

ABSTRACT
Pulmonary alveolar microlithiasis (PAM) is a rare autosomal recessive disease characterised by the deposition of calcium phosphate microliths in the alveoli. PAM has been reported in all continents and there is often a familial history. There is clinical-radiological dissociation as there is often a paucity of symptoms in contrast to the imaging findings. Patients often remain asymptomatic until the third or fourth decade of life, and dyspnea is the most common symptom. PAM is caused by a mutation within the solute carrier family 34 member 2 gene (the SLC34A2 gene) located on chromosome 4p15.2, which encodes a sodium/phosphate co-transporter. The imaging appearance of the disease is quite pathognomic with the high-resolution computed tomography (HRCT) demonstrating a diffuse micronodular appearance. Transbronchial lung biopsy also confirms the diagnosis. There is no effective therapy at present except lung transplantation. We herein, present a case of PAM along with clinical history, imaging study, histopathological study and genetic study of a 43-year-old female adult patient along with genetic analysis.
KEY WORDS: Flow-volume loop, restrictive ventilatory defect, upper airway obstruction
CASE
A 45-year-old female patient, a resident of Shimla, Himachal Pradesh, presented to Indira Gandhi Medical College, Shimla, India, with a history of shortness of breath for the last 3 years, which is persistent and progressive. She feels breathless on climbing four flights of stairs. She also complains of fatigue on walking uphill. She also had a history of cough, which is dry in nature. For the last 4 months, she had a low-grade fever (990F) and loss of appetite. She has no underlying comorbidities. She is a non-smoker and has a history of exposure to biomass fuel smoke. She has no cyanosis or pedal oedema. Her vitals parameters included an oxygen saturation of 95% at room air, pulse rate of 82/min and blood pressure of 126/78 mmHg. On physical examination, auscultation of the lungs revealed no abnormalities. Cardiovascular and other systems examination also revealed no abnormalities. Her chest X-ray revealed bilateral diffuse fine micronodules in the mid and lower zones of the lungs. High-resolution computed tomography (HRCT) of the thorax revealed bilateral diffuse micro-calcification within the lung parenchyma and pericardium [Figures 1 a and b and 2]. Fiberoptic bronchoscopy, bronchoalveolar lavage (BAL) and transbronchial lung biopsy were done. GeneXpert test of BAL fluid showed the presence of the Mycobacterium tuberculosis complex. The transbronchial lung biopsy revealed numerous lamellated calcified structures within alveolar spaces consistent with the diagnosis of pulmonary microlithiasis [Figure 3]. Whole exome sequencing revealed a novel homozygous variant c.T1136C (p.L380P) in the SLC34A2 gene responsible for pulmonary alveolar microlithiasis (PAM). The variant was classified as a likely pathogenic variant based on the American College of Medical Genetics and Genomics (ACMG)/ Association for Molecular Pathology (AMP) guidelines. The genomic study of the patient’s brother, son and daughter revealed the presence of the same mutation (heterozygous). She was treated with a full course of anti-tuberculous therapy (2RHZE/4RHE).
Figure 1.
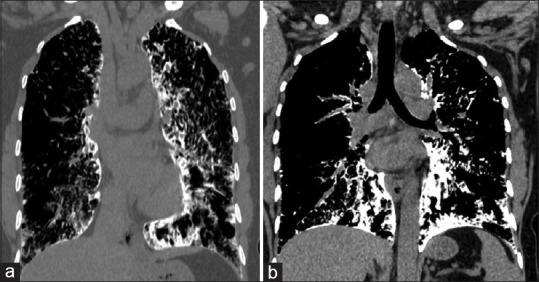
(a and b) HRCT thorax (mediastinal window, coronal view) showing pericardial calcification and calcification over the diaphragmatic pleura
Figure 2.
