

Abstract
Artificially improving persimmon (Diospyros kaki Thunb.), one of the most important fruit trees, remains challenging owing to the lack of reference genomes. In this study, we generated an allele-aware chromosome-level genome assembly for the autohexaploid persimmon ‘Xiaoguotianshi’ (Chinese-PCNA type) using PacBio CCS and Hi-C technology. The final assembly contained 4.52 Gb, with a contig N50 value of 5.28 Mb and scaffold N50 value of 44.01 Mb, of which 4.06 Gb (89.87%) of the assembly were anchored onto 90 chromosome-level pseudomolecules comprising 15 homologous groups with 6 allelic chromosomes in each. A total of 153,288 protein-coding genes were predicted, of which 98.60% were functionally annotated. Repetitive sequences accounted for 64.02% of the genome; and 110,480 rRNAs, 12,297 tRNAs, 1,483 miRNAs, and 3,510 snRNA genes were also identified. This genome assembly fills the knowledge gap in the autohexaploid persimmon genome, which is conducive in the study on the regulatory mechanisms underlying the major economically advantageous traits of persimmons and promoting breeding programs.
Subject terms: Genome duplication, Genome
Background & Summary
Persimmon (Diospyros kaki Thunb.), a member of the Ebenaceae, is an important fruit tree species that originated in East Asia and was successively introduced to Europe and America in the 18th–20th centuries1. The cultivated area of persimmon had reached 1.01 million ha, with a total yield of 4.24 million tons globally in 2020 (www.fao.org). The persimmon industry is negatively affected by labor-intensive artificial de-astringency treatment, transportation difficulties, short shelf life, and limited processing2.
To enhance the persimmon industry, superior cultivars suitable for fresh-eating and processing are urgently needed. Current persimmon cultivars are generally classified into either pollination-constant non-astringent (PCNA) or non-PCNA3,4, based on the natural de-astringency capacity of fruits at the commercial maturity stage (fruits with mature peel color but not soft). The PCNA contains the Japanese-type PCNA (JPCNA) and Chinese-type PCNA (CPCNA), which are edible without any artificial de-astringency treatment and have high market valuable5. The non-PCNA includes pollination-variant non-astringent (PVNA), pollination-constant astringent (PCA), and pollination-variant astringent (PVA)1.
In the PCNA breeding program, inbreeding repression limits the efficiency due to the high genetic similarity among the JPCNA cultivars6,7. Modern molecular refinement breeding methods, including CRISPR/Cas9, are potentially effective for breeding new superior cultivars. The molecular mechanisms regulating crucial traits should first be determined accurately when using these new methods. The natural de-astringency capacity of JPCNA is controlled by a recessive allele at a single locus of ASTRINGENCY (AST)8–10. A previous study suggested that the natural de-astringency capacity of CPCNA is controlled by dominant alleles11,12, indicating that the CPCNA type may be more effective for breeding new superior PCNA cultivars, although the precise natural de-astringency mechanism of CPCNAs remains unknown.
The most common persimmon cultivar is hexaploidy (2n = 6x = 90). Owing to a lack in genomic data, the principal molecular mechanisms underlying the natural de-astringency of CPCNA and other crucial traits, including fruit size, shape, and flavor, of hexaploidy persimmon remains challenging to understand. Fortunately, the publication of genomes of hexaploid persimmon close relatives Diospyros lotus (2n = 2x = 30) and Diospyros oleifera (2n = 2x = 30) provided help for the study of persimmon biology13–16. Hexaploid and diploid persimmons are different species with discrepant genomic information. Taking the diploid persimmon genome as a reference, data on the regulation mechanism of some traits in hexaploid persimmon are limited, which contributes to the reduction of breeding efficiency; hence, the desperate need for the hexaploid persimmon genome assembly, that will help in both basic and applied research.
The assembly of polyploid genomes is a major technical challenge hindered by repeat content, transposable elements, high heterozygosity, and gene content17. The assembly of autopolyploids with smaller genetic distances is more susceptible to the misassignment of sub-genome fragments than allopolyploids. With the advancement of sequencing and assembly technology, the autopolyploid genomes of some plants have been reported, such as Ipomoea batatas18, Saccharum spontaneum19,20, Medicago sativa21, and Solanum tuberosum22, which provide a reference for current genome assemblies.
This study uses PacBio circular consensus sequencing (CCS) and high-throughput chromosome conformation capture (Hi-C) technologies to generate an allele-aware chromosome-level genome assembly for D. kaki. The current genomic information will provide a molecular platform for future research and elaborate breeding programs.
Methods
Sampling and sequencing
‘Xiaoguotianshi’ persimmon is one of the five varieties of the CPCNA persimmon ‘Luotiantianshi’ with a good taste and higher soluble solids content than other CPCNA persimmons. The young leaves of D. kaki ‘Xiaoguotianshi’ and D. lotus (wild germplasm) were collected from the Persimmon Germplasm Resources Nursery of Research Institute of Non-timber Forestry, Chinese Academy of Forestry (Yuanyang County, Henan Province, China, 34°55′18″–34 °56′27″N, 113°46′14″–113°47′35″E).
Genomic DNA was extracted from the young leaf tissue of D. kaki using a DNAsecure Plant Kit (TIANGEN, Beijing, China). Sequencing libraries with insert sizes of 350 bp were constructed using a library construction kit, following manufacturer’s instructions (Illumina, San Diego, CA, USA). The libraries were sequenced using the Illumina HiSeq X platform.
For the PacBio library, the DNA was used to construct 15-kb-insert-size SMRTbell libraries using the SMRTbell Express Template Prep Kit 2.0, following manufacturer’s instructions (PacBio, CA). Then, libraries were sequenced using PacBio Sequel II, and HiFi reads were obtained using the CCS tool (https://github.com/PacificBiosciences/ccs; v6.0.0) by setting ‘min-passes = 3, min-rq = 0.99’.
For the Hi-C library, formaldehyde was used to fix the chromatin. Leaf cells were lysed, and HindIII endonuclease was used to digest the fixed chromatin. The 5 overhangs of the DNA were recovered with biotin-labeled nucleotides, and the resulting blunt ends were ligated to each other using DNA ligase. Proteins were removed with protease to release DNA molecules from the crosslinks. The purified DNA was sheared into 350-bp fragments and ligated to adaptors23. The biotin-labeled fragments were extracted using streptavidin beads; following PCR enrichment, the libraries were sequenced on an Illumina HiSeq X instrument.
