

Summary
Pancreatic islet cells derived from human pluripotent stem cells hold great promise for modeling and treating diabetes. Differences between stem cell-derived and primary islets remain, but molecular insights to inform improvements are limited. Here, we acquire single-cell transcriptomes and accessible chromatin profiles during in vitro islet differentiation and pancreas from childhood and adult donors for comparison. We delineate major cell types, define their regulomes, and describe spatiotemporal gene regulatory relationships between transcription factors. CDX2 emerged as a regulator of enterochromaffin-like cells, which we show resemble a transient, previously unrecognized, serotonin-producing pre-β-cell population in fetal pancreas, arguing against a proposed non-pancreatic origin. Furthermore, we observe insufficient activation of signal-dependent transcriptional programs during in vitro β-cell maturation and identify sex hormones as drivers of β-cell proliferation in childhood. Altogether, our analysis provides a comprehensive understanding of cell fate acquisition in stem cell-derived islets and a framework for manipulating cell identities and maturity.
Keywords: human pluripotent stem cells, islets, β-cell, pancreas, CDX2, serotonin, development, fetal pancreas, single-cell genomics, gene regulatory network, signals, transcription factors, ATAC-seq, RNA-seq
Graphical Abstract
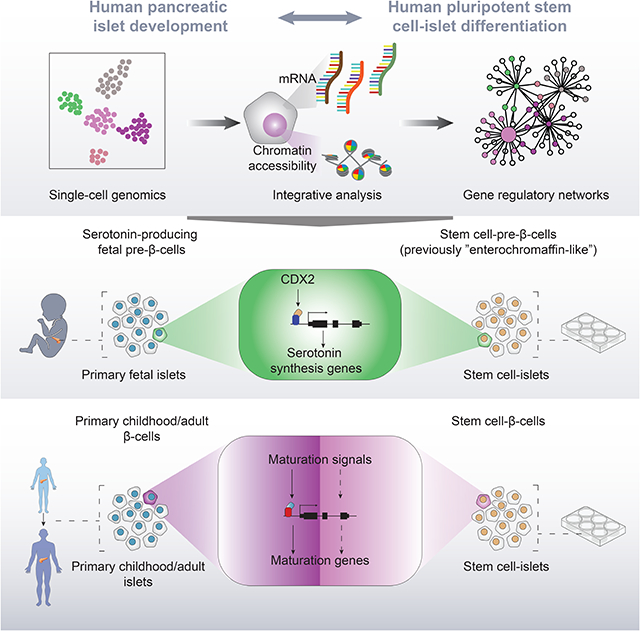
Zhu and Wang et al. compare regulomes of pancreatic endocrine cells from human stem cells and fetal, childhood and adult pancreas. They discovered a serotonin-producing fetal pre-β-cell population resembling stem cell-derived enterochromaffin-like cells, previously coined pancreas-aberrant, and identified insufficient activation of β-cell maturation signals during in vitro differentiation.
Introduction
The ability to generate pancreatic islet-like clusters from human pluripotent stem cells (hPSCs) holds great promise as a cell replacement therapy and in vitro disease model for diabetes. Current protocols mimic in vivo development by stepwise exposure of hPSCs to growth factors and small molecules1–8. Stem cell-derived islets (SC-islets) are comprised of insulin-producing β-cells, glucagon-producing α-cells, and somatostatin-producing δ-cells akin to the cell types found in pancreatic islets. SC-islets also contain cell types thought to be pancreas-aberrant, such as cells resembling enterochromaffin cells of the intestine7,8. Methodology to control cell type yields or eliminate unwanted populations is missing. Furthermore, despite protocol improvements, in vitro SC-β-cells are still functionally immature and respond differently to signals triggering insulin secretion compared to primary β-cells7. SC-β-cells acquire a more mature state when exposed to an in vivo environment by engraftment1,2,4,5,7,9,10, suggesting competence of SC-β-cells to respond to maturation signals but absence of these signals in vitro. In rodents and humans, β-cell functional maturation occurs postnatally11–19 and is driven by environmental cues17,20–23. However, a comprehensive understanding of the signals mediating β-cell maturation during postnatal life is still lacking.
Single-cell technologies can profile individual cells, allowing for detailed molecular characterization of developmental trajectories and cell states. Transcriptome analysis at single-cell level has defined signatures of cell populations during SC-islet differentiation and identified differentially expressed genes between SC-β-cells and primary β-cells7,8,10,24. However, gene expression alone provides a limited understanding of the regulatory features of each cell type or state which is determined by transcription factors (TFs) interacting with gene regulatory elements to enable precise gene expression control through gene regulatory networks (GRNs)25. The lack of GRN maps for SC-islets and primary islets hampers progress toward controlling cell fates and maturation states during SC-islet differentiation. By combining single-cell gene expression and chromatin accessibility profiling, it is possible to infer cell type- and cell state-specific GRNs to gain insight into the transcriptional programs driving cell fate acquisition and cell maturation.
Here, we built GRNs from single-cell transcriptome and chromatin accessibility data acquired throughout SC-islet differentiation and from primary childhood and adult islets. A major finding is that enterochromaffin-like cells (SC-ECs) produced during SC-islet differentiation resemble a pre-β-cell population in the fetal pancreas, suggesting a pancreatic rather than intestinal origin. Deletion of the SC-EC regulator CDX2 in hPSCs supports a close lineage relationship of SC-ECs and SC-β-cells. Comparison of regulatory programs from SC-β-cells and primary β-cells during postnatal maturation identified candidate signaling pathways involved in β-cell maturation and insufficiently activated in SC-β-cells. Together, the established GRNs provide a roadmap for understanding and manipulating SC-islet differentiation.
Results
Chromatin accessibility and gene expression during SC-islet differentiation
Pancreatic endocrine cell differentiation from hPSCs produces insulin+ β-cells, glucagon+ α-cells, and somatostatin+ δ-cells (Figure 1A and Figure S1A). Glucose-stimulated insulin secretion is acquired during a subsequent ~2-week period of SC-islet maturation (Figure S1B). To characterize gene regulatory programs governing SC-islet differentiation and maturation, we conducted single-nucleus ATAC-sequencing (snATAC-seq) and single-cell RNA-sequencing (scRNA-seq) at the pancreatic progenitor (day, D11), endocrine progenitor (D14), immature (D21), and maturing SC-islet cell stage (D32/39; Figure 1A, Table S1A). After quality control (see Methods; Figure S1C,D), we obtained chromatin accessibility profiles from 65,255 cells and transcriptomes from 25,686 cells across the four stages. Following UMAP dimensionality reduction, we defined ten distinct cell populations based on promoter chromatin accessibility or RNA expression using canonical genes (Figure 1B and Figure S1E,F): two pancreatic progenitor cell populations (PP1 and PP2), distinguished by NKX6-1 expression; NEUROG3high early endocrine progenitors (ENP1); α-like endocrine progenitors (ENP-α, ARX+); two late endocrine progenitor populations (ENP2 and ENP3), expressing LMX1A and RFX3, respectively; and differentiated cell types including α-cells (SC-α, GCG+), β-cells (SC-β, INS+ and IAPP+), and δ-cells (SC-δ, SST+). We also identified a previously described7,8 enterochromaffin cell-like SC-EC population (INS+ and SLC18A1+). Cultures at D11 and D14 were mostly comprised of pancreatic and endocrine progenitors, whereas at immature and maturing islet stages predominantly contained differentiated endocrine cell types (Figure S1G,H). We then integrated chromatin accessibility and gene expression data (Figure 1C) and generated in-silico pseudo-cells with matched epigenomic and transcriptomic information. The data integration revealed more cell type specificity in gene expression than in chromatin accessibility (Figure 1D), suggesting plasticity among cell populations.
Figure 1. Stem cell islet lineage trajectories based on integrated single-cell chromatin accessibility and transcriptome profiles.
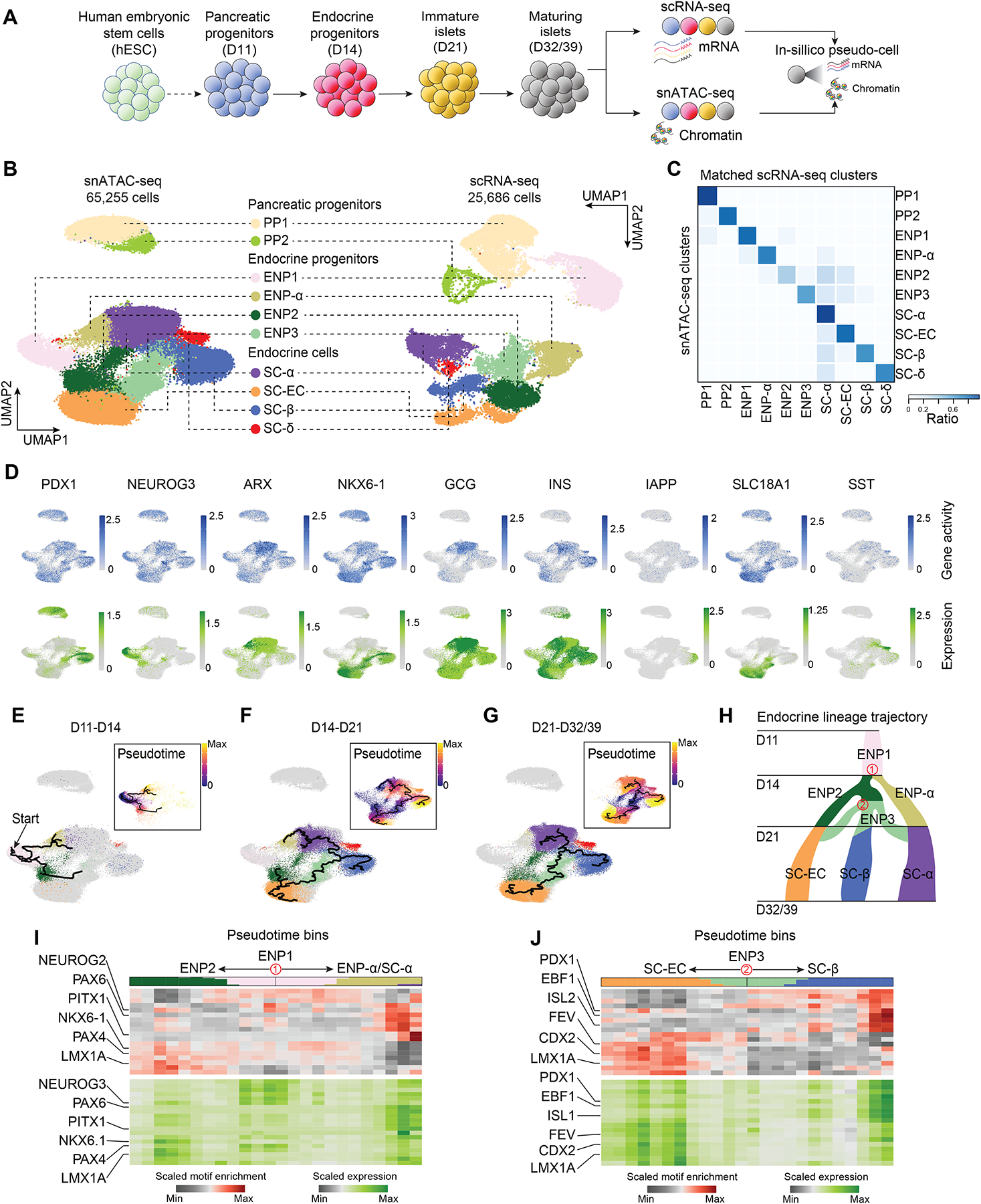
(A) Experimental design. scRNA-seq and snATAC-seq data were generated during SC-islet differentiation at day (D) 11, D14, D21, D32 and D39 and computationally integrated to generate “pseudo-cells”.
(B) UMAP embedding of chromatin accessibility (left) and transcriptome (right) data. Cluster identities were defined by promoter accessibility (snATAC-seq) or expression (scRNA-seq) of marker genes. PP, pancreatic progenitor; ENP, endocrine progenitor; SC-EC, stem cell-derived enterochromaffin cell-like cells.
(C) Heatmap showing ratio of cells with identities in scRNA-seq (column) data matching identities in snATAC-seq (row) data.
(D) Gene activity (top) and gene expression (bottom) for cell type marker genes.
(E-G) Trajectory analysis based on chromatin accessibility, showing trajectories from D11 and D14 (E), D14 and D21 (F), and D21 and D32/39 (G) data with ENP1, ENP2 and ENP3 set as the root, respectively. Cells were color-coded by either cluster identities or pseudotime values (insets). PP1 and PP2 cells were excluded from the analysis.
(H) Inferred endocrine lineage trajectory from e-g. Two branch points (in red) were used in analyses in (I) and (J).
(I, J) Heatmaps of transcription factor motif enrichment (top) and gene expression (bottom) along pseudotime bins downstream of trajectory branch points in (H). Top bar shows proportion of cell types in each pseudotime bin, using matching colors to cell type annotations in (B).
Given that chromatin accessibility signifies developmental potential beyond cell identity defined by gene expression26, we inferred lineage relationships between cell populations by trajectory analysis based on chromatin activity (Figure 1E–G and Figure S1I–K). This analysis identified ENP1 progenitors as a common precursor for all endocrine cell lineages. ENP1 progenitors were predicted to give rise to α-lineage-restricted ENP-α progenitors and ENP2 progenitors that generate SC-ECs as well as ENP3 progenitors producing SC-β-cells, SC-ECs, and SC-α-cells (Figure 1H). These results indicate that SC-α-cells can arise from two different progenitor populations, explaining findings from gene expression-based trajectories suggesting that SC-α-cells can form before or after the specification of SC-β-cells and SC-ECs8,24. Together, this analysis suggests close relatedness of SC-β-cells and SC-ECs.
To identify TFs governing lineage transitions, we analyzed lineage trajectories for TF binding motif enrichment and expression. We focused on two branch points in the lineage tree (Figure 1H): (i) separation of ENP-α and ENP2 progenitors from ENP1 progenitors (Figure 1I and Table S1C–E) and (ii) bifurcation between SC-β-cells and SC-ECs from ENP3 progenitors (Figure 1J and Table S1C–E). We found NEUROG3 motif enrichment and expression to be highest in ENP1, consistent with its function in endocrine lineage induction27,28, whereas PAX6 and PAX4 activity were highest in SC-α- and SC-β-cell precursors29–33, respectively (Figure 1I). Still uncharacterized regulators from this analysis included PITX1 predicted to specify SC-α-cells and LMX1A with a predicted role in non-α lineage choices (Figure 1I). Analysis of the SC-β-cell versus SC-EC lineage branch point confirmed PDX1 as a β-cell regulator34,35 and suggested a similar role for EBF1 (Figure 1J). Interestingly, FEV, LMX1A, and CDX2 exhibited motif enrichment and higher expression in SC-ECs compared to ENP3 progenitors or SC-β-cells (Figure 1J), indicating roles in SC-EC lineage specification.
Cell type-specific gene regulatory programs
To comprehensively characterize gene regulatory programs governing SC-islet cell type differentiation, we inferred GRNs for each cell population, linking TFs to candidate cis-regulatory elements (cCREs) and their target genes (Figure 2A and Figure S2A–C). This analysis yielded a GRN connecting 266 TFs, 51,281 cCREs and 11,997 target genes (Methods). On average each TF was predicted to bind to 1,053 cCREs (Figure S2F,G), each gene to be regulated by 5.6 cCREs and each cCRE to control 1.3 genes (Figure S2D,E). To characterize cell type-specific gene regulatory programs, we subset the GRN by clustering cCREs based on accessibility pattern across cell types (Methods; Figure 2B,C and Figure S2H). As expected, cCRE modules specific to related cell types (e.g., ENP-α and SC-α) were localized closely to each other on the cCRE UMAP (Figure 2B). Furthermore, target genes linked to cCREs within each module were cell type-specifically expressed (Figure 2C) and exhibited cell type-characteristic molecular functions (Figure S2I and Table S2A). Analysis of TFs regulating gene expression in each cell type revealed known gene regulatory roles for RFX3 and RFX6 in endocrine progenitors36,37 and ASCL2 and SCRT1 in SC-β-cells38,39 (Figure 2D, Figure S2J, and Table S2B). We also identified still uncharacterized candidate cell fate regulators, including ETV1 in SC-α-cells, EBF1 in SC-β-cells, and KLF10 in SC-δ-cells. CDX2, LMX1A, FEV, and HNF4G emerged as candidate TFs in SC-ECs and their precursors.
Figure 2. Gene regulatory network analysis of stem cell islet development.
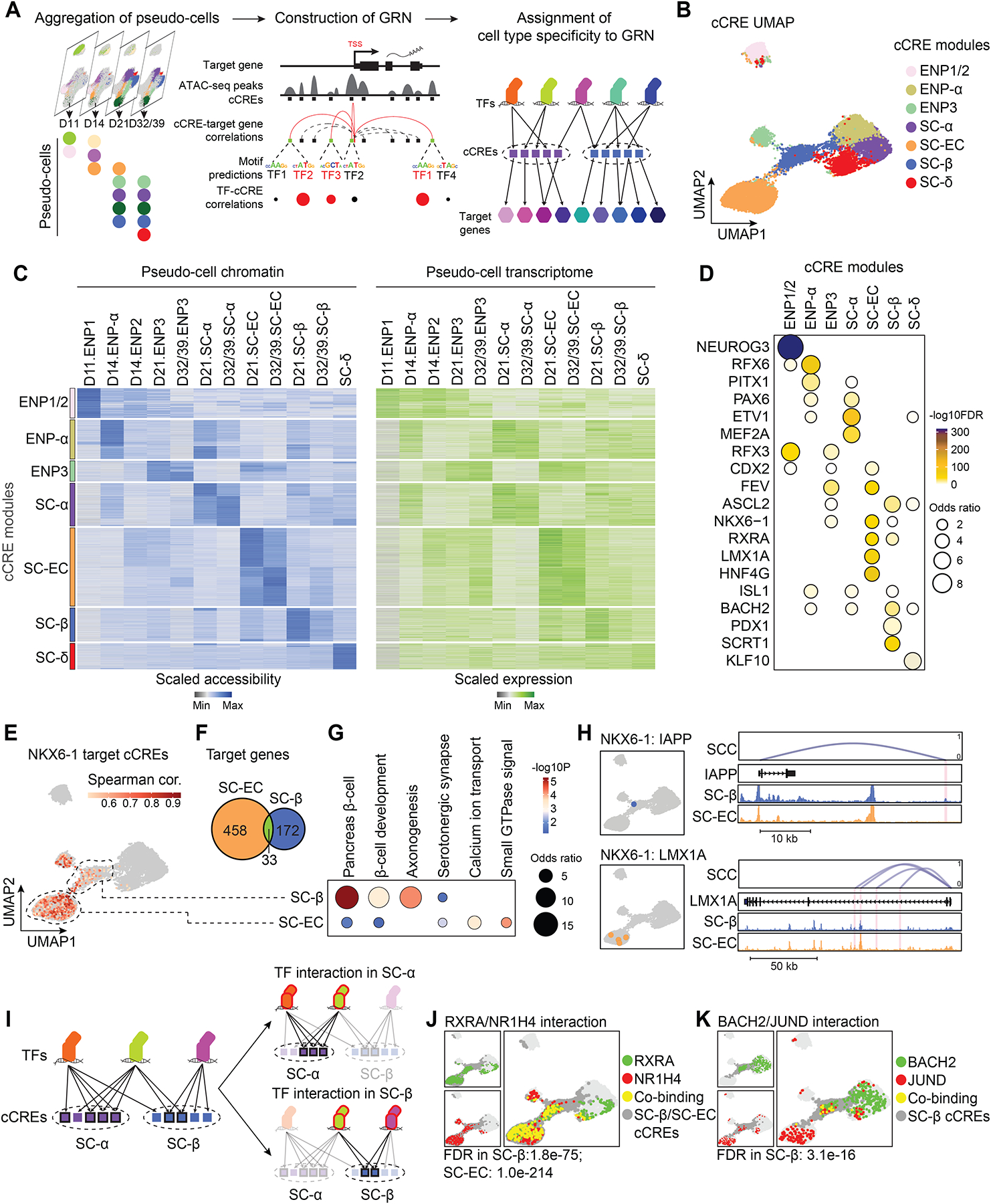
(A) Schematic of GRN inference framework and identification of cell type-specific transcriptional programs. cCRE, candidate cis regulatory element.
