

Abstract
Chickpea (Cicer arietinum L.) seeds are valued for their nutritional scores and limited information on the molecular mechanisms of chickpea fertilization and seed development is available. In the current work, comparative transcriptome analysis was performed on two different stages of chickpea ovules (pre- and post-fertilization) to identify key regulatory transcripts. Two-staged transcriptome sequencing was generated and over 208 million reads were mapped to quantify transcript abundance during fertilization events. Mapping to the reference genome showed that the majority (92.88%) of high-quality Illumina reads were aligned to the chickpea genome. Reference-guided genome and transcriptome assembly yielded a total of 28,783 genes. Of these, 3399 genes were differentially expressed after the fertilization event. These involve upregulated genes including a protease-like secreted in CO(2) response (LOC101500970), amino acid permease 4-like (LOC101506539), and downregulated genes MYB-related protein 305-like (LOC101493897), receptor like protein 29 (LOC101491695). WGCNA analysis and pairwise comparison of datasets, successfully constructed four co-expression modules. Transcription factor families including bHLH, MYB, MYB-related, C2H2 zinc finger, ERF, WRKY and NAC transcription factor were also found to be activated after fertilization. Activation of these genes and transcription factors results in the accumulation of carbohydrates and proteins by enhancing their trafficking and biosynthesis. Total 17 differentially expressed genes, were randomly selected for qRT-PCR for validation of transcriptome analysis and showed statistically significant correlations with the transcriptome data. Our findings provide insights into the regulatory mechanisms underlying changes in fertilized chickpea ovules. This work may come closer to a comprehensive understanding of the mechanisms that initiate developmental events in chickpea seeds after fertilization.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13205-023-03599-8.
Keywords: Chickpea, Differentially expressed genes, Fertilization, Ovule, Transcriptome sequencing
Introduction
The double fertilization process is a unique characteristic of flowering plants and a highly complicated regulatory event involving the fusion of one sperm cell with the egg cell and fusion of another sperm cell with the central cell to produce diploid zygote and triploid endosperm, respectively (Wu et al. 2011). The zygote grows into an embryo while the endosperm helps to nourish the growing embryo. During fertilization, many cell-to-cell communication events have also been reported to achieve maximum reproductive success (Dresselhaus and Franklin-Tong 2013). This process is double fertilization in angiosperms and that is expected to integrate multiple signals, and involves different metabolic pathways and molecular functions (Zhou and Dresselhaus 2019). Various plant hormones, gene families, and transcription factors, direct the double fertilization process and further seed development in plant species (Yang et al. 2002; Wu et al. 2007; Kang et al. 2013).
Fertilization triggers the activation of sets of genes for the proper development of the embryo to meet the corresponding demand at transcripts, proteins, and metabolite levels. Before fertilization, female gametes are in a quiescent stage and activated just after fertilization and transcription of zygote genome starts just immediately or soon after fertilization (Schulz and Harrison 2019). The non-equivalent contributions of parental and maternal genomes have been documented in the transcriptome of early plant embryos (Del Toro-De León et al. 2014). The epigenetic modifications in male and female genomes have also contributed to variation in expression profiles of their respective genes before and after fertilization events. The regulatory mechanism including epigenetic regulation, polycomb group genes, and RNA silencing also contribute transcription activation/deactivation during before and after fertilization processes (Gehring 2013). Genes associated with plant-fertilization have been documented in Arabidopsis thaliana (Takahashi et al. 2018; Adhikari et al. 2020). Deep sequencing studies of male and female reproductive organ(s) transcriptomes have been conducted in plants. These studies include pre- and post-meiotic samples, many on anthers, mature pollen and whole female organs in numerous plant species using various approaches for identification of especially expressed genes (Honys and Twell 2003; Yu et al. 2005; Deveshwar et al. 2011; Ma et al. 2018).
The female gamete (ovule) is the precursor of the seed and an organ of central importance (site of embryo sac development) during fertilization in flowers/life cycle of angiosperms (Endress 2011). In-fact, all important events including development of female gametophyte, fertilization process, and embryo and endosperm development occur in the ovule (Kelley and Gasser 2009). Before fertilization, the ovule consists of nucellus, integument (inner and outer) and micropyle and connected the placenta via funiculus and development of ovules has been well documented in Arabidopsis thaliana (Schneitz et al. 1995). In-fact, the ovule contains all the female tissues and functions for embryo development. The transcriptome analysis of abortive and developing ovules have been investigated during fruit development in many plant species including Solanum chacoense, Corylus heterophylla, and Xanthoceras sorbifolium (Tebbji et al. 2010; Cheng et al. 2015; Zhou and Cai 2021). Zhou and Cai (2021) documented the potential role of ethylene in the regulatory mechanism of ovule after fertilization in Xanthoceras sorbifolium. The gene regulatory network has also been elucidated in the determination of ovule development fate in Xanthoceras sorbifolium. The comparative expression profiling of pre-fertilized and post-fertilized ovules in S. chacoense has also identified 1024 genes that were regulated during these developmental stages. A valuable transcript framework of the ovule to understand the regulatory networks of ovule development has been curated in Jatropha curcas (Xu et al. 2019). The molecular mechanisms of ovule development have been studied in many other plant species including Arabidopsis thaliana, Brassica napus, and Oryza sativa (Baker et al. 1997; Whittle et al. 2010; Kubo et al. 2013). Kubo et al. (2013) analyzed the transcriptome of developing ovules using microarray gene expression data in rice (Oryza sativa). In Arabidopsis thaliana, genome-wide gene expression profiles during ovule development were analyzed and a large number of transcription factor genes having dynamic or enriched expression were identified (Schmidt et al. 2011). The transcriptome of mature female gametophyte has also been analyzed using RNA-Seq in maize (Chettoor et al. 2014).
The rapid progresses have been noticed toward understanding of the specific developmental processes of several plant species by providing publicly available transcriptome data set with detailed description. However, a few such studies have been performed in legumes including chickpea (Severin et al. 2010; Singh et al. 2013; Pradhan et al. 2014). Kalve and Tadege (2017) have been described various developmental stages of flowers including closed bud, hooded bud, half open flower, fully opened flower, fading flower of chickpea in comprehensive details. The fertilization has been documented after 3 days in half opened flower stage of chickpea (Kaleve and Tedege 2017). Singh et al (2013) analyzed the chickpea transcriptome in 11 selected samples (vegetative and flower tissues) has been analyzed to measure gene expression using RNA-Seq approach. A number of genes were found to be differentially expressed during flower development as compared to vegetative tissues. However, as far as is known, there is no report regarding the transcriptome analysis of male and female gametophytes in chickpea. Chickpea is an agriculturally important legume crop and ranks second after common beans worldwide in consumption. Despite increasing demand for consumption and potential scope for yield improvement, chickpea productivity is relatively low due to being affected by several biotic and abiotic interventions. Thus, there is an urgent need to pay attention toward understanding of reproductive processes using RNA-Seq in chickpea to gain insight for improvement of seed productivity.
To understand the fertilization event, RNA-Seq method was performed to investigate transcriptome profile in pre-fertilized and post-fertilized ovules of C. arietinum (desi chickpea cultivar, ICC4958). The identification of stage-specific genes and developmental process related genes will enrich our knowledge about the developmental clues of fertilization event in chickpea. The RNA-Seq has offered a throughout scenario after fertilization in chickpea transcriptome. High quality reads were obtained further mapped with reference genome has been screen out potential differentially expressed genes involve after fertilization. Functional annotation of differentially expressed genes was found in the enrichment of differentially expressed genes in carbohydrate metabolism and transport, and various binding and enzymatic activities. In addition, these differentially expressed genes were validated by quantitative real-time PCR. The comparative transcript profiling approach between pre-fertilized and post-fertilized ovules offers an insight into a subset of genes comprising this transcriptome. This work will deepen our knowledge of the complex transition process during fertilization and offer potential targets for improving yield and seed quality either by biotechnological or breeding means and both.
Materials and methods
Plant materials
Desi chickpea cultivar, ICC4958, was selected for the present study and the different samples were harvested at two different reproductive stages of flower pre-fertilized and post-fertilized. The different reproductive stages were identified based on specific measurements of floral bud according to Kalve and Tedege (2017), the pre-fertilized stage is represented by closed bud while fertilized stage is represented in or after half opened flower of same size. The interval duration between these two selected stages has been observed approximately 3 days. Only healthy and uniform size buds were used for the isolation of ovules.
Morphological and histological analysis of different stages of chickpea
Buds of pre- and post-fertilized were collected and ovary samples were isolated. Ovary samples were fixed in 3:1 ethanol-acetic acid (Farmer solution) for 24 h and then softened in 8 M sodium hydroxide (NaOH) for 8 h. Further, samples washed with distilled water, and stained with 0.1% aniline blue for 24 h. To visualize, stained samples were placed on glass slides and analyzed with the help of a light microscope. For histological analysis, ovary samples were fixed in Formaldehyde-acetic acid-alcohol (FAA) for 24 h. Further, dehydration was carried out using a series of ethanol and further with xylene series. Infiltration was carried out by paraffin wax and finally embedded in paraffin wax and sectioned with the help of microtome. Then, thin sections of samples were placed on slides and observed under light microscope.
Ovule isolation
The closed buds of pre-fertilization stage and half opened flowers of post-fertilization stage were collected in RNase free water. With the help of DEPC treated forceps and blade, sepals, petals and anthers were removed to disclose the female gametophyte. Then the ovules were collected by gently pressing the micropylar end of the female gametophyte with the help of a DEPC treated needle. Only healthy and unruptured ovules were taken for further isolation of RNA.
RNA isolation and quantification of RNA
Total RNA was isolated from pre- and post-fertilized ovules using TRI reagent (Sigma Life Science, St. Louis, MO) according to the manufacturer’s instructions with three replicates. The concentration of RNA and its purity were checked on the Nanodrop 2000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). RNA was isolated from individual samples in three independent replicates. The quality of RNA samples was also cross checked by RNase-free agarose gel electrophoresis and bioanalyzer (Bio-Rad, USA). Best quality RNA samples (two biological replicates of each stages) with a good 260/280 ratio and RNA integrity number (RIN > 7.0) were further selected for transcriptome sequencing.
