

Abstract
The mechanisms by which innate immune receptors mediate self-nonself discrimination are unclear. In this study, we discovered species-specific molecular determinants of self-DNA reactivity by GMP-AMP synthase (cGAS). Human cGAS contained a catalytic domain that was intrinsically self-DNA reactive and stimulated interferon responses in diverse cell types. This reactivity was prevented by an upstream N-terminal domain. The cGAS proteins from several nonhuman primate species exhibited a similar pattern of self-DNA reactivity in cells but chimpanzee cGAS was inactive even when its N-terminal domain was deleted. In contrast, the N-terminus of mouse cGAS promoted self-DNA reactivity. When expressed within tumors, only self-DNA reactive cGAS proteins protected mice from tumor-induced lethality. In vitro studies of DNA- or chromatin-induced cGAS activation did not reveal species-specific activities that correlate with self-DNA reactivity observed in macrophages. Cell biological analysis revealed that human, but not mouse, self-DNA reactivity correlated with localization to mitochondria. We found that epitope tag positions impacted self-DNA reactivity in cells and that DNA present in cell lysates undermines the reliability of cGAS biochemical fractionations. These studies reveal species-specific diversity of cGAS functions, even within the primate lineage, and highlight experimental considerations for the study of this innate immune receptor.
One Sentence Summary:
The regulation of self-DNA reactivity of cGAS is evolutionarily diverse in mammals.
INTRODUCTION
Mammalian cells are equipped with pattern recognition receptors (PRRs) that detect molecular evidence of infection or tissue injury. Among these PRRs is the enzyme cyclic GMP-AMP synthase (cGAS), which synthesizes the second messenger 2′3′-cyclic GMP-AMP (cGAMP) upon binding to double-stranded DNA (dsDNA) in the cytosol (1, 2). As healthy and non-infected cells should not contain cytosolic DNA, the detection of DNA by cGAS is an indicator of infection or cellular dysfunction. cGAS stimulates immune responses via the actions of the downstream cGAMP receptor STING. Upon binding cGAMP, STING traffics from the endoplasmic reticulum (ER) to the Golgi apparatus and oligomerizes into a scaffold that activates the kinase TANK-binding kinase 1 (TBK1) and the transcription factor IFN regulatory factor 3 (IRF3) (3). IRF3 then coordinates the expression of type I interferons (IFNs) and IFN-stimulated genes (ISGs) that promote inflammation and host defense. Diverse sources of DNA can activate cGAS, including those from infectious agents and host-derived nuclear and mitochondrial DNA (mtDNA) that have reached the cytosol (4–6). With the exception of differences in the length of DNA detected (7–9), it is thought that the studies of human cGAS reflect the function of this protein in other mammalian species. However, the symmetry of cGAS functions in nature have largely been explored in vitro, where cGAS access to DNA is not an experimental variable. Based on the common in vitro behaviors of cGAS, it is somewhat surprising that disparate findings have been made regarding the activities of cGAS within cells, even when studying human cGAS exclusively (10–13).
Human cGAS is a 522 amino acid protein consisting of an unstructured ~160-amino-acid N-terminal domain, and a ~360-amino-acid C-terminal domain that possesses DNA binding and nucleotidyltransferase (NTase) activities (1). Although the N-terminal domain was initially regarded as dispensable for DNA binding and IFN induction (1), recent studies reported that the N-terminal domain is required for cGAS functions. For example, several reports indicate that within cells, deletion of the N-terminus of cGAS renders this protein inactive and defective for DNA-induced IFN responses (10, 12, 13). However, our group found that the deletion of the N-terminus activates cGAS-dependent IFN responses to self-DNA (11). The reason for these disparate datasets has been unclear and was explored in detail herein.
In this study, we found that the N-terminus of human cGAS prevents the catalytic domain from inducing type I IFN responses against self-DNA in human and mouse cells. Evolutionary analysis of self-DNA reactivity revealed distinct species-specific mechanisms of cGAS self-DNA reactivity, with human, mouse and chimpanzee cGAS proteins (among others) displaying distinct mechanisms of self-DNA responsiveness. Intratumoral expression of self-DNA reactive cGAS transgenes promoted antitumor responses and improved mouse survival after tumor implantation. These findings reveal unexpectedly diverse functions of a single PRR in nature.
RESULTS
Human cGAS∆N induces type I IFN responses to self-DNA in human and mouse cells
The functions of the C-terminal DNA binding and catalytic domain of cGAS within cells are debatable. We have reported that within THP-1 monocytes, a human cGAS mutant lacking its N-terminal domain (hcGAS 160–522 a.a., hereafter hcGAS∆N) promotes type I IFN expression in the absence of exogenous dsDNA treatment (11). Others have concluded that hcGAS∆N is functionally defective within cells (10, 12). Our prior work relied on the stable expression of hcGAS∆N, which drives IFN responses constitutively and may cause aberrant cellular behaviors. To bypass this concern, we devised a distinct genetic system that enables inducible kinetic analysis of cGAS activities. We established a doxycycline (Dox)-inducible expression system for hcGAS full-length (FL) and hcGAS∆N in immortalized murine bone marrow-derived macrophages (iBMDMs). Using this system, we found that Dox-mediated hcGAS∆N expression induced the rapid expression of the gene Ifnb, which encodes IFN-β (Fig. 1A). In contrast, Dox-induced FL hcGAS expression did not trigger Ifnb expression (Fig. 1A). Near-coincident with Ifnb expression was the induced transcription of the ISG Rsad2 (radical SAM domain-containing 2) and its product viperin (virus inhibitory protein, endoplasmic reticulum-associated, interferon-inducible) (Fig. 1, B and C). Interferon-γ-inducible protein 10 (IP-10), another ISG, was also induced by hcGAS∆N (Fig. 1D). The IFN-stimulatory activities of hcGAS∆N occurred despite its lower abundance than FL hcGAS within cells (Fig. 1C).
Fig. 1. Human cGAS∆N induces aberrant type I IFN responses.
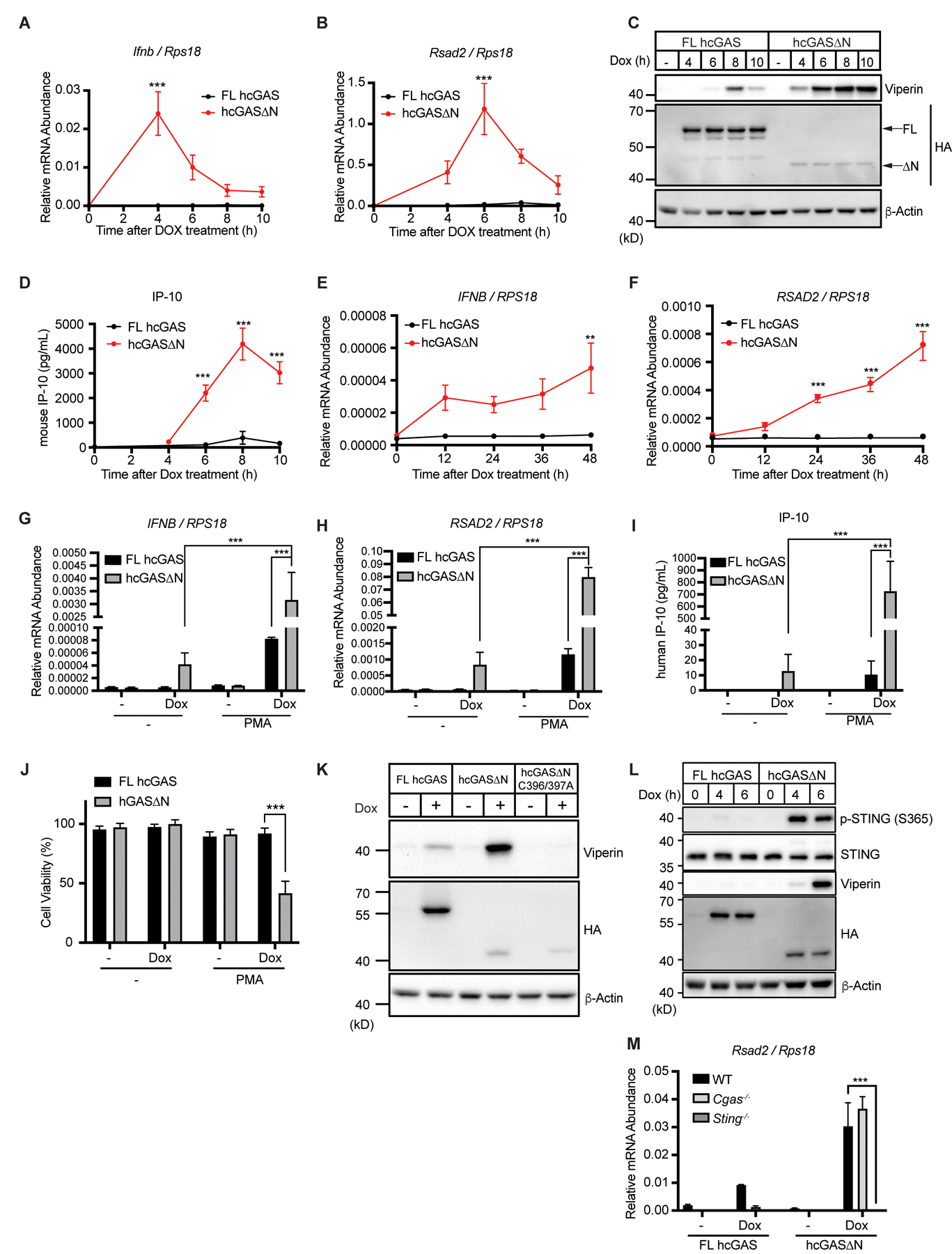
(A and B) Real-time quantitative reverse transcription (qRT-) PCR analysis of Ifnb (A) and Rsad2 (B) mRNAs in iBMDMs. Cells were treated with Dox, and mRNA expression levels were analyzed at the indicated time points. (C) Immunoblot analysis of iBMDM lysates after the same treatment as in (A) and (B). Arrows in the HA panel indicate FL human cGAS and human cGAS∆N. (D) IP-10 ELISA of supernatant of the cells in (A) and (B). (E and F) qRT-PCR analysis of IFNB (E) and RSAD2 (F) mRNAs in THP-1 monocytes treated with Dox for indicated time points. (G and H) Real-Time qRT-PCR analysis of IFNB and RSAD2 mRNAs in THP-1 monocytes treated with Dox together with PMA for 48 hours. (I) IP-10 ELISA analysis of THP-1 supernatant in (G and H). (J) Viability analysis of THP-1 cells treated as in (G) and (H). (K) Immunoblot analysis of lysates from iBMDMs treated with Dox for 8 hours. (L) Immunoblot analysis of STING phosphorylation in lysates from iBMDMs treated with Dox for indicated time points. (M) qRT-PCR analysis of Rsad2 mRNA in WT, Cgas−/−, or Sting−/− iBMDMs treated with Dox for 8 hours. Immunoblot data are the representative from three independent experiments. Graph data are means ± SEM of three (A, B, D, G, H, I, J, and M) or four (E and F) independent experiments. Statistical significance was determined by two-way ANOVA and Tukey’s multiple comparison test. Asterisks indicate the statistical significance between FL hcGAS and hcGAS∆N at each time point (A, B, D, E, and F) or connected two bars (G, H, I, J, and M). *P < 0.05; **P < 0.01; ***P < 0.001.