(B) Clustering of GRN cCREs highly variable across cell types and UMAP embedding. Cell identities were assigned to each cCRE module based on cell type with highest chromatin accessibility of the cCREs.
(C) Heatmaps showing scaled chromatin accessibility at cCREs (left) and expression levels (right) of target genes linked to the cCRE in each pseudo-cell.
(D) Dot plot showing enrichment of TFs predicted to bind to cCREs in each module against a background of all highly variable cCREs. Significance (−log10 FDR) and odds ratio of the enrichments are represented by color and dot size, respectively.
(E) UMAP projections of correlations between NKX6-1 expression and chromatin accessibility of predicted NKX6-1-bound cCREs. SC-β-cell- and SC-EC-specific cCRE modules are highlighted with dashed circles. Spearman cor., spearman correlation coefficient between NKX6-1 expression and cCRE accessibility.
(F) Venn diagram showing overlap between NKX6-1 target genes in SC-ECs and SC-β-cells. Cell type specificity of target genes was determined based on specificity of upstream cCREs. 33 genes are regulated by both SC-β-cell- and SC-EC-specific cCREs.
(G) Enriched gene ontology terms/pathways among SC-EC- or SC-β-cell-specific NKX6-1 target genes. Significance (−log10 p-value) and odds ratio of the enrichments are represented by color and dot size, respectively.
(H) UMAP locations (left) and genome browser snapshots (right) of predicted NKX6-1-bound cCREs at IAPP and LMX1A gene loci. Genome browser tracks show aggregated ATAC reads in SC-β-cells and SC-ECs. All tracks are scaled to uniform 1×106 read depth. SCC, spearman correlation coefficients for cCRE accessibility and target gene expression.
(I) Schematic of prediction method for cell type-specific TF interactions.
(J, K) UMAP projections of predicted TF-TF interactions. Green dots, cCREs bound by background TF; red dots, cCREs bound by test TF; yellow dots, cCREs co-bound by both TFs; dark grey dots, cCRE module(s) with predicted TF interaction(s).
The cell type-specific sub-GRNs allowed us to examine relationships between TFs in the regulation of individual genes as well as target gene specificity of individual TFs across different cell types. For example, we found that cCREs within the GCG locus were bound by PAX6 already in ENP-α whereas ETV1 bound to GCG cCREs mostly in SC-α-cells (Figure S2K), suggesting sequential actions of these TFs in GCG regulation during α-cell development. NKX6-1 and the retinoic acid X receptor A (RXRA) emerged as candidate regulators of SC-β-cells and SC-ECs; however, NKX6-1 bound to different cCREs and regulated different genes in SC-β-cells than in SC-ECs (Figure 2E,F and Figure S2L). Whereas NKX6-1 target genes in SC-β-cells were related to β-cell developmental and β-cell function, NKX6-1 controlled genes involved in serotonergic signaling in SC-ECs (Figure 2G and Table S2C), exemplified by NKX6-1 binding to cCREs in IAPP in SC-β-cells and cCREs in LMX1A, a TF controlling serotonin synthesis genes40, in SC-ECs (Figure 2H). These examples illustrate the power of this analysis for identifying distinct temporal and cell type-specific roles of individual TFs.
We further calculated the likelihood of cooperative gene regulation by TFs in each cell type (Methods; Figure 2I and Table S2D). We inferred ENP1-specific cooperativity between the known heterodimers NEUROG3 and TCF341 (Figure S2M) and cooperativity between MAFG and the cap’n’collar (CNC) family TF BACH2 in both SC-α-cells and SC-β-cells (Figure S2N), consistent with recruitment of CNC TFs by small MAF proteins42. RXRA was predicted to cooperate with the bile acid receptor NR1H4 in SC-β-cell and SC-EC gene regulation (Figure 2J), identifying a possible mechanism for the role of bile acids in insulin secretion43. Of interest is the SC-β-cell-specific interaction between BACH2 and JUND (Figure 2K), which contribute to β-cell dysfunction in type 2 diabetes44–46. Our GRN provides a resource for interrogating gene regulatory mechanisms in SC-islet cell types and their precursors.
SC-islet cell type lineage trajectories
The GRN identified candidate TFs involved in the specification of islet cell lineages. However, the analysis left unclear the order in which these TFs function to specify a lineage. To gain insight into temporal aspects of lineage specification, we ordered gene regulatory programs identified in the GRN along a lineage trajectory by assigning pseudotime values to a given TF, cCREs bound by the TF, and cCRE target genes (Methods; Figure S3A–I). Validating our method, cCREs with early activity in the lineage trajectory - reflected by low cCRE pseudotime values - projected to progenitor-specific cCRE modules in the cCRE UMAP (Figure 3A,B and Figure S3J).
Figure 3. Ordering of transcriptional programs along lineage trajectories.
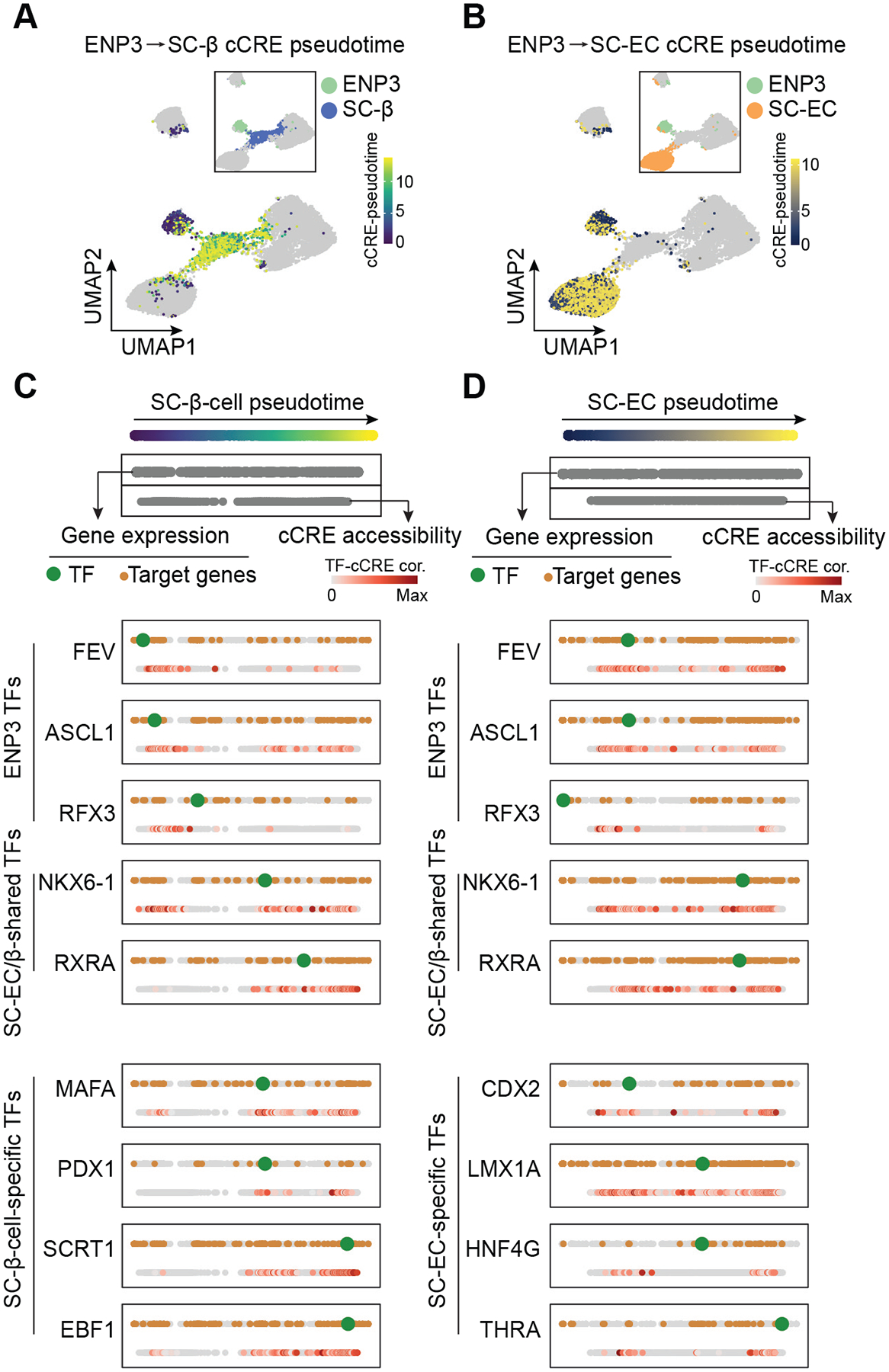
(A, B) UMAP projections of cCRE pseudotime on SC-β-cell (A) and SC-EC (B) lineage trajectories. Insets show cell type annotations of cCRE modules.
(C, D) Pseudotime ordering of transcriptional programs along SC-β-cell (C) and SC-EC (D) lineage trajectories from ENP3 progenitors. Gene expression and cCRE accessibility were assigned pseudotime values and plotted in two separate dotted lines (genes, top; cCREs, bottom). For each shown TF, the TF (green), TF-bound cCREs (colored based TF-cCRE correlations) and target genes (brown) are shown.
Integration of cCRE activity and gene expression into the pseudotemporal lineage trajectory (Figure 3C,D, Figure S3K, and Table S3), allowed us to quantify the temporal order of TF activity and their downstream target genes during SC-α-cell, SC-β-cell, and SC-EC development. In the SC-α-lineage trajectory, we identified ZNF414, NEUROG3, and PKNOX2 as the earliest TFs, followed by RFX6, PITX1, and PPARG in ENP-α progenitors and ETV1, PAX6, and MEF2C with later functions in SC-α-cell development (Figure S3K). While target genes of the early TFs ZNF414 and NEUROG3 remained expressed throughout the trajectory, PKNOX2 target genes were mostly transiently expressed (Figure S3K), suggesting distinct gene regulatory programs controlled by early α-cell lineage TFs. Analysis of SC-β-cell and SC-EC trajectories from ENP3 progenitors revealed FEV, ASCL1, and RFX3 as ENP3-active TFs with their target genes expressed throughout lineage development (Figure 3C,D). Later phase TFs in the SC-β-cell and SC-EC trajectories could be separated into two groups: (i) shared TFs between SC-β-cells and SC-ECs, exemplified by NKX6-1 and RXRA and (ii) cell type-specific TFs with MAFA, PDX1, SCRT1, and EBF1 regulating genes in SC-β-cells, and CDX2, LMX1A, HNF4G, and THRA regulating genes in SC-ECs. CDX2 was the earliest TF expressed during SC-EC development and CDX2-bound cCREs exhibited activity already in ENP3 progenitors (Figure 3D), suggesting an early role for CDX2 in SC-EC development.
Enterochromaffin cell-like resemble pre-β-cells in human fetal pancreas
Serotonin-producing SC-ECs are thought to be an erroneous cell type produced during SC-islet differentiation7,8. However, in both human fetal and adult pancreas, endocrine cells with serotonin granules have been reported using ultrastructural analysis47–49, raising the possibility that SC-ECs are a bona fide pancreatic endocrine cell type.
To test this, we assessed transcriptomic similarities between primary human fetal50 and SC-islet endocrine cells (Figure S4A,B). Interestingly, SC-ECs and SC-β-cells both co-localized with fetal β-cells on the UMAP (Figure 4A–C). To identify and characterize “EC-like” fetal β-cells, we isolated fetal β-cells and performed sub-clustering (Figure 4D). Among the five defined sub-clusters, fetal-β3 cells exhibited the highest expression of serotonin synthesis genes (Figure 4E). This population also expressed high levels of endocrine progenitor-characteristic TFs (e.g., FEV, PAX4, NEUROG3) and CDX2 (Figure 4E), suggesting fetal-β3 cells are a serotonin-producing pre-β-cell population. Immunofluorescence staining against serotonin (5HT) confirmed the presence of serotonin-producing β-cells (5HT+/INS+/PDX1+) in human fetal, neonatal, and infant pancreas (Figure 4F). This population gradually declined after birth and was rare in childhood (Figure 4G,H), indicating that serotonin-producing β-cells are a transient developmental β-cell population with progenitor characteristics.
Figure 4. A transient fetal pancreatic pre-β-cell population resembles stem cell-derived enterochromaffin cells.
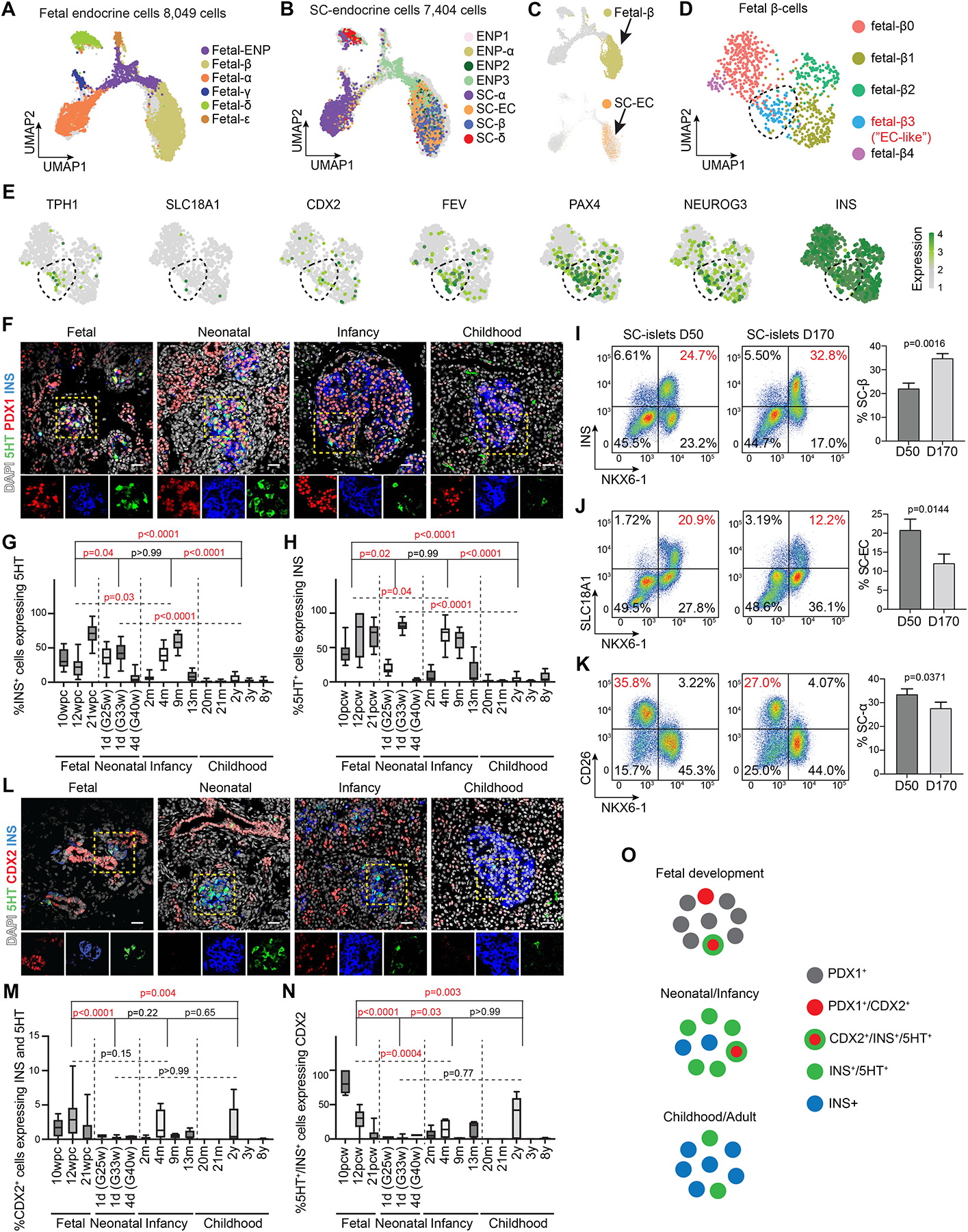
(A,B) UMAP co-embedding of single-cell transcriptomes from endocrine cells in fetal human pancreas (A) and during SC-islet differentiation (B). Cells are color-coded based on their annotated identities in Figure S4A and Figure 1B, respectively.
(C) Embedding of single-cell transcriptomes from fetal β-cells (top) and SC-ECs (bottom) on the same UMAP.
(D) UMAP embedding of fetal β-cells from the mSTRT-seq data. β-cell subclusters were defined by transcriptome similarities.
(E) Gene expression for fetal-β3 cell marker genes.
(F) Representative immunofluorescent images for 5HT, PDX1, and insulin (INS) on human pancreas at indicated development stages. Nuclei were labeled with DAPI. Scale bar, 20 μm.
(G,H) Quantification of INS+ cells expressing 5HT (G) and 5HT+ cells expressing INS (H) in fetal (10–21 wpc, n > 7 from each donor), neonatal (1–4 days postnatally, n > 6 from each donor, gestational week at birth is shown in the brackets), infant (2–13 months postnatally, n > 6 from each donor) and childhood (20 months to 8 years, n > 6 from each donor) human pancreas. Data are shown as mean ± S.D. Replicates (n) were obtained from randomly selected imaging regions. P-values were calculated using Tukey’s multiple comparisons test after one-way ANOVA.
(I-K) Representative flow cytometry plots (left, percentage of population of interest in red) and quantifications (right) of SC-β-cells (NKX6-1+/INS+, G), SC-ECs (NKX6-1+/SLC18A1+, H) and SC-α-cells (NKX6-1−/CD26+, I) in early (day (D) 50) and late (D170) SC-islet cultures. Data are shown as mean ± S.D. (n = 3 independent differentiations). P-values were calculated by unpaired two-tailed t-test.
(L) Representative immunofluorescent images for 5HT, CDX2, and insulin (INS) on human pancreas at indicated development stages. Nuclei were labeled with DAPI. Arrowheads in insets indicate CDX2 and INS co-positive cells. Scale bar, 20 μm.
(M,N) Quantification of CDX2+ cells expressing 5HT and INS (M) and 5HT+/INS+ cells expressing CDX2 (N) in samples from (G,H). Data are shown as mean ± S.D. Replicates (n) were obtained from randomly selected imaging regions. P-values were calculated using Tukey’s multiple comparisons test after one-way ANOVA.
(O) Illustration of CDX2 and 5HT expression in EC-like pre-β-cell.
See also Figure S4.
Long-term SC-islet culture induced β-cell functional maturation (Figure S4C), akin to postnatal β-cell maturation11–16. It also led to a decrease in the percentage of SC-ECs (NKX6-1+/SLC18A1+), an increase in SC-β-cells (NKX6-1+/INS+) but no change in SC-α-cell abundance (NKX6-1−/CD26+51; Figure 4I–K). Likewise, scRNA-seq data7 revealed a decrease in SC-ECs after long-term SC-islet culture and SC-islet engraftment (Figure S4D,E). Thus, like fetal serotonin-producing β-cells, SC-ECs are a transient, β-cell-related population.
Serotonin-producing fetal β-cells expressed CDX2 and endocrine progenitor cell markers (Figure 4E). We analyzed CDX2 expression together with PDX1 and 5HT in fetal and postnatal pancreas. In fetal pancreas, CDX2 was co-expressed with PDX1 in ductal pancreatic progenitors (Figure S4F,G). Among fetal CDX2+ cells, ~3% expressed insulin and 5HT, but CDX2+ cells co-expressing insulin and 5HT became rare postnatally (Figure 4L,M). In early fetal development, most serotonin-producing β-cells expressed CDX2; however, the percentage expressing CDX2 decreased at later fetal stages and remained low after birth (Figure 4N). Supporting the similarity between serotonin-producing fetal β-cells and SC-ECs, CDX2 was expressed in SC-derived pancreatic progenitors and SC-ECs and SC-EC CDX2 expression decreased during SC-islet differentiation (Figure S4H–J).
