

Significance
This study identifies the PRAK-NRF2 axis as a novel and relative unique antioxidant pathway in Th17 cells, which have the highest expression of PRAK among CD4+ effector T cells. The disruption of PRAK accelerates the degradation of NRF2 despite its decreased ubiquitin modification and causes oxidative stress in Th17 cells. Elevated cellular ROS disrupts the phosphorylation and tetramer isoform of PKM2, which mediates the phosphorylation of STAT3 and the differentiation of Th17. Moreover, this study demonstrates a protective role of Th17 in a MC38 tumor model and points out that ROS elimination is helpful for Th17-mediated antitumor immunity.
Keywords: PRAK, Th17, ROS, NRF2, glycolysis
Abstract
Oxidative stress is a key feature in both chronic inflammation and cancer. P38 regulated/activated protein kinase (PRAK) deficiency can cause functional disorders in neutrophils and macrophages under high oxidative stress, but the precise mechanisms by which PRAK regulates reactive oxygen species (ROS) elimination and its potential impact on CD4+ T helper subset function are unclear. The present study reveals that the PRAK-NF-E2-related factor 2(NRF2) axis is essential for maintaining the intracellular redox homeostasis of T helper 17(Th17) cells, thereby promoting Th17 cell differentiation and antitumor effects. Through mechanistic analysis, we identify NRF2 as a novel protein substrate of PRAK and find that PRAK enhances the stability of the NRF2 protein through phosphorylation NRF2 Serine(S) 558 independent of protein ubiquitination. High accumulation of cellular ROS caused by loss of PRAK disrupts both glycolysis and PKM2-dependent phosphorylation of STAT3, which subsequently impairs the differentiation of Th17 cells. As a result, Prak knockout (KO) mice display significant resistance to experimental autoimmune encephalomyelitis (EAE) but impaired antitumor immunity in a MC38 tumor model. This work reveals that the PRAK-NRF2-mediated antioxidant pathway is a metabolic checkpoint that controls Th17-cell glycolysis and differentiation. Targeting PRAK is a promising strategy for maintaining an active ROS scavenging system and may lead to potent Th17 cell antitumor immunity.
T helper 17 cells (Th17 cells) are a distinct T cell subset that expresses the transcription factors retinoic acid receptor-related orphan receptor-γt (RORγt) and secretes interleukin-17(IL-17). Th17 cells are critical components of the adaptive immune system, which contributes to host defense against extracellular pathogens and is important in autoimmune and inflammatory disease. In tumors, the effects of Th17 remain controversial (1–5). In contrast to the development of other helper T cell subsets, the development of Th17 cells is characterized mainly by co-stimulation with interleukin-6 (IL-6) and transforming growth factor-β (TGF-β) (6). Induced by these cytokines, naïve CD4 T cells initiate the expression of RORγt and promote the transcription of IL-17a/f (7). Recently, several studies demonstrated that metabolic stimuli in addition to cytokines, including lipid metabolites and redox status, can influence Th17-cell differentiation (8, 9). Whether these stimuli function independently or through signal crosstalk with TGF-β-SMAD or IL-6-STAT3-induced is unclear.
The differentiation of Th17 cells is a highly orchestrated event. Accompanying the activation of related genes and proteins, markedly enhanced aerobic glycolysis contributes to Th17 differentiation (9, 10). During glycolysis, pyruvate kinase (PK), a key rate-limiting enzyme, catalyzes the phosphorylation of phosphorpyruvate (PEP) and ADP to produce pyruvate and ATP. Four PK isoforms have been identified in different types of cells. In particular, PK isoforms M1(PKM1) and M2(PKM2) were detected in T cells, and two recent papers reported that PKM2 but not PKM1 plays a key role in Th17 cell differentiation (11, 12). Typically, PKM2 forms monomers, dimers or tetramers. Although the tetrameric form of PKM2 shows the highest catalytic activity, the dimeric form of PKM2 in the nuclear is a more profound glycolysis contributor (11, 13). Moreover, PKM2 binds and phosphorylate STAT3 directly, enhancing the nuclear accumulation of STAT3 and downstream transcription of RORγT, which is essential for the differentiation and functions of Th17 cells (12). Notably, compared with the aforementioned rate-limited glycolytic enzyme, PKM2 is more sensitive to reactive oxygen species (ROS), because cysteine 358(C358) could be oxidized by ROS, which impairs the tetramer formation and catalytic functions of PKM2 (14).
Growing evidence shows that low levels of ROS are beneficial and indispensable for the optimal activation of T cells, via mediating the activation of nuclear factor of activated T cells (NFAT) and subsequent IL-2 induction (15, 16). Excessive accumulation of ROS, also known as oxidative stress, inhibits T cell activation to a large extent (17). Oxidative stress is harmful to T cells in many ways, like damaging mitochondria activity or interfering with metabolism (18, 19). Recent work demonstrated that oxidative stress inhibited glycolysis, which is essential for the differentiation of Th17 cells (9, 20), in CD8+ T cells via the inhibition of mTOR pathway (19). However, how ROS influence Th17 cell differentiation (21, 22) and the influence dependent on glycolysis needs to be further discerned.
To maintain cellular redox homeostasis, several antioxidant responses are launched quickly upon exposure to excessive cellular ROS; among these responses, the NF-E2-related factor 2 (NRF2)-antioxidant response element (ARE) axis is the most crucial (23, 24). The stability of NRF2 varies due to its ROS-sensitive ubiquitin modification, which is mainly mediated by Kelch-like ECH associating protein 1 (Keap1), and the half-life time of NRF2 under quiescent conditions is as short as 20 min but is prolonged considerably upon exposure to high ROS levels (23–27). In addition, glycogen synthase kinase 3 beta (GSK3β) signaling has also been reported to be related to NRF2 degradation (28, 29).
Notably, several independent studies showed that the P38 pathway was associated with the activation of NRF2 under oxidative stress (30, 31). However, the molecular mechanism by which P38 signaling affects the NRF2 protein is unknown. p38-regulated/activated kinase (PRAK) is the p38 downstream kinase in the MAPK-activated protein kinase family. Our previous studies suggested that, upon phosphorylation and activation by p38MAPK, PRAK contributed to the regulation of a range of tumor biological processes, especially tumor metastasis (32). However, PRAK function in the immune response is of particular interest. Our previous work suggested PRAK deficiency led to interference with neutrophil extracellular traps (NETs) formation (33) and macrophage phagocytosis (34). Since PRAK is regarded sensitive to oxygen alterations and ROS concentration changes, it is a good time to explore the role played by P38 signaling on NRF2 expression and the subsequent effect of p38 signal on Th17 cell differentiation and function, which has also been proven to be sensitive to cellular ROS (21, 22).
In this research, we demonstrated that PRAK is a protein kinase that phosphorylates NRF2 on serine (S) 558. Forming a newly discovered ubiquitin-independent degradation pathway, phosphorylation by PRAK prolonged the half-life of NRF2 and enhanced the stability of this short-lived protein. S558 phosphorylation contributes the cellular accumulation and nuclear translocation of NRF2 upon exposure to high level of ROS in the tumor microenvironment. PRAK deficiency weakened NRF2-ARE axis and then caused disorders in the elimination of excessive cellular ROS. Therefore, excessive ROS further impaired glycolysis and disturbed the PKM2-dependent phosphorylation of STAT3, resulting in inhibited differentiation of Th17 and impaired Th17-mediated antitumor immunity.
Results
PRAK Is Essential for the Differentiation of Th17 Cells.
