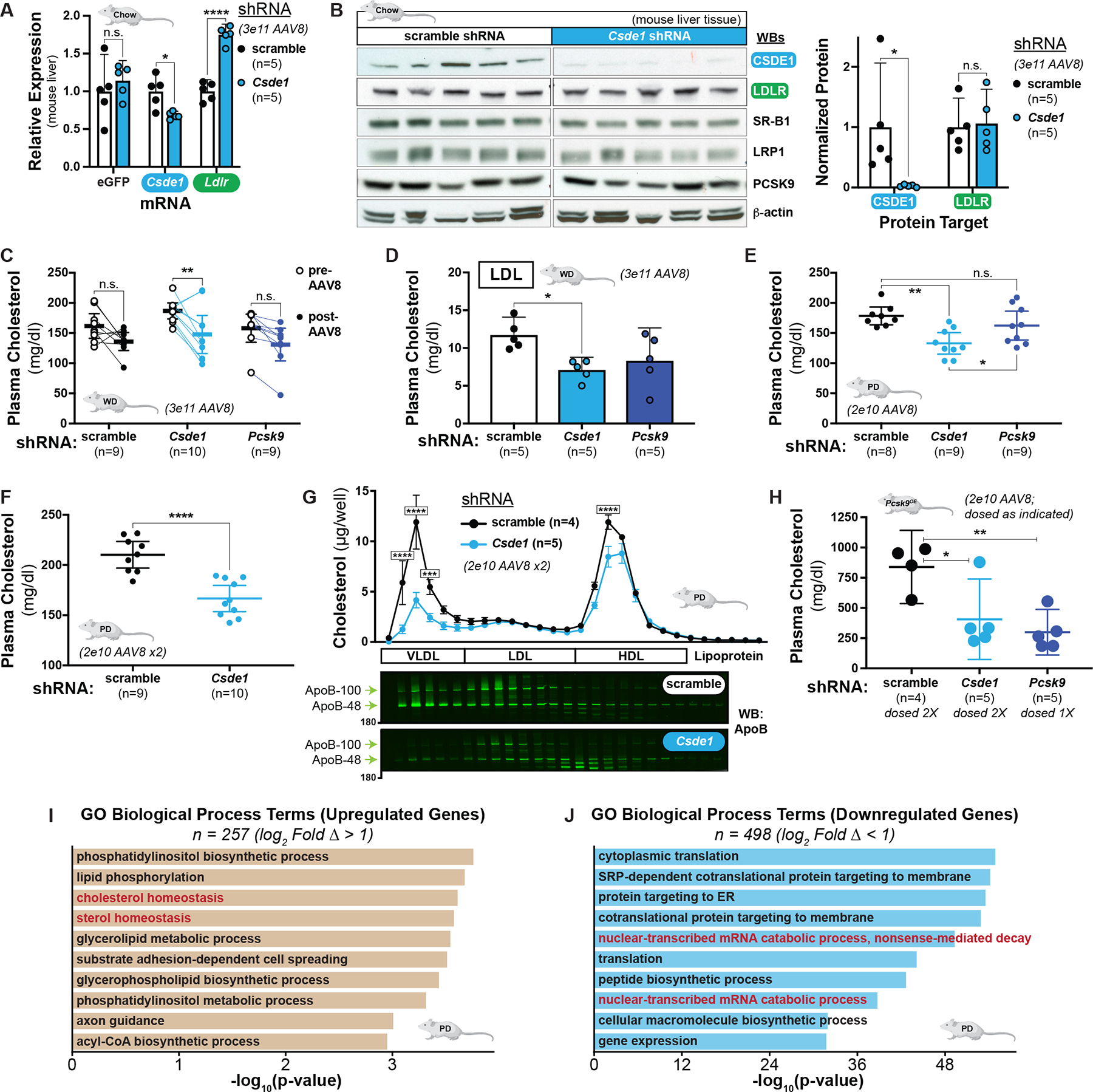
A) Relative expression of hepatic eGFP, Csde1, and Ldlr transcripts in chow-fed mice transduced with indicated moderate-dose AAV8-shRNA. Matched two-way ANOVA with Holm-Sidak multiple comparisons test shown. B) Immunoblots of liver extracts from mice in A. Each lane represents an individual mouse. Quantification of protein, normalized to loading control, shown at right. Unpaired t-tests shown. C) Mean plasma cholesterol concentrations of Western diet-fed mice before and 2 weeks after transduction with moderate-dose AAV8-shRNA. Matched two-way ANOVA with Sidak multiple comparisons test shown. D) Mean LDL cholesterol concentrations, fractionated by gel filtration, from individual mice from C. E) Mean plasma cholesterol concentrations of Paigen diet-fed mice 2 weeks after transduction with low-dose AAV8-shRNA. F) Mean plasma cholesterol concentration of mice in E 2 weeks after transduction with a second low-dose AAV8-shRNA. G) top: Mean cholesterol concentrations of fractions collected from gel filtration of plasma from individual mice in F. bottom: Immunoblots of fractions from representative mice against ApoB are shown. Note that fractions shown begin with the elution front from the size-exclusion column. Two-way ANOVA with Sidak’s multiple comparisons test shown to illustrate comparison between treatment arm within a given fraction. Error bars indicate standard error of the mean. H) Mean plasma cholesterol concentrations of Pcsk9-D377Y overexpressing and Paigen diet-fed mice 2 weeks after transduction with a second dose of low-dose AAV8-shRNA (8 weeks after first dose, for singly dosed mice). Note that Pcsk9-targeted mice were only given the first dose of AAV8. I and J) Leading upregulated (I) and downregulated (J) biological process GO terms in the differentially expressed genes (adj. P < 0.05, log2FC > |1|) in Csde1 knockdown mice on the Paigen diet. All panels) n.s. = non-significant (P ≥ 0.05), * = P < 0.05, ** = P < 0.01,*** = P < 0.001, and **** = P < 0.0001.
To evaluate the functional effect of Csde1 gene disruption, we turned to C57BL/6 mice on a Western-type (high-fat, high-cholesterol) diet (WD). We obtained baseline plasma samples on this diet and delivered a moderate dose (3 × 1011 genomes/mouse) of the AAV8-shRNA targeting either Csde1, Pcsk9, or a scramble control. Two weeks after transduction, we reassessed total plasma cholesterol. We observed a significant decrease, compared to baseline, only for the mice transduced with Csde1-targeting shRNA (21% reduction, P = 0.0027, Fig 5C). The decrease observed with Pcsk9-targeting shRNA fell just outside our prespecified cutoff for significance (P = 0.0592). Lipoprotein fractionation of the post-transduction samples (fig. S16A) revealed a 39% reduction in cholesterol in the LDL-containing fractions between the Csde1 knockdown mice and scramble controls (P = 0.0272, Fig. 5D), but no difference in the very low-density lipoprotein (VLDL) or high-density lipoprotein (HDL) fractions (fig. S16, B and C). Consistent with prior literature (23), we observed an increase in total hepatic LDLR protein with Pcsk9 knockdown (fig. S16D), but as with chow-fed mice, we observed neither differences in histologic appearance of the liver (fig. S17, A to D) nor in the levels of hepatic LDLR protein between the scramble control and Csde1 knockdowns (P = 0.744, figs. S16D and S17, E‘ to L‘).
To better assess the effects on non-HDL cholesterol, we also studied C57BL/6 mice on the atherogenic, cholate-rich Paigen diet (67, 68). Here we delivered low dose (2 × 1010 genomes/mouse) AAV8-shRNA targeting either Csde1, Pcsk9, or a scramble control. Two weeks later, we observed a 25% reduction in fasting plasma cholesterol in the Csde1 knockdown mice, which exceeded the effect of Pcsk9 knockdown (P = 0.0497, Fig. 5E). We then re-dosed the Csde1 and scramble AAV8-shRNA and, 2 weeks later, observed an even stronger phenotype (Fig. 5F). Despite the lower dose of AAV8 delivered, we observed the expected reduction in Csde1 mRNA and an increase in Ldlr mRNA in the livers of the Csde1-targeted mice (fig. S18A). Lipoprotein fractionation of the mouse plasma showed that Csde1 knockdown most strongly affected the VLDL-containing fractions (Fig. 5G), with immunoblots confirming a reduction in both apolipoprotein B100 (ApoB-100) and apoliproprotein B48 (ApoB-48), consistent with an increase in function of the murine LDLR on a cholate-rich dietary background (69, 70). As with the chow-fed mice, we observed no clear difference in the steady-state amount of LDLR protein in the liver (fig. S18B), nor did we observe hepatotoxicity from the Csde1-targeting shRNA (fig. S18C).
