

Abstract
Controlled polymerization methods are well-established synthetic protocols for the design and preparation of polymeric materials with a high degree of precision over molar mass and architecture. Exciting recent work has shown that the high end-group fidelity and/or functionality inherent in these techniques can enable new routes to depolymerization under relatively mild conditions. Converting polymers back to pure monomers by depolymerization is a potential solution to the environmental and ecological concerns associated with the ultimate fate of polymers. This perspective focuses on the emerging field of depolymerization from polymers synthesized by controlled polymerizations including radical, ionic, and metathesis polymerizations. We provide a critical review of current literature categorized according to polymerization technique and explore numerous concepts and ideas which could be implemented to further enhance depolymerization including lower temperature systems, catalytic depolymerization, increasing polymer scope, and controlled depolymerization.
1. Introduction
Polymeric materials are everywhere in modern-day life, representing one of the most important scientific advances of the previous century. However, polymers have (somewhat) justifiably been receiving increasing amounts of negative press in recent years as scientists discover that the ubiquity of polymers in modern life is extending to our water supplies, soil, oceans, wildlife, and even our own bodies. The consequences of this are either universally bad or else not fully known. Enhancing the sustainability of polymers is a topic at the forefront of the field of polymer science and engineering, and encompasses a wide range of approaches, from the development of biodegradable and biorenewable polymers,1 improving mechanical recycling,2 to degradation,3,4 chemical recycling and upcycling.5−8 Depolymerization is the process of reversing polymerization to regenerate monomers, and can be considered the most complete and ideal form of recycling. This overcomes the deterioration of properties associated with mechanical recycling, and thus is a highly attractive prospect that could enable a “circular economy” for plastics.9,10 In 2019 and 2020 IUPAC named depolymerization processes as one of the top ten most important emerging technologies in chemistry.11
However, in the 102 years since Hermann Staudinger demonstrated the existence of macromolecules12 the vast majority of research has been on making polymers and understanding their physical properties, with much less emphasis placed on unmaking them, as demonstrated in Figure 1. In fact, the low reactivity of polymers and their resilience to degradation is, in many cases, precisely why they are valued, with years of research aiming to make them as stable as possible. However, depolymerization is possible, and can potentially be a viable route to recycle plastic waste. Polymers prepared by polycondensation e.g. polyesters, can generally be considered to be highly depolymerizable due to the presence of heteroatoms such as oxygen and nitrogen along the polymer backbone which can be susceptible to chain-breaking reactions such as hydrolysis, aminolysis, glycolysis, methanolysis, etc.13 In fact, these methods are currently being commercialized by companies including Ioniqa, Garbo, Eastman, and Loop Industries to recycle waste PET and polyester fibers.14 Pioneering work in ring-opening polymerization (ROP) and ring-closing depolymerization, as reviewed by Coates and co-workers in 2020, is aiming to develop next-generation polymeric materials with physical properties similar to current commodity polymers, but with straightforward approaches to depolymerization for regeneration of monomer.9 More problematic are polymers with a backbone containing only carbon atoms, such as those produced from the polymerization of vinyl monomers, which pose much more of a challenge to depolymerize due to generally higher hydrolytic and thermal stability. Yet all-carbon backbone polymers, i.e. vinyl polymers, make up the vast majority of commercial plastics15 and almost all the monomers are derived from petrochemical feedstocks, meaning that effective depolymerization is of both commercial and environmental significance.
Figure 1.
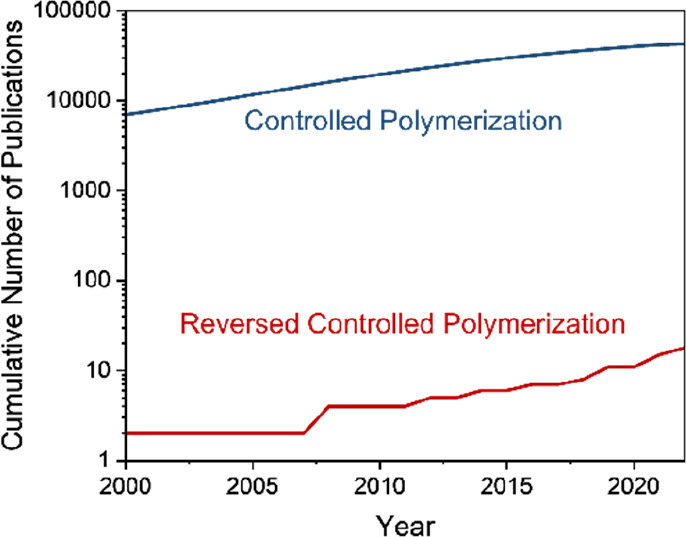
A comparison between the numbers of publications on controlled polymerization vs depolymerization since the year 2000.
The concept of “controlled polymerization” is a central tenet of modern synthetic polymer chemistry.16,17 In general, controlled polymerizations feature fast initiation, with termination either minimized (e.g., in reversible deactivation radical polymerization, RDRP)16,18 or eliminated (living anionic/cationic and selected transition metal mediated polymerizations). This means controlled polymerizations typically show first-order kinetic behavior and have a predeterminable degree of polymerization, narrow molar mass distributions, and long-lived chains with preserved end-group functionality.17 Controlled polymerization is allowing advancements in biomedical science, nanotechnology, and polymer electronics which would have been considered impossible just a few decades ago.18−20 Within the past few years polymers prepared by controlled polymerization have been demonstrated to be much more susceptible to depolymerization than analogs prepared by more conventional free radical polymerizations, which is enabling new routes to depolymerization and presents many exciting opportunities both from a fundamental, mechanistic chemistry perspective and for chemical recycling.21
This perspective will focus on the concept of reversed controlled polymerization (RCP), specifically for the depolymerization of polymers with an all-carbon backbone. We begin by introducing the thermodynamics and reversibility of polymerization. We then explain how the regeneration of active species in polymers prepared by controlled polymerization can induce depolymerization under much more mild conditions (section 2). In section 3 we present key advances from the growing body of literature around the subject and highlight commonalities that are starting to paint a clearer picture of the direction the field is heading. Finally, we discuss areas of research that could dramatically improve depolymerization (section 4) including retaining end-group fidelity, lower temperature systems, catalytic approaches, polymer scope, and controlled depolymerization.
2. Reversibility and Thermodynamics of Polymerization
2.1. Thermodynamics of Depolymerization
Reversibility of polymerization has been known since the early days of polymer science, but it was first fully described in thermodynamic terms by Dainton and Ivin in their seminal 1948 paper “Reversibility of the propagation reaction in polymerization processes and its manifestation in the phenomenon of a “ceiling temperature”” published in Nature.22,23
The Gibbs free energy of polymerization is shown in eq 1. Negative ΔG indicates that propagation (polymerization) is favored, whereas a positive ΔG indicates that depropagation (depolymerization) is predominant.24,25 The temperature at which ΔG = 0 is known as the ceiling temperature, Tc,26 where the rates of polymerization and depolymerization are equal. The enthalpic contributions (ΔH) of almost all known addition polymerization reactions are negative, meaning that the monomer is in a higher energy state than the polymer. For polymerization of a vinyl monomer, whereby a σ-bond is formed from a less stable π-bond, ΔH is typically around −20 kcal/mol (Figure 2).27 The change in entropy (ΔS) of almost all polymerization processes is negative as the number of molecules and the degrees of freedom both decrease as monomer is converted to polymer. The relationship between the Gibbs energy under standard conditions, ΔG° (usually pure monomer or a 1 M solution), and the equilibrium constant, Keq, is shown in eq 2, where Keq is defined as the ratio of the rate constant of propagation, kp, to the rate constant of depropagation, kdp, which is in turn related to the monomer concentration at equilibrium, [M]eq (eq 3). According to these equations, heating a polymerization above its Tc will make the rate of depropagation exceed the rate of polymerization and monomer will be generated until the new [M]eq is reached.
| 1 |
| 2 |
| 3 |
Figure 2.
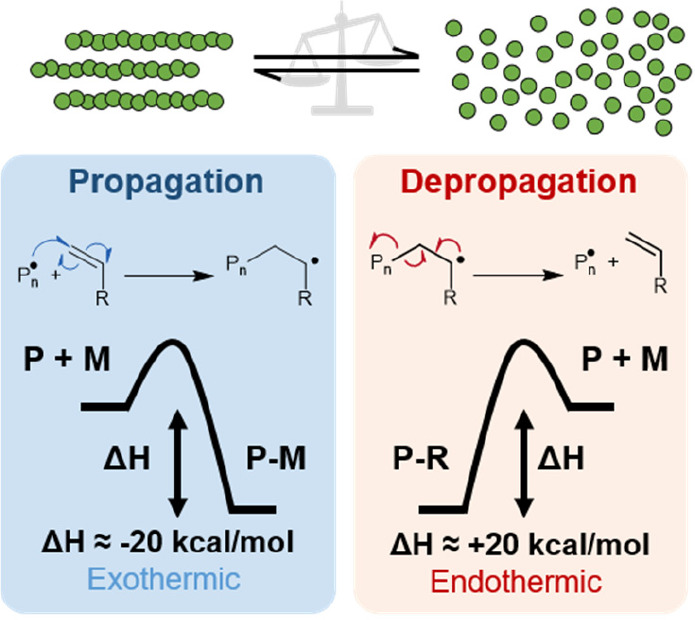
Thermodynamics of propagation vs depropagation for a typical vinyl monomer undergoing radical polymerization/depolymerization.
Indeed, the general trend of negative ΔH and −TΔS for almost all polymerizations could lead us to conclude that depolymerization can be induced by simply heating a polymer above its Tc, and the monomer is “removed” from the equilibrium (e.g., by evaporation) until no polymer remains. However, this does not necessarily paint an accurate picture of how depolymerization can practically be achieved; the ceiling temperature often tells only part of the story.
2.2. How is Ceiling Temperature related to “Depolymerizability”?
Ceiling temperatures are typically determined during polymer synthesis by performing a series of reactions with incremental temperature increases and then measuring the concentration of monomer postpolymerization.23,26 The temperature at which there is no net monomer conversion, i.e when the polymerization appears to cease, is defined as the ceiling temperature, and is often found by extrapolation/interpolation from the aforementioned experiments. Above the ceiling temperature, depolymerization is favored over polymerization and thus monomer is (re)generated. It is important to state clearly that the equilibrium invoked in polymerization is between a monomer and active propagating chains; chains that are terminated/capped are not part of the polymerization/depolymerization equilibrium and will not necessarily depolymerize above the Tc.
A necessary prerequisite for a terminated/capped chain to undergo depropagation is that sufficient energy must be supplied to break a bond and form an active species (a radical, anion, cation, etc.). However, in real terms, many terminated polymers are energetically “trapped”, where bonds cannot be readily cleaved to give a species capable of depropagation even above their ceiling temperature. Thus, polymers can exist at temperatures well in excess of their ceiling temperatures, meaning that depolymerization by simply heating the polymer and collecting the monomer can be incredibly energy intensive, or else will fail due to other degradation pathways being prevalent at the elevated temperatures required. An extreme example of this is observed with poly(olefin sulfones), which are known as ‘self-immolative’ polymers, used as electron beam resists.28 Such polymers are synthesized at low temperatures (−77 °C) as the rate drops to very low values at room temperature and above (the Tc is typically 5–30 °C depending on the olefin used).26 Thermal depolymerization requires a temperature of >200 °C, as a chain scission is required to break a bond, generating a radical that initiates the unzipping to generate monomer. However, scission induced by electron beam radiation causes rapid depolymerization at temperatures much lower than 200 °C.