Collectively, these findings identify a transient, CDX2+ serotonin-producing β-cell population in the human fetal pancreas (Figure 4O) that resembles SC-ECs produced during SC-islet differentiation.
CDX2 regulates serotonin synthesis genes
The GRN suggested that CDX2 is critical for gene regulation in endocrine progenitors and SC-ECs (Figure 2D). Candidate CDX2 target genes included serotonin pathway genes, such as tryptophan hydroxylase (TPH1) and the serotonin transporter SLC18A1. Both genes were expressed in endocrine progenitors and serotonin-producing fetal β-cells as well as in SC-derived ENP3 and SC-ECs (Figure 4E, Figure 5A, and Figure S5A). At the TPH1 locus, CDX2 bound to a distal cCRE active in SC-ECs and fetal endocrine cells52 but not in SC-β-cells or fetal pre-ductal/endocrine and pre-acinar cells (Figure 5B and Figure S5B). A similar chromatin activity pattern was found at the CDX2-bound SLC18A1 promoter (Figure 5C and Figure S5C). Thus, serotonin synthesis genes are expressed in a subset of human fetal endocrine progenitors and β-cells and these genes are predicted to be CDX2-regulated.
Figure 5. CDX2 regulates serotonin synthesis genes.
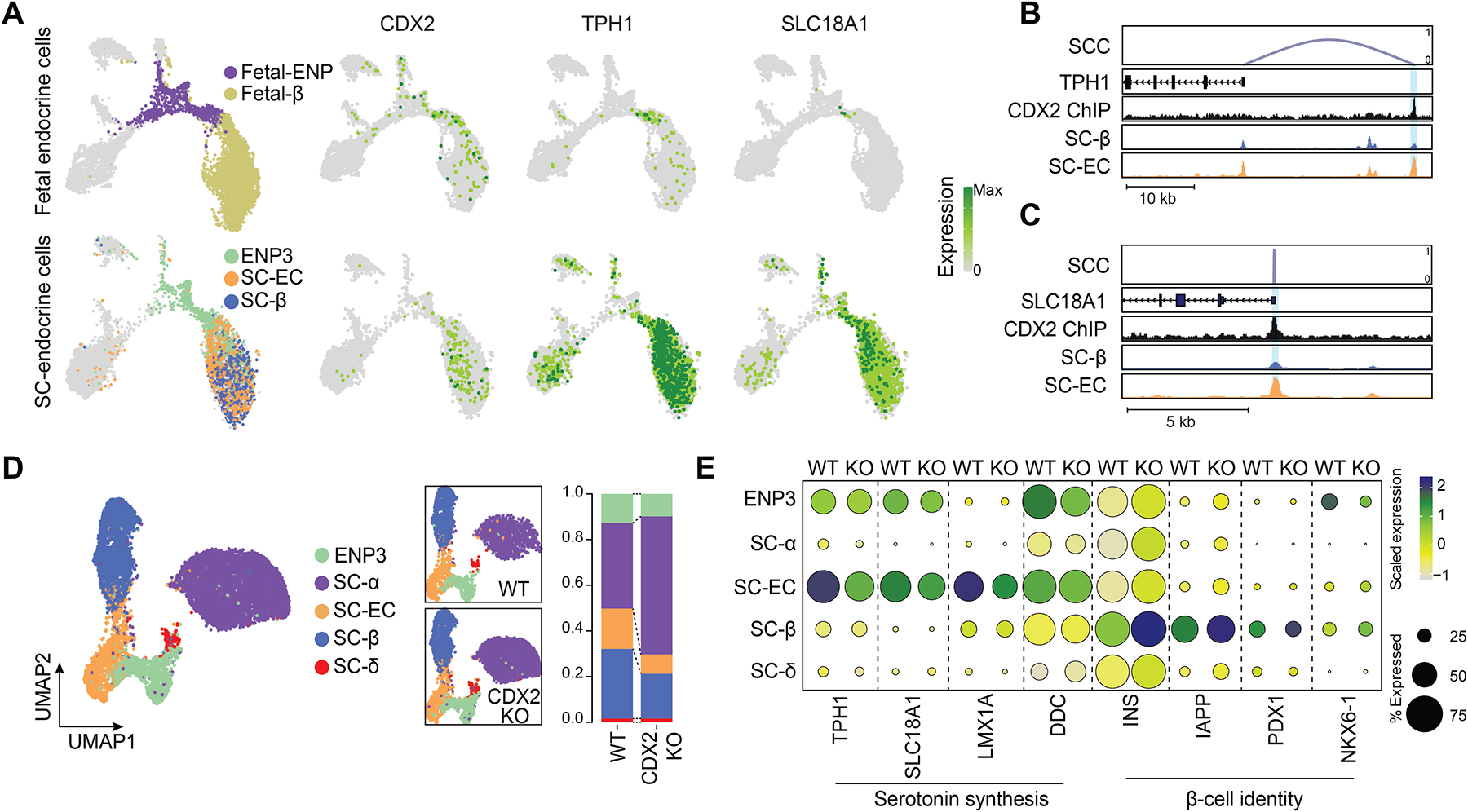
(A) Expression of CDX2, TPH1, and SLC18A1 in fetal (top) or stem cell-derived (bottom) endocrine cells. UMAPs on left indicate location of relevant cell types.
(B, C) Genome browser tracks showing CDX2 ChIP-seq reads in SC-islets and aggregated ATAC reads in SC-β-cells and SC-ECs at TPH1 (B) and SLC18A1 (C) gene loci. CDX2-bound cCREs are highlighted. All tracks are scaled to uniform 1×106 read depth. SCC, spearman correlation coefficients for cCRE accessibility and target gene expression.
(D) UMAP co-embedding of single cell transcriptomes from wild type (WT) and CDX2 knockout (KO) SC-islets. Cells are color-coded based by transferred identities from Figure 1b. The relative abundance of each cell type in WT and CDX2 KO SC-islets is shown on the right.
(E) Dot plot showing differentially expressed genes in WT and CDX2 KO SC-islet cell types. The color of each dot represents the expression level and the size the percentage of cells expressing the gene.
To examine CDX2 function during islet cell development, we deleted CDX2 in hESCs (CDX2-KO line) using CRISPR/Cas9-mediated genome editing (Figure S5D–F). We differentiated CDX2-KO hESCs and unedited wildtype (WT) hESCs into SC-islets and quantified endocrine cell type composition by flow cytometry and image analysis. CDX2 inactivation led to a significant reduction in SC-ECs, a slight reduction in SC-β-cells, but no change in SC-α-cells (Figure S5G–L). Single-cell RNA-seq analysis confirmed lower numbers of SC-ECs in CDX2-KO SC-islets and revealed a decrease in SC-β-cells and increase in SC-α-cells (Figure 5D), suggesting that CDX2 controls the lineage decisions at the SC-α-cell and SC-ECs/β-cell branchpoint. The discrepancy between effects of CDX2 deletion on cell type composition based on marker proteins and scRNA-seq likely reflects marker proteins only capturing a small aspect of cell identity.
Furthermore, expression of serotonin synthesis genes (TPH1, SLC18A1, LMX1A, DDC) was reduced in ENP3 endocrine progenitors and SC-ECs in the CDX2-KO, whereas β-cell identity genes (INS, IAPP, PDX1, NKX6-1) were more highly expressed in SC-ECs and SC-β-cells in the CDX2-KO (Figure 5E, Figure S5M, and Table S4). These findings suggest that CDX2 favors SC-EC over SC-β-cell identity. Interestingly, NKX6-1 expression was lower in CDX2-deficient ENP3 (Figure 5E) and the NKX6-1+ population decreased after CDX2 deletion (Figure S5I,J), possibly reflecting NKX6-1 regulation by CDX2 at an early phase of β-cell development in endocrine progenitors.
Together, our analysis suggests that CDX2 is transiently expressed in a bona fide fetal pre-β-cell population and that CDX2 regulates serotonin synthesis genes in pre-β-cells. Serotonin production is a feature of neonatal and adolescent β-cells53,54, and adult β-cells can activate serotonin synthesis during pregnancy55–58. Based on this evidence, we posit that SC-ECs are not an erroneous intestinal cell type of SC-islet differentiation.
Insufficient activation of signal-dependent gene regulatory programs in SC-islets
Next, we sought to determine how closely gene regulatory programs of SC-derived endocrine cells resemble those of corresponding cell types in postnatal pancreas. Toward this goal, we generated snATAC-seq, scRNA-seq, and single-nucleus RNA-sequencing (snRNA-seq) datasets from primary islets and pancreas from childhood (ages 13 months to 9 years) and adult donors (ages 20–66) complemented by publicly available islet scRNA-seq data59,60 (Table S1A,B). The inclusion of snRNA-seq data from frozen pancreas mitigated artifacts owing to induction of stress-response genes by the islet isolation procedure61,62.
To focus on endocrine cell types, we selected endocrine populations from each dataset (Figure 6A and Figure S6A–H) and integrated them into one UMAP for chromatin accessibility and gene expression, respectively (Figure 6B,C). In both types of data, we identified a single α-cell, δ-cell, and γ-cell cluster as well as β-cells comprised of four subclusters (Figure 6B,C and Figure S6I). Chromatin accessibility and gene expression data were highly concordant between clusters (Figure S6J) and cell type annotations in the integrated map largely corresponded to cell identities prior to data integration (Figure S6K,L). The α-cell and δ-cell clusters each comprised α-cells and δ-cells from stem cells, childhood, and adult pancreas, demonstrating similarity of SC-α- and SC-δ-cells with corresponding primary cells (Figure 6B,C and Figure S6K–M). A subset of SC-α-cells clustered with primary γ-cells in the integrated map (Figure 6B,C, dashed circles), consistent with developmental similarity between α-cells and γ-cells63. In contrast, SC-derived β-related cell types (ENP3, SC-β-cells and SC-ECs) clustered separately from primary β-cells (Figure 6B,C). This suggests that SC-β-cells are more distant to their primary counterparts than other SC-derived endocrine cell types, supported by correlation analysis of the transcriptomes (Figure S6N).
Figure 6. Insufficient activation of signal-dependent gene regulatory programs in stem cell β-cells.
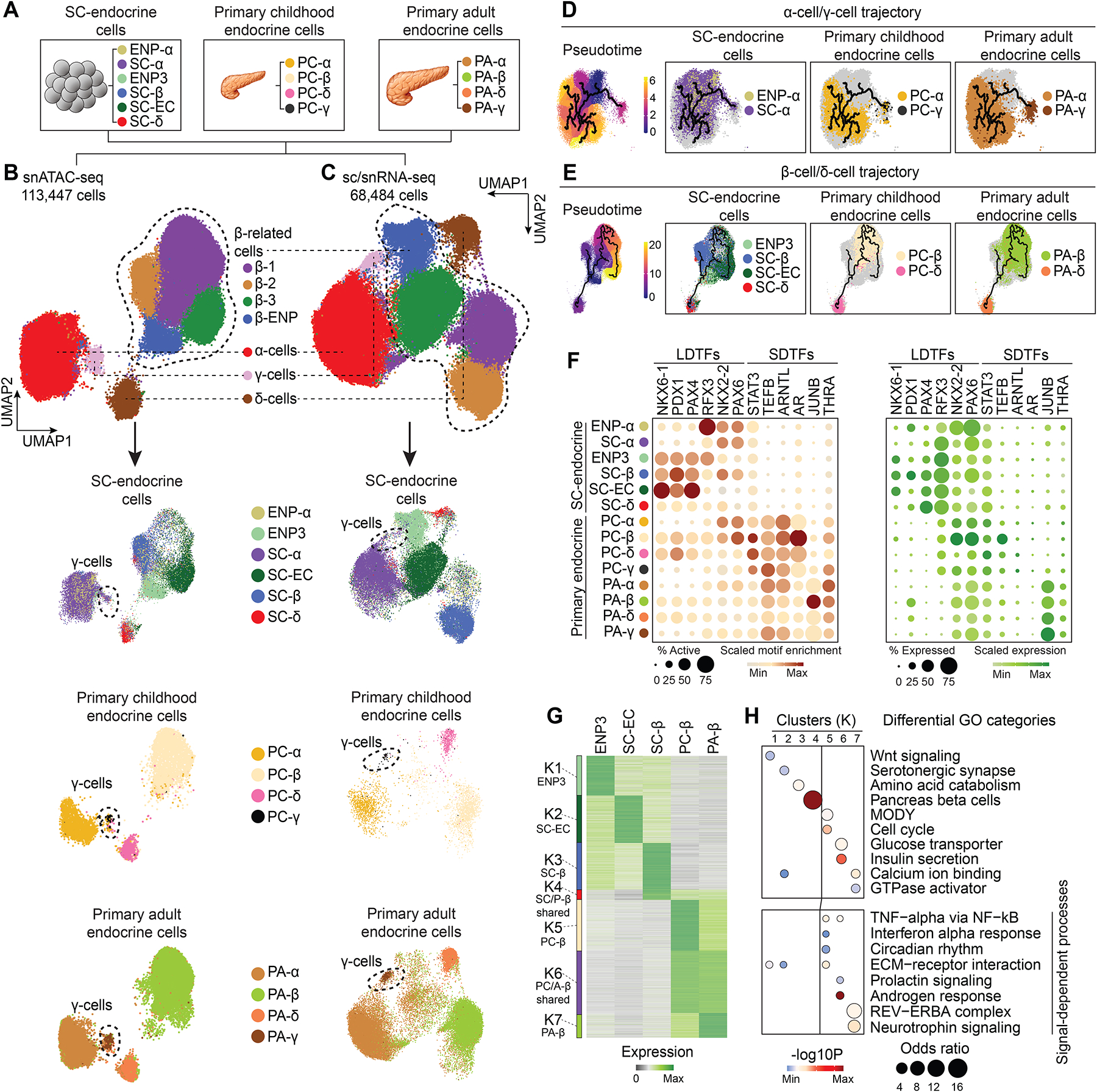
(A) Schematic showing cell types included into the integrative analysis of snATAC-seq and sc/snRNA-seq data.
(B, C) UMAP embedding of chromatin accessibility (B) and transcriptome (C) data from cell types detailed in (A). Cluster identities were defined by promoter accessibility (snATAC-seq) or expression (sc/snRNA-seq) of marker genes. The dashed line outlines β-cell-related cell types. Bottom panels: split UMAPs showing localization of stem cell, childhood and adult pancreatic endocrine cells. Cells were color-coded based on their identities from (A).
(D, E) Trajectory analysis based on chromatin accessibility, showing trajectories for α-cells/γ-cells (D) and β-cells/δ-cells (E) with ENP-α and ENP3 set as the root, respectively. Cells were color-coded by either original identities (A) or pseudotime values.
(F) Dot plots showing scaled average motif enrichment (left) or gene expression (right) of TFs. The color of each dot represents the average motif enrichment or expression level and the size of each dot the percentage of positive cells for each TF. LDTF, lineage-determining TF; SDTF, signal-dependent TF.
(G) K-means clustering of genes with variable expression across β-related cell types (ENP3, SC-ECs, SC-β-cells, PC-β-cells, and PA-β-cells). Clusters were annotated and color-coded based on gene expression patterns.
(H) Enriched gene ontology terms/pathways in each cluster. Significance (−log10 p-value) and odds ratio of the enrichments are represented by color and dot size, respectively.
We further analyzed the relatedness of SC-islet cells to primary endocrine cells by inferring lineage trajectories based on chromatin accessibility. We built two separate trajectories by grouping cell types with known lineage relationship63: (i) an α-cell/γ-cell trajectory with ENP-α, SC-α-cells, and primary childhood and adult α- and γ-cells (Figure 5D); and (ii) a β-cell/δ-cell trajectory with ENP3, SC-EC, SC-β-cells, SC-δ-cells, and primary childhood and adult β- and δ-cells (Figure 6E). In the α-cell/γ-cell trajectory, ENP-α progressed to SC-α-cells, to childhood α-cells, and finally to adult α-cells, suggesting immaturity of SC-α-cells but a correct differentiation path. In the β-cell/δ-cell trajectory, three trajectories each originated from ENP3 progenitors. One branch encompassed SC-δ-cells and primary δ-cells, one SC-β-cells, and a third primary β-cells with SC-ECs closely associated. This analysis confirms the relatedness of SC-ECs to β-cells, providing further support for SC-ECs resembling a unique β-cell state in human development.
To identify gene regulatory programs that distinguish primary from SC-derived endocrine cell types, we identified TF motifs with variable accessible chromatin enrichment across cell populations (Figure 6F and Table S5A). Motifs for lineage-determining TFs, such as NKX6-1, PDX1, NKX2–2, and PAX6, were enriched in SC-islet cell types, suggesting that gene regulatory programs driven by lineage-determining TFs are sufficiently active in SC-islet cells. By contrast, motifs for signal-dependent TFs were enriched in primary compared to SC-islet endocrine cell populations, consistent with lower expression of some of these TFs in SC-islet cells (Figure 6F). Signal-dependent TFs included STAT3 which is activated by signals from immune cells64,65, the circadian clock-dependent TF ARNTL23,66–68, and TFs activated by steroid hormones, such as the androgen receptor (AR) and thyroid hormone receptor (THRA)1,69,70. These findings suggest that SC-derived and primary endocrine cells are distinguished by insufficient activation of signal-dependent gene regulatory programs.
Next, we identified differentially expressed genes between related primary and SC-islet cell types and clustered them into gene modules based on their expression pattern (Figure 6G and Figure S6O,P). Genes in modules more highly expressed in primary β-cells than SC-β-related cells were associated with signaling pathways regulating β-cell function, including inflammatory, circadian, and neurotrophin signaling (Figure 6H and Table S5B). Signal-dependent genes were also lower expressed in SC-α-cells compared to primary α-cells (Figure S6Q and Table S5C). In addition, primary β-cells expressed higher levels of genes associated with insulin secretion, whereas genes involved in amino acid catabolism were more highly expressed in SC-β-cells (Figure 6H), consistent with a more pronounced insulin secretory response to amino acids in SC-β-cells compared to primary β-cells7,71. This phenocopies immature β-cells in newborn mammals, which utilize fat and amino acids as the major carbon source17,72,73. Together, these findings underscore that important signaling events are not sufficiently induced in SC-derived endocrine cells and suggest that insufficient activation of these signal-dependent processes could explain remaining functional differences between primary and SC-β-cells.
Steroid hormones stimulate β-cell proliferation
To comprehensively identify TFs and associated target genes with differential activity in primary and SC-β-cells, we constructed a GRN comprised of β-cell-related populations (approach see Figure 2A; Figure S7A–C and Figure S7F). We identified 377 TFs connected to 96,020 cCREs and 12,370 target genes. Each TF in the network bound an average of 2,698 cCREs, each cCRE regulated 1.55 genes, and each gene was regulated by 13.6 cCREs (Figure S7D,E,G).
We then subset the GRN by identifying cCRE modules specific to β-cell populations (ENP3, SC-EC, SC-β, and primary childhood and adult β-cells). In addition to population-specific modules, we identified cCRE modules shared between cell types, exemplified by a shared module between childhood and adult primary β-cells, SC-β-cells and primary β-cells, and SC-ECs and primary β-cells (Figure 7A and Figure S7H). The shared SC-β-cell/primary β-cell module lied between the SC-β-cell- and childhood β-cell-specific modules, suggesting that aspects of gene regulatory changes associated with β-cell maturation occur in SC-β-cells. Furthermore, presence of a SC-EC/primary β-cell module indicates that SC-ECs share gene regulatory features with primary β-cells, supporting their relatedness.
Figure 7. Gene regulatory network underlying β-cell maturation.
