

Abstract
Increasingly difficult-to-treat infections by antibiotic-resistant bacteria have become a major public health challenge. Rapid detection of common resistance mechanisms before empiric antibiotic usage is essential for optimizing therapeutic outcomes and containing further spread of resistance to antibiotics among other bacteria. Herein, we present a bioluminogenic probe, D-Bluco, for rapid detection of β-lactamase activity in viable pathogenic bacteria. D-Bluco is a pro-luciferin caged by a β-lactamase-responsive cephalosporin structure and further conjugated with a dabcyl quencher. The caging and quenching significantly decreased the initial background emission and increased the signal-to-background ratio by more than 1200-fold. D-Bluco was shown to detect a broad range of β-lactamases at the femtomolar level. An ultrasensitive RAPID bioluminescence assay using D-Bluco can detect 102 to 103 colony forming unit per milliliter (cfu/mL) of β-lactamase-producing Enterobacterales in urine samples within 30 min. The high sensitivity and rapid detection make the assay attractive for the use of point-of-care diagnostics for lactam-resistant pathogens.
Graphical Abstract
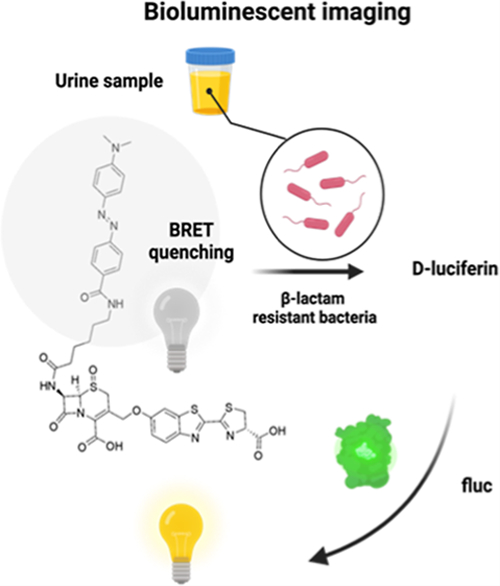
INTRODUCTION
Since the discovery of penicillin in 1928, β-lactam antibiotics (e.g., penicillins, cephalosporins, carbapenems, and mono-bactams) have retained a central role in treating bacterial infections, constituting 60% of worldwide antibiotic usage.1 However, bacterial resistance to β-lactam antibiotics has also emerged. This phenomenon has been further exacerbated by the over extensive use of antibiotics in veterinary and human medicine. To date, the most common resistance mechanism for this class of antibiotics is the production of β-lactamases such as penicillinases, extended-spectrum β-lactamases, AmpC-type β-lactamases, and carbapenemases. These enzymes can hydrolyze the amide bond of the β-lactam ring and inactivate the drugs.2 Over the past decades, the number of new β-lactamases has grown almost exponentially.3 The implementation of new diagnostic techniques for more rapid and sensitive detection of β-lactamase activity would be advantageous in optimizing the use of β-lactam antibiotics and promoting antimicrobial stewardship.
There are several clinically adopted methods for diagnosing β-lactam resistance. Culture-based methods such as the double-disk synergy test, combination disk tests, and automated liquid culture have been widely used as a clinical standard. In spite of good sensitivity and specificity (80−95%), these methods normally require 1−2 days to generate results.4,5 Molecular diagnostics such as fluorescence in situ hybridization and PCR have been developed for the detection of β-lactamase gene signatures with high sensitivity and specificity;6 however, they require pre-enrichment and isolation of pathogenic bacteria to generate a reliable readout. Resistance predicted by genotypic analysis does not always correlate with phenotypic results, and emerging new mutations may evade nucleic acid-based detection, giving false-negative results.3,7 Substrate-based enzyme function assays can directly reveal whether the bacteria possess the capability to destroy β-lactam antimicrobial activity. Colorimetric, fluorogenic, and Förster resonance energy transfer-based probes have been developed over the years for detecting β-lactamase activity in bacteria.8−17 The cefinase test and the Carba NP test, for example, have been approved for clinical use.18 However, these assays generally require overnight bacterial culture due to limitations in detection sensitivity or inability to work directly in complex clinical samples. Recently, Murthy’s group has developed a chemiluminescent probe for β-lactamase detection, yet the lowest detectable concentration of bacteria was 105 to 106 cfu/mL (colony forming unit per millimeter).19 Another study reports a carbapenem-caged chemiluminescent probe that required an even higher concentration of bacteria (107 cfu/mL) to monitor carbapenemase activity.20 Even with chemiluminescence detection, the sensitivity in both cases suffered from a high initial background.
The bioluminescence assay provides excellent detection sensitivity since it does not require excitation light and has low background. A portable ATP bioluminescence assay could detect as low as 10 cfu/mL viable bacteria in about 10 min21 Since the 6′-hydroxy or 6′-amino group of d-luciferin is critical for their bioluminescence emission, the design of bioluminescent probes often follows the “caged pro-luciferin” approach, which involves alkylating the 6′-hydroxyl group on d-luciferin or amidating the 6′amino group on aminoluciferin by a caging group.22−28 For example, we have previously developed a cefazolin analogue-caged bioluminescent probe (Figure 1), termed Bluco.29 Upon hydrolysis by β-lactamase, D-luciferin will be released for subsequent bioluminescence reactions. However, Bluco displayed a considerable amount of bioluminescence emission even in the absence of β-lactamase. Similar initial bioluminescence emission before uncaging has been reported with other caged pro-luciferins.24,30 It is often thought that this background emission was due to the existence of residual free D-luciferin. However, we discovered that caged pro-luciferins could also be processed by firefly luciferase and generate emission. While the emission efficiency was low, it led to a background signal that significantly compromised the target detection sensitivity. This background could be effectively suppressed via the bioluminescence resonance energy transfer (BRET) mechanism by attaching a broad-spectrum quenching moiety.31,32 We have demonstrated this approach by developing a cephalosporin-caged pro-luciferin with an attached quencher moiety (termed D-Bluco) (Figure 1). Only when activated by β-lactamases, the probe is cleaved to generate free d-luciferin for bioluminescence. Finally, we developed a bioluminescence assay for rapid detection of β-lactamase-expressing bacteria within 30 min at a concentration of 102 to 103 cfu/mL.
Figure 1.