For RNA sequencing, total RNA was extracted from the leaf, stem and fruit tissues using an RNAprep Pure Plant Kit (TIANGEN, Beijing, China), and genomic DNA contaminants were removed using RNase-Free DNase I (TIANGEN, Beijing, China). The RNA integrity was evaluated using 1.0% agarose gel stained with ethidium bromide (EB), while its quality and quantity were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). The integrated RNA was then used for cDNA library construction, Illumina and PacBio sequencing. The cDNA libraries were constructed using the NEBNext Ultra RNA Library Prep Kit (NEB, MA, USA) for Illumina and SMRTbell Express Template Prep Kit 2.0 (PacBio, CA, USA) for PacBio, following the manufacturers’ instructions. Prepared libraries were sequenced on the Illumina HiSeq X and PacBio Sequel platform.
Genome size estimation
K-mer frequency analysis was used to determine genome characteristics24. The genome size of D. kaki was calculated based on k-mer (k = 27) statistics using the modified Lander–Waterman algorithm. The total length of the sequence reads was divided by the sequencing depth; the peak value of the frequency curve represented the overall sequencing depth. We estimated the genome size using the following formula: (N × (L−K + 1) − B)/D = G, where N is the total number of the sequence reads, L is the average length of the sequence reads, K is the K-mer length (27 bp)25, B is the total number of low-frequency K-mers (frequency ≤ 1 in this analysis), G is the genome size, and D is the overall depth estimated via the K-mer distribution. Heterozygosity was reflected in the distribution of the number of distinct k-mers (k = 27). On the basis of a total of 222,144,314,592 27-mer and a peak 27-mer depth of 49, the estimated genome size was 4533.56 Mb (Fig. 1).
Fig. 1.
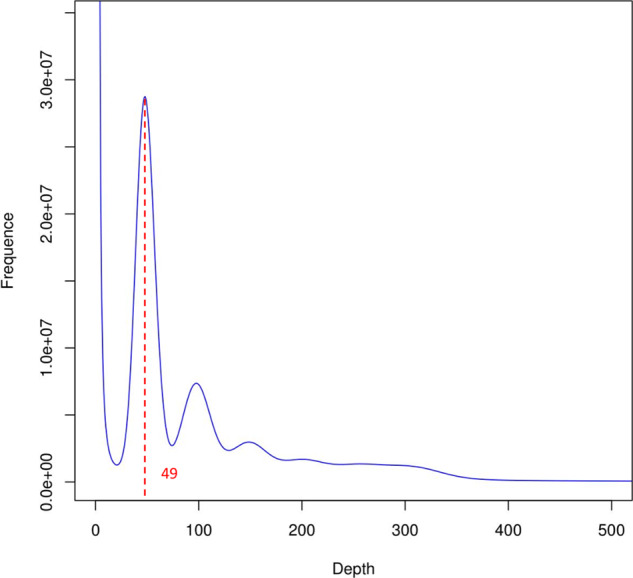
Results of 27-mer frequency analysis to estimate the D. kaki genome size. The haplotype genome size was calculated by dividing the total K-mer count by coverage-depth (222,144,314,592 /49 = 4,533,557,441).
The genome size of the sequenced individuals was confirmed using flow cytometry. Approximately 20–50 mg of fresh leaves of D. kaki and D. lotus were chopped using a razor blade in 1 ml of LB01 buffer (15 mM Tris, 2 mM Na2EDTA, 0.5 mM spermine tetrahydrochloride, 80 mM KCl, 20 mM NaCl, 0.1% (vol/vol) Triton X-100) adjusted to pH 7.5 with 1 M NaOH and b-mercaptoethanol to 15 mM. Cell culture was collected by gentle pipetting and filtered through a 400-mesh nylon strainer. The samples were stained with 100 μg/ml PI and 100 μg/ml RNase in an ice bath for 10 min before analysis using a MoFlo-XDP flow cytometer (Beckman Coulter Inc., USA).
Nuclear fluorescence was measured using a MoFlo-XDP high-speed flow cytometer with a 70 μm ceramic nozzle at a sheath pressure of 60 psi. PI fluorescence was detected with a solid-state laser (488 nm) and a 625-/26-nm HQ band-pass filter. The FL3-Height/SSC-Height gate method eliminated debris, cell fragments, and dead cells. Single and double cells were discriminated using FL3-Height /FL3-Area. The final results showed that the genome size of D. kaki was 4.61 Gb (Fig. 2).
Fig. 2.
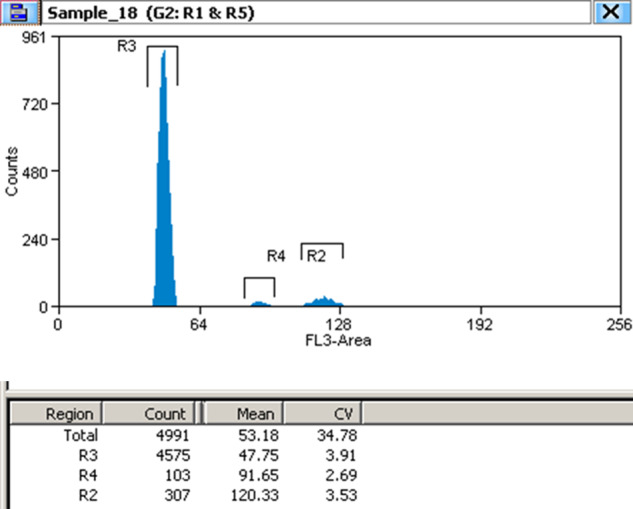
Results of flow cytometry analysis to estimate the D. kaki genome size. The D. lotus genome (2n = 2x = ~1.76 Gb) served as an internal reference standard. Peak R3 showed DNA amount of D. lotus. Peak R2 showed duplicated DNA amount of D. lotus. The ratio of peak mean was equal 2.52 (R2/R3) and 1.31 (R2/R4), hence the estimated genome size of D. kaki was 2n = 6x = ~4.44 Gb and 4.61 Gb. Due to the distance between peak R2 and peak R4 is less than the distance between peak R2 and peak R3, it is more accurate to estimate genome size of D. kaki 2n = 6x = ~4.61 Gb.