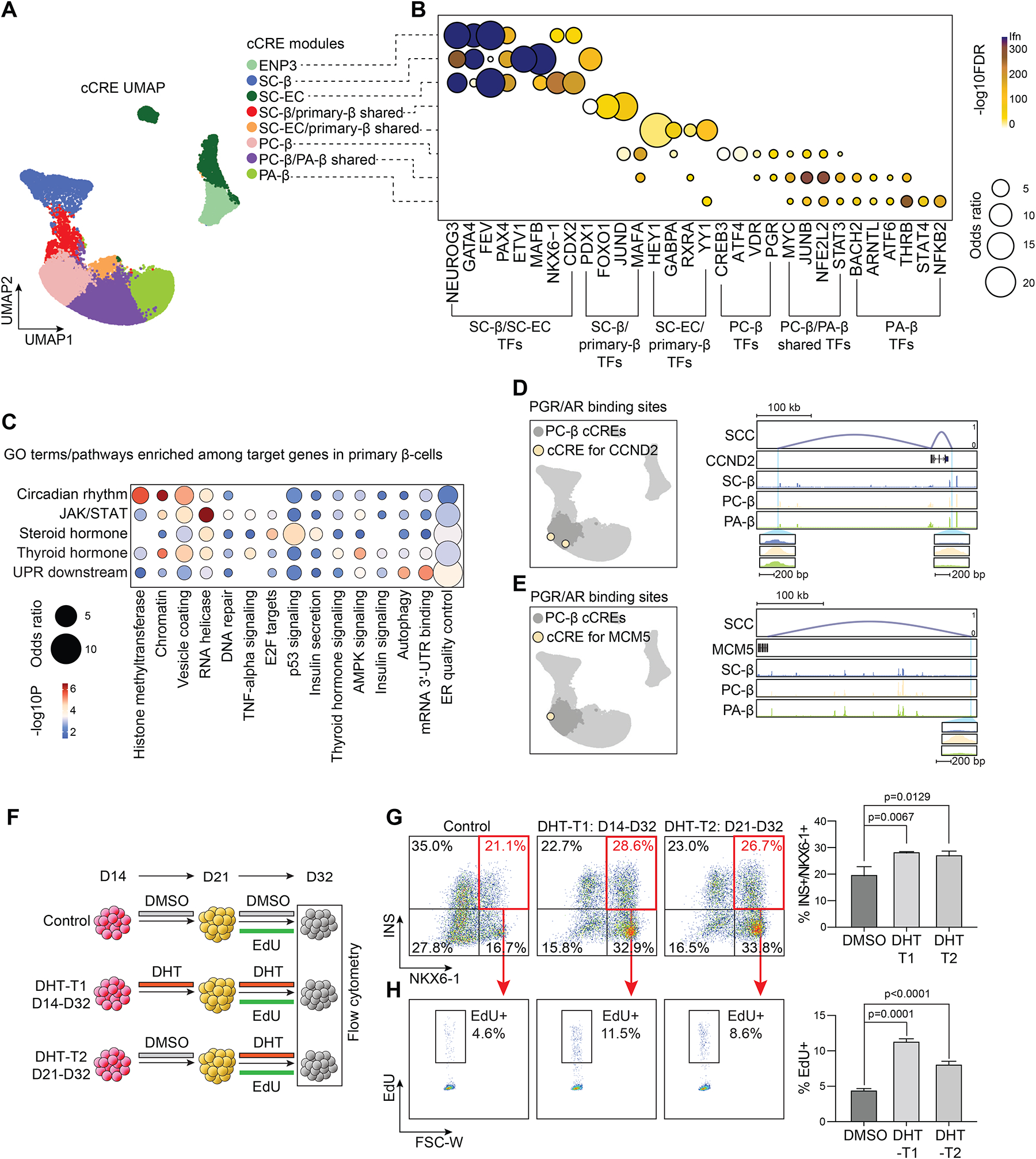
(A) Clustering of GRN cCREs highly variable across β-related cell types and UMAP embedding. Cell identities were assigned to each cCRE module based on cell type with highest chromatin accessibility of the cCREs. ENP, endocrine progenitor; SC, stem cell; EC, enterochromaffin-like cell; PC, primary childhood; PA, primary adult.
(B) Dot plot showing enrichment of TFs predicted to bind to cCREs in each module. Significance (−log10 FDR) and odds ratio of the enrichments are represented by color and dot size, respectively.
(C) Enriched gene ontology terms/pathways among target genes regulated by signals active in primary β-cells (PC-β and PA-β combined). Significance (−log10 p-value) and odds ratio of the enrichments are represented by color and dot size, respectively.
(D, E) UMAP locations (left) and genome browser snapshots (right) of predicted PGR/AR-bound cCREs at CCND2 (D) and MCM5 (E) gene loci. Genome browser tracks show aggregated ATAC reads in SC-β-cells, PC-β-cells, and PA-β-cells. PGR/AR-bound PC-β-cell-specific cCREs at CCND2 (D) and MCM5 (E) are highlighted. All tracks are scaled to uniform 1×106 read depth. SCC, spearman correlation coefficients for cCRE accessibility and target gene expression.
(F) Experimental design for dihydrotestosterone (DHT) treatment of SC-islets. EdU, nucleoside analogue 5-Ethynyl-2′-deoxyuridine.
(G) Representative flow cytometry plots (left, SC-β-cell percentage in red) and quantifications (right) of SC-β-cells (NKX6-1+/INS+) in D32 SC-islets with treatments shown in (F). Data are shown as mean ± S.D. (n = 3 independent differentiations). P-values were calculated by Dunnett’s multiple comparisons test after one-way ANOVA.
(H) Representative flow cytometry plots (left) and quantifications (right) of EdU+ SC-β-cells (NKX6-1+/INS+) in D32 SC-islets with treatments shown in (F). Data are shown as mean ± S.D. (n = 3 independent differentiations). P-values were calculated by Dunnett’s multiple comparisons test after one-way ANOVA.
Analysis of TFs with different activity across modules confirmed activation of programs downstream of lineage-determining TFs (e.g., FEV, PAX4, NKX6-1, CDX2, PDX1) and insufficient activation of programs regulated by signal-dependent TFs (e.g., PGR, VDR, STAT3, ARNTL, ATF6, THRA/B) in SC-derived β-cell populations (Figure 7B and Table S6A).
To catalog signal-dependent molecular processes insufficiently activated in SC-β-cells, we grouped signal-dependent TFs exclusively active in primary β-cells based on upstream signals regulating their activity and identified downstream target genes from the GRN. Primary β-cell-specific signaling pathways included circadian rhythm (ARNTL, NPAS2), interleukins (STAT1–4, STAT5A, STAT5B, STAT6), steroid hormones (AR, PGR, ESR1, ESR2), thyroid hormones (THRA, THRB), and the unfolded protein response (UPR; ATF6, ATF6B, ATF4) (Figure 7C and Table S6B). Validating the approach, thyroid hormone receptors were predicted to regulate genes involved in thyroid hormone signaling and TFs of the UPR genes involved in endoplasmic reticulum (ER) quality control (Figure 7C and Table S6B). The analysis predicted regulation of chromatin modifiers by circadian cues, suggesting a role for circadian signals in modulating the β-cell epigenome. Furthermore, identification of thyroid hormone as an upstream regulator of genes involved in AMPK signaling established a molecular link between thyroid hormone and AMPK signaling, which regulates β-cell maturation22,69,71. Consistent with temporarily distinct functions of THRA and THRB in murine β-cells74, the expression and activity of THRA and THRB differed between childhood and adult β-cells (Figure S7I–K). Furthermore, we found regulation of autophagy genes by UPR-activated TFs in β-cells, consistent with autophagy influencing β-cell function under ER stress75,76. The GRN provides a framework for understanding signal-dependent regulation of molecular processes in β-cells and identifies signal-dependent processes insufficiently activated during SC-β-cell differentiation.
The GRN predicted that steroid hormones regulate E2F target genes (Figure 7C and Table S6B), suggesting involvement of steroid hormones in β-cell proliferation. The childhood β-cell-specific module was enriched for genes regulated by the progesterone receptor (PGR) which shares a sequence motif with the androgen receptor (AR). We validated PGR/AR motif enrichment in childhood β-cells in H3K27ac ChIP-seq data from sorted childhood compared to adult human β-cells16 (Figure S7I). This finding indicates that sex hormones promote β-cell proliferation specifically during childhood. The GRN revealed the cell cycle genes CCND2 and MCM5 as targets of PGR/AR signaling in childhood β-cells (Figure 7D,E). To test whether AR receptor activation could induce β-cell proliferation, we treated SC-islets with dihydrotestosterone (DHT) during two different time windows of SC-islet differentiation and quantified relative β-cell numbers and proliferation rates (Figure 7F). During both treatment windows, DHT increased SC-β-cell numbers and proliferation assessed by EdU incorporation (Figure 7G,H). These results identify a role for AR signaling in β-cell proliferation, suggesting a connection between the surge in neonatal testosterone77,78 and early postnatal β-cell proliferation.
Discussion
It is still a major challenge to influence cell fate decisions during SC-islet differentiation and a roadmap for maturing in vitro-produced β-cells is missing. Here we integrated transcriptome and chromatin accessibility data from SC-islets and primary islets and inferred GRNs that describe cell type-specific gene regulatory programs. Our integrated GRN provides a framework for understanding gene regulatory mechanisms of islet cell fate acquisition and benchmarked gene regulatory programs of SC-islet cell types against those of primary islet cell types. This information provides a rich resource to design experiments for programming specific islet cell types and maturation states.
Previous work has described serotonin-producing cells during SC-islet differentiation7,8. Based on their similarity to intestinal enterochromaffin cells and absence of a similar cell type in adult islets, it was proposed that these cells lack a lineage relationship with β-cells8. We show that SC-ECs are similar to a transitory serotonin-producing pre-β-cell population in the human fetal pancreas, indicating that SC-ECs are not pancreas-aberrant. Serotonin-producing β-cells become rare later in development and are absent from adult pancreas. Likewise, SC-ECs decrease during in vitro maturation and after SC-islet engraftment. However, they persist even in prolonged culture and after engraftment8,10, indicating insufficient developmental progression of SC-ECs. A better understanding of the signals that trigger the transition from SC-ECs to SC-β-cells could help improve protocols for SC-β-cell production. Given that SC-ECs are a β-cell lineage intermediate, proposed depletion strategies8 might not be necessary for a SC-islet cell therapy.
Whether the identified pre-β-cell population represents a transitory state through which all progenitors progress or whether only a subset of adult β-cells arise from this population is still unclear. A subset of primary adult β-cells shares epigenomic features with SC-ECs (Figure S6K), which could indicate a distinct developmental origin of this β-cell subset. Lineage tracing studies will be necessary to determine the origin and fate of serotonin-producing pre-β-cells and the extent to which adult β-cell heterogeneity is developmentally determined. Another open question is whether reactivation of the fetal serotonin synthesis program during pregnancy55 is restricted to a subpopulation of β-cells or occurs in all β-cells.
Whereas β-cell differentiation occurs prenatally, the neonatal and early childhood period is characterized by the expansion, proliferation, and functional maturation of β-cells11–16. Postnatal changes in β-cells are thought to be driven by environmental cues20; however, the specific signals have remained poorly characterized. Our integrated GRN identified insufficient activation of circadian, JAK/STAT, steroid and thyroid hormone, as well as UPR signals in SC-β-cells. While circadian cues and thyroid hormone are known β-cell maturation signals23,69, the roles of the other signals remain to be studied. The GRN indicates that JAK/STAT-mediated regulation of stress response genes distinguishes primary from SC-β-cells, which could be due to the absence of islet-resident immune cells in SC-islets. Whether or not immune cells play a role in human β-cell maturation remains to be examined.
We identified sex hormone-mediated activation of proliferation genes as a program specific to childhood β-cells and showed that androgens stimulate SC-β-cell proliferation. Stimulation of sex hormone-dependent proliferation genes in β-cells could be linked to the neonatal testosterone surge77,78 or alternatively, be mediated by locally produced testosterone in islets79. Interestingly, a pro-proliferative effect of androgens has also been reported during neurogenesis in human brain organoids80, suggesting a shared mechanism between pancreatic and neuronal cells.
In summary, our GRN analysis provides a detailed understanding of the regulatory mechanisms defining SC-islet and primary islet cell types. The GRNs will be a valuable resource to inform strategies for producing precision cell therapy products.
Limitations of Study
One limitation of our study is the relatively small number of human pancreas samples we analyzed to characterize serotonin-producing pre-β-cells in primary pancreas and islets. Analysis of larger sample sizes should provide deeper insight into the population dynamics of pre-β-cells during human development, including the transition to β-cells. Furthermore, the cell type-specific GRNs in our study are based on the correlation between TF and target gene expression; however, TF activity can be regulated independently of TF expression levels.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Maike Sander (masander@ucsd.edu).
Material availability
CDX2 knockout H1 hESC line is available upon request.
Data and code availability
Single-nucleus ATAC sequencing (snATAC-seq), Single-nucleus RNA sequencing (snATAC-seq), Single-cell RNA sequencing (scRNA-seq), and CDX2 ChIP sequencing raw and processed data are available through the Gene Expression Omnibus under accession GSE202500. Other published datasets used in this study are summarized in Table S1. UCSC genome browser sessions of aggregated snATAC-seq, snRNA-seq, and scRNA-seq data are available at: https://genome.ucsc.edu/s/gaowei/hg19_islet.
Custom codes for main analysis used in this study have been deposited on GitHub: https://github.com/gaoweiwang/SCislet, and on Zendo. DOI is available in the Key Resource Table.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-insulin | DAKO | A0564 |
| Guinea pig anti-insulin | Invitrogen | PA1-26938 |
| Mouse pig anti-insulin | Abcam | ab9569 |
| Mouse anti-NKX6.1 | DSHB | F55A10 |
| Goat anti-PDX1 | R&D | AF2419 |
| Guinea pig anti-PDX1 | Abcam | ab47308 |
| Rabbit anti-CDX2 | Cell Signaling | 12306 |
| Rabbit anti-CDX2 | Abcam | ab76541 |
| Rabbit anti-SLC18A1 | Sigma | HPA063797 |
| Sheep anti-TPH1 | Sigma | AB1541 |
| Goat anti-5HT (serotonin) | Immunostar | 20079 |
| Goat anti-somatostatin | Santa Cruz | SC7819 |
| Mouse anti-glucagon | Sigma | G2654 |
| AlexaFluor® 647-conjugated anti-NKX6-1 | BD Biosciences | 563338 |
| PE-conjugated anti-insulin | Cell Signaling | 8508 |
| AlexaFluor® 488-conjugated donkey anti-rabbit IgG | Jackson Immunoresearch | 711-545-152 |
| Biotin-conjugated anti-CD26 | BioLegend | 302718 |
| Brilliant Violet 421™-conjugated Streptavidin | BioLegend | 405226 |
| Rabbit anti-CDX2 | Bethyl Laboratories | A300-691A |
| Biological Samples | ||
| Frozen childhood human pancreas | Pancreatic Organ Donors with Diabetes (nPOD) | HDL-052 HDL-067 HDL-077 HDL-015 HDL-019 HDL-021 |
| Frozen adult human pancreas | nPOD | 6229 6339 6366 6375 6479 6234 6401 |
| Isolated childhood human islets | ADI IsletCore | R394 |
| Fixed fetal human pancreas sections | MRC/Wellcome Trust-funded Human Developmental Biology Resource | N/A |
| Fixed fetal human pancreas sections | University of Washington Birth Defects Research Laboratory | N/A |
| Fixed neonatal human pancreas sections | nPOD | N/A |
| Fixed adult human pancreas sections | Prodo Labs | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Matrigel | Corning | 356238 |
| mTeSR1 media | Stem Cell Technologies | 85850 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140122 |
| Accutase | Thermo Fisher Scientific | 00-4555-56 |
| ROCK inhibitor Y-27632 | Stem Cell Technologies | 72307 |
| MCDB 131 medium | Thermo Fisher Scientific | 10372019 |
| NaHCO3 | Sigma | S6297 |
| GlutaMAX | Thermo Fisher Scientific | 35050061 |
| D-Glucose | Sigma | G8769 |
| Bovine Serum Albumin (BSA) | Lampire Biological Laboratories | 7500804 |
| Activin A | R&D Systems | 338-AC/CF |
| Wnt3a | R&D Systems | 5036-WN |
| L-Ascorbic Acid | Sigma | A4544 |
| FGF7 | R&D Systems | 251-KG |
| SANT-1 | Sigma | S4572 |
| Retinoic Acid | Sigma | R2625 |
| LDN193189 | Stemgent | 04-0074 |
| ITS-X | Thermo Fisher Scientific | 51500056 |
| TPB | Calbiochem | 565740 |
| T3 | Sigma | T6397 |
| ALK5i II | Cayman Chemicals | 14794 |
| ZnSO4 | Sigma | Z0251 |
| heparin | Sigma | H3149 |
| Gamma secretase inhibitor XX | Calbiochem | 565789 |
| Trace Element A | Corning | 89408-312 |
| Trace Element B | Corning | 89422-908 |
| MEM Non-Essential Amino Acids | Thermo Fisher Scientific | 11140076 |
| Dihydrotestosterone (DHT) | Sigma | D-073 |
| Hoechst 33342 | Invitrogen | H3570 |
| Horse serum | Invitrogen | 16050130 |
| Triton X-100 | Sigma | T8787 |
| Pierce Protease Inhibitor | Fischer | PIA32965 |
| DTT | Sigma | D9779 |
| Recombinant RNAsin RNase inhibitor | Promega | PAN2515 |
| EDTA | Invitrogen | 15575020 |
| DRAQ7 | Cell Signaling | 7406 |
| Critical Commercial Assays | ||
| XtremeGene 9 transfection reagents | Roche | 6365787001 |
| Cytofix/Cytoperm™ Plus Fixation/Permeabilizatio n Solution Kit | BD Biosciences | AB_2869009 |
| Click-iT™ EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit | Thermo Fisher | C10420 |
| VECTASHIELD® mounti ng media | Vector Laboratories | H-1300 |
| Dako Fluorescent Mounting Medium | Dako | S3023 |
| Tissue-Tek® O.C.T. Sakura® Finetek compound | VWR | 25608-930 |
| Superfrost Plus® Microscope Slides | Thermo Fisher | 22-037-246 |
| TSA blocking buffer | Perkin Elmer | NEL 701001KT |
| STELLUX® Chemi Human C-peptide ELISA | ALPCO | 80-CPTHU-CH01 |
| RNeasy Micro kit | QIAGEN | 74004 |
| iScript™ cDNA Synthesis Kit | Bio-Rad | 1708891 |
| iQ™ SYBR® Green Supermix | Bio-Rad | 1708880 |
| ChIP-IT High-Sensitivity kit | Active Motif | N/A |
| 10X Next GEM Single Cell 3’ v3.1 | 10X Genomics | PN-1000121 |
| 10X Chromium Chip E Single Cell ATAC Kit | 10X genomics | 1000086 |
| 10X Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.0 | 10X genomics | 1000175 |
| 10X Chromium Next GEM Chip H Single Cell Kit | 10X genomics | 1000161 |
| Deposited Data | ||
| snATAC-seq of SC-islet differentiation | Gene Expression Omnibus | GSE202500 |
| sc/snRNA-seq of SC-islet differentiation | Gene Expression Omnibus | GSE202500 |
| snATAC-seq of primary childhood human pancreas or pancreatic islet | Gene Expression Omnibus | GSE202500 |
| sc/snRNA-seq of primary childhood human pancreas or pancreatic islet | Gene Expression Omnibus | GSE202500 |
| CDX2 ChIP-seq of D21 SC-islet | Gene Expression Omnibus | GSE202500 |
| scRNA-seq of primary adult human pancreatic islets | Gene Expression Omnibus | GSE114297 |
| scRNA-seq of primary adult human pancreatic islets | HPAP | See Table S1A |
| snATAC-seq of primary fetal human pancreas | Domcke et al., 202052 (https://descartes.brotmanbaty.org/) | N/A |
| scRNA-seq of primary fetal human pancreas | OMIX | OMIX236 |
| Experimental Models: Cell Lines | ||
| Human: H1 ESC | WiCell Research Institute | WA01 |
| Oligonucleotides | ||
| List of primers used in this paper | See Table S7 for details | N/A |
| Recombinant DNA | ||
| PX458 | Addgene | 48138 |
| Software and Algorithms | ||
| HALO image analysis | Indica Lab | N/A |
| FlowJo V10 | FlowJo LLC. | N/A |
| GraphPad Prism (v8.1.2) | Dotmatics | N/A |
| R (v3.6.1) | CRAN | N/A |
| Cell Ranger ATAC v1.1.0 | 10X Genomics | N/A |
| Cell Ranger RNA v.3.0.2 | 10X Genomics | N/A |
| Scanpy (v.1.6.0) | Wolf et al., 201881 | N/A |
| Seurat | Stuart et al., 201982 | N/A |
| Monocle3 | Qiu et al., 201783 | N/A |
| Cicero | Pliner et al., 201884 | N/A |
| Enrichr | Kuleshov et al., 201685 | N/A |
| Bowtie2 | Langmead and Salzberg, 201286 | N/A |
| SAMtools | Li et al., 200987 | N/A |
| DeepTools | Ramirez et al., 201488 | N/A |
| MACS2 | Zhang et al., 200889 | N/A |
| chromVAR | Schep et al., 201790 | N/A |
| Custom codes | Zendo | DOI:10.5281/zenodo.7694211 |
Any additional information required to reanalyse the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human pancreata and pancreatic islets
Single-cell genomic assays were performed on snap frozen pancreas tissue or isolated islets obtained from 18 adult (20 to 61 years old) and 7 childhood (13-months to 9 years old) non-diabetic donors (HbA1c ≤ 5.6) through multiple sources including: Network for Pancreatic Organ Donors with Diabetes (nPOD), Integrated Islet Distribution Program (IIDP) and Alberta Diabetes Institute (ADI) IsletCore (see Table S1). Islet preparations were further enriched using zinc-dithizone staining followed by hand picking, and either directly processed for single-cell RNA sequencing (scRNA-seq) or snap frozen with liquid nitrogen or dry ice. Cryosections of fixed neonatal human pancreas were obtained from nPOD. Fixed human fetal pancreatic tissue samples were provided by the MRC/Wellcome Trust-funded Human Developmental Biology Resource (HDBR; https://www.hdbr.org; stages CS20, 10, 12, 20 and 21 wpc; gender not established) and by the University of Washington Birth Defects Research Laboratory (stages 13, 18, 19 wpc, gender not established). Lightly paraformaldehyde (PFA)-fixed pancreatic tissue from neonatal (1 and 4 days after birth), infant (2, 4, 13 months after birth), and childhood (20, 21 months, and 2, 3, 8 years old) stages was obtained for immunostaining through partnership with the International Institute for Advancement of Medicine (IIAM) as part of the Human Atlas of the Neonatal Development and Early Life Pancreas (HANDEL-P) program. Adult human pancreas tissue for immunostaining was obtained from Prodo Labs. All human tissues were obtained from de-identified donors, and protocols used in this study were approved by Institutional Review Board (IRB, protocol 091602XX) of the University of California San Diego or by the HDBR Steering Committee to the Spagnoli laboratory at King’s College London, UK (License #200523). The HDBR is a Research Ethics Committee (REC) approved and HTA licensed tissue bank. The Vanderbilt University Institutional Review Board does not consider studies on de-identified human pancreatic specimens to qualify as human subject research. For all human samples, informed consent was obtained for use of human tissue in research.