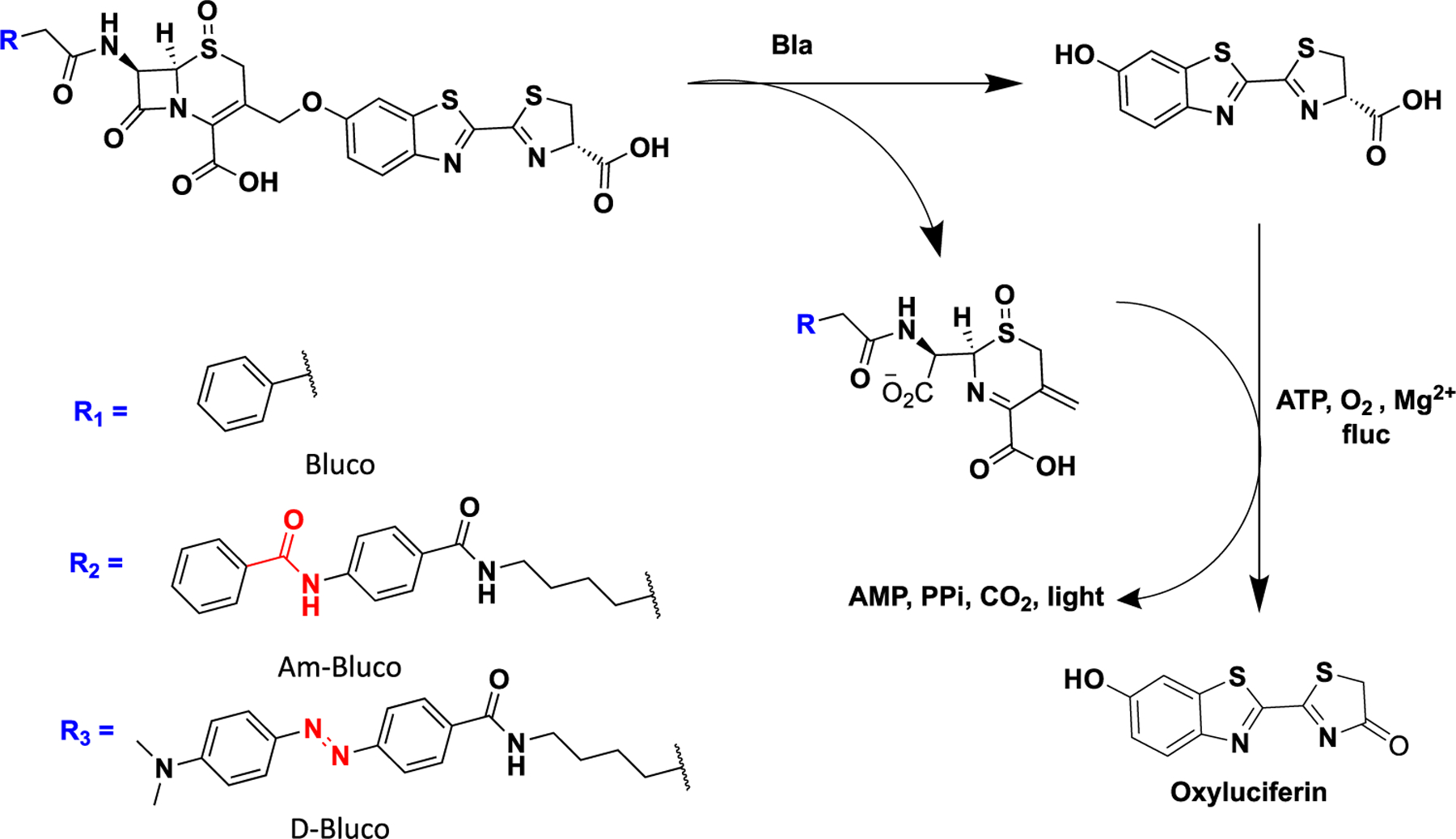
Bioluminogenic substrates for the detection of β-lactamase (Bla) activity.
EXPERIMENTAL SECTION
The synthetic routes for D-Bluco and Am-Bluco are depicted in Schemes S1 and S2 and described in the Supporting Information. The chemical structures of the probes were characterized by 1H NMR, 13C NMR, and HRMS spectra.
Bacterial Growth and Assay.
E. coli (BL21) transformed to express TEM-1 β-lactamase were grown in LB medium at 37 °C overnight and induced with 0.2% arabinose for 6−8 h at 30 °C, 205 rpm. Colony forming units per milliliter (cfu/mL) were determined by measuring the UV absorbance at OD600. Clinically isolated K. pneumoniaeexpressing KPC, E. cloacae expressing IMI, E. coli expressing TEM, E. coli expressing NDM, and E. cloacae expressing AmpC were cultured in BD Columbia agar plate containing 5% sheep blood. Resistant bacterial colonies near meropenem discs were further inoculated in nutrient broth to culture overnight before use. For D-Bluco incubation, working solutions of 10 μM were prepared freshly by diluting stock solutions (10 mM in pure dimethyl sulfoxide) in phosphate-buffered saline (PBS) (pH 7.4).
RAPID Bioluminescence Test with Engineered Bacterial or Clinic Isolates.
Bacterial solutions were incubated with 0.5% CHAPS in PBS (pH = 7.4) for 15 min (880 μL, buffer A). To each bacterial mixture was added D-Bluco (final concentration = 10 μM). After 15 min, the bioluminescence intensity of the mixture was determined by the addition of premixed bioluminescence imaging (BLI) assay reagents (Buffer B, 120 μL) containing luciferase (200 nM), ATP (2 mM), MgCl2 (4 mM), and coenzyme A (CoA) (5 μM). Bioluminescent signals were detected with a TurnerBiosystem luminometer or a SpectraMax iD3 multimode microplate reader (Molecular Device, San Jose, CA). The cfu/mL concentration of bacteria in each entry was further confirmed from the colonies on an agar plate after serial dilutions. Bioluminescence assays were carried out at 25 °C unless specified.
Inhibitor Test with Recombinant Bacteria.
The inhibitor test was adapted from a previous study.13 Briefly, one hundred microliters (100 μL) of freshly cultured E. coli/TEM (OD600 = 0.5) were pretreated with PBS, 2 mg/mL potassium clavulanate, or 2 mg/mL avibactam at 37 °C for 1 h. The bioluminescent signal was detected following the RAPID BLI test protocol.
Statistical Analysis.
GraphPad Prism 9 was used for plotting and statistical analysis. The statistical significance was calculated using the unpaired two-tailed Student’s t-test (**p < 0.0021 and ****p < 0.0001).
RESULTS AND DISCUSSION
Design and Characterization of D-Bluco.
D-Bluco is composed of a dabcyl quencher, a β-lactamase responsive cephalosporin structure, and a luciferin moiety (Figure 1). We first evaluated whether D-Bluco could be processed by β-lactamase to release D-luciferin with one of the most prevalent Ambler class A β-lactamases, TEM-1.33 We observed a rapid concentration-dependent fluorescence turn-on from released D-luciferin over a period of 40 min (Figures 2a and S2), and 1 μM of D-Bluco gave rise to a 92-fold increase in fluorescence (λmax excitation = 330 nm, λmax emission = 530 nm). In the absence of TEM-1, there was negligible emission signal intensity across these wavelengths (Figure 2b). D-Bluco was highly stable when incubating with E. coli or PBS (Figure S3). We next measured the bioluminescence emission from the released d-luciferin. The bioluminescence emission was dependent on the incubation time with TEM-1 and the concentration of D-Bluco (Figures S4 and S5). We also tested if there was interference between luciferase and β-lactamases in such a dual enzyme reaction system. No significant differences were noticed among the groups of d-luciferin with or without TEM-1 or IMP-1, a class B carbapenemase (Figure S6). Importantly, as shown in Figure 2c, D-Bluco produced a 1247-fold increase in bioluminescence emission upon TEM-1 treatment. Its initial bioluminescent signal was significantly lower than that of Bluco, which only generated a 59-fold increase in the bioluminescence emission after 15 min TEM-1 treatment.