Genome assembly
In total, 179.09 Gb PacBio HiFi long reads (8 SMRT cell; 39.53X coverage) and 445.72 Gb Hi-C paired-end reads (98.39X coverage) were obtained (Table 1). D. kaki genome was assembled with Hifisam (v0.13-r308)26 using PacBio HiFi reads with default parameter settings. After initial assembly, Hi-C sequencing data were aligned to the assembled contigs using the Burrows-Wheeler Aligner (BWA) mem option27, while pseudo-chromosomes were constructed based on ALLHIC (v0.9.8)20. We configured the parameter setting -K 90--minREs 50--maxlinkdensity 3--NonInformativeRabio 2. Finally, Hi-C scaffolding yielded 90 chromosome-length scaffolds. The final assembly contained 4.52 Gb with a contig N50 value of 5.28 Mb and scaffold N50 value of 44.01 Mb, respectively; 4.06 Gb (89.87%) of the assembly was anchored onto 90 chromosome-level pseudomolecules comprising 15 homologous groups, with six allelic chromosomes in each. The assignment to genome haplotypes was based on chromosome length (Tables 2, 3; Figs. 3, 4).
Table 1.
Statistics of data for genome assemblies of D.kaki.
| Read type | Read base (Gb) | Number of reads | Mean read length (bp) | Read length (N50) |
|---|---|---|---|---|
| HiFi reads | 179.09 | 12,077,194 | 14,828 | 14,875 |
| Hi-C reads | 445.72 | 1,485,733,937 | 150 | — |
Table 2.
Summary of D. kaki genome assembly.
| Genome assembly | Number | Size |
|---|---|---|
| Total contigs | 22,172 | 4.52 Gb |
| Contig N50 | 220 | 5.28 Mb |
| Contig N90 | 6,674 | 38.41 Kb |
| Total scaffolds | 12,715 | 4.52 Gb |
| Scaffold N50 | 45 | 44.01 Mb |
| Scaffold N90 | 97 | 751.00 Kb |
| Pseudo-chromosomes | 90 | 4.06 Gb |
Table 3.
Statistics of chromosome length in D. kaki genome.
| Chromosome | DkaA | DkaB | DkaC | DkaD | DkaE | DkaF |
|---|---|---|---|---|---|---|
| Length (bp) | Length (bp) | Length (bp) | Length (bp) | Length (bp) | Length (bp) | |
| chr1 | 61,854,060 | 56,088,851 | 55,416,202 | 53,982,736 | 53,941,820 | 46,325,742 |
| chr2 | 61,828,268 | 61,482,766 | 53,378,268 | 51,130,973 | 51,039,378 | 47,917,465 |
| chr3 | 59,673,878 | 58,299,181 | 56,577,420 | 55,427,238 | 52,248,968 | 50,631,600 |
| chr4 | 52,914,409 | 40,854,466 | 40,785,913 | 39,946,333 | 39,656,787 | 38,564,113 |
| chr5 | 48,795,537 | 48,609,808 | 48,061,808 | 46,857,559 | 45,974,615 | 27,067,684 |
| chr6 | 47,859,392 | 46,520,457 | 44,013,165 | 40,127,273 | 39,913,631 | 36,538,425 |
| chr7 | 44,533,068 | 43,838,789 | 41,532,886 | 41,183,422 | 39,021,685 | 37,333,795 |
| chr8 | 42,161,358 | 40,923,269 | 40,632,031 | 40,148,576 | 39,064,181 | 36,013,403 |
| chr9 | 56,056,215 | 54,404,229 | 43,939,775 | 42,977,330 | 42,624,317 | 41,332,283 |
| chr10 | 51,658,814 | 43,541,839 | 42,887,684 | 40,037,245 | 34,419,546 | 23,313,593 |
| chr11 | 47,898,209 | 46,534,985 | 41,284,961 | 37,036,693 | 36,825,070 | 32,340,415 |
| chr12 | 47,594,529 | 46,266,353 | 46,003,302 | 45,360,257 | 43,649,862 | 41,231,788 |
| chr13 | 43,497,036 | 42,187,849 | 40,963,846 | 40,947,171 | 40,100,542 | 38,578,397 |
| chr14 | 46,638,359 | 45,764,966 | 40,779,368 | 40,279,559 | 40,271,275 | 39,884,115 |
| chr15 | 50,207,220 | 50,127,302 | 49,236,925 | 48,601,615 | 46,818,767 | 44,634,995 |
| Total | 763,170,352 | 725,445,110 | 685,493,554 | 664,043,980 | 645,570,444 | 581,707,813 |
Fig. 3.

Overview of the D. kaki genome. From the outer ring to the inner ring are Chromosome, Gene density, TE density, GC content, and Synteny.
Fig. 4.

Overview of Hi-C contacts in the heat map visualization for assembled chromosomes.
Repetitive sequence annotation
Transposable elements (TEs) in the D. kaki genome were identified by combining de novo- and homology-based approaches. For the de novo-based approach, we used RepeatScout (v1.0.5; https://github.com/mmcco/RepeatScout)28, RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html), and LTR_FINDER (v1.0.7; https://github.com/xzhub/LTR_Finder)29 to build a de novo repeat library. For the homology-based approach, we used RepeatMasker (v3.3.0; http://www.repeatmasker.org/) against the Repbase TE library (http://www.girinst.org/server/RepBase/)30 with score cut-off of 225 and RepeatProteinMask (v4.0.5; http://www.repeatmasker.org/) against the TE protein database31 with a p-value cut-off of 1e-4. Tandem repeats were identified using Tandem Repeats Finder (v4.0.7; https://tandem.bu.edu/trf/)32 with parameters ‘matching weight: 2, mismatching penalty: 7, indel penalty: 7, minimum alignment score:50, maximum period size: 2000’. Ultimately, a total of 2.90 Gb of repetitive elements occupying 64.02% of the D. kaki genome were annotated (Table 4). Most of the repeats were long terminal repeats (LTRs) (51.28% of the genome; Table 5). The DNA, LINE, and SINE classes accounted for 5.93%, 2.66%, and 0.03% of the genome, respectively (Table 5).
Table 4.
Summary of repeat sequences in D. kaki genome.
| Type | Length (bp) | Percent (%) |
|---|---|---|
| Tandem Repeat Finder | 616,854,190 | 13.64 |
| RepeatMasker | 2,742,911,542 | 60.64 |
| RepeatProteinMask | 762,622,147 | 16.86 |
| Total | 2,896,122,867 | 64.02 |
Table 5.
