

Abstract
SINEUPs are natural and synthetic antisense long non-coding RNAs (lncRNAs) selectively enhancing target mRNAs translation by increasing their association with polysomes. This activity requires two RNA domains: an embedded inverted SINEB2 element acting as effector domain, and an antisense region, the binding domain, conferring target selectivity. SINEUP technology presents several advantages to treat genetic (haploinsufficiencies) and complex diseases restoring the physiological activity of diseased genes and of compensatory pathways. To streamline these applications to the clinic, a better understanding of the mechanism of action is needed. Here we show that natural mouse SINEUP AS Uchl1 and synthetic human miniSINEUP-DJ-1 are N6-methyladenosine (m6A) modified by METTL3 enzyme. Then, we map m6A-modified sites along SINEUP sequence with Nanopore direct RNA sequencing and a reverse transcription assay. We report that m6A removal from SINEUP RNA causes the depletion of endogenous target mRNA from actively translating polysomes, without altering SINEUP enrichment in ribosomal subunit-associated fractions. These results prove that SINEUP activity requires an m6A-dependent step to enhance translation of target mRNAs, providing a new mechanism for m6A translation regulation and strengthening our knowledge of SINEUP-specific mode of action. Altogether these new findings pave the way to a more effective therapeutic application of this well-defined class of lncRNAs.
Keywords: MT: Non-coding RNAs, SINEUP, lncRNA, RNA therapeutics, m6A, N6-methyladenosine, Nanopore direct RNA sequencing, translation
Graphical abstract
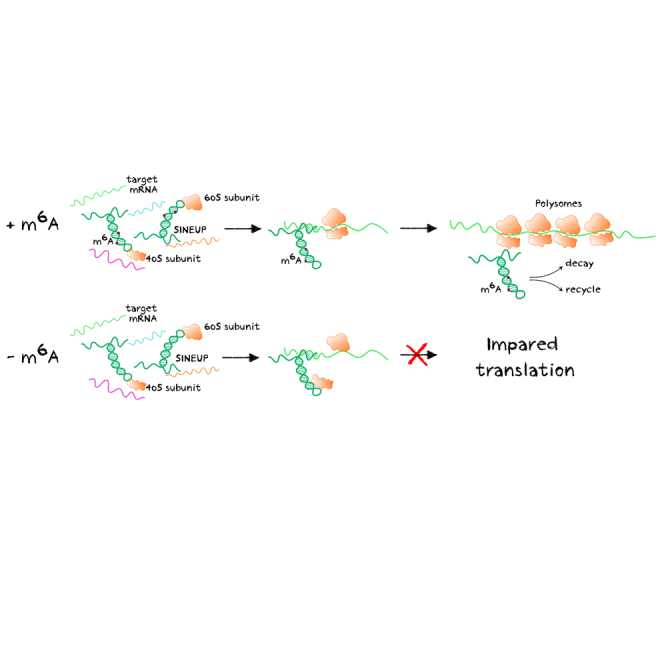
In this study, Pierattini and colleagues show that antisense long non-coding SINEUP RNAs require m6A-modified residues to exert their activity as enhancers of translation. These results are important to unveil the details of the molecular mechanism of SINEUPs to streamline their use as therapeutics to treat unmet medical needs.
Introduction
The majority of transcribed RNAs consist of non-coding RNAs (ncRNAs) whose function is scarcely known.1,2 Among them, long non-coding RNAs (lncRNAs) can play key regulatory functions in physiological and disease-associated processes and may represent a new class of candidate therapeutics or drug targets.3 About a third of lncRNAs overlap with protein-coding genes4 and many of them, being transcribed from the opposite strand, give rise to sense/antisense pairs.4,5 A lncRNA antisense (AS) to mouse Ubiquitin carboxy-terminal hydrolase L1 (Uchl1/Park5), called AS Uchl1, was previously studied for its ability to up-regulate UCHL1 protein synthesis by enhancing Uchl1 mRNA association to polysomes.6 AS Uchl1 and the sense-coding gene are organized in a 5′ head-to-head configuration in the genome, with an overlapping region located around Uchl1 translation initiation codon AUG.6,7 AS Uchl1 is the representative member of a functional class of natural and synthetic antisense lncRNAs, called SINEUPs, able to up-regulate translation through an embedded inverted SINEB2 (invSINEB2) element.7 SINEUPs have a modular architecture composed of two essential domains: in the 5′, the overlapping region, or binding domain (BD), confers target selection ability and specificity, while in the remaining part, the invSINEB2 element acts as an effector domain (ED), enhancing translation. Functional studies on various portions of natural AS Uchl1 have also revealed that the exclusive combination of BD and ED sequences, called miniSINEUPs, retain a comparable activity level while reducing SINEUP RNA length from ∼1,200 to ∼250 nt.8 From a structural point of view, the stem loop 1 (SL1) motif in the invSINEB2 sequence was proved to be crucial for SINEUP activity, as its deletion from AS Uchl1 causes a complete loss of UCHL1 induction.9,10,11 Designing antisense BD sequences complementary to an mRNA of interest, the translation-enhancing activity of SINEUPs can be directed to any endogenous or exogenous (e.g., co-transfected) gene. Synthetic SINEUPs and miniSINEUPs have been reported to be effective in several cell types on a wide range of targets in vitro, including EGFP, FLAG-tagged proteins, and recombinant antibodies,12,13,14 and in vivo on endogenous target mRNAs.15,16 These translation-enhancing properties have made synthetic SINEUPs a new platform for RNA therapeutics.14,15,16,17 As a roadmap for the use of SINEUP technology in the clinic, proof-of-concept experiments have been carried out to demonstrate the ability of SINEUPs to revert pathological phenotypes in different relevant biological models of human diseases: (1) a medaka fish model of an haploinsufficient disease,15 (2) patients’ cells from a genetic human disease,14 and (3) a mouse model of neurodegenerative disease.16
Despite all the knowledge accumulated so far on natural and synthetic SINEUPs, the molecular mechanism of their translation-enhancing activity remains unclear.
More than 100 post-transcriptional modifications (PTMs) have been reported in RNAs.18 N6-methyladenosine (m6A) is the most common modification found in mRNAs and ncRNAs. It is catalyzed post-transcriptionally in the cell nucleus and can exert regulatory functions in many cellular processes such as RNA splicing, stability, nuclear export, and translation.19,20,21,22,23,24,25,26 High-throughput transcriptome-wide approaches, often combined with antibody enrichment, have allowed the identification of DRACH (where D = G, A, or U; R = G or A; and H = C, A, or U), a consensus motif for m6A sites deposition.27,28 Although consensus sequences are quite common through the transcriptome, only a few of them are actually methylated, with site and transcript specificities that are still poorly understood.29 Several m6A-related proteins have been identified, displaying roles as “writers” (methyltransferases, mainly METTL3 and its adaptors),30,31 “erasers” (demethylases FTO and ALKBH5), and “readers” (YTHDF1, YTHDF2, YTHDF3, and others).32 Despite growing interest in understanding the function of m6A modification, most current technologies to map modified residues still rely on long and complex protocols. In this scenario, a major improvement is represented by Nanopore direct RNA sequencing (DRS), which allows us to sequence native full-length RNA molecules and retain information about RNA modifications.33,34 To date, most studies investigate the role of m6A modifications within mRNAs, but much less is known about their function in lncRNAs35 with the interesting exceptions of MALAT1,36 XIST,25 and chromatin-associated RNAs,37 including L1 TEs.26
m6A modification is an attractive candidate regulator of SINEUP function, as its incorporation in the chemical synthesis of SINEUP RNA molecules contributes to restore their activity,38 and it was reported to regulate mRNA translation.6,9,10
In this study, we show that natural and synthetic SINEUP RNAs are m6A modified and that a m6A-dependent step is required for their ability to enhance translation of their target mRNAs.
Results
Natural AS Uchl1 lncRNA and synthetic miniSINEUP RNAs are m6A methylated
To investigate whether m6A plays a role in SINEUP activity, we first studied the presence of modified residues in natural and synthetic SINEUPs. As representative candidates, we analyzed the natural mouse AS Uchl1 and the synthetic miniSINEUP-DJ-1 targeting endogenous human DJ-1/Park7 in human cells.
We performed a methyl-RNA immunoprecipitation (m6A-RIP) in untreated MN9D cells, showing that endogenous AS Uchl1 was m6A-modified in physiological conditions (Figure 1A). Upon transfection, an enrichment of overexpressed AS Uchl1 lncRNA was detected in m6A antibody-immunoprecipitated RNA (Figure 1A), at comparable level to the endogenous transcript and to Tlr3 mRNA, previously reported to be m6A modified.28 No significant enrichment was detected for Ndel1 mRNA, previously reported as non-modified,28 or IVT EGFP RNA spike-in, which were included as negative controls. We also monitored the ability of AS Uchl1 to increase endogenous UCHL1 protein levels post-transcriptionally, as confirmed using western blot and qRT-PCR analysis (Figures S1A and S1B).
