

Summary
Exponential accumulation of single-cell transcriptomes poses great challenge for efficient assimilation. Here, we present an approach entitled generative pretraining from transcriptomes (tGPT) for learning feature representation of transcriptomes. tGPT is conceptually simple in that it autoregressive models the ranking of a gene in the context of its preceding neighbors. We developed tGPT with 22.3 million single-cell transcriptomes and used four single-cell datasets to evalutate its performance on single-cell analysis tasks. In addition, we examine its applications on bulk tissues. The single-cell clusters and cell lineage trajectories derived from tGPT are highly aligned with known cell labels and states. The feature patterns of tumor bulk tissues learned by tGPT are associated with a wide range of genomic alteration events, prognosis, and treatment outcome of immunotherapy. tGPT represents a new analytical paradigm for integrating and deciphering massive amounts of transcriptome data and it will facilitate the interpretation and clinical translation of single-cell transcriptomes.
Subject areas: Automation in bioinformatics, Data processing in systems biology, Transcriptomics
Graphical abstract
Highlights
-
•
tGPT models gene rankings via autoregressive language modeling
-
•
tGPT works effectively on fundamental single-cell analysis tasks
-
•
tGPT captures distinctive features of different cell types
-
•
tGPT learns expression signatures linked to genomic and clinical features
Automation in bioinformatics; Data processing in systems biology; Transcriptomics
Introduction
Rapid advancement in single-cell RNA sequencing leads to dramatic drop in sequencing cost and allows for millions of single-cell transcriptomes to be digitized in a single experiment simultaneously. The whole human body is estimated to have 30 trillion cells. Single-cell transcriptome sequencing provided an unprecedented resolution to distinguish different cell type clusters, depict hierarchical cell arrangement and decipher transitional cell states. To achieve this goal, multiple single-cell atlasing projects have been established internationally, including Human Cell Atlas (HCA),1 Single Cell Expression Atlas (SCEA),2 COVID-19 Atlas,3 Tabula Muris Atlas4 and Mouse Cell Atlas.5 The HCA project1 aims to digitize all cells and create a reference map of the human body through community-driven initiative that researchers all around the world can contribute. SCEA2 compiles and annotates publicly available single-cell transcriptomes across multiple species and different studies. The COVID-19 Atlas3 aims at elucidating molecular mechanism and therapeutic target of COVID-19 by generating single-cell atlas of SARS-CoV-2 infection in COVID-19 patients. The Tabula Muris4 and MCA5 atlases constitute the single-cell reference maps of mouse with millions of cells obtained from different organs. These atlasing projects pose tremendous challenge in the integration of diverse transcriptomes from different projects. However, single-cell transcriptomes are generated by different platforms and experimental protocols. They are sparse, noise and prone to batch effect.6,7 Therefore, an analytical method to efficiently integrate ten millions of cells are urgently needed.
Over the past few years, deep learning approaches have led to seismic changes in image recognition and natural language understanding. The success of deep learning could largely attribute to the availability of big data, advancement in computational infrastructure, expressivity and scalability of the computational model. The deep learning model could adeptly handle super large-scale high dimensional data and assimilate real-world information. Owing to the exponential accumulation of millions of cell transcriptomes, elucidation of the reference map of single-cell transcriptomes with deep learning becomes an attractive application. Deep learning methods such as scVI,8 SAUCIE9 and INSCT10 have been developed for the analysis of single-cell transcriptomes.
The progress of artificial intelligence is undergoing a paradigm shift in computer vision and natural language processing. Deep neural networks based on transformer are becoming the de facto approach in wide variety of scenarios such as vision, language and reasoning.11 Transformer-based models pretrained on broad data at scale continues to achieve state-of-the-art progress in image classification12,13 and language understanding.14,15,16 The success of these pretrained models can be attributed to their high expressivity and scalability enabled by transformer to assimilate feature representation from massive amount of unlabeled data. However, the investigation of single-cell transcriptome pretraining at scale has not been well studied.
In this study, we present a deep learning approach entitled tGPT toward integration of unlimited number of cells. tGPT is built on transformer that has been widely used in natural language understanding and image recognition. The transformer is an essential component and key success of foundation models because of its high expressivity and scalability.11 tGPT takes as input the expression rankings of top-expressing genes rather than the actual expression levels. Rank-based methods for gene expression have been demonstrated to be insensitive to batch effects and data normalization.17,18,19,20 tGPT is conceptually simple and empirically efficient. It models the occurrence of a gene in the context of its preceding neighbors’ rankings. We developed tGPT with 22.3 million cells and systematically evaluated tGPT on four heterogeneous datasets for sensitivity to batch-effect, delineation of clustering performance and inference of developmental lineages. We applied tGPT to bulk cancer tissue sequencing samples and found that features obtained from tGPT are significantly associated with diverse genomic alteration events, patients’ prognosis and treatment outcome of immunotherapy. tGPT represents a new analytical paradigm to integrate and decipher large-scale single-cell transcriptomes. It will facilitate the integration and clinical translation of large volume of single-cell transcriptome data.
Results
An overview of tGPT and its downstream applications
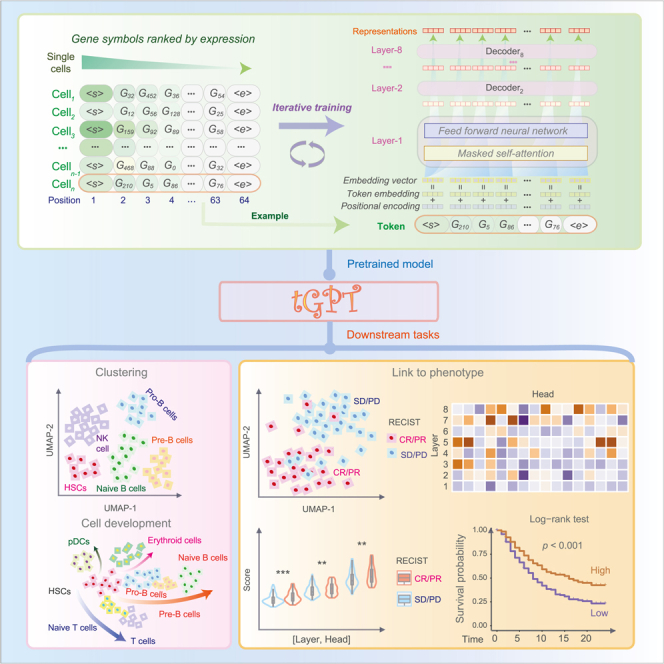
The analytical framework of tGPT (Figure 1) consists of three components: Development of tGPT, applications of tGPT for single-cell clustering, inference of developmental lineage, and interrogation of feature representation of bulk tissues in relation to genomic alterations, prognosis and treatment response of immunotherapy.