To validate our results, we applied our Dox-inducible system to human THP-1 monocytes. Dox treatment of THP-1 cells containing the hcGAS∆N transgene induced production of IFNB and RSAD2 mRNAs, whereas cells containing the FL cGAS were poorly immunostimulatory after Dox treatment (Fig. 1E and F). We reported that the treatment of THP-1 cells with phorbol 12-myristate 13-acetate (PMA) potentiated hcGAS∆N induced IFNB mRNA expression and ultimately caused cell death (11). In our Dox system, we observed that PMA enhanced the IFN-inducing ability of hcGAS∆N (Fig. 1, G to I), with IP-10 production by hcGAS∆N increasing over 70-fold after PMA+Dox treatment, as compared to Dox alone (Fig. 1I). This increase in IFN stimulatory activity by PMA correlated with lethality, specifically in cells expressing hcGAS∆N (Fig. 1J). The weak immunostimulatory activities of FL cGAS-expressing cells were marginally affected by PMA treatment (Fig. 1, G to I). We applied the same system to normal human oral keratinocytes. Dox treatment of keratinocytes containing the hcGAS∆N transgene induced the expression of IFNB and RSAD2 mRNAs followed by viperin and IP-10 production (fig. S1, A to D). Overall, these studies in diverse mouse and human cell types indicate that hcGAS∆N induces type I IFN responses upon expression.
To determine if hcGAS∆N requires DNA binding to induce IFN responses, we made alanine substitutions (C396A/C397A) in hcGAS that abolish DNA binding (14). Dox-mediated expression of hcGAS∆N C396A/C397A did not induce viperin expression in iBMDMs (Fig. 1K). These data indicate that the C-terminal catalytic domain of cGAS is intrinsically self-DNA reactive, and that this activity is prevented by its upstream N-terminal domain. Dox-mediated expression of hcGAS∆N led to STING phosphorylation at serine 365 at a time point that preceded viperin production, indicating STING activation (Fig. 1L). Genetic analysis revealed that hcGAS∆N did not induce Rsad2 mRNA or viperin in Sting−/− iBMDMs (Fig. 1M and fig. S1E). In contrast, iBMDMs derived from Cgas−/− mice induced comparable amounts of Rsad2 mRNA and viperin protein (Fig. 1M and fig. S1E). The roles of endogenous cGAS and STING in hcGAS∆N-induced IFN expression were confirmed using knockout (KO) cells generated by CRISPR-Cas9 gene editing (fig. S1, F to H). Altogether, these data indicate that the N-terminal deletion of human cGAS triggers self-DNA-induced type I IFN responses in a STING-dependent manner.
Amino acid determinants at the N-terminus of hcGAS∆N that govern self-DNA reactivity
Whereas our data have confirmed that hcGAS∆N induces DNA-dependent type I IFN responses, other studies have concluded that N-terminal deletion mutants of cGAS do not elicit IFN expression, even in the presence of exogenous dsDNA (10, 12, 13). To understand these discrepancies, we analyzed the reported cGAS constructs and noticed that each study has employed different epitope tag positions on their respective hcGAS∆N constructs. We have used C-terminally tagged hcGAS∆N constructs (11), while others have used N-terminally tagged alleles (10, 12, 13). Therefore, we hypothesized that the different tag positions led to the disparate results regarding hcGAS∆N signaling activities. To test this hypothesis, we engineered iBMDMs to express N-terminally or C-terminally hemagglutinin (HA)-tagged hcGAS∆N, as well as tag-free hcGAS∆N in a Dox-dependent manner. We found that hcGAS∆N-HA induced Ifnb mRNA transcripts and ISG expression, although not as strongly as tag-free hcGAS∆N. In contrast, HA-hcGAS∆N did not induce those responses (Fig. 2, A to D). To validate these results, we compared N-terminal or C-terminal green fluorescence protein (GFP)-tagged constructs and found that hcGAS∆N-GFP but not GFP-hcGAS∆N induced viperin expression (Fig. 2E). Moreover, the addition of an N-terminal FLAG tag to an otherwise active allele (hcGAS∆N-HA) inhibited the type I IFN responses induced by this protein (Fig. 2, F and G). These results therefore explain reported differences in cGAS function, as the position of the epitope tag determines self-DNA reactivity.
Fig. 2. Specific amino acids at the N-terminus of hcGAS ∆N determines self-DNA reactivity.