Tc and [M]eq values are also dependent on initial concentrations; hence, diluting a polymer can often result in depolymerization at temperatures significantly lower than common literature values of Tc. In Principles of Polymerization, Odian specifically cautions the reader that literature often refers to a singular Tc value and that this is misleading.24 Taking PMMA as an example, the Tc in bulk is 296 °C which lowers to 205 °C under dilute conditions (1 M).29 Depropagation of PMMA below this temperature has been linked to unsaturated chain-ends generated during termination and scission of “weak” bonds along the backbone which arise from head-to-head additions under conditions where the regenerated monomer is rapidly removed as a vapor.30−34 The complex nature of polymerization equilibria even results in a chain length dependence on [M]eq in certain cases, as shown by Tobolsky and Eisenberg,35,36 and Szwarc.37
2.3. Ceiling Temperature and Monomer Structure
Under real experimental conditions the nature of the active chain vs monomer equilibrium, together with the concentration dependence of ceiling temperature, means that depolymerization can often be achieved at lower temperatures than reported ceiling temperatures.24,38 Literature values for ceiling temperature do however offer a good measure of depolymerizability when comparing two different polymers (e.g., PMMA, 296 °C, versus polystyrene (PS), 397 °C39). In general, it can be said that polymers comprised of vinyl monomers which are disubstituted at the α-carbon will be significantly easier to depolymerize than monosubstituted equivalents. This is effectively demonstrated by comparing literature Tc values of polystyrene (397 °C)39 and poly-α-methylstyrene (PAMS) (65 °C) which differ only by methyl substitution on the α-carbon. Thermodynamically, polymerization of disubstituted monomers is less exothermic (ΔH is less negative) than for monosubstituted analogs. According to eq 1, this means that the temperature at which ΔG is 0 will be lower, and hence the ceiling temperature is lower. This phenomenon can also be thought of in terms of the kinetics of radical addition; more steric hindrance around the propagating radical, as in the case of disubstituted monomers, will make propagation less favorable. The influence of steric factors is also supported by a study of Tc’s of substituted poly(phenyl methacrylates), in which it was found that bulkier substituents depress ceiling temperature.40 Polycyanoacrylates and polyisobutylene are also disubstituted and have been successfully depolymerized (sections 3.4 and 3.5). Other disubstituted vinyl monomer classes resembling methacrylates include itaconates,41 ethacrylates,42 α-methoxyacrylates,43 and α-ethylsulfenyl acrylates,43 all of which are known to depolymerize back to monomer. Geminal captodative (i.e., an electronic “push–pull”) substitution is reported to significantly stabilize the resulting tertiary radical and decrease the strength of the C–C bond.43
2.4. The Potential of Reversing Controlled Polymerization
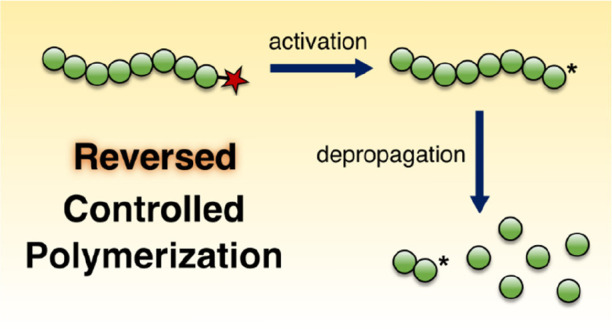
Since the equilibrium between monomer and polymer involves only active polymer chains, for a presynthesized polymer to be depolymerized we must first transform chains to an active state so that depropagation can occur (if temperature and concentration are amenable to the thermodynamics of this). As mentioned in section 2.2, this would normally mean having to supply enough energy for random scission of chains to occur. However, many “controlled” polymerizations (particularly RDRP techniques) rely on the presence of reactive/capped chain-ends, in contrast to the terminated chains found in uncontrolled polymers. High end-group fidelity is crucial to the formation of well-defined macromolecular architectures such as block copolymers in RDRP.44,45 As will be demonstrated in section 3, the ω chain-ends which make controlled polymerization possible (e.g., halogens or RAFT agent Z groups, etc.) can in many cases also be leveraged to overcome the energetic barrier to depolymerization at lower temperatures and initiate depropagation reactions (Figure 3). Even for techniques which do not explicitly require active end-groups, such as living anionic and living cationic polymerization, the presence of such end-groups has been found to be crucial in initiating depolymerization (see sections 3.4 and 3.5). Numerous examples now exist in the literature in which polymers prepared by controlled polymerization methods can depolymerize under conditions where analogous polymers synthesized by more conventional means are thermally stable.21 Coupled with dilute conditions, this has resulted in >90% monomer yields from common polymers such as polymethacrylates at temperatures much lower than what would perhaps be anticipated.46
Figure 3.

A cartoon representation of the concept of reversing controlled polymerization through activation of the ω chain-end to give an active species capable of depropagating to yield monomer.
While this underlying principle of reversing controlled polymerization has been known for many years, research efforts applied to depolymerization to regenerate monomer are relatively new. Thus, reversed controlled polymerization is an emerging field with many exciting possibilities, advancing chemical recycling, new polymer characterization techniques, and the revelation of hitherto unknown mechanistic pathways.
3. Depolymerization from Controlled Polymerization Methods
3.1. Reversed Atom Transfer Radical Polymerization
Atom transfer radical polymerization (ATRP) is a reversible deactivation radical polymerization (RDRP) that was independently developed by Matyjaszewski47 and Sawamoto48 in 1995. It has since grown to become one of the most widely used methods of controlled radical polymerization due to its wide scope and high tolerance to many functional groups.49 ATRP controls molecular weight through reversible termination facilitated by a transition metal catalyst (copper is ubiquitous, but other metals such as iron, ruthenium, and nickel are also commonly employed).50 The transition metal can abstract a halide from an initiating species (an alkyl halide) to give a carbon-centered propagating radical, while being simultaneously oxidized to a higher oxidation state. Propagating radicals are capped with halides by the oxidized complex to establish an equilibrium where the radical concentration is kept low, minimizing radical–radical termination reactions, Scheme 1.51
Scheme 1. Simplified Mechanism of ATRP Showing Conditions Favoring Polymerization and Depolymerization.
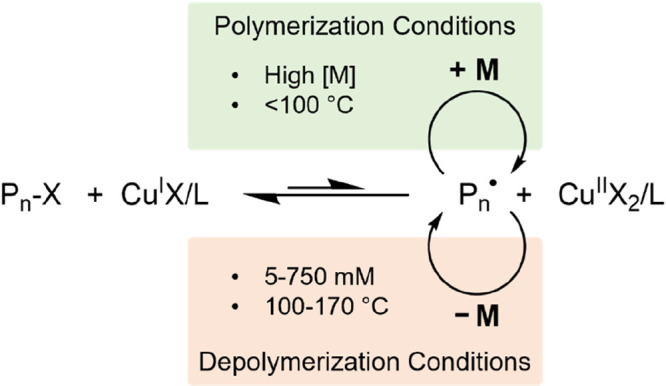
Methacrylic-type monomers with either polymeric side chains (macromonomers) or large, bulky groups have been central to the development of depolymerization protocols for ATRP. These large monomers typically exhibit less favorable polymerization thermodynamics than smaller methacrylates and have lower bulk repeat unit concentrations, making them more amenable to depolymerization.52 One of the first literature examples demonstrating depolymerization with ATRP was reported by Raus and co-workers in 2014.52 Raus found that the polymerization of commercially available methacrylate-functionalized bulky polyhedral oligomeric silsesquioxane, poly(dimethylsiloxane) methacrylate (POSS) monomer, iBuPOSSMA, had a very low ceiling temperature, to the extent that the expected linear pseudo-first-order kinetics of polymerization via ATRP was significantly perturbed at relatively low temperatures. After 24 h of polymerization, the reaction temperature was raised from 60 to 90 °C and polymerization was allowed to proceed for a further 24 h. Depolymerization was observed, with a 15% increase in the amount of monomer regenerated. This was a significant development, illustrating the first depolymerization of an ATRP polymer at particularly low temperature, but the limitations are certainly the uncommon monomer selected and that the polymerization and depolymerization were both performed in the same pot, without prepurification of the polymer prior to depolymerization.
In 2019 Ouchi and co-workers overcame both of these problems by reporting the radical depolymerization of PMMA via reversible activation of the chlorine end-group with a ruthenium catalyst.53 PMMA is a much more challenging polymer to depolymerize than polymethacrylate macromonomers due to its higher enthalpy of polymerization, more positive entropy, and higher repeat unit density. PMMA with a chloride end-group was prepared by ATRP with an indenyl-based ruthenium catalyst (Ru(Ind)) at 80 °C and isolated (Mn = 5600 Da, Đ = 1.16). Seven hours of heating this polymer at 100 °C with Ru(Ind) resulted in 4.5% of MMA monomer generated, increasing to 6.6% after 24 h (Figure 4A and B). Size exclusion chromatography (SEC) analysis showed a slight shift to lower molecular weight while retaining a narrow molar mass distribution (Đ = 1.18 and 1.21 at 7 and 24 h, respectively, Figure 4C). At 120 °C depolymerization was faster (24% in 24 h); however, the molecular weight distribution broadened to a bimodal trace with a dispersity of 1.28. Matrix-assisted laser desorption/ionization-time-of-flight (MALDI-ToF) analysis (Figure 4D) of this sample revealed that end-group fidelity had been compromised as a result of a side reaction, with olefin chain-end polymers being identified. The concentration of PMMA-Cl in the reaction was also found to affect the extent of depolymerization; concentrations between 10 and 0.5 mM were screened, and lower concentrations were found to give more depolymerization in line with the expected equilibrium between depolymerization and polymerization, Scheme 1. Once the equilibrium monomer concentration is reached, depolymerization cannot continue (see section 2.2). For depolymerizations at 100 °C, removing monomer (and solvent) periodically via vacuum evaporation and adding fresh solvent caused the molecular weight of PMMA-Cl to decrease from 5600 to 4500, albeit with a slight increase in low molecular weight tailing and associated dispersity value (Đ = 1.28). In this manner, higher total polymerization conversions could be attained. Despite the low depolymerization conversion reported by Ouchi, this work represents the first example of depolymerization of a presynthesized and purified polymethacrylate synthesized by ATRP.
Figure 4.

Depolymerization of PMMA-Cl with ruthenium catalyst: [PMMA–Cl]0/[Ru catalyst]0/[n-Bu3N]0 = 10/4.0/40 mM in toluene at 100 °C. A: the structure of PMMA–Cl and scheme of the depolymerization; B: 1H nuclear magnetic resonance (NMR) spectra for characterization of MMA generation and the terminal structures of PMMA; C and D: SEC and MALDI-ToF-MS traces during depolymerization. Reprinted from ref (53). Copyright 2019, with permission from Elsevier.
Matyjaszewski and co-workers have elegantly exploited the higher depolymerization propensity of methacrylic macromonomers to show that chlorine-capped poly(poly(dimethylsiloxane) methacrylate)) (P(PDMSMA)) bottlebrush polymers, in the presence of a copper(II) chloride/tris(2-pyridylmethyl)-amine (CuCl2/TPMA) catalyst system, depolymerizes at 170 °C.29 Thermogravimetric analysis (TGA) showed that P(PDMSMA) synthesized by conventional radical polymerization was slightly less thermally stable compared to a similar polymer synthesized by ATRP; however, the thermal stability of P(PDMSMA)-Cl decreased after being incubated at 170 °C, which was attributed to uncatalyzed loss of the −Cl ω-chain-end, giving a polymer more akin to its conventional radical polymerization counterpart. Despite being thermally stable at 170 °C, the presence of the CuCl2/TPMA catalytic system was found to dramatically increase the yield of monomer when P(PDMSMA)-Cl was heated in 1,2,4-trichlorobenzene, attributed to radical formation via atom transfer. Furthermore, depolymerization could be accelerated by increasing [TPMA]0/[CuCl2]0 from 1 to 6 to increase ligand-promoted reduction of CuII/L to CuI/L. These optimized conditions were able to generate up to 80% PDMSMA monomer. The reversibility of the system was demonstrated by repeated depolymerization and repolymerization reactions over as many as 4 cycles, albeit with loss of end-group functionality after each round (Figure 5). These results demonstrate a marked difference between polymers synthesized by conventional radical polymerization and ATRP. ATRP catalysts are able to “activate” the chain-ends and attain depolymerization under conditions which would be not feasible for conventional polymers.21
Figure 5.

Schematic of depolymerization/repolymerization cycling experiments with P(PDMSMA)-Cl. Reproduced from ref (29). Copyright 2021 American Chemical Society.
The Matyjaszewski group also reported a similar system for the more challenging depolymerization of poly(n-butyl methacrylate) (PnBMA), synthesized by activators regenerated by electron transfer (ARGET) ATRP.54 Depolymerizations were carried out with various polymer dilutions (8–21 wt %) in 1,2,4-trichlorobenzene at 170 °C (Figure 6A). Depolymerization was found to be negligible in the absence of an ATRP catalyst (Figure 6C, green), but addition of CuCl2/TPMA with an excess of ligand showed much more efficient depolymerizations, with up to 67% monomer being recovered under conditions with optimized Cu/L ratios at 750 mM repeat unit concentration (Figure 6B). The presence of ligand with no added copper also showed depolymerization (Figure 6C), attributed to reduction of the chloride chain-end leading to activation and unzipping. SEC analysis showed a slight shift toward lower molecular weight as depolymerization progresses, albeit with an increase in dispersity. This suggests that on the abstraction of the end-group halogen, the majority of polymer chains instantly depropagate back to monomer. A series of control experiments and determination of the theoretical [M]eq revealed that the depolymerizations ceased before [M]eq was reached, attributed to prematurely terminated chains. Incubation of model macroinitiators revealed that lactonization at the chain-end was responsible for loss of −Cl functionality, which increases the thermal stability and essentially makes depolymerization of the remaining chains impossible under the relatively mild conditions employed. This system shows the highest polymer concentration reported for reversed controlled polymerization to date but is limited by high temperatures utilized and associated lactonization, preventing further depolymerization from being achieved. More recently the Matyjaszewski group has expanded ATRP depolymerization to iron catalysts for both PMMA and PBMA.55 Utilizing Fe0 as a supplemental activator and reducing agent gave >70% depolymerization at a repeat unit concentration of 700 mM.