Library preparation and sequencing
Total RNA (~ 5 μg) from four samples (two best replicates from each category) was used for library preparation and sequencing. Pre-fertilized ovule samples were labeled as VBF (VBF-01 and VBF-02) and post-fertilized ovule samples were named as VAF (VAF-01 and VAF-02). Libraries were prepared using the Illumina TrueSeq RNA library method preparation guide (Illumina Technologies, San Diego, CA) and paired-end sequencing was performed for all 4 samples using Illumina Genome Analyzer II (Illumina) to obtain ≥ 25 million reads of 100 bp length.
Quality control and mapping on a reference genome
Raw data were processed through a quality check using the FastQC protocol (https://github.com/OpenGene/fastp), and high-quality (Phred score ≥ 30) reads by removing low quality reads (Phred score < 30), reads containing adaptors and poly-N. Further, the GC content of high-quality reads was estimated. Chickpea genome (Cicer arietinum) latest version (GCA_000331145.1) and its annotation file was used for mapping of high-quality reads using HISAT2 alignment tool (http://daehwankimlab.github.io/hisat2/main/) using the default parameters. The sequencing reads of all four samples were mapped individually on the genome.
Identification of differentially expressed genes
Fragments per kilobase of transcript per million (FPKM) was used for identification of differentially expressed (DE) genes between pre-fertilized ovules and post-fertilized ovules samples using Cuffdiff (a Cufflinks utility) (http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/) using default parameters. The FPKM value of each sample was also used to visualize their distribution in low dimension space using principal component analysis (PCA). Genes with |Log2 (Fold Change, FC)|> 1 and a false discovery rate (FDR) ≤ 0.05 were considered to be significant differentially expressed genes. Volcano plot was used to visualize the abundance and expression of differentially expressed genes. Identification of transcription factors (TFs) encoding genes from differentially expressed genes was searched from the chickpea annotation.
Gene coexpression network analysis for modules construction
Gene coexpression network analysis was performed using WGCNA package for differentially expressed genes. The TPM values more than one at least at time point in both post-fertilized and pre-fertilized stages were used. A convenient one-step network construction and module detection function were used for generation of co-expression gene network and recognition of each module with different functions. Heatmap of module 1 and module 3 was generated. Go- enrichment analysis was performed using R programming.
Gene ontology annotations and pathway analysis
Gene ontology (GO) terms differentially expressed genes were obtained by searching the Arabidopsis orthologous genes downloaded from TAIR (https://www.arabidopsis.org). Biological process and molecular functions GO terms were also downloaded from TAIR. The Fisher exact test with P-value ≤ 0.05 was used as a cutoff to find out the significantly enriched GO terms. Kyoto Encyclopedia of Genes and Genomes (KEGG) (available online: http://kobas.cbi.pku.edu.cn/) was used for pathway analysis.
Quantitative real-time PCR analysis
Quantitative real-time PCR (qRT-PCR) analysis was performed to validate the differentially expressed genes obtained from our sequencing analysis. A total of 17 differentially expressed genes from transcriptome analysis were selected for qRT-PCR. Gene specific primers were designed for each gene using Primer3 (version 0.4) software. A list of primers used for qRT-PCR has been provided in Table 1. The qRT-PCR was performed using Step One Real-Time PCR System (Applied Biosystem). The normalized expression of the transcripts obtained from qRT-PCR was plotted using Microsoft Excel. Pearson correlation of selected genes was also computed between the expression level of qRT-PCR and transcriptome analysis values.
Table 1.
List of primer pairs used for qRT-PCR
| Serial No | Gene name | Locus Id | Primer pairs (5′–3′) |
|---|---|---|---|
| 1 | Repetitive proline-rich cell wall protein 2 | LOC101489541 |
TGGCTTCCTTAAGCGTCCTA TTGGTACCCTGGTGGTTTGT |
| 2 | Glucose-6-phosphate/phosphate translocator 2 | LOC101492580 |
CTTGGTGGGCTTTGAATGTT TCAACTTTTGGGGAATCAGC |
| 3 | Homeobox-leucine zipper protein ATHB-40 | LOC101492395 |
AAGGCAAGACGGAGACGTAA ATCCAAACCAAGCTCCATTG |
| 4 | 1-aminocyclopropane-1-carboxylate oxidase homolog 1-like | LOC101512278 |
CAAAGGGCCAAGAGTTTCTG ACTTGAAAGGCTCCAAAGCA |
| 5 | Ribonuclease S-F11-like | LOC101495153 |
CACCACCACCACCTCCTTAT GGTTGCTGACCGGTTATGTT |
| 6 | MYB-related protein 305-like | LOC101493897 |
TAGCCAAAGCTGCTGGTCTT GCAATTTTGGACCACCTGTT |
| 7 | Pectinesterase-like | LOC101492300 |
AGCTGGGCCACATAAACATC AGTGCATTGCCAAACACAAA |
| 8 | Protein RADIALIS-like 3 | LOC101499379 |
TCAGCATCAGGTTCATGGAG GGGAATGGAACTTGACCTGA |
| 9 | Probable isoaspartyl peptidase/L-asparaginase 2 | LOC101510278 |
CATCACTGCTCTTCGTTCCA GGCGCCACATCTTCTCTTAG |
| 10 | L-ascorbate oxidase homolog | LOC101495562 |
TGATGTTGGACAGTGGTCGT TGAACAACGTGTGAGCCTTC |
| 11 | Pectate lyase-like | LOC101493043 |
TCATCCGACAAGACCATTGA CAGAGAGAGCATCACCGTCA |
| 12 | Pectate lyase-like | LOC101509352 |
CAAAAACATGCAGCAGGAGA GGGTACCAGGTTTTGGGTTT |
| 13 | L-ascorbate oxidase homolog | LOC101514053 |
CTGCTTCCAGGGTTAATGGA GTACGGAAAGTGGCGTTGAT |
| 14 | L-ascorbate oxidase homolog | LOC101514709 |
AAGGCCTAACCCACAAGGTT GCAACGTAACACTGCTTGGA |
| 15 | Putative pectinesterase/pectinesterase inhibitor 45 | LOC101499304 |
AATGGCGTTTCAAGATTTCG TTGTTGCTACCACCACCGTA |
| 16 | Uncharacterized | LOC101510421 |
GACTTGGGAAGGAACATCCA AGTGATGCCACAAATGTCCA |
| 17 | Fasciclin-like arabinogalactan protein 14 | LOC101499073 |
ACACCGTTTCGGTTCTTCAG AAAACAGCACCAACGAATCC |
Results
Morphological and histological analyses of different stages of chickpea buds
To capture the pre- and post-fertilization stages of chickpea, different stages (of flower buds) have been selected for the present work according to their size and morphological characters. The selected stages were found to have considerable differences, the flower buds (pre-fertilization) were identified as closed with green petals, while flower buds (post-fertilization) were identified as opened with pink petals (Fig. 1A). Similarly, many distinguished changes have also been noticed in gynoecium in selected stages. The pre-fertilized ovary has been found to be small, highly delicate and somewhat swollen in shape. In contrast, the post-fertilized ovary has been found to be elongated with fine hair on the surface (Fig. 1B). Pre-fertilized and post-fertilized ovary seen under the light microscope (Fig. 1C). Similar observations have also been documented in pre-fertilized ovules with small, transparent and highly delicate and post-fertilized ovules opaque, increased size due to rapid cell division (Fig. 1D). Histological analysis of pre- and post-fertilized ovary and ovule has been showing the formation of embryo sac in post-fertilized ovules as compared to pre-fertilized ovules (Fig. 1E).
Fig. 1.

Morphological differences between pre-fertilized and post-fertilized stages of chickpea. A Pre-fertilized and post-fertilized buds. B Pre-fertilized ovary with transparent, shiny, smooth surface and post-fertilized ovary with opaque, pale green, hairy surface. C Pre-fertilized and post fertilized transparent ovary with resolution. D The microstructure observation of post- fertilized stage ovules. E Histological analysis of pre-fertilized ovary and post-fertilized ovary
Transcriptome sequencing and reference-guided assembly
For identification of differential gene expression, the selected samples named verified before fertilization (VBF) and verified after fertilization (VAF) have been used for RNA-Seq analysis. A total of 211,002,516 raw reads were generated from Illumina sequencing platform. A total of 208,771,148 high-quality reads were obtained after filtering low quality reads (an average of 52,192,787 reads per sample) using FASTQ. The percentage of high-quality reads obtained range from 99 to 98.85% with an average of 98.94% per sample. The Q30 and GC content of different samples was reported as > 97.70% and 42.81% respectively. Only the high-quality reads having a phred score > 30 were used for further analysis. The high-quality reads of each sample (an average 48,478,117) were mapped on the reference genome of chickpea latest version (GCA_000331145). The percentage of mapped reads per sample ranged from 88.58 to 96.66% with an average of 92.88% per sample. The workflow of the whole analysis is also provided (Fig. 2A). The statistics of raw reads, high-quality reads, mapped reads and percentage of mapped reads are also given in Table 2. These results indicated that the sequenced data were high throughput, and the sequencing quality required further analysis. The high-quality reads were further assembled with a reference genome and generated a total of 28,783 genes (Supplementary Table 1). Normalized read counts in the form of FPKM were obtained after mapping the sequencing reads on the genome. This information was used further for reclassification of verified before fertilization (VBF) and verified after fertilization (VAF) samples using hierarchical clustering (HCL) and PCA analysis. The HCL clustering showed a clear differentiation of VBF and VAF samples (both replicates) (Fig. 2B). In addition, during PCA analysis, the VBF samples were found to be very close to each other (Fig. 2C). The correlation plot between two biological replicates of VBF and VAF deciphered that VBF samples are very much correlated (r2 = 0.97) as compared to VAF (r2 = 0.82) samples (Fig. 2D). The coorelation issue of VAF could be possible due to sampling of tissue in different condition of day. This analysis confirms that VBF and VAF samples are very distinct from each other and their transcriptional fingerprint is also uniquely different from each other. The transcript data generated in present investigation has also been submitted in the GEO database with accession number GSE217186.