To evaluate a potential role played by PRAK in T helper cells, we initially analyzed PRAK expression in various CD4+ helper T cell subsets. CD4+CD62L+CD44low T cells (naïve CD4+ T cells) from C57BL/6 mice were sorted and cultured to ensure proper polarization. We found that PRAK was constitutively expressed in freshly isolated naïve T cells and that PRAK level did not increase substantially in Th1 or Th2 cell subsets, whereas its expression was highly upregulated in regulatory T cell (Treg) and Th17 cell subsets (Fig. 1A and SI Appendix, Fig. S1A). Furthermore, in a kinetic analysis of Th17 cell differentiation, PRAK at both the mRNA and protein levels shown time-dependent increase, peaking on day 3 in culture (Fig. 1B and SI Appendix, Fig. S1B). Thereafter, to determine whether PRAK was essential for the differentiation of Th17 cells, we treated naïve CD4+ T cells with the reported PRAK inhibitor GLPG0259 during polarization. As expected, the inhibition of PRAK activity resulted in a 50% decline in the level of IL-17A+ cells compared with that in the control group (Fig. 1 C and D). To further explore precise mechanisms by which PRAK affects Th17 cells, we generated Prakflox/flox Cd4-cre mice (Prak knockout mice, KO) and Prakflox/flox mice (wild-type mice, WT). Specificity and efficacy of PRAK deletion were confirmed by Western Blotting (SI Appendix, Fig. S1C). We then polarized naïve CD4+ T cells into Th17 cells and compared the function of these cells in Prak KO and WT groups. Correspondingly, the frequency of IL-17A–producing cells generated from the cultured Prak KO T cells was much lower than that of the WT T cells (Fig. 1 E and F). On the other hand, Prak KO T cell’s differentiation into Th1 or Th2 cells was unaffected (SI Appendix, Fig. S1 D–G) and its differentiation into Treg cells was even slightly increased (SI Appendix, Fig. S1 H and I). These data correlated with a decreased Il-17a mRNA level (Fig. 1G) and IL-17A secretion level in the supernatant (Fig. 1H). Notably, the impaired capacity of Th17 differentiation was not due to their apoptosis and proliferation deficiency, which were relatively comparable in WT and KO cells (SI Appendix, Fig. S1 J, K, M, and N). To further exclude the interference of the slightly higher apoptosis on Prak KO T cells (SI Appendix, Fig. S1 J and K), we treated naïve T cells with pan-caspase inhibitor Z-VAD-FMK during the Th17 differentiation process. Z-VAD-FMK treatment significantly decreased the apoptosis level in both WT and KO Th17 and eliminated their apoptosis differences (SI Appendix, Fig. S1 J and K), whereas it had no effects on the Th17 differentiation divergency (SI Appendix, Fig. S1L). To figure out whether TGF-β-SMAD2 or IL-6-STAT3 signals were essential for the differentiation of Th17 cells, we examined the phosphorylation states of SMAD2 and STAT3. The phosphorylated SMAD2 had mild alteration (SI Appendix, Fig. S1O), whereas STAT3 phosphorylation and its nuclear translocation were both consistently abolished by Prak deficiency (Fig. 1I). To confirm this finding, we performed a genome-wide RNA sequencing (RNA-seq) analysis of cultured Th17 cells from WT and KO mice and found that JAK-STAT pathways activation was downregulated in the Prak KO groups, as expected (SI Appendix, Fig. S1P). Similarly, the expression of RorγT and Il-23r in Prak KO groups was downregulated concomitantly (SI Appendix, Fig. S1Q). Collectively, these results suggest that the PRAK signal was indispensable for the differentiation of Th17 cells in vitro.
Fig. 1.
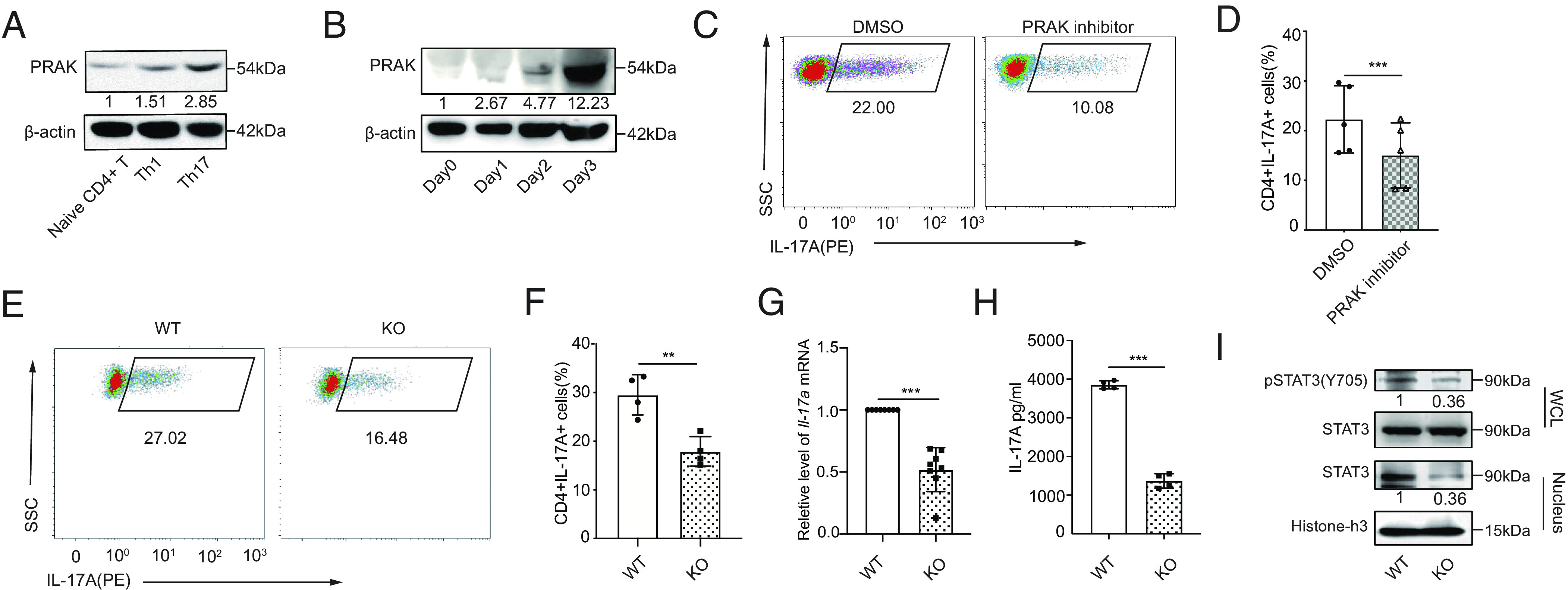
PRAK is essential for the differentiation of Th17 cells. Murine naïve CD4+ T cells were polarized into Th1(IFN-γ+IL-12) and Th17(TGF-β1+IL-6+anti-IL-4+anti-IFN-γ). (A) Immunoblot of PRAK protein in naïve CD4+ T cells and Th1, Th17 (n = 3). (B) PRAK expression in CD4+ T cells over the course of Th17 induction (n = 3). (C and D) Inhibition of Th17 cell differentiation by PRAK inhibitor GLPG0259. Representative dot plots are shown in C. Data from five independent experiments are presented as Mean ±SD (D). (E and F) Naïve CD4+ T cell from WT and Prak KO mice were cultured under Th17 polarization conditions for 3 days. IL-17A+ cells were detected by intracellular staining. Representative dot plots are shown in E. The percentage of IL-17A+ cells from four independent experiments are presented in F. Il-17a mRNA levels were assessed by quantitative RT-PCR (n = 8) (G). IL-17A in the supernatant was quantified by ELISA (n = 4) (H). Western botting was performed to detect total STAT3, nuclear STAT3 and Y705P STAT3 (n = 3). Representative blots were shown in (I). The numbers indicate the relative levels of pSTAT3 and nuclear STAT3 normalized against total STAT3 and histone-H3, respectively. The statistics was performed using Student’s t test. ∗∗P < 0.01; ∗∗∗P < 0.001.
T Cell-Specific Deletion of Prak Alleviates Experimental Autoimmune Encephalomyelitis (EAE).
Our observations led us to consider whether the expression of PRAK would affect a Th17-cell–mediated inflammatory disease model. We established models of EAE, a disease model similar to multiple sclerosis, to evaluate the influence of PRAK on disease severity. First, we immunized both Prak KO and WT mice with myelin oligodendrocyte glycoprotein MOG35–55 peptide as previously described (35). Supporting our hypothesis, the Prak KO mice exhibited significantly milder disease severity than the WT mice after day 14 (Fig. 2A). Moreover, histopathological analysis performed staining spinal cord lesions in EAE mice with hemoxylin and eosin (H&E) revealed less prominent demyelination and fewer inflammatory foci in the central nervous system (CNS) of the Prak KO mice (Fig. 2B). By flow cytometry analysis, gating on CD45+CD3+CD4+ cells, we also found less CD4+ T cell infiltration in the spinal cord of KO mice. In the meanwhile, both total CD4+ T cell and EAE-specific tetramer+ CD4+ T cell number in the spleen and lymph nodes of EAE mice were comparable (SI Appendix, Fig. S2 B–E). The damage to the frequency of CD4+IL-17A+ cells caused by the deficiency of PRAK was not owing to its effect on apoptosis since these cells had no alteration of viability (Fig. 2 C and D and SI Appendix, Fig. S2 F and G). And this less severe damage was relatively specific on the effect of PRAK on Th17 cells since Th1(CD4+IFN-γ+) cell and Treg (CD4+FOXP3+) cell frequency changed modestly (Fig. 2 E and F) and only the viability of Treg cells increased little in Prak KO mice (SI Appendix, Fig. S2 F and G). IL-17A secretion deficiency was also found in T cells obtained from the lymph nodes but not the spleen of the KO group mice (SI Appendix, Fig. S2 H–J), and no difference in the CD4+ IFNγ+ cell and CD4+FOXP3+ cell frequency in the lymph nodes or spleen were identified (SI Appendix, Fig. S2 G and J–M). Moreover, the IL-17A concentration in the serum was also clearly lower in the Prak KO mice (Fig. 2G). Hence, the expression of PRAK may have mediated the differentiation of in CD4+ T cells into Th17 cells in the IL-17–dependent EAE model, the absence of which would cause impaired recruitment of immune cells, regardless of sufficient storage in the peripheral immune organs.