Figure 1.

Natural AS Uchl1 lncRNA and synthetic miniSINEUP RNAs are m6A methylated
(A) m6A-RIP qPCR analysis on RNA from MN9D mouse dopaminergic cells expressing endogenous and overexpressed mouse natural AS Uchl1. Tlr3 and Ndel1 mRNAs were used as an endogenous positive and negative controls, respectively. (B) m6A-RIP qPCR analysis on RNA from miniSINEUP-DJ-1 transfected A549 cells. SON mRNA was used as an endogenous positive control. Eluates from IgG immunoprecipitation were used as negative controls. Unmodified IVT mRNA encoding a portion of EGFP was spiked in total RNA extract to assess the specificity of the m6A immunoprecipitation reaction. Data are mean ± SEM and are relative to n = 3 independent experiments. p values are calculated using one-way ANOVA and Dunnett’s multiple-comparisons test. ∗∗p < 0.01 ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001.
To assess the presence of m6A modifications in synthetic miniSINEUP-DJ-1 RNAs, an m6A-RIP was carried out on transfected A549 cells (Figure 1B). qRT-PCR analysis showed significant enrichment of miniSINEUP-DJ-1 RNA similar to SON mRNA, previously shown to harbor multiple m6A CLIP peaks,39 and compared with the negative control IVT EGFP RNA spike-in, proving that synthetic miniSINEUP-DJ-1 RNA was modified. To confirm that the modification was not exclusive to A549 cells, we also performed m6A-RIP on transfected HEK293T, obtaining similar results (Figure S1C). The ability of miniSINEUP-DJ-1 to increase endogenous DJ-1 protein levels (∼1.5-fold) with no changes in the target mRNA levels was confirmed (Figures S1D and S1E).
These results show that m6A methylation is a common feature of natural and synthetic SINEUP RNAs in mouse and human cells.
METTL3 expression regulates synthetic miniSINEUP activity without altering RNA subcellular distribution
As METTL3 is the major cellular methyltransferase responsible for m6A deposition, we carried out m6A-RIP experiments on METTL3-depleted human cells transfected with miniSINEUP-DJ-1. To this end, we used A549 cells stably transduced with shMETTL3 knockdown and shCtrl.40 To induce short hairpin RNA (shRNA) expression, cells were treated with 2 ng/mL doxycycline and transfected on day 7 of induction. A significantly lower enrichment of miniSINEUP RNA was observed upon shMETTL3 expression, confirming METTL3 enzyme as the m6A writer of miniSINEUP RNA modification (Figure 2A). A comparable decrease of enrichment was observed for CREBBP and SON mRNAs, previously reported to be m6A modified,39 while no change was observed for the endogenous negative control HPRT mRNA and the exogenous IVT EGFP RNA spike-in. Moreover, no significant enrichment was observed in DJ-1 mRNA levels in the eluates, proving its lack of m6A modifications (Figure 2A).
Figure 2.

METTL3 expression regulates synthetic miniSINEUP activity
(A) m6A-RIP qPCR analysis. miniSINEUP-DJ-1 was transfected in shCtrl and shMETTL3 A549 cells. CREBBP and SON mRNA were used as endogenous positive controls, while HPRT1 mRNA was used as a negative control. Eluates from IgG immunoprecipitation were used as negative controls. IVT EGFP mRNA was spiked in total RNA extract to assess the specificity of the immunoprecipitation reaction. Data are mean ± SEM and are relative to n = 3 independent experiments. p values are calculated using two-way ANOVA and Šidák’s multiple-comparisons test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. (B) Protein expression analysis. miniSINEUP-DJ-1 activity was assessed using western blotting with anti-METTL3 and anti-DJ-1 antibodies in A549 shCtrl and shMETTL3 cells. ΔBD (miniSINEUP-DJ-1 deprived of BD) was used as a negative control in each condition. One representative experiment is shown (left). SINEUP activity was calculated as an increase in protein quantities relative to the negative control (ΔBD) for each condition. First, band intensity was normalized to the relative β-actin band. Then, fold change values for miniSINEUP-DJ-1-transfected samples were calculated normalizing on negative control-transfected cells for each cell line (dotted line) and reported in the reassuming graph (right). Data indicate single replicate values and mean ± SEM relative to n = 3 independent experiments. p values are calculated using two-way ANOVA with Šidák’s multiple comparison. ∗p < 0.05 and ∗∗p < 0.01.
To study the functional consequences of METTL3-dependent m6A modifications of SINEUP RNAs, we assayed synthetic miniSINEUP-DJ-1 activity upon METTL3 knockdown. As expected, the transfection of miniSINEUP-DJ-1 in shCtrl cells caused a ∼1.4-fold increase in DJ-1 protein levels compared with cells transfected with the Ctrl plasmid (miniSINEUP-ΔBD, i.e., miniSINEUP-DJ-1 deprived of BD sequence). However, in shMETTL3 cells, miniSINEUP RNA expression led to a decrease of endogenous DJ-1 protein of 50% compared with the levels upon transfection of Ctrl plasmid (Figure 2B). No differences in endogenous DJ-1 levels were observed between shCtrl and shMETTL3 cells. These results show a dominant-negative effect of SINEUP RNA on endogenous DJ-1 protein levels when METTL3 expression is strongly reduced.
ShMETTL3 and shCtrl cells were then transfected with control, METTL3 wild-type (WT) or catalytically dead (CD) METTL3 expressing plasmids and, 24 h later, transfected again with miniSINEUP-DJ-1 along with a control plasmid to prove that SINEUP activity is dependent on a functional METTL3. Restoring physiological METTL3 levels in shMETTL3 knockdown cells led to the rescue of miniSINEUP activity (∼1.5-fold increase of protein amounts respect to endogenous physiological DJ-1 levels) while overexpression of METTL3 in shCtrl cells left unaltered the effects of miniSINEUP-DJ-1 RNA. Importantly, the overexpression of CD METTL3 enzyme in shMETTL3 knockdown cells failed to restore SINEUP activity and did not influence the dominant-negative effect of SINEUPs, with endogenous DJ-1 levels still ∼50% lower than in physiological conditions. In shCtrl cells METTL3 overexpression did not influence miniSINEUP-DJ-1 activity, maintaining an expected ∼1.6-fold increase of DJ-1 protein levels. Western blot analysis and qRT-PCR confirmed efficient knockdown and overexpression of METTL3 WT and CD mRNAs (Figure S2A, left). qRT-PCR also confirmed unaltered expression of DJ-1 mRNA in both shCtrl and shMETTL3 A549 cells (Figure S2A, center) and comparable transfection efficiency of SINEUP plasmids (Figure S2A, right).
To assess changes in total levels of m6A methylation upon manipulation of METTL3 expression, we then performed an RNA dot blot (Figure S2B). The experiment confirmed downregulation of m6A levels in poly-A+ enriched RNAs upon METTL3 knockdown induction, a relevant increase in shCtrl cells and a rescue in shMETTL3 cells upon METTL3 WT enzyme overexpression. On the other hand, the overexpression of CD METTL3 enzyme did not alter total m6A levels compared with control and knockdown cells transfected with control vector.
As m6A modification has been shown to influence subcellular RNA localization,41 and SINEUP distribution is a key factor in the regulation of target mRNA translation,6,9 we analyzed the nucleus/cytoplasm distribution of SINEUP RNA and DJ-1 target mRNA upon METTL3 depletion to investigate whether the dominant-negative effect on endogenous DJ-1 levels was caused by altered subcellular localization of SINEUP RNA and/or DJ-1 mRNA (Figure S2C). No difference was observed in the subcellular distribution between shCtrl and shMETTL3 A549 knockdown cells, with ∼20% of miniSINEUP-DJ-1 RNA localized in the cell nucleus and ∼80% in the cytoplasm, as previously reported for other synthetic SINEUP RNAs.6,9,10 No variation in DJ-1 mRNA subcellular distribution was observed (∼40% in the nucleus and ∼60% in the cytoplasm).
Taken together, these results show that METTL3 is the m6A writer enzyme of miniSINEUP-DJ-1 RNA and that its downregulation negatively affects SINEUP activity without altering its subcellular distribution. It is noteworthy that the upregulation of METTL3 enzymatic activity does not alter SINEUP effects, suggesting that the physiological deposition levels of m6A modification are sufficient to ensure the appropriate functionality of SINEUPs.