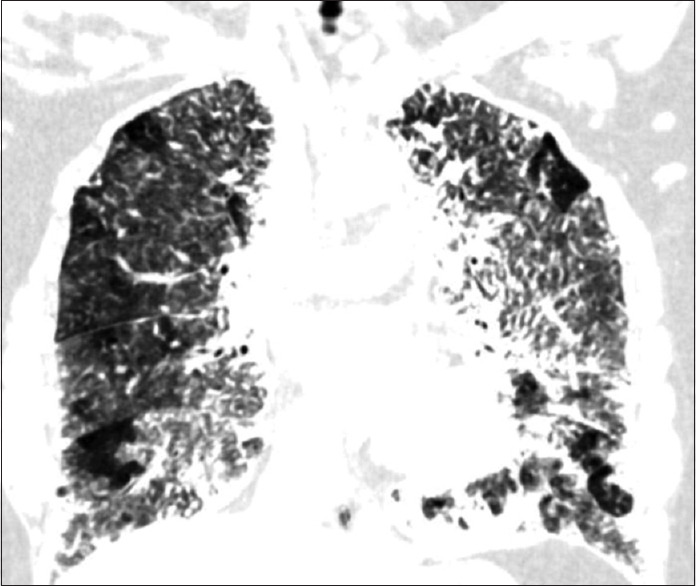
HRCT thorax (lung window, coronal view) showing innumerable calcified nodules within the lung parenchyma involving the alveoli and interlobular septa
Figure 3.
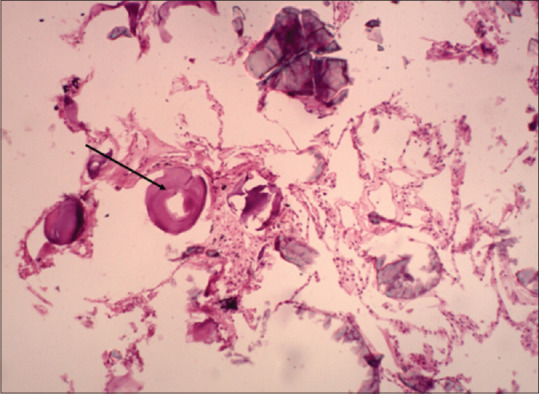
Laminated calcified bodies inside alveolar spaces on H and E staining
DISCUSSION
PAM is a rare hereditary lung disease characterised by the deposition of calcium phosphate microliths (calcospherites) within the alveolar spaces. PAM was first described in the literature by an Italian scientist, Marco Malpighi;[1] however, the disease was named ‘Microlithiasis Alveolaris Pulmonum’ by the Hungarian pathologist Ludwig Puhr in 1933.[2] Castellana et al.[3] published the largest survey of literature on PAM including 1,022 cases from 65 nations. They observed a familial link in 37% of patients. Although there is an overall male preponderance, cases from Italy, Spain and France had shown a female preponderance.[3] The majority of cases are diagnosed at the age of 30–40 years. PAM is caused by an inactivating mutation of the SCL34A2 gene. The SCL34A2 gene is located on chromosome 4p15.2. It has 13 exons and encodes a 2,280-nt mRNA and a 690-aa protein. The main function of the gene is maintaining homeostasis of inorganic phosphate via sodium phosphate IIb transporter protein (Npt2b transporter).[4] Npt2b transporter clears phosphate from alveolar spaces into type II pulmonary alveolar cells in the presence of sodium ions at a ratio of 3Na + 1:1HPO4-2.[5] The mutated SLC34A2 gene fails to clear phosphates from the alveolar space, resulting in a gradual accumulation of intra-alveolar calcium microliths. To further support the role of the Npt2b transporter, Saito et al.[4] observed in a mouse model that epithelial deletion of Npt2b results in a progressive pulmonary process characterised by diffuse alveolar microlith accumulation, radiographic opacification, restrictive physiology, inflammation, fibrosis and unexpected alveolar phospholipidosis. They also confirmed the potential therapeutic role of ethylene diamine tetraacetic acid (EDTA) lavage and a low phosphate diet. The SLC34A2 gene is expressed predominantly in type II alveolar epithelial cells. However, is also expressed in other organs such as mammary glands, the small intestine, kidneys, pancreas, ovaries, liver, testes, placenta and prostate.[6] Corut et al.[7] six homozygous exonic mutations in the seven unrelated patients with PAM. The types of mutations were the following; frameshifts (three), chain termination (one), amino acid substitution (one) and deletion spanning the minimal promoter and the first exon (one). They reported PAM as a recessive monogenic disease with full penetrance. There are also reports of variant localisation with a particular geographical area. Variants in exon 8 have been reported from China and Japan,[4] whereas, variant c. 1402_1404delACC in exon 12 has been reported in patients from Europe only.[8] Like many other rare autosomal recessive disorders, PAM is associated with consanguinity and consanguinity was always reported during vertical transmission.[3]
Patients with PAM typically show an indolent course. The majority of patients are asymptomatic at the time of diagnosis and they usually manifest in the third and fourth decades of life. With disease progression, patients develop exertional dyspnoea. Other symptoms are non-productive cough, chest pain and asthenia.[9] The majority of patients show a variable progression of the disease, although a few may remain stationary.[3] Patients with advanced disease may develop persistent hypoxemia, pulmonary fibrosis, respiratory failure and cor pulmonale.[10] Clinical–radiological dissociation is often seen and characterised by a paucity of physical signs/symptoms and extensive radiological involvement. Digital clubbing is seen in the advanced stages of the disease. Smoking and infection may accelerate disease progression.[7]