Last, we also employed a murine PCSK9 overexpression model to further assess the effects on atherogenic lipoproteins in a LDLR-depleted system. Again using C57BL/6 mice on the atherogenic, cholate-rich diet, we simultaneously delivered low-dose AAV8-Pcsk9-D377Y with low-dose AAV8-shRNA targeting either Csde1, Pcsk9, or a scramble control. We purposefully chose the sub-maximal dose (2 × 1010 genomes/mouse) for AAV8-Pcsk9-D377Y delivery (71) to reduce, but not completely ablate, the LDLR, given our hypothesis that CSDE1’s effects on cholesterol are LDLR-dependent. Two weeks after AAV8 delivery, we observed a marked reduction (73%, P = 0.0351) in total plasma cholesterol in the shRNA-Pcsk9 treated mice and a similar (57%) but nonsignificant (P = 0.1013) reduction in the shRNA-Csde1 treated mice, both when compared to the scramble controls (fig. S19). We therefore re-dosed the mice in the Csde1 and scramble treatment arms with the corresponding AAV8. Two weeks after this second dose, which was 8 weeks after the first dose, we again measured total plasma cholesterol and observed significant reductions for the Csde1 (52%, P = 0.0194) and the singly-dosed Pcsk9 (64%, P = 0.0052) knockdowns, both in comparison to the scramble controls (Fig. 5H). Taken together, the results of these four mouse models suggest that hepatic CSDE1 downregulates Ldlr mRNA expression and reduces clearance of non-HDL lipoproteins in vivo.
The in vivo effect of hepatic CSDE1 silencing on the mouse transcriptome
To gain further insight into the role of hepatic CSDE1, we performed bulk RNA sequencing (RNA-seq) on the liver tissue in the model with the strongest phenotype, the Paigen diet. We compared the Csde1 knockdown to control (scramble) mice (fig. S20, A to C), using the mice with the highest transcript counts of a vector-delivered eGFP reporter to control for variations in transduction efficiency. Because mice on the Paigen diet received a lower dose of AAV8 vector, we also filtered our results for the differentially expressed transcripts in the control mice at the extremes of eGFP expression to control for the effects of viral transduction alone. As expected, we found higher Ldlr expression in the Csde1 knockdown mice (log2FC = 0.43, P = 0.0029, Table S10). Consistent with our mechanistic hypothesis, GO enrichment analysis of the top differentially expressed genes (log2FC > |1|, 745 genes total) revealed upregulation of genes involved in cholesterol homeostasis (P = 2.5 × 10−4, Fig. 5I, fig. S20D, Table S11) and downregulation of genes involved in mRNA catabolic processes (P = 4.9 × 10−50, Fig. 5J, Table S11) or encoding RNA binding functions (P = 4.6 × 10−29, fig. S20E, Table S11). To remove the confounding effects of liver inflammation and steatosis induced by the cholate-containing diet (68), we repeated the bulk RNA-seq in chow-fed mice (fig. S21, A to C). Compared to the mice on the Paigen diet, we observed many fewer differentially expressed genes in the chow-fed Csde1 knockdowns (43 genes total, any log2FC, fig. S21, B to C, Table S12), consistent with less homeostatic perturbation. In agreement with the results from the Paigen diet model, we observed a positive enrichment for genes promoting mRNA stability (P = 4.9 × 10−4, fig. S21D, Table S13) and a negative enrichment for genes encoding RNA binding functions (P = 2.7 × 10−3, fig. S21E, Table S13).
Discussion
Since their introduction, statins, which upregulate the LDLR, have become a major public health success, and with the discovery of PCSK9 and the therapeutic antibodies targeting it, patients can safely reach much lower LDL than is achievable by statins alone (72). However, we may be able to push further on this LDL-LDLR axis to achieve greater clinical benefits.
In this study, we modeled a clinically relevant phenotype of LDLR abundance and function, complementing the independent investigations of other groups (73, 74). When synthesizing our screening and validation data together with large-scale genomics and additional pharmacologic perturbations, we produced an exploratory map of potential regulatory mechanisms for the LDLR (fig. S22). These data represent promising targets and also pathways likely to be impacted by therapies already in use in the clinic.
We have shown that CSDE1 regulates LDLR abundance in HepG2 cells by promoting LDLR mRNA decay via its 3’ UTR. These data lay in concert with CSDE1’s destabilizing effects on other transcripts, such as c-Fos (58). We have also shown that in vivo knockdown of Csde1 upregulates hepatic Ldlr mRNA expression and improves atherogenic lipid profiles in mice. This mimics the effect of deleting the 3’ UTR in vivo (75) and phenocopies a human variant with a large deletion in the LDLR 3’ UTR, the only gain-of-function LDLR mutation identified that markedly reduces LDL cholesterol (76). It is notable that several small molecules, including triciribine (62) and berberine (66, 77), have stabilizing effects on LDLR mRNA, though if their mechanisms directly involve CSDE1 remain to be elucidated. The magnitude of LDLR upregulation imparted by CSDE1 knockdown mirrored or exceeded that of HMGCR and PCSK9 in both tissue culture and mouse models, suggesting that a high-fidelity approach targeting CSDE1-mediated LDLR mRNA decay in the clinic could have similar effects. In addition, our mechanistic data suggest that targeting CSDE1 could be at least additive with the use of statins.
Although after CSDE1 knockdown we observed an increase in hepatic LDLR protein in our human tissue culture models, we observed no change in total hepatic LDLR protein in our mouse models despite a reduction in LDLR-cleared lipoproteins. Whether this difference between our models reflects hepatocyte physiology within an intact organism or instead a species-specific difference between the HDL-predominant mouse (69, 70) and the LDL-predominant human remains to be determined. Our data are consistent with metabolic labeling experiments demonstrating the homeostatic defense of steady-state hepatic LDLR in the mouse in response to cholesterol loading conditions and altered LDLR transcription (78). Our data are also consistent with mouse experiments that markedly perturb Ldlr mRNA but have comparatively muted effects on LDLR protein (66, 79). We hypothesize that inhibition of CSDE1 protects LDLR mRNA from CSDE1-mediated decay, upregulates LDLR synthesis, and induces in vivo homeostatic mechanisms that together increase LDLR function but maintain steady-state LDLR abundance in the mouse. The nature of these mechanisms will be important to uncover. Intriguingly, our tissue culture data showed that, compared to a statin, disrupting CSDE1 had a disproportionately larger effect on LDL uptake than surface LDLR amounts, supporting the possibility that CSDE1 could also affect LDLR function, in addition to its abundance. Further, these data also showed that CSDE1 knockdown also increased MYLIP transcripts. Because the gene target of MYLIP, IDOL, induces post-translational degradation of the LDLR, this effect might help explain our in vivo findings. In support of this possibility, the transcriptomic profiling of the chow-fed Csde1 knockdown mice revealed a negative enrichment in genes encoding ubiquitin ligase binding (P = 5.9 × 10−3, fig. S21D, Table S13), suggesting that ubiquitin-dependent regulators such as IDOL may be perturbed. Given our observation of increased LDL uptake with CSDE1 knockdown in the LDLR deficient background, a separate LDLR-independent effect of CSDE1 on lipoproteins also may contribute. Further work is needed to investigate the potential contributions of these and other mechanisms, such as increased endocytic turnover of existing LDLR and altered synthesis and secretion of non-HDL lipoproteins.
The degree to which CSDE1 inhibition affects other transcripts, or other tissues (57, 80), also remains an important question. As an RNA chaperone, CSDE1 can have a variety of effects, from mRNA stabilization (56) to promotion or inhibition of translation (81–83), dependent on the identity of the RNA it binds and the cofactors with which it interacts. Intriguingly, although CSDE1 was found to bind biotinylated LDLR 3’ UTR transcripts in HepG2 cell lysates (61), cross-linking immunoprecipitation approaches in both mouse brain and melanoma cells failed to identify LDLR mRNA as a CSDE1 binding partner (84, 85). This suggests that the CSDE1-LDLR interaction is context dependent. Advances in liver-specific delivery of gene-silencing agents (15, 86), gene editing technologies (87), and small molecules (88) offer the possibility that selectively targeting hepatic CSDE1 for cholesterol lowering could avoid systemic toxicities.
We acknowledge several limitations in our study. First, outside of CSDE1, the validation of gene targets affecting both LDL receptor abundance and function is limited to a single cell line. Additionally, of our validated hits in tissue culture, we only confirmed CSDE1 in an in vivo mammalian model. Third, the fundamental differences in lipoprotein metabolism in the mouse may limit the translation of our conclusions surrounding CSDE1 to humans (69, 70). Last, we have not directly assessed the effects of CSDE1 disruption in non-hepatic tissues.
Nevertheless, we observed no ill effects from CSDE1 disruption in either our tissue culture or in vivo models and our transcriptional profiling suggests only a small number (43) of differentially affected transcripts under normal feeding conditions. This suggests that potential toxicities of hepatic CSDE1 disruption may be low, which will be important to confirm in future studies that pursue CSDE1 as a therapeutic target for cholesterol lowering. Given that CSDE1 has such varied effects on other transcripts, further mechanistic dissection of the hepatic CSDE1-LDLR interaction could identify what makes this relationship unique and guide a potential therapeutic strategy. Our transcriptomic analyses may guide subsequent investigations of both possible toxicities and mechanistic details of hepatic CSDE1 disruption. Combination therapies targeting interconnected pathways to disease can provide increased benefits without inducing extreme side effects, with angiotensin receptor blockade and neprilysin inhibition in heart failure a prominent clinical example (89). To the extent that hepatic CSDE1 utilizes specific factors to downregulate LDLR mRNA, simultaneous tissue-specific drugging of both CSDE1 and these factors could widen the overall therapeutic window.