Identification of SINEUP m6A methylation sites
We then applied m6A prediction score algorithm (SRAMP)42 to identify m6A DRACH consensus sites within natural mouse AS Uchl1 and synthetic human miniSINEUP-DJ-1. The m6A consensus site contains D = G, A, or U; R = G or A; and H = C, A, or U. Applying SRAMP algorithm to AS Uchl1 sequence, nine putative consensus sites with different degrees of confidence along the full-length sequence were identified: two in the overlapping BD region, one adjacent to a partial Alu sequence, one in the invSINEB2 element (390, corresponding to A46 in the ED of miniSINEUP-DJ-1), and five in the downstream region. We then manually annotated three additional weaker putative consensus sites within the invSINEB2 sequence: 407, 425, and 455 nt (Figures S3A and S3C). Limiting the analysis to the invSINEB2 sequence of human miniSINEUP-DJ-1, only A46 was reported as a potential site for m6A deposition (Figure S3B).42 Three weaker putative consensus sites were then manually annotated in A61, A81, and A111 (Figures S3B and S3C), corresponding to A407, A425, and A455 in the full-length AS Uchl1 sequence (Figures S3A and S3C).
Most of the common methods to map post-transcriptional modifications are currently based on RNA immunoprecipitation,28 chemoenzymatic substitution of the modified base,43,44 or detection through specific reverse transcriptase (RT) enzyme efficiency alteration in correspondence of m6A modification sites.45,46 All these methods suffer from low sensitivity and specificity and are prone to artifacts introduced by the indirect probing. On the other hand, modified nucleotides can induce detectable signal deviations during the sequencing of a nucleic acid molecule through organic nanopores, making it possible to map DNA and RNA modifications with a resolution close to the single nucleotide.33,34 In the effort to push the boundaries of detection beyond m6A-RIP, and map m6A methylation sites on miniSINEUP RNA, we relied on Oxford Nanopore Technologies targeted direct RNA sequencing.
First, we compared the sequencing electric signals of an unmodified IVT miniSINEUP-DJ-1 RNA and of the same transfected molecule after undergoing cellular modifications. Then, to isolate the m6A-related differences, we compared the profiles in the presence and absence of METTL3 expression, taking advantage of A549 cell lines harboring inducible Ctrl and METTL3 shRNA constructs (Figure 3A). Thanks to this stringent experimental setup (see materials and methods for an extensive description of its design and rationale), we identified two high-confidence m6A modification sites within 5 nt distance from DRACH consensus in the invSINEB2 element: A46, exactly falling within a DRACH consensus sequence, and C108, slightly offset from the A111 site, residing in the next DRACH consensus (Figures 3B and S3D).
Figure 3.

Identification of SINEUP lncRNAs m6A methylation sites
(A) Nanopore direct RNA sequencing experimental design. Targeted sequencing of IVT miniSINEUP-DJ-1 RNA transfected in shCtrl and shMETTL3 A549 cells was compared to detect m6A methylation sites. Total RNA with 0.1% unmodified IVT miniSINEUP-DJ-1 RNA spike-in was used as a negative control. Differences in electrical signal between transfected Ctrl versus IVT-spiked samples and transfected Ctrl versus METTL3 knockdown cells were assessed. Putative m6A sites were then extracted in a stringent way by intersecting the statistically significant positions identified by both comparisons and retaining those contained within a DRACH motif. (B) Nanopore direct RNA sequencing analysis results. Statistically significant modified residues are highlighted along the invSINEB2 sequence structure (legend on the right). DRACH consensus sites are indicated within the structure in gray (not significantly modified) and red (significantly modified). Resulting hits derived from each comparison (shCtrl versus shMETTL3 and spiked-in versus transfected) and from final intersection are reported in Figure S3D. Two modification sites were identified within 5 nt distance from DRACH consensus: A46, exactly falling within a DRACH consensus sequence, and C108, likely accounting for A111 site that resides in the next DRACH consensus. (C and E) Validation of m6A sites with m6A-qRT-PCR. BstI reverse transcriptase efficiency is reduced in the presence of m6A-modified residues, while MRT enzyme efficiency is not altered by the presence of modification. Reverse transcription primers (+) were designed adjacent to each putative m6A site, and primers (−) were designed in regions with no m6A consensus site nearby. After real-time qPCR of the product, BstI reverse transcription efficiency is compared when using primers (+) and primer (−), and the resulting ratio is then compared with the one deriving from MRT enzyme reactions performed with the same primers. The absence of methylated residue in a putative m6A site results in a relative m6A value of 0.5 (dotted line). (C) m6A-qRT-PCR validation of Nanopore sequencing results. The RT assay was performed on the same samples using four reverse transcription primers (A46, A63, A81, and A111) located adjacent to putative m6A sites. A significant relative m6A signal was detected in sites A46 and A111 in the only IVT miniSINEUP-DJ-1 transfected shCtrl cells (black). No significant signal of m6A modification was detected in IVT miniSINEUP-DJ-1 spiked-in total RNA extract (gray) or transfected in shMETTL3 cells (purple). Data are mean ± SEM and are relative to n = 3 independent experiments. p values are calculated using one-sample t and Wilcoxon tests; ∗p < 0.05. (D) m6A-qRT-PCR on endogenously transcribed miniSINEUP-DJ-1 RNA. Plasmid-encoded miniSINEUP-DJ-1 RNA was transfected in shCtrl and shMETTL3 cells, total RNA was extracted and used for reverse transcription with four primers (A46, A63, A81, and A111) located adjacent to putative m6A sites. A significant relative m6A signal was detected in miniSINEUP-DJ-1 transfected shCtrl cells in A46 and A111 sites, while no relevant relative m6A signal was detected in miniSINEUP-DJ-1 transfected shMETTL3 cells. TUG1 and HPRT mRNAs were used as positive and negative controls, respectively. Data are mean ± SEM and are relative to n = 3 independent experiments. p values are calculated using two-way ANOVA with Šidák’s multiple comparison. ∗p < 0.05 and ∗∗p < 0.01. (E) m6A-qRT-PCR on endogenously transcribed mouse natural AS Uchl1 lncRNA. Plasmid-encoded AS Uchl1 was transfected in MN9D cells, total RNA was extracted and used for reverse transcription with ten primers adjacent to putative m6A sites. A significant relative m6A signal was detected in A275, A390 (i.e., A46 of invSINEB2) and A455 (i.e., A111 of invSINEB2). Data are mean ± SEM and are relative to n = 5 independent experiments. p values are calculated using one-sample t and Wilcoxon test; ∗p < 0.05 and ∗∗p < 0.01.
To validate Nanopore sequencing results, we took advantage of a reverse transcription m6A mapping assay.45 This technique is based on the BstI reverse transcriptase enzyme, whose efficiency is markedly reduced when used in combination with a primer adjacent to an m6A residue. We confirmed the presence of a low but significant m6A deposition signal at A46 and A111 sites in IVT miniSINEUP-DJ-1-transfected shCtrl cells, while we did not detect any modified site in IVT miniSINEUP-DJ-1 spiked-in RNA and in IVT miniSINEUP-DJ-1 transfected in shMETTL3 A549 cells. To strengthen these results, the same method was then used to map m6A sites in plasmid-encoded miniSINEUP-DJ-1 in shCtrl and shMETTL3 A549 cells. Indeed, a stronger m6A signal was detected in shCtrl plasmid-transfected cells (Figure 3D, black) compared with IVT-transfected ones (Figure 3C, black), probably accounting for co-transcriptional deposition of m6A modification. Conversely, in shMETTL3 knockdown cells no m6A deposition signal was detected upon plasmid-encoded miniSINEUP-DJ-1 transfection. TUG1 mRNA was used as a positive control, showing comparable m6A signal to miniSINEUP-DJ-1 in shCtrl cells and its significant reduction in shMETTL3 knockdown cells. Successful knockdown was assessed by qRT-PCR analysis of METTL3 mRNA levels (Figures S3E and S3F). The same method was then applied to AS Uchl1 RNA upon transfection into MN9D cells (Figure 3E). A strong m6A deposition signal was detected in sites A275, A390, and A455. Interestingly, A390 and 455 sites correspond to A46 and A111 nucleotides in the invSINEB2 sequence, confirming that their modification pattern is conserved in natural and synthetic SINEUPs (Figures 3 and S3).
m6A methylation sites regulate miniSINEUP activity
To gain more detailed insights into the role of m6A modification in SINEUP activity, we mutated the identified m6A sites in the invSINEB2 element of synthetic miniSINEUP-DJ-1. Since we previously reported that there is a strong correlation between the structure and the activity of the invSINEB2 sequence,11,47 we engineered a point mutation of A46 m6A site, by substitution of the A with a U, in the effort to perturb RNA secondary structure as little as possible. In the case of A111 m6A site, we had to perform a 3 nt mutation to avoid the possible formation of any cryptic consensus sites, substituting AAA109-111 with UUU sequence.