Figure 2.
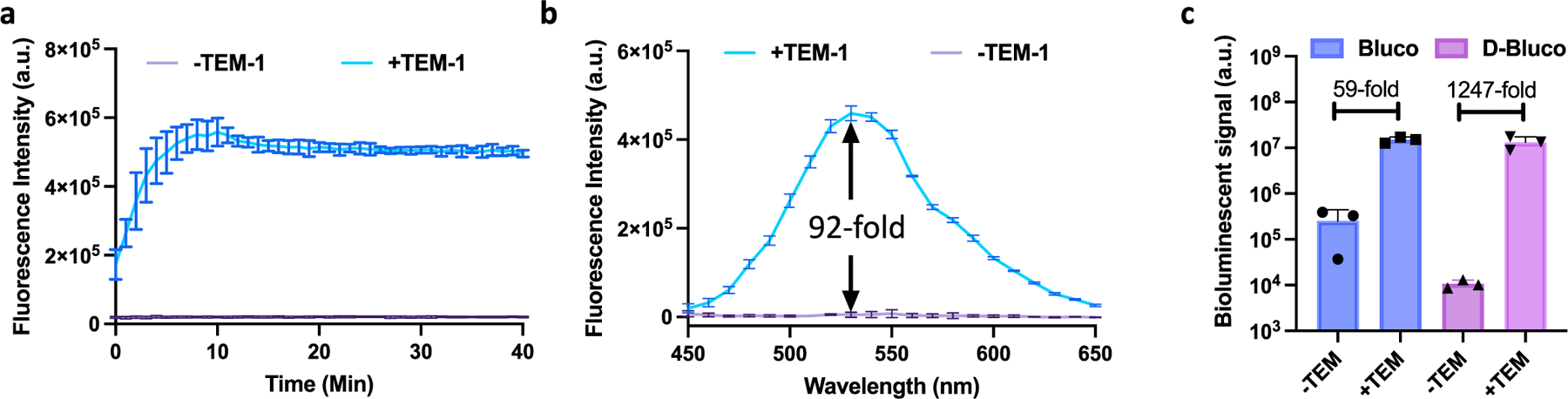
Characterization of D-Bluco with recombinant β-lactamase. (a) Longitudinal monitoring of fluorescence enhancement of D-Bluco (1 μM) with or without TEM-1 (100 nM) in PBS (pH = 7.4). (Ex-330/Em-530). (b) Fluorescence emission spectrum of D-Bluco (1 μM) incubated with TEM-1 (100 nM) for 40 min; a.u. indicates arbitrary unit. (c) Bioluminescent signal of Bluco or D-Bluco (10 μM) incubated with or without TEM-1 (20 nM) for 15 min in PBS (pH = 7.4). Experiments were conducted at 25 °C. Error bars indicate standard deviations (n = 3).
Origins of Bioluminescence Background of d-Bluco.
The initial bioluminescent background signal could partially stem from residual D-luciferin present in the sample. However, high-performance liquid chromatography analysis could not detect D-luciferin at a concentration less than 0.1 μM (Figure S7). When we added firefly luciferase sufficiently to consume residual D-luciferin as high as 1 μM (Figure S8), it was found that there was still bioluminescence emission when additional firefly luciferase (t2−t4) was added to the Bluco solution (Figure 3). In comparison, for D-Bluco, no further bioluminescence emission was detected after the first flash (t0) from the initial firefly luciferase addition. To further confirm the quenching effect of the dabcyl group, we prepared another control Am-Bluco, which is structurally similar to D-Bluco but with the dabcyl group replaced by a structure that has a different absorption spectrum (Figure 1). Like Bluco, Am-Bluco produced bioluminescence emission upon each addition of firefly luciferase (Figure 3). Mass spectrometry revealed that the pro-luciferin substrates Bluco, Am-Bluco, and D-Bluco were oxidized when incubated with firefly luciferase (Figure S9).
Figure 3.
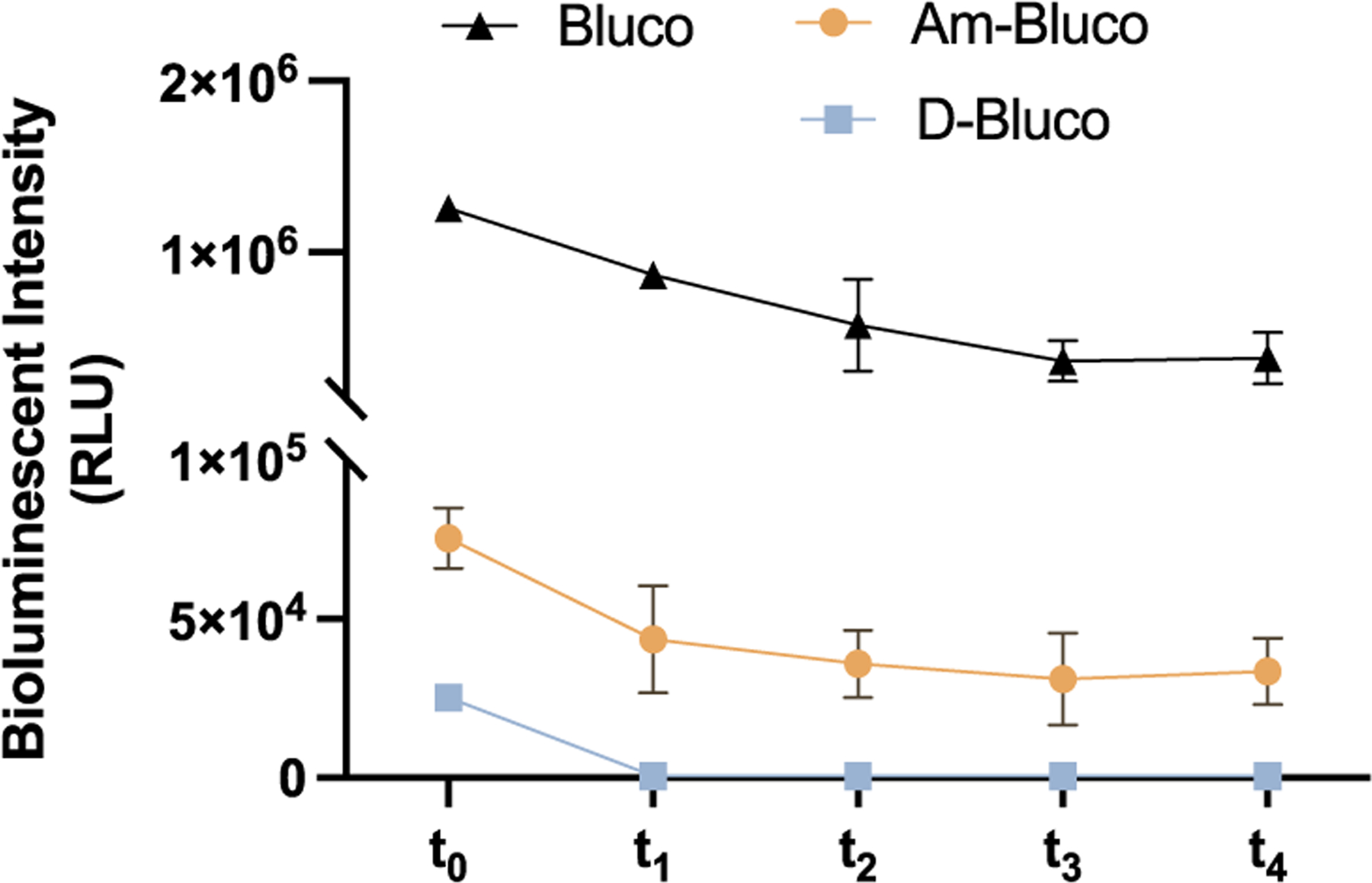
Verification of quenching ability with D-luciferin and Bluco analogues. Bluco, Am-Bluco, and D-Bluco (70 μM) were incubated with 4 μM of luciferase for 10 min (t0) before further luciferase addition at a 4 min interval (t1 to t4). Error bars indicate standard deviations (n = 3).