Figure 6.
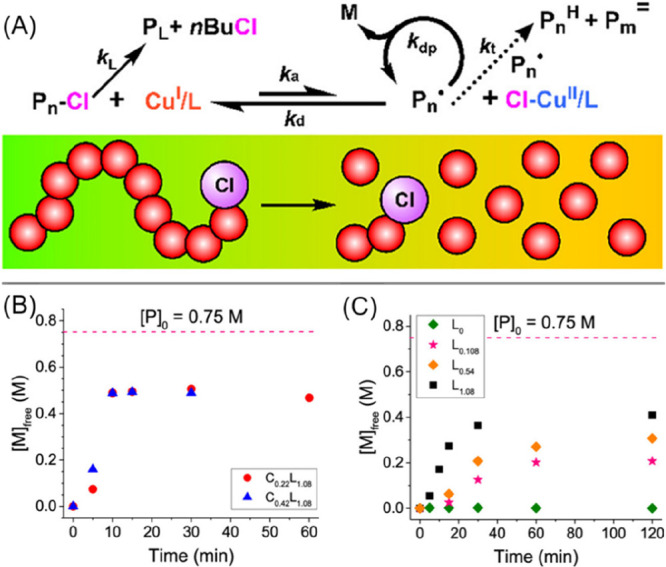
(A) activation of chlorine capped PBMA to give depropagating radical species, generated BMA monomer. (B) Free monomer concentration vs time for 750 mM PBMA solutions with Cu/TPMA. (C) Free monomer concentration vs time for 750 mM PBMA solutions with TPMA. Figure adapted from ref (54). Copyright 2021 American Chemical Society.
Reversing controlled polymerization via ATRP is a topic gaining more and more interest; the body of work available so far allows certain conclusions to be drawn about the potential limits of ATRP for depolymerization and the identification of problems which need to be overcome. The work of Ouchi and Matyjaszewski clearly demonstrates that depolymerization of nonbulky polymethacrylates is achievable to relatively high levels of monomer recovery and that catalysis is crucial to activate chain-ends. A limiting factor seems to be loss of chain-end fidelity at the higher temperatures employed rendering polymers with much higher thermal stability, indeed this has limited all the discussed examples to −Cl end-groups, precluding ATRP polymers employing bromine. The development of more robust chain-end chemistry and optimization of conditions to minimize side reactions could yield depolymerization systems capable of reaching theoretical monomer equilibrium values. While both Ouchi’s and Matyjaszewski’s examples show a decrease in molecular weight, the decrease is not proportional to the amount of monomer generated, suggesting that depolymerization is not “controlled”. Perhaps increasing the rate of deactivation by CuII species could improve this, allowing for depropagating radicals to be capped and all chains to decrease in length at similar rates.
3.2. Reversible Addition–Fragmentation Chain-Transfer (RAFT) Polymerization
RAFT polymerization was first reported in 1998 by Moad, Rizzardo, Thang, and co-workers56 and is mechanistically quite unlike other controlled polymerization systems in that the reversible deactivation of chains stems from reversible chain-transfer events rather than reversible termination (as in the case of ATRP and nitroxide-mediated polymerization).49 Nonetheless, RAFT polymerization is equally, if not more, effective at producing well-defined polymers and is arguably the most robust controlled radical polymerization technique in regard to tolerance to monomer functionality. Typically, the chain-transfer agent (CTA) is a thiocarbonylthio (Z–C(=S)S–R) compound whose Z-group primarily determines the reactivity of the C=S bond to a radical and the resulting stability of the intermediate radical, whereas the R-group is a good homolytic leaving group that is reactive enough to initiate propagation of new polymer chains.
Similar to ATRP-based depolymerization systems, RAFT polymethacrylates allow on-demand access to a chain-end radical which, under thermodynamically suitable conditions, depropagates to yield monomer. In 2018, Gramlich and co-workers first demonstrated the ex situ depolymerization (i.e., depolymerization of a purified polymer) of P(PDMSMA) and poly(oligo(ethylene glycol) methyl ether methacrylate) (POEGMA) bottlebrushes terminated with a trithiocarbonate end-group by heating the polymer in dioxane at 70 °C.57 For P(PDMSMA), under an initial repeat unit concentration of 28 mM, 35% depolymerization was achieved after 56 h. The initiation mechanism for depolymerization was not established, but the authors postulated it to be triggered by the polymer chain itself. Other potential sources of external radicals were not ruled out. Gramlich and co-workers conclude that successful polymerization of macromonomers such as PDMSMA requires careful selection of conditions to avoid depolymerization affecting the rate.
In 2022, Anastasaki and co-workers reported RAFT depolymerization of nonbulky polymethacrylates in solution.46 Up to 92% depolymerization could be achieved with a dithiobenzoate end-group at 120 °C under a repeat unit concentration of 5 mM in dioxane (Figure 7A). Notably, the RAFT agent could be reused after depolymerization, facilitating a controlled RAFT polymerization of the regenerated monomer at 70 °C in the presence of a free radical initiator. It was estimated that between 50% and 55% of the RAFT agent can be recovered in this way. Finally, the group also demonstrated the first depolymerization of an insoluble hydrogel synthesized by the copolymerization of OEGMA and its dimethacrylate analog (Figure 7B). Although the initiation mechanism could not be unequivocally established, successful chain-extensions in the absence of a free radical initiator perhaps imply radicals are directly formed at the polymer chain-ends. In a subsequent publication, the same group expanded the scope of the RAFT-based depolymerization methodology to various RAFT end-groups, solvents, and heat-sensitive polymers (i.e., polymers with thermally unstable side chains), as seen in Figure 7C and D.58 Notably, both the end-group and solvent had a significant effect on the final depolymerization conversion highlighting the importance of tailoring reaction conditions to minimize parallel side reactions and thereby maximize depolymerization.
Figure 7.

(a) 1H NMR spectra of the depolymerization reaction of poly(methyl methacrylate)-dithiobenzoate (5 mM of repeat unit and 120 °C in 1,4-dioxane). (b) Depolymerization of poly(oligo(ethylene glycol) methyl ether methacrylate), repolymerization into a hydrogel, and subsequent depolymerization of the hydrogel. (c) depolymerization of heat-sensitive polymers poly(tert-butyl methacrylate) and (d) poly(glycidyl methacrylate). (a) and (b) are adapted from ref (46). Copyright 2022 American Chemical Society. (c) and (d) are adapted from ref (58). Copyright 2022 American Chemical Society.
Reversing RAFT polymerization has thus far proven to be highly promising, particularly from the fact that the highest reported depolymerization conversions (92%) have been achieved. This suggests that at least 92% of chains possess the RAFT end-group prior to depolymerization. On the other hand, polymethacrylates synthesized by ATRP typically have lower chain-end fidelity and thus their maximum depolymerization conversions are inherently suppressed. For example, poly(n-butyl methacrylate) synthesized by ATRP had 81% chain-end fidelity54 (calculated from chain-extension experiments, so the actual livingness is likely to be higher) inherently restricting the maximum depolymerization to 81%.
One aspect that needs further investigation is the effect of the solvent on the degradation of the end-group. The great majority of studies have been performed using 1,4-dioxane as the solvent because the highest conversions could be achieved alongside minimal end-group degradation. In contrast, other solvents such as xylenes, toluene, and dimethylformamide had noticeably lower conversions and a faster end-group degradation at 120 °C. The dependence of solvent on the rate of end-group degradation is yet to be established although some hypotheses could be made, such as aminolysis by trace amounts of amines present in DMF. Solvent choice can also affect ΔH and ΔS,59−61 as has been shown for grafting-through radical polymerizations.62,63 Another aspect, perhaps the most important, is that the initiation mechanism for the depolymerization still needs to be unequivocally established. As the depolymerization reaction proceeds in the absence of a free radical initiator, an unconventional route must be involved in generating the chain-end radical. Due to the very low end-group concentration, a very low concentration of radicals is required to produce a chain-end radical. Thus, it is challenging to exclude the possibility of radical generation from an external source such as impurities in the solvent, impurities in the polymer sample, and the surface of the reactor especially since the reactions are under relatively high temperatures compared with that used in polymerization. Homolytic cleavage of the polymer-CTA C–S bond by heat has not been reported in literature and requires further investigation.
Sumerlin and co-workers recently reported photoassisted radical depolymerization of RAFT-synthesized polymers.64 This method relies on radical generation via direct photolysis of the CTA chain-end, similar to a photoiniferter polymerization process,65 instead of the thermal approach utilized by Gramlich and Anastasaki. Exploring depolymerization from PMMA with trithiocarbonate, dithiocarbamate, and p-substituted dithiobenzoate end-groups, it was found that CTAs which absorb light at lower wavelengths show dramatically increased rates of depolymerization. ∼ 70% depolymerization was achieved in just 1 h under optimized conditions. The photoactivation to generate depropagating radicals also seems to lower the temperature at which depolymerization can occur, with monomer generation seen at temperatures as low as 100 °C. The Tc of this system was experimentally determined to be 85 °C. Further discussion on lowering depolymerization temperature is included in section 4.2.
Simultaneously, Anastasaki and co-workers reported light-accelerated depolymerization of RAFT-synthesized polymethacrylates catalyzed by the photocatalyst Eosin Y at 100 °C.66 The synergetic effect of Eosin Y and visible light led to a significant acceleration of the depolymerization in the early stages of depolymerization, leading to a final conversion of up to 82% for dithiobenzoate-terminated PMMA. For trithiocarbonate and pyrazole carbodithioate chain-ends, a starker acceleration and enhancement of the final conversion could be seen, suggesting an efficient formation of chain-end radicals. Notably, this photothermal strategy was also highly compatible with a solvent other than dioxane, evidenced by the 82% conversion in DMSO which is remarkably higher than the 35% conversion obtained in the purely thermal system at 120 °C.46 The versatility of this method was further highlighted by the flexibility in the wavelength employed: green, blue, red, and white light irradiation led to highly comparable depolymerization conversions, greatly enhancing the versatility of the depolymerization methodology.
3.3. Reversed Iodine Transfer Polymerization
Iodine transfer polymerization (ITP) relies on reversible scission of a terminal C–I bond to form propagating radicals. This scission can be promoted in several ways, for example thermally or by organocatalysts such as amines.67 ITP has the advantage of being relatively simple to implement, as reflected by its commercialization in processes to synthesize surfactants, medical devices, and electronics.20 The C–I bond is more labile compared to C–Br or C–Cl commonly used in ATRP; thus, it can potentially be activated under milder conditions. As discussed in section 3.1, depolymerization of poly(butyl methacrylate) terminated by Cl using only TPMA as a catalyst resulted in the regeneration of 55% of monomer. Addition of 20% CuCl2 increased the reaction conversion by just 10%, suggesting that Lewis bases (e.g., amine-containing ligands) can be used solely for depolymerization on halogen-terminated polymers.
Recently, Liu and co-workers demonstrated the use of triaminocyclopropenium (TACP) iodides as an ion-pair catalyst in halogen-bonding catalyzed living radical polymerization.68 The iodine anion of the TACP iodide activates iodine-terminated polymers, resulting in the generation of propagating radicals. In addition to the reported polymerizations, the authors also showed that the same catalytic system can be employed for the depolymerization of PMMA with tetraglyme as a solvent, at 3.2 mM polymer concentration, at 120 °C for 24 h. Control experiments in the absence of catalyst showed no monomer generation; however, depolymerization was detected when 100 equiv of TACP catalyst were employed. NMR analysis showed the appearance of characteristic vinylic proton peaks and SEC chromatograms shifted to lower molecular weights, with increased dispersity. Depolymerization was not quantified via NMR, although SEC traces show a decrease in Mn from 10.2 kDa to 6.4 kDa which corresponds to 37% depolymerization (Figure 8). Upon increasing the temperature by 30 degrees (150 °C), a larger shift in SEC traces was observed suggesting that 120 °C is not the optimal temperature for this reaction.
Figure 8.