Human cell culture experiments
hESC research was approved by the University of California, San Diego (UCSD), Institutional Review Board and Embryonic Stem Cell Research Oversight Committee (protocol 090165ZX).
METHOD DETAILS
Maintenance and differentiation of H1 hESCs
H1 hESCs (male) were maintained as described by Geusz et al.91. In brief, hESCs were seeded onto Matrigel (Corning, 356238) coated tissue culture surfaces in mTeSR1 media (Stem Cell Technologies, 85850) supplemented with 1% Penicillin-Streptomycin (Thermo Fisher Scientific, 15140122), and propagated every 3 to 4 days. Accutase (Thermo Fisher Scientific, 00-4555-56) based enzymatic dissociation method was employed for passaging and 10 μM Y-27632 (Stem Cell Technologies, 72307) was supplied on the first day of each passage.
H1 hESCs were differentiated into SC-islets with a protocol we modified from previous publications by Rezania et al., Velazco-Cruz et al., and Hogrebe et al.1,4,6. After dissociation using Accutase, H1 cells were suspended with mTeSR1 media with %1 Penicillin-Streptomycin and 10 μM Y-27632 and plated using either a 3D culture or a 2D culture condition. For the 3D culture, cells were aggregated in 5.5mL medium at a concentration of 5.5 × 106 cells/well in a low attachment 6-well plate on an orbital shaker (100 rpm, 0.2 × g) in a 37 °C incubator. The following day (day 0), undifferentiated cells were washed in Stage 1/2 base medium (see below) and then differentiated using a seven-step protocol with stage-specific medium. Medium was refreshed daily until day 32. At day 8, the speed of the orbital shaker was increased to 110 rpm (0.3 × g). On day 21, cells were dissociated with Accutase, suspended in Stage 7 medium (see below) supplemented with 10 μM Y-27632 and re-aggregated at a concentration of 3 × 106 cells/well in a low attachment 6-well plate on an orbital shaker (100 rpm, 0.2 × g) in a 37 °C incubator. The speed of the shaker was increased to 110 rpm (0.3 × g) on the following day.
For a subset of experiments, a 2D differentiation protocol was used which is identical to the 3D protocol with the following exceptions: H1 hESCs cells were plated onto Matrigel coated tissue culture surfaces in base medium at a concentration of 5.7 × 105 cells/cm2. Stage 1 was extended to a total of 4 days (day 0–3). On day 29, cells were dissociated with Accutase, suspended in Stage 7 medium (see below) supplemented with 10 μM Y-27632, and re-aggregated at a concentration of 3 × 106 cells/well in a low attachment 6-well plate on an orbital shaker (100 rpm, 0.2 × g) in a 37 °C incubator. The speed of the shaker was increased to 110 rpm (0.3 × g) on the following day.
Base medium for all stage-specific media was comprised of MCDB 131 medium (Thermo Fisher Scientific, 10372019) supplemented with NaHCO3 (Sigma, S6297), GlutaMAX (Thermo Fisher Scientific, 35050061), D-Glucose (Sigma, G8769), and BSA (Lampire Biological Laboratories, 7500804) using the following concentrations:
Stage 1/2 base medium: MCDB 131 medium, 1.5 g/L NaHCO3, 1X GlutaMAX, 10 mM D-Glucose, 0.5% BSA
Stage 3/4 base medium: MCDB 131 medium, 2.5 g/L NaHCO3, 1X GlutaMAX, 10 mM D-glucose, 2% BSA
Stage 5/6 base medium: MCDB 131 medium, 1.5 g/L NaHCO3, 1X GlutaMAX, 20 mM D-glucose, 2% BSA
Stage 7 base medium: MCDB 131 medium, 1.5 g/L NaHCO3, 1X GlutaMAX, 2% BSA
Media compositions for each stage were as follows:
Stage 1 (days 0–2 for 3D culture and days 0–3 for 2D culture): base medium, 100 ng/mL Activin A (R&D Systems, 338-AC/CF), 25 ng/mL Wnt3a (R&D Systems, 5036-WN, only on day 0).
Stage 2 (days 3–5 for 3D culture and days 4–6 for 2D culture): base medium, 0.25 mM L-Ascorbic Acid (Sigma, A4544), 50 ng/mL FGF7 (R&D Systems, 251-KG)
Stage 3 (days 6–7 for 3D culture and days 7–8 for 2D culture): base medium, 0.25 mM L-Ascorbic Acid, 50 ng/mL FGF7, 0.25 μM SANT-1 (Sigma, S4572), 1 μM Retinoic Acid (Sigma, R2625), 100 nM LDN193189 (Stemgent, 04-0074), 1:200 ITS-X (Thermo Fisher Scientific, 51500056), 200 nM TPB (Calbiochem, 565740)
Stage 4 (days 8–10 for 3D culture and days 9–11 for 2D culture): base medium, 0.25 mM L-Ascorbic Acid, 2 ng/mL FGF7, 0.25 μM SANT-1, 0.1 μM Retinoic Acid, 200 nM LDN193189, 1:200 ITS-X, 100 nM TPB
Stage 5 (days 11–13 for 3D culture and days 12–14 for 2D culture): base medium, 0.25 μM SANT-1, 0.05 μM RA, 100 nM LDN-193189, 1 μM T3 (Sigma, T6397), 10 μM ALK5i II (Cayman Chemicals, 14794), 10 μM ZnSO4 (Sigma, Z0251), 10 μg/mL heparin (Sigma, H3149), 1:200 ITS-X
Stage 6 (days 14–20 for 3D culture and days 15–21 for 2D culture): base medium, 100nM LDN193189, 1 μM T3, 10 μM ALK5i II, 10 μM zinc sulfate, 100 nM gamma secretase inhibitor XX (Calbiochem, 565789), 10 μg/ml heparin, 1:200 ITS-X
Stage 7 (from day 21 for 3D culture and from day 22 for 2D culture): base medium, 10 μM zinc sulfate, 10 μg/ml heparin, 1:1000 Trace Element A (Corning, 89408-312), 1:1000 Trace Element B (Corning, 89422-908), 1:100 MEM Non-Essential Amino Acids (Thermo Fisher Scientific, 11140076)
Dihydrotestosterone treatment
H1 hESCs were differentiated as described above. 10nM dihydrotestosterone (DHT, Sigma, D-073) was added daily to the differentiation medium starting from either day 14 (end of Stage 5) or day 21 (end of Stage 6). Methanol was used as vehicle control.
Generation of CDX2 KO H1 hESC line
To generate a homozygous CDX2 deletion H1 hESC line, sgRNAs targeting the first exons of CDX2 were cloned into PX458 (Addgene, 48138). The plasmid was transfected into H1 hESCs with XtremeGene 9 (Roche, 6365787001), and 24 h later 5000 GFP+ cells were sorted into a well of six-well plate using mTeSR1 medium supplemented with 10 μM Y-27632. Individual colonies that emerged within 7 days were subsequently transferred manually into 48-well plates for expansion, genomic DNA extraction, PCR genotyping, and Sanger sequencing. A clone with a homozygous five base pair deletion in the CDX2 coding sequence was selected. For control clones, the PX458 plasmid was transfected into H1 hESCs, and cells were subjected to the same workflow as H1 hESCs transfected with sgRNAs. Sequence of sgRNA oligos used to generate CDX2 KO hESCs and PCR primers used to amply DNA after CDX2 gene editing can be found in Table S7.
Flow cytometry analysis
Cell aggregates derived from hESCs were allowed to settle in microcentrifuge tubes and washed with PBS. Cell aggregates were incubated with Accutase® at 37 °C until a single-cell suspension was obtained. Cells were washed with 1 mL ice-cold flow buffer comprised of 0.2% BSA in PBS and centrifuged at 200 × g for 5 min. BD Cytofix/Cytoperm™ Plus Fixation/Permeabilization Solution Kit was used to fix and stain cells for flow cytometry according to the manufacturer’s instructions. Briefly, cell pellets were resuspended in ice-cold BD Fixation/Permeabilization solution (300 μL per microcentrifuge tube). Cells were incubated for 20 min at 4 °C. Cells were washed twice with 1 mL ice-cold 1X BD Perm/Wash™ Buffer and centrifuged at 4 °C and 200 × g or 5 min. Cells were resuspended in 50 μL ice-cold 1X BD Perm/Wash™ Buffer containing diluted antibodies, for each staining performed. Cells were incubated at 4 °C in the dark for 1–3 h. If a secondary antibody staining was required, cells were washed twice with 1 mL ice-cold 1X BD Perm/Wash™ Buffer and centrifuged at 4 °C and 200 × g for 5 min. Cells were resuspended in 50 μL ice-cold 1X BD Perm/Wash™ Buffer containing diluted secondary antibodies. For EdU incorporation assays coupled with the flow analysis, cells were stained for EdU prior to primary antibody staining. Cells were washed with 1.25 mL ice-cold 1X BD Wash Buffer and centrifuged at 200 × g for 5 min. Cell pellets were resuspended in 300 μL ice-cold flow buffer and analyzed in a FACS LSRFortessa™ system (BD Biosciences). Antibodies used were AlexaFluor® 647-conjugated anti-NKX6-1 (1:5 dilution, BD Biosciences 563338); PE-conjugated anti-insulin (1:50 dilution, Cell Signaling 8508); rabbit anti-human SLC18A1 (1:300 dilution, Sigma HPA063797); AlexaFluor® 488-conjugated donkey anti-rabbit IgG (1:1000 dilution, Jackson Immunoresearch 711-545-152); Biotin-conjugated anti-CD26 (1:500 dilution, BioLegend 302718); and Brilliant Violet 421™-conjugated Streptavidin (1:500 dilution, BioLegend 405226). Data were processed using FlowJo software v10.
Nucleoside analog (EdU) incorporation assay
Proliferation in SC-β-cells was assayed using Click-iT™ EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit following the manufacturer’s instructions with modifications. In brief, nucleoside analog, EdU (5-ethynyl-2′-deoxyuridine, 10 μM), was added daily to SC-islets starting from day 21 until day 32 of 3D culture. Labeled cells were dissociated, fixed, and permeabilized using the same procedures as described in “Flow cytometry analysis”, and visualized with Alexa Fluor® 488 azide through “click” chemistry. To detect SC-β-cell-specific EdU incorporation, cells were stained with AlexaFluor® 647-conjugated anti-NKX6-1 and PE-conjugated anti-insulin (see “Flow cytometry analysis”), and analyzed in a FACS LSRFortessa™. Data were processed using FlowJo software v10.
Immunofluorescence analysis
SC-islets were washed twice with PBS and fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature. Fixed samples were washed twice with PBS and dehydrated in 30% (w/v) sucrose in PBS at 4 °C overnight. The following day, samples were embedded with Tissue-Tek® O.C.T. Sakura® Finetek compound (VWR) in disposable embedding molds (VWR), and frozen in a dry ice-ethanol bath. Tissue blocks were sectioned at 10 μm and sections were placed on Superfrost Plus® (Thermo Fisher) microscope slides and washed twice with PBS for 10 min. On neonatal and adult human pancreas sections, immunostaining was performed as described previously by Brissova et al. and Saunders et al.92,93. In brief we performed antigen retrieval by boiling sections in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 20 min. Sections were then permeabilized with 0.1% (v/v) Triton X-100 (Sigma-Aldrich) for 30 min, and blocked with blocking buffer, consisting of 0.1% (v/v) Triton X-100 (Sigma-Aldrich) and 1% (v/v) normal donkey serum (Jackson Immuno Research Laboratories, Cat 017-000-121) in PBS for 1h at room temperature. Primary antibody incubation was conducted in the same blocking buffer at 4 °C overnight. The following day, sections were washed three times with PBS and stained with diluted secondary antibodies and Hoechst 33342 (Invitrogen, H3570) for 1h at room temperature. Stained sections were washed five times with PBS before mounting with VECTASHIELD® (Vector Laboratories, H-1300). Images were obtained with a Zeiss Axio-Observer-Z1 microscope equipped with a Zeiss ApoTome and AxioCam digital camera and quantified using HALO image analysis (Indica Lab). Fetal human pancreas sections processed in the Spagnoli laboratory at King’s College London were stained with a similar procedure but using slightly different reagents, including a citrate buffer solution (Dako) for antigen retrieval, a TSA blocking buffer [0.5% TSA blocking powder (Perkin Elmer, Cat NEL 701001KT), 10% horse serum (Invitrogen, Cat 16050130) in 0.1% Triton 1x PBS] and Dako Fluorescent Mounting Medium (Dako, Cat S3023). Images were acquired on Zeiss LSM 700 laser scanning microscope or on FV3000 confocal laser scanning microscope (Olympus).
Dynamic glucose-stimulated insulin secretion (GSIS) assay
GSIS assays were carried out at 37°C using the Biorep perifusion system (Biorep v5), which allows a dynamic exchange of Krebs-Ringers-Bicarbonate-HEPES (KRBH) buffer (130 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 1.2 mM KH2PO4, 20 mM HEPES pH 7.4, 25 mM NaHCO3, and 0.1% BSA) with high (16.8 mM) and low (2.8 mM) glucose concentrations. 30–50 hand-picked SC-islets were loaded into the perifusion chamber and equilibrated with low glucose KRBH for 1h. SC-islets were then stimulated with KRBH containing indicated concentration of glucose for indicated duration.
Perifusate was collected every minute, and samples from indicated time points were analyzed using STELLUX® Chemi Human C-peptide ELISA (ALPCO). SC-islets from perifusion chambers were transferred to microcentrifuge tubes and lysed by sonication for total C-peptide content measurement.
RNA extraction and qRT-PCR
Approximately 500 SC-islets were collected and washed before RNA isolation using the RNeasy Micro kit (QIAGEN) according to the manufacturer’s instructions. RT-qPCR was performed as previously described by Wortham et al.94. In brief, 500 ng for total RNA was converted to cDNA using iScript™ cDNA Synthesis Kit (Bio-Rad). Gene expression was quantified with iQ™ SYBR® Green Supermix (Bio-Rad). Primers used for qPCR were listed in Table S7.
Single-cell RNA-sequencing (scRNA-seq)
Differentiating aggregates and hand-pick human islets were collected in microcentrifuge tubes and washed with PBS. Accutase® was used to dissociate aggregates into single cells, which were then stained with propidium iodide (Sigma) in a PBS solution containing 0.2% BSA. Approximately 200,000 live cells (propidium iodide-negative) were sorted with a FACSAriaTM Fusion Flow Sorter at a sorting speed lower than 3,000 events per second to minimize damage to the cells. Sorted cells were pelleted with 250 × g for 5 minutes at 4°C, and counted with a Scepter™ automated cell counter. 10,000 accurately counted cells per sample were loaded onto a 10X Chromium Controller for GEM formation and cell barcoding using Next GEM Single Cell 3’ v3.1 reagents. Barcoded single cells were subjected to cDNA synthesis and sequencing library construction using 10X Next GEM Single Cell 3’ v3.1 reagents according to manufacturer’s instructions. Final libraries were quantified using a Qubit fluorimeter (Life Technologies) and the fragmented cDNA was verified using a Tapestation (High Sensitivity D1000, Agilent). Libraries were sequenced on NextSeq 500, HiSeq 4000 or NovaSeq 6000 sequencers (Illumina) and reads were trimmed afterwards to fit into corresponding analysis pipeline.
Single-nucleus RNA-sequencing (snRNA-seq)
Nuclei were isolated from approximately 1,000 differentiated aggregates (~1,000 cells per aggregate) or approximately 35 mg of frozen human pancreas using a nuclei permeabilization buffer [0.1% Triton X-100 (Sigma-Aldrich, T8787), 1X Pierce Protease Inhibitor (Fischer, PIA32965), 1 mM DTT (Sigma-Aldrich, D9779), Recombinant RNase inhibitor (0.2 U/μl; Promega, 2% Fatty-acid-free BSA in PBS (Proliant, 7500804; Corning, 21-040-CV)]. For differentiated aggregates, nuclei extraction was done in a glass dounce, and for frozen human pancreas, samples were pulverized and resuspended in the nuclei permeabilization buffer.