We also evaluated potential contribution to the background signal from spontaneous hydrolysis of the Bluco probe and its variations. As shown in Table S1, we determined the spontaneous hydrolysis rate for each Bluco-based probe and their contributions to bioluminescence emission. The values of the bioluminescence signal from the spontaneously hydrolyzed Bluco and Am-Bluco are estimated to be very small (0.5−2.5% of the total signal at t1). On the other hand, while the value from spontaneously hydrolyzed D-Bluco (70 μM) is the lowest, it constituted approximately the total signal at t1. We also examined whether there was any intermolecular quenching by mixing D-Bluco and free d-luciferin, and the contribution of D-Bluco or the quenching molecule dabcyl acid to the d-luciferin emission was small and not statistically significant (Figure S10). These results together have established that caged pro-luciferin probes Bluco and Am-Bluco can be processed by firefly luciferase before uncaging to produce significant background bioluminescence emission and that the attached dabcyl group in D-Bluco can quench this background emission mainly through BRET mechanism.31,32
Detection of β-Lactamase Activities in Solution and in Bacteria.
We evaluated the effect of the attached dabcyl group on the hydrolysis kinetics of D-Bluco by β-lactamases. Several β-lactamases of clinical relevance were included: KPC-3, BlaC (Class A), IMP-1 (Class B), AmpC (Class C), and OXA-48 (Class D), in addition to TEM-1. These enzymes were recombinantly expressed and purified as previously described.14 They all triggered significant bioluminescence in hydrolyzing Bluco and D-Bluco. The hydrolysis kinetics of D-Bluco by these enzymes are characterized and summarized in Table 1 and Figure S11. The Michaelis constant (Km) of IMP-1 to D-Bluco was determined to be 9.88 ± 4.15 μM, and the catalytic constant (kcat) was calculated to be 52.19 ± 8.46 s−1, suggesting the enzyme has a high affinity to D-Bluco. This is probably due to its larger catalytic pocket than other class B enzymes and higher tolerability at 7-position modification.34 The kcat/Km for the hydrolysis by OXA-48 was low, which may be due to steric hindrance between the OXA-48 active site, located in a narrow crevice of ~5 × 10 × 20 Å (width, depth, length) and the bulky and hydrophobic side chain of D-Bluco. Kinetic analysis confirmed the substrate specificity of OXA-48, with a stronger preference for a flat side chain (e.g., nitrocefin, kcat/Km = 7.7 × 106 M−1 × s−1) compared with cefoxitin (2.6 × 102 M−1 × s−1). Other substrates with a bulky and flexible side chain, such as cefepime, ceftazidime, and cefotaxime, also have variable degrees of decrease in catalytic efficiency, ranging by approximately 10 to 10,000-fold.35,36
Table 1.
Michaelis−Menten Kinetic Data
| Enzyme | TEM-1 | KPC-3 | IMP-1 | BlaC | AmpC | OXA-48 |
|---|---|---|---|---|---|---|
| kcat (s−1) | 1.72 ± 0.13 | 9.23 ± 1.77 | 52.19 ± 8.46 | 0.22 ± 0.057 | 0.050 ± 0.028 | 0.0025 ± 0.00026 |
| Km (μM) | 2.97 ± 0.76 | 6.28 ± 3.55 | 9.88 ± 4.15 | 8.21 ± 1.97 | 20.25 ± 3.00 | 8.03 ± 2.8 |
| kcat/Km (M−1s−1) | 5.79 × 105 | 1.47 × 106 | 5.26 × 106 | 2.21 × 104 | 2.48 × 103 | 2.53 × 102 |
The limit of detection of D-Bluco for these β-lactamases was then quantified by calculating the bioluminescent signal of three times the standard deviation of the negative controls (D-Bluco in PBS). D-Bluco could detect OXA-48 and AmpC with a higher detection limit of 0.1 femtomole and for all other β-lactamases, the lower detection limit was at 0.01 femtomole (Figure 4). This sensitivity reflects a nearly 100-fold improvement over previously reported fluorescent cephalosporin probes.17,37 Transformed E. coli expressing TEM-1 (E. coli/ TEM-1) was tested with D-Bluco. Parental E. coli (BL21) was used as a negative control. The number of bacteria present in the assay was validated by the plating method (Figure S12). After one hour incubation of D-Bluco with E. coli/TEM-1, a positive correlation between the bioluminescent signal and concentration of bacteria (cfu/mL) was observed, with as low as 102 cfu/mL of bacteria detected within an hour −10 cfu in a 100 μL volume (Figure S13). In contrast, a 20 h incubation was required to detect 103 cfu/mL TEM-1 expressing bacteria with a recently reported 3,7-diester phenoxazine probe, CDA (Cephalosporin-caged Diester Amplex red analogue).37 The detection specificity of D-Bluco was confirmed by including β-lactamase inhibitors avibactam and clavulanate in the incubation. As shown in Figure 5a, the bioluminescence signals were abolished in the presence of inhibitors.
Figure 4.