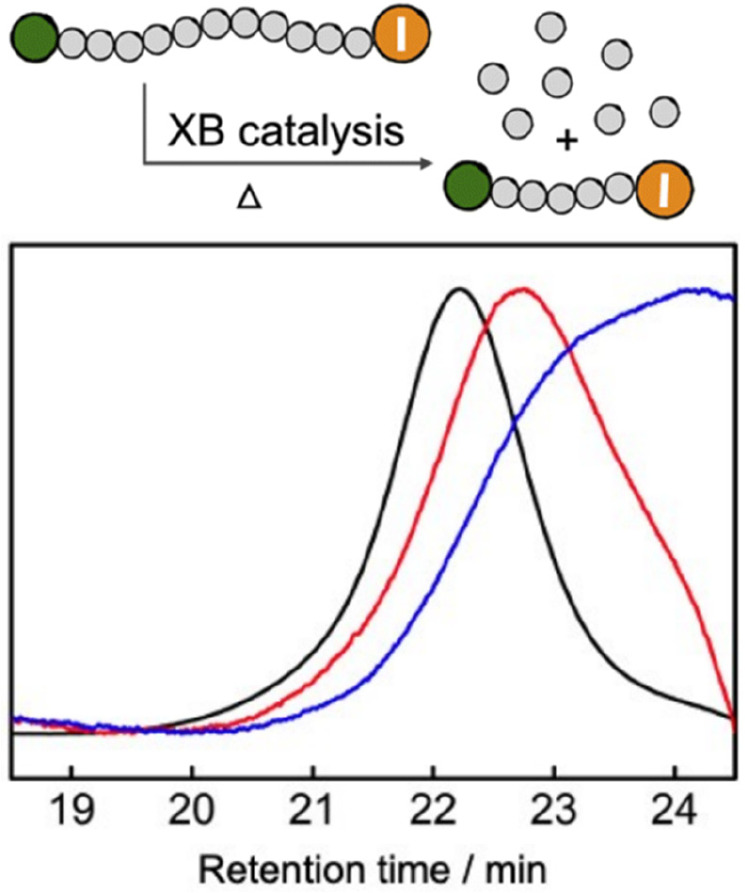
Depolymerization of [PMMA-I]0 (3.2 mM) in tetraglyme. GPC traces obtained from the samples: before reaction (black line); reacted at 120 °C for 24 h with TACP-I as catalyst (red line); reacted at 150 °C for 24 h with TACP-I as catalyst (blue line). Figure adapted with permission from Royal Society of Chemistry from ref (68), copyright 2022; permission conveyed through Copyright Clearance Center, Inc.
Noteworthy in this study is that the SEC chromatographs are shifted to significantly lower molecular weights as depolymerization progresses, in contrast to other reports46,54 in sections 3.1 and 3.2 where the polymer peak intensity decreases while any shift to lower molecular weights is limited. The different behavior exhibited by Liu’s system could be attributed to a more controlled depolymerization where the chains are simultaneously depolymerizing via multiple activation and deactivation cycles. The authors suggest that further optimization of the system may provide better results and a deeper understanding of the reaction mechanism.
3.4. Reversed Anionic Polymerization
Anionic polymerization was the first ever reported controlled or “living” polymerization, developed by Szwarc in 1956.69 Polymerization is typically initiated from an organometallic compound (e.g., an alkyl lithium) which adds to a monomer to give a propagating anion. Such polymerizations have no formal termination step unless the propagating chain reacts with a contaminant or specifically added termination agent (e.g., water, alcohol, etc.). Like radical polymerizations, anionic polymerizations are also equilibrium processes, as demonstrated by Szwarc and co-workers for α-methyl styrene.69 Polymers synthesized by anionic polymerization do not typically have labile end-groups; hence, anionic-synthesized PMMA is more thermally stable than PMMA synthesized via other methods and shows degradation (in bulk) at temperatures greater than 350 °C.30 However, in 1988 Chiantore and Gualta showed that cumyl cesium initiated anionic polymerization of MMA can result in polymers which are much less thermally stable, presenting a first stage of degradation attributed to the presence of a more labile cumyl α-end-group. Regular PMMA prepared by anionic polymerization showed degradation at temperatures above 320 °C, whereas multiple samples of PMMA from Cumyl Cesium initiation showed a degradation onset around 250 °C with MMA monomer being generated (Figure 9) when heated in bulk. In general, it does seem more challenging to incorporate labile end-groups on to polymer chains synthesized by anionic polymerization in comparison to alternate methods (sections 3.1–3.3) where polymer synthesis directly yields labile end-groups.
Figure 9.
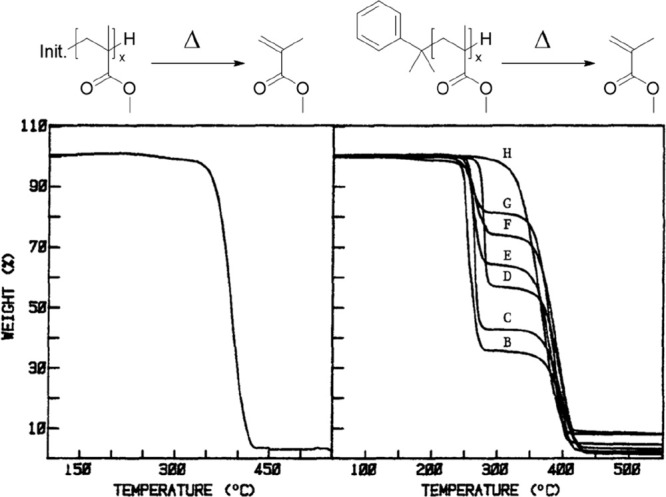
Left: Thermogravimetric analysis (TGA) of PMMA prepared by anionic polymerization. Right: TGA of PMMA prepared by anionic polymerizations with cumyl cesium as initiator. Figure is adapted with permission from Springer Nature from ref (30). Copyright 1988, Springer Nature.
One further interesting study by Ryan in 1996 reported a subceiling temperature rapid depolymerization (and subsequent repolymerization) of cyanoacrylate polymers by addition of a base to dilute polymer solutions at 21 °C.70 SEC analysis revealed that the intensity of the polymer peak decreased with time while a lower molecular weight “daughter polymer” peak increased in intensity, suggesting that monomer generated from depolymerization was being repolymerized. The authors propose that the addition of a base deprotonates the poly(butyl cyanoacrylate) chain-end to give depolymerization via an anionic mechanism, differing from all the examples discussed before which are radical in nature. A later study of poly(ethyl cyanoacrylate) revealed that heating the polymer resulted in degradation via a radical unzipping pathway to generate monomer, but also noted that depolymerization can also occur at much lower temperatures in basic media.71
3.5. Reversed Cationic Polymerization
Living cationic polymerization is a chain growth polymerization in which monomer adds to a cationic initiator to give a propagating cation. The process has no formal termination step, although chain-breaking reactions are more prevalent than in living anionic polymerization.72 Cationic chain growth is particularly useful for ring-opening polymerization of heterocycles although these polymers are beyond the scope of this perspective. For all-carbon backbone polymers, polymerization of electron-rich vinyl monomers such as isobutylene are particularly effective via a cationic pathway.
Polyisobutylene has a relatively low ceiling temperature, between 88 and 120 °C.73 However, thermal degradation of polyisobutylene is typically carried out at >300 °C to facilitate chain scissions initiating depropagation.74 A lower energy route to depolymerization via cationic depropagation has also been reported by treatment with Lewis acids.75 A cationic pathway was utilized by Bergbreiter and co-workers in 2019 to give efficient poly(isobutylene) depolymerization at room temperature.76 Poly(isobutylene) oligomers synthesized via living cationic polymerization with functionalized chain-ends were protonated with the strong Brønsted acid CF3SO3H to give a macromolecular cation capable of depropagation to give isobutylene. Unsaturated chain-ends were found to give the highest extent of depolymerization (Figure 10B). However, the process results in the transformation of the monomer into tert-butylbenzene. Isobutylene liberated from the chain-end is protonated and undergoes a Friedel–Crafts reaction with benzene (the solvent employed), keeping the concentration of isobutylene low and pushing depropagation further (Figure 10A).
Figure 10.

(A) Reaction via the equilibrium formation of some isobutylene by the depolymerization of the polyisobutyl cation. Reaction of this isobutylene with a Brønsted acid to form tert-butyl carbenium ions that, in a more enthalpically favorable step, form tert-butylbenzene by Friedel–Crafts chemistry. (B) Qualitative 1H NMR spectroscopic studies showing the extent of depolymerization of polyisobutylene oligomers with various end groups in toluene in the presence of 2.2 equiv of CF3SO3H at 25 °C for 20 h. Figure adapted from ref (76). Copyright 2019 American Chemical Society.
Poly(α-methylstyrene) prepared by cationic polymerization was successfully depolymerized by Choi and co-workers with ball-mill grinding.77 In this process, PAMS was milled with stainless steel balls in a vibratory ball-milling machine. Radical trapping experiments and simulations revealed that chain scission caused by mechanical force results in polymeric radicals capable of depropagation. Up to 64% depolymerization of PAMS with minimal amounts of side products was obtained after only 8 min of grinding. Bulk temperatures within the grinding vessel were reported to be much lower than the reported ceiling temperature of poly(α-methylstyrene), perhaps indicating localized “hot spots”. The report also demonstrates limited depolymerization of other polymers, as discussed further in section 4.5.
3.6. Reversed Ring-Opening Metathesis Polymerization
Ring-opening metathesis polymerization (ROMP) is a chain-growth polymerization whereby a transition metal catalyst sequentially ring-opens cyclic alkenes via an olefin metathesis reaction. Homogenous catalysts for ROMP such as the third generation Grubbs’ catalyst provide fast initiation rates with high functional group tolerance, thus enabling polymers and block copolymers to be synthesized with high precision and narrow molar mass distributions.
The fact that ring-opening metathesis is an equilibrium process between monomer and polymer was first demonstrated as early as the late 1960s, although utilizing and optimizing this for depolymerization was not investigated until the work of Badamshina and colleagues in the early 1980s.78 Polypentenamers synthesized by tungsten catalysis (Đ ∼ 2.1–5.1) containing unsaturated bonds were found to undergo depolymerization via tungsten coordination and cleavage at random double bonds along the chain. The coordinated transition metal then rapidly unzips the fragmented chain through ring-closing metathesis (RCM) to generate a linear macromonomer. In 2019 Kennemur and co-workers investigated RCM of bottlebrush polypentenamers (Đ ∼ 1.02–1.16) using Grubbs’ catalysts (Figure 11A).79 SEC data obtained during the depolymerization (Figure 11B) showed only chains of starting molecular weight and those that had fully depolymerized, suggesting that depolymerization is initiated through catalyst coordination and subsequent RCM at the chain-end, presumably due to steric hindrance of internal olefins with bulky side chains. Kennemur’s work demonstrates the potential of depolymerization of ROMP synthesized polymers; however, polypentenamers typically exhibit low Tg’s.
Figure 11.
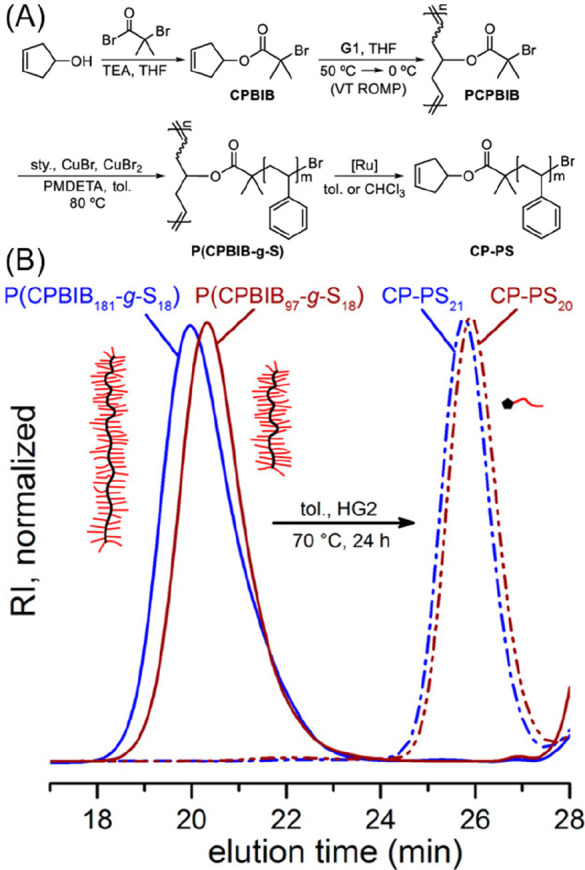
(A) Synthesis and depolymerization of bottlebrush polypentenamers, adapted from Scheme 1, ref (79). (B) SEC traces of bottlebrushes (blue solid trace, Mn = 248.6 kg mol–1, Đ = 1.35 and (red solid trace, Mn = 231.1 kg mol–1, Đ = 1.18) and their quantitative depolymerization to α-cyclopentenyl polystyrene grafts (blue dash-dot trace, Mn = 2.1 kg mol–1, Đ = 1.05 and red dash-dot trace, Mn = 1.9 kg mol–1, Đ = 1.16), respectively. Both depolymerizations were performed in toluene ([olefin]0 = 24 mM (5% (w/v)), [cat.]0 = 2.4 mM) at 70 °C for 24 h using HG2 catalyst. Figure adapted from ref (79). Copyright 2019 American Chemical Society.