Samples were incubated on a rotator for 5 min at 4°C and then centrifuged at 500g for 5 min (Eppendorf, 5920R; 4°C, ramp speed of 3/3). Supernatant was removed and pellet was resuspended in sort buffer [1mM EDTA (Invitrogen, 15575020), 0.2U/μL Recombinant RNAsin (Promega, PAN2515), 1% Fatty-acid-free BSA in PBS (Proliant, 7500804; Corning, 21-040-CV) and stained with DRAQ7 (1:150; Cell Signaling Technology, 7406). 60,000 nuclei were sorted using an SH800 sorter (Sony) into 50 μl of collection buffer [1.0U/μL Recombinant RNAsin (Promega, PAN2515), 5% Fatty-acid-free BSA in PBS (Proliant, 7500804; Corning, 21-040-CV)]. Sorted nuclei were then centrifuged at 1000 g for 15 min (Eppendorf, 5920R; 4°C, ramp speed of 3/3), and supernatant was removed. Nuclei were resuspended in reaction buffer [RNase inhibitor (0.2U/μL Recombinant RNAsin (Promega, PAN2515), 1% Fatty-acid-free BSA in PBS (Proliant, 7500804; Corning, 21-040-CV) and counted using a hemocytometer.
16,550 nuclei were loaded onto a Chromium controller (10x Genomics). Libraries were generated using the Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1 (10x Genomics, PN-1000121) according to the manufacturer specifications. Complementary DNA was amplified for 12 PCR cycles. SPRISelect reagent (Beckman Coulter) was used for size selection and cleanup steps. Final library concentration was assessed by the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific), and fragment size was checked using TapeStation High Sensitivity D1000 (Agilent) to ensure that fragment sizes were distributed normally around 500 bp. Libraries were sequenced using a NextSeq 500 or NovaSeq 6000 (Illumina).
Single-nucleus ATAC-sequencing (snATAC-seq)
Nuclei extraction and sorting were done using the same methodology as described in “Single nucleus RNA-seq”. Single nucleus ATAC-seq libraries were generated using either the Chromium Chip E Single Cell ATAC Kit (10x Genomics, 1000086) or Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.0 (10x Genomics, 1000175) with Chromium Next GEM Chip H Single Cell Kit (1000161) following the manufacturer’s instructions. Indexes used were Chromium i7 Multiplex Kit N, Set A (10x Genomics, 1000084) and Single Index Kit N Set A (1000212)10x Genomics, 1000084), respectively. Final libraries were quantified using a Qubit fluorimeter (Life technologies) and the nucleosomal pattern was verified using a Tapestation (High Sensitivity D1000, Agilent). Libraries were sequenced on NextSeq 500, HiSeq 4000 or NovaSeq 6000 sequencers (Illumina) and reads were trimmed afterwards to fit into corresponding analysis pipeline.
Chromatin immunoprecipitation sequencing (ChIP-seq)
ChIP-seq was performed using the ChIP-IT High-Sensitivity kit (Active Motif) according to the manufacturer’s instructions. Briefly, from day 21 SC-islets 5–10 × 106cells were harvested and fixed on a rocker for 15 min in an 11.1% formaldehyde solution. The reaction was quenched for 5 min in 0.125 M glycine, cells washed in DPBS containing 0.5% NP-40, then once again in DPBS supplemented with 0.5% NP-40 and 1 mM PMSF. Cells were lysed by sonication with a Bioruptor® Plus (Diagenode), on high for 3 × 5 min (30 s on, 30 s off). 30 μg of the resulting sheared chromatin was used for each immunoprecipitation. Equal quantities of sheared chromatin from each sample were used for immunoprecipitations carried out at the same time. 6 μg anti-CDX2 antibody (A300-691A, Bethyl Laboratories) was used for the ChIP-seq assay. Chromatin was incubated with primary antibody overnight at 4 °C on a rotator followed by incubation with Protein G agarose beads for 3 h at 4 °C on a rotator. Reversal of crosslinks and DNA purification were performed according to the ChIP-IT High-Sensitivity instructions, with the modification of incubation at 65 °C for 2–3 h, rather than at 80 °C for 2 h. Sequencing libraries were constructed using KAPA DNA Library Preparation Kits for Illumina® (Kapa Biosystems) and library sequencing was performed on either a HiSeq 4000 System (Illumina®) or NovaSeq 6000 System (Illumina®) with single-end reads of either 50 or 75 base pairs (bp). Sequencing was performed by the UCSD Institute for Genomic Medicine (IGM) core research facility. For the ChIP-seq experiment, replicates from two independent hESC differentiations were generated.
ChIP-seq data analysis
Bowtie286 (v2.3.4.1) was used for mapping of raw data to the human reference genome hg19 with a maximum of 2 mismatches allowed in the seed region, discarding reads aligning to multiple sites. Duplicate reads were removed using SAMtools87. DeepTools88 was used to generate bigwig format tracks for visualization in UCSC Genome Browser. Peak calling was performed using MACS289 with default setting for TF ChIP-seq and ChIP-seq input as the background control.
Single-cell raw data processing and quality control
Data processing using Cell Ranger software
Alignment to the hg19 genome and initial processing were performed using the 10x Genomics Cell Ranger ATAC v1.1.0 and Cell Ranger RNA v.3.0.2 pipelines. Sample information and a summary of the Cell Ranger ATAC-seq and RNA-seq quality metrics are provided in Table S1.
Filtering barcode doublets and low-quality cells for each individual donor
Cell barcodes from the 10x Chromium snATAC-seq assay may have barcode multiplets that have more than one oligonucleotide sequence95. We used ‘clean_barcode_multiplets_1.1.py’ script from 10x to identify barcode multiplets for each donor and excluded these barcodes from further analysis. We then filtered low quality snATAC-seq profiles by total UMIs (<1,000), fraction of reads overlapping TSS (<15%), fraction of reads overlapping called peaks (<30%), and fraction of reads overlapping mitochondrial DNA (>10%) according to the distribution of these metrics for all barcodes. We also excluded profiles that had extremely high unique nuclear reads (top 1%), fraction of reads overlapping TSS (top 1%) and called peaks (top 1%) to minimize the contribution of these barcodes to our analysis. For RNA-seq data, we used total UMIs (<1,000) and fraction of reads overlapping mitochondrial DNA (>10%) to filter cells with low quality RNA profiles. We also excluded profiles that had extremely high total UMIs (top 1%) to minimize the contribution of these barcodes to our analysis.
Cell clustering
After filtering low quality cells, we checked ATAC and RNA data quality from each sample by performing an initial clustering using Scanpy (v.1.6.0)81. For ATAC-seq data, we partitioned the hg19 genome into 5 kb sliding windows and removing windows overlapping blacklisted regions from ENCODE96,97 (https://www.encodeproject.org/annotations/ENCSR636HFF/). Using 5 kb sliding windows as features, we produced a barcode-by-feature count matrix consisting of the counts of reads within each feature region for each barcode. Detailed pipeline to process ATAC-seq data can be found in our previous work by Chiou et al.98. We normalized each barcode to a uniform read depth and extracted highly variable features. Then, we regressed out the total read depth for each cell, performed PCA, and extracted the top 50 principal components to calculate the nearest 30 neighbors using the cosine metric, which were subsequently used for UMAP dimensionality reduction with the parameters ‘min_dist=0.3’ and Leiden99 clustering with the parameters ‘resolution=0.8’.
We then performed initial cell clustering for cells from all donors using similar methods to cluster cells for each donor. Of note, we extracted highly variable features across cells from all experiments. Since read depth was a technical covariate specific to each experiment, we regressed this out on a per-experiment basis. We also used Harmony100 to adjust for batch effects across experiments. We identified clusters and subclusters (‘resolution’=1.5) with significantly different total UMIs, fraction of reads overlapping TSS, or fraction of reads overlapping called peaks compared to other clusters and subclusters. We excluded these clusters and subclusters and obtained final cell clusters by performing cell clustering using identical methods for initial clustering of all cells. We determined the cell type represented by each cluster by examining chromatin accessibility at the promoter regions of known marker genes.
Generating fixed-width and non-overlapping peaks that represent cCREs across cell types
We called peaks for each cell type using the MACS2 call peak command with parameters ‘--nomodel --extsize 200 –shift 0 --keep-dup all -q 0.05’ and filtered these peaks by the ENCODE hg19 blacklist. For each cell type, we generated fixed-width peaks (summits of these peaks from macs2 were extended by 250 bp on either side to a final width of 501 bp), as previously described by Satpathy et al.101. We quantified the significance of these fixed-width peaks in each cell type by converting the MACS2 peak scores (−log10(Q value)) to a ‘score quantile’. Then, fixed-width peaks for each cell type were combined into a cumulative peak set. As there are overlapping peaks across cell types, we retained the most significant peak and any peak that directly overlapped with that significant peak was removed. This process was iterated to the next most significant peak and so on until all peaks were either kept or removed due to direct overlap with a more significant peak. These fixed-width and non-overlapping peaks were defined as candidate cis-regulatory element, or cCREs.
Differential gene expression analysis
We used generalized linear regression model (glm function in R) to call differential expressed genes between different cell types or states from snRNA-seq data. Expression level of genes were normalized by total count for individual cells. In addition to cell types or states annotation, we also considered total count of individual cells as covariate in the model to calculate coefficient and p value. Adjusted p values (FDRs) were obtained using p.adjust function in R with a Benjamini & Hochberg method. Differentially expressed genes were selected using FDR<0.05 as cutoff.
K-means clustering
For α-cells, we used the cell type-by-genes count matrix and differentially expressed genes between α-cells from SC-islets, childhood, and adult primary islets (FDR<0.05) as input. We normalized the expression level of genes using total counts and performed K-means clustering analysis using kmeans function in R. We then repeated the same procedure for β-cells.
Integrating snATAC-seq and sc/snRNA-seq data
We used the Seurat package82 to integrate single modality snATAC-seq and sc/sn RNA-seq datasets (https://satijalab.org/seurat/articles/atacseq_integration_vignette.html). Raw count matrices of snATAC-seq and sc/snRNA-seq data, as well as cell clustering results were loaded into the Seurat package as input. To integrate and establish connections between transcriptome (sc/sn RNA-seq) and accessible chromatin (snATAC-seq) profiles, we first inferred gene activity scores from snATAC-seq data using the GeneActivity function and performed log-normalization. Gene activity scores from snATAC-seq data and gene expression from sc/snRNA-seq data were then compared and linked using FindTransferAnchors function with a Canonical Correlation Analysis (CCA) based dimension reduction method. Using anchors identified in the CCA space, cluster identities and mRNA counts of the snRNA-seq dataset were transferred to cells in snATAC-seq datasets. We applied the procedure to integrate snATAC- and sc/snRNA-seq data of SC-islets and endocrine cells from SC-islets and primary endocrine cells from human pancreas.
Integrating SC-islet endocrine cells with primary human pancreatic endocrine cells
We performed separate integration analyses for snATAC-seq data and sc/snRNA-seq data obtained from SC-islet endocrine cells and primary human pancreatic endocrine cells. For snATAC-seq, processed matrices from Scanpy were imported into the “Signac” R package (https://satijalab.org/signac/articles/pbmc_vignette.html). Data from SC- and primary endocrine cells were merged and normalized using a frequency-inverse document frequency (TF-IDF) method and dimension reduction was performed using singular value decomposition (SVD) followed by latent semantic indexing (LSI). Cells from the two datasets were compared in the LSI space and integration anchors were identified using the FindIntegrationAnchors function, and integrated with those integration anchors using IntegrateData. For sc/snRNA-seq datasets, integration was performed following “Seurat” data integration instructions (https://satijalb.org/seurat/articles/integration_introduction.html). In brief, SC- and primary endocrine cells were imported into “Seurat” package from “Scanpy” with original dimension reductions (PCA and UMAP) remaining the same. Integration anchors were found by comparing datasets in the PCA space using FindIntegrationAnchors before datasets were integrated using IntegrateData. Corrected snATAC-seq and sc/snRNA-seq matrices after integration were normalized and dimensionally reduced. Cells from SC- and primary endocrine cells were co-embedded on a same UMAP following the method described in “Single-cell raw data processing and quality control”. Both original cell identities and new identities obtained after integration were visualized using the first two UMAP components and compared.
TF motif enrichment analysis
Using the barcode-by-peaks (501 bp fixed-width peaks) count matrix as input, we inferred enrichment of TF motifs for each barcode using chromVAR90 (v.1.4.1). We filtered cells with minimal reads less than 1500 (min_depth=1500) and peaks with fraction of reads less than 0.15 (min_in_peaks=0.15) by using ‘filterSamplesPlot’ function from chromVAR. We also corrected GC bias based on ‘BSgenome.Hsapiens.UCSC.hg19’ using the ‘addGCBias’ function. Then, we used the TF binding profiles database JASPAR 2020 motifs102 and calculated the deviation z-scores for each TF motif in each cell by using the ‘computeDeviations’ function. High-variance TF motifs across all cell types were selected using the ‘computeVariability’ function with the cut-off 1.15 (n=315). For each of these variable motifs, we calculated the mean z-score for each cell type and normalized the values to 0 (minimal) and 1 (maximal).
Building pseudotime trajectories with Monocle3
For each developmental trajectory, cells from indicated lineages were selected in the snATAC-seq dataset Seurat object using the subset function and the subset object was imported into Monocle383 using the as.cell_data_set functions with default settings. snATAC-seq UMAP coordinates were used to estimate distance between cells and to identify the nearest neighbor cell. This process was combined with the establishment of a lineage trajectory using the learn_graph function with close_loop = F, and learn_graph_control=list(ncenter=500,minimal_branch_len=10). Pseudotime trajectory roots were chosen empirically based on prior knowledge of pancreas development, with an interactive interface using the order_cells function. To minimize computational noise introduced by the sparse nature of single-cell data, we created pseudo-bulk samples by cutting the entire pseudotime trajectory into 12 pseudotime bins and aggregated cells within each bin using the aggregate_by_cell_bin function. We then integrated chromatin accessibility, gene expression and TF motif enrichment data into each single-cell to compute CPM values of cCREs, genes, and mean motif enrichment scores in each pseudotime bin. Values were then scaled and plotted on a heatmap for visualization.
Inferring gene regulatory networks (GRNs)
Computing correlation between cCRE accessibility and target gene expression
To identify putative target genes of cCREs, we combined and modified previously published methods by Li et al.103. First, we identified cCRE-gene pairs with physical interaction with the following three methods: 1. cCREs within ± 1 kb of a TSS were defined as gene promoter cCREs. Promoter-gene pairs were established across all expressed genes in each cell type. 2. cCREs located outside ± 1 kb, but within ± 50 kb of a TSS were classified as proximal elements, and all proximal cCRE-gene interactions in each cell type were considered. 3. We used Cicero84 to calculate co-accessibility between long distance cCREs (see “Computing co-accessibility using Cicero”) and identified distal cCRE-gene pairs for individual cell types.
We then generated pseudo-bulk ATAC and mRNA profiles by aggregating single cells of the same cell type from different cell sources (stem cell-derived, endocrine cells from childhood or adult pancreas) and collection times during SC-islet differentiation (D11, D14, D21, D32, D39). In total there were 16 pseudo-bulk ATAC and RNA profiles. CPM (counts per million reads) values of cCRE accessibility and gene expression in each pseudo-bulk ATAC and RNA profile were calculated, cCREs with low accessibility (maximum CPM value across pseudo-bulk ATAC profile <1) and gene with low expression genes (maximum CPM value across pseudo-bulk RNA profile <3) were excluded from further analysis. Finally, we calculated the Spearman correlation coefficient (SCC) between cCRE accessibility and target gene expression across all cCRE-gene pairs identified above. To estimate background, we generated permuted pseudo-bulk ATAC and RNA profiles by randomly shuffling identities of pseudo-bulk profiles, cCREs, and genes. We estimated False-positive detection rates (FDR)104 based on the fraction of detected pairs from the shuffled group. Empirically defined cutoffs were used to identify the final lists of cCRE-gene pairs.
Computing co-accessibility using Cicero
We used Cicero84 (v.1.3.4.10) to calculate co-accessibility scores for pairs of peaks in each individual cell type. Using SC-β-cell as example, we started from the merged peak by cell sparse binary matrix, extracted SC-β-cells, and filtered out peaks that were not present in SC-β-cells. We used the ‘make_cicero_cds’ function to aggregate cells based on the 50 nearest neighbors. We then used Cicero to calculate co-accessibility scores using a window size of 1 Mb and a distance constraint of 250 kb. We then repeated the same procedure for other cell types. We used a co-accessibility threshold of 0.05 to define pairs of peaks as co-accessible. Peaks within and outside ± 5 kb of a TSS in GENCODE V19 were considered proximal and distal, respectively. Peaks within ± 500 bp of a TSS in GENCODE V19 were defined as promoter. Co-accessible pairs were assigned to one of three groups: distal-to-distal, distal-to-proximal and proximal-to-proximal. Distal-to-proximal co-accessible pairs were defined as potential enhancer-promoter connections. Genes linked to proximal or distal cCREs were identified.
Computing correlation between transcription factor (TF) expression and cCRE accessibility.
We used a position frequency matrix (PFMatrixList object) of TF DNA-binding preferences from the JASPAR 2020 database102 and width-fixed peaks as input to perform TF footprinting analysis. We used the ‘matchMotifs’ function in the R package motifmatchr to infer cCREs bound by TFs. This analysis established a preliminary set of TF-cCRE pairs. A matching set of aggregated pseudo-bulk ATAC and RNA profiles (see “Computing correlation between cCRE accessibility and target gene expression”) was used to quantify CPM values of TF expression and cCRE accessibility. We then calculated SCCs across all pseudo-bulk and permuted pseudo-bulk aggregates through randomization. The FDR was calculated using the same method as described in “Computing correlation between cCRE accessibility and target gene expression” and empirically defined cutoffs were used to define significantly correlated TF-cCRE pairs.
Establishment of cell type-specific GRNs
We identified highly variable cCREs across cell types based on cCRE-by-pseudo-bulk count matrices. We then performed k-means clustering of highly variable cCREs to identify cCREs modules, defined by cCREs exhibiting a similar accessibility pattern across cell types. Cell type-specific cCRE modules and cell type-shared cCRE modules were identified. Upstream TFs and downstream target genes of each cCRE from each module were used to define cell type-specific GRNs. To visualize features of cell type-specific GRNs, we performed a UMAP (umap function in “uwot” package in R) based dimension reduction analysis of the pseudocell-by-cCRE accessibility matrices used for the correlation-based GRN inference, and plotted individual cCREs using the first two UMAP components. Onto this cCRE UMAP, we plotted different features of cCREs used in the GRN analysis and retained cCRE module-specific information in the plots. Those cCRE features include: 1. Accessibility in each pseudocell; 2. cCRE module identity; 3. Correlation (SCC) between each cCRE and a given TF; 4. cCREs co-bound by two TFs; 5. cCRE pseudotime values.
Identification of cell type-specific transcriptional regulators
We used Fisher’s exact test to identify cell type-specific TFs. For each TF in query, we computed (fisher.test function in R) odds ratio and p value describing enrichment of cCREs bound by the TF within a cCRE module compared to TF-bound cCREs across all cCRE modules. This process was repeated for all TFs in all cCRE modules and adjusted p values (FDR) were obtained using p.adjust function in R with a Benjamini & Hochberg method. Significantly enriched TFs were selected if FDR<0.05.
Inferring cell type-specific TF interactions
TF interactions were inferred by one TF (TF1) binding to the same set of cCREs also bound by another TF (TF2) in a cell type-specific cCRE module. To test this, we focused on one cCRE module each time and estimated the enrichment of TF1-bound cCREs within TF2 binding sites compared to those in the entire cCRE module. This enrichment was summarized using odds ratios and p values calculated with fisher.test in R. This procedure was repeated for all TF pairs in all cCRE modules and adjusted p values (FDR) were obtained using p.adjust function in R with a Benjamini & Hochberg method. Significantly TF interactions were selected if FDR<0.05.
Pseudotime ordering of transcriptional programs
Transcriptional programs were ordered separately for α-cell, β-cell and SC-EC lineages. In each lineage, cells were ordered on a pseudotime trajectory established in Monocle3 (described in “Building pseudotime trajectories with Monocle3”). Both cCRE accessibility and gene expression levels were plotted in each single cell on the pseudotime trajectory.