Fig. 2.

Workflow and re-classification of samples. A Workflow of transcriptome analysis. B Phylogenetic relationship showing the similarities of replicates. C PCA plot showing the similarities and dissimilarities of samples. D Correlation of the expression value of gene expression among the different replicates of samples
Table 2.
Statistics of sequencing reads and their mapping status
| Sample | Raw reads | High quality (Phred score > 30) | Mapped reads on genome | % mapped reads on genome (%) |
|---|---|---|---|---|
| VAF-01 | 54,932,750 | 54,304,430 | 48,104,156 | 88.58 |
| VAF-02 | 57,471,612 | 56,898,900 | 53,998,848 | 94.90 |
| VBF-01 | 46,939,340 | 46,456,046 | 42,916,729 | 92.38 |
| VBF-02 | 51,658,814 | 51,111,772 | 48,892,732 | 95.66 |
Differentially expressed genes after fertilization
A total of 3399 differentially expressed genes (Supplementary Table 2) were identified in VAF. Among them, 2207 were found to be upregulated and 1192 were found to be downregulated. The gene, CO(2)-response secreted protease-like (LOC101500970) was found to be highly upregulated after fertilization and annotating for serine-type endopeptidase activity. Other genes including amino acid permease 4-like (LOC101506539), homeobox-leucine zipper protein ATHB-40 (LOC101492395), ribonuclease S-F11-like (LOC101495153), vacuolar-processing enzyme-like (LOC101497152), proline transporter 2-like (LOC101499779), protein LIGHT-DEPENDENT SHORT HYPOCOTYLS 10-like (LOC101497242), IAA-amino acid hydrolase ILR1-like 6 (LOC101510101), endoglucanase 11-like (LOC101489804), repetitive proline-rich cell wall protein 2 (LOC101489541), glucose-6-phosphate/phosphate translocator 2, chloroplastic-like (LOC101492580), 1-aminocyclopropane-1-carboxylate synthase 1-like (LOC101495414), asparagine synthetase (LOC101504705), 1-aminocyclopropane-1-carboxylate oxidase homolog 1-like (LOC101512278), heat shock protein 83 (LOC101511551), protein trichome birefringence-like 38 (LOC101491454), protein EXORDIUM-like (LOC101494486), F-box/kelch-repeat protein At1g67480 (LOC101495569), bidirectional sugar transporter N3-like (LOC101498095) were also found to be highly upregulated after fertilization.
In continuation, among downregulated expressed genes, the MYB-related protein 305-like (LOC101493897) was found to be highly downregulated after the fertilization event. Similarly, other downregulated expressed genes including receptor like protein 29 (LOC101491695), fasciclin-like arabinogalactan protein 14 (LOC101499073), protein RADIALIS-like 3 (LOC101499379), pectate lyase-like (LOC101509352), pectate lyase-like (LOC101493043), plasma membrane ATPase 4 (LOC101499920), probable isoaspartyl peptidase/L-asparaginase 2 (LOC101510278), putative pectinesterase/pectinesterase inhibitor 45 (LOC101499304), L-ascorbate oxidase homolog (LOC101495562), pectinesterase-like (LOC101492300), transcription factor bHLH18-like (LOC101515651), L-ascorbate oxidase homolog (LOC101514709), L-ascorbate oxidase homolog (LOC101514053), auxin-responsive protein SAUR78-like (LOC101493674), pectinesterase (LOC101513899), pectinesterase inhibitor 10 (LOC101489391) and ethylene-responsive transcription factor ERF086-like (LOC101500468) were also found to be downregulated after fertilization. A list of highly upregulated and downregulated differentially expressed gene with its log2-fold change value has been provided in Table 3. A list of genes/transcript identified which are involved in fertilization, embryo or seed development processes is also presented in Table 4.
Table 3.
List of highly upregulated and downregulated DE genes with Log2-fold change values
| Gene name | Locus ID | Log 2 fold change value | |
|---|---|---|---|
| Upregulated | CO(2)-response secreted protease-like | LOC101500970 | 11.4639 |
| Amino acid permease 4-like | LOC101506539 | 9.49148 | |
| Homeobox-leucine zipper protein ATHB-40 | LOC101492395 | 8.72617 | |
| Ribonuclease S-F11-like | LOC101495153 | 8.67134 | |
| Vacuolar-processing enzyme-like | LOC101497152 | 7.92538 | |
| Proline transporter 2-like | LOC101499779 | 7.75282 | |
| Protein LIGHT-DEPENDENT SHORT HYPOCOTYLS 10-like | LOC101497242 | 7.63213 | |
| IAA-amino acid hydrolase ILR1-like 6 | LOC101510101 | 7.38214 | |
| Endoglucanase 11-like | LOC101489804 | 7.36709 | |
| 1-aminocyclopropane-1-carboxylate synthase 1-like | LOC101495414 | 7.25615 | |
| Downregulated Genes | MYB-related protein 305-like | LOC101493897 | − 6.1669 |
| Receptor like protein 29 | LOC101491695 | − 5.51231 | |
| Fasciclin-like arabinogalactan protein 14 | LOC101499073 | − 5.42952 | |
| Protein RADIALIS-like 3 | LOC101499379 | − 5.40723 | |
| Pectate lyase-like | LOC101509352 | − 5.29702 | |
| Pectate lyase-like | LOC101493043 | − 5.21477 | |
| Plasma membrane ATPase 4 | LOC101499920 | − 5.13334 | |
| Probable isoaspartyl peptidase/L-asparaginase 2 | LOC101510278 | − 4.96111 | |
| Putative pectinesterase/pectinesterase inhibitor 45 | LOC101499304 | − 4.9541 | |
| L-ascorbate oxidase homolog | LOC101495562 | − 4.90468 |
Table 4.
List of genes/transcripts identified to be specifically involved in fertilization, embryo or seed development processes before in pre-fertilized and post-fertilized ovules of chickpea
| Sr. no | Gene name | Locus ID | Functional annotations |
|---|---|---|---|
| Upregulated genes/transcripts after fertilization | |||
| 1 | Seed maturation protein LEA 4 | LOC101506718 | Embryo development ending in seed dormancy |
| 2 | Protein HEADING DATE REPRESSOR 1 | LOC101499518 | Regulation of flower development |
| 3 | Phytosulfokines 2-like | LOC101514134 | Cell population proliferation |
| 4 | Uncharacterized | LOC101500771 | Recognition of pollen |
| 5 | G-type lectin S-receptor-like serine/threonine-protein kinase | LOC101514619 | |
| 6 | Expansin-like A2 | LOC101514490 | Sexual reproduction |
| 7 | Putative expansin-B2 | LOC101515268 | |
| 8 | Expansin-B3 | LOC101507266 | |
| Downregulated genes/transcripts after fertilization | |||
| 9 | Growth-regulating factor 9 | LOC101506579 | Developmental process |
| 10 | Growth-regulating factor 1 | LOC101497429 | |
In conclusion, the volcano plot has been presented to show the upregulated and downregulated genes in VAF and VBF (Fig. 3A). The heatmaps of differentially expressed (DE) genes displayed that VAF comprises the higher number of genes showing greater expression as compared to VBF (Fig. 3B). In conclusion, post-fertilization, the greater transcriptional activation was found to be higher as compared to pre-fertilization event.
Fig. 3.
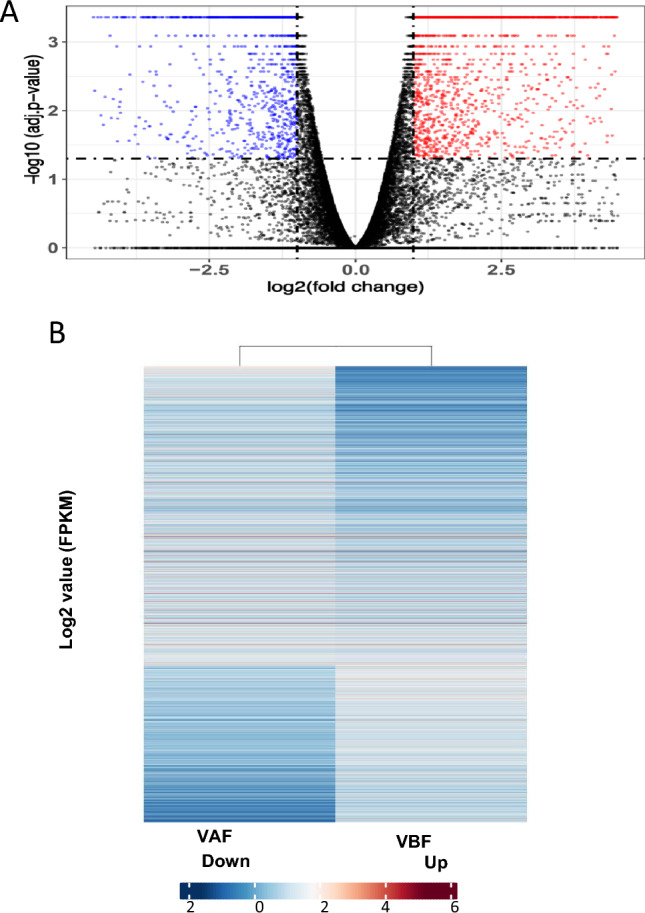
Expression of differentially expressed (DE) genes in both samples. A Volcano plot showing the upregulated and downregulated genes, Log2-fold change ≥ 1 or ≤ − 1 with FDR ≤ 0.01 was considered as DE genes. B Heatmap illustrating the differential expression of genes between pre-fertilized and post-fertilized stages of ovules in chickpea. The blue color represents the least expression whereas, red color showed the higher expression of genes
Weighted gene co-expression network analysis (WGCNA)
WGCNA analysis using (3399) differentially expressed genes after fertilization and pairwise comparison, successfully constructed four co-expression modules. Module 1 comprises of 1315 differentially expressed genes with VAF-01 and VAF-02 having high number of genes showing high expression as compared to VBF-01 and VBF-02. Module 2 having very less number of differentially expressed gene as shown (Fig. 4B). Module 3 has been constructed using 16 differentially expressed genes of VAF-01, VAF-02 comparatively VBF-01 and VBF-02 and among them, 10 are having high expression in after fertilized stage as compared to before fertilized stage (Fig. 4C). Module 4 also has very less number of differentially expressed genes (Fig. 4D). Heatmap of module 1 differentially expressed genes (1315 DE genes) displayed that VAF-01, VAF-02 comprises higher number of genes showing greater expression as compare to VBF-01 and VBF-02 (Fig. 4E). Heatmap of module 3 differentially expressed genes (16 genes) displayed that VAF-01, VAF-02 comprises the low number of genes showing great expression as compare to VBF-01 and VBF-02 (Fig. 4F). GO enrichment analysis on stage-specific modules revealed that majorly enrichment in polysaccharide catabolic process, cellulose catabolic process, glutamine metabolic process, biosynthetic processes, developmental processes, auxin activated signaling pathways, lipid transport, carbohydrate metabolic process, methylation, photosynthesis. Other minor enrichment in embryo development ending in seed dormancy, regulation of transcription, translation, transmembrane transport, and so on (Fig. 4G).