Fig. 2.

T cell-specific deletion of Prak alleviates experimental autoimmune encephalomyelitis (EAE). EAE model was established through MOG35-55 immunization. (A) Clinical scores were recorded from day 11. Data from 5 WT and 4 KO mice were analyzed by two-way ANOVA analysis. (B) Hematoxylin–eosin staining of spinal cord sections. (C–E) Intracellular staining of IL-17A and IFN-γ in the spinal cord infiltrating CD4+ T cells. Representative dot plots (C). Percentage of IL-17A+ (D) and IFN-γ+ (E) cells among CD4+ infiltrating T cells (n = 3). (F) Percentage of Foxp3+ cells in the spinal cord infiltrating CD4+ T cells (n = 3). (G) IL-17A levels in the serum of WT and KO mice, measured by ELISA (n = 3). The statistics was performed using Student’s t test. ∗P < 0.05; ∗∗P < 0.01; N.S not significant.
PRAK-Regulated Reactive Oxygen Species (ROS) Elimination Contributes to Th17 Cell Differentiation.
Our previous work shown that Prak deficiency resulted in elevated intracellular ROS levels in neutrophils, hindering the formation of NETs (33). Hence, in this study, we found that Prak deficiency disturbed redox homeostasis in Th17 cells. Indeed, Prak-deficient Th17 cells shown significantly higher ROS level (Fig. 3 A and B). We speculated that the higher level of ROS accounts for the decrease of Th17 abundance in Prak KO CD4+ T cells. To test this hypothesis, we treated WT CD4+CD62L+ cells with H2O2 or diamide during polarization. Both H2O2 and diamide treatment reduced the IL-17A+ cell frequency (Fig. 3 C and D and SI Appendix, Fig. S3A), and the IL-17A secretion in the supernatant of H2O2 treated cells declined by more than 70% (Fig. 3E). In contrast, N-acetylcysteine (NAC) treatment largely restored IL-17A+ T cell abundance, Il-17a mRNA and IL-17A secretion levels, and the rate of STAT3 nuclear translocation caused by Prak deficiency (Fig. 3 F–J and SI Appendix, Fig. S3B). The Prak-deficient Th17 cells benefited more than WT Th17 cells from ROS elimination. Moreover, treatment with tert-butylhydroquinone (tBHQ), another ROS scavenger, shown effects similar to NAC treatment (SI Appendix, Fig. S3C). Based on the observed ROS regulation, PRAK-mediated elimination of excessive ROS may be critical for the differentiation of Th17 cells.
Fig. 3.
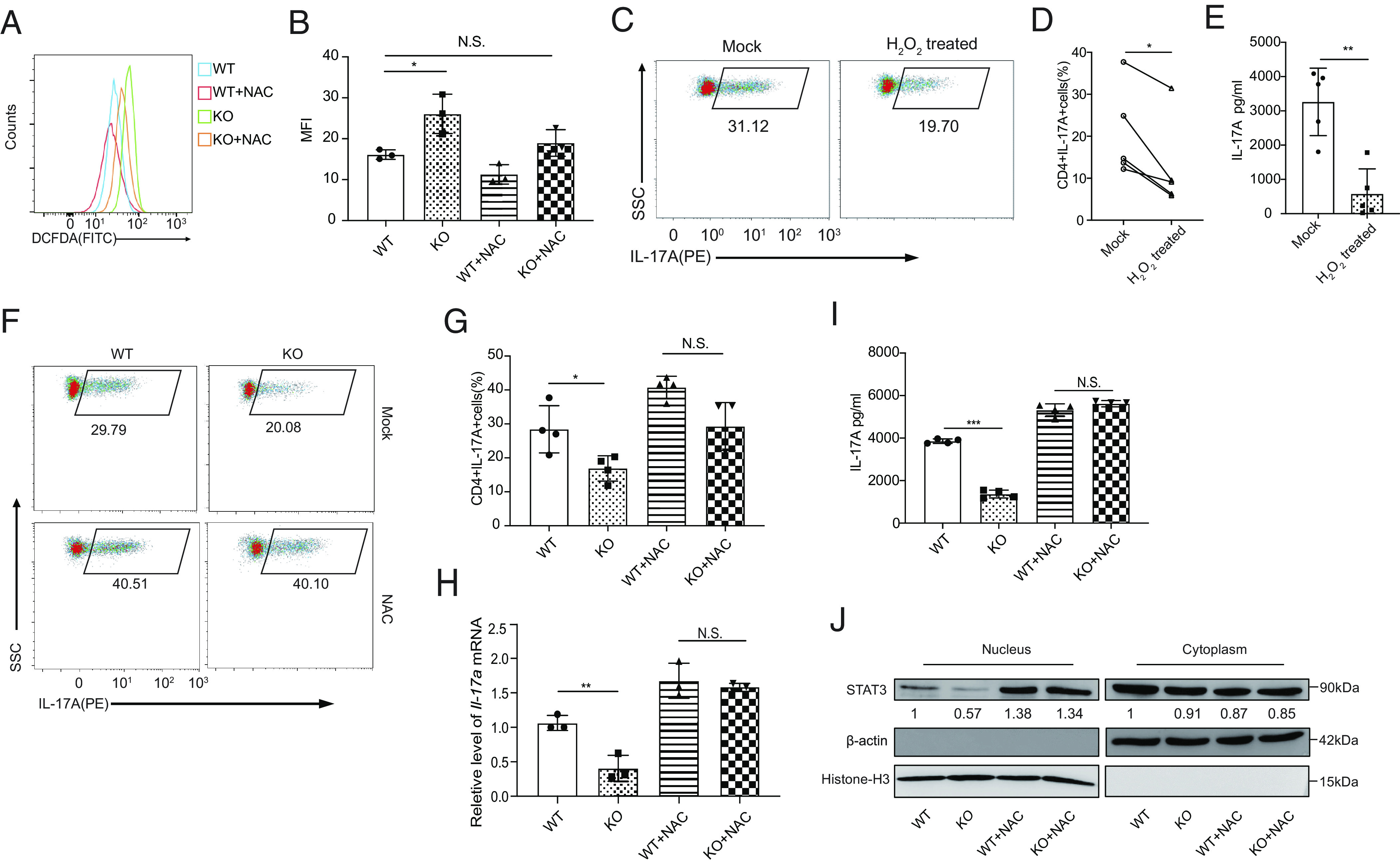
PRAK-regulated reactive oxygen species (ROS) elimination contributes to Th17 cell differentiation. Naïve CD4+T cells from WT and KO mice were cultured under Th17 polarizing conditions with or without the addition of 5mM N-acetyl-L-cysteine (NAC) or 4nM H2O2. All assays were performed at day 3. (A and B) Measurement of cellular ROS in WT and KO Th17 cells using the fluorescent probe DCFDA / H2DCFDA. Representative histogram (A) and mean fluorescence intensity (MFI) (B) from three independent experiments are shown. (C–E) Inhibition of WT Th17 cell differentiation by H2O2. Representative dot plots of intracellular IL-17A staining (C). Frequencies of IL-17A+ cells in the culture (n = 5) (D). IL-17A concentration in the supernatant as determined by ELISA (n = 5) (E). (F–J) Rescue of Th17 cell differentiation defect of Prak-deficient T cells by NAC. Representative dot plots of intracellular IL-17A staining (F). Frequencies of IL-17A+ cells the various cultures (n = 4) (G). Relative levels of Il-17a mRNA assessed by quantitative RT-PCR (n = 3) (H). IL-17A concentration in the culture supernatant (n = 4) (I). STAT3 in nuclear and cytoplasmic fractions of Th17 cells induced under various conditions. Representative blots from three independent experiments are shown (J). The numbers indicate the relative levels of expression normalized against histone H3 or β-actin. The statistics was performed using Student’s t test. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001; N.S. not significant.