Wang and co-workers examined the theoretical ring strain energies (RSEs) of many cyclic olefins in an attempt to find more robust polymers which are also capable of depolymerization.80 The RSE of cyclopentene is 5.2 kcal/mol, and its polymers can depolymerize, whereas the RSE of cyclooctene is 8.2 kcal/mol and its polymers cannot. Trans fused-ring monomers based on cyclooctene however were found to have very similar RSEs to cyclopentene but can yield polymers with Tg’s > 100 °C. Heating these polymers at 50 °C in chloroform with 1 mol % Grubbs’ catalyst (2nd generation) yielded up to 90% depolymerization in just 2 h (Figure 12).80 The thermal decomposition of these polymers was only achieved at >350 °C, an impressive demonstration of the potential of RCM depolymerization as a low energy pathway to depolymerization.
Figure 12.

(a–f) 1H NMR spectra for P1 (a), P2a (b), P3 (c), polycyclooctene (d), poly(cis-gDCC–CO) (e), and poly(trans-gDCC–CO) (f) before (in black) and after (in red) 2 h of heating the polymer solution (solvent, chloroform or deuterated chloroform; [olefin] = 25 mM) at 50 °C in the presence of Grubbs’s second-generation catalyst (G2). The 1H NMR spectra of the corresponding monomers are shown (in blue) as references. The red spectra (polymer treated with G2) and blue spectra (monomer) are nearly identical for tCBCO polymers, indicating complete depolymerization (a–c); however, the spectra are distinct for polycyclooctene, poly(cis-gDCC–CO), and poly(trans-gDCC–CO), suggesting no depolymerization occurred (d–f). Figure reproduced from ref (80) with permission from Springer Nature. Copyright 2021, Springer Nature.
4. Future Challenges in Depolymerization
4.1. Preserving End-Group Fidelity
One common feature in all of the work presented in section 3 is that depolymerization from controlled polymers relies heavily on the presence of reactive end-groups to trigger depropagation, a much lower energy pathway than inducing random chain scission to yield radicals capable of unzipping. Modern synthetic methods of controlling polymerizations have already been optimized to give maximum end-group fidelity, which is crucial to synthesizing high-order macromolecular architectures such as multiblock copolymers. However, it has been explicitly found (in the case of ATRP, section 3.1)54 or suggested (in the case of RAFT, section 3.2) that the extent of depolymerization is inhibited by loss of end-group fidelity during depolymerization. Thermolysis of chloride chain-ends in ATRP polymers under incubation was found to make poly(methacrylates) more thermally stable, meaning that this side reaction competes with chloride abstraction by copper catalysts and limits the number of chains that can undergo depolymerization.54 In RAFT, a thermal Chugaev-type elimination was suggested to compete with the proposed homolytic cleavage of the chain transfer agent, resulting in a methacrylic macromonomer instead of a macroradical capable of unzipping. Even in anionic depolymerization of cyanoacrylates70,71 the unique chain-end initiated depolymerization is attributed to deprotonation at the chain-end; an end-group with sufficient pKa is crucial.
Thus, to maximize depolymerization, experimental conditions must be further optimized to overcome the loss of end-group fidelity due to undesirable side reactions (Figure 13). For RAFT polymerization, CTAs could be selected according to their thermal stability and their propensity to undergo homolytic cleavage as opposed to Chugaev elimination, as investigated by He et al.81 Alternatively, catalytic approaches to activate chain-ends at lower temperatures could be explored (see section 4.3). In the case of ATRP, recent work has already identified that chloride-capped chains are expected to be more thermally stable than larger halides such as bromide and iodide. Regulation of controlled polymerization by external stimuli such as light is known to furnish polymers with high end-group fidelity.82 Such techniques might also be beneficial for preserving end-group fidelity during depolymerizations. Another option would be to develop new depolymerization methodologies able to operate at lower temperatures where deleterious thermally promoted reactions are minimized.
Figure 13.
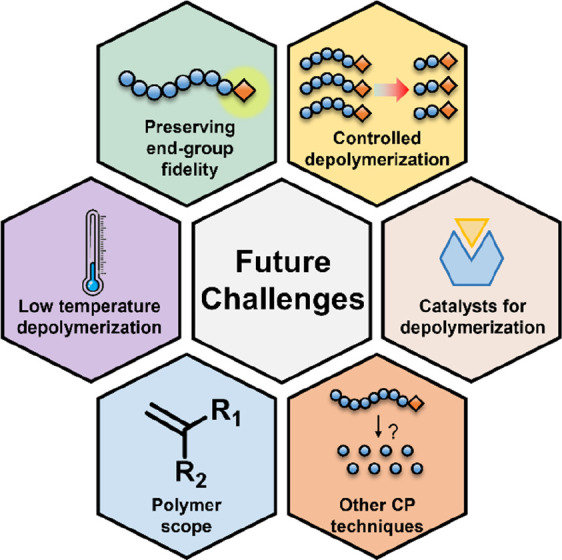
Future challenges for reversing controlled polymerization that could drastically improve current methodologies and expand the scope to many more materials.
4.2. Low Temperature Depolymerization
Generating monomers from polymers at low temperatures is a highly attractive prospect for many reasons. First and foremost, low temperature depolymerization could dramatically reduce the energy needs of reactions, making depolymerization much more attractive from an industrial and commercial point of view for the chemical recycling of commodity polymers. As mentioned in section 4.1, being able to depolymerize polymers at lower temperatures would also limit deleterious side reactions which compromise end-group fidelity.
One obvious approach to lower the temperature threshold at which depolymerization occurs would be to utilize reaction engineering to optimize conditions. Conducting depolymerization under reduced pressure with distillation apparatus would allow for continuous removal of monomer, pushing the equilibrium of depolymerization to produce more monomer. Removing monomer under reduced pressure has already been demonstrated by Ouchi and co-workers to re-establish [M]eq and drive depolymerization further, although the solvent (toluene) was also removed and had to be replenished.53 Utilizing even more dilute polymer solutions could also have the effect of lowering the effective Tc of the system further to enable low temperature depolymerization. These approaches could be particularly efficacious if used in combination. Polymer synthesis in continuous flow reactors has shown numerous benefits over batch approaches,83 and could also be of interest in depolymerization to give higher monomer yields at lower temperatures. Utilizing external stimuli such as light could also provide a route to activate chains at lower temperatures, as has recently been demonstrated for RAFT polymerization by the groups of Sumerlin64 and Anastasaki,66 and for ATRP by Yagci and co-workers.84
The poly(butyl cyanoacrylate) depolymerization reported by Ryan (see section 3.3) was unusual in that it was reported to occur at just 21 °C. In contrast to other reported depolymerizations it is also believed that the depropagation occurs via an anionic mechanism. Anionic depropagation could be a potential avenue of future research for low temperature depolymerization of other polymer classes.
4.3. Catalytic Approaches to Depolymerization
Catalysis is already playing an important role in some reversed controlled polymerizations. For example, in the ATRP-based depolymerization systems developed by Matyjaszewski and Ouchi, typical ATRP catalysts can abstract halides from polymer chain-ends to give a radical which then depropagates to yield monomer.29,53−55 In the absence of such catalysts the polymers do not undergo appreciable depolymerization under otherwise identical conditions. Depolymerization of ROMP-synthesized polymers is also achieved through catalysis, whereby the metathesis reaction is reversed to give an iterative ring-closing process that generates monomer.79,80 The ruthenium catalyst offers a much lower energy pathway than thermal degradation.80 These important examples raise the question of how catalysis could be further exploited to achieve more efficient depolymerization. Catalytic depolymerization is already established for polymers with heteroatoms within the backbone.13,85−87 A well-known example of this is the acid/base catalyzed hydrolysis of esters which can be used to repeatedly break poly(ester) chains to eventually regenerate monomer.13 A more state-of-the-art example is the work of Chen and co-workers on chemically recyclable polymers from γ-butyrolactone derivatives.88 Thermolysis of such polymers yielded pure monomer after 24 h at 300 °C, whereas adding a catalytic amount of ZnCl2 yielded pure monomer after 12 h at 120 °C.89
The RAFT depolymerizations reported by Gramlich and Anastasaki are catalyst-free processes, a feature which is potentially beneficial as catalysts introduce extra costs to the process. Photoinduced electron/energy transfer (PET) RAFT is a relatively new technique in which activation of the CTA chain-end is achieved with a catalyst or direct activation with light.90 Depolymerization via photoactivation of RAFT polymers has recently been reported by both the Sumerlin and Anastasaki groups, exhibiting both fast depolymerization and the possibility of depolymerization at temperatures as low as 100 °C.64,66 The former approach used direct photoactivation of the CTA, whereas the latter utilized Eosin Y as a photocatalyst to activate the end-group.
4.4. Controlled Depolymerization
The polymers discussed in section 3 are all synthesized in a controlled manner; initiation is fast, and termination is either absent or minimized allowing polymer chains to grow at the same rate, giving narrow molar mass distributions. Controlled polymerizations share a number of other common features such as a linear evolution of molecular weight with conversion, pseudo-first-order kinetics with respect to monomer consumption, and a decrease in dispersity with conversion. If polymerizations can exhibit these features, is it possible that depolymerization can show the direct opposite behavior? Depolymerization would have to occur by a fast loss of end-group to give active chain-ends which would then, in the presence of sufficient deactivation, depropagate at identical rates to show a linear decrease in molecular weight with monomer generation, as shown in Figure 14.
Figure 14.
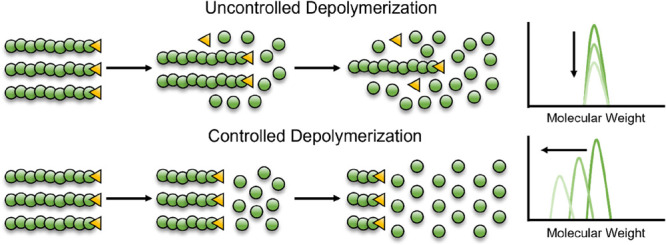
Cartoon representation of uncontrolled (top) and controlled (bottom) depolymerization. Uncontrolled gives little change in molecular weight with depolymerization conversion whereas controlled shows a decrease in molecular weight as chains depolymerize concurrently.
Liu and co-workers report that the depolymerization of iodo-capped PMMA shows clear shifts in molecular weight distributions to lower molecular weights, presenting a case for controlled depolymerization. From a mechanistic standpoint, controlled depolymerization with ATRP catalysts would require a fast initiation of the chain-end and relatively high rates of deactivation to ensure efficient capping of depropagating radicals, thus minimizing termination reactions to maintain end-group fidelity and depropagate all chains equally. Slow initiation and ineffective deactivation would cause some polymer chains to depolymerize rapidly and others remain at the initial DPn until later in the reaction. Thermolysis of chain-ends, as demonstrated by Matyjaszewski, could also perturb controlled depolymerization behavior through dead chains and possible changes to the ATRP equilibrium. The current examples of depolymerization from RAFT polymers in section 3.2 do not show controlled characteristics; namely the molecular weight shows only a slight decrease as monomer is continually generated, indicative of “unzipping” behavior, whereby depropagation is occurring faster than reversible fragmentation chain transfer. Linearization of depolymerization conversion, as demonstrated in the reports of Gramlich57 and Sumerlin,64 indicates a steady state concentration of radicals, but not necessarily a controlled process.49 In a similar fashion to reversed ATRP, improving the control of RAFT depolymerization would require more effective deactivation or more efficient chain transfer of depropagating radicals.
4.5. Polymer Scope of Reversed Controlled Polymerizations
In section 2.3 we discussed the relationship between ceiling temperature and monomer structure, concluding that 1,1 disubstituted monomers such as methacrylates are much more amenable to depolymerization due to their favorable sterics and thermodynamics. Comparing the many recent reports of depolymerization from controlled polymers in section 3, it is clear that a disproportionate number of examples focus on polymethacrylates, as summarized in Table 1. The high number of reports of depolymerization from polymethacrylates and the comparatively few examples of other polymers beg the question: what polymer scope is possible with reversed controlled polymerization?