Figure 1.

A flowchart illustrating the framework of tGPT and its downstream applications
It consists of three components: development of tGPT, applications of tGPT for single-cell and bulk tissue transcriptomes.
tGPT is formatted as an autoregressive language model in that the output from the previous step is used as input to the next step. The input to tGPT is a sequence of gene symbols that are ranked by their expression levels. The purpose is to predict the index of the next gene in the dictionary in the context of all previous genes. The dictionary consists of 20706 protein-coding genes. tGPT is trained as an unsupervised generative pretraining task.16 Specifically, for a given cell, let denote the gene symbols that are sorted in a descending order according to their expression levels. We use the standard language modeling objective to maximize the likelihood. Here, is the width of context window and are the parameters of tGPT that is used to model the conditional probability. The neural network consists of 8 transformer decoder blocks21 with 1024 hidden units and 16 attention heads.
Quantitative evaluations of tGPT on clustering
We systematically evaluated the clustering performance of tGPT on four heterogeneous single-cell datasets of different sizes (50-586k cells) from different species and two bulk tissue sequencing datasets (Tables S1 and S2). These four single-cell datasets include Human Cell Atlas Census of Immune Cells22 (HCA, n = 282,558), Human Cell Landscape23 (HCL, n = 586,135), Tabula Muris4 (n = 54,862) and Macaque Retina24 (n = 124,965) dataset (See STAR Methods for description). The two bulk tissue datasets are Geno-type-Tissue Expression25 (GTEx, n = 11,688) derived from 30 organs and The Cancer Genome Atlas26 (TCGA, n = 9,318) consisted of 33 cancer types.
We observed that tGPT is insensitive to batch effect as benchmarked against with the other methods that support batch-correction such as ComBat,27 MNN,28 Harmony,29 Seurat,30,31 BBKNN,32 Scanorama,33 Pegasus,34 scVI,8 scArches,35 iMAP36 and DESC37 as measured on the HCA dataset. tGPT achieved a comparable kBET acceptance rate38 of 0.87 among the aforementioned batch-correction methods (Figure S1L). The UMAP plots of these batch-correction methods and their clustering metrics and grid-search results are provided in Figures S1A–S1K, 2, and 3, respectively.
Figure 2.

The clustering performance of tGPT on four single-cell and two bulk tissue datasets
(A) Radar charts depicting clustering metrics of tGPT, Pegaus, scVI and Scanpy across these six datasets.
(B–G) UMAP visualization of feature representations learned by tGPT on the HCA (B), HCL (C), Tabula Muris (D), Macque Retina (E), GTEx (F) and TCGA (G).NMI, Normalized Mutual information; ARI, Adjusted Rand Index; FMI, Fowlkes-Mallows Index.
Figure 3.

Distinct features of different cell types from the HCA dataset learned by tGPT
(A) Heatmap representation of attention head entropy for different cell types, and hierarchical clustering plot clustered these attention heads.
(B) Heatmap representation of attention head importance for different cell types.
(C) Dot plot illustrating the attribution scores for cell type specific genes, gray cell types annotated the clusters of marker genes.
The clustering performance of tGPT is robust with respect to the numbers of top-expression genes being used. We found that the performance of tGPT pretrained on the ranking of top 62 and 126 genes were comparable across these six datasets (Figure S4). In addition, we observed that clustering performance on features extracted from different transformer layers [Layer-1, …, Layer-8] are comparable and better than features extracted from the embedding layer across all these six datasets (Figure S4). For each method, we reported the best performance via grid-search to identify optimal values of two parameters that are most relevant to clustering (see STAR Methods). The results from grid-search were provided in Figures S5–S10 and Data S1. Quantitatively, tGPT achieved an Normalized Mutual information (NMI) ranged from 0.75 on HCA to 0.90 on GTEx, Adjusted Rand Index (ARI) from 0.53 on HCL to 0.84 on Tabula Muris and Fowlkes-Mallows Index (FMI) from 0.55 on HCL to 0.85 on Tabula Muris (Figure 2A). The clustering performance achieved by tGPT are comparable to the other methods such as Scanpy,39 Pegasus34 and scVI8 (Figures 2A and S11–S13). Grid-search results of these methods were provided in Figure S14. Running time of these methods were provided in Table S3.
Across these datasets, tGPT was capable of grouping cells with the same or similar types (Figures 2B–2G). On the HCA dataset, tGPT was able to identify cells at different developmental phases. For example, it can delineate B cells of different types such as naive B cells, precursor B (pre-B) cells and progenitor B (pro-B) cells and homologous cells, such as conventional DCs (cDCs) and plasmacytoid DCs (pDC), CD14+ and CD16+ monocytes. Less represented cell types such as megakaryocytes (0.32%) and MSCs (0.10%) were also captured by tGPT (Figure 2B). On the HCL dataset, tGPT was able to distinguish between immune cells and nonimmune cells as well as different cell types from fetus and adult such as fetal enterocytes and adult enterocytes (Figures 2C and S12). On the Tabula Muris dataset, tGPT was also able to delineate 55 distinct cell types originated from 20 mouse organs (Figures 2D and S13). On the Macaque Retina dataset, distinctive cell clusters from foveal and peripheral regions of fascicularis retina defined by tGPT are well matched with cell types defined in the original literature24 (Figure 2E). On the GTEx dataset, tGPT is able to identify different tissues originated from lineage of organs (NMI = 0.90), and samples with similar histological structure are close together such as colon, small intestine and stomach (Figure 2F). On the TCGA dataset, different cancer types are well separated (NMI = 0.77). Cancer types with the same tissue of origin tend to clump together in the feature representation spaces captured by tGPT. For example, adenocarcinomas and squamous cell carcinomas are closely related in the UMAP plots, respectively. Different cancer subtypes originated from the same tissues are well separated such as lung cancer subtypes (e.g., LUAD and LUSC; Figure 2G), kidney cancer subtypes (e.g., KIRC, KIRP and KICH; Figure 2G) and breast cancer subtypes (e.g., Luminal A, Luminal B, HER+ and Basal cell carcinoma; Figure S15). In addition, tGPT achieved Bilingual Evaluation Understudy (BLEU) scores ≥0.69 four datasets examined in this study (Table S4). This suggested that tGPT obtained good quality in gene ranking generation.