PRAK Promotes Redox Homeostasis by Stabilizing Nuclear Factor Erythroid-Derived 2-like 2 (NRF2).
To explore the mechanisms through which PRAK mediate the elimination of excessive ROS, we first measured the expression of NRF2, which is a key mediator of the antioxidant system and has been reported to be associated with P38 activity (30, 31). Our data illustrated that Prak KO CD4+ T cells exhibited lower mRNA and protein expression of NRF2 (Fig. 4A) and NRF2 downstream target molecules, including HO-1 and Prdx2 (Fig. 4A). To verify whether the modulation of PRAK on NRF2 would hurt the ROS elimination and thereafter Th17 differentiation, we constructed Nrf2 overexpression on CD4+ T cells via retrovirus. As expected, our data demonstrated that Nrf2 overexpression could help elevate the Th17 differentiation both in WT and KO cells and diminish the differentiation divergence (Fig. 4 B and C). Given that NRF2 protein has a very short half-life and would accumulate immediately upon ROS exposure (25, 27), the failed accumulation of NRF2 protein under high oxidative stress in KO Th17 cells is a critical defect. Although the transcription of Nrf2 was also blocked (SI Appendix, Fig. S4A), we cannot exclude the possibility that it is due to the low autoregulation by NRF2 protein expression through an ARE-like element located in the proximal region of its promoter (36). Altogether, we speculated that PRAK had an influence on the stability of NRF2 protein under oxidative stress. As previously reported, Keap1 and GSK3β are the two master molecules regulating NRF2’s degradation (25–29), and therefore, we first measured their expressions in WT and Prak KO Th17 cells. However, the levels of Keap1, phosphorylated-GSK3β, and GSK3β were unchanged, regardless of whether PRAK was present or absent (Fig. 4D and SI Appendix, Fig. S4B). Later, we generated PRAK KO HEK293T cell lines through the CRISPR-Cas9 technique. Parent and PRAK-deficient cells were then transfected with FLAG-tagged NRF2 plasmid. Following the treatment with cyclohexane (CHX) to block protein synthesis, cells were harvested at different time points and the levels of FLAG-NRF2 were measured. As shown in Fig. 4E, NRF2 degradation was facilitated by PRAK knockout. Interestingly, despite the apparently accelerated degradation, ubiquitination of NRF2 was found to be less abundant in PRAK-deficient HEK293T cells (Fig. 4F). To confirm that NRF2 was degraded under these conditions, we treated Th17 cells with MG132 and lactacysin, two types of membrane-permeable proteasome inhibitors. Both inhibitors promoted the accumulation of NRF2 protein and eliminated the differences between WT and Prak-deficient Th17 cells (Fig. 4G). These data suggest that PRAK regulated NRF2 stability in a ubiquitin-independent but proteasome-dependent manner.
Fig. 4.
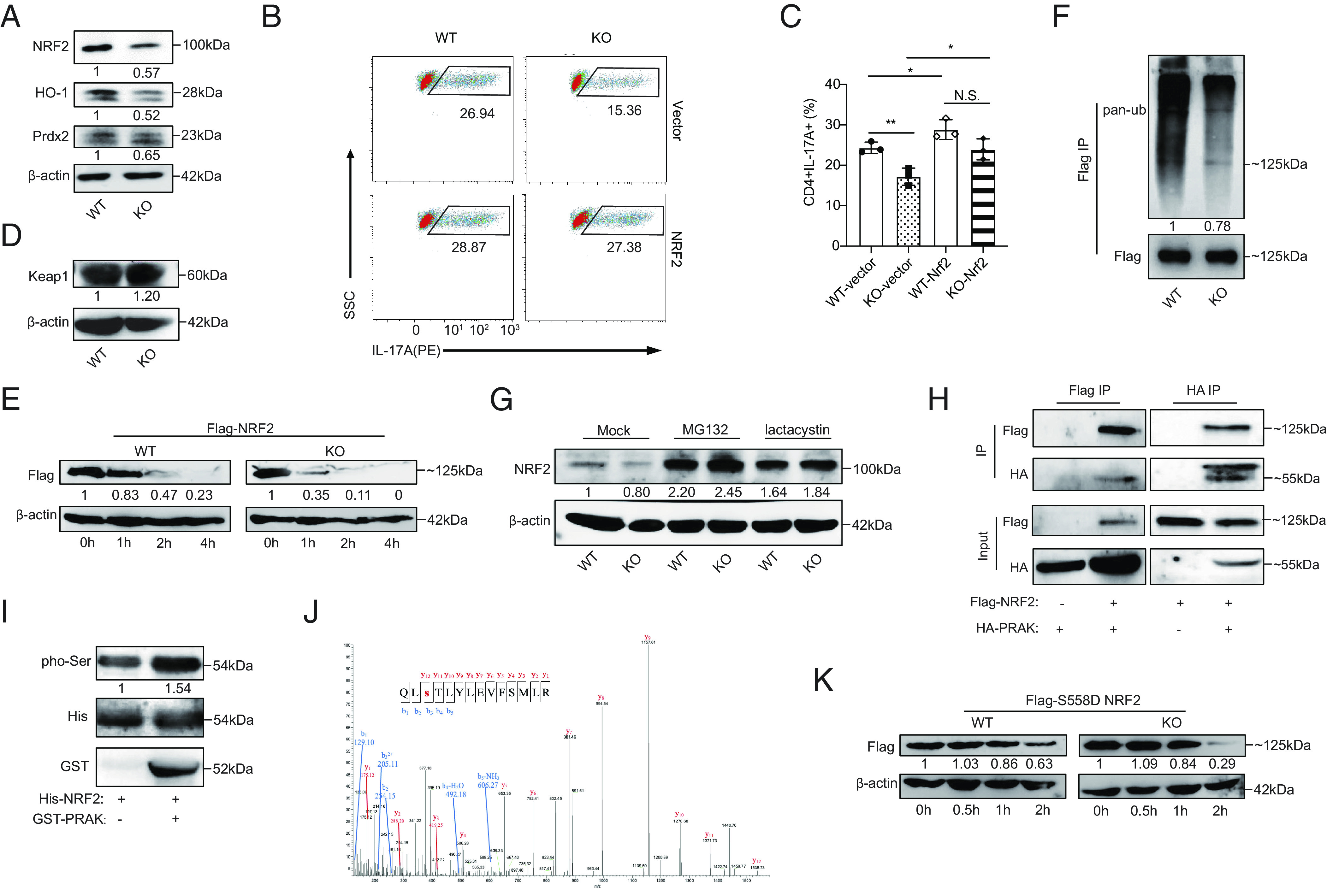
PRAK promotes redox homeostasis by stabilizing nuclear factor erythroid-derived 2-like 2 (NRF2). (A) Immunoblot of NRF2, HO-1, Prdx2 in WT and KO Th17 cells, densitometry performed using β-actin as loading control (n = 3). (B and C) WT and KO naïve CD4+ T cells were first transduced with Nrf2-expressing or empty vectors and then put into culture under Th17 polarizing conditions. IL-17A+ cells were detected by flow cytometry at day 3. Representative dot plots (B). Percentage of IL-17A+ cells (C) (n = 3). (D) Immunoblot of Keap1 in WT and KO Th17 cells (n = 3). (E) WT and PRAK KO HEK293T cells were transduced with equal amounts of Flag-tagged NRF2 plasmid. Cyclohexane (CHX) (200 μM) was added 24 h after transduction. Cells were harvested at various time points and examined for exogenous NRF2 expression by immunoblot. Densitometry was performed using β-actin as loading control (n = 3). (F) Ubiquitin modification of flag tagged NRF2 from WT and KO HEK293T, densitometry performed using Flag-NRF2 as control (n = 3). (G) Immunoblot of endogenous NRF2 in WT and KO Th17 cells after being polarized for 72 h with addition of 20 μM MG132, 5 μM lactacystin or isovolumetric DMSO in the last 12 h (n = 3). (H) Co-immunoprecipitation of HA-PRAK and Flag-NRF2 in HEK293T cells transduced with various combination of plasmids (n = 3). (I) In vitro kinase assay using purified His-NRF2 and GST-PRAK (n = 3). (J) Mass spectrum analysis of NRF2, purified from HEK293T, showing the phosphorylation on the Ser558 residual of NRF2. (K) Degradation of Flag-tagged S558D mutant of NRF2 in WT and PRAK KO HEK293T, assayed as in (D) (n = 3). The numbers indicate the relative protein levels normalized against β-actin and time 0.