Summary of TE sequences in D. kaki genome.
| Type | Length (bp) | Percent (%) |
|---|---|---|
| DNA | 268,202,039 | 5.93 |
| LINE | 120,230,805 | 2.66 |
| SINE | 1,375,715 | 0.03 |
| LTR | 2,319,894,281 | 51.28 |
| Unknown | 121,044,880 | 2.68 |
| Total | 2,823,554,370 | 62.42 |
Gene prediction and annotation
Homology-based, de novo, and transcriptome-based predictions were used to predict protein-coding genes in the D. kaki genome. Homologous proteins from five plant genomes (Arabidopsis thaliana, D. oleifera, D. lotus, Actinidia chinensis, and Camellia sinensis) were downloaded from Ensembl Plants (http://plants.ensembl.org/index.html) and NCBI (https://www.ncbi.nlm.nih.gov/). The protein sequences were then aligned to the D. kaki genome assembly using tblastN33, with an E-value cut-off of 1e-5. The BLAST hits were conjoined using a Solar software34. GeneWise (https://www.ebi.ac.uk/Tools/psa/genewise) was used to predict the exact gene structure of the corresponding genomic regions in each BLAST hit (Homo-set)35. The published RNA-seq data of female flowers and fruit at different developmental stages, and 0.33 Gb new sequencing RNA-seq data of the young leaves and stems of ‘Xiaoguotianshi’ (three biological replicates) were mapped to the D. kaki genome using HISAT2 (https://daehwankimlab.github.io/hisat2/, v2.2.1)36 and Cufflinks (http://cole-trapnell-lab.github.io/cufflinks/, v2.1.1)37 (Table 6). A total of 70.54 Gb Iso-seq data from PacBio transcriptome sequencing of mixed samples containing the young leaves, stems, and fruits of ‘Xiaoguotianshi’ (three biological replicates) were used to create several pseudo-ESTs. These pseudo-ESTs were mapped to the assembly, and gene models were predicted using PASA (http://pasapipeline.github.io/)38 (Table 6). This gene set was denoted as the PASA-T-set and used to train ab initio gene prediction programs. Five ab initio gene prediction programs, namely, Augustus (http://augustus.gobics.de/, v3.2.3), GENSCAN (http://genes.mit.edu/GENSCAN.html, v1.0), GlimmerHMM (http://ccb.jhu.edu/software/glimmerhmm/, v3.0.1), geneid (http://genome.crg.es/software/geneid/), and SNAP (http://korflab.ucdavis.edu/software.html) were used to predict coding regions in the repeat-masked genome39–42. Gene model evidence from homo-set, cufflinks-set, PASA-T-set, and ab initio programs were combined using EVidenceModeler (EVM) (http://evidencemodeler.sourceforge.net/) into a non-redundant set of gene structures43.
Table 6.
Statistics of RNA-seq and Iso-seq.
| Read type | Read base (Gb) | Number of reads | Mean read length (bp) | Read_length (N50) |
|---|---|---|---|---|
| RNA-seq | 0.33 | — | 150 | — |
| Iso-seq | 70.54 | 715,846 | 98,537 | 166,494 |
Functional annotation of protein-coding genes was performed using BLASTP (E-value: 1e-05) against two integrated protein sequence databases44: SwissProt (http://web.expasy.org/docs/swiss-prot_guideline.html) and NR (ftp://ftp.ncbi.nih.gov/blast/db/). Protein domains were annotated by searching against the InterPro (http://www.ebi.ac.uk/interpro/, v32.0) and Pfam (https://pfam-legacy.xfam.org/.org/, v27.0) databases using InterProScan (v4.8) and HMMER (http://www.hmmer.org/, v3.1), respectively45–48. Gene ontology (GO, http://www.geneontology.org/page/go-database) terms for each gene were obtained from the corresponding InterPro or Pfam entries. The pathways in which the genes might be involved were assigned using BLAST against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.kegg.jp/kegg/kegg1.html, release 53), with an E-value cut-off of 1e-05. Overall, a total of 153,288 protein-coding genes were predicted with an average sequence length of 7,397.94 bp and an average CDS length of 1,153.82 bp (Table 7). Of these, 135,446 genes are anchored to 90 chromosomes (Table 8). On average, each predicted gene contained 5.01 exons with an average sequence length of 230.33 bp (Table 7). 98.60% of the genes were functionally annotated via similarity searches against homologous sequences and protein domains (Table 9).
Table 7.
Summary of gene structure prediction in D. kaki genome.
| Gene set | Number | Average gene length (bp) | Average CDS length (bp) | Average exons per gene | Average exon length (bp) | Average intron length (bp) | |
|---|---|---|---|---|---|---|---|
| De novo | Augustus | 177,974 | 5,346.69 | 1,022.08 | 4.14 | 246.89 | 1,377.34 |
| GlimmerHMM | 321,260 | 10,295.63 | 601.95 | 3.09 | 194.89 | 4,641.03 | |
| SNAP | 319,132 | 7,138.48 | 610.14 | 3.33 | 183.25 | 2,802.42 | |
| Geneid | 390,239 | 3,850.38 | 577.88 | 3.24 | 178.29 | 1,460.13 | |
| Genscan | 230,229 | 10,666.94 | 976.81 | 5.22 | 187.04 | 2,294.9 | |
| Homolog | Dlo | 359,850 | 1,914.73 | 816.37 | 2.76 | 296.06 | 624.97 |
| Ath | 207,508 | 3,745.85 | 932.2 | 3.17 | 294.28 | 1,297.97 | |
| Dol | 227,166 | 4,359.26 | 895.42 | 3.6 | 248.67 | 1,331.78 | |
| Ach | 245,176 | 3,571.73 | 835.91 | 2.77 | 301.63 | 1,544.5 | |
| Csi | 145,462 | 5,893.18 | 1,125.04 | 4.23 | 266.16 | 1,477.65 | |
| RNA-seq | Cufflinks | 239,070 | 11,977.18 | 2,111.23 | 6.58 | 320.70 | 1,767.08 |
| PASA | 54,520 | 10,118.79 | 1,743.09 | 6.33 | 275.39 | 1,571.55 | |
| EVM | 190,809 | 6,293.70 | 1,010.92 | 4.37 | 231.18 | 1,566.28 | |
| PASA-update | 190,490 | 6,309.20 | 1,018.09 | 4.39 | 231.90 | 1,560.69 | |
| Final set | 153,288 | 7,397.94 | 1,153.82 | 5.01 | 230.33 | 1,557.38 | |
Table 8.