Distinct features learned by tGPT are connected to cell types
We observed that the head entropy and importance of different cell types from the HCA dataset (See STAR Methods) are distinctive from each other. Cells of similar lineages or functions such as T-lineage cells exhibited similar entropy patterns (Figure 3A). The head importance is varying considerably for different cell types, however, cells of similar types are alike as compared with the other cell types (Figure 3B). For each cell type, we calculated the contribution of each gene on the cell final feature representation (See STAR Methods). Celltype specific genes have higher attribution scores (Figure 3C). For example, NKG7, FGFBP2, PRF1, GNLY, GZMA and GZMB are highly represented in cytotoxic T cells and NK cells (Figure S16A). PPBP and PF4 are also highly represented in megakaryocytes (Figure S16B). B-lineage cells have high attribution scores for both CD79A and CD79B. Attribution scores of MS4A1 and MZB1 are relative higher in memory B cells and plasma cells, respectively (Figure S16C). The attribution score of CST3 is higher among CD14+ monocytes, CD16+ monocytes, cDCs and pDCs. In addition, each specific cell types can be defined by specific genes with high attribution scores, for instance plasmacytoid dendritic cells (pDCs, IRF7), conventional dendritic cells (cDC, FCER1A), CD14+ monocytes (CD14) and CD14+ monocytes (FCGR3A) (Figure S16D).
Inference of developmental lineage
We used the feature representations learned by tGPT to construct cell pseudo-temporal trajectories on HCA and HCL datasets (See STAR Methods). On the HCA dataset, the developmental trajectories originated from stem cells and differentiated toward multiple biologically functional cell branches (Figure 4A): HSCs to erythroid cells40 or DCs and monocytes (Figure 4B); naive T cells to cytotoxic T cells and NK cells41 (Figure 4C); pro-B cells to pre-B cells, then followed by matured naive B cells, and finally bifurcated into memory B cells or plasma cells42 (Figure 4D). In addition, we observed that the cell state signatures are aligned with cell developmental lineages (See STAR Methods). For instance, HSCs and pro-B cells are manifested by apparent progenitor signaling (Figure 4E). Naive and mature T cells are featured by distinguishable patterns (Figures 4F and 4G).
Figure 4.

Diffusion pseudo-time analysis on the HCA and HCL datasets
(A) The diffusion map of HCA dataset.
(B) Hematopoietic stem cells (HSCs) to erythroid cells or dendritic cells (DCs) and monocytes.
(C) Naive T cells to cytotoxic T cells and nature killer (NK) cells.
(D) Pro-B cells to plasma cells.
(E–G) Cell state signatures for progenitor signaling, naive signaling and cytotoxic signaling.
(H–K) The diffusion map of HCL dataset and its main branches. Token score is the norm of the learned token features extracted from tGPT.
On the HCL dataset, the developmental tree depicted three differential trajectories of fetal mesenchymal progenitor cells into different mature cell types (Figure 4H) with fetal cells at the center of the landscape. The fetal mesenchymal progenitor cells are differentiated into biologically functional fibroblasts (Figure 4I), enterocytes (Figure 4J), astrocytes and oligodendrocytes (Figure 4K).
Clinical significance of tGPT in bulk sequencing sample
Here, we demonstrated that tGPT is able to capture clinically significant patterns. On the TCGA dataset, we found that the importance scores are varying considerably for different attention heads among different layers. The importance score patterns can cluster different cancer types into distinct groups in that cancer of the same tissue-of-origin are closely related whereas cancers of different origins are well separated (See STAR Methods, Figure 5A). For example, skin cutaneous melanoma (SKCM) and uveal melanoma (UVM), glioblastoma multiforme (GBM) and brain lower grade glioma (LGG) are respectively located in the same clustering branches. In addition, we examined the association between attention head entropy and molecular alteration events (See STAR Methods). There are several attention heads exhibited significant association with tumor mutation burden (TMB) in the TCGA pan-cancer cohort and specifically in bladder urothelial carcinoma (BLCA), LUAD and LUSC (Figure 5B). We observed that attention heads also showed significant association with TP53 mutations at the pan-cancer level and across 9 cancer types (Figure 5C). There are also attention heads exhibited significant association with homologous recombination deficiency (HRD) and genome doubling (Figures 5D and 5E) at the pan-cancer level. The association of attention heads with HRD and genome doubling are statistically significant across 4 and 14 cancer types, respectively. Meanwhile, the attention heads exhibited prognostic significance at pan-cancer level (Figure 5E) and across 7 cancer types (Figure S17).
Figure 5.

The association of features learned by tGPT versus genomic alteration events and clinical phenotype
(A) Heatmap representation of attention head importance score across different cancer types on the TCGA dataset.
(B–F) Association of attention head entropy versus tumor mutation burden (B), TP53 mutation (C), homologous recombination deficiency (D), genome doubling (E) and overall survival (F) on the TCGA cohort.
(G and H) Heatmap representation of attention head importance and entropy on the urothelial carcinoma stratified by RECIST response. CR, complete response; PR, partial response; SD, stable disease; PD, progress disease.
(I) The varying entropy patterns from SD to PR to CR with PD as baseline.
(J) Exemplified violin plots depicting attention head entropy in SD/PD versus CR/PR.