Fig. 4.

Co-expression network analysis with curated modules. A Gene co-expression network of module 1. B Gene co-expression network of module 2. C Gene co-expression network of module 3. D Gene co-expression network of module 4. E Heatmap shows the expression profile of all the coexpressed genes in the module 1. F Heatmap shows the expression profile of all the coexpressed genes in the module 3. G Gene ontology enrichment of all the coexpressed genes of module 1
Differentially expressed genes encoded transcription factors
Transcription factors regulate the transcriptional regulation during the fertilization event. In present study, the expression dynamics of transcripts encoded different transcription factors were also analyzed. A total, 247 transcripts encoded the transcription factor have been identified to be differentially expressed after fertilization. Among them, 119 genes encoded transcription factors were found to be upregulated while 128 genes encoded transcription factors were found to be downregulated after fertilization (Fig. 5) In-fact, of total, approximately 48.18% gene encoded transcription factors were found to be upregulated, while approximately 51.82% gene encoded transcription factors were found to be downregulated post-fertilization process. Majority of transcription factors encoding differentially expressed genes have been reported from different transcription factor families including basic helix-loop-helix (bHLH), C2H2 zinc finger, ethylene responsive factor (ERF), MYB, MYB-related, and WRKY.
Fig. 5.
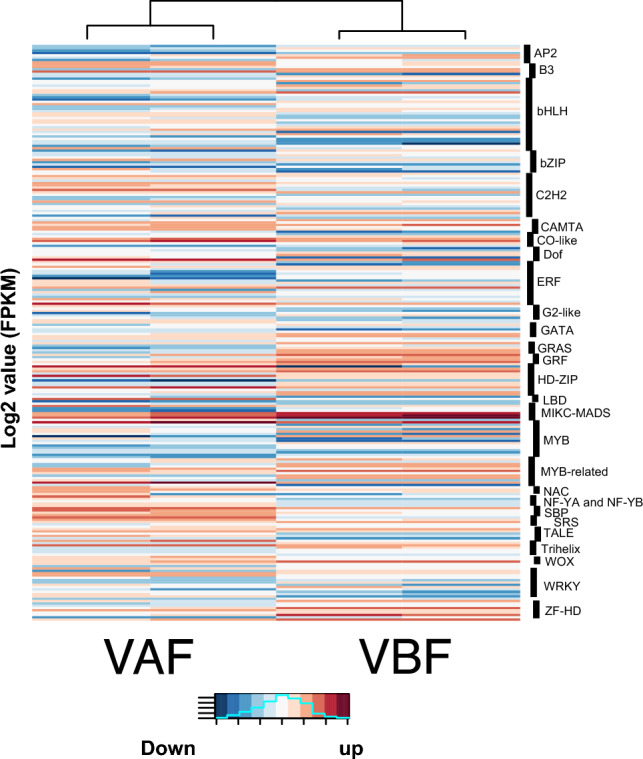
Expression of differentially expressed (DE) transcription factors. Heatmap illustrating the differential expression of transcription factor genes of pre-fertilized and post-fertilized chickpea ovule stages. Enriched transcription families are labelled. The color scale at the right shows high and low expressions as red and blue, respectively
A total of 31 genes encoding proteins from basic helix-loop-helix (bHLH) transcription factor family were found to be differentially expressed, among them, 14 were found to be upregulated and 17 were found to be downregulated in after fertilization. Similarly, a total of 19 genes encoding transcription factors of the C2H2 zinc finger family were found to be differentially expressed after fertilization. Among total, 10 C2H2 zinc finger were found to be upregulated and 19 were found to be downregulated after fertilization. The 19 genes encoding transcription factors of ethylene responsive factor (ERF) family were found to be differentially expressed, and among them, 9 were found to be upregulated and 10 were found to be downregulated. In the MYB transcription family, a total of 17 genes encoding this transcription factor, were found to be differentially expressed. Among them, 7 were found to be upregulated and 10 genes encoded by the MYB family were found to be downregulated after fertilization. Similarly, MYB-related transcription factor family, a total of 9 genes encoding transcription factor were found to be differentially expressed and among them 6 were reported to be upregulated and 3 are downregulated. In WRKY transcription factor family, 12 genes encoding transcription factor were found to be differentially expressed and among them 5 were reported to be upregulated and 7 were reported to be downregulated. Among, major differential expressed genes encoding transcription factors were found to be from bHLH (12.55%) and C2H2 zinc finger (7.69%) transcription factor families.
Other transcription factor families are also found to be differentially expressed including AP2, B3, bZIP, DBB, GATA, GRF, NAC, SBP, SRS, and WOX after fertilization.
Functional annotation
Gene ontology enrichment analysis was carried out for annotation of differential expression genes with corresponding processes and molecular functions. In present study, Arabidopsis orthologous genes have been used to discover their respective biological process and molecular function GO terms because this database is gold mine standard for plant-related research. Most differentially expressed genes were found to be involved in different biological processes categories including photosynthetic and light harvesting, methylation, carbohydrate metabolism, carbohydrate transport, biosynthetic processes, lipid metabolic processes and cell wall biosynthesis (Fig. 6A). Similarly, in the molecular functions category, most of differentially expressed genes were found to be involved in oxidoreductase activity, methyltransferase activity, catalytic activity, transferase activity, hydrolase activity, DNA binding, ATP binding (Fig. 6B).
Fig. 6.

Functional annotation of DE genes. Gene ontology (GO) enrichment of differentially expressed genes for biological processes (A) and molecular functions (B). The size of the circle represents the gene ratio. The P-value was computed using the Fisher exact test with Benjamini–Hochberg correction method. Only the GO terms with adjusted P-value < 0.05 were considered as significant
The differentially expressed genes including glycosyl hydrolase 5 family protein-like (LOC101490831), alpha-galactosidase-like (LOC101513035), polygalacturonase-like (LOC101509102), cysteine-rich receptor-like protein kinase 25 (LOC101500690), and probable beta-D-xylosidase 7 (LOC101497926) were found to be involved in carbohydrate metabolism and significantly upregulated after fertilization. Similarly, many differentially expressed genes involved in carbohydrate transport (sugar transport protein 5-like, sugar transporter ERD6-like 6, and bidirectional sugar transporter SWEET13-like) were also reported to be upregulated after fertilization. The differentially expressed genes involved in lipid related pathways including protein ECERIFERUM 1-like were also found to be upregulated after fertilization. Similarly, differential upregulated genes involved in lipid metabolic process after fertilization are documented as PI-PLC X-box domain-containing protein DDB_G0293730-like (LOC101497953) and PI-PLC X domain-containing protein At5g67130 (LOC101489369). The genes involved in lipid transport including non-specific lipid-transfer protein AP10-like (LOC101515705), GDSL esterase/lipase At4g10955-like (LOC101499273), non-specific lipid-transfer protein-like protein At5g64080 (LOC101508788), and non-specific lipid-transfer protein 1 (LOC101495412) were also found to be upregulated after fertilization. In continuation, the genes including oxygen-evolving enhancer protein 2 (LOC101515625), thylakoid lumenal 19 kDa protein (LOC1015051279) and photosystem I reaction cente r subunit III (LOC101506661) were found to be upregulated after fertilization process.
The various hormone related genes were also found to be differentially expressed after the fertilization process. The gene cytokinin riboside 5'-monophosphate phosphoribohydrolase LOG1-like (LOC101496786) identified as integral gene activity of cytokinin biosynthetic process was found to be upregulated after fertilization. Similarly, another gene, allene oxide cyclase (LOC101500382) involved in the jasmonic acid biosynthetic process was found to be upregulated after fertilization. The genes involved in abscisic acid biosynthetic pathway (zeaxanthin epoxidase: LOC101503567), and involved in regulation of abscisic acid biosynthetic pathway (nodulin-related protein 1-like: LOC101494085) were found to be upregulated after fertilization. The abscisic acid-activated signaling pathway gene (protein LlR18B-like: LOC101502390) and auxin acid-activated signaling pathways genes auxin-responsive protein IAA31, auxin-responsive protein IAA16, auxin efflux carrier component 4-like, auxin-induced protein 22E-like, auxin-induced protein 22B-like) were also found to be upregulated after fertilization.
Differential expression of genes related with KEGG metabolic pathway
The differential expression pattern of genes related to KEGG metabolic pathway was also identified and among 3399 DEGs, 1733 DEGs with pathway annotation were identified and further classified into three major categories: metabolism, cellular processes and signaling, and information storage and processing (Fig. 7).
Fig. 7.