Statistics of chromosome gene number in D. kaki genome.
| Chromosome | DkaA | DkaB | DkaC | DkaD | DkaE | DkaF |
|---|---|---|---|---|---|---|
| gene number | gene number | gene number | gene number | gene number | gene number | |
| chr1 | 2,633 | 2,509 | 2,486 | 2,420 | 2,259 | 2,235 |
| chr2 | 1,922 | 1,970 | 1,973 | 2,140 | 1,974 | 1,894 |
| chr3 | 2,084 | 2,018 | 2,117 | 1,967 | 2,172 | 2,028 |
| chr4 | 1,481 | 1,515 | 1,465 | 1,522 | 1,445 | 1,469 |
| chr5 | 1,717 | 1,656 | 1,667 | 1,638 | 1,675 | 1,114 |
| chr6 | 1,442 | 1,486 | 1,451 | 1,386 | 1,306 | 1,207 |
| chr7 | 1,466 | 1,355 | 1,265 | 1,296 | 1,363 | 1,309 |
| chr8 | 1,261 | 1,515 | 1,318 | 1,031 | 1,271 | 1,103 |
| chr9 | 1,189 | 1,264 | 1,235 | 1,349 | 1,204 | 1,146 |
| chr10 | 1,326 | 1,207 | 1,162 | 1,216 | 1,147 | 845 |
| chr11 | 1,036 | 1,057 | 1,049 | 962 | 1,114 | 961 |
| chr12 | 1,556 | 1,540 | 1,489 | 1,515 | 1,621 | 1,357 |
| chr13 | 1,144 | 1,167 | 1,123 | 1,193 | 1,137 | 1,179 |
| chr14 | 1,351 | 1,198 | 1,250 | 1,216 | 1,263 | 1,182 |
| chr15 | 1,766 | 1,762 | 1,822 | 1,768 | 1,687 | 1,695 |
| Total | 23,374 | 23,219 | 22,872 | 22,619 | 22,638 | 20,724 |
Table 9.
Statistics of gene function annotation in D. kaki genome.
| Database | Number | Percent (%) | |
|---|---|---|---|
| NR | 134,846 | 88.0 | |
| Swiss-Prot | 105,533 | 68.8 | |
| KEGG | 100,175 | 65.4 | |
| InterPro | All | 150,267 | 98.0 |
| Pfam | 103,027 | 67.2 | |
| GO | 136,771 | 89.2 | |
| Annotated | 151,088 | 98.6 | |
tRNA genes were identified using the tRNAscan-SE software49. The rRNA fragments were predicted by aligning the rRNA sequences using BlastN at an E-value of 1e-10. The miRNA and snRNA genes were predicted using the INFERNAL software50 against the Rfam database (release 9.1)51. As a result, 110,480 rRNA, 12,297 tRNA, 1,483 miRNA, and 3,510 snRNA genes were annotated (Table 10).
Table 10.
Statistics of non-coding RNA in D. kaki genome.
| Type | Copy | Average length (bp) | Total length (bp) | |
|---|---|---|---|---|
| miRNA | 1,483 | 122.95 | 182,342 | |
| tRNA | 12,297 | 75.59 | 929,561 | |
| rRNA | rRNA | 110,480 | 234.59 | 25,917,998 |
| 18 S | 8,496 | 1544.7 | 13,123,748 | |
| 28 S | 28,169 | 141.96 | 3,998,950 | |
| 5.8 S | 7,158 | 161.1 | 1,153,155 | |
| 5 S | 66,657 | 114.65 | 7,642,145 | |
| snRNA | snRNA | 3,510 | 112.12 | 393,524 |
| CD-box | 2,593 | 102.02 | 264,544 | |
| HACA-box | 237 | 132.64 | 31,435 | |
| splicing | 670 | 142.11 | 95,211 | |
Data Records
Raw data of genome sequencing and transcriptome sequencing of D. kaki are deposited in the NCBI SRA database under BioProject ID PRJNA810977. The SRA accession number of PacBio HiFi sequencing data are SRR1850047052, SRR1850047153, SRR1850047254, SRR1850047355 SRR1850047456, SRR1850047557, SRR1850047658, and SRR1850047759. The SRA accession number of Hi-C sequencing data are SRR1850048160, SRR1850048261, SRR1850048362, SRR1850048463, SRR1850048564, SRR1850048665, SRR1850048766 and SRR1850048867. The SRA accession number of Illumina sequencing data are SRR1850047968 and SRR1850048069. The SRA accession number of Iso-seq data SRA accession number is SRR1850046370. The SRA accession number of some RNA-seq data are SRR1850046471, SRR1850046572, SRR1850046673, SRR1850047874, SRR1850048975, SRR1850049076 and SRP15171577. The others RNA-seq data have been deposited in the NCBI SRA database under the SRR1637198478, SRR1637198579, SRR1637198680, SRR1637198781, SRR1637198882, SRR1637198983, SRR1637199084, SRR1637199185, SRR1637199286, SRR1637199387, SRR1637199488, SRR1637199589, SRR1637199690, SRR1637199791 and SRR1637199792, which is associated with the Bioproject ID PRJNA771936. The assembled genome sequence has been deposited at GenBank with accession number JAQSGO00000000093. Other data, such as gene structure annotation, predicted CDS and protein sequences, annotation of TEs, tandem repeat sequences, tRNA genes, miRNA genes, snRNA genes, and rRNA genes, are available at FigShare database94.
Technical Validation
Assessment of the completeness of the genome assembly using CEGMA indicated a 95.56% (Haplotype: DkaA 89.92%; DkaB 90.73%; DkaC 91.13%; DkaD 86.69%; DkaE 89.11%; DkaF 86.69%) coverage of the conserved core eukaryotic genes, while the BUSCO (v5.2.2; embryophyta odb10 database)95 results indicated that the genome and gene set was 99.50% (Haplotype: DkaA 92.70%; DkaB 93.50%; DkaC 92.10%; DkaD 90.10%; DkaE 90.20%; DkaF 84.60%) and 97.50 (Haplotype: DkaA 87.10%; DkaB86.60%; DkaC 86.10%; DkaD 83.90%; DkaE 84.60%; DkaF 79.80%) complete, respectively (Tables 11, 12), showing that the individual haplotypes lack genes present elsewhere in the genome. Additionally, 99.86% (Haplotype: DkaA 94.88%; DkaB 94.59%; DkaC 93.70%; DkaD 93.94%; DkaE 92.97%; DkaF 90.09%) of the high-quality short reads were mapped back to the assembly (Table 13). All in all, these results of these assessments indicate to us that the D. kaki genome assembly is complete and high quality.