The m6A modification levels of miniSINEUP-DJ-1 WT and its mutants A46U, AAA109-111UUU, and A46U; AAA109-111UUU transfected in A549 cells were assessed using m6A-RIP while simultaneously monitoring DJ-1 protein expression using western blot. A statistically significant reduction of enrichment by m6A antibody was detected for all mutants, indicating successful elimination of m6A modification sites (Figure 4A). Interestingly, transfection of single m6A sites mutants (A46 and AAA109-111UUU) failed to show any increase in endogenous DJ-1 protein levels. Importantly, when both m6A sites were killed in miniSINEUP-DJ-1 A46U; AAA109-111UUU, a decrease in endogenous DJ-1 protein levels of about 50% compared with control transfections, was observed (Figure 4B). These results are comparable with miniSINEUP-DJ-1 WT activity in shMETTL3 knockdown cells and upon their co-transfections with control and METTL3 CD overexpression (Figure 2B). Dominant-negative effects on endogenous DJ-1 protein levels were therefore observed when unmethylated SINEUP RNA is expressed, both in the absence of METTL3 activity and when lacking sites for m6A deposition.
Figure 4.

m6A methylation sites regulate miniSINEUP activity
miniSINEUP-DJ-1 mutants were designed to analyze the effect of m6A sites’ loss on SINEUP activity. miniSINEUP-DJ-1 WT and its mutants A46U, AAA109-111UUU, and A46U; AAA109-111UUU were transfected in A549 cells. Presence of m6A sites in each construct is reported on the x axis. ΔBD (miniSINEUP-DJ-1 deprived of BD) was used as a negative control. (A) m6A-RIP qPCR analysis. A549 cells were transfected with miniSINEUP-DJ-1 WT and A46U, AAA109-111UUU, A46U; AAA109-111UUU mutants. Eluates from IgG immunoprecipitation were used as negative controls. IVT EGFP mRNA was spiked in total RNA extract to assess the specificity of the immunoprecipitation reaction. Data are mean ± SEM and are relative to n = 3 independent experiments. p values are calculated using two-way ANOVA and Dunnett’s multiple-comparisons test. ∗∗∗∗p < 0.0001. (B) Protein expression analysis. miniSINEUP-DJ-1 mutants’ activity was assessed using western blotting with anti-DJ-1 antibodies A549 cells. ΔBD was used as a negative control. One representative experiment is shown (left). SINEUP activity was calculated as an increase in protein quantities relative to the negative control (ΔBD). First, band intensity was normalized to the relative β-actin band. Then, fold change values were calculated normalizing on negative control-transfected cells (dotted line). On the right, data indicate single replicate values and mean ± SEM and are relative to n = 4 independent experiments. p values are calculated using unpaired t test with Mann-Whitney correction. ∗p < 0.05, ∗∗p < 0.01
DJ-1 mRNA and miniSINEUP-DJ-1s RNA expression were analyzed using qRT-PCR to confirm that miniSINEUP activity occurs exclusively at the post-transcriptional level and to reveal unaltered expression of m6A site mutants (Figure S4A). As in the case of loss of METTL3 expression, the reduction of activity of miniSINEUP-DJ-1 A46; AAA109-111UUU was not due to altered subcellular distribution of DJ-1 target mRNA and miniSINEUP-DJ-1 RNA, as shown by qRT-PCR analysis of nuclear and cytosolic RNAs (Figure S4B).
In summary, the requirement of m6A methylation for miniSINEUP-DJ-1 activity was proved by two complementary experimental strategies: depletion of the methyltransferase enzyme activity responsible for their deposition and mutation of plasmid-encoded SINEUP RNA in the mapped modified sites.
The translation-enhancing activity of SINEUP RNA is impaired upon loss of m6A modification
Natural and synthetic SINEUPs increase the association of their sense target mRNAs to polysomes, enhancing protein synthesis.6,9 We therefore applied polysome fractionation analysis to investigate the molecular mechanism causing the reduction of endogenous DJ-1 protein expression upon transfection of unmethylated miniSINEUP-DJ-1, caused by METTL3 depletion or synthetic mutations. To this end, target DJ-1 mRNA and miniSINEUP RNA distribution were analyzed taking advantage of spiked-in IVT EGFP RNA as a normalizing factor and of GAPDH mRNA as a reference.
Polysome fractionation was first carried out on untransfected A549 shCtrl and shMETTL3 cells using a 15%–50% sucrose gradient separated into 26 fractions. In these experiments, the association of DJ-1 and GAPDH mRNAs to polysomes were independent from METTL3 expression (Figure S5D–S5E).
Experiments were then carried out on A549 shCtrl and shMETTL3 cells transfected with miniSINEUP-DJ-1 WT and miniSINEUP-ΔBD (as a negative control) as well as on A549 shCtrl cells transfected with miniSINEUP-DJ-1 WT, miniSINEUP-ΔBD (as a negative control) and miniSINEUP-DJ-1 A46U; AAA109-111UUU double mutant (Figure 5). Fractions were pulled two by two and analyzed for the presence of DJ-1 mRNA and SINEUP RNA.
Figure 5.

miniSINEUP-DJ-1 translation-enhancing activity is impaired upon loss of m6A modification
(A–C) Polysome fractionation analysis. Cell extracts from shCtrl cells transfected with ΔBD, miniSINEUP-DJ-1 WT or A46U; AAA109-111UUU and shMETTL3 cells transfected with ΔBD and miniSINEUP-DJ-1 WT were prepared and resolved on a 15%–50% sucrose gradient. The absorbance at 260 nm was continuously measured. (A) A representative ribosome profile is reported. The peaks corresponding to free RNA, 40S/60S subunits, 80S and polysomes are indicated. (B) SINEUP RNA and DJ-1 mRNA distribution in shCtrl and shMETTL3 cells transfected with miniSINEUP-DJ-1 WT or negative control (ΔBD). Data are mean ± SEM and are relative to n = 3 independent experiments. p values are calculated using two-way ANOVA and multiple-comparisons test relative to the negative control (ΔBD, shCtrl cells). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001; in black are relative to miniSINEUP-DJ-1 WT in shCtrl cells, in purple are relative to miniSINEUP-DJ-1 WT in shMETTL3 cells. (C) SINEUP RNA and DJ-1 mRNA distribution in shCtrl cells transfected with miniSINEUP-DJ-1 WT, A46U; AAA109-111UUU or negative control (ΔBD). Data are mean ± SEM and are relative to n = 3 independent experiments. p values are calculated using two-way ANOVA and multiple-comparisons test relative to the negative control (ΔBD). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001; in black are relative to miniSINEUP-DJ-1 WT in shCtrl cells, in light blue are relative to miniSINEUP-DJ-1 AAA109-111UUU.
First, SINEUP RNA was enriched in fractions containing 40S and 60S ribosomal subunits and absent in polysome-associated ones (Figures 5B and 5C), in line with previous studies.9 The analysis of all fractions proved that this distribution was maintained in absence of SINEUP BD and upon loss of METTL3 expression, showing that miniSINEUP RNA co-sediments with ribosomal subunits through the invSINEB2 sequence and that this pattern is independent of m6A modification. Upon miniSINEUP-DJ-1 WT transfection in A549 shCtrl cells, a statistically significant increase of DJ-1 mRNA association to polysomes was observed from ∼17% to ∼25%, as expected for SINEUP activity (Figures 5B and 5C). This increase underlies the induction of endogenous DJ-1 protein expression as evidenced on western blot analysis (Figure S5A). Upon miniSINEUP-DJ-1 WT transfection in A549 shMETTL3 cells, a marked shift of DJ-1 mRNA distribution toward non-actively translating ribosomal fractions and free RNA was observed (Figure 5B). In miniSINEUP-DJ-1 transfected cells, ∼6% of DJ-1 mRNA was associated with 40S/60S ribosomal subunit-containing fractions in shCtrl cells, while this percentage was increased to ∼13% upon shMETTL3 expression. Furthermore, DJ-1 mRNA accumulated in free RNA-containing fractions of shMETTL3 transfected cells in a statistically significant manner (15%). A concordant decrease in the amount of DJ-1 mRNA association with polysomes (from 25% to 13%) was observed.
When miniSINEUP-DJ-1 A46U; AAA109-111UUU was transfected in shCtrl cells, a statistically significant accumulation of endogenous DJ-1 mRNA in non-actively translating ribosomal fractions (from ∼4% to ∼10%) was observed. This occurred concomitantly with a strong decrease of DJ-1 mRNA association to polysomes (from 25% to 15%) (Figure 3C). These results phenocopied the effects of METTL3 knockdown on endogenous DJ-1 mRNA distribution in the presence of miniSINEUP RNA. Importantly, no changes in DJ-1 mRNA and SINEUP RNA expression were observed in total RNA analysis (Figure S5B).
Altogether, these results show that SINEUP RNA is enriched in sucrose gradient fractions containing 40S/60S ribosomal subunits complexes by the activity of the embedded invSINEB2 sequence and in an m6A-independent manner. However, an m6A-dependent step is required for SINEUP activity to enhance translation. Lack of m6A methylation inhibits the ability of SINEUP RNA to increase the association of its target mRNA to polysomes. Most important, unmethylated SINEUP RNA relegates its target mRNA to non-actively translating ribosomal fractions, depleting its association to polysomes, and therefore decreasing endogenous expression of the encoded protein acting as dominant-negative.
