

Abstract
Stimuli-responsive biomaterials show great promise for modelling disease dynamics ex vivo with spatiotemporal control over the cellular microenvironment. However, harvesting cells from such materials for downstream analysis without perturbing their state remains an outstanding challenge in 3D/4D culture and tissue engineering. In this manuscript, we introduce a fully enzymatic strategy for hydrogel degradation that affords spatiotemporal control over cell release while maintaining cytocompatibility. Exploiting engineered variants of the sortase transpeptidase evolved to recognize and selectively cleave distinct peptide sequences largely absent from the mammalian proteome, we sidestep many limitations implicit to state-of-the-art methods to liberate cells from gels. We demonstrate that evolved sortase exposure has minimal impact on the global transcriptome of primary mammalian cells and that proteolytic cleavage proceeds with high specificity; incorporation of substrate sequences within hydrogel crosslinkers permits rapid and selective cell recovery with high viability. In composite multimaterial hydrogels, we show that sequential degradation of hydrogel layers enables highly specific retrieval of single-cell suspensions for phenotypic analysis. We expect that the high bioorthogonality and substrate selectivity of the evolved sortases will lead to their broad adoption as an enzymatic material dissociation cue and that their multiplexed use will enable newfound studies in 4D cell culture.
Keywords: hydrogels, sortase, biomaterials, degradation, single-cell biology, bioorthogonal chemistry
Graphical Abstract
Using engineered sortase transpeptidases that orthogonally recognize and cleave peptide substrates largely absent from the mammalian proteome, multiplexed cell liberation from within 3D hydrogel biomaterials is uniquely achieved without significantly altering cell fate. Cell recovery is compatible with downstream single cell-based functional assays including flow cytometry.
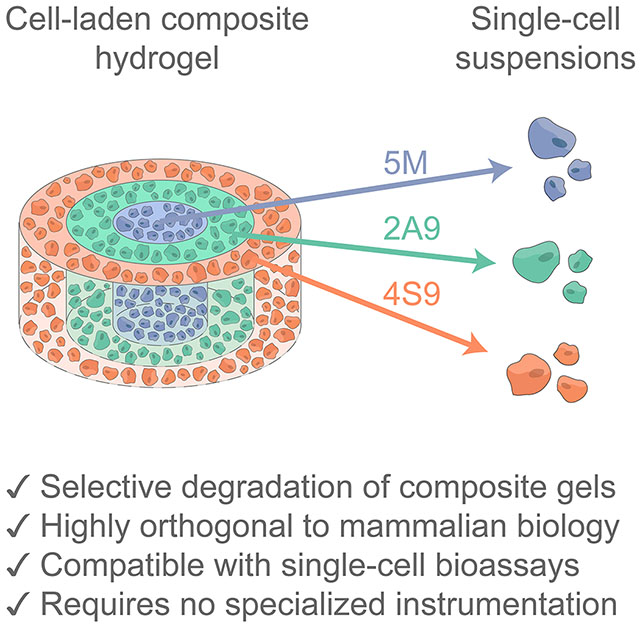
1. Introduction
Cell-laden 3D hydrogel biomaterials have become invaluable for the in vitro study of cellular pathways driving disease.[1–3] Improving on traditional 2D cell culture on plastic or glass, these 3D matrices offer physiologically relevant substrate mechanics and customizable biochemical ligand presentation in a flexible and easy-to-use format.[2,3] These emerging model systems are advantageous over in vivo experimentation for their reduced complexity, and allow for the isolated study of microenvironmental cues on cell state, helping to overcome issues of reproducibility and eventual translation to the clinic.[4,5] With the advent of dynamic hydrogel biomaterials and the resulting unprecedented control over the cellular microenvironment in vitro, the study of spatiotemporally complex disease phenotypes is now possible.[6,7]
Many gold-standard and emerging molecular biology techniques to assess biological fate require single-cell suspensions; uni- or multimodal single-cell omics,[8,9] flow cytometry,[10] fluorescence and magnetic cell sorting,[11,12] cytometry by time of flight,[13] and many others require a viable and unperturbed suspension. Deriving biological meaning from these techniques requires an assumption that the cell isolation method employed does not substantially alter cell state. Obtaining a cell suspension from a natural biomaterial matrix (e.g., native tissue, collagen, fibrin, Matrigel®, decellularized extracellular matrix) necessitates utilization of harsh proteases (e.g., trypsin, collagenase, Liberase™) to digest and dissociate the cell-surrounding substrate. Owing to the sequence promiscuity of such peptidases,[14] these enzymatic treatments have undesired and detrimental impacts on cell state.[15–17] Further complicating matters, natural biomaterials are often subject to batch-to-batch variability that obscures and confounds biological study.[18] To address these collective concerns, synthetic hydrogel matrices including those based on poly(ethylene glycol) (PEG) have been developed that offer reproducible structure and well-defined composition.[19–21] While most synthetic matrices are intrinsically nondegradable, scissile elements may be included within their backbone polymer that are responsive to highly specific inputs, either single or in Boolean YES/OR/AND logic-based combinations.[21–24]
Though degradable synthetic matrices have been well explored for drug delivery applications, there is a growing interest in the release of cells from 3D biomaterials using triggers that do not directly perturb their fate.[25] An ideal trigger should permit rapid material dissolution and bioorthogonal capture of the cell suspension for downstream phenotyping. To this end, photodegradable crosslinks have been extensively employed.[22,26–28] Light offers several advantages as a material degradation trigger, including that its presentation can be precisely controlled in time and space. While powerful, such photochemical strategies suffer drawbacks in their requirement of specialty equipment (e.g., high-intensity illumination lamps, photolithographic masks, multiphoton lithography setups) that are not mainstays in the average molecular biology laboratory. Moreover, photocleavable moieties attenuate light, practically restricting sample geometries.[22,29] Furthermore, the implementation of photolabile chemistries in 4D cell culture necessitates careful consideration of reaction intermediate/product cytotoxicity. These practical limitations underscore the need to develop new degradation chemistries that accommodate a wide range of bulk geometries in a fully biocompatible manner and without the need for niche instrumentation.
While materials that undergo uniform degradation in response to a single environmental trigger have proven powerful, many cell culture applications would benefit tremendously from the ability to confine degradation to specific sample subvolumes. Even within the reductionist environment of a synthetic matrix, cell phenotype is rarely uniform across space and time; paracrine, mechanical, and geometric cues all invoke dynamic changes in cell state that are critical to the pathological progression of any disease. Multiplexed triggers for material degradation enable researchers to extract cells from matrices on demand and with both spatial and temporal control. This has been previously accomplished using composite multimaterials whereby each region responds to an orthogonal degradation cue – most typically differently colored light.[23,28,30,31] Up until this point, multiplexed material dissolution using biologically invisible cues has not been demonstrated.
To address this challenge and overcome current limitations in externally triggered hydrogel degradation, we identified evolved sortases as a robust bioorthogonal tool for material dissolution. Sortase A is a gram-positive bacterial transpeptidase that natively cleaves the LPXTG peptide motif (where X is any amino acid) through transposition of nearby N-terminal polyglycine-containing species.[32] Since the substrate sequence is nearly absent from the mammalian proteome, sortase has become a widely used enzyme to C-terminally “sortag” proteins with noncanonical functionalities.[32–35] Given sortase’s unique functionality and broad utility in protein engineering and chemical biology, significant effort has been dedicated towards improving its activity and specificity: for example, pentamutant sortase (5M) has been engineered for rapid catalysis through a series of five individual point mutations.[36] These advances have been recently embraced by the materials community; a single sortase (i.e., 5M) was first used by the Griffith group to degrade uniform hydrogels crosslinked with peptides containing its LPETG recognition motif, liberating encapsulating cells while preserving their secretome.[37–40] To enable new applications in synthetic biology, two evolved sortase variants – eSrtA(2A9) and eSrtA(4S9) (respectively denoted 2A9 and 4S9) – have been created through directed evolution of 5M to recognize orthogonal peptide substrates: 2A9 acts on LAXTG while 4S9 binds LPXSG.[41] Building on 5M’s success in the biomaterials space, we hypothesized that the 2A9/4S9/5M substrate specificity could be exploited for multiplexed material degradation.
Here we demonstrate a versatile strategy to rapidly liberate individual cell suspensions from hydrogel biomaterials for downstream analysis (including single-cell methodologies) using substrate-selective variants of the enzyme sortase, 2A9 and 4S9. We show that these variants can be used with minimal perturbation to the transcriptomes of sensitive primary cell types, even more so than the 5M enzyme from which they are derived. In combination with open-microfluidic patterning of gel deposition, this strategy enables the construction of complex 3D cell culture models and subsequent spatiotemporal interrogation of the encapsulated cell state in a biologically faithful manner without specialized equipment. We expect that the simplicity of the staged degradation mechanism, compatibility with standard tissue culture practices and assays, and flexibility of this approach will prove useful to cell biologists seeking to uncover disease mechanisms in complex heterocellular models.