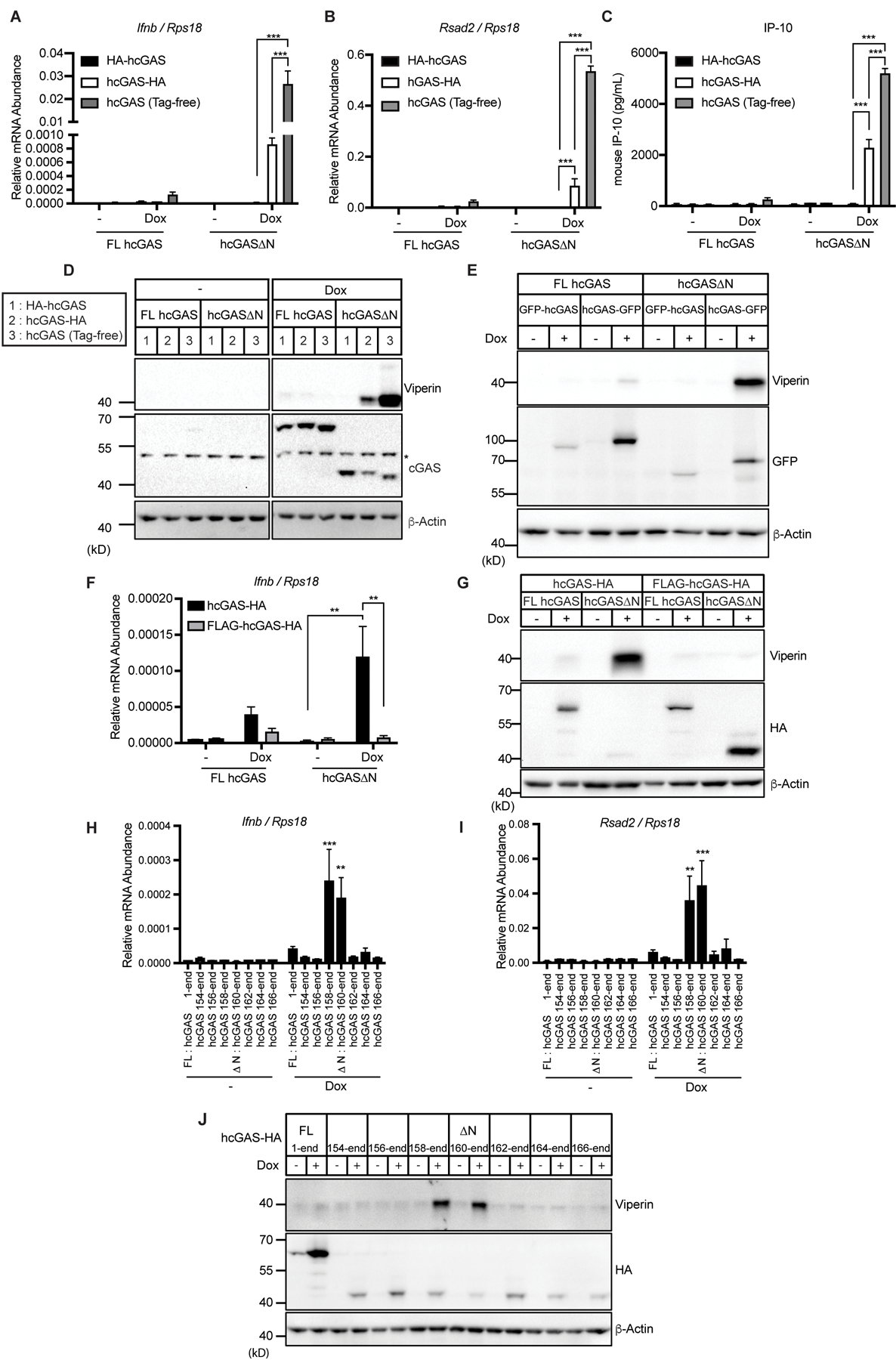
(A and B) qRT-PCR analysis of Ifnb (A) and Rsad2 (B) mRNAs in iBMDMs treated with Dox for 8 hours. (C) IP-10 ELISA analysis of iBMDM cell culture supernatant in (A) and (B). (D) Immunoblot analysis of iBMDMs treated with Dox as in (A) and (B). The asterisk indicates non-specific bands. (E) Immunoblot analysis of iBMDMs treated with Dox for 8 hours. (F) qRT-PCR analysis of Ifnb mRNAs in iBMDMs treated with Dox for 8 hours. (G) Immunoblot analysis of iBMDMs treated with Dox as in (F). (H and I) qRT-PCR analysis of Ifnb (H) and Rsad2 (I) mRNAs in iBMDMs treated with Dox for 8 hours. (J) Immunoblot analysis of lysates of iBMDMs treated as in (H) and (I). Immunoblot data are the representative from three independent experiments. Graph data are means ± SEM of three (A, B, and F) or five (C, H, and I) independent experiments. Statistical significance was determined by two-way ANOVA and Tukey’s multiple comparison test. Asterisks indicate the statistical significance between connected two bars (A, B, C, and F) or between untreated and Dox-treated conditions for indicated hcGAS mutants (H and I). *P < 0.05; **P < 0.01; ***P < 0.001.
To further understand the effect of N-terminal tags on hcGAS∆N activity, we generated differently truncated hcGAS-HA constructs and expressed each in a Dox-inducible manner in iBMDMs. Among all the hcGAS constructs tested, only hcGAS∆N (160–522 a.a.) and hcGAS 158–522 a.a. induced Ifnb and Rsad2 mRNAs, and viperin protein production (Fig. 2, H to J). Addition of 4 or more amino acids to the N-terminus of hcGAS∆N abolished all signaling activities of this protein.
Species-specific self-DNA reactivity by mammalian cGAS proteins
To assess whether cGAS∆N signaling activity is conserved in mice, we expressed mouse cGAS (mcGAS) FL and ∆N in iBMDMs. Whereas some studies have used mcGAS (148–507 a.a.) as the ∆N mutant of mcGAS, other groups have used differently truncated versions (1, 13). Since mcGAS 145–507 a.a. is more similar to hcGAS∆N, in terms of the N-terminal primary sequence (Fig. 3A), we tested both versions of mcGAS truncation mutants. Interestingly, both mcGAS∆N versions were unresponsive to self-DNA upon Dox-mediated expression in iBMDMs, leading to no IFN activities upon Dox-induction (Fig. 3, B to D). FL mcGAS, in contrast, induced type I IFN responses upon expression via Dox (Fig. 3, B to D). Thus, in contrast to our findings with hcGAS and its ∆N counterpart, mouse FL cGAS is self-DNA-reactive whereas both versions of mouse cGAS∆N are weakly immunostimulatory.
Fig. 3. cGAS∆N activities are diverse in mammalian species.
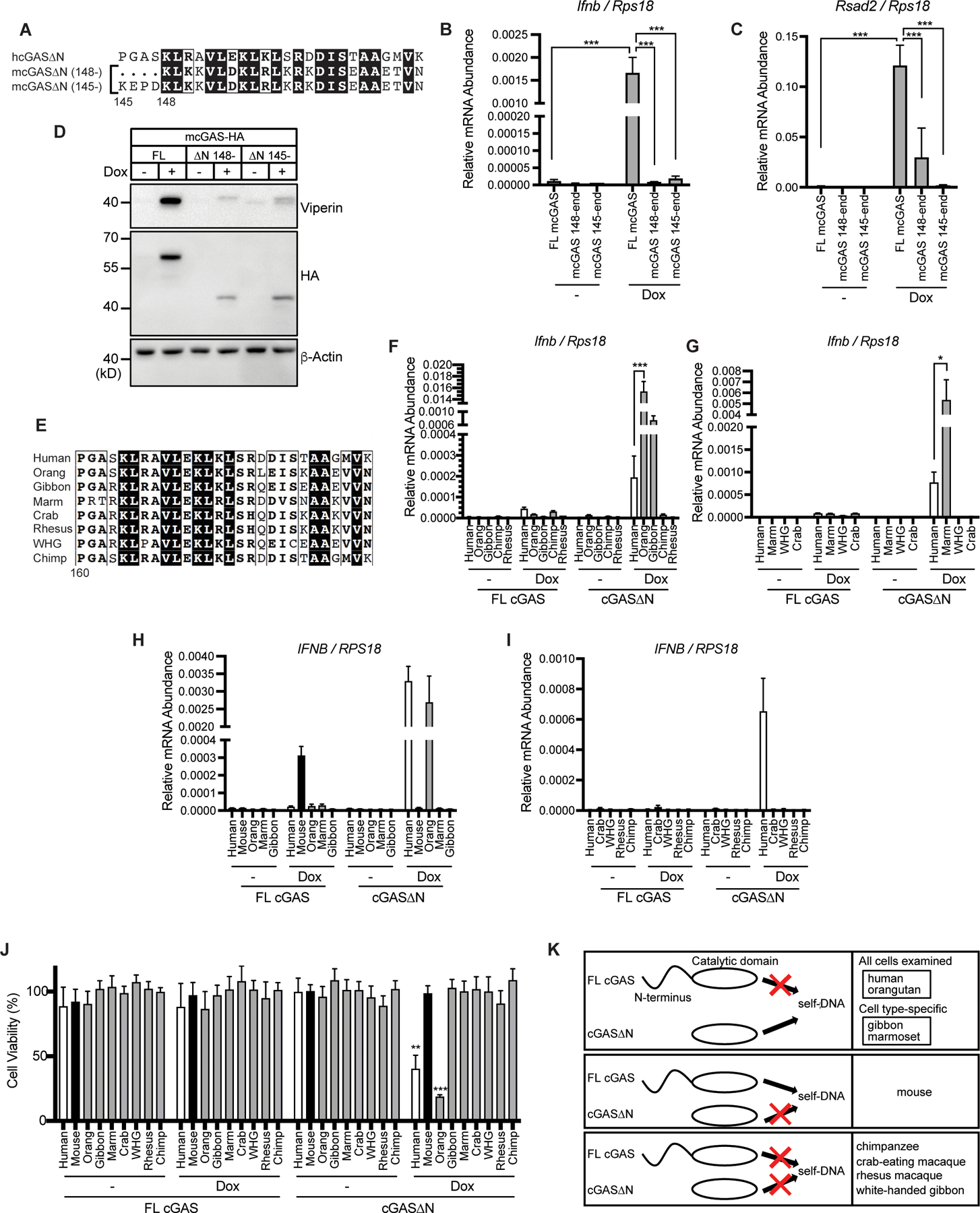
(A) N-terminal amino acid sequences of human cGAS∆N, mouse cGAS 145-, and mouse cGAS 148-. (B and C) qRT-PCR analysis of Ifnb (B) and Rsad2 (C) mRNAs in iBMDMs treated with Dox for 8 hours. (D) Immunoblot analysis of iBMDM lysates treated as in (B) and (C). (E) N-terminal amino acid sequences of human cGAS∆N and nonhuman primate (NHP) cGAS truncation mutants. Orang: orangutan, Marm: marmoset, Crab: crab-eating macaque, Rhesus: rhesus macaque, WHG: white-handed gibbon, Chimp: chimpanzee. (F and G) qRT-PCR analysis of Ifnb mRNAs in iBMDMs treated with Dox for 8 hours. (H and I) qRT-PCR analysis of IFNB mRNAs in THP-1 monocytes treated with PMA and Dox for 48 hours. (J) Viability of THP-1 cells treated with PMA and Dox as in (H) and (I). (K) Diverse self-DNA reactivity profiles of mammalian cGAS. Each box has different self-DNA reactivity patterns: only cGAS∆N has self-DNA reactivity; only FL cGAS has self-DNA reactivity; and neither reacts to self-DNA. Immunoblot data are the representative from three independent experiments. Graph data are means ± SEM of three independent experiments. Statistical significance was determined by two-way ANOVA and Tukey’s multiple comparisons test. Asterisks indicate the statistical significance between connected two bars (B, C, F, G, and H) or between untreated and Dox-treated conditions for indicated cGAS (J). *P < 0.05; **P < 0.01; ***P < 0.001
Human cGAS contains two amino acid substitutions that allow enhanced DNA-length specificity compared to mouse cGAS (9). Given human cGAS∆N reacts to self-DNA while mouse cGAS∆N does not, we hypothesized these two amino acids govern the self-DNA reactivity of cGAS∆N. These amino acids in human cGAS∆N (K187 and L195) were replaced with those in mouse cGAS (N172 and R181, respectively). Mutant hcGAS∆N K187N/L195R was expressed at slightly higher levels than WT hcGAS∆N, but no notable differences in IFN activity were observed when comparing these proteins (fig. S2, A and B). These results suggest that these two amino acids themselves do not affect self-DNA reactivity and that other structural differences determine cGAS∆N activities in these species. These findings prompted a broader evolutionary analysis of self-DNA reactivity by FL cGAS and cGAS∆N. We generated iBMDMs that encoded FL and cGAS∆N proteins from several nonhuman primates (NHPs), including orangutan, marmoset, gibbon, chimpanzee, white-handed gibbon, crab-eating macaque, and rhesus macaque (Fig. 3E), and assessed IFN responses upon Dox-induced transgene expression. First, we assessed the responsiveness of each FL cGAS NHP species to exogenous DNA. We expressed each NHP FL cGAS in cGAS-deficient