Diagnosis
Despite the calcium phosphate microliths deposition within the alveolar spaces, serum calcium and phosphates levels are normal and no evidence of any systemic disorder of calcium metabolism has been detected.[11,12] Surfactant protein (SP)-D, SP-A and monocyte chemotactic protein 1 (MCP-1) may be used as a biomarker in patients with PAM; however, their availability would be an issue. The pulmonary function tests (PFTs) typically show a restrictive defect with a reduction in diffusion capacity for carbon monoxide although it may be normal in early disease. Similar to other chronic diffuse lung diseases, they may demonstrate reduced exercise capacity and exercise-induced desaturation on 6-min walk testing.
PAM is diagnosed confidently by the characteristic imaging pattern and genetic testing showing the rare biallelic SCL34A2 gene variants.[13] If genetic testing is unavailable or negative, bronchoscopy-guided procedures such as bronchoalveolar lavage (BAL), transbronchial biopsy and open lung biopsy can be performed to confirm the diagnosis of PAM. The diagnostic yield with transbronchial cryobiopsies and surgical lung biopsies are higher with a higher risk of complications.[14] BAL and transbronchial biopsy may show microliths. Microliths show distinct calcareous concentric lamellae around a central nucleus with an amorphous or granular aspect on periodic acid–Schiff staining. The microliths have a diameter of 0.01 to 2.8 mm.[9] Microliths are mainly composed of calcium and phosphorus (phosphorus:calcium ratio of 1:2),[15] with varying amounts of iron, zinc, aluminium, silica, magnesium, potassium and copper.[9]
In the Castellana et al.’s[3] review of 1,022 PAM cases globally, tuberculosis was the comorbidity reported in five cases. The high prevalence of tuberculosis can explain this association in developing countries.
Imaging study
Chest radiography shows a diffuse distribution of innumerable, small, sand-like micronodules (also called microliths, calcipherites or calcospherites and <1 mm in diameter), within the air spaces, particularly in the lower parts of the lungs. This appearance is called ‘Sand-storm lung’. A few micronodules may be of bigger size (2–4 mm). Micronodules may obscure the mediastinal and diaphragmatic borders in the advanced stage. It preferentially involves the middle and lower zones due to greater blood supply.[16] HRCT should be the preferred modality as it detects minute parenchymal abnormalities.[17] HRCT features commonly seen are calcified pulmonary nodules, diffuse ground-glass opacity, sub pleural cysts and consolidation. Calcifications may also occur along the interlobular septa, bronchovascular bundles, fissures and pleura. Francisco et al.[18] reported ground glass opacities (GGO) and small parenchymal nodules as the predominant tomographic findings, present in 100% of cases. Other findings were small subpleural nodules (92.3%), sub pleural cysts (84.6%), sub pleural linear calcifications (69.2%), crazy-paving pattern (69.2%), fissure nodularity (53.8%), calcification along interlobular septa (46.2%) and dense consolidation (46.2%). The mosaic pattern has also been reported in the literature.[19] Marchiori et al.[20] reported a mosaic pattern in 40% of patients. Another characteristic imaging pattern is the black pleural line. It is a vertical stripe of peripheral hyperlucency between the ribs and the adjacent diffusely dense calcified lung parenchyma and is caused by subpleural cystic changes.