2. Results and Discussion
2.1. Evolved sortase treatment has minimal impact on the transcriptome of primary mammalian cells
Primary cell cultures derived from model organisms are a useful in vitro tool to study cellular mechanisms of disease but are particularly sensitive to culture conditions. For example, cardiac fibroblasts exhibit distinct transcriptional profiles and signaling networks that are not recapitulated in immortalized or embryonic fibroblast lines.[42] Due to the varied functions exhibited by cardiac fibroblasts – including responding to injury and tissue damage, aiding in the innate immune response, and engaging in crosstalk with the other cell types which compose the cardiac milieu – we employed them as a model primary cell type to assay whether sortase treatment can perturb a sensitive transcriptome.[42] In immortalized cancer cell lines, the effects of 5M have been evaluated on cell signaling at the proteome level using a small quantitative assay (31 total species) for various cytokines, growth factors, and matrix metalloproteinases (MMPs); unsurprisingly, proteins lacking the sortase recognition sequence were not enzymatically cleaved during treatment.[38] Though these were important findings, it is critical to note that proteolytic activity is not the only way such proteases could be impacting cell state, particularly given that the enzyme is bacterially derived. In Staphylococcus aureus, sortase A anchors proteins to the cell wall which aid in the infection of a host organism.[43] Many of these anchored proteins may be recognized by the innate immune response, a phenomena that has been used to immunize mice against streptococcal infections.[44,45] Therefore, there is an evolutionary basis to suspect that primary cells, in particular those with innate immune function, may recognize sortase in a manner leading to undesired biological outcomes. As such, to identify and broadly survey potential biological changes of state in an unbiased manner, we utilized RNAseq to evaluate the impacts of sortase treatment on the transcriptome of primary cardiac fibroblasts.
Primary murine cardiac fibroblasts cultured on tissue culture polystyrene were treated with each sortase (i.e., 5M, 2A9, or 4S9) (Figure S1) and triglycine (GGG) at literature-established concentrations for 45 minutes, then lysed in situ for global transcriptome quantitation by RNAseq which identified and quantified transcripts from over 13,000 genes across the Mus musculus transcriptome (Figure 1A, Method S1).[38] Hierarchical clustering of the sortase-treated conditions and controls based on Euclidean distance revealed that samples treated with the evolved 2A9 and 4S9 transpeptidases varied more in transcriptional state between technical replicates than between treatment conditions, indicating that treatment with 2A9/4S9 was not associated with any broad transcriptional changes to fibroblast state (Figure 1B). By principal component analysis (PCA), the first principal component (PC), accounting for a relatively small fraction (18%) of sample variance, distinguished the 5M-treated samples from other experimental groups. This first PC, however, did not resolve the transcriptome of 2A9- or 4S9-treated cells from one another or from the controls. The second PC accounted for 15% of variance and separated the 2A9- and 4S9-treated samples from the controls (Figure 1C). Altogether the PCA data indicates that 5M elicits a more distinct transcriptional response from cardiac fibroblasts than 2A9 or 4S9, but that the impacts were relatively minor overall. These results provide newfound and dramatically expanded credence in sortase’s use as a bioorthogonal tool for material actuation.
Figure 1 –
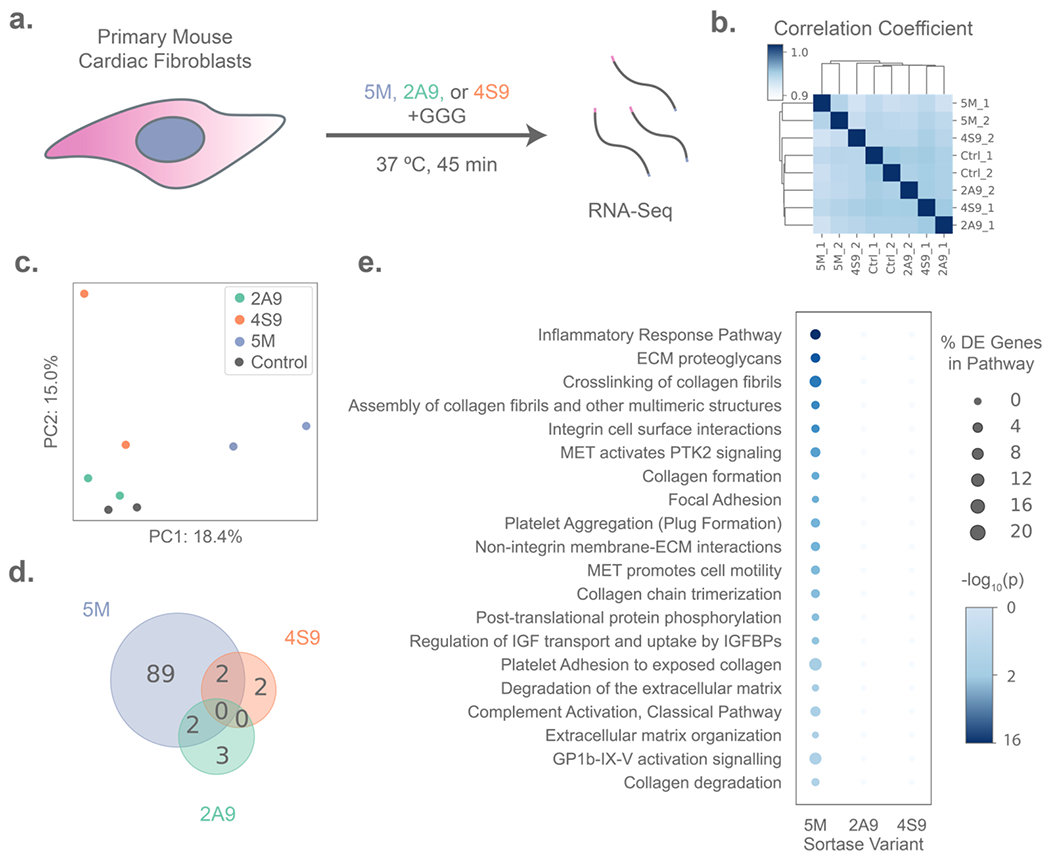
Transcriptomic analysis of primary cell responses to treatment with evolved sortase variants. (A) Experimental schematic in which primary mouse cardiac fibroblasts (n = 2 per condition) were treated with either 50 μM of a sortase variant (5M, 2A9, or 4S9) and 18 mM triglycine (GGG) peptide, or GGG-only control for 45 minutes before lysis for RNA isolation and sequencing. (B) Primary fibroblasts treated with 2A9 and 4S9 are more transcriptionally variable between technical replicates than controls, as illustrated by hierarchical clustering based on Euclidean distance of gene expression, whereas 5M-treated samples form a distinct cluster. (C) Principal Component Analysis (PCA) plot showing separation of sortase-treated samples on the first two principal components (PCs), with PC1 separating 5M-treated samples from all other groups and PC2 distinguishing 2A9 and 4S9 from controls. (D) Treatment with 2A9 and 4S9 results in lower numbers of differentially expressed genes than 5M versus the controls, as depicted by Venn diagram. (E) 5M (but not 2A9 or 4S9) treatment results in the differential regulation of genes significantly enriched in several biological pathways identified by G:Profiler analysis, with bubbles sized by the percentage of genes in a pathway that are differentially expressed in each sortase treatment, and shaded by p-value for enrichment (Fisher’s Exact Test).
To more thoroughly investigate the specific effects of sortase treatment on primary cardiac fibroblast state, we tested for differentially expressed genes between all experimental groups using EdgeR (Table S1). In samples treated with 5M – the only sortase variant which has been reported previously in literature to dissolve synthetic matrices – we found 89 differentially expressed genes associated with treatment. Of these, 19 named genes were upregulated and 26 downregulated relative to the control samples. In contrast, the evolved variants 2A9 and 4S9 only produced a significant response in the expression of four (one named) and five (two named) genes, respectively (Figure 1D). Intriguingly, genes associated with 5M treatment were predominantly related to the inflammation and innate immune response of primary fibroblasts (Figure 1E); enriched pathways included “Complement Activation” and “Inflammatory Response Pathway” comprising of downregulated genes including Clu, C3, Fn1, Thbs1, Col1a2, and Col1a1. Altogether, the altered transcription levels of these genes suggested downmodulation of the fibroinflammatory state of primary fibroblasts by 5M, also supported by downregulation of Egr1, a growth factor essential for fibrotic response to TGFβ1 that directly regulates Fn1.[46,47] Of note, only two of the affected genes encoded a protein bearing a sortase substrate motif.