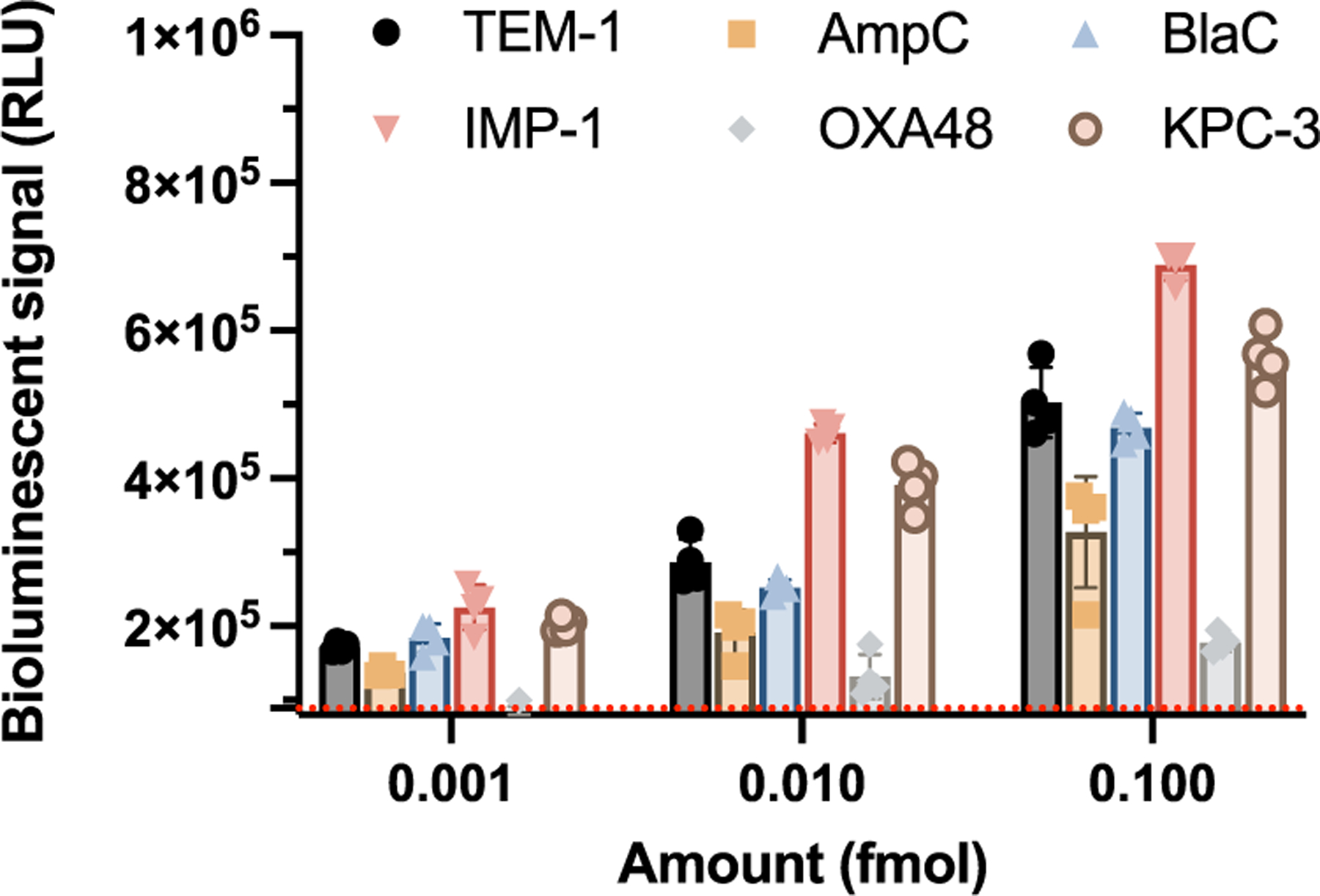
Sensitivity of D-Bluco toward different types of lactamase enzymes. Bioluminescent signal of D-Bluco incubated with different concentrations of enzyme for 2 h. The red dashed line indicates three times the standard deviation of the blank. RLU indicates relative light units. Experiments were conducted at 25 °C. Error bars indicate standard deviations (n = 4).
Figure 5.
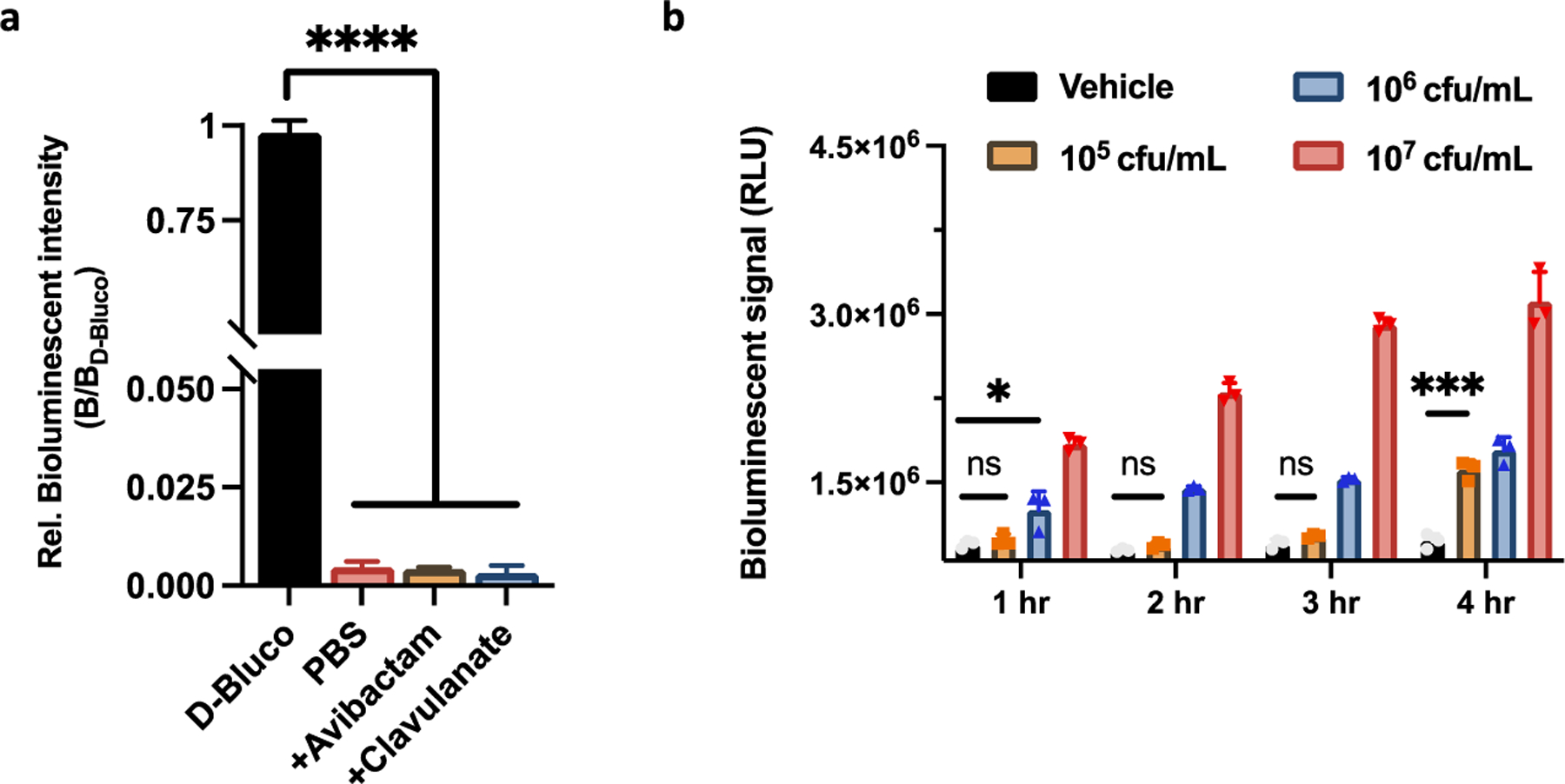
Bioluminescence detection of β-lactamase with clinic isolates by D-Bluco. (a) Inhibitory study with clavulanate (2 mg/mL) and avibactam (2 mg/mL) in the presence of D-Bluco (10 μM) with E. coli/TEM-1. E. coli/TEM-1 treated with D-Bluco exhibited a 260-fold increase of bioluminescent intensity over PBS, which was arbitrarily set as 1 to normalize the test samples and show percentage inhibition. (b) D-Bluco (10 μM) was incubated with different concentrations of E. coli or E. coli expressing TEM (E. coli/TEM). The BLI signal was monitored over 4 h in PBS (pH = 7.4). Statistical significance was calculated using the unpaired two-tailed Student’s t-test (*p < 0.0332, ***p < 0.0002, and ****p < 0.0001 ns: not significant). RLU indicates relative light units. Experiments were conducted at 25 °C. Error bars indicate standard deviations (n = 3).