(K) The varying of patterns of tumor evasion signature from SD to PR to CR with PD as baseline.
(L) The varying of patterns of T cell infiltration signature from SD to PR to CR with PD as baseline.
(M) Association between attention head entropy and overall survival on the urothelial carcinoma dataset.
(N) Exemplified survival curves stratified by attention head entropy.
In addition, we examined the attention head patterns in relation to immunotherapy in an immune checkpoint block (ICB) clinical trial of urothelial carcinoma consisted of 298 patients: 25 patients with CR, 43 with PR, 63 with SD and 63 with PD (See STAR Methods). We found that importance and entropy scores are distinguishable amongst patients with different therapeutic outcome (Figures 5G and 5H). We observed gradually varying entropy values from SD to PR to CR by taking the PD baseline (Figure 5I) and significant difference among 5 attention heads in patients with CR/PR versus SD/PD (Figure 5J). We quantified expression signatures such as tumor evasion and T cell immune infiltration attended by different attention heads (See STAR Methods). By taking PD as baseline, we observed a gradually decreasing patterns of tumor evasion and increasing patterns of T cell immune infiltration from SD to PR to CR (Figures 5K and 5L). The attention heads also exhibited prognostic significance in this clinical trial (Figures 5M and 5N).
Discussion
Efficient integration of accumulating large-scale single-cell transcriptomes is urgently needed. Here, we introduced a conceptually simple approach toward the integration of unlimited number of single-cell transcriptomes and examined its potential clinical translational relevance. The paradigm underpinning tGPT in essence is to predict the occurrence of a given gene with its previous context. We developed tGPT on a super large-scale single-cell transcriptome dataset that consists of 22.3 million cells and systematically evaluated its representation learning ability on different single-cell analysis tasks. We noted that tGPT was insensitive to batch effect and achieved competitive performance as compared with benchmark tools. The purpose of this study is to verify the validity of this new paradigm in deciphering large-scale transcriptome data, especially at the level of single-cell atlas. In addition, we showed that the pretrained tGPT model can be applied to bulk tissue sequencing samples to extract a variety of features exhibiting significant association with genomic alterations and response to immunotherapy treatment.
Artificial intelligence is undergoing a paradigm shift and the pretraining models based on transformer are becoming de facto standard in natural language processing and computer vision, achieving state-of-the-art across a wide range of tasks such as natural language understanding, image classification, video and audio recognition.11 Representative pretraining models include BERT14 and GPT.15 The advantage of these pretraining models lie in its ability to assimilate real-world information from super large-scale unlabeled and high-dimensional data. This advantage brings an attractive solution for deciphering single-cell transcriptomes as millions of cells have been sequenced, which exemplified by 22.3 million cells collected in our study. This number is expected to increase exponentially in years ahead. There is no analytical tool that is designed and evaluated on such large volume of data. The high expressivity and scalability of transformer enable tGPT to learn rich representation from transcriptomes in a self-supervised manner. The high clustering performance in single-cell cluster delineation is probably attributable to better feature representation learned by tGPT. In addition, feature representation from tGPT is insensitive to batch effect as the acceptance rate of kBET derived from tGPT is evenly distributed among the other tools that explicitly used batch information for batch-correction. This is probably because of the use of rankings of top expressing genes rather than actual expression levels by tGPT. tGPT is quite different from the other integration tools30,31,39 as the later use the actual expression levels of highly variable genes (HVGs) and the batch information. The independence of tGPT on batch information makes it attractive for integration of super large-scale transcriptomes because the batch information is not always available and often neglected by researchers.
The clustering performance in delineating single-cell clusters is robust with respect to the number of top expressing genes used and feature representation extracted from different tGPT transformer layers. The clustering metrics obtained from 62 top-expressing genes are comparable to the use of 126 top-expressing genes (Figure S4). This suggested that the rankings of 62 top-expressing genes are sufficient for cell cluster definition. The idea underpinning tGPT is to predict the occurrence of a gene in the context of the occurrences of its preceding neighbors. Thistype of pretraining is not directly related to cell clustering. This does not guarantee that feature representation from the last transformer layer could give rise to better clustering as compared with representation from its preceding layers. In our evaluation, the cluster metrics obtained from different transformer layers are comparable and consistently better than the embedding layer (Figure S4). In addition, we observed that cell-type specific genes have high attribution scores albeit only the rankings are used during pretraining. This finding can partially explain why features derived from tGPT could lead to high performance in cell clustering. Although this study also uses gene rankings as we did in our previous study,43 they are theoretically different. tGPT builds on autoregressive language modeling16 whereas the model developed in our previous study used masked language modeling.14 More importantly, we explored the feature patterns learned by tGPT in bulk tissues, which were not investigated in our previous work.
A new finding emerged from our study is that the pretrained tGPT model can be applied to bulk tissues. On the GTEx dataset, the feature representations of different organs extracted from tGPT can divide samples into distinct clusters, aligning with organs. On the TCGA dataset, we observed that different cancer types are well separated and cancers of the same origins are more closely related, which is consistent with previous report.44 In addition, the feature patterns of TCGA samples exhibited consistent and significant association with genomic alterations. This indicated that rankings of top-expressing genes carry information about alterations in tumor tissues. Meanwhile, the feature patterns derived from tGPT are distinctive among patients with different treatment outcomes for immunotherapy. Token together, our finding would facilitate translational research enabled by super large-scale transcriptomes.
We focused two main directions of tGPT for future development. First, tGPT can be used to generate large-scale reference mapping with the availability of large-scale disease reference datasets and phenotypes. Second, tGPT can be further investigated for clinical application such as treatment guiding and prognostic prediction.
Conclusion
In summary, we systematically verified a new, simple and effective analytical paradigm for integration of super large-scale transcriptomes and its implications in clinical translation.