Using Clu and Col1a2 as candidate genes identified as downregulated with 5M treatment, we performed reverse transcription-polymerase chain reaction (RT-PCR) for these gene transcripts from fibroblast cultures subjected to different sequential combination treatments of the sortase variants (4S9; 4S9 + 2A9; 4S9 + 2A9 + 5M) or controls involving only buffer treatment or that with proteolytic enzyme trypsin (Figure S2). In concordance with the RNAseq data, we observed downregulation of Clu after treatment with 5M, though this effect was nonsignificant and mean gene expression was not reduced any further than in samples sequentially treated with 2A9 and 4S9. Trypsin was the only treatment condition eliciting significant downregulation of Clu and C3, with gene expression shifts an order of magnitude greater than all three sortase treatments in sequence. A similar trend towards reduced Col1a2 expression was also observed, but with no statistical significance. Though the trends in gene expression matched our expectations from our RNAseq data, their lack of significance and relatively small effect size in comparison to trypsin treatment again support relatively mild effects of sortase treatment on the transcriptome.
In tandem with the suppression of inflammatory genes by 5M in cardiac fibroblasts, we also observed differential expression of genes related to injury response and fibrosis, owing to this cell type’s role as a sentinel for tissue damage. Among the top downregulated coding genes were fibronectin 1 (Fn1), which is a major constituent of the provisional matrix deposited by fibroblasts in the inflammatory phase following acute tissue damage, as well as collagen types 1 and 6 (Col6a1, Col1a1, Col1a2). Reduced transcription of these genes, comprising the “ECM Proteoglycans” and other matrix-related pathways associated with 5M treatment, underscores the link between inflammation and fibrosis in the cardiac fibroblast.
Collectively, these results suggest that 5M, but not 2A9 or 4S9, may suppress the fibroinflammatory state of fibroblasts and other cell types as an enzyme of bacterial origin. This finding is attributed to the variants’ deviation in sequence from the native transpeptidase; in addition to the original five mutations endowing 5M (which shares 97% sequence homology with wild-type sortase) with enhanced catalytic activity, 2A9 and 4S9 respectively possess 11 and 9 more point mutations from the wild-type transpeptidase (reducing their sequence homology with wild-type sortase to 89% and 91%). As such, these enzyme variants are likely more suitable as a bioorthogonal trigger for material degradation than 5M, particularly in sensitive signaling contexts involving primary cells. Despite some limitations in 5M’s bioorthogonality, we opted to continue its employment in our future experiments as it is already in use by the community, is far milder than many other chemistries to liberate cells from gels, and affords a third cue for multiplexed material modulation.
2.2. Evolved sortase-degradable crosslinks enable high viability encapsulation and selective release of cells from hydrogel culture
Heartened by the findings that sortase treatment can be conducted with minimal perturbation to the cellular transcriptome, we sought to develop materials that would degrade in response to these stimuli. To program sortase degradability into hydrogel biomaterials, we synthesized azide-flanked peptide crosslinks bearing the various sortase-recognition motifs, enabling the formation of idealized step-growth polymer-peptide hydrogels by a strain-promoted azide-alkyne cycloaddition (SPAAC) click reaction with a bicyclononyne-functionalized 4-arm PEG (PEG-tetraBCN Mn ~ 20 kDa) (Figures 2A–B, Figure S3, Method S2).[48–50] Hydrogel crosslinking kinetics and mechanics, as assessed using in situ shear rheology, were not affected by crosslink sequence (Figure 2C, Method S3). In each case, gelation occurred rapidly within the span of 5–10 minutes, a suitable timespan for cell encapsulation. When encapsulated in hydrogels functionalized with an azide-modified RGD cell adhesion peptide pendant,[33] model fibroblast-like HS5 human stromal cells exhibited high viability (91–96% mean viability across six gels per crosslink) in the sortase-degradable networks after a week of culture; viability of cells in gels comprised of the various degradable crosslinks was statistically indistinguishable (Figure 2D, Method S4). Furthermore, crosslinker identity afforded no differences in the size or morphology of viable cells at this extended timepoint (Figure 2E).
Figure 2 –
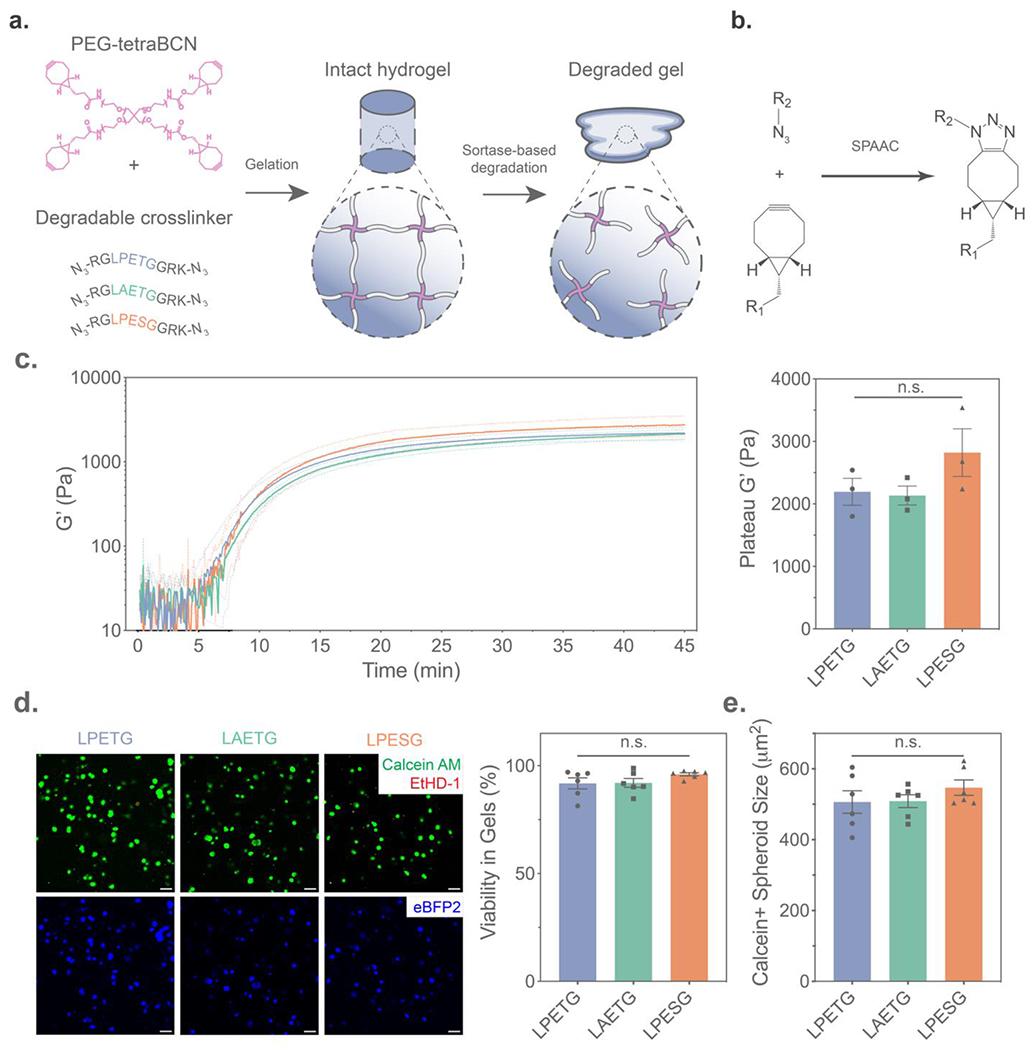
Synthesis and characterization of hydrogels crosslinked with orthogonal sortase motif sequences. (A) Hydrogels were formed by mixing PEG-tetraBCN with azide-flanked peptide crosslinkers bearing sortase substrate sequences to form a step-growth polymeric network, which could be subsequently degraded through exogenous substrate-matched sortase treatment. (B) Strain-promoted azide-alkyne cycloaddition (SPAAC) crosslinking of the gel precursors enabled cytocompatible hydrogel formation. (C) Gels with alternate sortase crosslinks formed networks of equivalent mechanics, as exemplified by time-sweep rheological characterization of the storage modulus (G’) during (left) and following complete gelation (right, n = 3 gels per condition). (D) Viability of encapsulated HS5-eBFP2 human stromal cells was verified by LIVE/DEAD staining with Calcein AM (Green) denoting viable cells and ethidium homodimer 1 (EtHD-1, Red) marking dead cells following one week of culture. (E) Spheroid size of cells staining positive for Calcein AM was also quantified after one week. Dashed lines and error bars indicate ±SEM for n = 6 technical replicates. “n.s.” indicates non-significance by one-way ANOVA with Tukey’s post-hoc test. Scale bars = 50 μm.