Discussion
SINEUP technology presents three major advantages as a new class of therapeutics: (1) it induces a 1.5- to 3-fold upregulation of the target protein, thus limiting side effects due to exaggerated overexpression (this is important when the overexpression of the protein of interest is toxic as in the case of those with pro-oncogenic properties or reciprocal copy number variation [CNVs]); (2) it acts on endogenous mRNAs, restricting translation enhancement to the time and space of endogenous gene expression avoiding ectopic protein synthesis in cells that normally do not present it and the need for cell type-specific promoters; and (3) it is scalable, in principle targeting any single mRNA isoform. Furthermore, it can induce the expression of large endogenous proteins, overcoming constrains in cDNA delivery of constructs not easily packaged into adeno-associated viral (AAV) particles because of their excessive size. SINEUPs can be the drug of choice for genetic diseases involving the loss of a single allele (haploinsufficiency and copy number variations) and in complex diseases to restore the activity of compensatory pathways. However, to streamline these applications to the clinics, a better understanding of the mechanism of action is needed.
Here we provide original evidence significantly advancing our understanding of the molecular mechanism of SINEUP activity. First, we show that SINEUP RNA is enriched in sucrose gradient fractions containing 40S/60S ribosomal subunits complexes, while it is remarkably absent in polysomes fractions. We also prove that this pattern depends on the presence of the invSINEB2 sequence, while the antisense BD is dispensable. Most important, we show that m6A modification is required for SINEUP activity.
So far, m6A modification has been shown to promote translation through a few different mechanisms: (1) METTL3 could exert a direct translation activation,48 remaining anchored to the methylated transcript after its export to the cytoplasm; (2) the reader protein YTHDF1 could bind eIF3, which in turn could recruit the 40S ribosome subunit onto the mRNAs, enhancing their translation49; and (3) an m6A-containing 5′ UTR could bind directly to eIF350 in an mRNA cap-binding protein-independent fashion. 5′ UTR m6A-dependent translation initiation seems to be especially important under selective stress conditions, such as heat shock.50 In the case of protein-coding circular RNAs (circRNAs), m6A modifications may play a relevant role in their biogenesis and cap-independent translation.51
In this study, we show that natural and synthetic SINEUPs are m6A modified and that METTL3 is the enzyme responsible for m6A deposition. Importantly, we show that in absence of m6A methylation, unmodified SINEUP RNA exerts a dominant-negative activity on its target mRNA, reducing the endogenous protein levels to about 50%, compared with controls. Unmodified SINEUP RNAs can be obtained with two different experimental strategies: (1) reducing METTL3 activity, through its knockdown or with overexpression of catalytically dead METTL3 enzyme in METTL3 knockdown cells, and (2) mutating the two DRACH consensus sites for m6A deposition in SINEUP RNA. Lack of m6A modification in both experimental systems neither influenced subcellular localization of SINEUP RNA nor its enrichment in 40S/60S ribosomal subunit-containing fractions. However, a striking effect was observed on the quantity of endogenous DJ-1 protein and on DJ-1 mRNA distribution upon polysome fractionation. When m6A modification was removed, endogenous DJ-1 protein levels were decreased to half of those in physiological conditions. Furthermore, DJ-1 mRNA’s association with polysomes strongly decreased while enriching in the 40S/60S ribosomal subunit-containing fractions or in free RNAs. The depletion of DJ-1 mRNA from active polysomes can account for the decrease of endogenous DJ-1 protein expression and therefore for the dominant-negative effect of unmodified miniSINEUP-DJ-1.
Building on these results, we can then speculate on the molecular mechanism of SINEUP activity. First, SINEUP RNA may associate to the 40S ribosomal subunit complex in an m6A-independent and invSINEB2-dependent mechanism. Then, an m6A-dependent step is required for the association of the target mRNA to polysomes. These observations are particularly intriguing given the experimental evidence that SINEUP RNA does not remain associated with polysomes. It is therefore tempting to hypothesize that m6A modifications may recruit an m6A reader that is essential for SINEUP activity. Its activity may be required to release target mRNAs from SINEUP binding and to trigger mRNA association to polysomes for translation. Future work will identify which of many candidate m6A readers are involved in SINEUP activity.
In conclusion, in this study we provide evidence that SINEUP activity depends on the combination of two RNA domains and on a specific pattern of m6A modification, exerting their functions during translation. This is important for the hypothesis that such antisense lncRNAs represent a specific functional class of natural and synthetic regulatory RNAs with their own peculiar mechanism of action. A better understanding of their molecular mechanism is also instrumental for their safe use as therapeutic RNAs to confront a plethora of unmet medical needs.17
Materials and methods
Constructs
miniSINEUP-DJ-1 plasmid was generated on the basis of SINEUP-DJ-1 and miniSINEUP-GDNF, as previously shown.8,16 AS Uchl1-expressing plasmid was previously described by Carrieri et al.6 m6A sites miniSINEUP-DJ-1 mutants were all synthesized by commercial preparation service (GeneScript).
As RNA secondary structure is strictly dependent on the sequence, every mutation can potentially lead to structural alterations. We therefore put our best effort into the design of miniSINEUP-DJ-1 m6A site mutants minimizing structural alterations. Taking advantage of RNAfold WebServer,52 we predicted the secondary structures of miniSINEUP-DJ-1 variants, selecting A46U and AAA109-111UUU as the most conservative mutations. Overexpression plasmids METTL3 WT and catalytically dead (carrying the D394A and W397A substitutions), used for rescue experiments, were obtained from Barbieri et al.40
Cell lines
MN9D cells were obtained from M. J. Zigmond and maintained in culture with high-glucose Dulbecco’s modified Eagle’s medium (catalog #41965069; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (catalog #10270106; Thermo Fisher Scientific) and 1% antibiotics (penicillin/streptomycin; catalog #15140122; Thermo Fisher Scientific). HEK293T cells were obtained from American Type Culture Collection (ATCC) (catalog #ATCC-CRL-11268 164 293T/17; HTB-96) and maintained in culture with Dulbecco’s modified Eagle’s medium (catalog #41965069; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (catalog #10270106; Thermo Fisher Scientific) and 1% antibiotics (penicillin/streptomycin, catalog #15140122; Thermo Fisher Scientific).
ShCtrl and shMETTL3 stably transduced A549 cells were generated as described by Barbieri et al.40 using the same PLKO-TETon-Puro lentiviral vectors and maintained in culture with high-glucose DMEM (catalog #41965069; Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (catalog #10270106; Thermo Fisher Scientific) and 1% antibiotics (penicillin/streptomycin; catalog #15140122; Thermo Fisher Scientific). To induce shRNA expression, cells were treated with 2 ng/mL doxycycline (catalog #D9891; Sigma-Aldrich) every other day and transfected on day 7 of induction in all experiments.
Transfections
MN9D, A549, and HEK293T cells were plated in 6-well plates, 10 mm dishes (subcellular fractionation), or 150 mm dishes (m6A-RIP and polysome fractionation experiments) and transfected respectively with 1 or 16 μg of control or miniSINEUP encoding plasmids using Lipofectamine 2000 (catalog #11668019; Thermo Fisher Scientific) and following the manufacturer’s instructions. Cells were harvested 48 h after transfections. For miniSINEUP activity experiments, RNA and proteins were obtained from the same transfection in each biological replicate. For METTL3 rescue experiment (Figure 2), two transfections were carried out one after the other in the same experiment: Ctrl (empty), METTL3 WT, and METTL3 CD plasmids were transfected after 7 days of shMETTL3 induction by administration of doxycycline, followed by transfection of SINEUP-expressing plasmids after additional 24 h. This experimental strategy allowed the restoration of optimal enzyme levels prior to SINEUP synthesis. Cells were harvested on day 10 of induction as previously described for miniSINEUP activity experiments and processed for WB, RNA, and dot-blot analysis.
Western blot
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer with the addition of protease inhibitor cocktail (catalog #P83490; Sigma-Aldrich), briefly sonicated, and boiled with 1X Laemmli buffer for 5 min at 95°C. 5 μg of total lysate were resolved by 10% SDS-PAGE TGX pre-cast gels (Bio-Rad) and transferred to nitrocellulose membrane using Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked with 5% non-fat dry milk in TBS/0.1% Tween 20 and incubated with the following primary antibodies: anti-β-actin-HRP 1:10,000 (catalog #A3854; Sigma-Aldrich), anti-UCHL1 1:1,000 (catalog #3524S; Cell Signaling Technology), anti-DJ-1 1:8,000 (catalog #ADI-KAM-SA100-E; Enzo Lifesciences). Proteins of interest were visualized with the SuperSignal West Pico PLUS Chemiluminescent Substrate (catalog #34579; Thermo Fisher Scientific). Western blotting images were acquired with ChemiDoc MP Imaging System (Bio-Rad), and band intensity was calculated using ImageJ software.