iBMDMs and found that all responded to calf thymus DNA (CT-DNA) transfection, as indicated by Ifnb mRNA transcription and IFN-β protein secretion (fig. S2, C and D). However, similar to the behaviors of the human proteins, no self-DNA responsiveness was observed for FL cGAS proteins from any NHP examined (Fig. 3, F and G). Also similar to human, cGAS∆N from orangutan, marmoset, and gibbon all induced Ifnb mRNA and viperin protein production, to an extent even greater than what was observed for hcGAS∆N (Fig. 3, F and G and fig. S2, E to H). In contrast, several other NHP cGAS∆N proteins, including those from chimpanzee and crab-eating macaque did not trigger type I IFN responses upon expression in iBMDMs (Fig. 3, F and G and fig. S2, E to H). Most cGAS activities in iBMDMs were also observed in PMA-treated THP-1 cells. For example, hcGAS∆N and FL mcGAS induced IFNB transcripts, while FL hcGAS and mcGAS∆N did not (Fig. 3H). Among NHP cGAS tested, orangutan cGAS∆N was also active in iBMDMs and THP-1 cells (Fig. 3, H and I). PMA-mediated potentiation of ISG expression and cell death was also induced in THP-1 cells expressing hcGAS∆N and orangutan cGAS∆N, which induced IFNB transcripts (Fig. 3J and fig. S2, I to L). However, marmoset and gibbon cGAS∆N were only active in iBMDMs, not in THP-1 cells (Fig. 3H). These collective data reveal species-specific activities of cGAS that relate to self-DNA reactivity (Fig. 3K).
All cGAS enzymes are intrinsically active in vitro
We considered the possibility that each class of cGAS proteins may display intrinsic differences in DNA-induced cGAMP production. To address this possibility, we incubated recombinant cGAS with DNA in vitro and measured cGAMP synthesis. We examined self-DNA reactive proteins (human and orangutan cGAS∆N and mouse FL cGAS) and the non-self-reactive chimpanzee cGAS. We also included N-terminally FLAG-tagged hcGAS∆N and mouse and chimpanzee cGAS∆N, all of which were inactive in self-DNA-induced IFN expression within cells. Despite differences in self-DNA reactivity observed within cells, all the cGAS proteins examined behaved similarly in in vitro assays (Fig. 4A). All cGAS proteins were able to synthesize cGAMP in response to synthetic dsDNA, but not to nucleosomal DNA that was isolated from iBMDMs (Fig. 4A). Protease treatment of nucleosomes rendered the resulting DNA samples capable of stimulating cGAMP production by cGAS proteins examined (Fig. 4A). The inability of cGAS to produce cGAMP in response to nucleosomal DNA was reported for human cGAS (15–21), but our findings suggest that this inability extends across the mammalian species we have examined. Consistent with previous findings, FL mcGAS synthesized cGAMP in response to DNA at a lower concentration than hcGAS, suggesting FL mcGAS has a stronger reactivity to DNA (9). However, despite different activities of orangutan and chimpanzee cGAS∆N in cells, the sensitivities of these proteins to DNA in vitro were comparable to those of hcGAS∆N (fig. S3, A and B). Therefore, the diversity in cGAS∆N signaling activities is not due to protein intrinsic activities.
Fig. 4. Mitochondrial localization and signaling activities of cGAS.
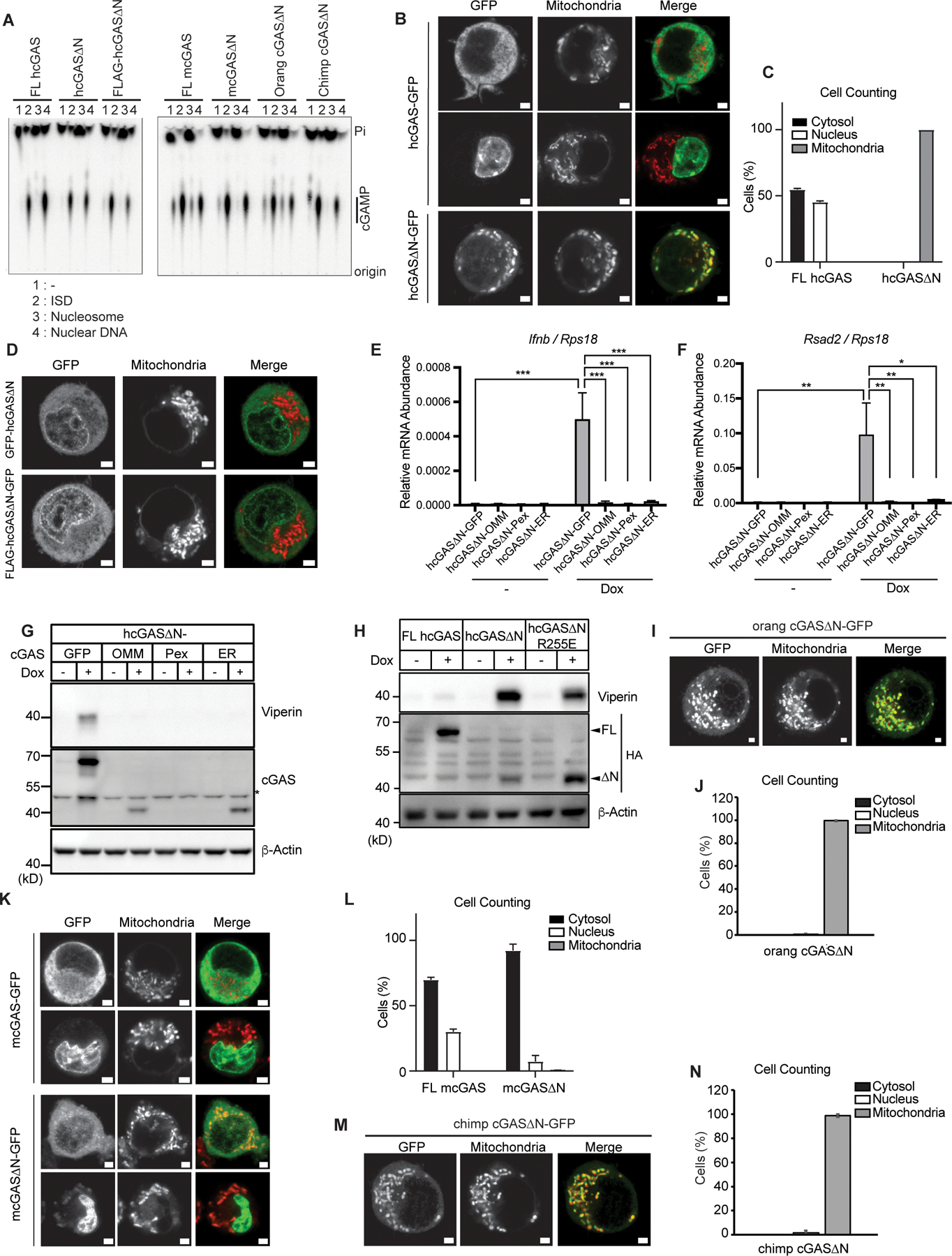
(A) cGAS production of cGAMP in vitro with purified components consisting of 45 bp interferon stimulatory DNA (ISD), nucleosomal DNA, or nuclear dsDNA. Recombinant cGAS proteins included FL hcGAS, hcGAS∆N (160–522 a.a.), FLAG-hcGAS∆N, FL mcGAS, mcGAS∆N (148–507 a.a.), orangutan (orang) cGAS∆N, and chimpanzee (chimp) cGAS∆N. (B) Live confocal micrographs of C-terminally GFP-tagged human FL cGAS or cGAS∆N. (C) Cell counting in (B). Cells with nuclear, cytosolic, and mitochondrial cGAS localization were counted and the ratio was calculated. (D) Live confocal micrographs of human cGAS∆N with indicated tags. Representative cell images are shown. (E and F) qRT-PCR analysis of Ifnb (E) and Rsad2 (F) mRNAs in iBMDMs treated with Dox for 8 hours. OMM: outer mitochondrial membrane; Pex: peroxisomes; ER: endoplasmic reticulum. (G) Immunoblot analysis of iBMDM lysates treated as in (E) and (F). The asterisk indicates non-specific bands. (H) Immunoblot analysis of iBMDM lysates treated with Dox for 8 hours. (I) Live confocal micrographs of C-terminally GFP-tagged orangutan (orang) FL cGAS or cGAS∆N. Representative cell images are shown. (J) Cell counting in (I). (K) Live confocal micrographs of C-terminally GFP-tagged mouse FL cGAS or cGAS∆N. Representative cell images are shown. (L) Cell counting in (K). Cells with nuclear, cytosolic, and mitochondrial cGAS localization were counted and the percentages of each localization pattern were calculated. (M) Live confocal micrographs of C-terminally GFP-tagged chimpanzee (chimp) FL cGAS or cGAS∆N. Representative cell images are shown. (N) Cell counting in (M). Scale bars in all the images indicate 2 µm. Green signals indicate GFP, and red signals indicate mitochondria in all the merged images. Images and immunoblot data are representative from three independent experiments. Graph data are means ± SEM of three independent experiments. Statistical significance was determined by two-way ANOVA and Tukey’s multiple comparison test. Asterisks indicate the statistical significance between connected two bars. *P < 0.05; **P < 0.01; ***P < 0.001.