In summary, we leveraged a clinically relevant phenotype in an unbiased, genome-wide forward genetics screen to identify a panel of previously unrecognized regulators of the LDLR. Together, our mechanistic experiments in tissue culture and physiologic models in mice suggest that CSDE1 regulates hepatic LDLR function by promoting LDLR mRNA decay and may be a potential therapeutic target for heart disease.
Materials and Methods
Study design
We designed the study as a discovery biology experiment to identify new regulators of the LDL receptor. We used an established tissue culture model, HepG2 cells, to evaluate for LDL receptor regulation. We used wild-type zebrafish (Ekwill) and wild-type mice (C57BL/6) to validate the contribution of our top hit, CSDE1, to LDL receptor regulation in vivo. We evaluated sufficient cells for the LDLR and TFR screens to provide adequate coverage for transduction and downstream sequencing of each sgRNA in the genome-wide library. Sample sizes for animal experiments were estimated to provide 80% power (two-tailed α = 0.05) for a 25% difference in cholesterol compared to controls, based on effects in these models in the existing literature. The numbers of animals used in each experiment are noted in the figures and manuscript. Unless otherwise noted, all in vitro data are representative of multiple (≥ 3) biological replicates to ensure robust outcomes. Experimental and control arms were randomly assigned at the outset of the experiment, with no exclusion criteria predefined and animal samples selected randomly for analyses requiring only a subset of the treatment arm. Experiments were not performed in a blinded fashion. All animal studies were performed in accordance with institutional animal care and use committee (IACUC) approved protocols at the University of California, San Francisco as well as the NIH Guide for the Care and Use of Laboratory Animals. Reporting of the animal studies is in compliance with ARRIVE guidelines 2.0 listed in the EQUATOR Network library.
Generation of CRISPRi cell lines
All cell lines were transduced using virus-containing supernatant in the presence of 8 μg/ml polybrene (MilliporeSigma). HepG2 cells expressing dCas9-KRAB were derived by transduction with lentivirus harboring SFFV-dCas9-BFP-KRAB, followed by two rounds of FACS for blue fluorescent protein (BFP) positive cells on a Becton Dickinson (BD) FACSAria II. dCas9-KRAB HepG2 with individual targeting sgRNAs were derived by transduction with lentivirus harboring the desired sgRNA, followed by 48 hrs of puromycin selection (2 μg/ml, InvivoGen), before experiments.
Quantitative reverse transcription PCR (RT-qPCR)
dCas9-KRAB HepG2 cells stably expressing an appropriate sgRNA were harvested, lysed, and total RNA was extracted via the RNeasy Mini Kit (Qiagen). RNA was converted into complementary DNA (cDNA) using qScript cDNA SuperMix (QuantaBio) following the manufacturer’s instructions. RT-qPCR was performed against indicated targets with PrimeTime qPCR primers (Integrated DNA Technologies) using the SYBR Select Master Mix (Thermo Fisher Scientific) according to the manufacturer’s instructions on a CFX96 Touch Real-Time PCR Detection System (BioRad). Fold changes were calculated using ΔΔCt analysis, normalizing each sample to B2M controls, using CFX Maestro software (BioRad). RNA extracted from mouse liver from in vivo experiments was processed similarly but with additional B2m, Actb, and Gapdh housekeeping controls.
Genome-wide CRISPRi screen
The screen was conducted similarly to prior descriptions (12–14). Approximately 200 × 106 dCas9-KRAB HepG2 were transduced with hCRISPRi-v2 top 5 sgRNAs/gene lentivirus at an MOI of ~0.5, and with polybrene at 8 μg/ml, on day 1. Cells were grown on 15-cm dishes, subdivided into four replicates immediately upon transduction (biological duplicate for each screen), and reseeded every 3–4 days as necessary to avoid over-confluence. Cells were selected with puromycin (2 mg/ml) from day 2 through day 6. On day 5, cells for the LDLR sort were placed in DMEM with 5% LPDS. On day 7, approximately 50 × 106 cells from 2 replicates were live-dead stained and stained for LDLR as described in the supplementary materials and methods, and then two-way sorted on a BD FACSAria II for the top and bottom 33% by LDLR abundance. Cells were spun down, washed in PBS and frozen at −80 °C. On day 8, the sort was repeated except in one replicate, cells were stained for TFR instead of LDLR and then sorted as per above. Genomic DNA was isolated using a NucleoSpin Blood DNA extraction kit (Macherey-Nagel). The sgRNA-containing region was PCR-amplified with NEBNext Ultra II Q5 MasterMix (New England Biolabs), acrylamide gel-purified, and size-selected by SPRI beads (Beckman Coulter), all as previously described, prior to sequencing on an Illumina HiSeq 4000.
Human genomic analysis
Protein coding variants for hits validated with individual sgRNAs were assayed in the UK Biobank (90) for associations with LDL-C. In the setting of a statin medication, LDL-C was divided by 0.7 as done previously (31). Genotyping and imputation was performed in the UK Biobank as previously described (33), and nonsynonymous protein coding variants with minor allele frequencies greater than 0.001 were considered. Efficient linear mixed models adjusting for age, sex, genotyping array, and principal components of ancestry were employed, using BOLT-LMM (91). Statistical significance was assigned at α = 0.05/117 = 0.000427 to account for multiple hypothesis testing.
Overexpression experiments
HepG2 or engineered dCas9-HepG2 cell lines were seeded into 96 well plates at 5 × 104 cells per well in HepG2 growth medium. After 24 hrs, cells were washed and changed into low-glucose DMEM with 5% LPDS. Each well was transfected with 100 ng of the appropriate CSDE1 overexpression construct, or vector control, in a total of 10 μL OptiMEM (Thermo Fisher Scientific) using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were incubated at 37°C with 5% CO2 for 72 hrs, and then harvested for LDL receptor expression analysis as described in the supplementary materials and methods.
mRNA Decay Experiments
Engineered dCas9-HepG2 cell lines harboring appropriate sgRNAs were seeded into 12 well plates at 5 × 105 cells per well in HepG2 growth medium. After 24 hrs, cells were washed and changed into sterol-depleting media (low-glucose DMEM with 5% LPDS) supplemented with 6 μM simvastatin. After an additional 24 hrs, actinomycin D (MilliporeSigma) was added at 5 μg/ml, and cells were harvested at the indicated timepoints.
siRNA and AAV8-shRNA knockdowns in tissue culture
Appropriate cell types were transfected with Silencer Select siRNA (Thermo Fisher Scientific) against CSDE1 (Assay ID s15373) or control (Negative Control No. 1) at a final concentration of 25 μM with RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. For HepG2 cells, reverse siRNA transfections were performed, and for Huh7 cells and primary hepatocytes, forward transfections were performed. For primary hepatocytes, the transfection agent was removed and replaced with fresh media after 4 h. For AAV8-delivered shRNA, primary mouse hepatocytes were transduced with custom-generated AAV8 (Vector Biolabs) at approximately 1 × 106 genomes/cell with 4 mM polybrene. Cells were incubated at 37°C with 5% CO2 for 72 h, with daily media changes for primary hepatocytes, prior to downstream analyses (RT-qPCR, LDL receptor abundance, and LDL uptake as described above and in the supplementary materials and methods).
Dual-luciferase assays
Engineered dCas9-HepG2 cells were seeded into opaque white 96 well plates, at 2.2 × 104 cells per well, in 100 μL growth medium the day prior to transfection. On the day of transfection, medium was replaced or changed to sterol-depleted medium (low-glucose DMEM with 5% LPDS) with or without 6 μM simvastatin as appropriate. Each well was transfected with 100 ng of Luc2-PromLDLR based construct and 1 ng of secreted nanoluciferase control construct (pSS-NLuc) in a total of 10 μL OptiMEM using Lipofectamine 3000 according to the manufacturer’s instructions. 6 replicates were transfected per construct per experiment. After 48 hours at 37°C and 5% CO2, 10 μL of medium was removed from each plate and aliquoted into a separate 384 well plate. Firefly luciferase activity was evaluated in the plates containing the cells by adding an equal volume of a 2× firefly lytic assay buffer [100 mM Tris-HCl pH 7.7, 50 mM NaCl, 2 mM MgCl2, 0.17% Triton X-100, 10 mM dithiothreitol (DTT), 0.4 mM coenzyme A, 0.3 mM adenosine triphosphate (ATP), and 0.28 mg/ml luciferin (Goldbio)] (92). Nanoluciferase activity was evaluated from the conditioned medium using a non-lytic 2× coelenterazine (Goldbio) reagent as previously described (93, 94). Raw luminescence was obtained on a SPARK plate reader (Tecan) with 1 second integration time. Readout of firefly luciferase in each well was normalized to the corresponding secreted nanoluciferase control and data were visually inspected and cleaned to remove values from poorly transfected wells (formally defined by ROUT = 1%) during analysis.
Zebrafish handling, maintenance, and Cas9-ribonucleoprotein knockdowns
All zebrafish studies were performed as previously described (65) with minor modifications. Briefly, wild type zebrafish embryos were injected at the one-cell stage with Cas9-RNP complexes and raised at 28 °C. Cas9-RNP complexes were prepared as previously described (65) using custom oligonucleotides against the indicated targets (Elim Biopharmaceuticals). Targeting of tyrosinase, which results in larval albinism, was used as an injection control. Larvae were fed a high-cholesterol diet (64) of Golden Pearls (5–50 micron, Brine Shrimp Direct,) supplemented (4% w/w) with cholesterol (MilliporeSigma) 3× daily from 4 dpf, fasted on 7 dpf to clear intestinal cholesterol, and harvested at 8 dpf. Larvae were collected, extensively washed, anesthetized in tricaine, and collected in groups of 10 per sample prior to storage at −80 °C.