Table 1. Summary of Reported Conditions for Reversed Controlled Polymerizations of (Meth)Acrylic Monomers.
| Monomer | Polym Technique | Depolym Conv | Depolym Conditionsa | ref |
|---|---|---|---|---|
| Methacrylates | ||||
| MMA | ATRP | 8% | 510 mM, Toluene, Ru(Ind), 100 °C, 24 h | (53) |
| ATRP | 76% | 700 mM, TEGDME, Fe0, 170 °C, 20 min | (55) | |
| RAFT | 86% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (46) | |
| RAFT | 80% | 5 mM, Dioxane, Eosin Y, 100 °C, green light, 8 h | (66) | |
| RAFT | 70% | 5 mM, Dioxane, 100 °C, UV light, 24 h | (64) | |
| ITP | –b | 320 mM, Tetraglyme, TACP-I, 120°C, 24 h | (68) | |
| nBMA | ATRP | 67% | 750 mM, Trichlorobenzene, CuCl2/TPMA, 170 °C, 10 min | (54) |
| ATRP | 72% | 700 mM, TEGDME, Fe0, 170 °C, 15 min | (55) | |
| RAFT | 92% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (46) | |
| BzMA | RAFT | 92% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (46) |
| RAFT | 78% | 5 mM, Dioxane, Eosin Y, 100 °C, green light, 8 h | (66) | |
| DMAEMA | RAFT | 74% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (46) |
| RAFT | 66% | 5 mM, Dioxane, Eosin Y, 100 °C, green light, 8 h | (66) | |
| HEMA | RAFT | 72% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (46) |
| TFEMA | RAFT | 80% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (46) |
| PEGMA | RAFT | 87% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (46) |
| RAFT | 80% | 5 mM, Dioxane, Eosin Y, 100 °C, green light, 8 h | (66) | |
| GMA | RAFT | 84% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (58) |
| tBMA | RAFT | 85% | 5 mM, Dioxane, no catalyst, 120 °C, 8 h | (58) |
| PDMSxMA | ATRP | 78% | 275 mM, Trichlorobenzene, CuCl2/TPMA, 170 °C, 10 min | (29) |
| RAFT | 30% | 100 mM, Dioxane, no catalyst, 70 °C, 56 h | (57) | |
Depolymerization conditions in the following order: concentration of monomer units, solvent, catalyst, temperature, and reaction time.
Entry left blank as the percentage depolymerization was not reported.
Polyacrylates, analogous to polymethacrylates without methyl groups along the backbone, exhibit much higher kp values and are often so thermodynamically stable that they do not exhibit ceiling temperatures, instead degrading via other mechanisms at elevated temperatures (e.g., formation of midchain radicals).91 The improbable thermodynamics of polyacrylate depolymerization suggests that radical unzipping is not a viable option. This would also be the case for polyacrylamides which are reported to undergo degradation before they can depropagate.92
Polystyrene (PS) is a very common commodity polymer, but it has a relatively high Tc and there is currently a limited number of reports on depolymerization of PS made by a controlled polymerization. There is however a large body of literature describing pyrolysis of polystyrene at high temperatures93,94 and numerous routes to upcycle and obtain useful chemical building blocks from the commodity polymer.95−98 The low temperature depolymerization of polystyrene reported in a ball-milling process by Balema and co-workers is particularly interesting, as it generates up to 7% styrene monomer from a commercial PS sample at less than 60 °C.98 A more recent report by Choi and co-workers,77 discussed in section 3.5, also reported ball-mill grinding depolymerization of polystyrene (1% depolymerization) prepared by anionic polymerization. These results are intriguing and suggest that ball-mill grinding could be a route to efficient depolymerization of more “challenging” polymers such as PS in bulk. Pyrolysis of PS prepared by nitroxide mediated polymerization has recently been reported to be enhanced (increased monomer yield) compared to PS from conventional radical polymerization, presumably due to thermally promoted homolytic cleavage of the chain-end nitroxide species enabling more efficient radical formation than relying on chain scission.99,100
Depolymerization of polyolefins, such as polyethylene (PE) and polypropylene (PP), in a low energy and cost-effective manner would be a highly valuable development from an environmental standpoint, as polyolefins make up the majority of commodity polymers; yet their highly stable nature makes depolymerization very challenging. Recent pioneering work by Buchwald has shown the generation of propylene from commercial PE by employing a dual catalytic system whereby PE was first partially dehydrogenated to give unsaturated segments which then underwent catalytic isomerization by metathesis with ethylene.101 A similar dual catalytic approach was first used by Guan to generate liquid fuels from polyolefins.102 From a controlled polymerization perspective, olefin polymerization is typically controlled by catalysts which undergo a Cossee–Arlman type coordination–insertion process. The reverse of this process, β-alkyl elimination, has recently been demonstrated for quasi-depolymerizations of polydiolefins103 with a zirconium metallocene catalyst, but has not yet been shown to be capable of regenerating high yields of olefinic monomers from polyolefins.
4.6. Enabling Depolymerization from Other Controlled Polymerizations
As can be seen from section 3 of this perspective, depolymerization from controlled polymerizations is still in its relative infancy, and because of this there are still many controlled polymerization techniques which have not yet been demonstrated to be capable of efficient depolymerization. In this section we discuss the possibility and potential of some of these techniques in depolymerization (Figure 15), focusing on methodologies which could show advantages or unique mechanistic features compared to those in section 3. Just as each polymerization methodology has its advantages, they could offer various benefits to depolymerization as well.
Figure 15.
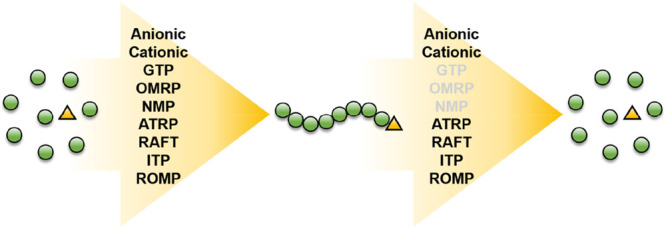
Common controlled polymerization techniques, showing those which have not yet been explored for their depolymerization potential.
Nitroxide-mediated polymerization (NMP) was the first reported RDRP techniques capable of yielding well-defined polymers with high end-group fidelity.104 Control over the polymerization is attained by a reversible termination reaction between carbon centered propagating radicals and a stable free radical nitroxide. The homolytic cleavage of nitroxide species intrinsic to the NMP mechanism could serve as an ideal route to depolymerization as radicals would readily be formed upon heating polymer solutions. Under suitable conditions (low polymer concentration, high temperatures, etc.), monomer would be expected to be generated from depropagation. However, the biggest barrier NMP faces is its limited monomer scope, particularly toward methacrylate monomers, which are known to undergo side reactions.105,106 Styrenics and acrylates are inherently more suited to NMP, yet are difficult to depolymerize from a thermodynamic standpoint. Future efforts could also focus on the synthesis of alkoxyamine components that would permit C–O bond scission under milder conditions, allowing for fast and simple depolymerization procedures. Organometallic-mediated radical polymerization (OMRP), mediated by cobalt complexes reversibly terminating propagating radicals, has also not yet been explored in depolymerizations. In a similar manner to NMP, homolytic cleavage is usually thermally promoted, meaning that OMRP could be particularly well-suited to depolymerization reactions as heat will likely form polymeric radicals capable of depropagation with fewer potential side reactions than ATRP, for example.
As mentioned in section 3 depolymerization via ionic mechanisms, as opposed to radical unzipping, could be a potential avenue to achieving regeneration of monomer at room temperature for a range of polymers. This would not only be cost-efficient but would also prevent the thermally promoted side reactions during depolymerization, increasing the yields of monomer generation. For example, group transfer polymerization,107 an industrially relevant technique to polymerize methacrylates, operates under an ionic mechanism that could be explored for new depolymerization reactions. It does not require a metal catalyst and the polymer chains are terminated with easily functionalized acetals, which could be advantageous for potential depolymerization trigger points. Other polymerization methodologies, such as classical or frustrated Lewis pair polymerization, may also have great potential in depolymerization reactions.108 Their zwitterionic nature and unique kinetics, alongside the versatility of polymerization conditions, may allow the development of new and powerful depolymerization approaches.109
5. Summary and Conclusions
Reversed controlled polymerization is an emerging field which is enabling depolymerization under conditions which are incredibly challenging to implement for polymers made by more conventional means. The pioneering reports of depolymerization for ATRP, RAFT, and ITP-synthesized polymers rely on radical generation at the chain-end preceding depropagation reactions promoted by thermodynamically favorable dilution and elevated temperatures. The results of this early work are very promising, yet it can be envisaged that many advances will be made over the coming years as the field matures. Increasing the effective polymer concentration to make depolymerization a more effective and viable approach to polymer recycling will certainly be one of the first steps in this process. Polymerization techniques such as NMP and OMRP have still not been investigated for depolymerization potential. The development of catalysts for enhanced depolymerization can also be expected to progress, potentially allowing broader polymer scope and lower temperature systems.
As the field develops further, it will be particularly interesting to see how the techniques, catalysts, and fundamental mechanistic understanding of reversed controlled polymerization could be potentially applied to the large volume of plastics produced each year with limited end-of-life possibilities.
Acknowledgments
A.A. gratefully acknowledges ETH Zurich for financial support. N.P.T. acknowledges the award of a DECRA Fellowship from the ARC (DE180100076). H.S.W. acknowledges the award of the Swiss Government Excellence Scholarship (ESKAS No. 2020.0324). K.P. thanks The Onasis Foundation as this scientific paper was partially supported by the Onassis Foundation - Scholarship ID: FZQ051-1/2020-2021. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (DEPO: Grant No. 949219).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c00589.
The authors declare no competing financial interest.