Limitations of the study
First, tGPT uses only the top-expressing genes; therefore, it may miss the information that is specifically represented within the low-expressing genes. Second, tGPT uses gene expression rankings but not actual expression levels. Thus, the fold changes among genes are neglected and this can affect biological interpretation. Third, the language modeling objective function used by tGPT is to predict the gene rankings, which is not directly related to biological issues. Therefore, further study is required to investigate associations between prediction of gene rankings and biological functions.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and materials should be directed to andwill be fulfilled by the lead contact, Xiangchun Li (lixiangchun2014@foxmail.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
We collected the transcriptomes of 22.3 million single-cells (Table S1), 9318 bulk tissue transcriptomes of TCGA cohort from the supplemental data of pan-cancer immune landscape study,26 11,688 bulk tissue transcriptomes from GTExdatabase25 and 298 bulk tissue transcriptomes from the clinical trial study on immunotherapy for urothelial carcinoma.45 We discarded mitochondrial genes, ribosomal genes and non-protein coding genes for the single-cell data. Four single-cell and two bulk tissue sequencing datasets are used in downstream evaluation of tGPT. Annotated cell labels provided by the original studies are used as the ground truth label (Table S2).
Human Cell Atlas Census of Immune Cells (HCA)
Bone marrow cells (n = 282,588) from 64 healthy donors in Human Cell Atlas (HCA) project. The data are subjected to 10x sequencing protocol22 and contained 18 cell types such as hematopoietic stem cells (HSCs), mesenchymal stem cells (MSCs), erythrocytes, megakaryocytes and different kinds of immune cells.
Human cell Landscape (HCL)
HCL dataset includes 586,135 human cells obtained from a Chinese Han population,23 the dataset encompasses samples of fetal and adult tissue and covered 60 human tissue types, and are subjected to Microwell-seq protocol.
Tabula Mursi
The Tabula Muris dataset (n = 54,865) is consisted of single-cells sorted by FACS from Mouse Cell Atlas4 across 20 different organs subjected to 10x and Smart-seq2 sequencing protocols.
The Cancer Genome Atlas (TCGA)
The TCGA dataset is consisted of 9,318 bulk samples with primary cancer and matched normal samples spanning 33 cancer types.
Genotype-Tissue Expression Project (GTEx)
The GTEx dataset includes 11,688 bulk samples across 30 organs obtained from healthy donors.
Known marker genes of different cell types are curated from CellMarker database46: plasma cell (MZB1), DCs and monocytes (CST3, FCER1A, IRF7, CD14 and FCGR3A), megakaryocyte (PPBP and PF4), B cell (CD79A, CD79B and MS4A1) NK and cytotoxic T cell (NKG7, FGFBP2, GNLY, GZMA, GZMB and PRF1).
Cell state signatures are curated from CellMarker database,46 including progenitor signaling (STMN1, TUBA1B and HIST1H4C), naïve signaling (CCR7, LEF1 and SELL) and cytotoxic signaling (GZMA, CD8A, CD8B, GZMB, PRF1, IL2, GNLY, GAMK, IFNG and NKG7).
T cellinfiltration signature is obtained from CellMarker database46; it consists of CD3D, CD3E and CD8A.
Tumor evasion signature is curated from the Figure 1 of a previous study.47
Method details
Input preprocessing
The input sequence list of top-expressing genes was obtained via descending sorting. The input to tGPT was formulated as [<s>, G1, G2, G3, …,<e>], where G1, G2 and G3 are gene symbols and<s>and<e>are two special tokens respectively added to the start and end of the input sequence. The input sequence is padded with special token<pad>if its length is less than a predefined value. The input sequence list is truncated if its length exceeds the predefined value. We evaluated a length of 64 and 128 in this study. The dictionary used by tGPT consists of 20706 protein-coding genes.
The architecture of tGPT
Embedding layer transforms the input gene symbols into a real-value matrix that carries the information on gene token embedding and position encoding. The gene token embedding was obtained via an embedding layer (parameterized as ) that maps the indices of input genes obtained from the gene symbol dictionary to real-value space. The position encoding (parameterized as carries information on the sorted gene rankings. For an input sequence , where k is the width of context window, the embedding layer injects position encoding onto gene token embedding as:
Transformer decoder blocks applies multi-headed masked self-attention over the input embeddings followed by position-wise feed-forward layers, then through a softmax layer. tGPT use a multi-layer of transformer decoder.21
where is the transformer decoder block and is the softmax layer, and is the embedding matrix of the th decoder block.
Masked Self-Attention is a variant self-attention mechanism.48 Each attention head adopts the scale dot-product attention to map a query and a set of key-value pairs to an output. The input consists of query and key of dimension , and value of dimensions Self-attention is calculated as dot products of the query () with key () divided each by and multiply with value () after sofmax transformed49:
Masked self-attention is implemented with the aid of attention mask. It basically always scores the future tokens as 0 so tGPT cannot pick from future. The multi-head self-attention is formulated as:
where denotes the learned output projection matrix.
Position-wise FeedForward neural network is a layer with fully-connected feed-forward layer. This layer consists of two linear transformations with a ReLu activation function in between:
where W1 and W2 are weight matrices and and are the bias.
Training scheme
tGPT was pretrained with a batch-size of 64 for 100 epochs. We used Adam with β1 = 0.9, β2 = 0.95, weight decay of 0.01 and a learning rate of 0.003. The learning rate is warmed up for four epochs, and then decays to 0 following a cosine schedule.12 tGPT was trained with PyTorch (version 1.7.1) and transformers (version 4.10.0) on NVIDIA DGX A100 with 8 GPUs each with 40 Gb memory.
Clustering on feature representation from tGPT
We respectively extracted the feature representations from the embedding layer and 8 different transformer layers. The extracted features were used to construct K-Nearest Neighbors (KNN) graphs for subsequent community detection by Leiden algorithm50 implemented in Scanpy (version 1.8.1). We performed grid-search to identify optimal values of two parameters n_neighbors and resolution that are the most relevant for clustering. Batch-correction was not applied in clustering. The value of n_neighbors examined was ranged from 5 to 100 with step of 5. The value of resolution examined was ranged from 0.1 to 2 with step of 0.2. The uniform manifold approximation and projection (UMAP) visualization51 is used.
Features derived from self-attention
Entropy of the self-attention matrices for a given input sequence is calculated as52:
where α is the self-attention matrix and is the attention weight between the ith and jth tokens. We averaged the entropy of all cells in a cluster to derive a cluster-level entropy.