Mitochondrial localization of self-reactive cGAS∆N proteins correlate with signaling activity
We considered the possibility that access to intracellular DNA may explain self-DNA reactivity. To test this idea, we examined the subcellular localization of cGAS. Consistent with our prior work in THP-1 cells (11), differential sedimentation of THP-1 or iBMDM homogenates revealed that cGAS was not detected in cytosolic S100 fractions (fig. S4, A and B). Also consistent with prior work (11), membrane flotation assays of cell homogenates revealed cGAS migration from dense to light membrane fractions (fig. S4, C and D), and cGAS flotation was resistant to Triton X-100, but sensitive to sodium dodecyl sulfate (SDS) (fig. S4E). However, we found that cGAS migrated to different fractions than the membrane proteins Na+/K+ ATPase, STING, and TIRAP (fig. S4, A to E). The differential migration of cGAS from other membrane proteins prompted further examination of cGAS localization.
Recent studies have highlighted that cGAS undergoes biochemical changes upon DNA binding, leading to monomers of this PRR assembling into large aggregates that display liquid droplet properties (10). The size of these aggregates (>1 micron) would be expected to impact biochemical behaviors in fractionation-based analyses. We considered this issue because DNA is unavoidably released into cell homogenates during the lysis step that precedes biochemical fractionations. It was therefore possible that biochemical analyses of endogenous cGAS localization have been hampered by post-lysis DNA binding events, which would result in cGAS aggregates that do not reflect the pre-lysis state of this protein.
A simple means to test if post-lysis changes in cGAS behavior were possible is to add recombinant cGAS protein to cell extracts prior to fractionation and ask if this exogenous protein displayed properties similar to its endogenous counterpart. We added recombinant human cGAS to post-nuclear supernatants (PNS) of iBMDMs and performed membrane flotation assays. We found that exogenous recombinant cGAS migrated to the same fractions as endogenous cGAS (fig. S4F). The flotation of recombinant cGAS was independent of its endogenous counterpart, as the use of PNS from cGAS-deficient macrophages yielded the same results (fig. S4G). These results indicate that cGAS can interact with cellular structures post-lysis, a finding that complicates biological interpretation of subcellular fractionations.
To test whether DNA induces membrane-like cGAS protein flotation, we incubated recombinant cGAS protein and 100 bp immunostimulatory DNA (ISD), in the absence of any PNS or cellular material, and then performed membrane flotation assays. We found that the presence of ISD-induced cGAS flotation, even in the absence of PNS (fig. S4H). Moreover, this flotation of cGAS was resistant to 1% Triton X-100 but sensitive to 2% SDS (fig. S4I), which was consistent with the behavior of endogenous cGAS in PNS (fig. S4E). DNA-induced cGAS oligomers are inhibited by the high concentration of sodium chloride (NaCl) (10). The same high concentrations of NaCl are reported to extract cGAS from being tethered onto chromatin, where cGAS has been proposed to reside in resting cells (12). We found that NaCl prevented recombinant cGAS from floating in the presence of ISD (fig. S4J). Notably, cGAS-DNA flotation was resistant to the highly efficient DNase Benzonase, even at concentrations that degrade free DNA (fig. S4, K and L). These collective data indicate that commonly used methods of cGAS biochemical analysis, within cell lysates, are complicated by the possibility of post-lysis cGAS-DNA interactions. These post-lysis interactions can result in cGAS behaviors that give the appearance of chromatin or membrane associations. For these reasons, we shifted our focus from biochemical analysis of cGAS localization to cGAS visualization in live cells.
Live cell confocal imaging of C-terminally GFP-tagged hcGAS in iBMDMs revealed that hcGAS∆N localized to mitochondria, while FL hcGAS-GFP was not detected in these organelles (Fig. 4, B and C and movie S1). Interestingly, when an N-terminal GFP or HA tag was placed onto hcGAS∆N, mitochondrial localization was abolished (Fig. 4D). Similarly, N-terminal tagging hcGAS∆N abolished IFN activities, as described in Figure 2E. These findings are consistent with a recent study by Chen and colleagues (22). We reasoned that if mitochondrial localization was important for hcGAS∆N signaling, forcing its localization to distinct subcellular locations should prevent IFN activities. We therefore engineered hcGAS∆N to contain C-terminal membrane localization sequences that direct this protein to the outer mitochondrial membrane (OMM), peroxisomes or the ER. This was accomplished by appending onto hcGAS∆N transmembrane domains from MAVS (OMM), Pex13 (peroxisomes) or VAMP2 (ER). When expressed via Dox in iBMDMs, none of the membrane-targeted hcGAS∆N proteins induced type I IFN responses (Fig. 4, E to G). Interestingly, these membrane-targeted hcGAS∆N did not respond to transfected non-self-DNA, suggesting that membrane anchoring prevents cGAS activation not only to mitochondrial DNA but also to cytosolic DNA (fig. S5). Taken together, these data indicate that restricting hcGAS∆N from access to mitochondria prevents self-DNA reactivity.
As we did not detect hcGAS∆N within the nucleus, we considered it unlikely that this PRR was driving IFN responses to intranuclear DNA. To address this possibility directly, we introduced R255E mutation in hcGAS∆N, which has been structurally defined to regulate cGAS interactions with chromatin in the nucleus (17–21). If hcGAS∆N were signaling from the nucleus, then this mutation should amplify IFN responses. We found that R255E mutations did not enhance signaling activity of hcGAS∆N, suggesting that hcGAS∆N is not reacting to self-DNA in the nucleus (Fig. 4H). Based on these findings, and the primary location to mitochondria, we propose that hcGAS∆N is reacting to intra-mitochondrial mtDNA.