Supplementary Material
References
- Miller S. A. Sustainable Polymers: Opportunities for the Next Decade. ACS Macro Lett. 2013, 2, 550–554. 10.1021/mz400207g. [DOI] [PubMed] [Google Scholar]
- Vogt B. D.; Stokes K. K.; Kumar S. K. Why is recycling of postconsumer plastics so challenging?. ACS Appl. Polym. Mater. 2021, 3, 4325–4346. 10.1021/acsapm.1c00648. [DOI] [Google Scholar]
- Adili A.; Korpusik A. B.; Seidel D.; Sumerlin B. S. Photocatalytic Direct Decarboxylation of Carboxylic Acids to Derivatize or Degrade Polymers. Angew. Chem., Int. Ed. 2022, 61, e202209085 10.1002/anie.202209085. [DOI] [PubMed] [Google Scholar]
- Garrison J. B.; Hughes R. W.; Sumerlin B. S. Backbone Degradation of Polymethacrylates via Metal-Free Ambient-Temperature Photoinduced Single-Electron Transfer. ACS Macro Lett. 2022, 11, 441–446. 10.1021/acsmacrolett.2c00091. [DOI] [PubMed] [Google Scholar]
- Korley L. T. J.; Epps T. H.; Helms B. A.; Ryan A. J. Toward polymer upcycling—adding value and tackling circularity. Science 2021, 373, 66–69. 10.1126/science.abg4503. [DOI] [PubMed] [Google Scholar]
- Jehanno C.; Alty J. W.; Roosen M.; De Meester S.; Dove A. P.; Chen E. Y.-X.; Leibfarth F. A.; Sardon H. Critical advances and future opportunities in upcycling commodity polymers. Nature 2022, 603, 803–814. 10.1038/s41586-021-04350-0. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Romain C.; Williams C. K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. 10.1038/nature21001. [DOI] [PubMed] [Google Scholar]
- Rosenboom J.-G.; Langer R.; Traverso G. Bioplastics for a circular economy. Nat. Rev. Mater. 2022, 7, 117–137. 10.1038/s41578-021-00407-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates G. W.; Getzler Y. D. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 2020, 5, 501–516. 10.1038/s41578-020-0190-4. [DOI] [Google Scholar]
- Hong M.; Chen E. Y.-X. Chemically recyclable polymers: a circular economy approach to sustainability. Green Chem. 2017, 19, 3692–3706. 10.1039/C7GC01496A. [DOI] [Google Scholar]
- IUPAC . Top Ten Emerging Technologies in Chemistry. https://iupac.org/what-we-do/top-ten/ (accessed 10/12/2022). [Google Scholar]
- Staudinger H.On Polymerization. A Source Book in Chemistry, 1900–1950; Harvard University Press: 2013; pp 259–264. [Google Scholar]
- Thiyagarajan S.; Maaskant-Reilink E.; Ewing T. A.; Julsing M. K.; Van Haveren J. Back-to-monomer recycling of polycondensation polymers: opportunities for chemicals and enzymes. RSC Adv. 2021, 12, 947–970. 10.1039/D1RA08217E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ügdüler S.; Van Geem K. M.; Denolf R.; Roosen M.; Mys N.; Ragaert K.; De Meester S. Towards closed-loop recycling of multilayer and coloured PET plastic waste by alkaline hydrolysis. Green Chem. 2020, 22, 5376–5394. 10.1039/D0GC00894J. [DOI] [Google Scholar]
- Geyer R.; Jambeck J. R.; Law K. L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782 10.1126/sciadv.1700782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkatzidis K.; Wang H. S.; Truong N. P.; Anastasaki A. Recent developments and future challenges in controlled radical polymerization: a 2020 update. Chem. 2020, 6, 1575–1588. 10.1016/j.chempr.2020.06.014. [DOI] [Google Scholar]
- Grubbs R. B.; Grubbs R. H. 50th Anniversary Perspective: Living Polymerization: Emphasizing the Molecule in Macromolecules. Macromolecules 2017, 50, 6979–6997. 10.1021/acs.macromol.7b01440. [DOI] [Google Scholar]
- Corrigan N.; Jung K.; Moad G.; Hawker C. J.; Matyjaszewski K.; Boyer C. Reversible-deactivation radical polymerization (Controlled/living radical polymerization): From discovery to materials design and applications. Prog. Polym. Sci. 2020, 111, 101311. 10.1016/j.progpolymsci.2020.101311. [DOI] [Google Scholar]
- Destarac M. Controlled radical polymerization: industrial stakes, obstacles and achievements. Macromol. React. Eng. 2010, 4, 165–179. 10.1002/mren.200900087. [DOI] [Google Scholar]
- Destarac M. Industrial development of reversible-deactivation radical polymerization: is the induction period over?. Polym. Chem. 2018, 9, 4947–4967. 10.1039/C8PY00970H. [DOI] [Google Scholar]
- Martinez M. R.; Matyjaszewski K. Degradable and Recyclable Polymers by Reversible Deactivation Radical Polymerization. CCS Chem. 2022, 4, 2176. 10.31635/ccschem.022.202201987. [DOI] [Google Scholar]
- Ivin K. Thermodynamics of addition polymerization processes. Angew. Chem., Int. Ed. 1973, 12, 487–494. 10.1002/anie.197304871. [DOI] [Google Scholar]
- Dainton F.; Ivin K. Reversibility of the propagation reaction in polymerization processes and its manifestation in the phenomenon of a ‘Ceiling Temperature. Nature 1948, 162, 705–707. 10.1038/162705a0. [DOI] [Google Scholar]
- Odian G.Principles of polymerization; John Wiley & Sons: 2004; Chapter 3–9, pp 279–281. [Google Scholar]
- Young R. J.; Lovell P. A.. Introduction to polymers; CRC Press: 2011; Chapter 4, pp 89–90. [Google Scholar]
- Snow R.; Frey F. The reaction of sulfur dioxide with olefins: the ceiling temperature phenomenon. J. Am. Chem. Soc. 1943, 65, 2417–2418. 10.1021/ja01252a052. [DOI] [Google Scholar]
- Roberts D. E. Heats of polymerization. A summary of published values and their relation to structure. J. Res. Natl. Bur. Stand 1950, 44, 221–232. 10.6028/jres.044.021. [DOI] [Google Scholar]
- Yardley R. E.; Kenaree A. R.; Gillies E. R. Triggering depolymerization: progress and opportunities for self-immolative polymers. Macromolecules 2019, 52, 6342–6360. 10.1021/acs.macromol.9b00965. [DOI] [Google Scholar]
- Martinez M. R.; Dadashi-Silab S.; Lorandi F.; Zhao Y.; Matyjaszewski K. Depolymerization of P(PDMS11MA) Bottlebrushes via Atom Transfer Radical Polymerization with Activator Regeneration. Macromolecules 2021, 54, 5526–5538. 10.1021/acs.macromol.1c00415. [DOI] [Google Scholar]
- Chiantore O.; Gualta M. Chain end initiated depolymerization in anionic poly(methylmethacrylate)s. Polym. Bull. 1988, 20, 201–206. 10.1007/BF00256116. [DOI] [Google Scholar]
- Manring L. E. Thermal degradation of saturated poly (methyl methacrylate). Macromolecules 1988, 21, 528–530. 10.1021/ma00180a046. [DOI] [Google Scholar]
- Manring L. E. Thermal degradation of poly (methyl methacrylate). 4. Random side-group scission. Macromolecules 1991, 24, 3304–3309. 10.1021/ma00011a040. [DOI] [Google Scholar]
- Manring L. E.; Sogah D. Y.; Cohen G. M. Thermal degradation of poly (methyl methacrylate). 3. Polymer with head-to-head linkages. Macromolecules 1989, 22, 4652–4654. 10.1021/ma00202a048. [DOI] [Google Scholar]
- Manring L. E. Thermal degradation of poly (methyl methacrylate). 2. Vinyl-terminated polymer. Macromolecules 1989, 22, 2673–2677. 10.1021/ma00196a024. [DOI] [Google Scholar]
- Tobolsky A.; Eisenberg A. Transition phenomena in equilibrium polymerization. J. Colloid Sci. 1962, 17, 49–65. 10.1016/0095-8522(62)90075-2. [DOI] [Google Scholar]
- Tobolsky A.; Eisenberg A. A general treatment of equilibrium polymerization. J. Am. Chem. Soc. 1960, 82, 289–293. 10.1021/ja01487a009. [DOI] [Google Scholar]
- Vrancken A.; Smid J.; Szwarc M. Equilibria between low-molecular-weight poly-α-methyl styrenes and their monomer. Trans. Faraday Soc. 1962, 58, 2036–2048. 10.1039/tf9625802036. [DOI] [Google Scholar]
- Penczek S.; Kaluzynski K.. Thermodynamic and Kinetic Polymerizability. Polymer Science: A Comprehensive Reference; Matyjaszewski K., Möller M., Eds.; Elsevier Science: 2012; Vol. 4, Chapter 2, pp 5–20. [Google Scholar]
- Mark J.Polymer Data Handbook; Polymer Data Handbook; Oxford University Press: 1999; p 829. [Google Scholar]
- Otsu T.; Yamada B.; Sugiyama S.; Mori S. Effects of ortho-substituents on reactivities, tacticities, and ceiling temperatures of radical polymerizations of phenyl methacrylates. J. Polym. Sci.: Polym. Chem. Ed. 1980, 18, 2197–2207. 10.1002/pol.1980.170180715. [DOI] [Google Scholar]
- Szablan Z.; Stenzel M. H.; Davis T. P.; Barner L.; Barner-Kowollik C. Depropagation kinetics of sterically demanding monomers: A pulsed laser size exclusion chromatography study. Macromolecules 2005, 38, 5944–5954. 10.1021/ma050444l. [DOI] [Google Scholar]
- Kennemur J. G.; Bates F. S.; Hillmyer M. A. Revisiting the Anionic Polymerization of Methyl Ethacrylate. Macromol. Chem. Phys. 2018, 219, 1700282. 10.1002/macp.201700282. [DOI] [Google Scholar]
- Tanaka H.; Hall H. K. Jr. Thermal degradation of polymers of captodative substituted olefins. Macromol. Chem. Phys. 1994, 195, 2073–2081. 10.1002/macp.1994.021950616. [DOI] [Google Scholar]
- Dau H.; Jones G. R.; Tsogtgerel E.; Nguyen D.; Keyes A.; Liu Y.-S.; Rauf H.; Ordonez E.; Puchelle V.; Basbug Alhan H. Linear block copolymer synthesis. Chem. Rev. 2022, 122, 14471–14553. 10.1021/acs.chemrev.2c00189. [DOI] [PubMed] [Google Scholar]
- Antonopoulou M.-N.; Whitfield R.; Truong N. P.; Wyers D.; Harrisson S.; Junkers T.; Anastasaki A. Concurrent control over sequence and dispersity in multiblock copolymers. Nat. Chem. 2022, 14, 304–312. 10.1038/s41557-021-00818-8. [DOI] [PubMed] [Google Scholar]
- Wang H. S.; Truong N. P.; Pei Z.; Coote M. L.; Anastasaki A. Reversing RAFT Polymerization: Near-Quantitative Monomer Generation Via a Catalyst-Free Depolymerization Approach. J. Am. Chem. Soc. 2022, 144, 4678–4684. 10.1021/jacs.2c00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-S.; Matyjaszewski K. Controlled/“living” radical polymerization. atom transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. 10.1021/ja00125a035. [DOI] [Google Scholar]
- Kato M.; Kamigaito M.; Sawamoto M.; Higashimura T. Polymerization of methyl methacrylate with the carbon tetrachloride/dichlorotris-(triphenylphosphine) ruthenium (II)/methylaluminum bis (2, 6-di-tert-butylphenoxide) initiating system: possibility of living radical polymerization. Macromolecules 1995, 28, 1721–1723. 10.1021/ma00109a056. [DOI] [Google Scholar]
- Truong N. P.; Jones G. R.; Bradford K. G.; Konkolewicz D.; Anastasaki A. A comparison of RAFT and ATRP methods for controlled radical polymerization. Nat. Rev. Chem. 2021, 5, 859–869. 10.1038/s41570-021-00328-8. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K.; Xia J. Atom transfer radical polymerization. Chem. Rev. 2001, 101, 2921–2990. 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- Lorandi F.; Fantin M.; Matyjaszewski K. Atom Transfer Radical Polymerization: A Mechanistic Perspective. J. Am. Chem. Soc. 2022, 144, 15413–15430. 10.1021/jacs.2c05364. [DOI] [PubMed] [Google Scholar]
- Raus V.; Čadová E.; Starovoytova L.; Janata M. ATRP of POSS monomers revisited: Toward high-molecular weight methacrylate–POSS (co) polymers. Macromolecules 2014, 47, 7311–7320. 10.1021/ma501541g. [DOI] [Google Scholar]
- Sano Y.; Konishi T.; Sawamoto M.; Ouchi M. Controlled radical depolymerization of chlorine-capped PMMA via reversible activation of the terminal group by ruthenium catalyst. Eur. Polym. J. 2019, 120, 109181. 10.1016/j.eurpolymj.2019.08.008. [DOI] [Google Scholar]
- Martinez M. R.; De Luca Bossa F.; Olszewski M.; Matyjaszewski K. Copper (II) Chloride/Tris (2-pyridylmethyl) amine-Catalyzed Depolymerization of Poly (n-butyl methacrylate). Macromolecules 2022, 55, 78–87. 10.1021/acs.macromol.1c02246. [DOI] [Google Scholar]
- Martinez M. R.; Schild D.; De Luca Bossa F.; Matyjaszewski K. Depolymerization of Polymethacrylates by Iron ATRP. Macromolecules 2022, 55, 10590–10599. 10.1021/acs.macromol.2c01712. [DOI] [Google Scholar]
- Chiefari J.; Chong Y. K.; Ercole F.; Krstina J.; Jeffery J.; Le T. P. T.; Mayadunne R. T. A.; Meijs G. F.; Moad C. L.; Moad G.; et al. Living Free-Radical Polymerization by Reversible Addition–Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. 10.1021/ma9804951. [DOI] [Google Scholar]
- Flanders M. J.; Gramlich W. M. Reversible-addition fragmentation chain transfer (RAFT) mediated depolymerization of brush polymers. Polym. Chem. 2018, 9, 2328–2335. 10.1039/C8PY00446C. [DOI] [Google Scholar]