Having demonstrated the gels’ ability to support long-term 3D primary cell culture, we next assessed the suitability of the evolved sortases to selectively release cells from hydrogels containing each recognition sequence. Gel degradation was accomplished by incubation with the respective sortase variant (50 μM) and a free triglycine peptide (GGG; 18 mM) (Figure 3A, Method S5). To monitor and quantify degradation through changes in supernatant fluorescence, an azide-modified fluorophore (AFDye 568 azide) was sparsely tethered throughout each gel (1:200 molar ratio with the PEG backbone) (Figure 3B). Diffusion of the sortase variants, which are just under 18kDa, into the sortase-degradable networks occurred over a timespan of hours (Figure S4, Method S6). Therefore, to promote rapid bulk material degradation, sortase was diffused into the gels for an hour prior to GGG addition. As expected, minimal sortase-mediated hydrolysis was observed during diffusive enzyme gel loading. Upon triglycine addition to the gel supernatant, we found that each sortase variant was capable of rapidly and selectively degrading hydrogels containing its respective recognition site; all hydrogels degraded within one hour and with similar profiles in response to their corresponding sortase variant (Figure 3C). Gels crosslinked with the alternative recognition sequences remained intact and largely unperturbed following divergent variant treatment; the 4S9/2A9 evolved sortases exhibited near-perfect orthogonality with one another, while 5M displayed some nonspecific activity towards the 4S9 recognition sequence. Pre-incubation with sortase prior to GGG addition was not necessary; selective degradation proceeds effectively when both essential components were added simultaneously to gels, albeit with somewhat slower kinetics (Figure S5). As typical media concentrations of calcium (1.8 mM in DMEM) deviate from the optimized sortase reaction conditions described by literature (10 mM), we varied the Ca2+ concentration present during material dissolution in a set of parallel experiments. We found that each of the three sortase variants were active in calcium concentrations typical to media. Surprisingly, we also observed that 2A9 (but not 4S9 or 5M) was functionally active even in the absence of Ca2+ (Figure S6); this calcium insensitivity has not been previously reported and may prove useful in a variety of biological contexts. Intriguingly, both the LPXTG-recognizing calcium-independent sortase heptamutant (7M)[51] and 2A9 share a mutated amino acid (E105K for 7M, E105D for 2A9) in the enzyme’s calcium-binding pocket that is unique to these variants, potentially explaining the shared calcium independence. As expected, HS5 cells released with all sortase variants maintained high viability after material dissolution, which can be accomplished under physiological conditions in cell culture media or Ca2+-supplemented phosphate-buffered saline (Figure 3D–E, Figure S7).
Figure 3 –
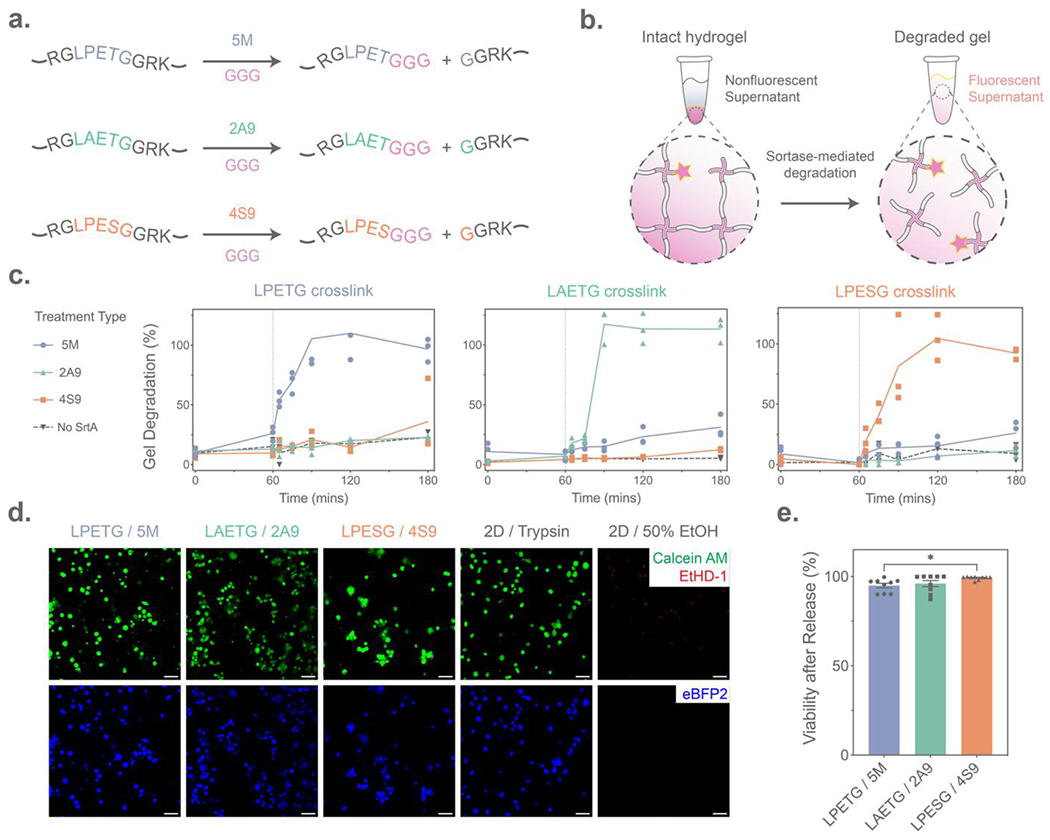
Evolved sortase-mediated hydrogel degradation. (A) Sortase-susceptible crosslinks are cleaved by their corresponding sortase variant (5M, 2A9, 4S9) through a transpeptidation reaction with added polyglycine (GGG) peptide. (B) Hydrogel degradation due to sortase transpeptidation was monitored through the supernatant release of a pendant AFDye 568 azide (pink star) initially crosslinked into the hydrogel network. (C) Each sortase-degradable crosslink exhibited enzyme-specific hydrogel degradation (n = 3 per condition), as illustrated by an increase in supernatant fluorescence following treatment with evolved sortase variants for 60 minutes, followed by addition of the GGG trigger (dashed line). Y-axis corresponds to percentage gel degradation, quantified through relative supernatant fluorescence. (D) HS5 human stromal cell viability was maintained 60 minutes following sortase-mediated hydrogel degradation, as assessed by LIVE/DEAD staining with Calcein AM (Green) indicating live cells and EtHD-1 (Red) staining dead cell nuclei. (E) Flow cytometry quantification of HS5 cells stained with LIVE/DEAD immediately after release from hydrogels. Error bars indicate ±SEM across n = 9 technical replicates. “*” indicates p < 0.05 by one-way ANOVA with Tukey’s post-hoc test. Scale bars = 50 μm.
2.3. Open-microfluidic rail patterning of orthogonally degradable sortase gels enables spatial control over hydrogel patterning and degradation
Encouraged that the sortase variants could be used to orthogonally trigger homogenous material dissolution, we hypothesized that the 2A9/4S9/5M enzymes could be multiplexed to spatially control material degradation and enable phenotypic analysis of HS5 cells from different regions of an engineered tissue. To demonstrate spatially controlled cell release, we constructed a hydrogel “bullseye” pattern consisting of three concentric and orthogonally degradable layers (250 μm height); each differently sortase-sensitive layer contained encapsulated HS5 cells constitutively expressing one of three fluorescent proteins to assess the specificity of cell release via confocal microscopy and flow cytometry (Figure S8): the inner core contained cells expressing mTagBFP2 with a crosslinker sensitive to 5M; the middle layer housed GFP+ cells and responded to 2A9; the outer layer encapsulated mCherry-expressing cells and was responsive to 4S9 (Figure 4A). Hydrogel geometry was specified through recently reported open-microfluidic rail patterning methodologies, a chemistry-agnostic approach able to create structurally complex 3D gel multimaterials (Figures 4B, Figure S9, Method S7).[52–55] Using this method, SPAAC-based gel precursors were pipetted through a 3D-printed well-plate insert that employs capillary pinning to shape the solution prior to and throughout gelation; following individual layer gelation and device removal, the process was repeated iteratively to create multi-layered 3D structures. Since this technique is indiscriminate to hydrogel type and formulation chemistry, we find this to be a particularly appealing approach for the construction of heterogenous engineered tissues.