KEGG Classification analysis. KEGG Classification analysis of the differentially expressed genes of pre-fertilized and post fertilized chickpea ovule stages
In category of metabolism, total annotated genes with gene frequency was found to be involved in different pathways including secondary metabolites biosynthesis/transport and catabolism (8.02%), inorganic ion transport and metabolism (5.40%), lipid transport and metabolism (5.42%), coenzyme transport and metabolism (0.92%), nucleotide transport and metabolism (1.10%), amino acid transport and metabolism (6.69%), carbohydrate transport and metabolism (8.66%) and energy production and conversion (5.48%).
In cellular processes and signaling, differential expressed genes with frequency was found to be involved in cell cycle control, cell division and chromosome partitioning (2.13%), nuclear structure (0.06%), defence mechanism (1.04%), signal transduction mechanism (10.62%), cell wall/membrane biogenesis (1.33%), cell motility (0.06%), cytoskeleton (2.42%), extracellular structure (0.40%), intracellular trafficking, secretion and vesicular transport (2.13%), post-translational modification, protein turnover and chaperone (12.52%).
In the category of information storage and processing, total annotated genes with gene frequency were found to be involved in pathways including translation, ribosomal structure and biogenesis (2.42%), RNA processing and modification (2.25%), transcription (6.92%), replication recombination, and repair (3.63%), chromatin structure and dynamics (0.92%).
Among 1733 DEG, the majority of DEGs (18.98%) was documented with general function prediction with gene function. The highest number of differentially expressed genes, 12.52% were found to be involved post-translational modification, protein turnover, and chaperones related cellular processes.
Verification of differentially expressed genes through qRT-PCR
To verify the result of transcriptomic analysis, quantitative real-time PCR (qRT-PCR) was performed using 17 randomly selected differentially expressed genes as identified by transcriptomic data analysis. Among 17, five genes were found to be upregulated and 12 were found to be downregulated after the fertilization process in silico analysis. Five upregulated genes were identified as repetitive proline-rich cell wall protein 2 (LOC101489541), glucose-6-phosphate/phosphate translocator 2 (LOC101492580), homeobox-leucine zipper protein ATHB-40 (LOC101492395), 1-aminocyclopropane-1-carboxylate oxidase homolog 1-like (LOC101512278), ribonuclease S-F11-like (LOC101495153). Another 12 downregulated genes were MYB-related protein 305-like (LOC101493897), pectinesterase-like (LOC101492300), protein RADIALIS-like 3 (LOC101499379), probable isoaspartyl peptidase/L-asparaginase 2 (LOC101510278), L-ascorbate oxidase homolog (LOC101495562), pectate lyase-like (LOC101493043), pectate lyase-like (LOC101509352) L-ascorbate oxidase homolog (LOC101514053), L-ascorbate oxidase homolog (LOC101514709), putative pectinesterase/pectinesterase inhibitor 45 (LOC101499304), uncharacterized LOC101510421, and fasciclin-like arabinogalactan protein 14 (LOC101499073). The expression of all selected genes was verified using real time PCR in VBF and VAF samples. A scatterplot was also made by comparing the log2-fold change values obtained by the transcriptome analysis and qRT-PCR (Fig. 8A). The correlation among these analyses was examined and the results revealed that expression patterns of all these genes obtained by qRT-PCR analysis were found to be well correlated with those by transcriptome analysis with R2 = 0.95, thus validating the reliability of transcriptome analysis. Similarly, the expression values of the 17 genes for RNA-Seq and qRT-PCR are depicted in graphical representation (Fig. 8B). In conclusion, the patterns obtained from real-time PCR analysis were found to be highly correlated with those from transcriptomic analysis.
Fig. 8.

Validation of the RNA-Seq using the qRT-PCR. A Close correlation (r2 = 0.95) of randomly 17 selected genes between RNA-Seq dataset and qRT-PCR’s result. B Comparison of the RNA-Seq and the qRT-PCR considering the expression of the 17 selected genes
Discussion
Chickpea seeds are a rich source of proteins for a large number of people in developing countries and have drawn much more attention from researchers worldwide (Agarwal et al. 2012). A lot of chickpea genomic and transcriptomic resources have also been developed in the last ten years, and all these resources have been utilized for the improvement of quantity and quality of chickpea seed with elucidation of complexity of seed development and key differences in desi and kabuli chickpea seed (Hiremath et al. 2011; Jhanwar et al. 2012; Kudapa et al. 2014). The seed-size related traits have been found to play a crucial role in determining seed quality, appearance, and yield in chickpea and other legumes (Cobos et al. 2007; Chen et al. 2021). Similarly, in depth understanding of seed development has also been performed in various plant species including Arabidopsis, soybean and oat using different high throughput methods (Le et al. 2010; Jones and Vodkin 2013; Gutierrez-Gonzalez et al. 2013). The seed development process just initiates after double fertilization, and fertilized ovules develop into a seed and the ovary differentiates into fruit and fully developed components of seeds nourish and protect the developing embryo. Just after fertilization, intensive cross-talk has been established between different components of seeds. Thus, already a number of key genes and their functions in double fertilization have been investigated in many plant species including Arabidopsis, rice etc. (Takahashi et al. 2018; You et al. 2021). However, similar transcriptomic information in chickpea is very limited and only a few reports have been published related to different stages of whole flower buds (Singh et al. 2013; Bajaj et al. 2015). In other plant species including Solanum melongena, Solanum aethiopicum, transcriptomics analysis of fertilization events has been reported (Li et al. 2020). In-fact, the ovule has been proposed as a model system for the investigation of genetic regulation during fertilization in many plant species (Li et al. 2020). However, no such comprehensive information about the transcriptomic analysis of fertilized chickpea ovules has been yet not carried out. In continuation, this in-depth re-examination of the influence of fertilization on ovule is necessary to unravel the early development and early structural aspects of seed development and seed size related traits in chickpea. The whole transcriptome sequencing strategy was used to study the expression of transcripts and define their putative function in fertilized ovules. A comparative gene expression data set of pre-fertilized and post-fertilized chickpea ovules (208 million high-quality reads, total) has been generated using Illumina sequencing strategy. The generated high-quality reads were mapped with the chickpea genome latest version (GCA_000331145) and 92.23% high-quality reads mapped with reference genome with curation of more than 28,000 genes.
Different developmental stages of chickpea flower including closed bud (immature stigma), hooded bud (receptive stigma), half opened flower (self pollination take place), fully opened flower (fertilization has been occurred) and fading flower (ovary elongated) have been well defined (Kalve and Tadege 2017). Earlier, several flower buds have been used to capture the different stages of fertilization and seed development (Singh et al. 2013; Pradhan et al. 2014). However, based upon the morphological and histological analysis, the closed bud and half opened flowers have been found to be closely related to pre-fertilization and post-fertilization events in present work. The approximately time in-between these two stages have been estimated as 3 days. In-fact, the fertilization has been found to initiate many morphological changes in ovary and ovule of chickpea. The ovary is found to be highly delicate, shiny, and somewhat swollen pre-fertilization and post-fertilization, it was found to be elongated, pale in color, with surface covered with fine hairs like structures. The ovule was found to be transparent and highly delicate pre-fertilized and size of ovule was found to be increased due to rapid cell division in post-fertilized stage. These easily distinguished changes proved that these two selected stages of chickpea ovule offered a clear different molecular and biological set of information and investigation of comparative outputs about these two different biological samples is useful to assess the effect of fertilization event on the early stages of seed development. A list of genes/transcripts in Table 4 also reflected transcript behavior of genes specifically related to fertilization and seed developmental related processes. The seed maturation protein LEA 4 related to embryo development and expansin-like A2, putative expansin-B2, expansin-B3 related to sexual reproduction have been found to be upregulated after fertilization event. Two genes/transcripts related to developmental process named as growth-regulating factor 9 and factor 1 were found to be downregulated after fertilization event.
In flowering plants, fertilization activates the transition of ovule to seed and this process is highly energy consuming that depends on efficient utilization of sucrose as an energy source (Ruan et al. 2012). It is reported that, after fertilization, development of seed is greatly influenced by alternation in starch metabolism related enzyme activity (Kumar et al. 2022). Starch concentration in the ovule at specific stages of development is also found to be closely associated with fertility (Rodrigo and Herrero 1998). It has been documented that low hexose-to-sucrose ratio leads to increase in starch biosynthesis and cease cell division in embryo during early seed development stage, in contrast high hexose-to-sucrose ratio suppresses accumulation of starch and initiate cell division in embryo (Weber et al. 1995). In present study, majority of differentially expressed and upregulated genes including glycosyl hydrolase 5 family protein-like (LOC101490831), alpha-galactosidase-like (LOC101513035), polygalacturonase-like (LOC101509102), probable beta-D-xylosidase 7 (LOC101497926) etc. were found to be involved in carbohydrate metabolism processes after fertilization. Sugar transporter plays a very important role in providing signaling and nutritional sugar into developing embryo and endosperm after fertilization. Role of sugar transporter encoding transcript OsSWEET11 has been investigated during early stages of grain filling of rice and found strong expression of OsSWEET11 in developing caryopsis. Defective grain filling was reported due to decreased concentration of sucrose by CRISPR-Cas9 mediated knockdown of OsSWEET11 (Ma et al. 2017). Genes related to carbohydrate transport bidirectional sugar transporter N3-like (LOC101498095), sugar transport protein 5-like (LOC101488983), bidirectional sugar transporter SWEET3 (LOC101504169), bidirectional sugar transporter SWEET13-like (LOC101491054) were also reported to be upregulated after fertilization. To meet the sufficient demand for energy source, carbohydrate metabolism and carbohydrate transport related genes were found to be upregulated post-fertilized stage. Previously, the effect of pollination and fertilization on sugar transporters has been documented in the styles and ovaries of tomato (Shen et al. 2019). Thus, this report dissects the effect of fertilization on carbohydrate metabolism, carbohydrate transport and its related genes in ovules of chickpea and could be a good source for further investigation.