To determine if mitochondrial localization was also linked to self-DNA responses induced by other cGAS proteins, we examined the localization of orangutan cGAS∆N and FL mcGAS, both of which are self-DNA reactive, and the inactive chimpanzee cGAS∆N protein. Like its human counterpart, orangutan cGAS∆N localized to mitochondria, suggesting that IFN activities and mitochondrial localization are a common feature of self-reactive primate cGAS proteins (Fig. 4, I and J). Interestingly, we found that mouse FL cGAS, which is self-DNA reactive, was not localized to mitochondria (Fig. 4, K and L) and chimpanzee cGAS∆N, which is not self-DNA reactive, was localized to mitochondria (Fig. 4, M and N). These results suggest species-specific regulation of self-DNA reactivity by cGAS.
cGAS N-terminus cannot prevent self-DNA reactivity in trans
Our studies have exclusively examined cells that were engineered to express either the N- or C-terminal domains of cGAS (not both). To overcome this limitation, we generated a hcGAS transgene that contains a T2A ribosome skip sequence (23, 24) between the N-terminal domain (hcGAS N) and hcGAS∆N (Fig. 5A). This gene encodes a bicistronic mRNA that produces hcGAS N and hcGAS∆N upon translation. We found that Dox-mediated expression of this engineered cGAS within iBMDMs produced cGAS N and hcGAS∆N, indicating T2A functionality, and also led to the production of the ISG viperin (Fig. 5B). These results suggest that the N-terminal domain cannot inhibit cGAS∆N signaling activity when these domains are separate polypeptides. These data therefore support the idea that the activities present in the C-terminal catalytic domain are sufficient to stimulate IFN responses, and that a central function of the N-terminal domain may be to prevent localization of the C-terminus to self-DNA.
Fig. 5. User-defined cGAS∆N responses to a viral protease and in vivo tumor transplant.
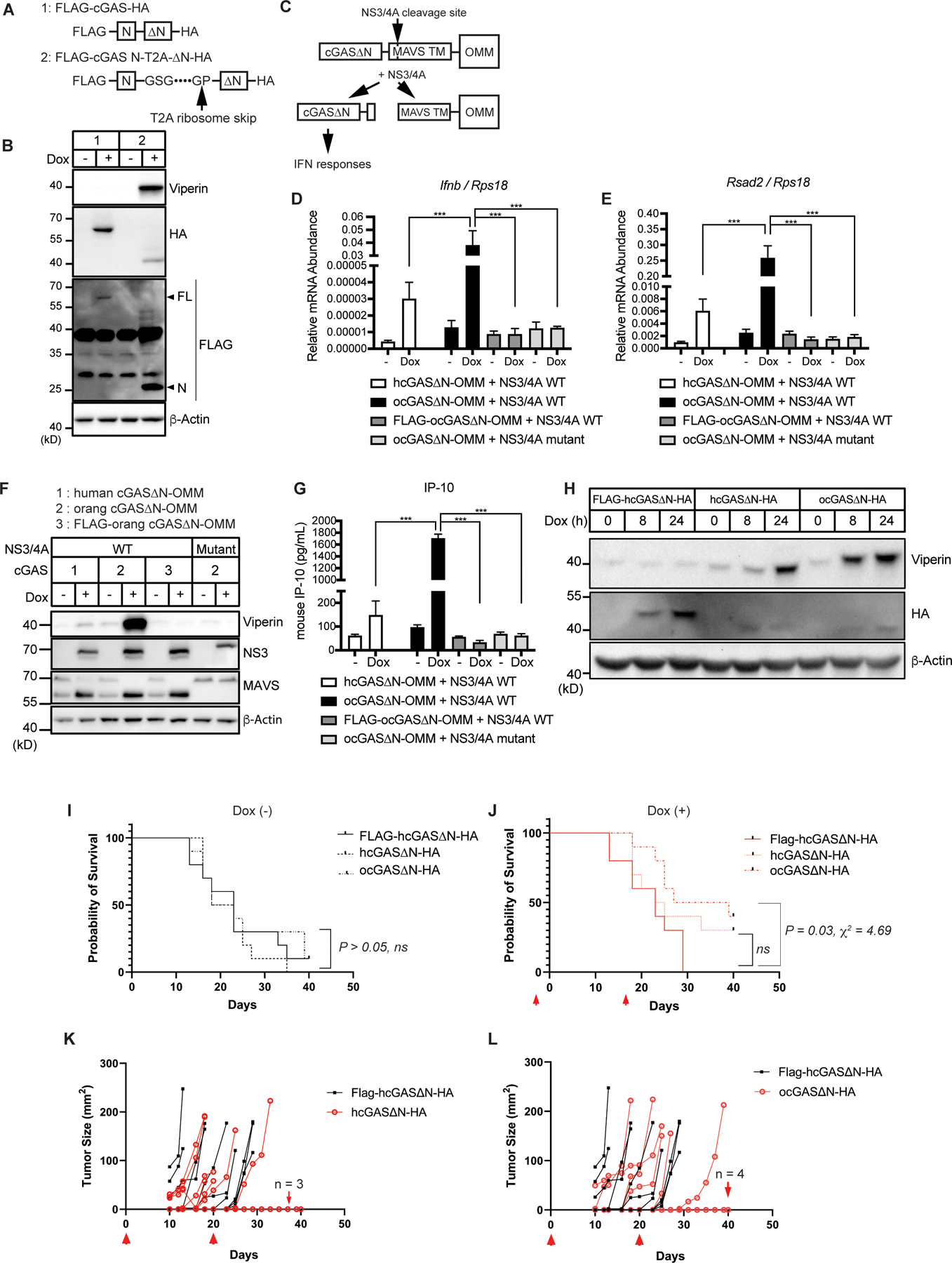
(A) Schematic designs of cGAS constructs with T2A consensus sequences. FLAG-cGAS N-T2A-∆N-HA is separated upon translation at the indicated arrow. (B) Immunoblot analysis of lysates of iBMDMs treated with Dox for 8 hours to induce expression of cGAS alleles shown in (A). (C) Schematic design of cGAS-outer mitochondrial membrane (OMM) construct and the cleavage by NS3/4A protease. (D and E) Real-Time qRT-PCR analysis of Ifnb (D) and Rsad2 (E) mRNAs in cGAS-expressing iBMDMs treated with Dox for 24 hours to induce expression of WT or mutant NS3/4A proteases. Cells stably expressed indicated human cGAS (hcGAS) or orangutan cGAS (ocGAS) constructs. (F) Immunoblot analysis of iBMDMs treated as in (D) and (E). (G) IP-10 ELISA analysis of iBMDM cell culture supernatant in (D) and (E). (H) Immunoblot analysis of lysates from B16OVA treated with Dox for indicated times to induce expression of cGAS∆N constructs. (I and J) Kaplan-Meier survival of mice s.c.-transplanted with B16OVA with indicated transgenes without (I) or with (J) Dox administration. Red arrows in (J) indicate the timing of three-day administration of Dox in their drinking water. n = 10 mice per group. (K and L) Tumor area in the mice s.c.-transplanted with B16OVA and administered with Dox in the drinking water. Comparison between mice with FLAG-hcGAS∆N-expressing tumors and hcGAS∆N-expressing tumors (K) and mice with FLAG-hcGAS∆N-expressing tumors and orangutan cGAS∆N-expressing tumors (L) are shown. Red arrows indicate the timing of three-day administration of Dox in their drinking water. The number of mice that did not develop tumors are indicated in each graph. n = 10 mice per group. Immunoblot data are representative from three independent experiments. Graph data (D, E, and G) are means ± SEM of three independent experiments. In vivo experiments (K to L) were performed once. Statistical significance was determined by two-way ANOVA and Tukey’s multiple comparisons test (D, E, and G) or calculated by the Gehan-Breslow-Wilcoxon method following Kaplan-Meier analysis (I and J). Asterisks indicate the statistical significance between connected two bars. *P < 0.05; **P < 0.01; ***P < 0.001. ns: non-significant.
Viral protease-mediated release of self-DNA reactive cGAS∆N induces type I IFN responses
Our ability to induce self-DNA reactivity by hcGAS∆N, even within cells that contain cGAS N, raised the possibility that other means of dissociating these domains would stimulate IFN production. In this regard, we considered the protein NLRP1, a sensor of viral and bacterial proteases (25–28). Cleavage of NLRP1 by pathogen proteases leads to the induction of pyroptosis. NLRP1 is considered a guard protein, as opposed to a PRR, with the latter detecting conserved microbial products and the former detecting virulence factor activities (29, 30). Given that the T2A-mediated separation of the cGAS N- and C-termini was sufficient to trigger IFN responses, it was possible that cGAS can be engineered to operate as an IFN-inducing sensor of viral protease activity. Hepatitis C virus (HCV) is a hepatotropic virus that possesses the protease NS3/4A. This protease is immune-evasive, based on its ability to cleave the RIG-I like receptor (RLR) signaling adaptor MAVS off membranes (31). In Figures 4E–G, we anchored hcGAS∆N to the outer mitochondrial membrane (OMM) using the transmembrane domain of MAVS, which contains the NS3/4A cleavage site. Therefore, we hypothesized that this hcGAS∆N-OMM protein would be cleaved by NS3/4A, leading to the release of hcGAS∆N to the cytoplasm and subsequent IFN induction. To test this hypothesis, hcGAS∆N-OMM was expressed stably in iBMDMs that encoded a Dox-inducible NS3/4A transgene (Fig. 5C). Expression of hcGAS∆N-OMM in the absence of NS3/4A did not lead to any IFN activities, but Dox-mediated induction of NS3/4A stimulated some expression of Ifnb and Rsad2 mRNAs (Fig. 5. D and E). Endogenous MAVS in these cells was cleaved upon Dox treatment, confirming the proteolytic function of NS3/4A (Fig. 5F). Notably, the induction of Ifnb and Rsad2 mRNAs was enhanced substantially when orangutan cGAS was used instead of hcGAS in these assays (Fig. 5, D and E). Orangutan cGAS∆N-OMM stimulated viperin production and IP-10 secretion upon expression of NS3/4A, which hcGAS∆N-OMM barely induced (Fig. 5, F and G). Furthermore, and consistent with the importance of mitochondrial localization for self-DNA reactivity, the addition of a FLAG-tag to these cGAS constructs abolished IFN responses upon NS3/4A expression (Fig. 5, D to G). All Dox-inducible activities were mediated by the protease activity of NS3/4A, as no changes in IFN activities were observed in cells that produced a catalytically inactive NS3/4A mutant (Fig. 5, D to G). These data validate the model that the N-terminal domain of human and orangutan cGAS proteins prevents self-DNA reactivity and reveal a synthetic biology-based strategy to redesign this PRR into a PRR-guard hybrid, which operates as an IFN-inducing sensor of viral protease activities.