To evaluate the possibility that NRF2 is a substrate of PRAK, we performed coimmunoprecipitation (Co-IP) with HEK293T cells overexpressing Flag-tagged NRF2 and hemagglutinin (HA)-tagged PRAK plasmids. As shown in Fig. 4H, anti-HA and anti-Flag can reciprocally precipitate Flag-tagged NRF2 and HA-tagged PRAK. Later, in vitro kinase assay was performed using purified His-tagged NRF2 and GST-tagged PRAK (Fig. 4I). To further study how PRAK functions after binding to NRF2, we performed mass spectrometry (MS) analysis of the NRF2 protein. The MS results showed that the phosphorylation of NRF2 at S558 was only detected in the presence of PRAK (Fig. 4J). To determine the functional relevance of this modification, S558 was replaced with an aspartic acid (D) residue to mimic a persistent phosphorylated state. In line with our speculation, the S558D mutation prolonged NRF2 half-life both in parent and PRAK-deficient HEK293T cells and reduced the difference in lifespan between them (Fig. 4K). Collectively, these data indicated that NRF2 was a substrate of PRAK and that phosphorylation at S558 induced by PRAK enhanced its stability in a ubiquitin-independent manner.
ROS Accumulation in Prak-Deficient Th17 Cells Inhibits Glycolysis and STAT3 Phosphorylation in a PKM2-Dependent Way.
To gain insight into the mechanism underlying the inhibition of ROS on Th17 cell differentiation, RNA-seq was employed to compare the transcriptional profiles of polarized WT and Prak KO Th17 cells. Our results demonstrated that compared to WT, there were 891 genes upregulated and 1242 genes downregulated in Prak KO Th17 cells (SI Appendix, Fig. S5A). Gene ontology (GO) analysis and Gene set enrichment analysis (GSEA) both revealed that glycolysis, which is highly elevated during Th17-polarizaiton, was dramatically downregulated in the Prak KO Th17 cells (Fig. 5 A–C). Consistently, the intake of 2-NBDG in the Prak KO Th17 cells was lower but had no statistical difference (SI Appendix, Fig. S5 B and C) and the remaining glucose concentration in the supernatants shown a little increase in Prak KO group (SI Appendix, Fig. S5D), indicating there may be a slight blockade in its glucose uptake. Otherwise, we did not observe any significant change in the level of total neutral lipids (SI Appendix, Fig. S5 E and F) and lipids intake (SI Appendix, Fig. S5 G and H) between WT and KO Th17 cells. Later, a reduction in the gene and protein levels of several key enzymes in glycolysis was confirmed by real-time PCR (SI Appendix, Fig. S5I) and western blotting (Fig. 5D). In addition, an extracellular acidification rate (ECAR) analysis was performed to confirm that aerobic glycolysis was inhibited by Prak deficiency (Fig. 5 E–G). In contrast, treatment with NAC rescued the expression of glycolysis enzymes (SI Appendix, Fig. S5I), and significantly elevate ECAR in Prak KO Th17 cells (Fig. 5H).
Fig. 5.
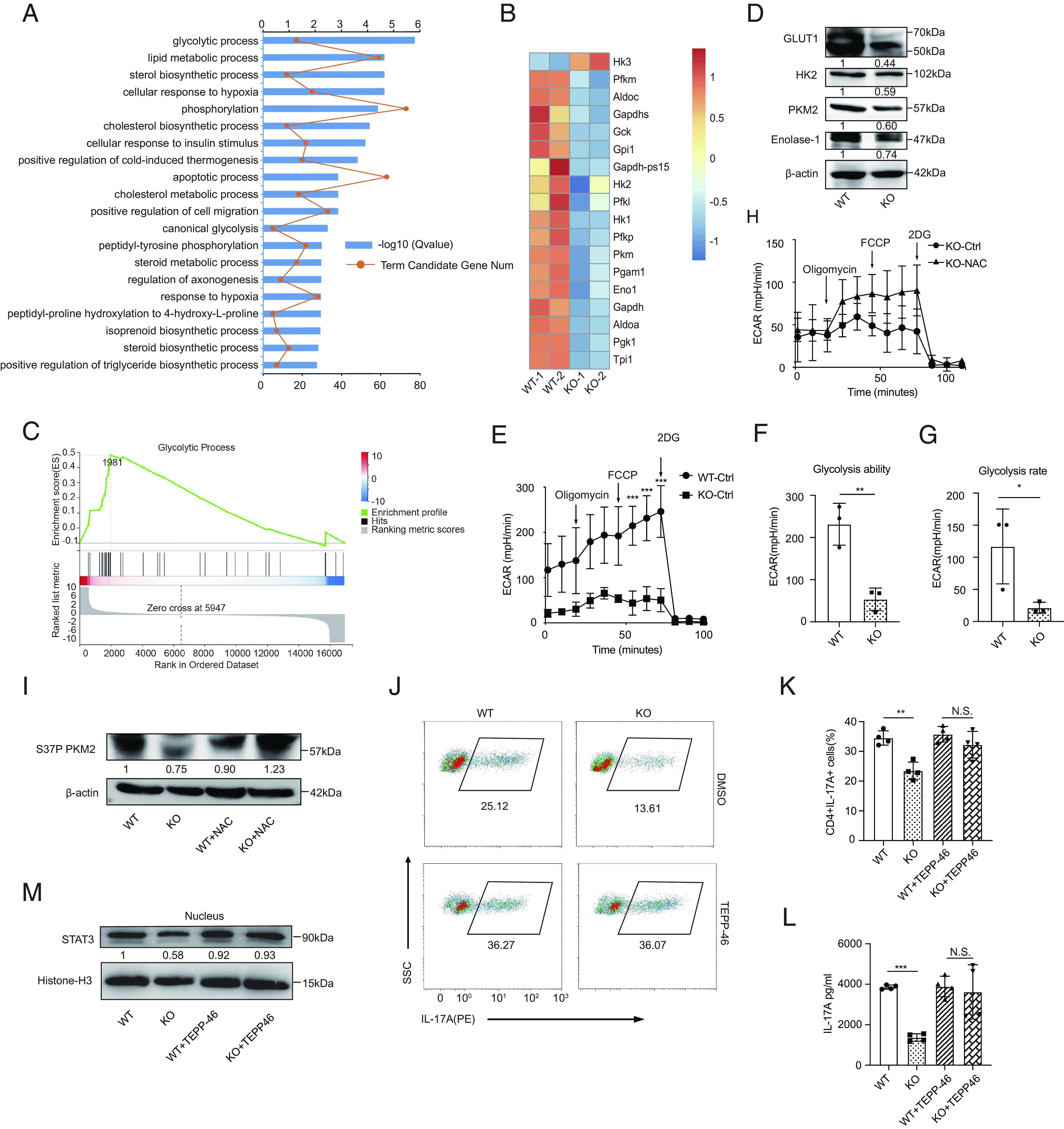
ROS accumulation in Prak-deficient Th17 cells inhibits glycolysis and STAT3 phosphorylation in a PKM2-dependent way. (A–C) RNA-seq was performed with WT and KO Th17 cells (n = 2). Gene oncology (GO) analysis of differentially expressed genes (A). Heatmap of glycolysis-related, differentially expressed genes (B). GSEA analysis of genes involved in glycolytic process (C). (D) Verification of the differential expression of HK2, Glut1, PKM2, and Enolase 1 at protein level by Western blotting (n = 3). (E–G) Extracellular acidification rate (ECAR) assay was performed with WT and KO Th17 cells (E). Glycolysis ability and glycolysis rate are shown in F and G, respectively. (H) ECAR assay of KO Th17 cells following treatment with NAC or solvent (n = 3). (I) Immunoblot of S37P PKM2 in WT and KO Th17 cells generated in the presence or absence of NAC (n = 3). (J–M) Th17 cells were generated with the addition of TEPP-46 or isovolumetric DMSO. Representative dot plots showing intracellular staining of IL-17A (J). Frequencies of IL-17A+ cells (J) (n = 4). Protein level of IL-17A in the culture supernatant (L) (n = 4). Immunoblot of nuclear STAT3, densitometry performed using histone-H3 as loading control (n = 3). The statistics was performed using Student’s t test. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001; N.S. not significant.