In plants, the elevated carbon dioxide levels as environmental stimuli has been reported to be involved in stomatal development. Engineer et al. (2014) reported the involvement of CO(2)-response secreted protease (CRSP) encoded protein as negative regulation of stomatal development elevated CO2 level in Arabidopsis thaliana. The expression of this gene has been reported in many tissues including meristemoids- and pavement-cell-enriched samples (Pillitteri et al. 2011). In present work, the expression of CO(2)-response secreted protease-like (LOC101500970) gene was found to be highly upregulated in the fertilized ovule of chickpea. This observation provides evidence that extracellular signaling in response to CO2 has been managed by this protease during cell fate specification in developing embryo and endosperm. The another highest expressed gene, amino acid permeases 4-like was documented after fertilization and its expression precedes storage protein synthesis by amino acid supply in developing seeds (Miranda et al. 2001). Hirner et al. (1998) documented the potential role of AtAAP in amino acid import in embryo as well as in endosperm. Similarly, higher expression of PsAAP1 in the transfer cell layer of cotyledon supports the function in uptake of a wide range of amino acids from seed coat (Tegeder et al. 2000). Thus upregulation of amino acid transport after fertilization has been documented as their role in storage protein accumulation. Similarly, homeobox-leucine zipper protein ATHB-40 (LOC101492395) was found to be highly expressed in fertilized ovules. The encoded protein by this gene was reported to have a potential role during embryogenesis in plants (Roodbarkelari and Groot 2017). The expression of this gene has been reported in regulating apical-basal and adaxial-abaxial patterning during post embryonic development of plants (Ariel et al. 2007; Harris et al. 2011). The higher expression of this gene indicates the active determination of apical-basal and adaxial-abaxial pattern during rapid cell division after fertilization. In contrast, many genes have been found to be downregulated after fertilization including MYB-related protein 305-like and receptor-like protein 29. The cDNA encoding the ornamental tobacco MYB305 has been isolated, characterized and documented as a transcription factor. The knockdown lines of MYB305 has also resulted in reduced expression of both nectarines and flavonoids biosynthetic genes and concluded as regulator of netarin genes in the floral nectary. The expression of MYB305 has also been abruptly downregulated after fertilization (Liu et al. 2009). This previous observation is highly correlated with findings of our research work in which downregulation of MYB305 has been reported in fertilized ovules. Thus, the highly upregulated and downregulated documented genes in fertilized ovule showed significant outcomes in line of previous findings.
Previously, the gene regulatory circuitry of seed maturation mechanism has been well refined on the bases of transcriptome profiling of various part of seed in Arabidopsis (Belmonte et al. 2013). Transcriptional modules discovery can leads to recognition of gene regulatory network that regulate biological mechanism related with seed development (Becker et al. 2014). Recently, WGCNA analysis has been approved to an effective method for recognizing key modules and genes associated with target trait. In chickpea, WGCNA analysis has been conducted to identify the modules and hub gene associated with seed size/weight during seed development (Garg et al. 2017). In present study, four gene co-expression network of differentially expressed genes has been curated. Module 1 was found to be having higher number of differentially expressed genes and reflected high expressions in post fertilized ovule stage. This result provided that lot of genes have been upregulated after fertilization and involve in different developmental processes.
The coordination of regulatory complex events required for development of three different seed compartments, has been carried out using various molecular factors including hormones. Numerous studies have reported that hormones are associated with fertilization, embryo and seed development in plants (Alabadí et al. 2009; Locascio et al. 2014; Matilla 2020). Abscisic acid plays a very critical task in regulating various plant developmental processes after fertilization including embryo morphogenesis, seed maturation and desiccation (Kanno et al. 2010). It has been reported that, in ABA-deficient mutant, maternal abscisic acid transfer from vegetative tissue to increase to embryo growth suggesting maternally derived abscisic acid regulates seed development in Nicotiana plumbaginifolia after fertilization (Frey et al. 2004). Subsequently, biosynthesis of ABA also initiates in the embryo after fertilization (Cheng et al. 2014). Spatiotemporal expression of abscisic acid biosynthesis gene suggested that abscisic acid is generated in all seed compartments during seed development (Seo and Marion-Poll 2019). In the present study, the gene, nuclear transcription factor Y subunit C-3-like (LOC101503561) that is involved in the abscisic acid biosynthetic process was found to be significantly upregulated after fertilization. Similarly, genes protein LlR18B-like (LOC101502390) and MLP-like protein 423 (LOC101499631) that are reported to be involved in abscisic-activated signaling pathway were also found to be upregulated after fertilization. Similarly, in present work, gene, allene oxide cyclase (LOC101500382) has been found to be significantly upregulated after fertilization and this is reported to be involved in the jasmonic acid biosynthetic process (Li et al. 2019). The jasmonic acid has been already reported to be involved in regulating reproductive development in plants including Arabidopsis thaliana (Yuan and Zhang 2015). Another genes, cytokinin riboside 5'-monophosphate phosphoribohydrolase LOG1-like (LOC101496786) and cytokinin dehydrogenase 3-like (LOC101493024) and cytokinin dehydrogenase 1 (LOC101493173) have also been found to be upregulated and involved in the cytokinin biosynthetic/metabolic process after fertilization. Thus, genes related to jasmonic acid and cytokinin biosynthetic/metabolic processes have been found to significantly contribute to early seed development after fertilization. Auxin, phytohormone, has already been documented to play a critical role during the fertilization of ovule, further embryogenesis, embryo polarity determination, and seed development (Ribalta et al. 2019). The genes, auxin-responsive protein IAA16 (LOC101498926), auxin efflux carrier component 4-like (LOC101502756), auxin-induced protein 22E-like (LOC101503044), auxin-responsive protein IAA31 (LOC101504852), and auxin-induced protein 22B-like (LOC101512178) were found to be upregulated in fertilized ovule and proved significant correlation of auxin related molecular activity with early seed development of chickpea. In conclusion, the molecular activity of different phytohormones including auxin, cytokinin, abscisic acid and jasmonic acid biosynthesis and their related signaling pathways significantly upregulated in post-fertilized ovules of chickpea.
The transcription factors play a very significant role in the various developmental and biochemical processes to stimulate or repress different metabolic processes during different plant developmental stages (Tolosa and Zhang 2020). In present investigation, a total of 247 transcription factor encoding genes were found to be differentially expressed after fertilization and most of these belong to transcription factor families such as bHLH (basic helix-loop-helix), C2H2 zinc finger, ERF (ethylene responsive factor), MYB, NAC, WRKY. In Arabidopsis, bHLH-encoding gene SPATULA was documented to regulate flowers and fruit development (Groszmann et al. 2008). In Brassica rapa, bHLH transcription factor has been reported to determine seed coat color (Li et al. 2012). In present study, bHLH transcription factor encoding gene were found to be upregulated in after fertilization. In plants, WRKY transcription factors are one of the most numerous families of transcriptional regulators and make the integral component of signaling webs that regulate various plant processes. WRKY gene has been implicated in various gene expression studies during seed development (Rhuston et al. 2011). In Solanum chacoense, ScWRKY1, a group of WRKY transcription factor was reported to be transiently and strongly expressed in fertilized ovules at torpedo stage (Lagacé and Matton 2004). In present study, five genes encoding WRKY transcription factor were found to be upregulated after fertilization. Similarly, MYB transcription factor family is reported to be involve in various plant mechanism including development, metabolism, differentiation, biotic and abiotic stresses, defence and so on (Ambawat et al. 2013). In Arabidopsis thaliana, MYB56 transcription factor was found to be dominantly expressed in developing seeds regulating the expression of genes that involve in cell division and expansion, controlling seed size (Zhang et al. 2013). In this study, seven genes encoding the MYB transcription factor were found to be upregulated after fertilization. Ethylene responsive factors (ERFs) have a very important regulatory role in various developmental processes in plants. In Rice, OsERF101, an ERF encoding transcription factor gene was reported to be predominantly expressed in reproductive tissues and having a vital role in the improvement in seed setting rate under drought conditions (Jin et al. 2018). In present study, nine genes encoding the ERF transcription factor were found to be upregulated after fertilization. It has been already reported the involvement of the NAC transcription factors in various developmental processes in plants (Sun et al. 2018). The transcription factors of the NAC family, like CUC gene, has been already reported to be involved in ovule primordial development (Galbiati et al. 2013). Xu et al. (2019) also documented the significant contribution of the NAC family in the development of ovule and 11 NAC transcription factors were found to upregulated during ovule development in the Jatropha curcas. Similarly, members of the NAC family, also reported to have played a vital role in embryo and seed development after fertilization in plants (Souer et al. 1996; Agarwal et al. 2011). In Arabidopsis thaliana, NAC family members have been documented to regulate cell division and also mediate cytokinin signaling during cell division (Kim et al. 2006). Thus, upregulation of NAC family members after the fertilization event in present work is correlated with all previously reported research and plays a significant role in regulating cell division after the fertilization process.
Conclusions and future prospective
In conclusion, the comparative transcriptome analysis of pre-fertilized and post-fertilized ovules after the morphological study of ovules in C. aretinium revealed the genes and transcription factors upregulated after the fertilization event. This investigation has also revealed a large set of genes that contribute essential roles during the early stage of seed development and could be further used to find out their significant roles after the fertilization process and could be useful targets for crop improvement via breeding and genetic engineering approaches. The data can serve as a better understanding of plant reproductive biology and gene regulation to assess the impact of fertilization of an ovule in chickpea and could be further use for minimize the ovule abortion in agriculture crops.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Authors would like to express gratitude to the Vice chancellor of Central University of Punjab, India for unconditional support during this course of work. RS acknowledged UGC for JRF fellowship for her doctoral work.
Author contributions
VK and SKY conceived and designed present work. VK, RS and RS, analyzed the data, RS and VK wrote the manuscript. All authors read and approved the manuscript.
Funding
VK acknowledged the UGC-BSR research Start-Up-Grant for sponsoring this work.
Data availability
The transcriptome data deposited into the Gene Expression Omnibus database under accession number GSE217186 and are available at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE217186.
Declarations
Conflict of interests
The authors declare that they have no competing interests.
Ethical standard
Authors declared that the whole manuscript is strictly complied with ethical standards of 3Biotech.