Assay Development for Clinic Isolates.
We further tested a clinically isolated E. coli strain producing TEM-type β-lactamase (E. coli/TEM).38 The bioluminescent signal increased in a time- and probe concentration-dependent manner (Figures 5b and S14). However, clinically isolated E. coli/TEM was detected at a concentration of 106 cfu/mL after an hour of incubation and 105 cfu/mL within 4 h of incubation with D-Bluco. In comparison to the transformed strain expressing recombinant β-lactamase, the clinical isolate has less copies of plasmids, and the degree of gene amplification within plasmids and the promoter efficiency may also vary, resulting in significant difference in the level of β-lactamases expressed.39
To evaluate whether releasing β-lactamases from the periplasm could improve detection sensitivity, we sought to lyse the bacteria. 3-[(3-Cho-l-amidopropyl)-dimethylammonio]-1-propanesul-fonate (CHAPS) is a non-denaturing zwitterionic detergent and has been used for bacterial lysis.12 We tested different concentrations and found that 0.5% CHAPS significantly enhanced the bioluminescence signal in 106 cfu/mL E. coli/TEM without a negative impact on the luciferase activity (Figure S15). In addition to E. coli, Klebsiella pneumoniae (K. pneumoniae) is among the most common nosocomial Enterobacterales capable of developing lactam-resistance.38,40 We tested two clinically significant isolates, K. pneumoniae expressing KPC carbapenemase (K. pneumoniae/ KPC) and E. coli expressing New Delhi metallo β-lactamase (E. coli/NDM).38 Compared to intact bacteria, incubating D-Bluco with E. coli/TEM, K. pneumoniae/KPC, or E. coli/NDM lysate generated 52-, 141-, and 145-fold signal enhancement, respectively (Figure S16). Moreover, to further improve the sensitivity of our assay, we increased the assay volume to 1 mL, a 10-fold increase, and attempted to introduce CoA to the assay. CoA has been suggested to help enhance the bioluminescent signal by carrying out a thiolytic reaction to block the inhibitory side product, dehydroluciferyl adeny-late.41,42 Interestingly, in our case, a stabilized signal was observed but no significant bioluminescent signal enhancement was achieved (Figure S17). These results led us to develop an optimized assay for rapid, highly sensitive detection of β-lactamase activity, as shown in Figure 6a. Bacteria were mixed with 0.5% CHAPS lysis buffer for 15 min, and then incubated with D-Bluco at room temperature for another 15 min. The resulted mixture was combined with an assay buffer which contained MgCl2, CoA, ATP, and luciferase for immediate bioluminescence reading.
Figure 6.
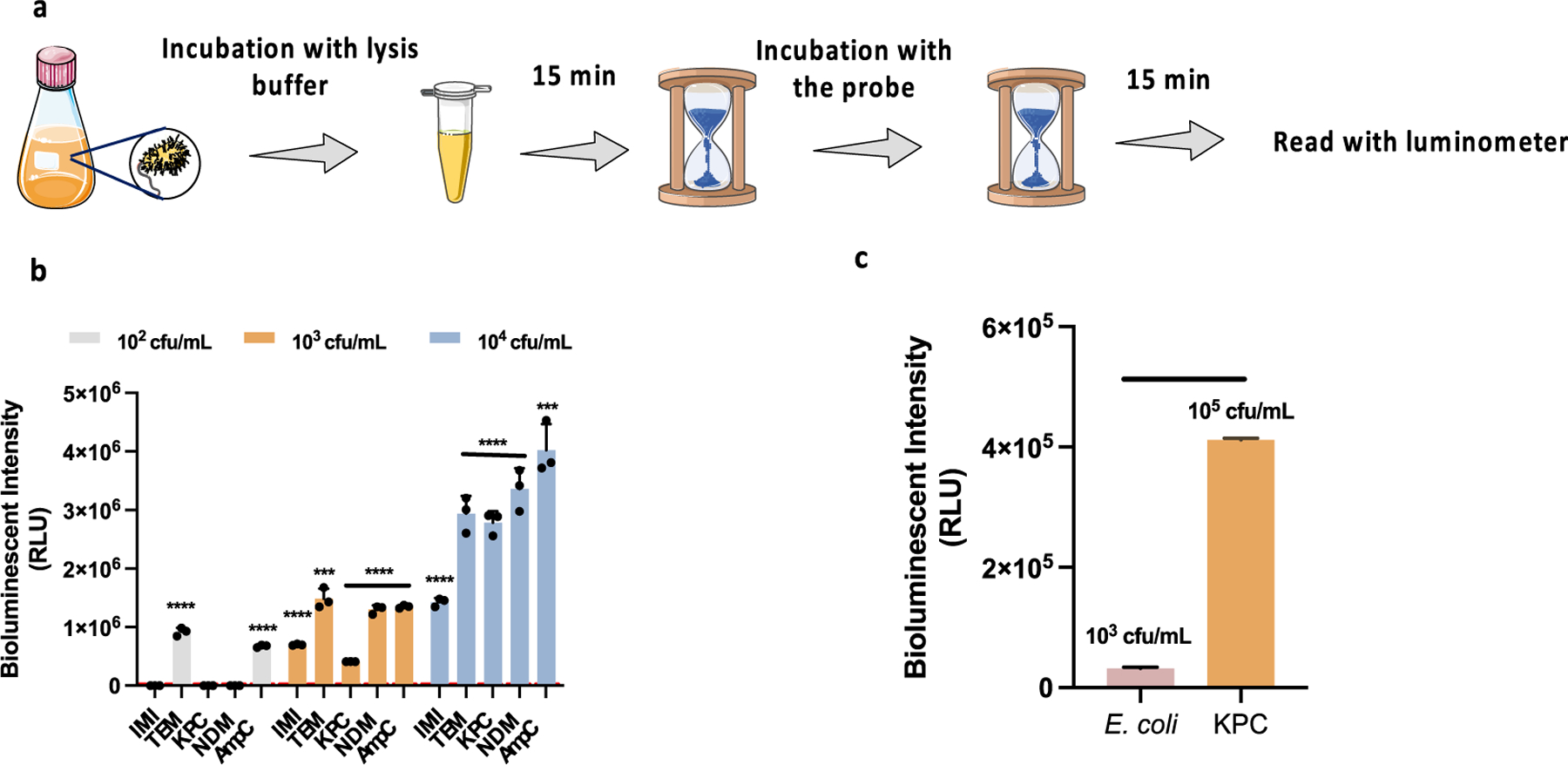
Development of RAPID BLI test for clinical isolates of UTI. (a) RAPID BLI assay workflow for clinical sample detection. (b) Bioluminescent intensity of 102, 103, 104 cfu/mL of β-lactamases-expressing clinic isolates after incubation with D-Bluco (10 μM) in diluted urine. From left to right: (1) E. cloacae/IMI, (2) E. coli/TEM, (3) K. pneumoniae/KPC, (4) E. coli/NDM, (5) E. cloacae/AmpC. (c) Bioluminesecent intensity of 105 cfu/mL E. coli and 103 cfu/mL K. pneumoniae/KPC. The modified rapid BLI protocol is applied. The signal of D-Bluco in PBS was subtracted before plotting. The working concentration of D-Bluco was 10 μM. Statistical significance was calculated using the unpaired two-tailed Student’s t-test (***p < 0.0002 and ****p < 0.0001, for comparison with 105 cfu/mL E. coli) RLU indicates relative light units. Dot line represents 3SD of the negative control. Part of the image is adapted from Servier Medical Art. Error bars indicate standard deviations (n = 3).