Cholesterol analysis of zebrafish homogenates
Total cholesterol was analyzed as previously described (64) with minor modifications. Briefly, frozen larvae were homogenized in PBS with a plastic pestle, and then clarified at 18,000 × g for 15 min. Supernatants were recovered and total protein content was analyzed by bicinchoninic acid (BCA) assay. Homogenates were then analyzed, in duplicate, at the appropriate dilution (typically 1:12 in PBS) for total cholesterol content using a commercial fluorometric assay (Cayman Chemical). Fluorescence outputs were measured on a Tecan SPARK plate reader, and cholesterol concentrations were interpolated from a regression line calculated from a standard curve. Cholesterol was normalized to total protein content for analysis and subsequently to the scramble control for comparison between experiments.
Mouse handling, maintenance, and shRNA knockdowns
8–10 week old male C57BL/6 mice (The Jackson Laboratory) were maintained on a normal chow diet and then placed on the Western diet (0.15% cholesterol, 21% fat, D12079Bi, Research Diets) or atherogenic “Paigen” diet (1.25% cholesterol, 15% fat, 0.5% cholate, D12336i, Research Diets) (67) at the beginning of the appropriate experiment (week 0). After 2 to 4 weeks on the appropriate diet, AAV8-packaged expression vector encoding Pcsk9-D377Y or eGFP, or AAV8-packaged shRNA against mouse Csde1 (NM_144901), Pcsk9 (NM_153565), or scramble control (Vector Biolabs), were diluted in sterile PBS to a concentration of either 2 × 1011 (low dose) or 3 × 1012 (moderate dose) genomes/ml. 100 μL of diluted AAV8 (2 × 1010 or 3 × 1011 genomes/mouse) harboring the appropriate construct shRNA was administered to each mouse via tail vein injection. Two weeks after AAV8 injection, mice were fasted overnight and then underwent blood sampling via submandibular vein puncture. Approximately 50 μL of blood was collected into an EDTA-coated tube, centrifuged at 2000 × g for 10 min at 4 °C, and the plasma recovered and stored at −20 °C until further analysis. Total plasma cholesterol, after approximately 1:200 to 1:400 dilution in assay buffer, or 1:4000 dilution for Pcsk9-D377Y boosted mice, was evaluated by commercial fluorometric cholesterol assay (Cayman) according to the manufacturer’s instructions. Mouse plasma was evaluated for alanine and aspartate aminotransferase activity using commercial assays (Cayman) according to the manufacturer’s instructions. For mice on the Paigen diet, 6 weeks after initial AAV8 injection, mice from the same exposure arm were re-dosed with AAV8-shRNA targeting either Csde1 or scramble control. At the time of sacrifice, the mice were again fasted overnight and then euthanized after CO2 narcosis followed by cervical dislocation. The abdominal cavity was opened with a ventral midline incision, the inferior vena cava was cannulated, and plasma was collected as described above. The liver and vasculature were perfused with PBS, and the samples of the liver were harvested. Tissue samples for RNA evaluation were placed in TRIzol (Thermo Fisher Scientific) and those for protein analysis were flash frozen in liquid N2 and stored at −80 °C.
RNA-seq library preparation
Total RNA was extracted from frozen liver samples using the Qiagen RNeasy Plus Universal mini kit followed by Manufacturer’s instructions (Qiagen). RNA samples were quantified using Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and RNA integrity was checked using Agilent TapeStation 4200 (Agilent Technologies). Purified RNA was used for mouse RT-qPCR experiments as described above. RNA sequencing libraries were prepared via polyA selection using the NEBNext Ultra RNA Library Prep Kit for Illumina using manufacturer’s instructions (New England Biolabs). Briefly, mRNAs were initially enriched with Oligod(T) beads. Enriched mRNAs were fragmented for 15 minutes at 94 °C. First strand and second strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3’ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by PCR with limited cycles. The sequencing library was validated on the Agilent TapeStation (Agilent) and quantified by using Qubit 2.0 Fluorometer (Invitrogen) as well as by quantitative PCR (KAPA Biosystems). The sequencing libraries were clustered on a single lane of a flowcell. After clustering, the flowcell was loaded on the Illumina HiSeq instrument (4000 or equivalent) according to manufacturer’s instructions. The samples were sequenced using a 2 × 150 bp Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification. RNA library preparation and sequencing were conducted by GENEWIZ, LLC.
RNA-seq analysis
All raw sequencing data underwent quality control checks with FastQC (v 0.11.8). Reads were mapped to the mm10 mouse reference genome using Rsubread (v 2.4.3) and assigned to Ensembl gene IDs. Ensembl gene IDs were then mapped to org.Mm.eg.db (v3.12.0) gene symbols using AnnotationDBI (v1.52.0). Gene expression was quantified using raw counts and differential expression gene testing was performed on the scramble-shRNA samples comparing the groups (n=3 in each group) at the highest and lowest amounts of raw eGFP expression in the Paigen diet model with EdgeR (95, 96) (v.3.32.1) using the glmQLFit method, default settings (97). Statistical significance was set at 5% false discovery rate (FDR; Benjamini-Hochberg). Differential expression gene testing was then performed on the Csde1-shRNA and scramble-shRNA at the highest amounts of eGFP expression with the overlap of differentially expressed genes identified between these two analyses subsequently removed. Functional enrichment gene-set analysis for GO (Gene Ontology) terms was performed using Enrichr (98) via the enrichR R package (v.3.0). Heatmaps were generated using the Bioconductor package ComplexHeatmap (99) (v.2.6.2) using log2-transformed CPM values (counts-per-million; values shown are log2-transformed and row-normalized). Volcano plots were generated using the Bioconductor package EnhancedVolcano (v.1.8.0).
Statistical analysis
Fluorescence values from gated populations in flow cytometry experiments were background corrected by unstained controls and normalized to the values of the cell line harboring negative control sgRNA when appropriate. Normalized data were then grouped by the Cochrane method (100), and values for cell lines transduced with individual sgRNAs were compared those of the negative control by t-test with Holm-Sidak correction. For direct comparison of flow cytometry populations, the Tχ metric was also used (101). For comparison of one-phase decay regression curves in mRNA decay experiments, the extra sum-of-squares F test was used. Pairwise testing to controls was performed in all other experiments using Welch’s t-test with Holm-Sidak correction unless otherwise noted. For comparison across more than two groups, one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test or two-way ANOVA with Sidak’s multiple comparisons test were used unless otherwise noted. When parametric tests were used, data was tested for normality by the D’Agostino-Pearson or Kolmogorov-Smirnov tests. Adjusted P values < 0.05 (two-sided testing) were considered significant. Unless otherwise noted, error bars indicate 95% confidence intervals. In figures, n.s. = non-significant at P > 0.05, * = P < 0.05, ** = P < 0.01, *** = P < 0.001, and **** = P < 0.0001. Statistical analysis was performed using Prism 7 (GraphPad). All experiments were biologically replicated thrice unless otherwise noted.
Supplementary Material
Table 1: Association of nonsynonymous variants in CRISPRi screen hits with serum LDL cholesterol in the UK Biobank.
BETA indicates the linear regression standardized effect size, and P_BOLT_LMM indicates the linear mixed model P value using BOLT-LMM (91).
| GENE | Variant rsID | BETA | P_BOLT_LMM | Consequence | IMPACT |
|---|---|---|---|---|---|
| HNF4A | rs1800961 | 0.0564144 | 0 | missense_variant | MODERATE |
| BCAM | rs28399659 | −0.0174111 | 7.70E-29 | missense_variant | MODERATE |
| BCAM | rs200398713 | −0.0803165 | 1.80E-28 | splice_region_variant,intron_variant | LOW |
| BCAM | rs199922856 | −0.342179 | 6.20E-28 | missense_variant | MODERATE |
| BCAM | rs28399654 | 0.220592 | 6.10E-10 | missense_variant | MODERATE |
| BCAM | rs3810141 | 0.020077 | 5.50E-07 | stop_gained | HIGH |
| TIMELESS | rs2291738 | 0.00388393 | 0.00014 | splice_region_variant,intron_variant | LOW |
| BCAM | rs149302547 | −0.147327 | 0.005 | missense_variant | MODERATE |
| BCAM | rs1135062 | −0.0213642 | 0.0074 | missense_variant | MODERATE |
| C6orf132 | rs55772414 | 0.0116856 | 0.013 | missense_variant | MODERATE |
| MSMO1 | rs142496142 | 0.0432195 | 0.015 | missense_variant | MODERATE |
Acknowledgments
We thank M. Horlbeck for guidance with the CRISPRi system, J.D. Brown for helpful discussion on in vivo experimental designs, R. Yang for assistance with histologic imaging, and P. Cheng and R. Baylis for helpful discussion on histologic insights and in vivo experimental planning. Plasmids for the CRISPRi system were a generous gift from L. Gilbert and J. Weissman. We thank the Gladstone Institutes Flow Cytometry core facility for their assistance with flow cytometry experiments. UK Biobank analyses were conducted using the UK Biobank resource under application 7089.