Since chromatin accessibility signals are binary, for each cCRE, we estimated the density of accessible cCREs along pseudotime using the density function in R, and identified pseudotime points with highest cCRE density. For each gene, we fitted gene expression along pseudotime with the smooth.spline function, and identified pseudotime points with maximum gene expression. cCRE accessibility and gene expression pseudotime values were defined as time points with highest density of accessible cCREs and maximum gene expression, respectively. Based on these pseudotime values, cCREs and genes were aligned and ordered for each lineage. Using the established GRN that connects TFs to cCREs and target genes, we were able to plot entire transcriptional programs downstream of each TF in a lineage-specific manner.
Gene ontology (GO) enrichment analysis
We performed gene ontology and pathway enrichment analysis using R package Enrichr85. Libraries “GO_Biological_Process_2018”, “GO_Cellular_Component_2018”, “GO_Biological_Process_2018”, “KEGG_2019_Human”, “MSigDB_Hallmark_2020”, “Reactome_2016” were used with default parameters. To compare enrichment among multiple gene sets, GO and pathway terms significantly enriched (p value<0.05) in at least one gene set were merged. Odds ratios and p values of those terms in each gene set were summarized in a dot plot.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism (v8.1.2), and R (v3.6.1). Statistical parameters such as the value of n, mean, standard deviation (SD), p values, and the statistical tests used are reported in the figures and figure legends. In H1 hESC differentiation experiments, the “n” refers to the number of independent hESC differentiation experiments analyzed (biological replicates, Figure 4I–K, and Figure 7G,H). In human pancreas immunofluorescence staining in Figure S4G, “n” indicates the number of donors from which samples were obtained. In human pancreas immunofluorescence staining in Figure 4G,H,M,N, “n” indicates the number of randomly selected imaging regions (technical replicates) from each donor. At each human developmental stage, more than four donors were quantified (biological replicates). All bar graphs and line graphs are displayed as mean ± SD. Paired (if observations were related, Figure S5M) or unpaired (observations were independent, Figure 4I–K and Figure S4I–L) student’s t-tests were used for two-sample comparisons. For multiple-sample comparisons and comparisons done between multiple types of variables, one-way and two-way ANOVA was used, respectively. ANOVAs were coupled with one of the three multiple-comparisons tests: Šidák-Holm’s test (for two column data in two-way ANOVA, Figure S4C and S5S); Tukey (if every column compared with every other column, Figure 4G,H,M,N, Figure S1B, and Figure S4G); or Dunnett’s test (if every column compared with a control column, Figure 7G,H).
Supplementary Material
Table S1. Summary of single-cell datasets, samples, and pseudotime analysis of transcription factors, related to Figure 1. (A) Summary of data sets used in this study.
(B) Summary of donor features.
(C) Expression of key TFs in each pseudotime bin downstream of branch point 1 and branch point 2 from Figure 1H. CPM (counts per million reads) values from pseudocells aggregated in each pseudotime bin are shown.
(D) Motif enrichment score of key TFs in each pseudotime bin downstream of branch point 1 and branch point 2 from Figure 1H.
(E) Proportion of cell types in each pseudotime bin downstream of branch point 1 and branch point 2 from Figure 1H.
Table S2. Features of gene regulatory networks governing stem cell islet development, related to Figure 2. (A) GO terms/pathways enriched among target genes in cell type-specific cCRE module.
(B) TFs regulating SC-δ cCRE module.
(C) GO terms/pathways enriched among NKX6-1 target genes in SC-β-cells and SC-ECs.
(D) Predicted interactions TFs in cell type-specific cCRE modules.
Table S3. Temporal order of transcription factors and their downstream regulatory programs, related to Figure 3. (A) Temporal order of TFs in SC-α, SC-β and SC-EC lineages.
(B) Temporal order of cCREs in SC-α, SC-β and SC-EC lineages.
Table S4. Genes differentially expressed between control and CDX2 KO SC-islet cell types, related to Figure 5. (A) Genes differentially expressed between control and CDX2 KO SC-ECs.
(B) Genes differentially expressed between control and CDX2 KO SC-β-cells.
(C) Genes differentially expressed between control and CDX2 KO ENP3 cells. Gene expression is shown in CPM (counts per million reads).
Table S5. Differential gene regulatory programs between stem cell islets and primary islets, related to Figure 6. (A) TF motifs with variable enrichment score in SC- and primary- endocrine cell types.
(B) GO terms/pathways enriched among genes specific to different β-related cell types.
(C) GO terms/pathways enriched among genes specific to different α-related cell types.
Table S6. Features of gene regulatory network regulating β-cell maturation, related to Figure 7. (A) TFs regulating cell type-specific cCRE module.
(B) GO terms/pathways enriched among target genes of signal-dependent TFs.
Highlights:
Cell type-specific gene regulatory programs governing SC-islet differentiation
SC-derived enterochromaffin-like cells resemble fetal pancreatic β-cell-like cells
Signal-dependent transcriptional programs are insufficiently activated in SC-islets
Sex hormones promote β-cell proliferation in childhood
Acknowledgements
We acknowledge support of the UCSD IGM Genomic Center and P30 DK064391 for sequencing. This work was supported by grants from the National Institutes of Health U01 DK120429 (M.S. and K.J.G.), U01 DK105541 (M.S. and K.J.G.), R01 DK068471 (M.S.), UH3 DK122639 (M.S.), and postdoctoral fellowships from the California Institute for Regenerative Medicine EDUC4-12804 (H.Z.), the Diabetes Research Connection (H.Z.), and Juvenile Diabetes Research Foundation (K.V.N.). Work at the Center for Epigenomics was supported in part by the UC San Diego School of Medicine. We thank Ryan Geusz and Medhavi Mallick for discussions. We thank the organ donors and their families for their donations and the International Institute for Advancement of Medicine as well as organ procurement organizations for their partnership in studies of human pancreatic tissue for research. This study used pediatric pancreas samples from the Human Atlas of the Neonatal Development and Early Life Pancreas and associated Immune organs, a project supported by The Leona M. and Harry B. Helmsley Charitable Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
K.J.G. does consulting for Genentech and holds stock in Vertex Pharmaceuticals.
REFERENCE
- 1.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32, 1121–1133. 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- 2.Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, and Melton DA (2014). Generation of functional human pancreatic β cells in vitro. Cell 159, 428–439. 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nostro MC, Sarangi F, Yang C, Holland A, Elefanty AG, Stanley EG, Greiner DL, and Keller G (2015). Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Reports 4, 591–604. 10.1016/j.stemcr.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, and Millman JR (2019). Acquisition of Dynamic Function in Human Stem Cell-Derived β Cells. Stem Cell Reports 12, 351–365. 10.1016/j.stemcr.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, Juang C, Li ML, Nguyen VQ, Giacometti S, et al. (2019). Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat Cell Biol 21, 263–274. 10.1038/s41556-018-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L, and Millman JR (2020). Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat Biotechnol 38, 460–470. 10.1038/s41587-020-0430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balboa D, Barsby T, Lithovius V, Saarimäki-Vire J, Omar-Hmeadi M, Dyachok O, Montaser H, Lund PE, Yang M, Ibrahim H, et al. (2022). Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat Biotechnol 40, 1042–1055. 10.1038/s41587-022-01219-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veres A, Faust AL, Bushnell HL, Engquist EN, Kenty JH, Harb G, Poh YC, Sintov E, Gürtler M, Pagliuca FW, et al. (2019). Charting cellular identity during human in vitro β-cell differentiation. Nature 569, 368–373. 10.1038/s41586-019-1168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vegas AJ, Veiseh O, Gürtler M, Millman JR, Pagliuca FW, Bader AR, Doloff JC, Li J, Chen M, Olejnik K, et al. (2016). Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nat Med 22, 306–311. 10.1038/nm.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Augsornworawat P, Maxwell KG, Velazco-Cruz L, and Millman JR (2020). Single-Cell Transcriptome Profiling Reveals β Cell Maturation in Stem Cell-Derived Islets after Transplantation. Cell Rep 32, 108067. 10.1016/j.celrep.2020.108067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jermendy A, Toschi E, Aye T, Koh A, Aguayo-Mazzucato C, Sharma A, Weir GC, Sgroi D, and Bonner-Weir S (2011). Rat neonatal beta cells lack the specialised metabolic phenotype of mature beta cells. Diabetologia 54, 594–604. 10.1007/s00125-010-2036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aguayo-Mazzucato C, Koh A, El Khattabi I, Li WC, Toschi E, Jermendy A, Juhl K, Mao K, Weir GC, Sharma A, and Bonner-Weir S (2011). Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 54, 583–593. 10.1007/s00125-010-2026-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otonkoski T, Andersson S, Knip M, and Simell O (1988). Maturation of insulin response to glucose during human fetal and neonatal development. Studies with perifusion of pancreatic isletlike cell clusters. Diabetes 37, 286–291. 10.2337/diab.37.3.286. [DOI] [PubMed] [Google Scholar]
- 14.Rorsman P, Arkhammar P, Bokvist K, Hellerström C, Nilsson T, Welsh M, Welsh N, and Berggren PO (1989). Failure of glucose to elicit a normal secretory response in fetal pancreatic beta cells results from glucose insensitivity of the ATP-regulated K+ channels. Proc Natl Acad Sci U S A 86, 4505–4509. 10.1073/pnas.86.12.4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henquin JC, and Nenquin M (2018). Immaturity of insulin secretion by pancreatic islets isolated from one human neonate. J Diabetes Investig 9, 270–273. 10.1111/jdi.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, Spitale RC, Dai C, Gu X, Qu K, et al. (2016). Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human β Cell Function. Cell Metab 23, 909–920. 10.1016/j.cmet.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, and Dor Y (2015). Weaning triggers a maturation step of pancreatic β cells. Dev Cell 32, 535–545. 10.1016/j.devcel.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Guo L, Inada A, Aguayo-Mazzucato C, Hollister-Lock J, Fujitani Y, Weir GC, Wright CV, Sharma A, and Bonner-Weir S (2013). PDX1 in ducts is not required for postnatal formation of β-cells but is necessary for their subsequent maturation. Diabetes 62, 3459–3468. 10.2337/db12-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, and Melton DA (2012). Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol 30, 261–264. 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wortham M, and Sander M (2021). Transcriptional mechanisms of pancreatic β-cell maturation and functional adaptation. Trends Endocrinol Metab 32, 474–487. 10.1016/j.tem.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacovetti C, Matkovich SJ, Rodriguez-Trejo A, Guay C, and Regazzi R (2015). Postnatal β-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nat Commun 6, 8084. 10.1038/ncomms9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaafar R, Tran S, Shah AN, Sun G, Valdearcos M, Marchetti P, Masini M, Swisa A, Giacometti S, Bernal-Mizrachi E, et al. (2019). mTORC1 to AMPK switching underlies β-cell metabolic plasticity during maturation and diabetes. J Clin Invest 129, 4124–4137. 10.1172/jci127021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rakshit K, Qian J, Gaonkar KS, Dhawan S, Colwell CS, and Matveyenko AV (2018). Postnatal Ontogenesis of the Islet Circadian Clock Plays a Contributory Role in β-Cell Maturation Process. Diabetes 67, 911–922. 10.2337/db17-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weng C, Xi J, Li H, Cui J, Gu A, Lai S, Leskov K, Ke L, Jin F, and Li Y (2020). Single-cell lineage analysis reveals extensive multimodal transcriptional control during directed beta-cell differentiation. Nat Metab 2, 1443–1458. 10.1038/s42255-020-00314-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleck JS, Jansen SMJ, Wollny D, Seimiya M, Zenk F, Santel M, He Z, Gray Camp J, and Treutlein B (2021). Inferring and perturbing cell fate regulomes in human cerebral organoids. bioRxiv, 2021.2008.2024.457460. 10.1101/2021.08.24.457460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beerman I, and Rossi DJ (2015). Epigenetic Control of Stem Cell Potential during Homeostasis, Aging, and Disease. Cell Stem Cell 16, 613–625. 10.1016/j.stem.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gradwohl G, Dierich A, LeMeur M, and Guillemot F (2000). neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A 97, 1607–1611. 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGrath PS, Watson CL, Ingram C, Helmrath MA, and Wells JM (2015). The Basic Helix-Loop-Helix Transcription Factor NEUROG3 Is Required for Development of the Human Endocrine Pancreas. Diabetes 64, 2497–2505. 10.2337/db14-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sander M, Neubüser A, Kalamaras J, Ee HC, Martin GR, and German MS (1997). Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev 11, 1662–1673. 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- 30.Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, and Gruss P (1997). The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 386, 399–402. 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- 31.Ramond C, Beydag-Tasöz BS, Azad A, van de Bunt M, Petersen MBK, Beer NL, Glaser N, Berthault C, Gloyn AL, Hansson M, et al. (2018). Understanding human fetal pancreas development using subpopulation sorting, RNA sequencing and single-cell profiling. Development 145. 10.1242/dev.165480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sean d.l.O., Liu Z, Sun H, Yu SK, Wong DM, Chu E, Rao SA, Eng N, Peixoto G, Bouza J, et al. (2022). Single-Cell Multi-Omic Roadmap of Human Fetal Pancreatic Development. bioRxiv, 2022.2002.2017.480942. 10.1101/2022.02.17.480942. [DOI] [Google Scholar]
- 33.Gonçalves CA, Larsen M, Jung S, Stratmann J, Nakamura A, Leuschner M, Hersemann L, Keshara R, Perlman S, Lundvall L, et al. (2021). A 3D system to model human pancreas development and its reference single-cell transcriptome atlas identify signaling pathways required for progenitor expansion. Nature Communications 12, 3144. 10.1038/s41467-021-23295-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holland AM, Hale MA, Kagami H, Hammer RE, and MacDonald RJ (2002). Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci U S A 99, 12236–12241. 10.1073/pnas.192255099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, Yang C, Pannikar A, Doliba N, Zhang T, et al. (2014). Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab 19, 259–271. 10.1016/j.cmet.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith SB, Qu HQ, Taleb N, Kishimoto NY, Scheel DW, Lu Y, Patch AM, Grabs R, Wang J, Lynn FC, et al. (2010). Rfx6 directs islet formation and insulin production in mice and humans. Nature 463, 775–780. 10.1038/nature08748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ait-Lounis A, Bonal C, Seguín-Estévez Q, Schmid CD, Bucher P, Herrera PL, Durand B, Meda P, and Reith W (2010). The transcription factor Rfx3 regulates beta-cell differentiation, function, and glucokinase expression. Diabetes 59, 1674–1685. 10.2337/db09-0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanheer L, Schiavo AA, Van Haele M, Haesen T, Janiszewski A, Chappell J, Roskams T, Cnop M, and Pasque V (2020). Revealing the Key Regulators of Cell Identity in the Human Adult Pancreas. bioRxiv, 2020.2009.2023.310094. 10.1101/2020.09.23.310094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sobel J, Guay C, Elhanani O, Rodriguez-Trejo A, Stoll L, Menoud V, Jacovetti C, Walker MD, and Regazzi R (2021). Scrt1, a transcriptional regulator of β-cell proliferation identified by differential chromatin accessibility during islet maturation. Sci Rep 11, 8800. 10.1038/s41598-021-88003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gross S, Garofalo DC, Balderes DA, Mastracci TL, Dias JM, Perlmann T, Ericson J, and Sussel L (2016). The novel enterochromaffin marker Lmx1a regulates serotonin biosynthesis in enteroendocrine cell lineages downstream of Nkx2.2. Development 143, 2616–2628. 10.1242/dev.130682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tiedemann HB, Schneltzer E, Beckers J, Przemeck GKH, and Hrabě de Angelis M (2017). Modeling coexistence of oscillation and Delta/Notch-mediated lateral inhibition in pancreas development and neurogenesis. J Theor Biol 430, 32–44. 10.1016/j.jtbi.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Kannan MB, Solovieva V, and Blank V (2012). The small MAF transcription factors MAFF, MAFG and MAFK: current knowledge and perspectives. Biochim Biophys Acta 1823, 1841–1846. 10.1016/j.bbamcr.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 43.Vettorazzi JF, Ribeiro RA, Borck PC, Branco RC, Soriano S, Merino B, Boschero AC, Nadal A, Quesada I, and Carneiro EM (2016). The bile acid TUDCA increases glucose-induced insulin secretion via the cAMP/PKA pathway in pancreatic beta cells. Metabolism 65, 54–63. 10.1016/j.metabol.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 44.Good AL, Cannon CE, Haemmerle MW, Yang J, Stanescu DE, Doliba NM, Birnbaum MJ, and Stoffers DA (2019). JUND regulates pancreatic β cell survival during metabolic stress. Mol Metab 25, 95–106. 10.1016/j.molmet.2019.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Son J, Ding H, Farb TB, Efanov AM, Sun J, Gore JL, Syed SK, Lei Z, Wang Q, Accili D, and Califano A (2021). BACH2 inhibition reverses β cell failure in type 2 diabetes models. J Clin Invest 131. 10.1172/jci153876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang K, Cui Y, Lin P, Yao Z, and Sun Y (2021). JunD Regulates Pancreatic β-Cells Function by Altering Lipid Accumulation. Front Endocrinol (Lausanne) 12, 689845. 10.3389/fendo.2021.689845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deconinck JF, Potvliege PR, and Gepts W (1971). The ultrasturcture of the human pancreatic islets. I. The islets of adults. Diabetologia 7, 266–282. 10.1007/bf01211879. [DOI] [PubMed] [Google Scholar]
- 48.Like AA, and Orci L (1972). Embryogenesis of the human pancreatic islets: a light and electron microscopic study. Diabetes 21, 511–534. 10.2337/diab.21.2.s511. [DOI] [PubMed] [Google Scholar]
- 49.Pictet RL, Clark WR, Williams RH, and Rutter WJ (1972). An ultrastructural analysis of the developing embryonic pancreas. Dev Biol 29, 436–467. 10.1016/0012-1606(72)90083-8. [DOI] [PubMed] [Google Scholar]