To further understand how ROS influence glycolysis, we hypothesized that PKM2 functions as a hinge that link ROS and PRAK-mediated Th17 cell differentiation. PKM2 is a rate-limiting enzymes in glycolysis, and more importantly, PKM2 can be oxidized at C358 when exposed to high level of ROS (14), and it plays a part in STAT3 activation (12, 37). Our data illustrated that the levels of total PKM2, monomeric PKM2, and tetrameric PKM2 were much lower in Prak KO Th17 cells (Fig. 5D and SI Appendix, Fig. S5 I–K). In addition, treatment with NAC eliminated the phosphorylation differences in PKM2 at two reported sites Ser37(S37) or Thy105(Y105) between WT and Prak KO groups (Fig. 5I and SI Appendix, Fig. S5L). We found that treatment of these cells with TEPP-46, a type of PKM2 activator that could promote the formation of tetrameric isoform in Th17 cells (SI Appendix, Fig. S5M), rescued the Th17 differentiation in a significantly increased and dose-dependent manner in Prak KO Th17 cells, but this treatment exerted negligible effects on the differentiation of WT Th17 cells (Fig. 5 J–L and SI Appendix, Fig. S5 N and O). Mechanistical consistently, TEPP-46 treatment promoted the nuclear translocation of STAT3(Fig. 5M) by facilitating the phosphorylation of PKM2 S37 and Y105 (SI Appendix, Fig. S5L), which has been reported to be essential for the PKM2 regulation of STAT3 activity (11). Hence, our data indicated that excessive intracellular ROS levels inhibit glycolysis and STAT3 activation in Th17 cells by inhibiting both tetramer formation and phosphorylation of PKM2.
PRAK Regulates the Antitumor Immunity of Th17 Cells in Colon Cancer.
Th17 cells exert a marked effect on the secretion of proinflammatory cytokines; thus, the Th17 cell contribution to autoimmunity and infectious diseases are readily acknowledged (38). However, the relationship between Th17 cells and tumor immunopathology is still debated (1–5, 39). By analyzing transcriptome data obtained from the Gene Expression Profiling Interactive Analysis (GEPIA) database (40), we found that colon cancer patients with higher expression of IL-17A or PRAK exhibited a longer survival period (Fig. 6A and SI Appendix, Fig. S6A). To investigate the function of PRAK-mediated Th17 differentiation in tumor development, we subcutaneously inoculated WT and Prak KO mice with MC38 colon cancer cells. Compared with WT mice, the Prak KO mice exhibited accelerated tumor growth, leading to a greater tumor weight and shorter survival period (Fig. 6 B–D and SI Appendix, Fig. S6B). At the end of observation, we analyzed tumor-infiltrating T cells in the tumor-bearing mice, with a gating strategy shown in SI Appendix, Fig. S6C. While total CD4+ T cells and the IFN-γ+Th1, IL-4+Th2, and Foxp3+Treg subsets (SI Appendix, Fig. S6 D–G) were equally represented in WT and KO mice, a lower frequency of IL-17A+ cells was detected in the tumor tissues of Prak KO mice (Fig. 6 E and F). Besides, we found less CD8+ T cell infiltration in Prak KO mice (Fig. 6 G and H), although they appeared to be functionally intact as indicated by the comparable levels of cytokine production (IFN-γ and TNF-α), and granzyme B and Ki67 expression (SI Appendix, Fig. S6 H–K). To dissect the role of IL-17A+CD4+ T cells in the control of tumor growth in this particular model, we first treated mice with the anti-CD4 antibody. Indeed, CD4+ T cell depletion (SI Appendix, Fig. S6L) resulted in a much enhanced tumor growth in WT mice while showing no impact in KO mice (Fig. 6 I–K). Next, we attempted to antagonize the action of IL-17A using a neutralizing antibody, which had previously been used in similar studies (2). As with anti-CD4, anti-IL-17A promoted tumor growth in WT but not in KO mice (Fig. 6 I–K). The irresponsiveness of KO mice is most likely due to the already low levels of tumor-infiltrating IL-17A+ cells in Prak KO mice. Of note, IL-17A neutralizing antibody treatment also reduced the infiltration of CD8+ T cells in WT mice, to a level similar to that in KO mice (SI Appendix, Fig. S6M), indicative of an important role of IL-17A in recruiting CD8+ T cells. To verify that the reduction in excessive ROS levels could rescue Th17-mediated antitumor immunity in Prak KO mice, we treated tumor-bearing mice with 67 mg/kg NAC via intraperitoneal injection. As expected, NAC treatment substantially slowed the tumor growth rate in the Prak KO mice but exerted only a mild effect on the WT mice (Fig. 6B). In addition, after the NAC treatment, the weight of the tumors in the Prak KO mice was similar to that in the WT mice, which was significantly different from that in the untreated Prak KO group (Fig. 6 B–D). We attributed the reversibility of the tumor formation in the Prak KO mice to NAC-induced elimination of excessive ROS and a subsequent increase in the frequency of IL-17A+ tumor-infiltrated Th17 cells (Fig. 6 E and F) but not its effect on other Th subsets (SI Appendix, Fig. S6 E–G). Enhanced IL-17A then helps recruit more CD8+ T cells to tumor environment (Fig. 6 G and H). However, NAC treatment did not influence the function of CD8 cells directly, such as their cytokine secretion, granzyme B expression and proliferation (SI Appendix, Fig. S6 H–K). In summary, our results suggested that the impaired antitumor immunity in the Prak KO mice was due to defective Th17 cell production and that eliminating excessive ROS greatly ameliorated this effect.
Fig. 6.
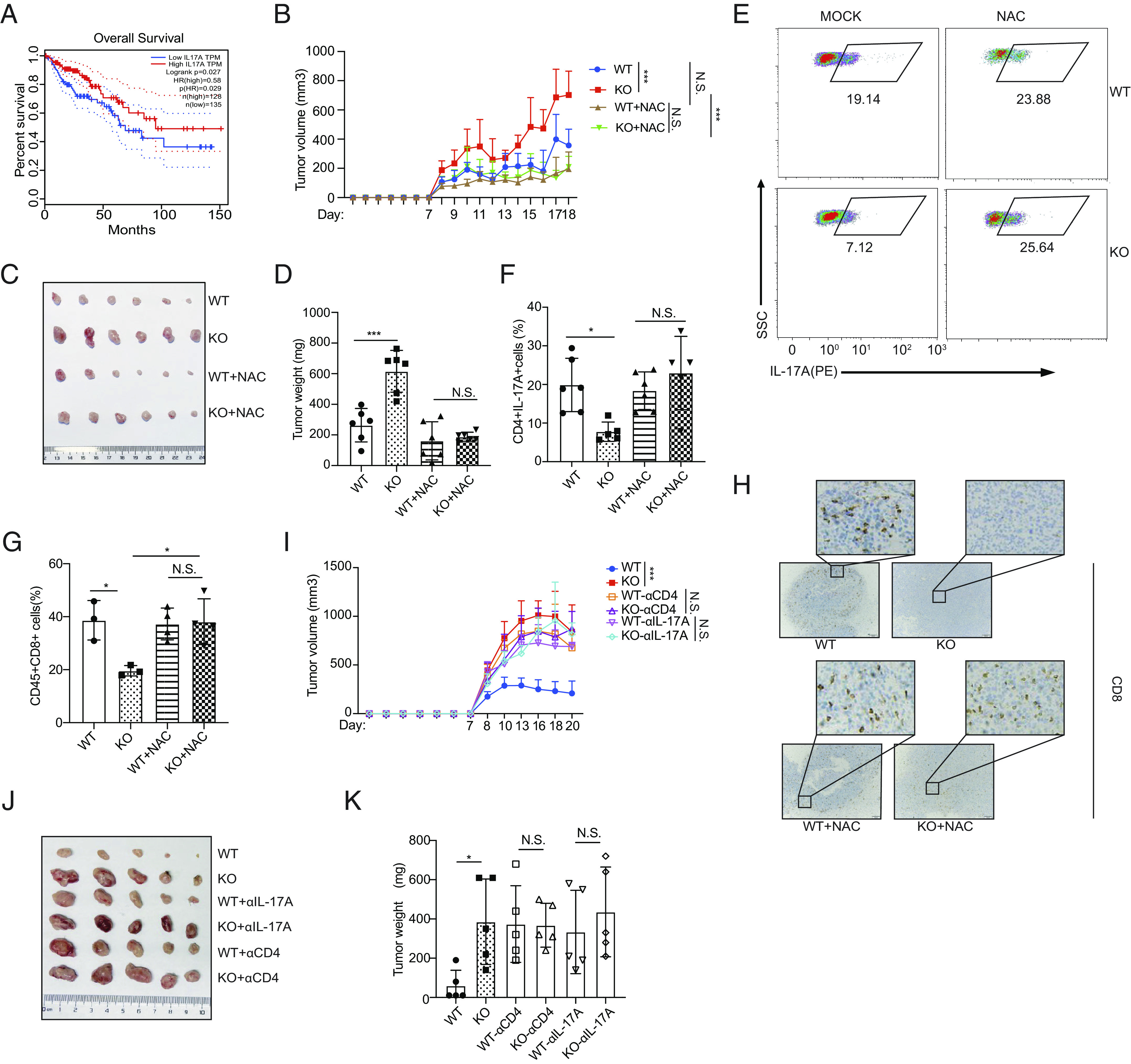
PRAK regulates the antitumor immunity of Th17 cells in colon cancer. (A) The survival curve of colon cancer patients grouped according to IL-17A expression. Data were collected from Gene Expression Profiling Interactive Analysis (GEPIA). (B–H) WT or KO mice were subcutaneously injected with 1 × 106 MC38 cells, with or without daily treatment of NAC (67 mg/kg). Tumor volume, calculated as (L, length, W, width), was recorded daily after tumor inoculation (n = 6 for each group) (B). At the end of observation (day 18), tumors were removed and weighted before the isolation of tumor-infiltrating lymphocytes. Tumor size (C), tumor weight (D). Representative dot plots showing intracellular of IL-17A staining of infiltrating CD4+ T cells (E). Frequency of tumor-infiltrating CD4+IL-17A+ cells (n = 5) (F). Frequencies of tumor-infiltrating CD8+ T cells (G). IHC staining for CD8 of tumor sections (H). (I–K) WT and KO mice were treated with anti-CD4 or anti-IL-17A prior to the inoculation of MC38. Growth curve of tumors in different groups (n = 5 for each) (I). Tumor size (J) and weight (K) at the end of observation. Statistics was performed using Student’s t test for paired comparison and two-way ANOVA analysis for tumor growth over time. ∗P < 0.05; ∗∗∗P < 0.001; N.S. not significant.