Urinary tract infection (UTI) is one of the most common infections, affecting almost 50% of the population at least once in their lifetime, and one of the largest groups for routine antibiotic administration.43 Overuse of antibiotics in UTI has raised a major concern in developing resistance. Many broad-spectrum antimicrobials are prescribed before an antibiotic susceptibility test report is available, especially in rapidly progressing infections. Subsequently, as many as 40% of patients may expose to unnecessary or inappropriate antibiotics.44 These broad-spectrum antibiotics can adversely affect the natural gut microbiota, thus exposing individuals to Clostridium difficile colitis and favoring resistance in specific bacterial strains. We assessed the clinical value of the RAPID BLI test for detecting β-lactamase expressing bacterial pathogens in UTI.
Clinic isolates representing different classes (class A: E. cloacae/IMI, E. coli/TEM, E. coli/KPC; class B: E. coli/NDM; and class C: E. cloacae/AmpC) were spiked into synthetic urine samples. As shown in Figure 6b,c, clinic isolates can be detected directly without culture at a concentration of 102 cfu/mL for E. coli/TEM and E. cloacae/AmpC and 103 cfu/mL for all other bacteria. According to the UTI criteria defined by CDC (Centers for Disease Control and Prevention), a positive urine culture has ≥105 cfu/mL of one but not more than two bacterial species.45 Therefore, the sensitivity of the RAPID BLI assay is significantly below this threshold. Compared to conventional methods, our assay is rapid, ultrasensitive, and easy to use, and thus holds great potential to be used for point-of-care diagnostics.
CONCLUSIONS
In this work, we have investigated the source of the bioluminescence background emission of bioluminogenic pro-luciferin probes. Caged pro-luciferin probes can be oxidized by luciferases due to the promiscuity of the firefly luciferase enzyme pocket. We demonstrated that this background could be suppressed by the attached broad-spectrum quenching moiety via the BRET mechanism. By combining BRET quenching and chemical caging, we developed an ultrasensitive bioluminescent probe D-Bluco that could detect as low as 10−18 moles of β-lactamase. We demonstrated a D-Bluco based bioluminescence assay (RAPID BLI) could detect β-lactamase activity in clinical bacterial isolates in urine samples in 30 min with a superior sensitivity (102 to 103 cfu/mL). The strategy of combining BRET quenching and chemical caging to suppress background and achieve high sensitivity may serve as a general approach for the development of other bioluminogenic sensors.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH grants R01AI125286 and R37AI051622. T.D. thanks the support from the Center for Molecular Analysis and Design (CMAD) at Stanford. J.B. thanks the support from the NIH-NIGMS Postdoctoral Fellowship (1F32GM134689).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.3c00478.
Additional experimental details, including the synthesis of D-Bluco and Am-Bluco, 1H NMR spectra of final products and intermediates, the emission spectrum, characterization of stability of the compounds, enzymatic kinetics, and optimization of assay conditions (PDF)
Notes
The authors declare the following competing financial interest(s): C.R.B. is a cofounder of Redwood Biosciences (a subsidiary of Catalent), Enable Biosciences, Palleon Pharmaceuticals, InterVenn Bio, Lycia Therapeutics, and OliLux Biosciences, and a member of the Board of Directors of Eli Lilly. All other authors declare no competing financial interest.
Contributor Information
Tingting Dai, Department of Chemistry, Stanford University, Stanford, California 94305, United States.
Jinghang Xie, Department of Radiology, Molecular Imaging Program at Stanford, Stanford University School of Medicine, Stanford, California 94305, United States;.
Joseph A. Buonomo, Department of Chemistry, Stanford University, Stanford, California 94305, United States; Sarafan ChEM-H, Stanford University, Stanford, California 94305, United States
Angel Moreno, Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, United States.
Niaz Banaei, Department of Pathology and Division of Infectious Diseases and Geographic Medicine, Department of Medicine, Stanford University School of Medicine, Stanford, California 94305, United States; Clinical Microbiology Laboratory, Stanford University Medical Center, Palo Alto, California 94304, United States.
Carolyn R. Bertozzi, Department of Chemistry and Howard Hughes Medical Institute, Stanford University, Stanford, California 94305, United States; Sarafan ChEM-H, Stanford University, Stanford, California 94305, United States;
Jianghong Rao, Department of Chemistry, Stanford University, Stanford, California 94305, United States; Department of Radiology, Molecular Imaging Program at Stanford, Stanford University School of Medicine, Stanford, California 94305, United States; Sarafan ChEM-H, Stanford University, Stanford, California 94305, United States;.