Figure 4 –
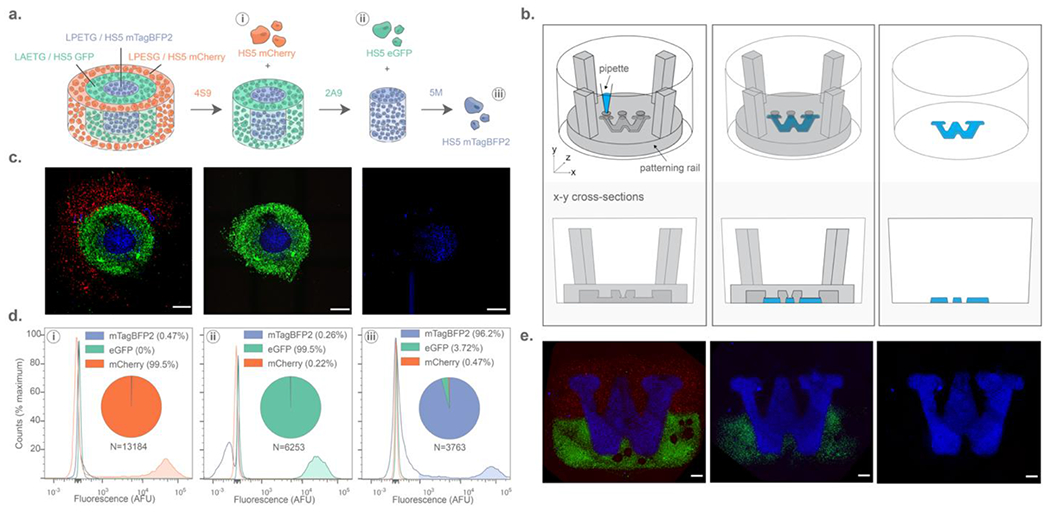
Spatiotemporally controlled liberation of single-cell suspensions of HS5 human stromal cells from complex trilayered materials through multiplexed sortase-based degradation. (A) Three distinctly fluorescent cell types were released from different sortase-degradable hydrogel fractions, with release fidelity was quantified by flow cytometry. (B) Arbitrary hydrogel geometries were constructed using open-microfluidic gel patterning through well-plate inserts. (C) Maximum intensity projections (MIPs) of 10x confocal images showing sortase-degradable bullseyes prior to degradation (left), following 4S9 treatment (center), and after 2A9 treatment (right). (D) Flow cytometry quantification of liberated cells following sequential treatment of 4S9 (left), 2A9 (center), and 5M (right) reveals that highly specific cell capture can be obtained from three different gel subregions with minimal off-target release. (E) Degradation of an arbitrary pattern as illustrated by MIPs of hydrogels patterned in the University of Washington Logo prior to degradation (left), following 4S9 treatment (center), and 2A9 treatment (right). Scale bars = 1 mm.
With the cell-laden bullseyes successfully constructed, we tested all possible enzyme-treatment orders to liberate cells from different composite gel compartments. Sortase-released cells were analyzed by flow cytometry to quantify capture fidelity (Method S8). Extremely specific spatial control over cell recovery was achieved through sequential dissolution, with successive treatments yielding a distinct cell collection matching the expected color composition (Figure 4C–D). Consistent with acellular gel experiments showing that 5M could partially recognize the evolved sortase variants’ substrate sequences (Figure 3C), specificity was found to be dependent on sortase treatment order (Figure S10). Between 2A9 and 4S9, treatment order did not majorly affect the specificity of material degradation; either evolved variant may be used first to release cells with cumulative accuracies exceeding 92% from all three gel layers. These strategies are readily extended to support sequential cell release from more complex gel multimaterial geometries with high fidelity (Figure 4E).
2.4. Evolved sortase degradable hydrogels enable the study of spatially dependent patterns in cardiac fibroblast proliferation and activation
Armed with a uniquely powerful strategy for region-selective biomaterial degradation, we sought to exploit multiplexed sortase-based gel dissolution to investigate complex and evolving disease phenotypes in vitro. For this, we turned our attention to cardiac fibroblast activation – a disease phenotype modulated through positive feedback that drives the expansion of myocardial scar tissue, leading to maladaptive remodeling, heart failure, and patient death.[42,56,57] Seeking to better understand the paracrine effects underlying this phenomenon, we activated primary cardiac fibroblasts using transforming growth factor-beta (TGFβ1) and encapsulated those cells within the center region (5M-degradable) of a three-layer bullseye gel, with middle and outer layers containing quiescent fibroblasts (untreated with TGFβ1) at a common seeding density and respectively degradable by 2A9 and 4S9 (Figure 5A, Method S9). To support the fibroblast-mediated matrix remodeling, gel layers were formed using modified sortase-degradable crosslinkers that also included an MMP-cleavable peptide sequence (GPQGIWGQ; Figure 5B, Figure S11, Method S2). The resultant materials functioned as Boolean OR gates, responding to inputs of either endogenous cell-secreted MMPs or exogenous user-administered sortase, respectively facilitating fibroblast spreading within the bulk material and on-demand dissolution of individual gel regions.
Figure 5 –
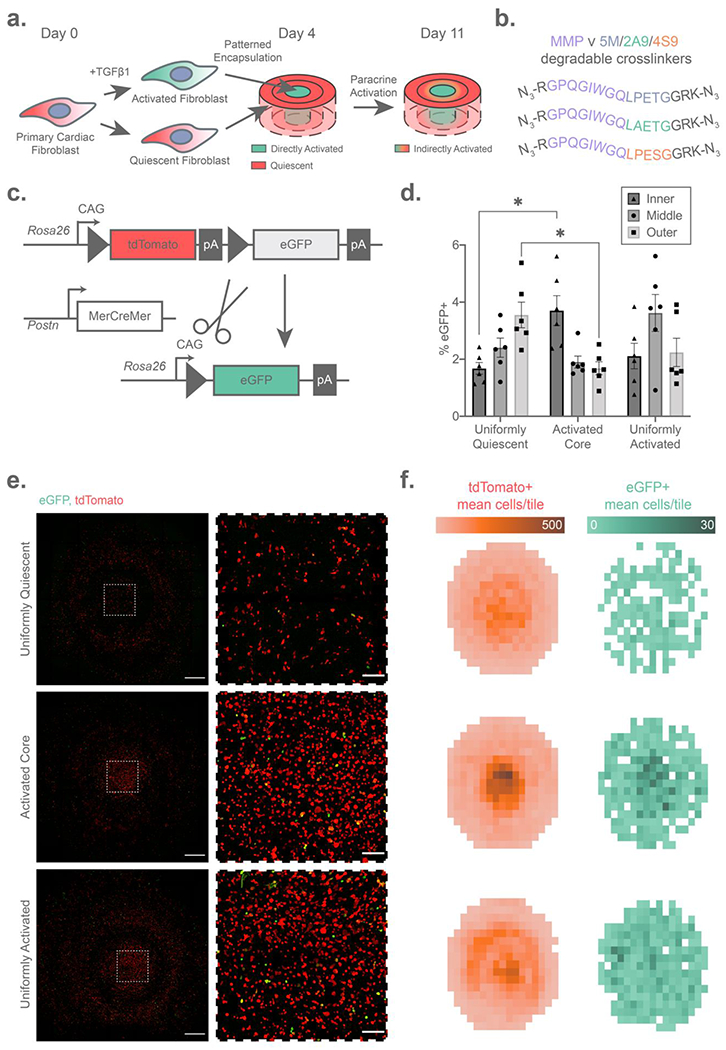
Paracrine activation of cardiac fibroblasts dissected using composite sortase-degradable hydrogels. (A) Schematic illustrating the fibroblast activation bullseye experiment. (B) Schematic of cell-cleavable MMP ∨ 5M/2A9/4S9 (Boolean OR-type) crosslinks used in this study. (C) Genetics of the dual-color periostin (Postn) reporter mice, in which fibroblasts expressing the activation marker Postn undergo Cre-mediated exision of a tdTomato cassette, resulting in an irreversible red-to-green fluorescence switch. (D) Flow cytometry quantification reveals a higher rate of activation in fibroblasts liberated from the activated cores of bullseye gels following iterative sortase degradation, but a lower rate of fibroblast activation in the outer regions measured by the percentage of eGFP+ fibroblasts on the y-axis. (E) Elevated cell densities in the cores of activated hydrogels are visible by max intensity projection confocal images at day 7 of hydrogel culture shown in whole on the left (scale bars = 1 mm), with regions of interest from the center of the bullseye shown to the right (scale bars = 200 μm). (F) Fibroblast activation in the bullseye center leads to increased cell density, visualized by 2D histograms representing mean cell density (n = 3 bullseyes) of quiescent (left) and activated (right) fibroblasts. Error bars represent ±SEM across n = 5 hydrogel bullseyes. “*” indicates p < 0.05 by two-way ANOVA with Tukey’s post-hoc test.
To track the activation state of fibroblasts seeded throughout all the gel, we exploited transgenic mouse cardiac fibroblasts featuring an inducible dual-color reporter of the periostin (Postn) promoter in conjunction with fluorescent imaging and flow cytometry of cell fractions liberated by sortase (Figure 5C). Postn is a matricellular protein expressed at early stages of fibroblast activation following a myocardial infarction; its promoter activity has been widely used in the field as a readout for fibroblast activation.[58–60] In the employed model, all cells fluoresce red until they express Postn, which drives Cre-mediated excision of tdTomato gene and expression of the downstream eGFP cassette, resulting in an irreversible red-to-green fluorescence switch marking those cells which have activated. As the Cre-recombinase utilized here is tamoxifen-inducible, the start of lineage tracing was specified through controlled administration of the small molecule 4-hydroxytamoxifen (4-OHT) in culture media following encapsulation. The advantage of tracking fibroblast activation using this mouse model is that activation may be easily quantified with a binary fluorescent readout from a single-cell suspension via flow cytometry. In the context of our bullseye model of fibroblast activation driven by paracrine feedback, we hypothesized that initially quiescent fibroblasts neighboring an activated core would become proximally activated, resulting in higher rates of eGFP positivity relative to quiescent controls lacking an activated core.