Table 11.
Assessment of the completeness of the genome assembly.
| Genome | BUSCO | CEGMA |
|---|---|---|
| The whole genome | C:99.50% [S:0.90%, D:98.60%], F:0.20%, M:0.30%, n:1614 | 95.56% |
| DkaA | C:92.70% [S:89.70%, D:3.00%], F:2.00%, M:5.30%, n:1614 | 89.92% |
| DkaB | C:93.50% [S:91.00%, D:2.50%], F:1.70%, M:4.80%, n:1614 | 90.73% |
| DkaC | C:92.10% [S:89.70%, D:2.40%], F:1.70%, M:6.20%, n:1614 | 91.13% |
| DkaD | C:90.10% [S:87.00%, D:3.10%], F:2.00%, M:7.90%, n:1614 | 86.69% |
| DkaE | C:90.20% [S:87.60%, D:2.60%], F:2.70%, M:7.10%, n:1614 | 89.11% |
| DkaF | C:84.60% [S:82.20%, D:2.40%], F:2.10%, M:13.30%, n:1614 | 86.69% |
Table 12.
Assessment of the completeness of the gene set.
| Gene set | BUSCO |
|---|---|
| The whole gene set | C:97.50% [S:2.50%, D:95.00%], F:1.90%, M:0.60%, n:1614 |
| DkaA | C:87.10% [S:83.50%, D:3.60%], F:5.30%, M:7.60%, n:1614 |
| DkaB | C:86.60% [S:83.10%, D:3.50%], F:6.20%, M:7.20%, n:1614 |
| DkaC | C:86.10% [S:82.90%, D:3.20%], F:6.60%, M:7.30%, n:1614 |
| DkaD | C:83.90% [S:79.70%, D:4.20%], F:6.50%, M:9.60%, n:1614 |
| DkaE | C:84.60% [S:81.70%, D:2.90%], F:6.60%, M:8.80%, n:1614 |
| DkaF | C:79.80% [S:76.10%, D:3.70%], F:5.70%, M:14.50%, n:1614 |
Table 13.
Coverage statistics of D. kaki genome.
| The whole genome | Reads | Mapping rate (%) | 99.86 |
| Average sequencing depth | 27.29X | ||
| Coverage (%) | 99.72 | ||
| Genome | Coverage at least 4X (%) | 99.45 | |
| Coverage at least 10X (%) | 97.2 | ||
| DkaA | Reads | Mapping rate (%) | 94.88 |
| Average sequencing depth | 306.75X | ||
| Coverage (%) | 99.85 | ||
| Genome | Coverage at least 4X (%) | 99.79 | |
| Coverage at least 10X (%) | 99.73 | ||
| DkaB | Reads | Mapping rate (%) | 94.59 |
| Average sequencing depth | 325.76X | ||
| Coverage (%) | 99.93 | ||
| Genome | Coverage at least 4X (%) | 99.89 | |
| Coverage at least 10X (%) | 99.84 | ||
| DkaC | Reads | Mapping rate (%) | 93.7 |
| Average sequencing depth | 341.1X | ||
| Coverage (%) | 99.97 | ||
| Genome | Coverage at least 4X (%) | 99.95 | |
| Coverage at least 10X (%) | 99.93 | ||
| DkaD | Reads | Mapping rate (%) | 93.34 |
| Average sequencing depth | 346.92X | ||
| Coverage (%) | 99.99 | ||
| Genome | Coverage at least 4X (%) | 99.97 | |
| Coverage at least 10X (%) | 99.95 | ||
| DkaE | Reads | Mapping rate (%) | 92.97 |
| Average sequencing depth | 356.66X | ||
| Coverage (%) | 99.99 | ||
| Genome | Coverage at least 4X (%) | 99.98 | |
| Coverage at least 10X (%) | 99.96 | ||
| DkaF | Reads | Mapping rate (%) | 90.09 |
| Average sequencing depth | 375.72X | ||
| Coverage (%) | 99.99 | ||
| Genome | Coverage at least 4X (%) | 99.99 | |
| Coverage at least 10X (%) | 99.97 |
Inter-genomic comparison analysis revealed a distinct 6-to-1 syntenic relationship between D. kaki and D. oleifera (Fig. 5), which further supported the high quality of the D. kaki assembly.
Fig. 5.

The syntenic dot plots of D. kaki and D. oleifera.
Acknowledgements
This work was supported by the National Key R & D Program of China (2018YFD1000606).
Author contributions
Fu, J. and Li, F. designed the project, and they are the corresponding authors of this paper. Han, W., Suo, Y., and Diao, S. collected the samples. Li, H., Sun P., Wang, Y. and Zhang, Z. conducted genome assembly, analyze data, and wrote the manuscript, and they contributed equally to this study. Yang, J. provided intellectual insights. All authors critically commented and approved the manuscript.
Code availability
All software used in this work are in the public domain, with parameters described in the Methods section. The commands used in the processing were all executed according to the manuals and protocols of the corresponding bioinformatics software.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Fangdong Li, Email: lifangdong66@163.com.
Jianmin Fu, Email: fjm371@163.com.