Treatment
There is no specific therapy for PAM and the management is mainly supportive. Therapy with corticosteroids, calcium-binding agents and whole-lung lavage has been tried and has shown no benefit. Supplemental oxygen may be advised in hypoxemic patients. All PAM patients should be advised of influenza and pneumococcal vaccination. In advanced disease, lung transplantation is the only effective therapy and bilateral lung transplant is preferred to unilateral lung transplantation as persistent shunting of blood through the lung may cause persistent intrapulmonary shunts.[21] Jackson et al.[22] in single lung transplantation for PAM patients reported post-lung transplantation survival without recurrence for 15 years. Bisphosphonates such as etidronate decrease bone resorption by inhibiting osteoclastogenesis.[23] Moreover, etidronate interferes with phosphate incorporation into hydroxyapatite, culminating in reduced crystal formation.[24] The response to etidronate treatment has shown variable results.[25] Saito et al.[4] had shown a beneficial effect of a low-phosphate diet in mice with epithelial deletion of Npt2b as the reduced formation of microliths but human studies are lacking.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Author contribution
MS conceived and designed the article, made critical revisions, and approved the final version. SKS and AJ performed the histopathological examination of the lung biopsy sample. AJ reported the imaging study. All authors reviewed and approved the final manuscript.
Acknowledgment
We are thankful to Dr. Vinod Scaria MBBS, PhD Principal Scientist CSIR Institute of Genomics and Integrative Biology (CSIR-IGIB), South Campus, Mathura Road, New Delhi 110 025. India for providing us the facility for genetic testing of the patient and her relatives.
REFERENCES
- 1.Saito A, McCormack FX. Pulmonary alveolar microlithiasis. Clin Chest Med. 2016;37:441–8. doi: 10.1016/j.ccm.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puhr L. Mikrolithiasis alveolaris pulmonum. Virchows ArchPatholAnatPhysiolKlinMed. 1933;290:156–60. [Google Scholar]
- 3.Castellana G, Castellana G, Gentile M, Castellana R, Resta O. Pulmonary alveolar microlithiasis:Review of the 1022 cases reported worldwide. Eur Respir Rev. 2015;24:607–20. doi: 10.1183/16000617.0036-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saito A, Nikolaidis NM, Amlal H, Uehara Y, Gardner JC, LaSance K, et al. Modeling pulmonary alveolar microlithiasis by epithelial deletion of the Npt2b sodium phosphate cotransporter reveals putative biomarkers and strategies for treatment. Sci Transl Med. 2015;7:313ra181. doi: 10.1126/scitranslmed.aac8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bacconi A, Virkki LV, Biber J, Murer H, Forster IC. Renouncing electroneutrality is not free of charge:Switching on electrogenicity in a Naþ-coupled phosphate cotransporter. Proc Natl Acad Sci U S A. 2005;102:12606–11. doi: 10.1073/pnas.0505882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu A, Shaharyar S, Chokshi B, Bhardwaj N. Pulmonary alveolar microlithiasis “stone lungs”:A case of clinico-radiological dissociation. Cureus. 2016;8:e749. doi: 10.7759/cureus.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corut A, Senyigit A, Ugur SA, Altin S, Ozcelik U, Calisir H, et al. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am J Hum Genet. 2006;79:650–6. doi: 10.1086/508263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jönsson ÅLM, Bendstrup E, Mogensen S, Kopras EJ, McCormack FX, Campo I, et al. Eight novel variants in the SLC34A2 gene in pulmonary alveolar microlithiasis. Eur Respir J. 2019;55:1900806. doi: 10.1183/13993003.00806-2019. [DOI] [PubMed] [Google Scholar]
- 9.Mariotta S, Ricci A, Papale M, De Clementi F, Sposato B, Guidi L, et al. Pulmonary alveolar microlithiasis:Report on 576 cases published in the literature. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21:173–81. [PubMed] [Google Scholar]