Following one week of culture in bullseye gels and cell collection through multiplexed sortase-mediated dissolution, we examined region-specific fibroblast activation via flow cytometry. To establish the suitability of flow cytometry for analysis of recovered cell-suspensions from sortase-degradable hydrogels, we verified that the percentage of cytometric singlet events did not significantly vary with fibroblast activation state, bullseye region, or seeded cell density (Figure S12). As anticipated, we observed that cell populations encapsulated within the activated core retained elevated eGFP positivity, while outer regions exhibited less activation (Figure 5D). Intriguingly, the percentage of eGFP+ cells found in the outermost layer of the core-activated bullseye gels was depressed relative to those in control gels uniformly seeded with quiescent fibroblasts (i.e., those where the core was not preactivated using TGFβ1), and region-specific activation was observed in the uniformly quiescent gels. To resolve whether these differences were associated with variable cell densities spanning gel conditions and subvolumes, we compared flow cytometry findings with those obtained via fluorescent confocal microscopy. Full-gel images of the bullseye multimaterials revealed that those with an activated core had dramatically increased cell density in the center relative to controls and those that were uniformly activated (Figure 5E–F). Concordant with flow cytometry data from released cell suspensions, we also observed via imaging a concentrated region of Postn+ cells in the center of gels with activated cells seeded into the core, whereas in uniformly activated gels pockets of Postn+ cells were observed throughout. To test whether increased fibroblast proliferation was causing the activation-dependent change in cell density, we imaged gels stained for Ki67 at an earlier timepoint one day after encapsulation. In contrast to the week-long timepoint, after one day cell densities remained consistent throughout bullseyes with an activated core (Figure S13A). Ki67-expressing fibroblasts were observed throughout all regions of all bullseyes, confirming that fibroblasts may indeed proliferate within the sortase-OR-MMP-degradable gels (Figure S13B). Surprisingly however, uniformly quiescent bullseyes contained higher densities of Ki67+ cells, suggesting that a mechanism other than proliferation may be at play. Although the mechanisms for increased cell density in the gels with an activated core – such as cell migration towards the center – remain under investigation, these experiments demonstrate the utility of multiplexed sortase-based degradation to study spatiotemporally dependent cell signaling in an engineered biomaterial.
Though activation can also be quantified in this experimental setup by fluorescent imaging, we used flow cytometry experiments on single-cell suspensions liberated from gel regions as a model single cell technique that may be later adapted for more in-depth phenotypic analysis in the future. For example, fluorescent populations may be sorted and transcriptomically compared by RNAseq to identify genes and pathways that distinguish directly activated fibroblasts from those activated by spatial/paracrine effects within the gel.
Though combined utilization of hydrogel multimaterials with sequential sortase degradation is powerful in assaying heterogenous cell phenotype, it is not without limitations. Since material degradation is implicitly coupled with bulk softening, encapsulated cells may sense and experience mechanobiological changes throughout their release. To capture phenotypes which require a tighter time fidelity than can be accommodated with this strategy (typically on the order of tens of minutes), paraformaldehyde fixation may by employed prior to gel degradation.[38] Additionally, since sortase treatment will not degrade cell-secreted nascent extracellular matrix that hold multicellular aggregates together, subsequent processing may be required to further disaggregate into single cells. While a potential drawback for some applications, this is likely a useful attribute in liberating intact multicellular structures including organoids from synthetic gels.
3. Conclusion
In summary, we have developed a versatile strategy for iterative enzymatic degradation of cell-laden multimaterials that offers unique compatibility with the arsenal of bioassays requiring single-cell suspensions. Utilizing engineered sortases that recognize distinct peptide substrates encoded within hydrogel crosslinkers as a biologically compatible cue for material degradation, the reported system permits single-cell populations to be liberated from gels with minimal perturbation to their transcriptomic state. Employing materials chemistry-agnostic open microfluidics to specify heterogenous gel geometry and compositional arrangement of state-of-the-art primary reporter cells, we have demonstrated spatiotemporal control over gel degradation and its utility in examining disease phenotypes in vitro. We anticipate that these materials-based strategies will enable biomedical researchers to more easily dissect pathological signaling networks processes in 3D tissue-mimetic microenvironments.
4. Experimental Procedures
Complete experimental procedures are provided in the online data supplement.
Materials
Sortase A pentamutant (5M) in pET29, eSrtA(2A-9) (2A9) in pET29b, and eSrtA(4S-9) (4S9) in pET29b were gifts from David Liu (Addgene Plasmids #75144, 75145, 75146). Poly(ethylene glycol) tetra-bicyclononyne (PEG-tetraBCN, Mn = 20 kDa), 4-azidobutanoic acid (N3-COOH), and N3-GRGDS-NH2 (RGD-azide) were synthesized and characterized as previously described.[33,49] HS5 immortalized human bone marrow stromal cells were a gift from Dr. Brian Hayes at the Fred Hutchison Cancer Research Center. Fibroblast isolations – described in the Supplemental Methods – were conducted under a protocol (#4376-01) approved by the University of Washington Institutional Animal Care and Use Committee, an AAALAC accredited institution (#000523). Postn lineage reporter (Postn-mT/mG) mice were generated by crossing mice bearing a tamoxifen-inducible Cre cassette knocked into the Postn locus (PostnMerCreMer) with mice expressing a membrane targeted conditional dual color fluorescent reporter (mT/mG) knocked in to Rosa26 genomic locus.[58,61]
Sortase Expression
Plasmid-expressing BL21 colonies grown in 500 mL Luria Broth (LB) were induced using isopropyl β-d-1-thiogalactopyranoside (IPTG, 0.5 mM) at an OD600 of 0.6, after which expression was conducted at 18°C overnight. Cells were lysed by sonication and clarified by centrifugation. Sortases were then purified by immobilized metal affinity chromatography from the clarified lysate using an ÄKTA Pure 25 L FPLC (Cytiva; Marlborough, MA), spin concentrated, and stored at −80 °C in reaction buffer (20 mM tris, 50 mM NaCl; pH 7.56), phosphate buffered saline (PBS), or DMEM, all containing 20% glycerol.
RNA Sequencing
Cardiac fibroblasts at 80% confluency on a six-well plate were treated with either 50 μM sortase and 18 mM triglycine (GGG; Sigma Aldrich; St. Louis, MO) in DMEM with 20% glycerol, or control containing GGG and glycerol but no sortase. After 45 minutes at 37 °C, RNA was isolated from TRIZol (Thermo Fisher Scientific) lysed samples with a Direct-Zol RNA microprep kit (Zymo Research; Irvine, CA). Samples were transferred to BGI Genomics for library prep and sequencing on the DNBseq platform (20 million clean paired-end reads per sample). Reads were aligned using RNA STAR on the Galaxy Web Server[62,63], counted using featurecounts, and evaluated for differential expression using EdgeR.[64,65] Differentially expressed genes were input into G:Profiler for pathway enrichment analysis against the Reactome and Wikipathways databases.[66–68]
Peptide Synthesis
Peptides were synthesized on rink amide resin (0.5 mmol scale) using standard Fluorenylmethyloxycarbonyl (Fmoc) microwave-assisted solid-phase peptide synthesis techniques on a Liberty1 Peptide Synthesizer (CEM; Matthews, NC), then azide functionalized and purified by methods previously described.[23] All peptide sequences, synthesis details, and purified product mass spectra are included in the supplementary information.
Rheometry
Gel formation kinetics and plateau modulus were measured at 37 °C using 8 mm parallel plate geometry (Gap: 0.5 mm; Strain: 1%; Frequency: 1 Hz) on a Physica MCR301 rheometer (Anton Paar; Graz, AT) for 30 μL hydrogels containing 4 mM PEG-tetraBCN and 8 mM di-azide peptide crosslinker.
Cell Encapsulation and Viability
HS5 cells were encapsulated at 107 cells mL−1 in 5 μL droplet hydrogels containing 4 mM PEG-tetraBCN, 8 mM di-azide peptide crosslinker, and 1 mM RGD-azide. After gelation for 30 mins at 37 °C, media was added and gels were cultured for 7 days. DMEM with 50 μM sortase and 18mM GGG was added to each gel to induce dissolution. One hour following dissolution, cell viability was analyzed using a LIVE/DEAD assay (Thermo Fisher; Waltham, MA) and imaged on a Leica SP8 confocal microscope at 10x magnification, using cells trypsinized from 2D culture as a positive control and cells treated with 50% ethanol as a negative control. LIVE/DEAD staining was quantified via flow cytometry with a BD CantoRUO flow cytometer (BD Bioscience; San Jose, CA).