Head importance score53 is defined as the influence of input on head output. It is calculated via gradient backpropagation, formulated as:
where x is the input sequence and is the corresponding loss given the input. is high score while is liable to have a large effect on the model.
Token attribution score is defined as the norm of the learned token features () extracted from tGPT, which is defined as:
Attention analysis in relation to signaling
We define an attention-based pathway signaling score in a similar way as54:
where is the attention weight between the ith and jth gene. For a given gene signature, we set = 1 if the ith gene or jth gene occurs in that gene set.
Diffusion pseudo-time maps construction
We constructed the diffusion pseudo-time maps using package Pegasus34 (v1.4.3), and the cell trajectory was visualized with force-directed layout embedding (FLE) algorithm.55 We set δ and nδ to its default values: δ = 2.0 and nδ =5,000.
Firstly, we used the features obtained from last transformer decoder blocks to construct affinity matrix of cells , and the top-k nearest neighbor cells were find by community detection algorithm56 and the HNSW algorithm,57 and the formula of affinity matrix is define as:
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
The Equation 1 represented the distance between cell-x and cell-y, is the x’s local kernel width, x and y are features of last transformer decoder block for cell-x and cell-y. The affinity matrix W was calculated as the density-normalized kernel according to Equation 3.
We then calculated the Markov chain transition matrix P and the symmetric transition matrix Q as the formula:
The symmetrical matrix Q can be decomposed as UAUT. Let . A family with parameter timescale of t for approximated diffusion maps is defined as:
Benchmark methods
We also performed single-cell analysis using Scanpy (version 1.6.0), Pegasus (version 1.4.3) and scVI (version 0.13.0). Batch-correction was performed with MNN (version 1.8.0),28 Combat (version 1.8.0),58 Harmony (version 0.1.6),29Seurat (version 3.1.5),30,31 Pegasus (version 1.4.3),34 Scanorama (version 1.7.1),33 DESC (version 2.1.1),37 iMAP (version 1.0.0),36 scVI (version 0.13.0),8 scArches (version 1.7.0),35 BBKNN (version 1.7.1).32
Scanpy is a comprehensive toolkit for analyzing single-cell transcriptome. We first filtered out cells with the number of expressing genes <200 or mitochondrial counts> 30%. We used the function scanpy.pp.highly_variable_genes to selected highly variable genes by setting max_mean to 3 and min_mean to 0.0125, which are the default values. We then applied clustering pipeline and grid-search to perform single-cell clustering on KNN graph. The UMAP is used for visualizing clustering result.
scVI is a deep generative model for mining the single-cell omics data. We filtered out cells with the number of expressing genes <200 or mitochondrial counts> 30%, and selected HVGs with scanpy.pp.highly_variable_genes by setting max_mean to 3 and min_mean to 0.0125. We used the default parameter of scVI to extract the 10 latent features. These latent features were used to construct KNN graphs for community detection by Leiden algorithm.50
Pegasus is complete single-cell analysis pipline that is efficient on large datasets. We used the recommended parameters: min_genes of 500, max_genes of 6000, and percent_mito of 10. We identified the robust genes with the default percent_cells of 0.05. Single-cell clustering was performed on KNN graph followed by Leiden algorithm50 for community detection.
Quantification and statistical analysis
Clustering and batch-effect metrics
We used Adjusted Rand Index (ARI), Normalized Mutual information (NMI) and Fowlkes-Mallows Index (FMI) to measure clustering performance. We used the kBET acceptance rate38 as a measurement of batch-effect. The clustering metrics of ARI, NMI and FMI were calculated with sklearn (version 0.21.2). kBET acceptance rate is computed with Pegasus (version 1.4.3).
ARI is calculated based on the contingency table summarizing the truth labels and clustering, and the rows and columns represent truth and clustering labels in the contingency table, respectively. The formula is as follows:
where denoted the numbers of cell in common between clustering labels and truth labels, the sum of row and the sum of column of the contingency table.
NMI is also used to measure the similarity between the clustering labels and actual labels. We assumed that the clustering labels and actual labels of N cells are U and V, and the entropy of U and V is as the following formula:
where is the probability that a cell picked at random from U falls into , is the probability that a cell picked at random from V falls into . We then calculated the mutual information (MI) between U and V, and normalized the mutual information:
where is the probability that a cell picked at random falls into classes and .
Fowlkes-Mallows Index (FMI) is used to measure the consistency between clustering results and real category, and the range of index is from 0 to 1. The FMI metric is denfined as the geometric mean between of the precision and recall:
where TP is true positive, FP false positive, FN false negiative.
kBET acceptance rate is a measurement of batch effect. We assumed that the dataset of cells with batches of m, and there are cells in batch j. The batch mixing frequency denotes as:
where . Then, we calculated the number of neighbors of cell-i belonging to batch j is . Its test statistic and p-value with degrees of (m-1) are defined as follows:
where represents the cumulated density function. The kBET acceptance rate is defined as the percentage of cells that accept the null hypothesis at significance level α as follows:
I(x) is the indicator function where I(x) = 1 if x> 0 otherwise I(x) = 0. We used Pegasus (v1.4.3) to calculate the kBET acceptance rate by setting K and α to 5 and 0.01, respectively.
Bilingual Evaluation Understudy (BLEU) is an algorithm for evaluating match variable length phrases between output and the reference sequence.59 The basic metric requires the calculation of a brevity penalty as:
Where r is the length of the reference sequence, and the length of predicted sentence is c.
BLEU score is calculated as:
are the positive weights summing to one. is the n-gram precision and it is calculated using n-grames with a maximum length of N.
Additional resources
This study did not generate additional data.
Acknowledgments
We are grateful for researchers for their generosity to made their data publicly available. This work was supported by National Key Research and Development Program of China (Grant No. 2021YFC2500400 to K.C.), National Natural Science Foundation of China (Grant No. 32270688 and 31801117 to X.L., 31900471 to M.Y. and 82073287 to Q.Z.) and Tianjin Municipal Health Commission Foundation (Grant No. RC20027 to Y.L). This work was funded by Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-009A).