Funding:
This work was supported by grants from the Tobacco-Related Disease Research Program (578649 to Ar.P.), the A.P. Giannini Foundation (P0527061 to Ar.P.), the Michael Antonov Charitable Foundation Inc. to Ar.P., the Sarnoff Cardiovascular Research Foundation to Ar.P., the Japan Society for the Promotion of Science Overseas Research Fellowship to T.N., the Burroughs Wellcome Foundation to R.J., a Hassenfeld Scholar Award from the Massachusetts General Hospital to P.N., the Fondation Leducq (TNE-18CVD04 to P.N.), the Roddenberry Foundation to D.S., the L.K. Whittier Foundation to D.S., the Younger Family Fund to D.S., the Howard Hughes Medical Institute to K.M.S., a Pfizer ASPIRE Cardiovascular Award to J.S.C., the Harris Fund and Research Evaluation and Allocation Committee of the UCSF School of Medicine to J.S.C., the NIH/NCRR (C06 RR018928 to the Gladstone Institute and D.S.), and the NIH/NHLBI (K08 HL157700 to Ar.P., R01 HL139783 to R.J., R01 HL127564 to P.N., R01 HL142711 to P.N., R01 HL148565 to P.N., R01 HL148050 to P.N., R01 DK119621 to B.L.B., P01 HL146366 to B.L.B. and D.S., P01 HL098707 to D.S., R01 HL057181 to D.S., R01 HL127240 to D.S., K08 HL124068 to J.S.C., R03 HL145259 to J.S.C., R01 HL146404 to J.S.C., and R01 HL159457 to J.S.C.)
Footnotes
Competing Interests: P.N. reports investigator-initiated grant support from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Novartis, and Roche/Genentech, is a co-founder of TenSixteen Bio, is a shareholder of geneXwell and TenSixteen Bio, and spousal employment at Vertex, all unrelated to the present work. R.S.W. is an employee of Amgen, Inc. D.S. is the scientific cofounder, shareholder, and director of Tenaya Therapeutics, unrelated to the present work. K.M.S. has consulting agreements for the following companies involving cash and/or stock compensation: Black Diamond Therapeutics, BridGene Bioscences, Denali Therapeutics, Dice Molecules, eFFECTOR Therapeutics, Erasca, Genentech/Roche, Janssen Pharmaceuticals, Kumquat Biosciences, Kura Oncology, Merck, Mitokinin, Petra Pharma, Revolution Medicines, Type6 Therapeutics, Venthera, Wellspring Biosciences (Araxes Pharma). J.S.C. has received consulting fees from Gilde Healthcare and is an uncompensated scientific advisor to Eko, both unrelated to this work.
Data and Materials Availability:
All data associated with this study are in the paper or supplementary materials. All requests for materials, including plasmids or cell lines, generated in this study are available from the corresponding author via material transfer agreement. UK Biobank data is available by application to the UK Biobank. The raw and processed sequencing data from the RNA-seq experiments have been deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE206846.
References
- 1.Brown MS, Goldstein JL, A receptor-mediated pathway for cholesterol homeostasis, Science 232, 34–47 (1986). [DOI] [PubMed] [Google Scholar]
- 2.Goldstein JL, Brown MS, A century of cholesterol and coronaries: From plaques to genes to statins, Cell 161, 161–172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, Braunwald E, Sabatine MS, Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions, JAMA 316, 1289–1297 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Kochanek KD, Xu J, Arias E, Mortality in the United States, 2019, NCHS Data Brief 395, 1–8 (2020). [PubMed] [Google Scholar]
- 5.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR, FOURIER Steering Committee and Investigators, Evolocumab and clinical outcomes in patients with cardiovascular disease, N. Engl. J. Med. 376, 1713–1722 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Ma C, Gurol ME, Huang Z, Lichtenstein AH, Wang X, Wang Y, Neumann S, Wu S, Gao X, Low-density lipoprotein cholesterol and risk of intracerebral hemorrhage, Neurology 93, e445–e457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabatine MS, Wiviott SD, Im K, Murphy SA, Giugliano RP, Efficacy and safety of further lowering of low-density lipoprotein cholesterol in patients starting with very low levels: A meta-analysis, JAMA Cardiol. 3, 823–828 (2018),. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garg A, Fazio S, Duell PB, Baass A, Udata C, Joh T, Riel T, Sirota M, Dettling D, Liang H, Garzone PD, Gumbiner B, Wan H, Molecular characterization of familial hypercholesterolemia in a North American cohort, J. Endocr. Soc 4, bvz015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klarin D, Damrauer SM, Cho K, V Sun Y, Teslovich TM, Honerlaw J, Gagnon DR, DuVall SL, Li J, Peloso GM, Chaffin M, Small AM, Huang J, Tang H, Lynch JA, Ho Y-L, Liu DJ, Emdin CA, Li AH, Huffman JE, Lee JS, Natarajan P, Chowdhury R, Saleheen D, Vujkovic M, Baras A, Pyarajan S, Di Angelantonio E, Neale BM, Naheed A, V Khera A, Danesh J, Chang K-M, Abecasis G, Willer C, Dewey FE, Carey DJ, Concato J, Gaziano JM, O’Donnell CJ, Tsao PS, Kathiresan S, Rader DJ, Wilson PWF, Assimes TL, G. L. G. Consortium, M. I. G. (MIGen) Consortium, T. G.-R. D. Collaboration, T. V. A. M. V. Program, Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program, Nat. Genet. 50, 1514–1523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, Harrison SC, Li K, Drenos F, Karpe F, Neil HAW, Descamps OS, Langenberg C, Lench N, Kivimaki M, Whittaker J, Hingorani AD, Kumari M, Humphries SE, Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study, Lancet 381, 1293–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Oetjens MT, Kelly MA, Sturm AC, Martin CL, Ledbetter DH, Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders, Nat. Commun. 10, 4897 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS, Genome-scale CRISPR-mediated control of gene repression and activation, Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, Weissman JS, Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation, Elife 5, e19760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, Pak RA, Gray AN, Gross CA, Dixit A, Parnas O, Regev A, Weissman JS, A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response, Cell 167, 1867–1882.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray KK, Wright RS, Kallend D, Koenig W, Leiter LA, Raal FJ, Bisch JA, Richardson T, Jaros M, Wijngaard PLJ, Kastelein JJP, Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol, N. Engl. J. Med. 382, 1507–1519 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Knowles BB, Howe CC, Aden DP, Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen, Science 209, 497–499 (1980). [DOI] [PubMed] [Google Scholar]
- 17.Scharnagl H, Schinker R, Gierens H, Nauck M, Wieland H, März W, Effect of atorvastatin, simvastatin, and lovastatin on the metabolism of cholesterol and triacylglycerides in HepG2 cells, Biochem. Pharmacol. 62, 1545–1555 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Zhang F, Li C, Lin M, Briggs MR, Synergistic activation of human LDL receptor expression by SCAP ligand and cytokine oncostatin M, Arterioscler. Thromb. Vasc. Biol. 23, 90–96 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, Prat A, Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia, Arterioscler. Thromb. Vasc. Biol. 24, 1454–1459 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD, Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice, J. Clin. Invest. 116, 2995–3005 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown MS, Faust JR, Goldstein JL, Kaneko I, Endo A, Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase, J. Biol. Chem. 253, 1121–1128 (1978). [PubMed] [Google Scholar]