Intratumoral activation of self-DNA reactive cGAS∆N promotes antitumor immunity in vivo
There is an increasing interest in the cGAS-STING pathway for cancer therapeutics, yet this pathway has dichotomous roles in cancer (32). While cGAS-STING signaling can promote tumor progression (5, 32–34), this pathway can also induce protective immunity (32, 35–40). Our ability to engineer cells to express active cGAS alleles provided an opportunity to determine if cGAS signaling in a single cell type would impact tumor progression. We engineered OVA-expressing B16.F10 melanoma cells (B16OVA) to induce expression of several cGAS∆N transgenes (FLAG-hcGAS∆N, hcGAS∆N, or orangutan cGAS∆N) in a Dox-dependent manner (Fig. 5H). Consistent with our findings in other cells, Dox-mediated expression of hcGAS∆N and orangutan cGAS∆N in vitro induced viperin production in B16OVA cells, with the latter displaying strongest IFN-inducing activities, whereas FLAG-hcGAS∆N did not (Fig. 5H). These engineered B16OVA cells were transplanted subcutaneously into mice. After the tumor implantation, mice were treated with Dox in their drinking water to induce cGAS∆N expression in tumor cells. Regardless of genotype, almost all mice that did not receive Dox in drinking water experienced significant tumor growth and did not survive (Fig. 5I). Dox-induced expression of human or orangutan cGAS∆N in tumor cells improved the survival of mice, whereas Dox did not impact disease outcome in mice containing FLAG-hcGAS∆N-expressing tumors (Fig. 5J). Interestingly, orangutan cGAS∆N was most effective at protecting mice from tumor growth and lethality, even when compared to human cGAS∆N (Fig. 5, J to L). These results demonstrate that 1) cGAS signaling from within melanoma cells is sufficient to protect mice from tumor-induced lethality, and 2) that stronger cGAS signaling activities lead to more robust protective immunity.
DISCUSSION
Because the protein architecture of cGAS is highly conserved throughout mammalian evolution, studies have been largely based on the assumption that the regulatory mechanisms of cGAS in one species hold true for another. In this study, while we confirmed the self-DNA reactivity of the human cGAS catalytic domain (11), we also identified the evolutionary diversity of cGAS regulation. The cGAS species we examined were studied in human and mouse cells, as opposed to their native counterparts, in order to distill experimental analysis down to a single variable—the amino acid sequence of each cGAS protein. The study of different cGAS proteins in different cell types or species is an important consideration for future studies. One of the notable aspects of the species-specificity of self-DNA reactivity is the potential impact on clinical strategies to target cGAS. Indeed, animal models including mouse, chimpanzee, and rhesus macaque are often components of therapeutic development pipelines. The studies described herein should be considered when undertaking such endeavors, as not all cGAS proteins behave the same.
Our mechanistic analysis revealed an exquisite sensitivity of the N-terminus of human cGAS∆N for self-DNA responsiveness, as adding greater than two amino acids onto the N-terminus was sufficient to abolish IFN activities. These findings impact interpretations of studies using N-terminal epitope tags to study FL and cGAS∆N functions. Future studies should consider the position of the epitope tags to be as important an experimental variable as the species of the cGAS protein under investigation. Similar considerations apply to the use of biochemical fractionation as a tool of cGAS cell biology. The aggregation of cGAS that occurs upon binding to DNA, while key for protein function, may hinder the reliable study of cGAS present in cell lysates. Indeed, we have not identified a condition of cell lysis where DNA is not released into the PNS.
Finally, it is noteworthy that the self-DNA reactivity of select cGAS∆N proteins can be leveraged to induce immunity to infection and cancer. Viral proteases, in particular NS3/4A, are naturally immune-evasive because they cleave host signaling proteins to inactivate PRR-induced responses to infection. Classic therapeutic strategies to target viral proteases involve inhibition, which results in selective pressure for viral escape mutants. Rather than inhibiting viral proteases, our synthetic PRR-guard cGAS has forced the normally immune-evasive NS3/4A protease to operate as an immunostimulant. These findings provide a model to consider synthetic biology-based strategies that force immune evasion mechanisms of pathogens to stimulate host immune responses. When combined with our finding that intratumor expression of self-DNA reactive cGAS∆N transgenes protects mice from tumor-induced lethality, these findings provide a mandate to explore the impact of species-specific diversity of cGAS self-DNA reactivity in various contexts of health and disease.
MATERIALS AND METHODS
Study design
The aim of this study was to investigate the self-DNA reactivity of cGAS and its domains in human and other mammals. We investigated the activities of these cGAS and mutants using doxycycline-mediated transient expression system in mouse and human cells. Sample sizes and experimental replicates for each experiment are indicated in the figure captions.
Cell culture
Immortalized murine bone marrow-derived macrophages (iBMDMs), HEK293Ts, and Plat-GP cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Gibco), referred to as complete DMEM, at 37°C in 5% CO2. For passage, iBMDMs were lifted using PBS (Gibco) supplemented with 2.5 mM EDTA (Invitrogen) and plated at a 1:10 dilution. HEK293T, L929, and B16OVA cells were grown under the same conditions as iBMDMs but were passaged by washing with PBS and lifting with 0.25% Trypsin-EDTA (Gibco) with a 1:10 dilution. THP-1 cells were grown in suspension culture using RPMI-1640 media (Lonza) supplemented with 10% FBS, referred to as complete RPMI-1640, at 37°C in 5% CO2. For passage, cells were split at a dilution of 1:5. For experiments examining the effects of PMA-induced differentiation of THP-1 cell lines, cells were treated for 72 hours with PMA (MilliporeSigma) at a concentration of 50 ng/mL. Normal oral keratinocytes were cultured in keratinocyte SFM (Gibco) supplemented with Human Keratinocyte Growth Supplement (Gibco) and passaged by washing with PBS and lifting with 0.25% Trypsin-EDTA at a 1:10 dilution.
Generating cells with stable or doxycycline-inducible gene expression
cDNAs of wild type and truncation mutant cGAS were amplified by polymerase chain reaction (PCR) using oligonucleotide primers containing restriction enzyme digestion sites. For the amplification of nonhuman primate (NHP) cGAS, cDNAs in the pcDNA6 vector (41) were used as PCR templates. For Dox-induced gene expression, cGAS cDNAs were inserted in the BamHI and NotI restriction sites in pRetroX-TRE3G (Takara Bio) using In-Fusion Snap Assembly Master Mix (Takara Bio). For stable gene expression, cGAS DNAs were inserted in pLenti CMV GFP Puro in replacement of GFP using XbaI and SalI. For fusion mutants and the T2A insertion mutant of cGAS cDNAs, In-Fusion Snap Assembly Master Mix was used to incorporate cGAS and the other fragments together in the vectors. All constructs generated here were sequence-confirmed by Sanger sequencing.
To generate lentiviral particles for the stable expression of transgenes, HEK293T cells were transfected with the packaging plasmids psPAX2 and pCMV-VSV-G along with the transgene in pLenti CMV GFP Puro using Lipofectamine 2000 (Invitrogen). pCMV-VSV-G was a gift from Bob Weinberg (Addgene plasmid # 8454 ; http://n2t.net/addgene:8454 ; RRID:Addgene_8454) (42). psPAX2 was a gift from Didier Trono (Addgene plasmid # 12260; http://n2t.net/addgene:12260; RRID: Addgene_12260). pLenti CMV GFP Puro (658–5) was a gift from Eric Campeau & Paul Kaufman (Addgene plasmid # 17448; http://n2t.net/addgene:17448; RRID: Addgene 17448) (43). All genes of interest were subcloned into the GFP site. Plasmids were transfected into 10 cm2 dishes of HEK293Ts at 50–80% confluency using Lipofectamine 2000 (Invitrogen) by mixing DNA and Lipofectamine 2000 ratio at 1:2. Media was changed on transfected HEK293Ts 16–24 hours after transfection, and virus-containing supernatants were harvested 24 hours following the media change. Viral supernatants were passed through a 0.45 µm filter to remove any cellular debris. Filtered viral supernatants were mixed with 5 µg/mL polybrene (MilliporeSigma) and placed directly onto target cells, followed by spinfection (centrifugation at 1,250 × g, 30°C for 1 hour). Cell culture media were replaced with the appropriate complete media and cells were incubated for 24 hours. Spinfection with the viral supernatants was repeated on the following day, and cells were used for indicated assays.