Fluorophore release experiments
PEG-tetraBCN was pre-reacted with AFDye 568 azide (Click Chemistry Tools; Scottsdale, AZ) at a molar ratio of 40:1 for incorporation into 10 μL hydrogels with a final PEG-tetraBCN concentration of 4 mM and 2:1 molar ratio of crosslink to PEG-tetraBCN (8 mM peptide). Following gelation at 37 °C for 60 mins, gels were swollen overnight in sortase reaction buffer. To initiate dissolution, 200 μL of buffer containing 50 μM sortase was added for one hour, then replaced by 200 μL of 50 μM sortase and 18 mM GGG (Sigma Aldrich; St. Louis, MO), both at 37 °C under agitation. At each timepoint, 5 μL of the supernatant was transferred to 50 μL buffer in a 96-well plate for fluorescence measurements on a plate reader (Molecular Devices; San Jose, CA). Measurements were normalized per-experiment to the mean fluorescence of fully degraded gels.
Patterning device design and fabrication
Open microfluidic patterning devices were designed in Solidworks 2017 (Dassault Systémes; Vélizy-Villacoublay, FR) and 3D printed using a Form 2 stereolithography 3D printer (Formlabs; Somerville, MA). Design files for all patterning devices are included in the Supporting Information and a schematic for the devices are found in Figure S7.
Multilayer hydrogel encapsulation and release
Multilayer hydrogels containing layers of HS5-mTagBFP2, HS5-eGFP, and HS5-mCherry cells were formed either by sequential casting of hydrogels between glass slides to form a bullseye pattern (Flow Cytometry Experiments) or by pipetting gel precursor into the patterning devices for sequential casting steps (Imaging Experiments). Composite gels were then sequentially treated with sortases as described above, with each dissolution step proceeding for 1 hr. Released cells were collected and fixed in 4% paraformaldehyde in PBS (10 mins, RT) prior to analysis on a FACSCanto RUO cytometer (BD Bioscience; San Jose, CA). Fixed multilayer gels were also imaged after sequential dissolution steps on a Nikon Ti microscope with a Yokogawa W1 spinning disk head under 10x magnification.
Dual color cardiac fibroblast bullseyes
Three days prior to encapsulation, cardiac fibroblasts isolated from Postn-mT/mG mice to be used for activated regions were switched to activation media (DMEM containing 2% FBS) containing 10 ng mL−1 bovine TGFβ1 (R&D Systems; Minneapolis, MN), which was replenished daily prior to encapsulation (10 million cells mL−1) in gels containing 3 mM PEG tetraBCN, 1.5 mM sortase/MMP degradable crosslink, and 1 mM N3-GRGDS-NH2. After 30 mins of polymerization, DMEM containing 2% FBS and 2.5 μM 4-hydroxytamoxifen (Tocris Bioscience; Bristol, UK) was added and changed every other day for 7 days. Gels were imaged live on a Leica Stellaris 5 confocal microscope under 10x magnification. Cells expressing tdTomato and eGFP were segmented using LAS-X (Leica Microsystems), and a custom Python script was then used to generate histograms displaying mean cell count across 3 technical replicates per condition. For flow cytometry, gels were sequentially treated with 4S9, 2A9, and 5M, the supernatant and one 1X PBS rinse was collected per treatment. Released cells were fixed in 4% PFA for 10 mins, then rinsed twice with 1X PBS prior to cytometry for tdTomato and eGFP on a BD FACSAria (BD Bioscience; San Jose, CA).
Statistical Analysis
Unless otherwise stated, data were plotted and statistically analyzed using GraphPad Prism 7.0. Flow cytometry data were analyzed and plotted using FlowJo 10 software (BD Biosciences; San Jose, CA), with additional graph shading in Illustrator CC (Adobe; San Jose, CA). Images were analyzed using the FIJI distribution of ImageJ.[69,70] For RNAseq data, statistical significance was assessed using EdgeR, using a false discovery rate of 0.05 as threshold for significance. Three group comparisons in Figures 2 and 3 were by one-way ANOVA with Tukey’s post-hoc testing and a significance threshold of p < 0.05. Flow cytometry data for Figure 5 was evaluated using two-way repeated measures ANOVA with Tukey’s post-hoc testing and significance threshold of p < 0.05. For all data, error bars present mean ± SEM, with sample sizes given in figure legends.
Supplementary Material
Acknowledgements
The authors would like to thank Alexander Prossnitz for useful feedback on and discussions regarding the manuscript, Jared Shadish and Barry Badeau for assistance with protein and peptide methods, Brian Hayes for providing the fluorescent HS5 cell lines, Dale Hailey for technical assistance with microscopy, and Kyle Herstad for flow cytometry assistance. This research was supported by the Cell Analysis Facility Flow Cytometry and Imaging Core in the Department of Immunology, the Mass Spectrometry Center in the Department of Medicinal Chemistry, the Garvey Imaging Core in the Institute for Stem Cell and Regenerative Medicine at the University of Washington. The authors acknowledge support from the National Science Foundation (NSF) in the form of an unsolicited grant (DMR 1807398, to C.A.D.), a Faculty Early Career Development Program (CAREER) (DMR 1652141, to C.A.D.), two Graduate Research Fellowships (DGE 1762114 individually to R.C.B. and I.K.), and through a seed grant through the NSF-sponsored University of Washington Materials Research Science and Engineering Center (DMR 1719797 to A.B.T. and C.A.D.). The authors further acknowledge support from the National Institutes of Health in the form of two Maximizing Investigators’ Research Awards (R35GM138036 to C.A.D.; R35GM128648 to A.B.T.), a Biological Mechanisms of Healthy Aging Training Grant (T32AG066574 to D.B.), Research Project Grants (R01HL141187 and R01HL142624 to J.D.), and a Ruth L. Kirschstein National Research Service Award (NRSA) F30 (F30HL158030, to A.J.H.). The authors acknowledge additional funding support from the Arnold and Mabel Beckman Foundation (Beckman Young Investigator Award, to A.B.T.), the Mary Gates Endowment for Students (Research Scholarship, to J.D.K.), and the Washington NASA Space Grant Consortium (Summer Undergraduate Research Program, to J.D.K.).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Ross C. Bretherton, Department of Bioengineering, University of Washington; Seattle, WA 98105, USA; Institute for Stem Cell & Regenerative Medicine, University of Washington; Seattle, WA 98109, USA; Center for Cardiovascular Biology, University of Washington; Seattle, WA 98109, USA
Amanda J. Haack, Department of Chemistry, University of Washington; Seattle, WA 98105
Irina Kopyeva, Department of Bioengineering, University of Washington; Seattle, WA 98105, USA; Institute for Stem Cell & Regenerative Medicine, University of Washington; Seattle, WA 98109, USA.
Fariha Rahman, Department of Bioengineering, University of Washington; Seattle, WA 98105, USA; Institute for Stem Cell & Regenerative Medicine, University of Washington; Seattle, WA 98109, USA.
Jonah D. Kern, Department of Bioengineering, University of Washington; Seattle, WA 98105, USA; Institute for Stem Cell & Regenerative Medicine, University of Washington; Seattle, WA 98109, USA
Darrian Bugg, Center for Cardiovascular Biology, University of Washington; Seattle, WA 98109, USA.
Ashleigh B. Theberge, Department of Urology, University of Washington; Seattle, WA 98105, USA
Jennifer Davis, Department of Bioengineering, University of Washington; Seattle, WA 98105, USA; Institute for Stem Cell & Regenerative Medicine, University of Washington; Seattle, WA 98109, USA; Center for Cardiovascular Biology, University of Washington; Seattle, WA 98109, USA.