Author contributions
X.L. and K.C. designed and supervised the study; X.L., H.S., J.L., and J.H. performed data analysis and wrote the manuscript; X.L. developed the model; X.L., H.S., X.S., C.Z., D.W., M.F., J.H., J.L., Y.Y., Y.L., M.Y., W.W., and Q.Z. collected data; H.S., X.L., K.C., and J.Y. revised the manuscript.
Declaration of interests
The authors declare that they have no conflict of interest.
Published: April 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106536.
Contributor Information
Kexin Chen, Email: chenkexin@tmu.edu.cn.
Xiangchun Li, Email: lixiangchun2014@foxmail.com.
Supplemental information
Data and code availability
-
•
All the gene expression matrices were downloaded from public databases. The source list of these datasets was provided in the key resources table and Table S1. Source code is available at https://github.com/deeplearningplus/tGPT.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M., et al. The human cell atlas. Elife. 2017;6 doi: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papatheodorou I., Moreno P., Manning J., Fuentes A.M.-P., George N., Fexova S., Fonseca N.A., Füllgrabe A., Green M., Huang N., et al. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2020;48:D77–D83. doi: 10.1093/nar/gkz947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabula Muris Consortium. Overall coordination. Logistical coordination. Organ collection and processing. Library preparation and sequencing. Computational data analysis. Cell type annotation. Writing group. Supplemental text writing group. Principal investigators Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562:367–372. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F., et al. Mapping the mouse cell atlas by microwell-seq. Cell. 2018;173:1307. doi: 10.1016/j.cell.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Tung P.Y., Blischak J.D., Hsiao C.J., Knowles D.A., Burnett J.E., Pritchard J.K., Gilad Y. Batch effects and the effective design of single-cell gene expression studies. Sci. Rep. 2017;7:39921. doi: 10.1038/srep39921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hicks S.C., Townes F.W., Teng M., Irizarry R.A. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics. 2018;19:562–578. doi: 10.1093/biostatistics/kxx053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amodio M., van Dijk D., Srinivasan K., Chen W.S., Mohsen H., Moon K.R., Campbell A., Zhao Y., Wang X., Venkataswamy M., et al. Exploring single-cell data with deep multitasking neural networks. Nat. Methods. 2019;16:1139–1145. doi: 10.1038/s41592-019-0576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simon L.M., Wang Y.-Y., Zhao Z. Integration of millions of transcriptomes using batch-aware triplet neural networks. Nat. Mach. Intell. 2021;3:705–715. [Google Scholar]
- 11.Bommasani R., Hudson D.A., Adeli E., Altman R., Arora S., von Arx S., Bernstein M.S., Bohg J., Bosselut A., Brunskill E. On the opportunities and risks of foundation models. arXiv. 2021 doi: 10.48550/arXiv:2108.07258. Preprint at. [DOI] [Google Scholar]
- 12.Chen M., Radford A., Child R., Wu J., Jun H., Luan D., Sutskever I. PMLR; 2020. Generative Pretraining from Pixels; pp. 1691–1703. [Google Scholar]
- 13.Bao H., Dong L., Wei F. BEiT: BERT pre-training of image transformers. arXiv. 2021 doi: 10.48550/arXiv:2106.08254. Preprint at. [DOI] [Google Scholar]
- 14.Devlin J., Chang M.-W., Lee K., Toutanova K. Bert: pre-training of deep bidirectional transformers for language understanding. arXiv. 2018 doi: 10.48550/arXiv:1810.04805. Preprint at. [DOI] [Google Scholar]
- 15.Radford A., Narasimhan K., Salimans T., Sutskever I. 2018. Improving Language Understanding by Generative Pre-training. [Google Scholar]
- 16.Brown T.B., Mann B., Ryder N., Subbiah M., Kaplan J., Dhariwal P., Neelakantan A., Shyam P., Sastry G., Askell A. Language models are few-shot learners. arXiv. 2020 doi: 10.48550/arXiv:2005.14165. Preprint at. [DOI] [Google Scholar]
- 17.Wang H., Sun Q., Zhao W., Qi L., Gu Y., Li P., Zhang M., Li Y., Liu S.L., Guo Z. Individual-level analysis of differential expression of genes and pathways for personalized medicine. Bioinformatics. 2015;31:62–68. doi: 10.1093/bioinformatics/btu522. [DOI] [PubMed] [Google Scholar]
- 18.Qi L., Li T., Shi G., Wang J., Li X., Zhang S., Chen L., Qin Y., Gu Y., Zhao W., Guo Z. An individualized gene expression signature for prediction of lung adenocarcinoma metastases. Mol. Oncol. 2017;11:1630–1645. doi: 10.1002/1878-0261.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng F., Wang R., Zhang Y., Zhao Z., Zhou W., Chang Z., Liang H., Zhao W., Qi L., Guo Z., Gu Y. Differential expression analysis at the individual level reveals a lncRNA prognostic signature for lung adenocarcinoma. Mol. Cancer. 2017;16:98. doi: 10.1186/s12943-017-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng F., Zhang Y., Wang R., Zhou W., Zhao Z., Liang H., Qi L., Zhao W., Wang H., Wang C., et al. Identification of differentially expressed miRNAs in individual breast cancer patient and application in personalized medicine. Oncogenesis. 2016;5:e194. doi: 10.1038/oncsis.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu P.J., Saleh M., Pot E., Goodrich B., Sepassi R., Kaiser L., Shazeer N. Generating wikipedia by summarizing long sequences. arXiv. 2018 doi: 10.48550/arXiv:1801.10198. Preprint at. [DOI] [Google Scholar]
- 22.Regev, A., Teichmann, S., Rozenblatt-Rosen, O., Stubbington, M., Ardlie, K., Amit, I., Arlotta, P., Bader, G., Benoist, C., and Biton, M. (2018). The human cell atlas white paper. Preprint at arXiv. 10.48550/arXiv.1810.05192. [DOI]
- 23.Han X., Zhou Z., Fei L., Sun H., Wang R., Chen Y., Chen H., Wang J., Tang H., Ge W., et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581:303–309. doi: 10.1038/s41586-020-2157-4. [DOI] [PubMed] [Google Scholar]