References
- 1.Yonemori K, Sugiura A, Yamada M. Persimmon Genetics and Breeding. Plant. Breed. Rev. 2000;19:191–225. [Google Scholar]
- 2.Itamura H, Zheng Q, Akaura K. Industry and research on persimmon in Japan. III. Int. Sym. Persimmon. 2004;685:37–44. [Google Scholar]
- 3.Akagi T, Katayama-Ikegami A, Yonemori K. Proanthocyanidin biosynthesis of persimmon (Diospyros kaki Thunb.) fruit. Sci. Hortic. 2011;130:373–380. doi: 10.1016/j.scienta.2011.07.021. [DOI] [Google Scholar]
- 4.Xu J, Zhang Q, Xu L, Guo D, Luo Z. Recent developments in deastringency mechanism of persimmon fruit. Acta. Hortic. Sinica. 2016;43:1653. [Google Scholar]
- 5.Sato A, Yamada M. Persimmon breeding in Japan for pollination-constant non-astringent (PCNA) type with marker-assisted selection. Breeding. Sci. 2016;66:60–68. doi: 10.1270/jsbbs.66.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamada M, Yamane H, Ukai Y. Genetic analysis of Japanese persimmon fruit weight. J. Am. Soc. Hortic. Sci. 1994;119:1298–1302. doi: 10.21273/JASHS.119.6.1298. [DOI] [Google Scholar]
- 7.Pei X, Zhang Q, Guo D, Liu J, Luo Z. Development of genetic improvement in Chinese PCNA persimmon. J. Fruit. Sci. 2015;32:313–321. [Google Scholar]
- 8.Akagi T, Tao R, Tsujimoto T, Kono A, Yonemori K. Fine genotyping of a highly polymorphic ASTRINGENCY-linked locus reveals variable hexasomic inheritance in persimmon (Diospyros kaki Thunb.) cultivars. Tree. Genet. Genomes. 2012;8:195–204. doi: 10.1007/s11295-011-0432-0. [DOI] [Google Scholar]
- 9.Ikeda I, Yamada M, Kurihara A, Nishida T. Inheritance of astringency in Japanese persimmon. J. Jpn. Soc. Hortic. Sci. 1985;54:39–45. doi: 10.2503/jjshs.54.39. [DOI] [Google Scholar]
- 10.Yamada M, Sato A. Segregation for fruit astringency type in progenies derived from crosses of ‘Nishimurawase’× pollination constant non-astringent genotypes in oriental persimmon (Diospyros kaki Thunb.) Sci. Hortic. 2002;92:107–111. doi: 10.1016/S0304-4238(01)00285-0. [DOI] [Google Scholar]
- 11.Ikegami A, et al. Segregations of astringent progenies in the F1 populations derived from crosses between a chinese pollination-constant nonastringent (PCNA) ‘Luo tian tian shi’, and japanese PCNA and pollination-constant astringent (PCA) cultivars of Japanese origin. HortScience. 2006;41:561–563. doi: 10.21273/HORTSCI.41.3.561. [DOI] [Google Scholar]
- 12.Ikegami A, Yonemori K, Sugiura A, Sato A, Yamada M. Segregation of astringency in F1 progenies derived from crosses between pollination-constant, nonastringent persimmon cultivars. HortScience. 2004;39:371–374. doi: 10.21273/HORTSCI.39.2.371. [DOI] [Google Scholar]
- 13.Sun P, et al. Genomics-based discrimination of 2n gamete formation mechanisms in polyploids: a case study in nonaploid Diospyros kaki ‘Akiou’. G3-Genes. Genom. Genet. 2021;11:jkab188. doi: 10.1093/g3journal/jkab188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akagi T, et al. The persimmon genome reveals clues to the evolution of a lineage-specific sex determination system in plants. PLoS. Genet. 2020;16:e1008566. doi: 10.1371/journal.pgen.1008566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suo Y, et al. A high-quality chromosomal genome assembly of Diospyros oleifera Cheng. GigaScience. 2020;9:giz164. doi: 10.1093/gigascience/giz164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Q, et al. The persimmon (Diospyros oleifera Cheng) genome provides new insights into the inheritance of astringency and ancestral evolution. Hortic. Res. 2019;6:1–15. doi: 10.1038/s41438-019-0227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kyriakidou M, Tai HH, Anglin NL, Ellis D, Strömvik MV. Current strategies of polyploid plant genome sequence assembly. Front. Plant. Sci. 2018;9:1660. doi: 10.3389/fpls.2018.01660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, et al. Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants. 2017;3:696–703. doi: 10.1038/s41477-017-0002-z. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018;50:1565–1573. doi: 10.1038/s41588-018-0237-2. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Zhang S, Zhao Q, Ming R, Tang H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants. 2019;5:833–845. doi: 10.1038/s41477-019-0487-8. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 2020;11:1–11. doi: 10.1038/s41467-020-16338-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun H, et al. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat. Genet. 2022;54:342–348. doi: 10.1038/s41588-022-01015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 2011;43:1059–1065. doi: 10.1038/ng.947. [DOI] [PubMed] [Google Scholar]
- 24.Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv. preprint. arXiv:1308.2012 (2013).
- 25.Zhang Q, et al. The genome of Prunus mume. Nat. Commun. 2012;3:1–8. doi: 10.1038/ncomms2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat.Methods. 2021;18:170–175. doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–2851. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 29.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic. Acids. Res. 2007;35:W265–W268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC. Bioinformatics. 2006;7:1–7. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. 2004;5:4.10. 1–4.10. 14. doi: 10.1002/0471250953.bi0410s05. [DOI] [PubMed] [Google Scholar]
- 32.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic. Acids. Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Yu X, Zheng H, Wang J, Wang W, Su B. Detecting lineage-specific adaptive evolution of brain-expressed genes in human using rhesus macaque as outgroup. Genomics. 2006;88:745–751. doi: 10.1016/j.ygeno.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 35.Cook CE, Bergman MT, Cochrane G, Apweiler R, Birney E. The European Bioinformatics Institute in 2017: data coordination and integration. Nucleic. Acids. Res. 2018;46:D21–D29. doi: 10.1093/nar/gkx1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh, S. & Chan, C.-K.K. Analysis of RNA-Seq data using TopHat and Cufflinks. Plant. Bioinformatics: Methods. Protoc. 339–361 (2016). [DOI] [PubMed]