Cole A. DeForest, Department of Bioengineering, University of Washington; Seattle, WA 98105, USA; Department of Chemical Engineering, University of Washington; Seattle, WA 98105, USA; Institute for Stem Cell & Regenerative Medicine, University of Washington; Seattle, WA 98109, USA Molecular Engineering & Sciences Institute, University of Washington; Seattle, WA 98109; Institute for Protein Design, University of Washington; Seattle, WA 98105
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Caliari SR, Burdick JA, Nat. Methods 2016, 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bretherton RC, DeForest CA, ACS Biomater. Sci. Eng 2021, 7, 3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Griffith LG, Swartz MA, Nat. Rev. Mol. Cell Biol 2006, 7, 211. [DOI] [PubMed] [Google Scholar]
- [4].Kola I, Landis J, Nat. Rev. Drug Discov 2004, 3, 711. [DOI] [PubMed] [Google Scholar]
- [5].Bart van der Worp H, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, Macleod MR, PLoS Med. 2010, 7, e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brown TE, Anseth KS, Chem. Soc. Rev 2017, 46, 6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Burdick JA, Murphy WL, Nat. Commun 2012, 3. [DOI] [PubMed] [Google Scholar]
- [8].Zhu C, Preissl S, Ren B, Nat. Methods 2020, 17, 11. [DOI] [PubMed] [Google Scholar]
- [9].Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA, Mol. Cell 2015, 58, 610. [DOI] [PubMed] [Google Scholar]
- [10].Tung JW, Heydari K, Tirouvanziam R, Sahaf B, Parks DR, Herzenberg LA, Herzenberg LA, Clin. Lab. Med 2007, 27, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Radbruch A, Mechtold B, Thiel A, Miltenyi S, Pflüger E, Methods Cell Biol 1994, 42, 387. [DOI] [PubMed] [Google Scholar]
- [12].Bonner WA, Hulett HR, Sweet RG, Herzenberg LA, Rev. Sci. Instrum 2003, 43, 404. [DOI] [PubMed] [Google Scholar]
- [13].Bendall SC, Simonds EF, Qiu P, Amir EAD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP, Science 2011, 332, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD, Nucleic Acids Res. 2018, 46, D624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fakhri O, Tan RSH, Cell. Immunol 1975, 15, 452. [DOI] [PubMed] [Google Scholar]
- [16].O’Flanagan CH, Campbell KR, Zhang AW, Kabeer F, Lim JLP, Biele J, Eirew P, Lai D, McPherson A, Kong E, Bates C, Borkowski K, Wiens M, Hewitson B, Hopkins J, Pham J, Ceglia N, Moore R, Mungall AJ, McAlpine JN, Shah SP, Aparicio S, Genome Biol. 2019, 20, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Denisenko E, Guo BB, Jones M, Hou R, De Kock L, Lassmann T, Poppe D, Poppe D, Clément O, Simmons RK, Simmons RK, Lister R, Forrest ARR, Genome Biol. 2020, 21, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aisenbrey EA, Murphy WL, Nat. Rev. Mater 2020, 5, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Deforest CA, Anseth KS, Annu. Rev. Chem. Biomol. Eng 2012, 3, 421. [DOI] [PubMed] [Google Scholar]
- [20].Tibbitt MW, Anseth KS, Biotechnol. Bioeng 2009, 103, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lutolf MP, Hubbell JA, Nat. Biotechnol 2005, 23, 47. [DOI] [PubMed] [Google Scholar]
- [22].Kloxin AM, Kasko AM, Salinas CN, Anseth KS, Science 2009, 324, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Badeau BA, Comerford MP, Arakawa CK, Shadish JA, DeForest CA, Nat. Chem 2018, 10, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Badeau BA, DeForest CA, Annu. Rev. Biomed. Eng 2019, 21, 241. [DOI] [PubMed] [Google Scholar]
- [25].Kharkar PM, Kiick KL, Kloxin AM, Chem. Soc. Rev 2013, 42, 7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lunzer M, Shi L, Andriotis OG, Gruber P, Markovic M, Thurner PJ, Ossipov D, Liska R, Ovsianikov A, Angew. Chemie Int. Ed 2018, 57, 15122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Griffin DR, Schlosser JL, Lam SF, Nguyen TH, Maynard HD, Kasko AM, Biomacromolecules 2013, 14, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Truong VX, Li F, Forsythe JS, ACS Appl. Mater. Interfaces 2017, 9, 32441. [DOI] [PubMed] [Google Scholar]
- [29].DeForest CA, Anseth KS, Nat. Chem 2011, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Azagarsamy MA, Anseth KS, Angew. Chemie - Int. Ed 2013, 52, 13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rapp TL, Wang Y, Delessio MA, Gau MR, Dmochowski IJ, RSC Adv. 2019, 9, 4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Popp MW-L, Ploegh HL, Angew. Chemie Int. Ed 2011, 50, 5024. [DOI] [PubMed] [Google Scholar]
- [33].Shadish JA, Benuska GM, DeForest CA, Nat. Mater 2019, 18, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gawade PM, Shadish JA, Badeau BA, DeForest CA, Adv. Mater 2019, 31, 1902462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Batalov I, Stevens KR, DeForest CA, Proc. Natl. Acad. Sci. U. S. A 2021, 118, DOI 10.1073/pnas.2014194118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen I, Dorr BM, Liu DR, Proc. Natl. Acad. Sci. U. S. A 2011, 108, 11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Below CR, Kelly J, Brown A, Humphries JD, Hutton C, Xu J, Lee BY, Cintas C, Zhang X, Hernandez-Gordillo V, Stockdale L, Goldsworthy MA, Geraghty J, Foster L, O’Reilly DA, Schedding B, Askari J, Burns J, Hodson N, Smith DL, Lally C, Ashton G, Knight D, Mironov A, Banyard A, Eble JA, Morton JP, Humphries MJ, Griffith LG, Jørgensen C, Nat. Mater 2022, 21, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Valdez J, Cook CD, Ahrens CC, Wang AJ, Brown A, Kumar M, Stockdale L, Rothenberg D, Renggli K, Gordon E, Lauffenburger D, White F, Griffith L, Biomaterials 2017, 130, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hernandez-Gordillo V, Kassis T, Lampejo A, Choi GH, Gamboa ME, Gnecco JS, Brown A, Breault DT, Carrier R, Griffith LG, Biomaterials 2020, 254, 120125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chrisnandy A, Blondel D, Rezakhani S, Broguiere N, Lutolf MP, Nat. Mater 2022, 21, 479. [DOI] [PubMed] [Google Scholar]
- [41].Dorr BM, Ham HO, An C, Chaikof EL, Liu DR, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bretherton RC, Bugg D, Olszewski EO, Davis J, Matrix Biol. 2020, 91–92, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mazmanian SK, Liu G, Ton-That H, Schneewind O, Science 1999, 285, 760. [DOI] [PubMed] [Google Scholar]
- [44].Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S, Immunity 1999, 11, 443. [DOI] [PubMed] [Google Scholar]
- [45].Fan X, Wang X, Li N, Cui H, Hou B, Gao B, Cleary PP, Wang B, PLoS One 2014, 9, e107638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Baron V, Adamson ED, Calogero A, Ragona G, Mercola D, Cancer Gene Ther. 2006, 13, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shannan B, Seifert M, Leskov K, Willis J, Boothman D, Tilgen W, Reichrath J, Cell Death Differ. 2006, 13, 12. [DOI] [PubMed] [Google Scholar]
- [48].DeForest CA, Polizzotti BD, Anseth KS, Nat. Mater 2009, 8, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].DeForest CA, Tirrell DA, Nat. Mater 2015, 14, 523. [DOI] [PubMed] [Google Scholar]
- [50].Arakawa CK, Badeau BA, Zheng Y, DeForest CA, Adv. Mater 2017, 29, 1703156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jeong H-J, Abhiraman GC, Story CM, Ingram JR, Dougan SK, PLoS One 2017, 12, e0189068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lee UN, Day JH, Haack AJ, Bretherton RC, Lu W, DeForest CA, Theberge AB, Berthier E, Lab Chip 2020, 20, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee SH, Helnz AJ, Shln S, Jung YG, Chol SE, Park W, Roe JH, Kwon S, Anal. Chem 2010, 82, 2900. [DOI] [PubMed] [Google Scholar]
- [54].Casavant BP, Berthier E, Theberge AB, Berthier J, Montanez-Sauri SI, Bischel LL, Brakke K, Hedman CJ, Bushman W, Keller NP, Beebe DJ, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Berry SB, Zhang T, Day JH, Su X, Wilson IZ, Berthier E, Theberge AB, Lab Chip 2017, 17, 4253. [DOI] [PubMed] [Google Scholar]
- [56].Zeigler AC, Nelson AR, Chandrabhatla AS, Brazhkina O, Holmes JW, Saucerman JJ, Matrix Biol. 2020, 91–92, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Elson EL, Qian H, Fee JA, Wakatsuki T, Prog. Biophys. Mol. Biol 2019, 144, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J Lin S-C, Aronow BJ, Tallquist MD, Molkentin JD, Nat. Commun 2016, 7, 12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis J, Circulation 2017, 136, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ivey MJ, Tallquist MD, Circulation 2016, 80, 2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Muzumdar MD, Tasic B, Miyamichi K, Li N, Luo L, genesis 2007, 45, 593. [DOI] [PubMed] [Google Scholar]
- [62].Afgan E, Baker D, Béŕ B, Batut B, Van Den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Bj¨ B, Grüninggr¨grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, Blankenberg D, Nucleic Acids Res. 2018, 46, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, Bioinformatics 2013, 29, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Liao Y, Smyth GK, Shi W, 2014, 30, 923. [DOI] [PubMed] [Google Scholar]
- [65].Robinson MD, McCarthy DJ, Smyth GK, Bioinformatics 2010, 26, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J, Nucleic Acids Res. 2019, 47, W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, Loney F, May B, Milacic M, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P, Nucleic Acids Res. 2020, 48, D498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, Miller RA, Digles D, Lopes EN, Ehrhart F, Dupuis LJ, Winckers LA, Coort SL, Willighagen EL, Evelo CT, Pico AR, Kutmon M, Nucleic Acids Res. 2021, 49, D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Nat. Methods 2012, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW, BMC Bioinformatics 2017, 18, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.