RNA isolation, reverse transcription, and qRT-PCR real-time analysis
RNA extraction was performed with QIAzol reagent (catalog #79306; Qiagen) or RNeasy Mini Kit (catalog #74106; Qiagen) with DNase I treatment to remove DNA contamination (catalog #79256; Qiagen). Up to 1 μg of RNA was retrotranscribed with iScript cDNA synthesis kit (Bio-Rad, catalog #1708891) following manufacturer’s protocol.
qRT-PCR was carried out using SYBR green fluorescent dye (iTaq Universal SYBR Green Super Mix; catalog #1725124; Bio-Rad) and an iCycler IQ Real-Time PCR System (Bio-Rad). Human or mouse GAPDH mRNAs were used as normalizing control in all qRT-PCR experiments. The amplified transcripts were quantified using the comparative Ct method and the differences in gene expression are presented as normalized fold expression using the ΔΔCt method. The complete list of oligonucleotides used for quantitative real-time PCR experiments is included in Table S1.
m6A dot blot
As m6A is abundant on ribosomal RNA and its deposition derives from the activity of writer enzymes other than METTL3,53 we performed poly-A enrichment before dot blotting to allow the specific detection of METTL3-dependent m6A modification levels. Poly-A enrichment was performed according to NEB “Isolation of mRNA from Mammalian Cells” protocol (https://international.neb.com/protocols/2019/11/18/isolation-of-mrna-from-mammalian-cells) with some modification. Briefly, total RNA was extracted as described under RNA isolation, reverse transcription, and qRT-PCR real-time analysis in materials and methods. Fifty micrograms of total RNA extract was then used to selectively purify RNA molecules longer than 200 nt using the RNA Clean & Concentrator-25 kit (catalog #R1018; Zymo Research). Ten micrograms of total RNA >200 nt was then used as input for poly-A enrichment through two rounds of oligo-dT magnetic beads purification (catalog #S1419S; NEB). mRNA was then further cleaned and concentrated using the RNA Clean & Concentrate-5 kit (catalog #R1016; Zymo Research). The quality and concentration of obtained samples were determined using the Agilent 2100 Bioanalyzer. Fifteen and 7.5 ng of poly-A+ RNA was then used for each condition for dot blotting. Briefly, RNA was incubated at 70°C for 3′ and kept on ice to denature secondary structures before spotting on Amersham Hybond-N+ membrane (catalog #RPN203B; VWR). The membrane was left to dry and RNA was UV crosslinked to the membrane at 254 nm and 120 mJ/cm2. Blocking was performed by incubating the membrane in Denhart solution (1% Ficoll type 400, 1% polyvinylpyrrolidone, and 1% bovine serum albumin) for 1 h at room temperature (RT). m6A Ab (catalog #202111; SySy) was then incubated O/N at 1:1,000 dilution at 4°C. After incubation with appropriate secondary antibody, the membrane was visualized with the SuperSignal West Pico PLUS Chemiluminescent Substrate (catalog #34579; Thermo Fisher Scientific). Images were acquired with ChemiDoc MP Imaging System (Bio-Rad).
Methyl-RNA immunoprecipitation
m6A-RIP was performed as previously described with some modifications.27 Briefly, cells were harvested 48 h post-transfection, and total RNA was extracted with QIAzol reagent or RNeasy Mini Kit, and DNA contamination was removed by treatment with DNaseI following the manufacturer’s instructions. One hundred micrograms total RNA extract, diluted with immunoprecipitation (IP) buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.1% NP-40, RNase inhibitor supplemented) was then incubated with 10 μg m6A Ab (catalog #202111; SySy) antibody for 4 h at 4°C on a rotating wheel. The mixture was then immunoprecipitated with 50 μL G-coupled Dynabeads (catalog #10003D; Thermo Fisher Scientific) at 4°C for additional 3 h on a rotating wheel. Beads were washed 5 times for 5′ at RT on a rotating wheel with IP buffer and resuspended in QIAzol reagent for elution. RNA was extracted, concentrated using the RNA Clean & Concentrate-5 kit (catalog #R1016; Zymo Research) according to the manufacturer’s protocol and analyzed using qRT-PCR in real-time. Normal mouse IgG antibody (catalog #2025; Santa Cruz), IVT EGFP mRNA fragment and beads-only samples were used as negative controls.
In vitro transcription
For the production of unmodified miniSINEUP-DJ-1 RNA, synthetic double stranded DNA miniSINEUP template was cloned downstream a T7 promoter. 1.4 μg linearized DNA template was used to transcribe miniSINEUP-DJ-1 RNAs with MEGAscript T7 Transcription Kit (catalog #AM1333; Thermo Fisher Scientific) following manufacturer’s protocol. In the same way, IVT EGFP RNA was produced. In vitro transcription products were DNAseI-treated, and on-column purified with RNeasy Mini Kit as previously described under RNA isolation, reverse transcription, and qRT-PCR real-time analysis in materials and methods.
Nanopore targeted direct RNA sequencing
RNA sequencing was performed following Oxford Nanopore Technologies (Oxford, UK) instructions, on FLO-MIN106D flow cells (R9.4.1 chemistry) and using direct-RNA sequencing kit (SQK-RNA002). For library preparation, 2 μg total RNA from each replicate were used with custom reverse transcription adapters complementary to the 3′ end of miniSINEUP-DJ-1 RNA following ONT sequence specific DRS protocol. (DSS_9081_v2_revM_14Aug2019). Total RNA with 0.1% unmodified IVT miniSINEUP-DJ-1 RNA spike-in was used as negative control. The amount of IVT spiked-in RNA to be added was determined by comparison with expression levels of transfected miniSINEUP-DJ-1 through qRT-PCR in order to maintain the amount of reads within the same range for each sample and replicate.
Nanopore DRS experimental rationale and data analysis
The detection of RNA modifications via ONT direct RNA sequencing is still in its early days, and the most common strategies to identify chemical marks are based on the comparison of either the raw signals or the error rates between biologically distinct groups of reads. This approach may suffer from a number of unrelated confounding factors including nucleotide mismatches, RNA structural features, difficult to base-call kmers, strand damages, and sugar puckering, possibly resulting in potential artifacts. In order to collect highly reliable m6A information at the expense of other, lesser known modifications, we devised a conservative strategy on the basis of the collection of multiple lines of evidence.
We first compared the differences in electrical signal between transfected versus IVT-spiked samples to extract all the differential signals possibly due to RNA post-transcriptional modifications. Then, we analyzed the profiles of SINEUP RNAs transfected in WT versus METTL3 knockdown cells, in order to focus on METTL3-dependent signals, representing bona fide m6A modifications.
Sites were then extracted in a stringent way by intersecting the statistically significant positions (p < 0.05) identified by both comparisons. As a final layer of specificity, we retained those contained within a DRACH motif (see also Figures 5B and S3D), a known consensus for METTL3-dependent m6A modifications.
Statistically significant electrical differences between kmer groups were identified using the xPore pipeline.54 Briefly, fast5 files were base-called using Guppy 4.4.1 (Oxford Nanopore Technologies), then reads were aligned to SINEUP reference sequence using minimap2.55 Kmer-level data were obtained by resquiggling using the nanopolish eventalign command56 and processed with the xPore python utilities using default settings. RNA structures showing superimposed significance values were plotted using R2R software.57
Raw sequencing data have been deposited in the European Nucleotide Archive (ENA) database with accession code PRJEB55645.
Reverse transcription m6A mapping assay
This technique is based on the BstI reverse transcriptase enzyme, whose efficiency was reported to be markedly reduced when used in combination with a primer adjacent to an m6A residue, in contrast with M-MuLV RT (MRT), whose efficiency is not affected by the proximity of m6A residues and was previously described by Castellanos-Rubio et al.45 We performed the BstI RT followed by qPCR of the product with reverse transcription primers located adjacent to putative m6A sites (primer [+]), and one primer with no m6A consensus site nearby (primer [−]). In parallel, we did the same reaction with the MRT enzyme instead of BstI. After qPCR of the product, we compared BstI reverse transcription efficiency when using primers (+) to the one of primer (−) and we then compared the resulting ratio to the one deriving from MRT enzyme reactions. We then applied the following formula to calculate relative m6A level with qRT-PCR, as previously reported:
| Relative m6A = 2−[Ctprimer(−)BstI−Ctprimer(−)MRT/Ctprimer(+)BstI−Ctprimer(+)MRT]. |
Total RNA was extracted from cells and treated with DNaseI as described under RNA isolation, reverse transcription, and qRT-PCR real-time analysis in materials and methods. Reactions were performed as previously reported by Castellanos-Rubio et al.45 Briefly, 100 ng RNA, 100 nM of each primer, 50 μM dNTPs, and 0.1 U of BstI (NEB) or 0.8 U MRT were used for each reaction. One microliter of the RT reaction was then amplified with qRT-PCR as described under RNA isolation, reverse transcription, and qRT-PCR real-time analysis in materials and methods. The complete list of oligonucleotides used for reverse transcription m6A mapping experiments is included in Table S1.