Discussion
Prak-deficiency causes functional disorders in neutrophils and macrophages by inducing oxidative stress, but the precise mechanisms by which PRAK regulates ROS elimination and whether it affects CD4+ T helper cell subsets differentiation remain unclear. Here, we demonstrated that the PRAK-NRF2-ROS axis was essential for maintaining the intracellular redox homeostasis in Th17 cells and that activation of this axis promoted Th17 cell differentiation and induced antitumor effects. NRF2 was found to be a novel substrate of PRAK, and PRAK was shown to modulate the stability of the NRF2 protein independent of ubiquitination, extending the NRF2 half-life. Therefore, cellular ROS production induced to excess via NRF2 disrupted both glycolysis and PKM2-dependent phosphorylation of STAT3 and thus hindered Th17 cell differentiation. The deficiency on Th17 cell differentiation led to less severe symptoms in Prak KO mouse models of EAE and impaired antitumor immunity in Prak KO mouse models carrying MC38 tumors.
The tumor microenvironment consists of various types of stress factors, including but not limiting to hypoxia, low pH, high pressure, and high level of lactate, all of which can be potent ROS inducer (41). Therefore, extreme oxidative stress in tumor-infiltrating lymphocytes (TILs) creates a high barrier for immune antitumor therapies. As by-product of mitochondrial metabolism, ROS are strictly regulated to maintain balanced levels and would be removed efficiently under physiologic conditions. Quantitative accumulation of cellular ROS induced by pathological factors, as mentioned above, is harmful, and the effects are not limited to a tendency toward apoptosis. Cytosolic ROS accompanied by TCR or CD28 signals may act as a signal passenger19 and are essential for the optimal activation of T cells, mechanistically promoting the transcription of IL-2 and the activation of NFκB as well as downstream effector genes (16, 42). Nevertheless, the effects of ROS on Th17 cells are still debated. For example, a study showed that high-glucose-diet-induced ROS production promoted the activation of TGF-β signaling and the differentiation of Th17 cells, while other studies have shown that elevated ROS activated misshapen/NIK-related kinase 1 (MINK1) and thus negatively regulated Th17 differentiation (21, 22). In this study, we demonstrated that PRAK was important for Th17 cells to maintain redox homeostasis and that PRAK acted in a NRF2-dependent manner. High levels of ROS can disrupt glycolysis and PKM2-dependent phosphorylation of STAT3, hindering Th17 cell differentiation. Of course, failure to remove ROS that had been produced due to Prak-deficiency was clearly evident in all T cell subsets, and further studies are needed to explore the role played by PRAK in other T cell subsets, especially Tregs, with high expression of PRAK.
The NRF2-mediated antioxidant system plays a master role in eliminating excessive cytosolic ROS. NRF2 consists of six conserved domains, including those numbered from NRF2-ECH homology domain-1(Neh1) to Neh618. Under normal resting conditions, through the ETGE motif in the Neh2 domain, most NRF2 binds with Keap1, which forms a ubiquitin E3 ligase complex with CULLIN3 (CUL3) to polyubiquitinate NRF2, promoting NRF2 degradation. Once exposed to high level of ROS, Keap1 is oxidized and NRF2 is released and accumulates, leading to increased transcription (23). Many tumor cells have been identified to carry missense point mutations nearby ETGE motif, which disrupt the interaction between NRF2 and Keap1, so accumulated NRF2 endows tumor cells with powerful antioxidants weapon that alleviates their own oxidative stress (43). Besides, several independent studies demonstrated that GSK3β could phosphorylate NRF2 in the Neh6 domain and promote the polyubiquitination of NRF2 in a Keap1-independent way (44, 45). Hence, until now, it has been widely accepted that the degradation of NRF2 was mediated mainly through ubiquitination. Nevertheless, it remains unknown whether the other Neh domains influence the degradation of NRF2 in a ubiquitin-independent manner. In this study, we demonstrated that PRAK expression was upregulated when NRF2 was activated. These two proteins showed direct interactions, and upon their combination, PRAK facilitated NRF2 phosphorylation at a previously undiscovered site, S558. Phosphorylation of this newly identified site prolonged half-life of NRF2 protein even when the ubiquitination rate was increased. Further study is needed to identify the precise mechanism of this ubiquitin-independent degradation pathway and to determine whether it competitively occupies Neh domain to inhibit NRF2 ubiquitination. In every experiment, our findings provide a new possibility for endowing Th cells with stronger resistance to oxidation stress.
Glycolysis is a key metabolic feather of T cells in a tumor environment. During glycolysis, PKM2, a splice variant of PKM gene, is a rate-limiting enzyme. Several studies have proved the key role played by PKM2 in CD4+ Th17 cell differentiation through phosphorylate STAT3 on Y705, which promote the nuclear translocation of STAT3 (12). PKM2 has been reported to form monomers, dimers, and tetramers, and these different isoforms show different functions. For example, the tetramer-forming isoform exhibits catalytic functions and is located in cell cytoplasm, and only the dimeric isoform could be phosphorylated, enter the nuclear and regulate a series of gene transcription through direct phosphorylation of histone H3 (46). Although it shows negligible catalytic activity (47), dimeric PKM2 but not tetrameric PKM2 promote glycolysis by enhancing the transcriptional activity of certain key factors (11). Whether the monomeric or tetrameric isoforms contribute to STAT3 activation remains unclear. The formation and nuclear translocation of the PKM2 isoform has been reported to be mediated mainly by phosphorylation at two sites, namely S37 and Y105. S37 is phosphorylated by ERK1/2, mediating the nuclear translocation of PKM2. Y105 is phosphorylated by PKC, promoting the formation of dimeric isoform (48). Notably, PKM2 protein is clearly one of the glycolysis targets of ROS. ROS oxidize C358 in PKM2, preventing the formation of the tetrameric isoforms and destroying the catalytic functions of PKM2 (14). Hence, our results showed that exposure to high ROS levels or Prak-deficiency hampered the gene expression of PKM2, and the abundance of all PKM2 isoforms, including monomers, dimers (phosphorylated isoforms) and tetramers, was obviously decreased in Prak KO Th17 cells, but this reduced abundance would be rescued by NAC treatment. Our data are consistent with those of a study reporting PKM2 transactivated a key regulator of aerobic glycolysis hypoxia-inducible factor-1α (HIF1α) (49). Interestingly, the nuclear STAT3 level, which may be regulated by PKM2 dimer, was found to be lower. However, even in the cytoplasm, the phosphorylation of STAT3 was clearly inhibited after ROS regulation. In this case, the tetramer isoform of PKM2, which is the main isoform in cell cytoplasm, may facilitate the phosphorylation of STAT3 before its translocation into nuclear. This hypothesis was supported data from the effect of TEPP-46 treatment experiments. The precise function of PKM2 tetramer need to be confirmed by further studies, the data from which may help us determine whether ROS influence the formation of tetramers or intervene in the function of tetrameric isoforms and whether the ROS regulation of tetramer activity depends on S37 or Y105 PKM2 phosphorylation.