- 24.Peng Y.R., Shekhar K., Yan W., Herrmann D., Sappington A., Bryman G.S., van Zyl T., Do M.T.H., Regev A., Sanes J.R. Molecular classification and comparative taxonomics of foveal and peripheral cells in primate retina. Cell. 2019;176:1222–1237.e22. doi: 10.1016/j.cell.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.GTEx Consortium Erratum: genetic effects on gene expression across human tissues. Nature. 2018;553:530. doi: 10.1038/nature25160. [DOI] [PubMed] [Google Scholar]
- 26.Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A., et al. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 28.Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L., Yu X., Zheng L., Zhang Y., Li Y., Fang Q., Gao R., Kang B., Zhang Q., Huang J.Y., et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 32.Polański K., Young M.D., Miao Z., Meyer K.B., Teichmann S.A., Park J.-E. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics. 2020;36:964–965. doi: 10.1093/bioinformatics/btz625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hie B., Bryson B., Berger B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 2019;37:685–691. doi: 10.1038/s41587-019-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X., Zhang F., Hou Z., Wang Z., Mian L., Zhang J., Tang J. Self-supervised learning: generative or contrastive. arXiv. 2020 doi: 10.48550/arXiv:2006.08218. Preprint at. [DOI] [Google Scholar]
- 35.Lotfollahi M., Naghipourfar M., Luecken M.D., Khajavi M., Büttner M., Wagenstetter M., Avsec Ž., Gayoso A., Yosef N., Interlandi M., et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 2022;40:121–130. doi: 10.1038/s41587-021-01001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang D., Hou S., Zhang L., Wang X., Liu B., Zhang Z. iMAP: integration of multiple single-cell datasets by adversarial paired transfer networks. Genome Biol. 2021;22:63. doi: 10.1186/s13059-021-02280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X., Wang K., Lyu Y., Pan H., Zhang J., Stambolian D., Susztak K., Reilly M.P., Hu G., Li M. Deep learning enables accurate clustering with batch effect removal in single-cell RNA-seq analysis. Nat. Commun. 2020;11:2338. doi: 10.1038/s41467-020-15851-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Büttner M., Miao Z., Wolf F.A., Teichmann S.A., Theis F.J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods. 2019;16:43–49. doi: 10.1038/s41592-018-0254-1. [DOI] [PubMed] [Google Scholar]
- 39.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klimchenko O., Mori M., DiStefano A., Langlois T., Larbret F., Lecluse Y., Feraud O., Vainchenker W., Norol F., Debili N. A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell–derived primitive hematopoiesis. Blood. 2009;114:1506–1517. doi: 10.1182/blood-2008-09-178863. [DOI] [PubMed] [Google Scholar]
- 41.Trinchieri G. Biology of natural killer cells. Adv. Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LeBien T.W., Tedder T.F. B lymphocytes: how they develop and function. Blood. The Journal of the American Society of Hematology. 2008;112:1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen H., Shen X., Feng M., Wu D., Zhang C., Yang Y., Yang M., Hu J., Liu J., Wang W., et al. A universal approach for integrating super large-scale single-cell transcriptomes by exploring gene rankings. Briefings Bioinf. 2022;23:bbab573. doi: 10.1093/bib/bbab573. [DOI] [PubMed] [Google Scholar]
- 44.Hoadley K.A., Yau C., Wolf D.M., Cherniack A.D., Tamborero D., Ng S., Leiserson M.D.M., Niu B., McLellan M.D., Uzunangelov V., et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., III, Koeppen H., Astarita J.L., Cubas R., et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M., et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawson K.A., Sousa C.M., Zhang X., Kim E., Akthar R., Caumanns J.J., Yao Y., Mikolajewicz N., Ross C., Brown K.R., et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature. 2020;586:120–126. doi: 10.1038/s41586-020-2746-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser Ł., Polosukhin I. 2017. Attention Is All You Need; pp. 5998–6008. [Google Scholar]
- 49.Cheng J., Dong L., Lapata M. Long short-term memory-networks for machine reading. arXiv. 2016 doi: 10.48550/arXiv:1601.06733. Preprint at. [DOI] [Google Scholar]
- 50.Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becht E., McInnes L., Healy J., Dutertre C.-A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 52.Ghader H., Monz C. What does attention in neural machine translation pay attention to? arXiv. 2017 doi: 10.48550/arXiv:1710.03348. Preprint at. [DOI] [Google Scholar]
- 53.Michel P., Levy O., Neubig G. Are sixteen heads really better than one? arXiv. 2019 doi: 10.48550/arXiv:1905.10650. Preprint at. [DOI] [Google Scholar]
- 54.Vig J., Madani A., Varshney L.R., Xiong C., Socher R., Rajani N.F. Bertology meets biology: interpreting attention in protein language models. arXiv. 2020 doi: 10.48550/arXiv:2006.15222. Preprint at. [DOI] [Google Scholar]
- 55.Schiebinger G., Shu J., Tabaka M., Cleary B., Subramanian V., Solomon A., Gould J., Liu S., Lin S., Berube P., et al. Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell. 2019;176:1517–1943. doi: 10.1016/j.cell.2019.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reichardt J., Bornholdt S. Statistical mechanics of community detection. Phys. Rev. 2006;74:016110. doi: 10.1103/PhysRevE.74.016110. [DOI] [PubMed] [Google Scholar]
- 57.Malkov Y.A., Yashunin D.A. Efficient and robust approximate nearest neighbor search using hierarchical navigable small world graphs. IEEE Trans. Pattern Anal. Mach. Intell. 2020;42:824–836. doi: 10.1109/TPAMI.2018.2889473. [DOI] [PubMed] [Google Scholar]
- 58.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 59.Wołk K., Marasek K. Enhanced bilingual evaluation understudy. arXiv. 2015 doi: 10.48550/arXiv:1509.09088. Preprint at. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All the gene expression matrices were downloaded from public databases. The source list of these datasets was provided in the key resources table and Table S1. Source code is available at https://github.com/deeplearningplus/tGPT.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.