References
- Adhikari PB, Liu X, Wu X, et al. Fertilization in flowering plants: an odyssey of sperm cell delivery. Plant Mol Biol. 2020;103:9–32. doi: 10.1007/s11103-020-00987-z. [DOI] [PubMed] [Google Scholar]
- Agarwal P, Kapoor S, Tyagi AK. Transcription factors regulating the progression of monocot and dicot seed development. BioEssays News Rev Mol Cell Dev Biol. 2011;33:189–202. doi: 10.1002/bies.201000107. [DOI] [PubMed] [Google Scholar]
- Agarwal G, Jhanwar S, Priya P, et al. Comparative analysis of kabuli chickpea transcriptome with desi and wild chickpea provides a rich resource for development of functional markers. PLoS ONE. 2012;7:e52443. doi: 10.1371/journal.pone.0052443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabadí D, Blázquez MA, Carbonell J, et al. Instructive roles for hormones in plant development. Int J Dev Biol. 2009;53:1597–1608. doi: 10.1387/ijdb.072423da. [DOI] [PubMed] [Google Scholar]
- Ambawat S, Sharma P, Yadav NR, Yadav RC. MYB transcription factor genes as regulators for plant responses: an overview. Physiol Mol Biol Plants. 2013;19:307–321. doi: 10.1007/s12298-013-0179-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariel FD, Manavella PA, Dezar CA, Chan RL. The true story of the HD-Zip family. Trend Plant Sci. 2007;12:419–426. doi: 10.1016/j.tplants.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Bajaj D, Upadhyaya HD, Khan Y, et al. A combinatorial approach of comprehensive QTL-based comparative genome mapping and transcript profiling identified a seed weight-regulating candidate gene in chickpea. Sci Rep. 2015;5:9264. doi: 10.1038/srep09264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SC, Robinson-Beers K, Villanueva JM, et al. Interactions among genes regulating ovule development in Arabidopsis thaliana. Genetics. 1997;145:1109–1124. doi: 10.1093/genetics/145.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker MG, Hsu SW, Harada JJ, Belmonte MF. Genomic dissection of the seed. Front Plant Sci. 2014;5:464. doi: 10.3389/fpls.2014.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte MF, Kirkbride RC, Stone SL, et al. Comprehensive developmental profiles of gene activity in regions and subregions of the Arabidopsis seed. Proc Natl Acad Sci USA. 2013;110:E435–E444. doi: 10.1073/pnas.1222061110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Lancon-Verdier V, Le Signor C, et al. Genome-wide association study identified candidate genes for seed size and seed composition improvement in M. truncatula. Sci Rep. 2021;11:4224. doi: 10.1038/s41598-021-83581-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng ZJ, Zhao XY, Shao XX, et al. Abscisic acid regulates early seed development in Arabidopsis by ABI5-mediated transcription of SHORT HYPOCOTYL UNDER BLUE1. Plant Cell. 2014;26:1053–1068. doi: 10.1105/tpc.113.121566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Liu J, Zhang H, et al. Transcriptome analysis and gene expression profiling of abortive and developing ovules during fruit development in hazelnut. PLoS ONE. 2015;10:e0122072. doi: 10.1371/journal.pone.0122072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chettoor AM, Givan SA, Cole RA, et al. Discovery of novel transcripts and gametophytic functions via RNA-seq analysis of maize gametophytic transcriptomes. Genome Biol. 2014;15:414. doi: 10.1186/s13059-014-0414-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos MJ, Rubio J, Fernández-Romero MD, et al. Genetic analysis of seed size, yield and days to flowering in a chickpea recombinant inbred line population derived from a Kabuli × Desi cross. Ann Appl Biol. 2007;151:33–42. doi: 10.1111/j.1744-7348.2007.00152.x. [DOI] [Google Scholar]
- Del Toro-De LG, García-Aguilar M, Gillmor CS. Non-equivalent contributions of maternal and paternal genomes to early plant embryogenesis. Nature. 2014;514:624–627. doi: 10.1038/nature13620. [DOI] [PubMed] [Google Scholar]
- Deveshwar P, Bovill WD, Sharma R, et al. Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice. BMC Plant Biol. 2011;11:78. doi: 10.1186/1471-2229-11-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresselhaus T, Franklin-Tong N. Male-female crosstalk during pollen germination, tube growth and guidance, and double fertilization. Mol Plant. 2013;6:1018–1036. doi: 10.1093/mp/sst061. [DOI] [PubMed] [Google Scholar]
- Endress PK. Angiosperm ovules: diversity, development, evolution. Ann Bot. 2011;107:1465–1489. doi: 10.1093/aob/mcr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engineer CB, Ghassemian M, Anderson JC, Peck SC, Hu H, Schroeder JI. Carbonic anhydrases, EPF2 and a novel protease mediate CO2 control of stomatal development. Nature. 2014;513:246–250. doi: 10.1023/nature13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey A, Godin B, Bonnet M, et al. Maternal synthesis of abscisic acid controls seed development and yield in Nicotiana plumbaginifolia. Planta. 2004;218:958–964. doi: 10.1007/s00425-003-1180-7. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Sinha Roy D, Simonini S, et al. An integrative model of the control of ovule primordia formation. Plant J Cell Mol Biol. 2013;76:446–455. doi: 10.1111/tpj.12309. [DOI] [PubMed] [Google Scholar]
- Garg R, Singh VK, Rajkumar MS, Kumar V, Jain M. Global transcriptome and coexpression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 2017;91:1088–1107. doi: 10.1111/tpj.13621. [DOI] [PubMed] [Google Scholar]
- Gehring M. Genomic imprinting: insights from plants. Annu Rev Genet. 2013;47:187–208. doi: 10.1146/annurev-genet-110711-155527. [DOI] [PubMed] [Google Scholar]
- Groszmann M, Paicu T, Smyth DR. Functional domains of SPATULA, a bHLH transcription factor involved in carpel and fruit development in Arabidopsis. Plant J. 2008;55:40–52. doi: 10.1111/j.1365-313X.2008.03469.x. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Gonzalez JJ, Tu ZJ, Garvin DF. Analysis and annotation of the hexaploid oat seed transcriptome. BMC Genomics. 2013;14:471. doi: 10.1186/1471-2164-14-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JC, Hrmova M, Lopato S, Langridge P. Modulation of plant growth by HD-Zip class I and II transcription factors in response to environmental stimuli. New Phytol. 2011;190:823–837. doi: 10.1111/j.1469-8137.2011.03722.x. [DOI] [PubMed] [Google Scholar]
- Hiremath PJ, Farmer A, Cannon SB, et al. Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol J. 2011;9:922–931. doi: 10.1111/j.1467-7652.2011.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirner B, Fischer WN, Rentsch D, Kwart M, Frommer WB. Developmental control of H+/amino acid permease gene expression during seed development of Arabidopsis. Plant J. 1998;14:535–544. doi: 10.1046/j.1365-313x.1998.00151.x. [DOI] [PubMed] [Google Scholar]
- Honys D, Twell D. Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiol. 2003;132:640–652. doi: 10.1104/pp.103.020925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhanwar S, Priya P, Garg R, et al. Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant Biotechnol J. 2012;10:690–702. doi: 10.1111/j.1467-7652.2012.00712.x. [DOI] [PubMed] [Google Scholar]
- Jin Y, Pan W, Zheng X, Cheng X, Liu M, Ma H, Ge X. OsERF101, an ERF family transcription factor, regulates drought stress response in reproductive tissues. Plant Mol Biol. 2018;98:51–65. doi: 10.1007/s11103-018-0762-5. [DOI] [PubMed] [Google Scholar]
- Jones SI, Vodkin LO. Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS ONE. 2013;8:e59270. doi: 10.1371/journal.pone.0059270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalve S, Tadege M. A comprehensive technique for artificial hybridization in Chickpea (Cicer arietinum) Plant Methods. 2017;13:1–9. doi: 10.1186/s13007-017-0202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X, Li W, Zhou Y, Ni M. A WRKY transcription factor recruits the SYG1-like protein SHB1 to activate gene expression and seed cavity enlargement. PLoS Genet. 2013;9:e1003347. doi: 10.1371/journal.pgen.1003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno Y, Jikumaru Y, Hanada A, et al. Comprehensive hormone profiling in developing Arabidopsis seeds: examination of the site of ABA biosynthesis, ABA transport and hormone interactions. Plant Cell Physiol. 2010;51:1988–2001. doi: 10.1093/pcp/pcq158. [DOI] [PubMed] [Google Scholar]
- Kelley DR, Gasser CS. Ovule development: genetic trends and evolutionary considerations. Sex Plant Reprod. 2009;22:229–234. doi: 10.1007/s00497-009-0107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-S, Kim S-G, Park J-E, et al. A membrane-bound NAC transcription factor regulates cell division in Arabidopsis. Plant Cell. 2006;18:3132–3144. doi: 10.1105/tpc.106.043018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Fujita M, Takahashi H, et al. Transcriptome analysis of developing ovules in rice isolated by laser microdissection. Plant Cell Physiol. 2013;54:750–765. doi: 10.1093/pcp/pct029. [DOI] [PubMed] [Google Scholar]
- Kudapa H, Azam S, Sharpe AG, et al. Comprehensive transcriptome assembly of Chickpea (Cicer arietinum L.) using sanger and next generation sequencing platforms: development and applications. PLoS ONE. 2014;9:e86039. doi: 10.1371/journal.pone.0086039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Thakur M, Mitra R, et al. Sugar metabolism during pre- and post-fertilization events in plants under high temperature stress. Plant Cell Rep. 2022;41:655–673. doi: 10.1007/s00299-021-02795-1. [DOI] [PubMed] [Google Scholar]
- Lagacé M, Matton DP. Characterization of a WRKY transcription factor expressed in late torpedo-stage embryos of Solanum chacoense. Planta. 2004;219:185–189. doi: 10.1007/s00425-004-1253-2. [DOI] [PubMed] [Google Scholar]
- Le BH, Cheng C, Bui AQ, et al. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci. 2010;107:8063–8070. doi: 10.1073/pnas.1003530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chen L, Hong M, Zhang Y, et al. A large insertion in bHLH transcription factor BrTT8 resulting in yellow seed coat in Brassica rapa. PLoS ONE. 2012;7:e44145. doi: 10.1371/journal.pone.0044145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Guang Y, Yang Y, Zhou Y. Identification and expression analysis of two allene oxide cyclase (AOC) genes in watermelon. Agriculture. 2019;9:225. doi: 10.3390/agriculture9100225. [DOI] [Google Scholar]