- 22.Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin M-C, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chrétien M, Prat A, Seidah NG, NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol, J. Biol. Chem. 279, 48865–48875 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD, Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9, Proc. Natl. Acad. Sci. U. S. A. 102, 5374–5379 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan JC, Piper DE, Cao Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J, Xia Z, Di Y, Shetterly S, Arimura Z, Salomonis H, Romanow WG, Thibault ST, Zhang R, Cao P, Yang XP, Yu T, Lu M, Retter MW, Kwon G, Henne K, Pan O, Tsai MM, Fuchslocher B, Yang E, Zhou L, Lee KJ, Daris M, Sheng J, Wang Y, Shen WD, Yeh WC, Emery M, Walker NP, Shan B, Schwarz M, Jackson SM, A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates, Proc. Natl. Acad. Sci. U. S. A. 106, 9820–9825 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aisen P, Transferrin receptor 1, Int. J. Biochem. Cell Biol. 36, 2137–43 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Coffey R, Ganz T, Iron homeostasis: An anthropocentric perspective, J. Biol. Chem. 292, 12727–12734 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zelcer N, Hong C, Boyadjian R, Tontonoz P, LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor, Science 325, 100–104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshinaga M, Nakatsuka Y, Vandenbon A, Ori D, Uehata T, Tsujimura T, Suzuki Y, Mino T, Takeuchi O, Regnase-1 maintains iron homeostasis via the degradation of transferrin receptor 1 and prolyl-hydroxylase-domain-containing protein 3 mRNAs, Cell Rep. 19, 1614–1630 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD, PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools, Nucleic Acids Res. 47, D419–D426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y, Lee J-Y, Park T, Kim K, Sim X, Twee-Hee Ong R, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Hua Zhao J, Yuan X, Luan J, Lamina C, Ziegler A, Zhang W, Zee RYL, Wright AF, Witteman JCM, Wilson JF, Willemsen G, Wichmann H-E, Whitfield JB, Waterworth DM, Wareham NJ, Waeber G, Vollenweider P, Voight BF, Vitart V, Uitterlinden AG, Uda M, Tuomilehto J, Thompson JR, Tanaka T, Surakka I, Stringham HM, Spector TD, Soranzo N, Smit JH, Sinisalo J, Silander K, Sijbrands EJG, Scuteri A, Scott J, Schlessinger D, Sanna S, Salomaa V, Saharinen J, Sabatti C, Ruokonen A, Rudan I, Rose LM, Roberts R, Rieder M, Psaty BM, Pramstaller PP, Pichler I, Perola M, Penninx BWJH, Pedersen NL, Pattaro C, Parker AN, Pare G, Oostra BA, O’Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, Meitinger T, McPherson R, McCarthy MI, McArdle W, Masson D, Martin NG, Marroni F, Mangino M, Magnusson PKE, Lucas G, Luben R, Loos RJF, Lokki M-L, Lettre G, Langenberg C, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, Kronenberg F, König IR, Khaw K-T, Kaprio J, Kaplan LM, Johansson Å, Jarvelin M-R, Cecile J A Janssens W, Ingelsson E, Igl W, Kees Hovingh G, Hottenga J-J, Hofman A, Hicks AA, Hengstenberg C, Heid IM, Hayward C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Gyllensten U, Guiducci C, Groop LC, Gonzalez E, Gieger C, Freimer NB, Ferrucci L, Erdmann J, Elliott P, Ejebe KG, Döring A, Dominiczak AF, Demissie S, Deloukas P, de Geus EJC, de Faire U, Crawford G, Collins FS, Chen YI, Caulfield MJ, Campbell H, Burtt NP, Bonnycastle LL, Boomsma DI, Boekholdt SM, Bergman RN, Barroso I, Bandinelli S, Ballantyne CM, Assimes TL, Quertermous T, Altshuler D, Seielstad M, Wong TY, Tai E-S, Feranil AB, Kuzawa CW, Adair LS, Taylor HA Jr, Borecki IB, Gabriel SB, Wilson JG, Holm H, Thorsteinsdottir U, Gudnason V, Krauss RM, Mohlke KL, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJP, Schadt EE, Rotter JI, Boerwinkle E, Strachan DP, Mooser V, Stefansson K, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, van Duijn CM, Peltonen L, Abecasis GR, Boehnke M, Kathiresan S, Biological, clinical and population relevance of 95 loci for blood lipids, Nature 466, 707–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Natarajan P, Peloso GM, Zekavat SM, Montasser M, Ganna A, Chaffin M, Khera AV, Zhou W, Bloom JM, Engreitz JM, Ernst J, O’Connell JR, Ruotsalainen SE, Alver M, Manichaikul A, Johnson WC, Perry JA, Poterba T, Seed C, Surakka IL, Esko T, Ripatti S, Salomaa V, Correa A, Vasan RS, Kellis M, Neale BM, Lander ES, Abecasis G, Mitchell B, Rich SS, Wilson JG, Cupples LA, Rotter JI, Willer CJ, Kathiresan S, NHLBI TOPMed Lipids Working Group, Deep-coverage whole genome sequences and blood lipids among 16,324 individuals, Nat. Commun. 9, 3391 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, Saleheen D, Emdin C, Alam D, Alves AC, Amouyel P, Di Angelantonio E, Arveiler D, Assimes TL, Auer PL, Baber U, Ballantyne CM, Bang LE, Benn M, Bis JC, Boehnke M, Boerwinkle E, Bork-Jensen J, Bottinger EP, Brandslund I, Brown M, Busonero F, Caulfield MJ, Chambers JC, Chasman DI, Chen YE, Chen Y-DI, Chowdhury R, Christensen C, Chu AY, Connell JM, Cucca F, Cupples LA, Damrauer SM, Davies G, Deary IJ, Dedoussis G, Denny JC, Dominiczak A, Dubé M-P, Ebeling T, Eiriksdottir G, Esko T, Farmaki A-E, Feitosa MF, Ferrario M, Ferrieres J, Ford I, Fornage M, Franks PW, Frayling TM, Frikke-Schmidt R, Fritsche LG, Frossard P, Fuster V, Ganesh SK, Gao W, Garcia ME, Gieger C, Giulianini F, Goodarzi MO, Grallert H, Grarup N, Groop L, Grove ML, Gudnason V, Hansen T, Harris TB, Hayward C, Hirschhorn JN, Holmen OL, Huffman J, Huo Y, Hveem K, Jabeen S, Jackson AU, Jakobsdottir J, Jarvelin M-R, Jensen GB, Jørgensen ME, Jukema JW, Justesen JM, Kamstrup PR, Kanoni S, Karpe F, Kee F, V Khera A, Klarin D, Koistinen HA, Kooner JS, Kooperberg C, Kuulasmaa K, Kuusisto J, Laakso M, Lakka T, Langenberg C, Langsted A, Launer LJ, Lauritzen T, Liewald DCM, Lin LA, Linneberg A, Loos RJF, Lu Y, Lu X, Mägi R, Malarstig A, Manichaikul A, Manning AK, Mäntyselkä P, Marouli E, Masca NGD, Maschio A, Meigs JB, Melander O, Metspalu A, Morris AP, Morrison AC, Mulas A, Müller-Nurasyid M, Munroe PB, Neville MJ, Nielsen JB, Nielsen SF, Nordestgaard BG, Ordovas JM, Mehran R, O’Donnell CJ, Orho-Melander M, Molony CM, Muntendam P, Padmanabhan S, Palmer CNA, Pasko D, Patel AP, Pedersen O, Perola M, Peters A, Pisinger C, Pistis G, Polasek O, Poulter N, Psaty BM, Rader DJ, Rasheed A, Rauramaa R, Reilly DF, Reiner AP, Renström F, Rich SS, Ridker PM, Rioux JD, Robertson NR, Roden DM, Rotter JI, Rudan I, Salomaa V, Samani NJ, Sanna S, Sattar N, Schmidt EM, Scott RA, Sever P, Sevilla RS, Shaffer CM, Sim X, Sivapalaratnam S, Small KS, V Smith A, Smith BH, Somayajula S, Southam L, Spector TD, Speliotes EK, Starr JM, Stirrups KE, Stitziel N, Strauch K, Stringham HM, Surendran P, Tada H, Tall AR, Tang H, Tardif J-C, Taylor KD, Trompet S, Tsao PS, Tuomilehto J, Tybjaerg-Hansen A, van Zuydam NR, Varbo A, V Varga T, Virtamo J, Waldenberger M, Wang N, Wareham NJ, Warren HR, Weeke PE, Weinstock J, Wessel J, Wilson JG, Wilson PWF, Xu M, Yaghootkar H, Young R, Zeggini E, Zhang H, Zheng NS, Zhang W, Zhang Y, Zhou W, Zhou Y, Zoledziewska M, Howson JMM, Danesh J, McCarthy MI, Cowan CA, Abecasis G, Deloukas P, Musunuru K, Willer CJ, Kathiresan S, Abecasis G, Deloukas P, Musunuru K, Willer CJ, Kathiresan S, Exome-wide association study of plasma lipids in >300,000 individuals, Nat. Genet. 49, 1758–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P, Marchini J, The UK Biobank resource with deep phenotyping and genomic data, Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loregger A, Nelson JK, Zelcer N, Assaying low-density-lipoprotein (LDL) uptake into cells, Methods Mol. Biol. 1583, 53–63 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Mazein A, Watterson S, Hsieh WY, Griffiths WJ, Ghazal P, A comprehensive machine-readable view of the mammalian cholesterol biosynthesis pathway, Biochem. Pharmacol. 86, 56–66 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose RB, Bayle JH, Endrizzi JA, Cronk JD, Crabtree GR, Alber T, Structural basis of dimerization, coactivator recognition and MODY3 mutations in HNF-1α, Nat. Struct. Biol. 7, 744–748 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Delaforest A, Di Furio F, Jing R, Ludwig-Kubinski A, Twaroski K, Urick A, Pulakanti K, Rao S, Duncan SA, HNF4A regulates the formation of hepatic progenitor cells from human iPSC-derived endoderm by facilitating efficient recruitment of RNA pol II, Genes (Basel). 10, 21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang K, Holterman AX, Pathophysiologic role of hepatocyte nuclear factor 6, Cell. Signal. 24, 9–16 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, Zur Hausen A, Brunton VG, Morton J, Sansom O, Schüler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T, The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs, Nat. Cell Biol. 11, 1487–1495 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Gao CL, Zhu JG, Zhao YP, Chen XH, Ji CB, Zhang CM, Zhu C, Xia ZK, Peng YZ, Guo XR, Mitochondrial dysfunction is induced by the overexpression of UCP4 in 3T3-L1 adipocytes, Int. J. Mol. Med. 25, 71–80 (2010). [PubMed] [Google Scholar]