Subcellular fractionation
For subcellular fractionation experiments, cells were transfected as described under transfections in materials and methods. Nucleo-cytoplasmic fractionation was performed as previously described by Mayer and Churchman.58 Nuclear and cytoplasmic RNAs were extracted using the RNeasy Mini Kit (catalog #74106; Qiagen) with DNase I treatment to remove DNA contamination. The purity of nuclear and cytoplasmic fractions was confirmed by qRT-PCR as on pre-ribosomal RNA 45S and GAPDH mRNA respectively. qRT-PCRs were performed as described under RNA isolation, reverse transcription, and qRT-PCR real-time analysis in materials and methods.
Polysome fractionation
Polysome fractionation was performed as previously described by Panda et al.59 Briefly, A549 shCtrl and shMETTL3 were plated in 15 cm plates and transfected as previously reported after 7 days of doxycycline induction with control, miniSINEUP-DJ-1 WT or miniSINEUP-DJ-1 A46U; AAA109-111UUU vectors. Forty-eight hours after transfection, cells were incubated with 0.1 mg/mL cycloheximide (CHX; catalog #01810-5G; Sigma-Aldrich) for 10 min at 37°C. Cells were then washed with 0.1 mg/mL CHX-supplemented PBS and harvested by scraping. Collected cells were centrifuged at 400 x g for 5 min at 4°C. Cell pellets were resuspended in 400 μL ice-cold lysis buffer (20 mM Tris-HCl [pH 7.5], 100 mM KCl, 5 mM MgCl2, 0.5% Nonidet P-40) supplemented with 0.1 mg/mL CHX and RNase inhibitors. The cell lysate was incubated for 15 min on ice followed by centrifugation at 16,000 × g at 4°C for 10 min to separate the nuclei. The cytoplasmic lysate was layered onto a 15%–50% sucrose gradient and centrifuged in an SW41Ti Beckman rotor at 41,000 × g at 4°C for 90 min. The sucrose gradient was separated into 26 fractions calculated by Triax flow cell (Biocomp). Four hundred microliters of each fraction was then extracted by sodium acetate overnight precipitation. Briefly, 170 μL 3 M sodium acetate (pH 5.3) was added to each fraction together with 1 mL of 100% EtOH, 3 μL Glycoblue co-precipitant (catalog #AM9515; Thermo Fisher Scientific) and 300 ng IVT EGFP spike-in, used for qRT-PCR real-time normalization of target RNA expression levels. The solution was incubated overnight at −20°C for precipitation. The following day, samples were centrifuged at 13,000 × g at 4°C for 30 min. Pellets were pulled two by two at this point, washing with 1 mL 100% EtOH. Samples were then centrifuged 13,000 × g at 4°C for 15 min and EtOH was removed. Pellets were then resuspended in H2O with subsequent cleanup using the RNeasy Mini Kit (catalog #74106; Qiagen) and DNase I treatment to remove DNA contamination. As a control, GAPDH mRNA analysis was performed, according to previous publications. IVT EGFP RNA spiked-in was used to normalize for RNA precipitation efficiency. qRT-PCRs were performed as previously described.
Statistical analysis
All data are expressed as mean ± SEM on n ≥ 3 replicas. Statistical analysis was performed using GraphPad Prism software. Statistically significant differences were assessed using one-sample t and Wilcoxon tests, one-way ANOVA, or two-way ANOVA, as specified in the figure legend for each experiment.
Data availability
Raw sequencing data have been deposited in the ENA database with accession code PRJEB55645.
Acknowledgments
S.G. was supported by intramural funding of the Istituto Italiano di Tecnologia. We are indebted to all the members of the SG laboratory and of the RNA Initiative@IIT for thought-provoking discussions. We are grateful to the technical and administrative staff of IIT (especially to Eva Ferri, Rosa Maria Cossu, and Alessandra Sanna), SISSA, and Università del Piemonte Orientale.
Author contributions
B.P. conceived and performed experiments, analyzed results, and wrote the manuscript. S.D. performed experiments and analyzed results. C.B. carried out preliminary experiments. O.P. carried out experiments and analyzed results. A.A. established inducible shCtrl and shMETTL3 A549 cell lines. A.C. and G.R. analyzed experiments. F.P., R.S., P.C., C.S., and E.S. analyzed results and reviewed the manuscript. P.V. conceived Nanopore experiments. L.P. conceived, supervised the study and performed Nanopore experiment analysis, and wrote the manuscript. S.G. conceived and supervised the study and wrote the manuscript.
Declaration of interests
S.G., C.S., and P.C. are co-founders of Transine Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.04.002.
Contributor Information
Paola Valentini, Email: paola.valentini@iit.it.
Luca Pandolfini, Email: luca.pandolfini@iit.it.
Stefano Gustincich, Email: stefano.gustincich@iit.it.
Supplemental information
References
- 1.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.FANTOM Consortium and the RIKEN PMI and CLST DGT. Forrest A.R.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J.L., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M., et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hon C.C., Ramilowski J.A., Harshbarger J., Bertin N., Rackham O.J.L., Gough J., Denisenko E., Schmeier S., Poulsen T.M., Severin J., et al. An atlas of human long non-coding RNAs with accurate 5’ ends. Nature. 2017;543:199–204. doi: 10.1038/nature21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G., et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katayama S., Tomaru Y., Kasukawa T., Waki K., Nakanishi M., Nakamura M., Nishida H., Yap C.C., Suzuki M., Kawai J., et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 6.Carrieri C., Cimatti L., Biagioli M., Beugnet A., Zucchelli S., Fedele S., Pesce E., Ferrer I., Collavin L., Santoro C., et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491:454–457. doi: 10.1038/nature11508. [DOI] [PubMed] [Google Scholar]
- 7.Zucchelli S., Cotella D., Takahashi H., Carrieri C., Cimatti L., Fasolo F., Jones M.H., Sblattero D., Sanges R., Santoro C., et al. SINEUPs: a new class of natural and synthetic antisense long non-coding RNAs that activate translation. RNA Biol. 2015;12:771–779. doi: 10.1080/15476286.2015.1060395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zucchelli S., Fasolo F., Russo R., Cimatti L., Patrucco L., Takahashi H., Jones M.H., Santoro C., Sblattero D., Cotella D., et al. SINEUPs are modular antisense long non-coding RNAs that increase synthesis of target proteins in cells. Front. Cell. Neurosci. 2015;9:174. doi: 10.3389/fncel.2015.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toki N., Takahashi H., Sharma H., Valentine M.N.Z., Rahman F.U.M., Zucchelli S., Gustincich S., Carninci P. SINEUP long non-coding RNA acts via PTBP1 and HNRNPK to promote translational initiation assemblies. Nucleic Acids Res. 2020;48:11626–11644. doi: 10.1093/nar/gkaa814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fasolo F., Patrucco L., Volpe M., Bon C., Peano C., Mignone F., Carninci P., Persichetti F., Santoro C., Zucchelli S., et al. The RNA-binding protein ILF3 binds to transposable element sequences in SINEUP lncRNAs. Faseb. J. 2019;33:13572–13589. doi: 10.1096/fj.201901618RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Podbevšek P., Fasolo F., Bon C., Cimatti L., Reißer S., Carninci P., Bussi G., Zucchelli S., Plavec J., Gustincich S., et al. Structural determinants of the SINE B2 element embedded in the long non-coding RNA activator of translation AS Uchl1. Sci. Rep. 2018;8:3189. doi: 10.1038/s41598-017-14908-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patrucco L., Chiesa A., Soluri M.F., Fasolo F., Takahashi H., Carninci P., Zucchelli S., Santoro C., Gustincich S., Sblattero D., Cotella D. Engineering mammalian cell factories with SINEUP noncoding RNAs to improve translation of secreted proteins. Gene. 2015;569:287–293. doi: 10.1016/j.gene.2015.05.070. [DOI] [PubMed] [Google Scholar]