- 50.Yu XX, Qiu WL, Yang L, Wang YC, He MY, Wang D, Zhang Y, Li LC, Zhang J, Wang Y, and Xu CR (2021). Sequential progenitor states mark the generation of pancreatic endocrine lineages in mice and humans. Cell Res 31, 886–903. 10.1038/s41422-021-00486-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molakandov K, Berti DA, Beck A, Elhanani O, Walker MD, Soen Y, Yavriyants K, Zimerman M, Volman E, Toledo I, et al. (2021). Selection for CD26(−) and CD49A(+) Cells From Pluripotent Stem Cells-Derived Islet-Like Clusters Improves Therapeutic Activity in Diabetic Mice. Front Endocrinol (Lausanne) 12, 635405. 10.3389/fendo.2021.635405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, Aldinger KA, Pokholok D, Zhang F, Milbank JH, et al. (2020). A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612. doi: 10.1126/science.aba7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moon JH, Kim H, Kim H, Park J, Choi W, Choi W, Hong HJ, Ro HJ, Jun S, Choi SH, et al. (2020). Lactation improves pancreatic β cell mass and function through serotonin production. Sci Transl Med 12. 10.1126/scitranslmed.aay0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Castell AL, Goubault C, Ethier M, Fergusson G, Tremblay C, Baltz M, Dal Soglio D, Ghislain J, and Poitout V (2022). β Cell mass expansion during puberty involves serotonin signaling and determines glucose homeostasis in adulthood. JCI Insight 7, e160854. 10.1172/jci.insight.160854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, Fujitani Y, Kawamori R, Miyatsuka T, Kosaka Y, et al. (2010). Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 16, 804–808. 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohara-Imaizumi M, Kim H, Yoshida M, Fujiwara T, Aoyagi K, Toyofuku Y, Nakamichi Y, Nishiwaki C, Okamura T, Uchida T, et al. (2013). Serotonin regulates glucose-stimulated insulin secretion from pancreatic β cells during pregnancy. Proc Natl Acad Sci U S A 110, 19420–19425. 10.1073/pnas.1310953110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baeyens L, Hindi S, Sorenson RL, and German MS (2016). β-Cell adaptation in pregnancy. Diabetes Obes Metab 18 Suppl 1, 63–70. 10.1111/dom.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moon JH, Kim YG, Kim K, Osonoi S, Wang S, Saunders DC, Wang J, Yang K, Kim H, Lee J, et al. (2020). Serotonin Regulates Adult β-Cell Mass by Stimulating Perinatal β-Cell Proliferation. Diabetes 69, 205–214. 10.2337/db19-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xin Y, Dominguez Gutierrez G, Okamoto H, Kim J, Lee AH, Adler C, Ni M, Yancopoulos GD, Murphy AJ, and Gromada J (2018). Pseudotime Ordering of Single Human β-Cells Reveals States of Insulin Production and Unfolded Protein Response. Diabetes 67, 1783–1794. 10.2337/db18-0365. [DOI] [PubMed] [Google Scholar]
- 60.Kaestner KH, Powers AC, Naji A, and Atkinson MA (2019). NIH Initiative to Improve Understanding of the Pancreas, Islet, and Autoimmunity in Type 1 Diabetes: The Human Pancreas Analysis Program (HPAP). Diabetes 68, 1394–1402. 10.2337/db19-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, Robin C, and van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat Methods 14, 935–936. 10.1038/nmeth.4437. [DOI] [PubMed] [Google Scholar]
- 62.Wang YJ, and Kaestner KH (2019). Single-Cell RNA-Seq of the Pancreatic Islets--a Promise Not yet Fulfilled? Cell Metab 29, 539–544. 10.1016/j.cmet.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, et al. (2016). A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst 3, 346–360 e344. 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Linnemann AK, Blumer J, Marasco MR, Battiola TJ, Umhoefer HM, Han JY, Lamming DW, and Davis DB (2017). Interleukin 6 protects pancreatic β cells from apoptosis by stimulation of autophagy. Faseb j 31, 4140–4152. 10.1096/fj.201700061RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Groef S, Renmans D, Cai Y, Leuckx G, Roels S, Staels W, Gradwohl G, Baeyens L, Heremans Y, Martens GA, et al. (2016). STAT3 modulates β-cell cycling in injured mouse pancreas and protects against DNA damage. Cell Death Dis 7, e2272. 10.1038/cddis.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alvarez-Dominguez JR, Donaghey J, Rasouli N, Kenty JHR, Helman A, Charlton J, Straubhaar JR, Meissner A, and Melton DA (2020). Circadian Entrainment Triggers Maturation of Human In Vitro Islets. Cell Stem Cell 26, 108–122.e110. 10.1016/j.stem.2019.11.011. [DOI] [PubMed] [Google Scholar]
- 67.Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, Peek CB, Hong H, Huang W, Omura C, et al. (2015). Pancreatic β cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 350, aac4250. 10.1126/science.aac4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vieira E, Marroquí L, Batista TM, Caballero-Garrido E, Carneiro EM, Boschero AC, Nadal A, and Quesada I (2012). The clock gene Rev-erbα regulates pancreatic β-cell function: modulation by leptin and high-fat diet. Endocrinology 153, 592–601. 10.1210/en.2011-1595. [DOI] [PubMed] [Google Scholar]
- 69.Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, Hollister-Lock J, El Khattabi I, Marsili A, Weir GC, Sharma A, Larsen PR, and Bonner-Weir S (2013). Thyroid hormone promotes postnatal rat pancreatic β-cell development and glucose-responsive insulin secretion through MAFA. Diabetes 62, 1569–1580. 10.2337/db12-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bruin JE, Asadi A, Fox JK, Erener S, Rezania A, and Kieffer TJ (2015). Accelerated Maturation of Human Stem Cell-Derived Pancreatic Progenitor Cells into Insulin-Secreting Cells in Immunodeficient Rats Relative to Mice. Stem Cell Reports 5, 1081–1096. 10.1016/j.stemcr.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Helman A, Cangelosi AL, Davis JC, Pham Q, Rothman A, Faust AL, Straubhaar JR, Sabatini DM, and Melton DA (2020). A Nutrient-Sensing Transition at Birth Triggers Glucose-Responsive Insulin Secretion. Cell Metab 31, 1004–1016.e1005. 10.1016/j.cmet.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grasso S, Messina A, Saporito N, and Reitano G (1968). Serum-insulin response to glucose and aminoacids in the premature infant. Lancet 2, 755–756. 10.1016/s0140-6736(68)90954-9. [DOI] [PubMed] [Google Scholar]
- 73.Pildes RS, Hart RJ, Warrner R, and Cornblath M (1969). Plasma insulin response during oral glucose tolerance tests in newborns of normal and gestational diabetic mothers. Pediatrics 44, 76–83. 10.1542/peds.44.1.76. [DOI] [PubMed] [Google Scholar]
- 74.Aguayo-Mazzucato C, Lee TB Jr., Matzko M, DiIenno A, Rezanejad H, Ramadoss P, Scanlan T, Zavacki AM, Larsen PR, Hollenberg A, et al. (2018). T(3) Induces Both Markers of Maturation and Aging in Pancreatic β-Cells. Diabetes 67, 1322–1331. 10.2337/db18-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bugliani M, Mossuto S, Grano F, Suleiman M, Marselli L, Boggi U, De Simone P, Eizirik DL, Cnop M, Marchetti P, and De Tata V (2019). Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells Under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front Endocrinol (Lausanne) 10, 52. 10.3389/fendo.2019.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bachar-Wikstrom E, Wikstrom JD, Ariav Y, Tirosh B, Kaiser N, Cerasi E, and Leibowitz G (2013). Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes 62, 1227–1237. 10.2337/db12-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corbier P, Edwards DA, and Roffi J (1992). The neonatal testosterone surge: a comparative study. Arch Int Physiol Biochim Biophys 100, 127–131. 10.3109/13813459209035274. [DOI] [PubMed] [Google Scholar]
- 78.Clarkson J, and Herbison AE (2016). Hypothalamic control of the male neonatal testosterone surge. Philos Trans R Soc Lond B Biol Sci 371, 20150115. 10.1098/rstb.2015.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ogishima T, Mitani F, and Suematsu M (2008). Cytochrome P-450(17alpha) in beta-cells of rat pancreas and its local steroidogenesis. J Steroid Biochem Mol Biol 111, 80–86. 10.1016/j.jsbmb.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 80.Kelava I, Chiaradia I, Pellegrini L, Kalinka AT, and Lancaster MA (2022). Androgens increase excitatory neurogenic potential in human brain organoids. Nature 602, 112–116. 10.1038/s41586-021-04330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15. 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982. 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, et al. (2018). Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol Cell 71, 858–871 e858. 10.1016/j.molcel.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ramirez F, Dundar F, Diehl S, Gruning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–191. 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978. 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Geusz RJ, Wang A, Lam DK, Vinckier NK, Alysandratos KD, Roberts DA, Wang J, Kefalopoulou S, Ramirez A, Qiu Y, et al. (2021). Sequence logic at enhancers governs a dual mechanism of endodermal organ fate induction by FOXA pioneer factors. Nat Commun 12, 6636. 10.1038/s41467-021-26950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, Bottino R, Campbell-Thompson M, Aramandla R, Poffenberger G, et al. (2018). α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep 22, 2667–2676. 10.1016/j.celrep.2018.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saunders DC, Brissova M, Phillips N, Shrestha S, Walker JT, Aramandla R, Poffenberger G, Flaherty DK, Weller KP, Pelletier J, et al. (2019). Ectonucleoside Triphosphate Diphosphohydrolase-3 Antibody Targets Adult Human Pancreatic β Cells for In Vitro and In Vivo Analysis. Cell Metab 29, 745–754.e744. 10.1016/j.cmet.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wortham M, Liu F, Fleischman JY, Wallace M, Mulas F, Vinckier NK, Harrington AR, Cross BR, Chiou J, Patel NA, et al. (2019). Nutrient regulation of the islet epigenome controls adaptive insulin secretion. bioRxiv, 742403. 10.1101/742403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lareau CA, Ma S, Duarte FM, and Buenrostro JD (2020). Inference and effects of barcode multiplets in droplet-based single-cell assays. Nat Commun 11, 866. 10.1038/s41467-020-14667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Consortium, E.P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chiou J, Zeng C, Cheng Z, Han JY, Schlichting M, Miller M, Mendez R, Huang S, Wang J, Sui Y, et al. (2021). Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk. Nat Genet 53, 455–466. 10.1038/s41588-021-00823-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 9, 5233. 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 37, 925–936. 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92. 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li YE, Preissl S, Hou X, Zhang Z, Zhang K, Qiu Y, Poirion OB, Li B, Chiou J, Liu H, et al. (2021). An atlas of gene regulatory elements in adult mouse cerebrum. Nature 598, 129–136. 10.1038/s41586-021-03604-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhu C, Zhang Y, Li YE, Lucero J, Behrens MM, and Ren B (2021). Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat Methods 18, 283–292. 10.1038/s41592-021-01060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of single-cell datasets, samples, and pseudotime analysis of transcription factors, related to Figure 1. (A) Summary of data sets used in this study.
(B) Summary of donor features.
(C) Expression of key TFs in each pseudotime bin downstream of branch point 1 and branch point 2 from Figure 1H. CPM (counts per million reads) values from pseudocells aggregated in each pseudotime bin are shown.
(D) Motif enrichment score of key TFs in each pseudotime bin downstream of branch point 1 and branch point 2 from Figure 1H.
(E) Proportion of cell types in each pseudotime bin downstream of branch point 1 and branch point 2 from Figure 1H.
Table S2. Features of gene regulatory networks governing stem cell islet development, related to Figure 2. (A) GO terms/pathways enriched among target genes in cell type-specific cCRE module.
(B) TFs regulating SC-δ cCRE module.
(C) GO terms/pathways enriched among NKX6-1 target genes in SC-β-cells and SC-ECs.
(D) Predicted interactions TFs in cell type-specific cCRE modules.
Table S3. Temporal order of transcription factors and their downstream regulatory programs, related to Figure 3. (A) Temporal order of TFs in SC-α, SC-β and SC-EC lineages.
(B) Temporal order of cCREs in SC-α, SC-β and SC-EC lineages.
Table S4. Genes differentially expressed between control and CDX2 KO SC-islet cell types, related to Figure 5. (A) Genes differentially expressed between control and CDX2 KO SC-ECs.
(B) Genes differentially expressed between control and CDX2 KO SC-β-cells.
(C) Genes differentially expressed between control and CDX2 KO ENP3 cells. Gene expression is shown in CPM (counts per million reads).
Table S5. Differential gene regulatory programs between stem cell islets and primary islets, related to Figure 6. (A) TF motifs with variable enrichment score in SC- and primary- endocrine cell types.
(B) GO terms/pathways enriched among genes specific to different β-related cell types.
(C) GO terms/pathways enriched among genes specific to different α-related cell types.
Table S6. Features of gene regulatory network regulating β-cell maturation, related to Figure 7. (A) TFs regulating cell type-specific cCRE module.
(B) GO terms/pathways enriched among target genes of signal-dependent TFs.
Data Availability Statement
Single-nucleus ATAC sequencing (snATAC-seq), Single-nucleus RNA sequencing (snATAC-seq), Single-cell RNA sequencing (scRNA-seq), and CDX2 ChIP sequencing raw and processed data are available through the Gene Expression Omnibus under accession GSE202500. Other published datasets used in this study are summarized in Table S1. UCSC genome browser sessions of aggregated snATAC-seq, snRNA-seq, and scRNA-seq data are available at: https://genome.ucsc.edu/s/gaowei/hg19_islet.
Custom codes for main analysis used in this study have been deposited on GitHub: https://github.com/gaoweiwang/SCislet, and on Zendo. DOI is available in the Key Resource Table.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-insulin | DAKO | A0564 |
| Guinea pig anti-insulin | Invitrogen | PA1-26938 |
| Mouse pig anti-insulin | Abcam | ab9569 |
| Mouse anti-NKX6.1 | DSHB | F55A10 |
| Goat anti-PDX1 | R&D | AF2419 |
| Guinea pig anti-PDX1 | Abcam | ab47308 |
| Rabbit anti-CDX2 | Cell Signaling | 12306 |
| Rabbit anti-CDX2 | Abcam | ab76541 |
| Rabbit anti-SLC18A1 | Sigma | HPA063797 |
| Sheep anti-TPH1 | Sigma | AB1541 |
| Goat anti-5HT (serotonin) | Immunostar | 20079 |
| Goat anti-somatostatin | Santa Cruz | SC7819 |
| Mouse anti-glucagon | Sigma | G2654 |
| AlexaFluor® 647-conjugated anti-NKX6-1 | BD Biosciences | 563338 |
| PE-conjugated anti-insulin | Cell Signaling | 8508 |
| AlexaFluor® 488-conjugated donkey anti-rabbit IgG | Jackson Immunoresearch | 711-545-152 |
| Biotin-conjugated anti-CD26 | BioLegend | 302718 |
| Brilliant Violet 421™-conjugated Streptavidin | BioLegend | 405226 |
| Rabbit anti-CDX2 | Bethyl Laboratories | A300-691A |
| Biological Samples | ||
| Frozen childhood human pancreas | Pancreatic Organ Donors with Diabetes (nPOD) | HDL-052 HDL-067 HDL-077 HDL-015 HDL-019 HDL-021 |
| Frozen adult human pancreas | nPOD | 6229 6339 6366 6375 6479 6234 6401 |
| Isolated childhood human islets | ADI IsletCore | R394 |
| Fixed fetal human pancreas sections | MRC/Wellcome Trust-funded Human Developmental Biology Resource | N/A |
| Fixed fetal human pancreas sections | University of Washington Birth Defects Research Laboratory | N/A |
| Fixed neonatal human pancreas sections | nPOD | N/A |
| Fixed adult human pancreas sections | Prodo Labs | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Matrigel | Corning | 356238 |
| mTeSR1 media | Stem Cell Technologies | 85850 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140122 |
| Accutase | Thermo Fisher Scientific | 00-4555-56 |
| ROCK inhibitor Y-27632 | Stem Cell Technologies | 72307 |
| MCDB 131 medium | Thermo Fisher Scientific | 10372019 |
| NaHCO3 | Sigma | S6297 |
| GlutaMAX | Thermo Fisher Scientific | 35050061 |
| D-Glucose | Sigma | G8769 |
| Bovine Serum Albumin (BSA) | Lampire Biological Laboratories | 7500804 |
| Activin A | R&D Systems | 338-AC/CF |
| Wnt3a | R&D Systems | 5036-WN |
| L-Ascorbic Acid | Sigma | A4544 |
| FGF7 | R&D Systems | 251-KG |
| SANT-1 | Sigma | S4572 |
| Retinoic Acid | Sigma | R2625 |
| LDN193189 | Stemgent | 04-0074 |
| ITS-X | Thermo Fisher Scientific | 51500056 |
| TPB | Calbiochem | 565740 |
| T3 | Sigma | T6397 |
| ALK5i II | Cayman Chemicals | 14794 |
| ZnSO4 | Sigma | Z0251 |
| heparin | Sigma | H3149 |
| Gamma secretase inhibitor XX | Calbiochem | 565789 |
| Trace Element A | Corning | 89408-312 |
| Trace Element B | Corning | 89422-908 |
| MEM Non-Essential Amino Acids | Thermo Fisher Scientific | 11140076 |
| Dihydrotestosterone (DHT) | Sigma | D-073 |
| Hoechst 33342 | Invitrogen | H3570 |
| Horse serum | Invitrogen | 16050130 |
| Triton X-100 | Sigma | T8787 |
| Pierce Protease Inhibitor | Fischer | PIA32965 |
| DTT | Sigma | D9779 |
| Recombinant RNAsin RNase inhibitor | Promega | PAN2515 |
| EDTA | Invitrogen | 15575020 |
| DRAQ7 | Cell Signaling | 7406 |
| Critical Commercial Assays | ||
| XtremeGene 9 transfection reagents | Roche | 6365787001 |
| Cytofix/Cytoperm™ Plus Fixation/Permeabilizatio n Solution Kit | BD Biosciences | AB_2869009 |
| Click-iT™ EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit | Thermo Fisher | C10420 |
| VECTASHIELD® mounti ng media | Vector Laboratories | H-1300 |
| Dako Fluorescent Mounting Medium | Dako | S3023 |
| Tissue-Tek® O.C.T. Sakura® Finetek compound | VWR | 25608-930 |
| Superfrost Plus® Microscope Slides | Thermo Fisher | 22-037-246 |
| TSA blocking buffer | Perkin Elmer | NEL 701001KT |
| STELLUX® Chemi Human C-peptide ELISA | ALPCO | 80-CPTHU-CH01 |
| RNeasy Micro kit | QIAGEN | 74004 |
| iScript™ cDNA Synthesis Kit | Bio-Rad | 1708891 |
| iQ™ SYBR® Green Supermix | Bio-Rad | 1708880 |
| ChIP-IT High-Sensitivity kit | Active Motif | N/A |
| 10X Next GEM Single Cell 3’ v3.1 | 10X Genomics | PN-1000121 |
| 10X Chromium Chip E Single Cell ATAC Kit | 10X genomics | 1000086 |
| 10X Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.0 | 10X genomics | 1000175 |
| 10X Chromium Next GEM Chip H Single Cell Kit | 10X genomics | 1000161 |
| Deposited Data | ||
| snATAC-seq of SC-islet differentiation | Gene Expression Omnibus | GSE202500 |
| sc/snRNA-seq of SC-islet differentiation | Gene Expression Omnibus | GSE202500 |
| snATAC-seq of primary childhood human pancreas or pancreatic islet | Gene Expression Omnibus | GSE202500 |
| sc/snRNA-seq of primary childhood human pancreas or pancreatic islet | Gene Expression Omnibus | GSE202500 |
| CDX2 ChIP-seq of D21 SC-islet | Gene Expression Omnibus | GSE202500 |
| scRNA-seq of primary adult human pancreatic islets | Gene Expression Omnibus | GSE114297 |
| scRNA-seq of primary adult human pancreatic islets | HPAP | See Table S1A |
| snATAC-seq of primary fetal human pancreas | Domcke et al., 202052 (https://descartes.brotmanbaty.org/) | N/A |
| scRNA-seq of primary fetal human pancreas | OMIX | OMIX236 |
| Experimental Models: Cell Lines | ||
| Human: H1 ESC | WiCell Research Institute | WA01 |
| Oligonucleotides | ||
| List of primers used in this paper | See Table S7 for details | N/A |
| Recombinant DNA | ||
| PX458 | Addgene | 48138 |
| Software and Algorithms | ||
| HALO image analysis | Indica Lab | N/A |
| FlowJo V10 | FlowJo LLC. | N/A |
| GraphPad Prism (v8.1.2) | Dotmatics | N/A |
| R (v3.6.1) | CRAN | N/A |
| Cell Ranger ATAC v1.1.0 | 10X Genomics | N/A |
| Cell Ranger RNA v.3.0.2 | 10X Genomics | N/A |
| Scanpy (v.1.6.0) | Wolf et al., 201881 | N/A |
| Seurat | Stuart et al., 201982 | N/A |
| Monocle3 | Qiu et al., 201783 | N/A |
| Cicero | Pliner et al., 201884 | N/A |
| Enrichr | Kuleshov et al., 201685 | N/A |
| Bowtie2 | Langmead and Salzberg, 201286 | N/A |
| SAMtools | Li et al., 200987 | N/A |
| DeepTools | Ramirez et al., 201488 | N/A |
| MACS2 | Zhang et al., 200889 | N/A |
| chromVAR | Schep et al., 201790 | N/A |
| Custom codes | Zendo | DOI:10.5281/zenodo.7694211 |
Any additional information required to reanalyse the data reported in this work paper is available from the Lead Contact upon request.