- 41.Quazi F, Molday RS, Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants, J. Biol. Chem. 288, 34414–34426 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kung JE, Jura N, The pseudokinase TRIB 1 toggles an intramolecular switch to regulate COP 1 nuclear export, EMBO J. 38, e99708 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soubeyrand S, Martinuk A, McPherson R, TRIB1 is a positive regulator of hepatocyte nuclear factor 4-Alpha, Sci. Rep. 7, 5574 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bauer RC, Sasaki M, Cohen DM, Cui J, Smith MA, Yenilmez BO, Steger DJ, Rader DJ, Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPα, J. Clin. Invest. 125, 3809–3818 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burkhardt R, Toh SA, Lagor WR, Birkeland A, Levin M, Li X, Robblee M, Fedorov VD, Yamamoto M, Satoh T, Akira S, Kathiresan S, Breslow JL, Rader DJ, Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice, J. Clin. Invest. 120, 4410–4414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Motley A, Bright NA, Seaman MNJ, Robinson MS, Clathrin-mediated endocytosis in AP-2-depleted cells, J. Cell Biol. 162, 909–918 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parsons SF, Mallinson G, Holmes CH, Houlihan JM, Simpson KL, Mawby WJ, Spurr NK, Warne D, Barclay AN, Anstee DJ, The Lutheran blood group glycoprotein, another member of the immunoglobulin superfamily, is widely expressed in human tissues and is developmentally regulated in human liver, Proc. Natl. Acad. Sci. U. S. A. 92, 5496–5500 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mishra SK, Keyel PA, Edeling MA, Dupin AL, Owen DJ, Traub LM, Functional dissection of an AP-2 β2 appendage-binding sequence within the autosomal recessive hypercholesterolemia protein, J. Biol. Chem. 280, 19270–19280 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Scandinavian Simvastatin Survival Study Group, Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S), Lancet 344, 1383–1389 (1994). [PubMed] [Google Scholar]
- 50.Lintner NG, McClure KF, Petersen D, Londregan AT, Piotrowski DW, Wei L, Xiao J, Bolt M, Loria PM, Maguire B, Geoghegan KF, Huang A, Rolph T, Liras S, Doudna JA, Dullea RG, Cate JHD, Selective stalling of human translation through small-molecule engagement of the ribosome nascent chain, PLoS Biol. 15, e2001882 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ, Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9, J. Lipid Res. 49, 394–398 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Suzuki T, Terasaki M, Takemoto-Hori C, Hanada T, Ueda T, Wada A, Watanabe K, Structural compensation for the deficit of rRNA with proteins in the mammalian mitochondrial ribosome. Systematic analysis of protein components of the large ribosomal subunit from mammalian mitochondria, J. Biol. Chem. 276, 21724–21736 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson A, Sjöstedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Pontén F, Von Feilitzen K, Lilley KS, Uhlén M, Lundberg E, A subcellular map of the human proteome, Science 356, eaal3321 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Li H, Dong B, Park SW, Lee H-S, Chen W, Liu J, Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine, J. Biol. Chem. 284, 28885–95 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo A-X, Cui J-J, Wang L-Y, Yin J-Y, The role of CSDE1 in translational reprogramming and human diseases, Cell Commun. Signal. 18, 14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dinur M, Kilav R, Sela-Brown A, Jacquemin-Sablon H, Naveh-Many T, In vitro evidence that upstream of N-ras participates in the regulation of parathyroid hormone messenger ribonucleic acid stability, Mol. Endocrinol. 20, 1652–1660 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Moore KS, Yagci N, Van Alphen F, Paolini NA, Horos R, Held NM, Houtkooper RH, Van Den Akker E, Meijer AB, T’Hoen PAC, Von Lindern M, Csde1 binds transcripts involved in protein homeostasis and controls their expression in an erythroid cell line, Sci. Rep. 8, 2628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang TC, Yamashita A, Chen CYA, Yamashita Y, Zhu W, Durdan S, Kahvejian A, Sonenberg N, Bin Shyu A, UNR, a new partner of poly(A)-binding protein, plays a key role in translationally coupled mRNA turnover mediated by the c-fos major coding-region determinant, Genes Dev. 18, 2010–2023 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson GM, Vasa MZ, Deeley RG, Stabilization and cytoskeletal-association of LDL receptor mRNA are mediated by distinct domains in its 3’ untranslated region, J. Lipid Res. 39, 1025–1032 (1998). [PubMed] [Google Scholar]
- 60.Horlbeck MA, Xu A, Wang M, Bennett NK, Park CY, Bogdanoff D, Adamson B, Chow ED, Kampmann M, Peterson TR, Nakamura K, Fischbach MA, Weissman JS, Gilbert LA, Mapping the genetic landscape of human cells, Cell 174, 953–967.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Chen W, Zhou Y, Abidi P, Sharpe O, Robinson WH, Kraemer FB, Liu J, Identification of mRNA binding proteins that regulate the stability of LDL receptor mRNA through AU-rich elements, J. Lipid Res. 50, 820–831 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bjune K, Wierød L, Naderi S, Triciribine increases LDLR expression and LDL uptake through stabilization of LDLR mRNA, Sci. Rep. 8, 16174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bakheet T, Frevel M, Williams BRG, Greer W, Khabar KSA, ARED: Human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins, Nucleic Acids Res. 29, 246–254 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu C, Kim YS, Kim J, Pattison J, Kamaid A, Miller YI, Modeling hypercholesterolemia and vascular lipid accumulation in LDL receptor mutant zebrafish, J. Lipid Res 59, 391–399 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu RS, Lam II, Clay H, Duong DN, Deo RC, Coughlin SR, A Rapid Method for Directed Gene Knockout for Screening in G0 Zebrafish., Dev. Cell 46, 112–125.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Singh AB, Li H, Kan CFK, Dong B, Nicolls MR, Liu J, The critical role of mRNA destabilizing protein heterogeneous nuclear ribonucleoprotein D in 3′ untranslated region-mediated decay of low-density lipoprotein receptor mRNA in liver tissue, Arterioscler. Thromb. Vasc. Biol. 34, 8–16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P, Variation in susceptibility to atherosclerosis among inbred strains of mice, Atherosclerosis 57, 65–73 (1985). [DOI] [PubMed] [Google Scholar]
- 68.Lichtman AH, Clinton SK, Iiyama K, Connelly PW, Libby P, Cybulsky MI, Hyperlipidemia and atherosclerotic lesion development in LDL receptor–deficient mice fed defined semipurified diets with and without cholate, Arterioscler. Thromb. Vasc. Biol. 19, 1938–1944 (1999). [DOI] [PubMed] [Google Scholar]
- 69.Getz GS, Reardon CA, Diet and murine atherosclerosis, Arterioscler. Thromb. Vasc. Biol. 26, 242–249 (2006). [DOI] [PubMed] [Google Scholar]
- 70.von Scheidt M, Zhao Y, Kurt Z, Pan C, Zeng L, Yang X, Schunkert H, Lusis AJ, Applications and limitations of mouse models for understanding human atherosclerosis, Cell Metab. 25, 248–261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bjørklund MM, Hollensen AK, Hagensen MK, Dagnæs-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF, Induction of atherosclerosis in mice and hamsters without germline genetic engineering, Circ. Res. 114, 1684–1689 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Giugliano RP, Pedersen TR, Park J-G, De Ferrari GM, Gaciong ZA, Ceska R, Toth K, Gouni-Berthold I, Lopez-Miranda J, Schiele F, Mach F, Ott BR, Kanevsky E, Pineda AL, Somaratne R, Wasserman SM, Keech AC, Sever PS, Sabatine MS, FOURIER Investigators, Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial, Lancet 390, 1962–1971 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Emmer BT, Sherman EJ, Lascuna PJ, Graham SE, Willer CJ, Ginsburg D, Genome-scale CRISPR screening for modifiers of cellular LDL uptake, PLOS Genet. 17, e1009285 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trinh MN, Brown MS, Goldstein JL, Han J, Vale G, McDonald JG, Seemann J, Mendell JT, Lu F, Last step in the path of LDL cholesterol from lysosome to plasma membrane to ER is governed by phosphatidylserine, Proc. Natl. Acad. Sci. U. S. A. 117, 18521–18529 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Knouff C, Malloy S, Wilder J, Altenburg MK, Maeda N, Doubling expression of the low density lipoprotein receptor by truncation of the 3′-untranslated region sequence ameliorates type III hyperlipoproteinemia in mice expressing the human ApoE2 isoform, J. Biol. Chem. 276, 3856–3862 (2001). [DOI] [PubMed] [Google Scholar]