In summary, our data reveal an important role played by PRAK in maintaining the appropriate level of NRF2 expression and regulation Th17 cell differentiation as well as its antitumor immunity. We demonstrate that increased PRAK abundance is a molecular feature of Th17 cells under oxidative stress in tumor-bearing hosts. This increase in PRAK expression protects the NRF2 protein from degradation. NRF2-mediated ROS elimination strongly promotes glycolysis and STAT3 activation, resulting in Th17 differentiation and antitumor function. We suggest that excessive ROS form a barrier to the development of effector Th17 cells. Targeting PRAK holds promise for maintaining an active ROS scavenging system and is meaningful for sustained potent Th17 cell antitumor immunity.
Materials and Methods
Briefly, the differentiation efficacy of CD4 subsets was analyzed in polarized condition in vitro. Frequencies or functions of specific T cell subsets were examined in EAE model and MC38 tumor model. RNA-seq, western blot, and real-time PCR were used to identify and confirm the pathway alteration. More detailed information is available in the SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Lanfen Chen (Xiamen University) for the gift of NRF2 plasmid; Xiuyuan Sun for technical support of flow cytometry sorting and analysis and all members in our laboratory for insightful discussion and technical assistance. This work was supported by grants from the National Natural Sciences Foundation of China (31872735, 31970840, and 32330037), Beijing Municipal Natural Science Foundation (7212058) and Natural Science Foundation of Shanxi Province (20210302124092) and Clinical Medicine Plus X - Young Scholars Project, Peking University, the Fundamental Research Funds for the Central Universities (71006Y2823).
Author contributions
Z.Z., Y.W., Y. Zhang, and W.W. designed research; Z.Z., Y.W., Y.J., Y. Zhao, X.P., and W.W. performed research; S.G. contributed new reagents/analytic tools; Z.Z., Y.G., Z.W., and B.Z. analyzed data; and Z.Z., Y.W., and W.W. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. W.Z. is a guest editor invited by the Editorial Board.
Contributor Information
Yu Zhang, Email: zhangyu007@hsc.pku.edu.cn.
Wei Wang, Email: wangwei83427@bjmu.edu.cn.
Data, Materials, and Software Availability
The raw data of bulk RNA-seq used in this study has been deposited in the Gene Expression Omnibus (GEO) database (GSE229887), https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE229887 (50). All other study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Zou W., Restifo N. P., T(H)17 cells in tumour immunity and immunotherapy. Nat. Rev. Immunol. 10, 248–256 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qiu W., Targeting histone deacetylase 6 reprograms Interleukin-17-producing helper T cell pathogenicity and facilitates immunotherapies for hepatocellular carcinoma. Hepatology 71, 1967–1987 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Kryczek I., et al. , Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 114, 1141–1149 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez L. G., et al. , TGF-beta signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat. Commun. 11, 2608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benchetrit F., et al. , Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood 99, 2114–2121 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Weaver C. T., Harrington L. E., Mangan P. R., Gavrieli M., Murphy K. M., Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity 24, 677–688 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Korn T., Bettelli E., Oukka M., Kuchroo V. K., IL-17 and Th17 Cells. Annu. Rev. Immunol. 27, 485–517 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Wang C., et al. , CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 163, 1413–1427 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi L. Z., et al. , HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu L., et al. , Niche-selective inhibition of pathogenic Th17 cells by targeting metabolic redundancy. Cell 182, 641–654.e620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Angiari S., et al. , Pharmacological activation of pyruvate kinase M2 inhibits CD4(+) T cell pathogenicity and suppresses autoimmunity. Cell Metab. 31, 391–405.e398 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damasceno L. E. A., et al. , PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J. Exp. Med. 217, e20190613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo W., et al. , Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anastasiou D., et al. , Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaminski M. M., et al. , Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: Mechanism of ciprofloxacin-mediated immunosuppression. J. Immunol. 184, 4827–4841 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Sena L. A., et al. , Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franchina D. G., Dostert C., Brenner D., Reactive oxygen species: involvement in T cell signaling and metabolism. Trends Immunol. 39, 489–502 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Wang L., et al. , Selective oxidative stress induces dual damage to telomeres and mitochondria in human T cells. Aging Cell 20, e13513 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Z., et al. , SENP7 senses oxidative stress to sustain metabolic fitness and antitumor functions of CD8+ T cells. J. Clin. Inves. 132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dang E. V., et al. , Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang D., et al. , High glucose intake exacerbates autoimmunity through reactive-oxygen-species-mediated TGF-beta cytokine activation. Immunity 51, 671–681.e675 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu G., et al. , Suppression of Th17 cell differentiation by misshapen/NIK-related kinase MINK1. J. Exp. Med. 214, 1453–1469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto M., Kensler T. W., Motohashi H., The KEAP1-NRF2 System: A Thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169–1203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sies H., Jones D. P., Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Cullinan S. B., Gordan J. D., Jin J., Harper J. W., Diehl J. A., The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell Biol. 24, 8477–8486 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furukawa M., Xiong Y., BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell Biol. 25, 162–171 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi A., et al. , Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 24, 7130–7139 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chowdhry S., et al. , Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32, 3765–3781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain A. K., Jaiswal A. K., GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 282, 16502–16510 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Yao P., et al. , Quercetin protects human hepatocytes from ethanol-derived oxidative stress by inducing heme oxygenase-1 via the MAPK/Nrf2 pathways. J. Hepatol. 47, 253–261 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Hourihan J. M., Moronetti Mazzeo L. E., Fernandez-Cardenas L. P., Blackwell T. K., Cysteine sulfenylation directs IRE-1 to activate the SKN-1/Nrf2 antioxidant response. Mol. Cell 63, 553–566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y., et al. , The essential role of PRAK in tumor metastasis and its therapeutic potential. Nat. Commun. 12, 1736 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu T., et al. , Cyclooxygenase-1 regulates the development of follicular Th cells via prostaglandin E2. J. Immunol. 203, 864–872 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Mi L., et al. , PRAK promotes the pathogen clearance by macrophage through regulating autophagy and inflammasome activation. Front. Immunol. 12, 618561 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J. Y., et al. , Serum amyloid a proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell 180, 79–91.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwak M. K., Itoh K., Yamamoto M., Kensler T. W., Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell Biol. 22, 2883–2892 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao X., Wang H., Yang J. J., Liu X., Liu Z. R., Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 45, 598–609 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stockinger B., Omenetti S., The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 17, 535–544 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Vitiello G. A., Miller G., Targeting the interleukin-17 immune axis for cancer immunotherapy. J. Exp. Med. 217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang Z., et al. , GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–w102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu K., et al. , Hypoxia-induced ROS promotes mitochondrial fission and cisplatin chemosensitivity via HIF-1alpha/Mff regulation in head and neck squamous cell carcinoma. Cell Oncol. (Dordr.) 44, 1167–1181 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Los M., et al. , IL-2 gene expression and NF-kappa B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 14, 3731–3740 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayes J. D., McMahon M., NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 34, 176–188 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Rada P., et al. , SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 31, 1121–1133 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rada P., et al. , Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell Biol. 32, 3486–3499 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang W., et al. , PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150, 685–696 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lunt S. Y., et al. , Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol. Cell 57, 95–107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prakasam G., Iqbal M. A., Bamezai R. N. K., Mazurek S., Posttranslational modifications of pyruvate kinase M2: Tweaks that benefit cancer. Front Oncol. 8, 22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ouyang X., et al. , Digoxin suppresses pyruvate kinase M2-promoted HIF-1alpha transactivation in steatohepatitis. Cell Metab. 27, 339–350.e333 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Z., Wang Y., Wang W., The PRAK-NRF2 axis promotes the differentiation of Th17 cells by mediating the redox homeostasis and glycolysis. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE229887. Deposited 14 April 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
The raw data of bulk RNA-seq used in this study has been deposited in the Gene Expression Omnibus (GEO) database (GSE229887), https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE229887 (50). All other study data are included in the article and/or SI Appendix.