- Li D, Li S, Li W, et al. Comparative transcriptome analysis provides insights into the molecular mechanism underlying double fertilization between self-crossed Solanum melongena and that hybridized with Solanum aethiopicum. PLoS ONE. 2020;15:e0235962. doi: 10.1371/journal.pone.0235962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Ren G, Guirgis A, Thronburg RW. The MYB305 transcription factor regulates expression of nectarin genes in the ornamental tobacco floral nectary. Plant Cell. 2009;21:2672–2687. doi: 10.1105/tpc.108.060079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locascio A, Roig-Villanova I, Bernardi J, Varotto S. Current perspectives on the hormonal control of seed development in Arabidopsis and maize: a focus on auxin. Front Plant Sci. 2014;5:412. doi: 10.3389/fpls.2014.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zhang D, Miao Q, et al. Essential role of sugar transporter OsSWEET11 during the early stage of rice grain filling. Plant Cell Physiol. 2017;58:863–873. doi: 10.1093/pcp/pcx040. [DOI] [PubMed] [Google Scholar]
- Ma Q, Chen C, Zeng Z, et al. Transcriptomic analysis between self- and cross-pollinated pistils of tea plants (Camellia sinensis) BMC Genomics. 2018;19:289. doi: 10.1186/s12864-018-4674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla AJ. Auxin: hormonal signal required for seed development and dormancy. Plants. 2020;9:705. doi: 10.3390/plants9060705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M, Borisjuk L, Tewes A, Heim U, Sauer N, Wobus U, Weber H. Amino acid permeases in developing seeds of Vicia faba L.: expression precedes storage protein synthesis and is regulated by amino acid supply. Plant J. 2001;28:61–71. doi: 10.1046/j.1365-313x.2001.01129.x. [DOI] [PubMed] [Google Scholar]
- Pillitteri LJ, Peterson KM, Horst RJ, Torii KU. Molecular profiling of stomatal meristemoids reveals new component of asymmetric cell division and commonalities among stem cell populations in Arabidopsis. Plant Cell. 2011;23:3260–3275. doi: 10.1105/tpc.111.088583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan S, Bandhiwal N, Shah N, et al. Global transcriptome analysis of developing chickpea (Cicer arietinum L.) seeds. Front Plant Sci. 2014;5:698. doi: 10.3389/fpls.2014.00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribalta FM, Pazos-Navarro M, Edwards K, et al. Expression patterns of key hormones related to pea (Pisum sativum L.) embryo physiological maturity shift in response to accelerated growth conditions. Front Plant Sci. 2019;10:1154. doi: 10.3389/fpls.2019.01154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert HS, Park C, Gutièrrez CL, et al. Maternal auxin supply contributes to early embryo patterning in Arabidopsis. Nat Plants. 2018;4:548–553. doi: 10.1038/s41477-018-0204-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo J, Herrero M. Influence of intraovular reserves on ovule fate in apricot (Prunus armeniaca L.) Sex Plant Reprod. 1998;11:86–93. doi: 10.1007/s004970050124. [DOI] [Google Scholar]
- Roodbarkelari F, Groot EP. Regulatory function of homeodomain-leucine zipper (HD-ZIP) family proteins during embryogenesis. New Phytol. 2017;213:95–104. doi: 10.1111/nph.14132. [DOI] [PubMed] [Google Scholar]
- Ruan Y-L, Patrick JW, Bouzayen M, et al. Molecular regulation of seed and fruit set. Trends Plant Sci. 2012;17:656–665. doi: 10.1016/j.tplants.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Rushton PJ, Somssich IE, Ringler P, Shen QJ. WRKY transcription factors. Trends Plant Sci. 2010;15:247–258. doi: 10.1016/j.tplants.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Wuest SE, Vijverberg K, et al. Transcriptome analysis of the Arabidopsis megaspore mother cell uncovers the importance of RNA helicases for plant germline development. PLoS Biol. 2011;9:e1001155. doi: 10.1371/journal.pbio.1001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneitz K, Hülskamp M, Pruitt RE. Wild-type ovule development in Arabidopsis thaliana: a light microscope study of cleared whole-mount tissue. Plant J. 1995;7:731–749. doi: 10.1046/j.1365-313X.1995.07050731.x. [DOI] [Google Scholar]
- Schulz KN, Harrison MM. Mechanisms regulating zygotic genome activation. Nat Rev Genet. 2019;20:221–234. doi: 10.1038/s41576-018-0087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo M, Marion-Poll A. Chapter One - Abscisic acid metabolism and transport. In: Seo M, Marion-Poll A, editors. Advances in botanical research. Academic Press; 2019. pp. 1–49. [Google Scholar]
- Severin AJ, Woody JL, Bolon Y-T, et al. RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol. 2010;10:160. doi: 10.1186/1471-2229-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Ma S, Liu Y, et al. Cell wall invertase and sugar transporters are differentially activated in tomato styles and ovaries during pollination and fertilization. Front Plant Sci. 2019;10:506. doi: 10.3389/fpls.2019.00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh VK, Garg R, Jain M. A global view of transcriptome dynamics during flower development in chickpea by deep sequencing. Plant Biotechnol J. 2013;11:691–701. doi: 10.1111/pbi.12059. [DOI] [PubMed] [Google Scholar]
- Souer E, van Houwelingen A, Kloos D, et al. The no apical meristem gene of Petunia is required for pattern formation in embryos and flowers and is expressed at meristem and primordia boundaries. Cell. 1996;85:159–170. doi: 10.1016/s0092-8674(00)81093-4. [DOI] [PubMed] [Google Scholar]
- Sun H, Hu M, Li J, et al. Comprehensive analysis of NAC transcription factors uncovers their roles during fiber development and stress response in cotton. BMC Plant Biol. 2018;18:150. doi: 10.1186/s12870-018-1367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Mori T, Ueda K, et al. The male gamete membrane protein DMP9/DAU2 is required for double fertilization in flowering plants. Dev Camb Engl. 2018;145:dev170076. doi: 10.1242/dev.170076. [DOI] [PubMed] [Google Scholar]
- Tebbji F, Nantel A, Matton DP. Transcription profiling of fertilization and early seed development events in a solanaceous species using a 7.7 K cDNA microarray from Solanum chacoenseovules. BMC Plant Biol. 2010;10:174. doi: 10.1186/1471-2229-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegeder M, Offler CE, Frommer WB, Patrick JW. Amino acid transporters are localized to transfer cells of developing pea seeds. Plant Physiol. 2000;122:319–326. doi: 10.1104/pp.122.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosa LN, Zhang Z. The Role of major transcription factors in solanaceous food crops under different stress conditions: current and future perspectives. Plants Basel Switz. 2020;9:E56. doi: 10.3390/plants9010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber H, Borisjuk L, Heim U, et al. Seed coat-associated invertases of fava bean control both unloading and storage functions: cloning of cDNAs and cell type-specific expression. Plant Cell. 1995;7:1835–1846. doi: 10.1105/tpc.7.11.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle CA, Malik MR, Li R, Krochko JE. Comparative transcript analyses of the ovule, microspore, and mature pollen in Brassica napus. Plant Mol Biol. 2010;72:279–299. doi: 10.1007/s11103-009-9567-x. [DOI] [PubMed] [Google Scholar]
- Wu X, Chory J, Weigel D. Combinations of WOX activities regulate tissue proliferation during Arabidopsis embryonic development. Dev Biol. 2007;309:306–316. doi: 10.1016/j.ydbio.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-C, Diggle PK, Friedman WE. Female gametophyte development and double fertilization in Balsas teosinte, Zea mays subsp. parviglumis (Poaceae) Sex Plant Reprod. 2011;24:219–229. doi: 10.1007/s00497-011-0164-1. [DOI] [PubMed] [Google Scholar]
- Xu G, Huang J, Lei S-K, et al. Comparative gene expression profile analysis of ovules provides insights into Jatropha curcas L. ovule development. Sci Rep. 2019;9:15973. doi: 10.1038/s41598-019-52421-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang J, Huang Z, et al. Correlation of cytokinin levels in the endosperms and roots with cell number and cell division activity during endosperm development in rice. Ann Bot. 2002;90:369–377. doi: 10.1093/aob/mcf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L, Yu L, Liang R, et al. Identification and analysis of genes involved in double fertilization in Rice. Int J Mol Sci. 2021;22:12850. doi: 10.3390/ijms222312850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H-J, Hogan P, Sundaresan V. Analysis of the female gametophyte transcriptome of Arabidopsis by comparative expression profiling. Plant Physiol. 2005;139:1853–1869. doi: 10.1104/pp.105.067314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Zhang D. Roles of jasmonate signalling in plant inflorescence and flower development. Curr Opin Plant Biol. 2015;27:44–51. doi: 10.1016/j.pbi.2015.05.024. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liang W, Shi J, Xu J, Zhang D. MYB 56 encoding a R2 R 3 MYB transcription factor regulates seed size in Arabidopsis thaliana. J Integ Plant Biol. 2013;55:1166–1178. doi: 10.1111/jipb.12094. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Cai Q. Role of ethylene in the regulatory mechanism underlying the abortion of ovules after fertilization in Xanthoceras sorbifolium. Plant Mol Biol. 2021;106:67–84. doi: 10.1007/s11103-021-01130-2. [DOI] [PubMed] [Google Scholar]
- Zhou L-Z, Dresselhaus T. Friend or foe: signaling mechanisms during double fertilization in flowering seed plants. Curr Top Dev Biol. 2019;131:453–496. doi: 10.1016/bs.ctdb.2018.11.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The transcriptome data deposited into the Gene Expression Omnibus database under accession number GSE217186 and are available at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE217186.