- 38.Haas BJ, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic. Acids. Res. 2003;31:5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr. Protoc. Bioinformatics. 2007;18:4.3. 1–4.3. 28. doi: 10.1002/0471250953.bi0403s18. [DOI] [PubMed] [Google Scholar]
- 40.Burge CB, Karlin S. Finding the genes in genomic DNA. Curr. Opin. Struc. Biol. 1998;8:346–354. doi: 10.1016/S0959-440X(98)80069-9. [DOI] [PubMed] [Google Scholar]
- 41.Keller O, Kollmar M, Stanke M, Waack S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics. 2011;27:757–763. doi: 10.1093/bioinformatics/btr010. [DOI] [PubMed] [Google Scholar]
- 42.Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 43.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:1–22. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic. Acids. Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RD, et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic. Acids. Res. 2017;45:D190–D199. doi: 10.1093/nar/gkw1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finn, R.D. et al. HMMER web server: 2015 update. Nucleic. Acids. Res. 30–8 (2015). [DOI] [PMC free article] [PubMed]
- 47.Finn RD, et al. Pfam protein families database. Nucleic. Acids. Res. 2010;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zdobnov, E.M. & Rolf, A. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 847–848 (2001). [DOI] [PubMed]
- 49.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic. Acids. Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Bateman A. Rfam: Annotating Non-Coding RNAs in Complete Genomes. Nucleic. Acids. Res. 2005;33:D121–4. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.2023. NCBI Sequence Read Archive. SRR18500470
- 53.2023. NCBI Sequence Read Archive. SRR18500471
- 54.2023. NCBI Sequence Read Archive. SRR18500472
- 55.2023. NCBI Sequence Read Archive. SRR18500473
- 56.2023. NCBI Sequence Read Archive. SRR18500474
- 57.2023. NCBI Sequence Read Archive. SRR18500475
- 58.2023. NCBI Sequence Read Archive. SRR18500476
- 59.2023. NCBI Sequence Read Archive. SRR18500477
- 60.2023. NCBI Sequence Read Archive. SRR18500481
- 61.2023. NCBI Sequence Read Archive. SRR18500482
- 62.2023. NCBI Sequence Read Archive. SRR18500483
- 63.2023. NCBI Sequence Read Archive. SRR18500484
- 64.2023. NCBI Sequence Read Archive. SRR18500485
- 65.2023. NCBI Sequence Read Archive. SRR18500486
- 66.2023. NCBI Sequence Read Archive. SRR18500487
- 67.2023. NCBI Sequence Read Archive. SRR18500488
- 68.2023. NCBI Sequence Read Archive. SRR18500479
- 69.2023. NCBI Sequence Read Archive. SRR18500480
- 70.2023. NCBI Sequence Read Archive. SRR18500463
- 71.2023. NCBI Sequence Read Archive. SRR18500464
- 72.2023. NCBI Sequence Read Archive. SRR18500465
- 73.2023. NCBI Sequence Read Archive. SRR18500466
- 74.2023. NCBI Sequence Read Archive. SRR18500478
- 75.2023. NCBI Sequence Read Archive. SRR18500489
- 76.2023. NCBI Sequence Read Archive. SRR18500490
- 77.2019. NCBI Sequence Read Archive. SRP151715
- 78.2022. NCBI Sequence Read Archive. SRR16371984
- 79.2022. NCBI Sequence Read Archive. SRR16371985
- 80.2022. NCBI Sequence Read Archive. SRR16371986
- 81.2022. NCBI Sequence Read Archive. SRR16371987
- 82.2022. NCBI Sequence Read Archive. SRR16371988
- 83.2022. NCBI Sequence Read Archive. SRR16371989
- 84.2022. NCBI Sequence Read Archive. SRR16371990
- 85.2022. NCBI Sequence Read Archive. SRR16371991
- 86.2022. NCBI Sequence Read Archive. SRR16371992
- 87.2022. NCBI Sequence Read Archive. SRR16371993
- 88.2022. NCBI Sequence Read Archive. SRR16371994
- 89.2022. NCBI Sequence Read Archive. SRR16371995
- 90.2022. NCBI Sequence Read Archive. SRR16371996
- 91.2022. NCBI Sequence Read Archive. SRR16371997
- 92.2022. NCBI Sequence Read Archive. SRR16371998
- 93.2023. NCBI GenBank. JAQSGO000000000
- 94.Li H, Sun P, Wang Y, Zhang Z. 2023. Persimmon genome. Figshare. [DOI]
- 95.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2023. NCBI Sequence Read Archive. SRR18500470
- 2023. NCBI Sequence Read Archive. SRR18500471
- 2023. NCBI Sequence Read Archive. SRR18500472
- 2023. NCBI Sequence Read Archive. SRR18500473
- 2023. NCBI Sequence Read Archive. SRR18500474
- 2023. NCBI Sequence Read Archive. SRR18500475
- 2023. NCBI Sequence Read Archive. SRR18500476
- 2023. NCBI Sequence Read Archive. SRR18500477
- 2023. NCBI Sequence Read Archive. SRR18500481
- 2023. NCBI Sequence Read Archive. SRR18500482
- 2023. NCBI Sequence Read Archive. SRR18500483
- 2023. NCBI Sequence Read Archive. SRR18500484
- 2023. NCBI Sequence Read Archive. SRR18500485
- 2023. NCBI Sequence Read Archive. SRR18500486
- 2023. NCBI Sequence Read Archive. SRR18500487
- 2023. NCBI Sequence Read Archive. SRR18500488
- 2023. NCBI Sequence Read Archive. SRR18500479
- 2023. NCBI Sequence Read Archive. SRR18500480
- 2023. NCBI Sequence Read Archive. SRR18500463
- 2023. NCBI Sequence Read Archive. SRR18500464
- 2023. NCBI Sequence Read Archive. SRR18500465
- 2023. NCBI Sequence Read Archive. SRR18500466
- 2023. NCBI Sequence Read Archive. SRR18500478
- 2023. NCBI Sequence Read Archive. SRR18500489
- 2023. NCBI Sequence Read Archive. SRR18500490
- 2019. NCBI Sequence Read Archive. SRP151715
- 2022. NCBI Sequence Read Archive. SRR16371984
- 2022. NCBI Sequence Read Archive. SRR16371985
- 2022. NCBI Sequence Read Archive. SRR16371986
- 2022. NCBI Sequence Read Archive. SRR16371987
- 2022. NCBI Sequence Read Archive. SRR16371988
- 2022. NCBI Sequence Read Archive. SRR16371989
- 2022. NCBI Sequence Read Archive. SRR16371990
- 2022. NCBI Sequence Read Archive. SRR16371991
- 2022. NCBI Sequence Read Archive. SRR16371992
- 2022. NCBI Sequence Read Archive. SRR16371993
- 2022. NCBI Sequence Read Archive. SRR16371994
- 2022. NCBI Sequence Read Archive. SRR16371995
- 2022. NCBI Sequence Read Archive. SRR16371996
- 2022. NCBI Sequence Read Archive. SRR16371997
- 2022. NCBI Sequence Read Archive. SRR16371998
- 2023. NCBI GenBank. JAQSGO000000000
- Li H, Sun P, Wang Y, Zhang Z. 2023. Persimmon genome. Figshare. [DOI]
Data Availability Statement
All software used in this work are in the public domain, with parameters described in the Methods section. The commands used in the processing were all executed according to the manuals and protocols of the corresponding bioinformatics software.