- 10.Terada T. Pulmonary alveolar microlithiasis with cor pulmonale:An autopsy case demonstrating a marked decrease in pulmonary vascular beds. Respir Med. 2009;103:1768–71. doi: 10.1016/j.rmed.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Francisco FA, Pereira e Silva JL, Hochhegger B, Zanetti G, Marchiori E. Pulmonary alveolar microlithiasis. State-of-the-art review. Respir Med. 2013;107:1–9. doi: 10.1016/j.rmed.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 12.Krishnakurup J, Abdelsayed G. The calcareous lung. Mayo Clin Proc. 2011;86:85. doi: 10.4065/mcp.2010.0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bendstrup E, Jönsson ÅLM. Pulmonary alveolar microlithiasis:No longer in the stone age. ERJ Open Res. 2020;6:00289–2020. doi: 10.1183/23120541.00289-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johannson KA, Marcoux VS, Ronksley PE, Ryerson CJ. Diagnostic yield and complications of transbronchial lung cryobiopsy for interstitial lung disease:A systematic review and metaanalysis. Ann Am Thorac Soc. 2016;13:1828–38. doi: 10.1513/AnnalsATS.201606-461SR. [DOI] [PubMed] [Google Scholar]
- 15.Barnard NJ, Crocker PR, Blainey AD, Davies RJ, Ell SR, Levison DA. Pulmonary alveolar microlithiasis. A new analytical approach. Histopathology. 1987;11:639–45. doi: 10.1111/j.1365-2559.1987.tb02674.x. [DOI] [PubMed] [Google Scholar]
- 16.Delic JA, Fuhrman CR, Trejo Bittar HE. Pulmonary alveolar microlithiasis:AIRP best cases in radiologic-pathologic correlation. Radiographics. 2016;36:1334–8. doi: 10.1148/rg.2016150259. [DOI] [PubMed] [Google Scholar]
- 17.Deniz O, Ors F, Tozkoparan E, Ozcan A, Gumus S, Bozlar U, et al. High resolution computed tomographic features of pulmonary alveolar microlithiasis. Eur J Radiol. 2005;55:452–60. doi: 10.1016/j.ejrad.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Francisco FAF, Rodrigues RS, Barreto MM, Escuissato DL, Araujo Neto CA, Silva JL, et al. Can chest high-resolution computed tomography findings diagnose pulmonary alveolar microlithiasis? Radiol Bras;2015;48:205–10. doi: 10.1590/0100-3984.2014.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sigari N, Nikkhoo B. First presentation of a case of pulmonary alveolar microlithiasis with spontaneous pneumothorax. Oman Med J. 2014;29:450–3. doi: 10.5001/omj.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marchiori E, Gonçalves CM, Escuissato DL, Teixeira KI, Rodrigues R, Barreto MM, et al. Pulmonary alveolar microlithiasis:High-resolution computed tomography findings in 10 patients. J Bras Pneumol. 2007;33:552–7. doi: 10.1590/s1806-37132007000500010. [DOI] [PubMed] [Google Scholar]
- 21.Jindal A, Rahulan V, Balasubramani G, Dutta P, Attawar S. Pulmonary alveolar microlithiasis:A rare disease treated with lung transplantation, first case from India. Lung India. 2019;36:546–9. doi: 10.4103/lungindia.lungindia_50_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson KB, Modry DL, Halenar J, L’abbe J, Winton TL, Lien DC, et al. Single lung transplantation for pulmonary alveolar microlithiasis. J Heart Lung Transplant. 2001;20:226. doi: 10.1016/s1053-2498(00)00500-3. [DOI] [PubMed] [Google Scholar]
- 23.Xu X-L, Gou W-L, Wang A-Y, Wang Y, Guo Q-Y, Lu Q, et al. Basic research and clinical applications of bisphosphonates in bone disease:What have we learned over the last 40 years? J Transl Med. 2013;11:303. doi: 10.1186/1479-5876-11-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw BM, Shaw SD, McCormack FX. Pulmonary alveolar microlithiasis. Semin Respir Crit Care Med. 2020;41:280–7. doi: 10.1055/s-0040-1702211. [DOI] [PubMed] [Google Scholar]
- 25.Cakir E, Gedik AH, Özdemir A, Buyukpınarbasili N, Bilgin M, Ozgen IT. Response to disodium etidronate treatment in three siblings with pulmonary alveolar microlithiasis. Respiration. 2015;89:583–6. doi: 10.1159/000375464. [DOI] [PubMed] [Google Scholar]