- 13.Sasso E., Latino D., Froechlich G., Succoio M., Passariello M., De Lorenzo C., Nicosia A., Zambrano N. A long non-coding SINEUP RNA boosts semi-stable production of fully human monoclonal antibodies in HEK293E cells. mAbs. 2018;10:730–737. doi: 10.1080/19420862.2018.1463945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bon C., Luffarelli R., Russo R., Fortuni S., Pierattini B., Santulli C., Fimiani C., Persichetti F., Cotella D., Mallamaci A., et al. SINEUP non-coding RNAs rescue defective frataxin expression and activity in a cellular model of Friedreich’s Ataxia. Nucleic Acids Res. 2019;47:10728–10743. doi: 10.1093/nar/gkz798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Indrieri A., Grimaldi C., Zucchelli S., Tammaro R., Gustincich S., Franco B. Synthetic long non-coding RNAs [SINEUPs] rescue defective gene expression in vivo. Sci. Rep. 2016;6 doi: 10.1038/srep27315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Espinoza S., Scarpato M., Damiani D., Managò F., Mereu M., Contestabile A., Peruzzo O., Carninci P., Santoro C., Papaleo F., et al. SINEUP non-coding RNA targeting GDNF rescues motor deficits and neurodegeneration in a mouse model of Parkinson’s disease. Mol. Ther. 2020;28:642–652. doi: 10.1016/j.ymthe.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Espinoza S., Bon C., Valentini P., Pierattini B., Matey A.T., Damiani D., Pulcrano S., Sanges R., Persichetti F., Takahashi H., et al. SINEUPs: a novel toolbox for RNA therapeutics. Essays Biochem. 2021;65:775–789. doi: 10.1042/EBC20200114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis C.J.T., Pan T., Kalsotra A. RNA modifications and structures cooperate to guide RNA-protein interactions. Nat. Rev. Mol. Cell Biol. 2017;18:202–210. doi: 10.1038/nrm.2016.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miano V., Codino A., Pandolfini L., Barbieri I. The non-coding epitranscriptome in cancer. Brief. Funct. Genomics. 2021;20:94–105. doi: 10.1093/BFGP/ELAB003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prats A.-C., David F., Diallo L.H., Roussel E., Tatin F., Garmy-Susini B., Lacazette E. Circular RNA, the key for translation. Int. J. Mol. Sci. 2020;21:8591. doi: 10.3390/IJMS21228591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wen S., Wei Y., Zen C., Xiong W., Niu Y., Zhao Y. Long non-coding RNA NEAT1 promotes bone metastasis of prostate cancer through N6-methyladenosine. Mol. Cancer. 2020;19:171. doi: 10.1186/S12943-020-01293-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.M., Li C.J., Vågbø C.B., Shi Y., Wang W.L., Song S.H., et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/J.MOLCEL.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y., Li Y., Toth J.I., Petroski M.D., Zhang Z., Zhao J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014;16:191–198. doi: 10.1038/NCB2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim G.-W., Siddiqui A. N6-methyladenosine modification of HCV RNA genome regulates cap-independent IRES-mediated translation via YTHDC2 recognition. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/PNAS.2022024118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patil D.P., Chen C.K., Pickering B.F., Chow A., Jackson C., Guttman M., Jaffrey S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature (Lond.) 2016;537:369–373. doi: 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang S.Y., Jung H., Mun S., Lee S., Park K., Baek S.C., Moon H.C., Kim H., Kim B., Choi Y., et al. L1 retrotransposons exploit RNA m6A modification as an evolutionary driving force. Nat. Commun. 2021;12:880. doi: 10.1038/S41467-021-21197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dominissini D., Moshitch-Moshkovitz S., Salmon-Divon M., Amariglio N., Rechavi G. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013;8:176–189. doi: 10.1038/nprot.2012.148. [DOI] [PubMed] [Google Scholar]
- 28.Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaccara S., Ries R.J., Jaffrey S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019;20:608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 30.Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X., et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Śledź P., Jinek M. Structural insights into the molecular mechanism of the m(6)A writer complex. Elife. 2016;5 doi: 10.7554/eLife.18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao B.S., Roundtree I.A., He C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu L., Seki M. Recent advances in the detection of base modifications using the Nanopore sequencer. J. Hum. Genet. 2020;65:25–33. doi: 10.1038/s10038-019-0679-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furlan M., Delgado-Tejedor A., Mulroney L., Pelizzola M., Novoa E.M., Leonardi T. Computational methods for RNA modification detection from nanopore direct RNA sequencing data. RNA Biol. 2021;18:31–40. doi: 10.1080/15476286.2021.1978215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacob R., Zander S., Gutschner T. The dark side of the epitranscriptome: chemical modifications in long non-coding RNAs. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18112387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T. N6 -methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J., Dou X., Chen C., Chen C., Liu C., Xu M.M., Zhao S., Shen B., Gao Y., Han D., He C. N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science. 2020;367:580–586. doi: 10.1126/SCIENCE.AAY6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valentini P., Pierattini B., Zacco E., Mangoni D., Espinoza S., Webster N.A., Andrews B., Carninci P., Tartaglia G.G., Pandolfini L., Gustincich S. Towards SINEUP-based therapeutics: design of an in vitro synthesized SINEUP RNA. Mol. Ther. Nucleic Acids. 2022;27:1092–1102. doi: 10.1016/J.OMTN.2022.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., et al. m6A-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/NATURE12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbieri I., Tzelepis K., Pandolfini L., Shi J., Millán-Zambrano G., Robson S.C., Aspris D., Migliori V., Bannister A.J., Han N., et al. Promoter-bound METTL3 maintains myeloid leukaemia via m6A-dependent translation control. Nature. 2017;552:126–131. doi: 10.1038/NATURE24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathlin J., Le Pera L., Colombo T. A census and categorization method of epitranscriptomic marks. Int. J. Mol. Sci. 2020;21:4684. doi: 10.3390/IJMS21134684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91. doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu N., Parisien M., Dai Q., Zheng G., He C., Pan T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA. 2013;19:1848–1856. doi: 10.1261/rna.041178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shu X., Dai Q., Wu T., Bothwell I.R., Yue Y., Zhang Z., Cao J., Fei Q., Luo M., He C., Liu J. N 6-allyladenosine: a new small molecule for RNA labeling identified by mutation assay. J. Am. Chem. Soc. 2017;139:17213–17216. doi: 10.1021/jacs.7b06837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castellanos-Rubio A., Santin I., Olazagoitia-Garmendia A., Romero-Garmendia I., Jauregi-Miguel A., Legarda M., Bilbao J.R. A novel RT-QPCR-based assay for the relative quantification of residue specific m6A RNA methylation. Sci. Rep. 2019;9:4220. doi: 10.1038/s41598-019-40018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harcourt E.M., Ehrenschwender T., Batista P.J., Chang H.Y., Kool E.T. Identification of a selective polymerase enables detection of N(6)-methyladenosine in RNA. J. Am. Chem. Soc. 2013;135:19079–19082. doi: 10.1021/ja4105792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohyama T., Takahashi H., Sharma H., Yamazaki T., Gustincich S., Ishii Y., Carninci P. An NMR-based approach reveals the core structure of the functional domain of SINEUP lncRNAs. Nucleic Acids Res. 2020;48:9346–9360. doi: 10.1093/nar/gkaa598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choe J., Lin S., Zhang W., Liu Q., Wang L., Ramirez-Moya J., Du P., Kim W., Tang S., Sliz P., et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–560. doi: 10.1038/S41586-018-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/J.CELL.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meyer K.D., Patil D.P., Zhou J., Zinoviev A., Skabkin M.A., Elemento O., Pestova T.V., Qian S.B., Jaffrey S.R. 5’ UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Timoteo G., Dattilo D., Centrón-Broco A., Colantoni A., Guarnacci M., Rossi F., Incarnato D., Oliviero S., Fatica A., Morlando M., Bozzoni I. Modulation of circRNA Metabolism by m. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107641. [DOI] [PubMed] [Google Scholar]
- 52.Gruber A.R., Lorenz R., Bernhart S.H., Neuböck R., Hofacker I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–W74. doi: 10.1093/NAR/GKN188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lei K., Lin S., Yuan Q. N6-methyladenosine (m6A) modification of ribosomal RNAs (rRNAs): critical roles in mRNA translation and diseases. Genes Dis. 2023;10:126–134. doi: 10.1016/J.GENDIS.2021.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pratanwanich P.N., Yao F., Chen Y., Koh C.W.Q., Wan Y.K., Hendra C., Poon P., Goh Y.T., Yap P.M.L., Chooi J.Y., et al. Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore. Nat. Biotechnol. 2021;39:1394–1402. doi: 10.1038/s41587-021-00949-w. [DOI] [PubMed] [Google Scholar]
- 55.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/BIOINFORMATICS/BTY191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loman N.J., Quick J., Simpson J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods. 2015;12:733–735. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 57.Weinberg Z., Breaker R.R. R2R - software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinf. 2011;12:3–9. doi: 10.1186/1471-2105-12-3/FIGURES/7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayer A., Churchman L.S. A detailed protocol for subcellular RNA sequencing (subRNA-seq) Curr. Protoc. Mol. Biol. 2017;120 doi: 10.1002/cpmb.44. 4-29.1-4.29.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Panda A.C., Martindale J.L., Gorospe M. Polysome fractionation to analyze mRNA distribution profiles. Bio. Protoc. 2017;7 doi: 10.21769/BIOPROTOC.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data have been deposited in the ENA database with accession code PRJEB55645.