- 76.Bjornsson E, Gunnarsdottir K, Halldorsson GH, Sigurdsson A, Arnadottir GA, Jonsson H, Olafsdottir EF, Niehus S, Kehr B, Sveinbjörnsson G, Gudmundsdottir S, Helgadottir A, Andersen K, Thorleifsson G, Eyjolfsson GI, Olafsson I, Sigurdardottir O, Saemundsdottir J, Jonsdottir I, Magnusson OT, Masson G, Stefansson H, Gudbjartsson DF, Thorgeirsson G, Holm H, V Halldorsson B, Melsted P, Norddahl GL, Sulem P, Thorsteinsdottir U, Stefansson K, Lifelong reduction in LDL (low-density lipoprotein) cholesterol due to a gain-of-function mutation in LDLR, Circ. Genomic Precis. Med. 14, e003029 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C, Wang Y, Wang Z, Si S, Pan H, Wang S, Wu J, Wang Y, Li Z, Liu J, Jiang JD, Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins, Nat. Med. 10, 1344–1351 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Dandan M, Han J, Mann S, Kim R, Mohammed H, Nyangau E, Hellerstein M, Turnover rates of the low-density lipoprotein receptor and PCSK9: Added dimension to the cholesterol homeostasis model, Arterioscler. Thromb. Vasc. Biol. 41, 2866–2876 (2021). [DOI] [PubMed] [Google Scholar]
- 79.Singh AB, Kan CFK, Shende V, Dong B, Liu J, A novel posttranscriptional mechanism for dietary cholesterol-mediated suppression of liver LDL receptor expression, J. Lipid Res. 55, 1397–1407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ju Lee H, Bartsch D, Xiao C, Guerrero S, Ahuja G, Schindler C, Moresco JJ, Yates JR, Gebauer F, Bazzi H, Dieterich C, Kurian L, Vilchez D, A post-transcriptional program coordinated by CSDE1 prevents intrinsic neural differentiation of human embryonic stem cells, Nat. Commun. 8, 1456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boussadia O, Niepmann M, Créancier L, Prats A-C, Dautry F, Jacquemin-Sablon H, Unr is required in vivo for efficient initiation of translation from the internal ribosome entry sites of both rhinovirus and poliovirus, J. Virol. 77, 3353–3359 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dormoy-Raclet V, Markovits J, Jacquemin-Sablon A, Jacquemin-Sablon H, Regulation of Unr expression by 5′- and 3′-untranslated regions of its mRNA through modulation of stability and IRES mediated translation, RNA Biol. 2, e27–35 (2005). [PubMed] [Google Scholar]
- 83.Duncan KE, Strein C, Hentze MW, The SXL-UNR corepressor complex uses a PABP-mediated mechanism to inhibit ribosome recruitment to msl-2 mRNA, Mol. Cell 36, 571–582 (2009). [DOI] [PubMed] [Google Scholar]
- 84.Guo H, Li Y, Shen L, Wang T, Jia X, Liu L, Xu T, Ou M, Hoekzema K, Wu H, Gillentine MA, Liu C, Ni H, Peng P, Zhao R, Zhang Y, Phornphutkul C, Stegmann APA, Prada CE, Hopkin RJ, Shieh JT, McWalter K, Monaghan KG, van Hasselt PM, van Gassen K, Bai T, Long M, Han L, Quan Y, Chen M, Zhang Y, Li K, Zhang Q, Tan J, Zhu T, Liu Y, Pang N, Peng J, Scott DA, Lalani SR, Azamian M, Mancini GMS, Adams DJ, Kvarnung M, Lindstrand A, Nordgren A, Pevsner J, Osei-Owusu IA, Romano C, Calabrese G, Galesi O, Gecz J, Haan E, Ranells J, Racobaldo M, Nordenskjold M, Madan-Khetarpal S, Sebastian J, Ball S, Zou X, Zhao J, Hu Z, Xia F, Liu P, Rosenfeld JA, de Vries BBA, Bernier RA, Xu ZQD, Li H, Xie W, Hufnagel RB, Eichler EE, Xia K, Disruptive variants of CSDE1 associate with autism and interfere with neuronal development and synaptic transmission, Sci. Adv. 5, eaax2166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wurth L, Papasaikas P, Olmeda D, Bley N, Calvo GT, Guerrero S, Cerezo-Wallis D, Martinez-Useros J, García-Fernández M, Hüttelmaier S, Soengas MS, Gebauer F, UNR/CSDE1 drives a post-transcriptional program to promote melanoma invasion and metastasis, Cancer Cell 30, 694–707 (2016). [DOI] [PubMed] [Google Scholar]
- 86.Gennemark P, Walter K, Clemmensen N, Rekić D, Nilsson CAM, Knöchel J, Hölttä M, Wernevik L, Rosengren B, Kakol-Palm D, Wang Y, Yu RZ, Geary RS, Riney SJ, Monia BP, Isaksson R, Jansson-Löfmark R, Rocha CSJ, Lindén D, Hurt-Camejo E, Crooke R, Tillman L, Rydén-Bergsten T, Carlsson B, Andersson U, Elebring M, Tivesten A, Davies N, An oral antisense oligonucleotide for PCSK9 inhibition, Sci. Transl. Med. 13, eabe9117 (2021). [DOI] [PubMed] [Google Scholar]
- 87.Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, Wang K, Iyer S, Dutta C, Clendaniel V, Amaonye M, Beach A, Berth K, Biswas S, Braun MC, Chen H-M, V Colace T, Ganey JD, Gangopadhyay SA, Garrity R, Kasiewicz LN, Lavoie J, Madsen JA, Matsumoto Y, Mazzola AM, Nasrullah YS, Nneji J, Ren H, Sanjeev A, Shay M, Stahley MR, Fan SHY, Tam YK, Gaudelli NM, Ciaramella G, Stolz LE, Malyala P, Cheng CJ, Rajeev KG, Rohde E, Bellinger AM, Kathiresan S, In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates, Nature 593, 429–434 (2021). [DOI] [PubMed] [Google Scholar]
- 88.McClure KF, Piotrowski DW, Petersen D, Wei L, Xiao J, Londregan AT, Kamlet AS, Dechert-Schmitt A-M, Raymer B, Ruggeri RB, Canterbury D, Limberakis C, Liras S, DaSilva-Jardine P, Dullea RG, Loria PM, Reidich B, Salatto CT, Eng H, Kimoto E, Atkinson K, King-Ahmad A, Scott D, Beaumont K, Chabot JR, Bolt MW, Maresca K, Dahl K, Arakawa R, Takano A, Halldin C, Liver-targeted small-molecule inhibitors of proprotein convertase subtilisin/kexin type 9 synthesis, Angew. Chemie Int. Ed 56, 16218–16222 (2017). [DOI] [PubMed] [Google Scholar]
- 89.V McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR, Angiotensin–neprilysin inhibition versus enalapril in heart failure, N. Engl. J. Med. 371, 993–1004 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, Liu B, Matthews P, Ong G, Pell J, Silman A, Young A, Sprosen T, Peakman T, Collins R, UK Biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age, PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, Patterson N, Price AL, Efficient Bayesian mixed-model analysis increases association power in large cohorts, Nat. Genet. 47, 284–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baker JM, Boyce FM, High-throughput functional screening using a homemade dual-glow luciferase assay, J. Vis. Exp. 88, e50282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chorba JS, Galvan AM, Shokat KM, Stepwise processing analyses of the single-turnover PCSK9 protease reveal its substrate sequence specificity and link clinical genotype to lipid phenotype, J. Biol. Chem. 293, 1875–1886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chorba JS, Galvan AM, Shokat KM, A high-throughput luciferase assay to evaluate proteolysis of the single-turnover protease PCSK9, J. Vis. Exp. 138, e58265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lun ATL, Chen Y, Smyth GK, It’s DE-licious: A recipe for differential expression analyses of RNA-seq experiments using quasi-likelihood methods in edgeR, Methods Mol. Biol. 1418, 391–416 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A, Enrichr: a comprehensive gene set enrichment analysis web server 2016 update, Nucleic Acids Res. 44, W90–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gu Z, Eils R, Schlesner M, Complex heatmaps reveal patterns and correlations in multidimensional genomic data., Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 100.Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions version 5.1.0, The Cochrane Collaboration; (2011). Available from www.handbook.cochrane.org. [Google Scholar]
- 101.Roederer M, Moore W, Treister A, Hardy RR, Herzenberg LA, Probability binning comparison: A metric for quantitating multivariate distribution differences, Cytometry 45, 47–55 (2001). [DOI] [PubMed] [Google Scholar]
- 102.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO, Enzymatic assembly of DNA molecules up to several hundred kilobases, Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 103.Kim JH, Lee S-R, Li L-H, Park H-J, Park J-H, Lee KY, Kim M-K, Shin BA, Choi S-Y, High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell Lines, zebrafish and mice, PLoS One 6, e18556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gibson DG, Smith HO, Hutchison CA, Venter JC, Merryman C, Chemical synthesis of the mouse mitochondrial genome, Nat. Methods 7, 901–903 (2010). [DOI] [PubMed] [Google Scholar]
- 105.Consortium Uniprot, UniProt: A worldwide hub of protein knowledge, Nucleic Acids Res. 47, D506–D515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Padmanabhan A, Alexanian M, Linares-Saldana R, González-Terán B, Andreoletti G, Huang Y, Connolly AJ, Kim W, Hsu A, Duan Q, Winchester SAB, Felix F, Perez-Bermejo JA, Wang Q, Li L, Shah PP, Haldar SM, Jain R, Srivastava D, BRD4 (bromodomain-containing protein 4) interacts with GATA4 (GATA binding protein 4) to govern mitochondrial homeostasis in adult cardiomyocytes, Circulation 142, 2338–2355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or supplementary materials. All requests for materials, including plasmids or cell lines, generated in this study are available from the corresponding author via material transfer agreement. UK Biobank data is available by application to the UK Biobank. The raw and processed sequencing data from the RNA-seq experiments have been deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE206846.