REFERENCES
- (1).Fleming A Br. J. Exp. Pathol 1929, 10, 226–236. [Google Scholar]
- (2).Tooke CL; Hinchliffe P; Bragginton EC; Colenso CK; Hirvonen VHA; Takebayashi Y; Spencer J J. Mol. Biol 2019, 431, 3472–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bush K Antimicrob. Agents Chemother 2018, 62, No. e01076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Aruhomukama D Afr. Health Sci 2020, 20, 1090–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gazin M; Paasch F; Goossens H; Malhotra-Kumar SJ Clin. Microbiol 2012, 50, 1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jamal W; Al Roomi E; AbdulAziz LR; Rotimi VO J. Clin. Microbiol 2014, 52, 2487–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Paterson DL; Bonomo RA Clin. Microbiol. Rev 2005, 18, 657–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang J; Shen Y; May SL; Nelson DC; Li S Angew. Chem., Int. Ed 2012, 51, 1865–1868. [DOI] [PubMed] [Google Scholar]
- (9).Thai HBD; Yu JK; Park BS; Park Y-J; Min S-J; Ahn D-R Biosens. Bioelectron 2016, 77, 1026–1031. [DOI] [PubMed] [Google Scholar]
- (10).Aw J; Widjaja F; Ding Y; Mu J; Liang Y; Xing B Chem. Commun 2017, 53, 3330–3333. [DOI] [PubMed] [Google Scholar]
- (11).Khan S; Sallum UW; Zheng X; Nau GJ; Hasan T BMC Microbiol 2014, 14, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ma C-W; Ng KK-H; Yam BH-C; Ho P-L; Kao RY-T; Yang DJ Am. Chem. Soc 2021, 143, 6886–6894. [DOI] [PubMed] [Google Scholar]
- (13).Dai T; Xie J; Zhu Q; Kamariza M; Jiang K; Bertozzi CR; Rao J J. Am. Chem. Soc 2020, 142, 15259–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Xie H; Mire J; Kong Y; Chang M; Hassounah HA; Thornton CN; Sacchettini JC; Cirillo JD; Rao J Nat. Chem 2012, 4, 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Song A; Cheng Y; Xie J; Banaei N; Rao J Chem. Sci 2017, 8, 7669–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cheng Y; Xie J; Lee K-H; Gaur RL; Song A; Dai T; Ren H; Wu J; Sun Z; Banaei N; Akin D; Rao J Sci. Transl. Med 2018, 10, No. eaar4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shi H; Cheng Y; Lee KH; Luo RF; Banaei N; Rao J Angew. Chem., Int. Ed 2014, 53, 8113–8116. [DOI] [PubMed] [Google Scholar]
- (18).M100Ed31 Performance Standards for Antimicrobial Susceptibility Testing, 31st ed; Clinical & Laboratory Standards Institute. https://clsi.org/standards/products/microbiology/documents/m100/(accessed on April 02, 2021).
- (19).Maity S; Wang X; Das S; He M; Riley LW; Murthy N Chem. Commun 2020, 56, 3516–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Das S; Ihssen J; Wick L; Spitz U; Shabat D Chem.—Eur. J 2020, 26, 3647–3652. [DOI] [PubMed] [Google Scholar]
- (21).He B; Liu X; Yue W; Zhou A; Luo J; Cai X Afr. J. Microbiol. Res 2009, 3, 575–580. [Google Scholar]
- (22).Miska W; Geiger R Biol. Chem. Hoppe Seyler 1988, 369, 407–412. [DOI] [PubMed] [Google Scholar]
- (23).Li J; Chen L; Du L; Li M Chem. Soc. Rev 2013, 42, 662− 676. [DOI] [PubMed] [Google Scholar]
- (24).Sharma DK; Adams ST; Liebmann KL; Miller SC Org. Lett 2017, 19, 5836–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Takakura H; Kojima R; Kamiya M; Kobayashi E; Komatsu T; Ueno T; Terai T; Hanaoka K; Nagano T; Urano YJ Am. Chem. Soc 2015, 137, 4010–4013. [DOI] [PubMed] [Google Scholar]
- (26).Takakura H; Kojima R; Urano Y; Terai T; Hanaoka K; Nagano T Chem. − Asian J 2011, 6, 1800–1810. [DOI] [PubMed] [Google Scholar]
- (27).Van de Bittner GC; Bertozzi CR; Chang CJ J. Am. Chem. Soc 2013, 135, 1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yang Y; Shao Q; Deng R; Wang C; Teng X; Cheng K; Cheng Z; Huang L; Liu Z; Liu X; Xing B Angew. Chem., Int. Ed 2012, 51, 3125–3129. [DOI] [PubMed] [Google Scholar]
- (29).Yao H; So M; Rao J Angew. Chem., Int. Ed 2007, 46, 7031–7034. [DOI] [PubMed] [Google Scholar]
- (30).Ke B; Chen H; Cui Y; Ma L; Liu Y; Hu X; Bai Y; Du L; Li M Talanta 2019, 194, 925–929. [DOI] [PubMed] [Google Scholar]
- (31).Xu Y; Piston DW; Johnson CH Proc. Natl. Acad. Sci. U.S.A 1999, 96, 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Adamczyk M; Moore JA; Shreder K Org. Lett 2001, 3, 1797–1800. [DOI] [PubMed] [Google Scholar]
- (33).Robin F; Delmas J; Schweitzer C; Tournilhac O; Lesens O; Chanal C; Bonnet R Antimicrob. Agents Chemother 2007, 51, 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hu L; Liu R; Ma Z; Yu T; Li Z; Zou Y; Yuan C; Chen F; Xie H Chem. Commun 2021, 57, 13586–13589. [DOI] [PubMed] [Google Scholar]
- (35).Akhtar A; Pemberton OA; Chen Y ACS Infect. Dis 2020, 6, 261–271. [DOI] [PubMed] [Google Scholar]
- (36).Docquier J-D; Calderone V; De Luca F; Benvenuti M; Giuliani F; Bellucci L; Tafi A; Nordmann P; Botta M; Rossolini GM; Mangani S Chem. Biol 2009, 16, 540–547. [DOI] [PubMed] [Google Scholar]
- (37).Xie J; Mu R; Fang M; Cheng Y; Senchyna F; Moreno A;Banaei N; Rao J Chem. Sci 2021, 12, 9153–9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Senchyna F; Gaur RL; Sandlund J; Truong C; Tremintin G; Kultz D; Gomez CA; Tamburini FB; Andermann T; Bhatt A; Tickler I; Watz N; Budvytiene I; Shi G; Tenover FC; Banaei N Diagn. Microbiol. Infect. Dis 2019, 93, 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Livermore DM Clin. Microbiol. Infect 1997, 3, 4S10–4S19. [PubMed] [Google Scholar]
- (40).Flores-Mireles AL; Walker JN; Caparon M; Hultgren SJ Nat. Rev. Microbiol 2015, 13, 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fraga H; Fernandes D; Fontes R; da Silva JCGE FEBS J 2005, 272, 5206–5216. [DOI] [PubMed] [Google Scholar]
- (42).Wang L; Li Y; Guo R; Li S; Chang A; Zhu Z; Tu P PLoS One 2019, 14, No. e0223096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Foxman B Nat. Rev. Urol 2010, 7, 653–660. [DOI] [PubMed] [Google Scholar]
- (44).McIsaac WJ; Low DE; Biringer A; Pimlott N; Evans M; Glazier R Arch. Intern. Med 2002, 162, 600–605. [DOI] [PubMed] [Google Scholar]
- (45).Wilson ML; Gaido LL Clin. Infect. Dis 2004, 38, 1150–1158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.