- Wang H. S.; Truong N. P.; Jones G. R.; Anastasaki A. Investigating the Effect of End-Group, Molecular Weight, and Solvents on the Catalyst-Free Depolymerization of RAFT Polymers: Possibility to Reverse the Polymerization of Heat-Sensitive Polymers. ACS Macro Lett. 2022, 11, 1212–1216. 10.1021/acsmacrolett.2c00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywater S. Evaluation of heats and entropies of polymerization from measurements of equilibrium monomer concentration in solution. Macromol. Chem. Phys. 1962, 52, 120–124. 10.1002/macp.1962.020520110. [DOI] [Google Scholar]
- Leonard J. Thermodynamics of equilibrium polymerization in solution. Effect of polymer concentration on the equilibrium monomer concentration. Macromolecules 1969, 2, 661–666. 10.1021/ma60012a019. [DOI] [Google Scholar]
- Wang W.; Hutchinson R. A.; Grady M. C. Study of butyl methacrylate depropagation behavior using batch experiments in combination with modeling. Ind. Eng. Chem. Res. 2009, 48, 4810–4816. 10.1021/ie900060x. [DOI] [Google Scholar]
- Martinez M. R.; Cong Y.; Sheiko S. S.; Matyjaszewski K. A Thermodynamic Roadmap for the Grafting-through Polymerization of PDMS11MA. ACS Macro Lett. 2020, 9, 1303–1309. 10.1021/acsmacrolett.0c00350. [DOI] [PubMed] [Google Scholar]
- Martinez M. R.; Krys P.; Sheiko S. S.; Matyjaszewski K. Poor solvents improve yield of grafting-through radical polymerization of OEO19MA. ACS Macro Lett. 2020, 9, 674–679. 10.1021/acsmacrolett.0c00245. [DOI] [PubMed] [Google Scholar]
- Young J. B.; Bowman J. I.; Eades C. B.; Wong A. J.; Sumerlin B. S. Photoassisted Radical Depolymerization. ACS Macro Lett. 2022, 11, 1390–1395. 10.1021/acsmacrolett.2c00603. [DOI] [PubMed] [Google Scholar]
- Hartlieb M. Photo-Iniferter RAFT Polymerization. Macromol. Rapid Commun. 2022, 43, 2100514. 10.1002/marc.202100514. [DOI] [PubMed] [Google Scholar]
- Bellotti V.; Parkatzidis K.; Wang H. S.; Watuthanthrige N. D. A.; Orfano M.; Monguzzi A.; Truong N. P.; Simonutti R.; Anastasaki A. Light-Accelerated Depolymerization Catalyzed by Eosin Y. Polym. Chem. 2023, 14, 253–258. 10.1039/D2PY01383E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y.; Zhang L.; Cheng Z.; Zhu X. Iodine-mediated reversible-deactivation radical polymerization: A powerful strategy for polymer synthesis. Polym. Chem. 2019, 10, 2504–2515. 10.1039/C9PY00091G. [DOI] [Google Scholar]
- Huang S.; Su X.; Wu Y.; Xiong X.-G.; Liu Y. Promoting Halogen-Bonding Catalyzed Living Radical Polymerization through Ion-Pair Strain. Chem. Sci. 2022, 13, 11352–11359. 10.1039/D2SC04196K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szwarc M. ‘Living’polymers. Nature 1956, 178, 1168–1169. 10.1038/1781168a0. [DOI] [Google Scholar]
- Ryan B.; McCann G. Novel sub-ceiling temperature rapid depolymerization-repolymerization reactions of cyanoacrylate polymers. Macromol. Rapid Commun. 1996, 17, 217–227. 10.1002/marc.1996.030170404. [DOI] [Google Scholar]
- Han M. G.; Kim S.; Liu S. X. Synthesis and degradation behavior of poly (ethyl cyanoacrylate). Polym. Degrad. Stab. 2008, 93, 1243–1251. 10.1016/j.polymdegradstab.2008.04.012. [DOI] [Google Scholar]
- Aoshima S.; Kanaoka S. A Renaissance in Living Cationic Polymerization. Chem. Rev. 2009, 109, 5245–5287. 10.1021/cr900225g. [DOI] [PubMed] [Google Scholar]
- Deák G.; Pernecker T.; Kennedy J. P. Carbocationic polymerization in suprecritical CO 2: III. The ceiling temperature of and the effect of temperature on the polymerization of isobutylene. Polym. Bull. 1994, 33, 259–265. 10.1007/BF00314261. [DOI] [Google Scholar]
- Kunal K.; Paluch M.; Roland C.; Puskas J.; Chen Y.; Sokolov A. Polyisobutylene: A most unusual polymer. J. Polym. Sci., Part B: Polym. Phys. 2008, 46, 1390–1399. 10.1002/polb.21473. [DOI] [Google Scholar]
- Berlin A. A.; Minsker K.; Sangalov Y. A.; Prochukhan Y. A. Kinetic features of the cationic degradation of polyisobutylene. Polym. Sci. U.S.S.R. 1983, 25, 1684–1694. 10.1016/0032-3950(83)90281-2. [DOI] [Google Scholar]
- Watson C. B.; Tan D.; Bergbreiter D. E. Enthalpy-Driven Polyisobutylene Depolymerization. Macromolecules 2019, 52, 3042–3048. 10.1021/acs.macromol.9b00313. [DOI] [Google Scholar]
- Jung E.; Yim D.; Kim H.; Peterson G. I.; Choi T. L. Depolymerization of poly (α-methyl styrene) with ball-mill grinding. J. Polym. Sci. 2023, 61, 553–560. 10.1002/pol.20220578. [DOI] [Google Scholar]
- Badamshina E.; Timofeyeva G.; Korshak Y. V.; Berlin A. A.; Vdovin V.; Kutepov D.; Pavlova S.-S. Investigation of the mechanism of polypentenamer degradation in the presence of metathesis catalysts. Polym. Sci. U.S.S.R. 1982, 24, 164–170. 10.1016/0032-3950(82)90092-2. [DOI] [Google Scholar]
- Neary W. J.; Isais T. A.; Kennemur J. G. Depolymerization of bottlebrush polypentenamers and their macromolecular metamorphosis. J. Am. Chem. Soc. 2019, 141, 14220–14229. 10.1021/jacs.9b05560. [DOI] [PubMed] [Google Scholar]
- Sathe D.; Zhou J.; Chen H.; Su H.-W.; Xie W.; Hsu T.-G.; Schrage B. R.; Smith T.; Ziegler C. J.; Wang J. Olefin metathesis-based chemically recyclable polymers enabled by fused-ring monomers. Nat. Chem. 2021, 13, 743–750. 10.1038/s41557-021-00748-5. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; He J.; Li C.; Hong L.; Yang Y. Dependence of thermal stability on molecular structure of RAFT/MADIX agents: A kinetic and mechanistic study. Macromolecules 2011, 44, 8446–8457. 10.1021/ma201570f. [DOI] [Google Scholar]
- Leibfarth F. A.; Mattson K. M.; Fors B. P.; Collins H. A.; Hawker C. J. External regulation of controlled polymerizations. Angew. Chem., Int. Ed. 2013, 52, 199–210. 10.1002/anie.201206476. [DOI] [PubMed] [Google Scholar]
- Zaquen N.; Rubens M.; Corrigan N.; Xu J.; Zetterlund P. B.; Boyer C.; Junkers T. Polymer synthesis in continuous flow reactors. Prog. Polym. Sci. 2020, 107, 101256. 10.1016/j.progpolymsci.2020.101256. [DOI] [Google Scholar]
- Arslan Z.; Kiliclar H. C.; Yagci Y. Dimanganese decacarbonyl catalyzed visible light induced ambient temperature depolymerization of poly (methyl methacrylate). Des. Monomers Polym. 2022, 25, 271–276. 10.1080/15685551.2022.2135730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsigny L.; Berthet J.-C.; Cantat T. Depolymerization of waste plastics to monomers and chemicals using a hydrosilylation strategy facilitated by Brookhart’s iridium (III) catalyst. ACS Sustainable Chem. Eng. 2018, 6, 10481–10488. 10.1021/acssuschemeng.8b01842. [DOI] [Google Scholar]
- McGuire T. M.; Deacy A. C.; Buchard A.; Williams C. K. Solid-State Chemical Recycling of Polycarbonates to Epoxides and Carbon Dioxide Using a Heterodinuclear Mg (II) Co (II) Catalyst. J. Am. Chem. Soc. 2022, 144, 18444–18449. 10.1021/jacs.2c06937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer F. N.; Deacy A. C.; McGuire T. M.; Williams C. K.; Buchard A. Chemical Recycling of Poly (Cyclohexene Carbonate) Using a Di-MgII Catalyst. Angew. Chem. 2022, 61, e202201785 10.1002/anie.202201785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M.; Chen E. Y.-X. Towards Truly Sustainable Polymers: A Metal-Free Recyclable Polyester from Biorenewable Non-Strained γ-Butyrolactone. Angew. Chem., Int. Ed. 2016, 55, 4188–4193. 10.1002/anie.201601092. [DOI] [PubMed] [Google Scholar]
- Zhu J.-B.; Watson E. M.; Tang J.; Chen E. Y.-X. A synthetic polymer system with repeatable chemical recyclability. Science 2018, 360, 398–403. 10.1126/science.aar5498. [DOI] [PubMed] [Google Scholar]
- Phommalysack-Lovan J.; Chu Y.; Boyer C.; Xu J. PET-RAFT polymerisation: towards green and precision polymer manufacturing. Chem. Commun. 2018, 54, 6591–6606. 10.1039/C8CC02783H. [DOI] [PubMed] [Google Scholar]
- Vandenbergh J.; Junkers T. Synthesis of Macromonomers from High-Temperature Activation of Nitroxide Mediated Polymerization (NMP)-made Polyacrylates. Macromolecules 2013, 46, 3324–3331. 10.1021/ma400477t. [DOI] [Google Scholar]
- Kitahara Y.; Okuyama K.; Ozawa K.; Suga T.; Takahashi S.; Fujii T. Thermal decomposition of acrylamide from polyacrylamide: time-resolved pyrolysis with ion-attachment mass spectrometry. J. Therm. Anal. Calorim. 2012, 110, 423–429. 10.1007/s10973-012-2544-7. [DOI] [Google Scholar]
- Maafa I. M. Pyrolysis of polystyrene waste: A review. Polymers 2021, 13, 225. 10.3390/polym13020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Pan F.; Sun M.; Xu J.; Munyaneza N. E.; Croft Z. L.; Cai G.; Liu G. Cascade degradation and upcycling of polystyrene waste to high-value chemicals. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2203346119 10.1073/pnas.2203346119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Khan A.; Rabnawaz M. Efficient Depolymerization of Polystyrene with Table Salt and Oxidized Copper. ACS Sustainable Chem. Eng. 2022, 10, 6493–6502. 10.1021/acssuschemeng.1c08400. [DOI] [Google Scholar]
- Oh S.; Stache E. E. Chemical Upcycling of Commercial Polystyrene via Catalyst-Controlled Photooxidation. J. Am. Chem. Soc. 2022, 144, 5745–5749. 10.1021/jacs.2c01411. [DOI] [PubMed] [Google Scholar]
- Yeung C. W.; Teo J. Y.; Loh X. J.; Lim J. Y. Polyolefins and polystyrene as chemical resources for a sustainable future: challenges, advances, and prospects. ACS Mater. Lett. 2021, 3, 1660–1676. 10.1021/acsmaterialslett.1c00490. [DOI] [Google Scholar]
- Balema V. P.; Hlova I. Z.; Carnahan S. L.; Seyedi M.; Dolotko O.; Rossini A. J.; Luzinov I. Depolymerization of polystyrene under ambient conditions. New J. Chem. 2021, 45, 2935–2938. 10.1039/D0NJ05984F. [DOI] [Google Scholar]
- Monroy-Alonso A.; Ordaz-Quintero A.; Ramirez J. C.; Saldívar-Guerra E. Thermal Pyrolysis of Polystyrene Aided by a Nitroxide End-Functionality Improved Process and Modeling of the Full Molecular Weight Distribution. Polymers 2022, 14, 160. 10.3390/polym14010160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordaz-Quintero A.; Monroy-Alonso A.; Saldívar-Guerra E. Thermal Pyrolysis of Polystyrene Aided by a Nitroxide End-Functionality. Experiments and Modeling. Processes 2020, 8, 432. 10.3390/pr8040432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höcker H. Thermodynamic recycling—On ring-opening polymerization and ring-closing depolymerization. J. Macromol. Sci., Part A: Pure Appl. Chem. 1993, 30, 595–601. 10.1080/10601329308021248. [DOI] [Google Scholar]
- Jia X.; Qin C.; Friedberger T.; Guan Z.; Huang Z. Efficient and selective degradation of polyethylenes into liquid fuels and waxes under mild conditions. Sci. Adv. 2016, 2, e1501591 10.1126/sciadv.1501591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y.; Lin Y.; Jiang H.; Li B.; Wu C.; Liu J.; Liu J.; Wang Y.; Cui D.; Tang T. Mechanism for Coordination Quasi-Depolymerization of Polydiolefins with Cp2ZrHCl. Macromolecules 2022, 55, 7732–7739. 10.1021/acs.macromol.2c01000. [DOI] [Google Scholar]
- Harth E.; Bosman A.; Hawker C. New polymer synthesis by nitroxide mediated living radical polymerization. Chem. Rev. 2001, 101, 3661–3688. 10.1021/cr990119u. [DOI] [PubMed] [Google Scholar]
- Ballard N.; Aguirre M.; Simula A.; Agirre A.; Leiza J. R.; Asua J. M.; van Es S. New class of alkoxyamines for efficient controlled homopolymerization of methacrylates. ACS Macro Lett. 2016, 5, 1019–1022. 10.1021/acsmacrolett.6b00547. [DOI] [PubMed] [Google Scholar]
- Guillaneuf Y.; Gigmes D.; Marque S. R.; Tordo P.; Bertin D. Nitroxide-mediated polymerization of methyl methacrylate using an SG1-based alkoxyamine: how the penultimate effect could lead to uncontrolled and unliving polymerization. Macromol. Chem. Phys. 2006, 207, 1278–1288. 10.1002/macp.200600125. [DOI] [Google Scholar]
- Webster O. W. Group transfer polymerization: a critical review of its mechanism and comparison with other methods for controlled polymerization of acrylic monomers. New Synth. Methods 2003, 167, 1–34. 10.1007/b12303. [DOI] [Google Scholar]
- McGraw M. L.; Chen E. Y.-X. Lewis pair polymerization: perspective on a ten-year journey. Macromolecules 2020, 53, 6102–6122. 10.1021/acs.macromol.0c01156. [DOI] [Google Scholar]
- Chen E. Y.-X. Polymerization by classical and frustrated Lewis pairs. Frustrated Lewis Pairs II 2012, 334, 239–260. 10.1007/128_2012_372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.