Doxycycline (Dox)-inducible gene-expressing cell lines were generated using Retro-X™ Tet-On 3G Inducible Expression System (Takara Bio). Plat-GP cells were transfected with the pRetroX-Tet3G plasmid together with the packaging plasmid pCMV-VSV-G using Lipofectamine 2000. Using viral supernatant of Plat-GP cells, iBMDMs or THP-1 cells were subjected to spinfection in a similar manner as above. After consecutive spinfections for two days, Tet3G-transduced cells were selected using G418 (InvivoGen) and single cell clones were isolated. Tet3G-containing cell clones were transduced with TRE3G virus prepared from Plat-GP cells transfected with pRetroX-TRE3G containing genes of interest and packaging plasmid pCMV-VSV-G. TRE3G-transduced cells were selected using puromycin (Gibco) and pooled cell culture was used in each assay. Cells were treated with 1 µg/mL Dox (MilliporeSigma) were subjected to each analysis at the indicated time points in each figure caption.
Generation of CRISPR-Cas9-mediated knockout (KO) cells
For cloning, the sense and antisense oligonucleotides containing guide RNA (gRNA) sequences and AfeI/SbfI restriction sites were purchased from Integrated DNA Technologies, and equal molar ratios of sense and antisense oligonucleotides were annealed in water on PCR block (95ºC for 1 minute, followed by a drop of 5°C every minute to 10°C). Oligonucleotide duplexes were then subcloned into AfeI/SbfI-digested pRRL-Cas9-Puro vector (kindly provided by Dr. D. Stetson (44)) using In-Fusion Snap Assembly Master Mix (Takara Bio). gRNAs targeting mouse genes used in this study were as follows: Cgas #1: GAGGCGCGGAAAGTCGTAAG, Cgas #2: GGCAGCCCAGAGCGCCGCGA, Sting #1: GGCCAGCCTGATGATCCTTT, Sting #2: GCTGGCCACCAGAAAGATGA.
To generate lentiviral particles for the stable expression of Cas9 and gRNAs, HEK293T cells were transfected with the packaging plasmids psPAX2 and pCMV-VSV-G along with the gRNAs-containing pRRL-Cas9-Puro vector using Lipofectamine 2000 (Invitrogen). Plasmids were transfected into 10 cm2 dishes of HEK293Ts at 50%–80% confluency using Lipofectamine 2000 by mixing DNA and Lipofectamine 2000 ratio at 1:2. Media was changed on transfected HEK293Ts 16–24 hours after transfection, and virus-containing supernatants were harvested 24 hours following the media change. Viral supernatants were passed through a 0.45 µm filter to remove any cellular debris. Filtered viral supernatants were mixed with polybrene (Millipore) and placed directly onto target cells, followed by the spinfection (centrifugation at 1,250 x g for 1 hour). Cell culture media were replaced with the appropriate complete media and cells were incubated for 24 hours. Spinfection with the viral supernatants was repeated on the following day, and cells were used for indicated assays.
Real-Time quantitative reverse transcription (qRT-) PCR
RNA was isolated from cells using QIAshredder (QIAGEN) homogenizers and the PureLink RNA Mini Kit (Life Technologies) and subsequently treated with DNase I (Invitrogen) to remove genomic DNA. Relative mRNA expression was analyzed using the TaqMan RNA-to-Ct 1-Step Kit (Thermo Fisher Scientific) with indicated TaqMan probes (Thermo Fisher Scientific) on a CFX384 Real-Time Cycler (Bio-Rad Laboratories). Each CT value was normalized with the mRNA expression of the control genes (RPS18 for human and Rps18 for mouse) and the relative mRNA abundance was calculated by the ∆∆CT method. TaqMan probes used in this study are as follows: Ifnb (mouse): Mm00439552_s1, Rsad2 (mouse): Mm00491265_m1, Rps18 (mouse): Mm02601777_g1, IFNB (human): Hs01077958_s1, RSAD2 (human): Hs00369813_m1, RPS18 (human): Hs01375212_g1.
Cell viability assay
Cell viability was measured using CellTiter-Glo (Promega Corporation), a luminescent assay for ATP in living cells. Untreated cells were used as a positive control for 100% cell viability and subjected to serial dilution for the standard curve. Luminescent outputs were read on a Tecan plate reader and viability was calculated using the standard curve.
Immunoblotting and ELISA analysis
Cells were lysed with RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), which was supplemented with cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail [Roche] and Benzonase [Millipore] before use) and the lysates were centrifuged at 4°C, 16,000 × g for 10 minutes. Supernatants were mixed with 6 × SDS sample buffer supplemented with Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Thermo Fisher Scientific) and boiled at 100°C for 5 minutes. Samples were separated by SDS-PAGE and transcribed to PVDF membrane by immunoblotting. PVDF was blocked with 5% skim milk for 1 hour and probed with indicated primary antibodies overnight at 4°C, followed by secondary antibodies (1:2000 dilution) for 1 hour. Primary antibodies used in this study include viperin (1:1000 dilution, MilliporeSigma), HA (1:1000 dilution, MilliporeSigma), b-Actin (1:1000 dilution, Cell Signaling Technology), cGAS (1:1000 dilution, MilliporeSigma), STING (1:1000 dilution, Cell Signaling Technology), and NS3 (1:1000, GeneTex). For the detection of cGAS in Figure S1, a primary antibody purchased from Cell Signaling Technology was used (1:1000 dilution). Secondary antibodies for human, mouse, and rat immunoglobulins (IgGs) were purchased from Rockland Immunochemicals. Culture supernatants from treated cells were collected and applied to the IP-10 antibody-coated plate for ELISA analysis (R&D Systems) following the manufacturer’s instructions.
In vitro 2′3′-cGAMP assay
FL cGAS and cGAS∆N recombinant proteins were prepared as previously described (9, 45). Briefly, cGAS mutants were cloned into a custom pET vector for expression of an N-terminal 6 x His-SUMO2 fusion protein in E. coli. E. coli BL21-RIL DE3 (Agilent) bacteria harboring a pRARE2 tRNA plasmid were transformed with a pET cGAS plasmid, and 6 × His-SUMO2-cGAS recombinant proteins were purified from clarified E. coli lysate by binding to Ni-NTA (QIAGEN) and gravity chromatography. The His-SUMO2 tags were removed by dialyzed overnight at 4°C in dialysis buffer (20 mM HEPES-KOH pH 7.5, 300 mM NaCl, 1 mM DTT) after supplementing ~250 µg of human SENP2 protease (fragment D364–L589 with M497A mutation).
Nucleosomal DNA was isolated from untreated iBMDMs using EpiScope Nucleosome Preparation Kit (Takara Bio) and the counterpart naked double-stranded DNA (dsDNA) was obtained by removing histones using Proteinase K following the manufacturer’s instructions. cGAS activation and cGAMP synthesis was performed in vitro using purified components and measured with thin-layer chromatography as previously described (9). Briefly, 0.2–5 µM cGAS recombinant proteins were incubated with the DNA above or 45 bp interferon stimulatory DNA (ISD) (45) in a reaction buffer containing 10 mM Tris-HCl pH 7.5, 62.5 mM KCl, 10 mM MgCl2, 1 mM DTT, 6.25 µM ATP, 6.25 µM GTP, and [ɑ−32P] ATP (~1 µCi) at 37°C for 2 hours. Reactions were terminated by heating at 95°C for 3 min, and subsequently incubated with 4 U of alkaline phosphatase (New England Biolabs) at 37°C for 30 min to hydrolyze unreacted NTPs. 1 µL of each reaction was spotted on a PEI-Cellulose F thin-layer chromatography plate (EMD Biosciences) developed with 1.5 M KH2PO4 (pH 3.8) as a running buffer.
Live imaging confocal microscopy
GFP-tagged cGAS-expressing iBMDMs were plated on uncoated 35 mm dishes (MatTek Corporation). Cells were treated with MitoTracker Deep Red (Invitrogen) in Opti-MEM (Gibco) and incubated at 37°C, 5% CO2 for 60 minutes. Cells were washed with PBS and imaged using a 63× oil immersion objective on the LSM 880 with Airyscan (Zeiss). Images were processed using ZEN software (Zeiss) and ImageJ (NIH). Dox-untreated cells or non-GFP-expressing cells were used for the negative controls of GFP signaling, and negative signals were subtracted from GFP signals of cGAS. For counting, at least 100 cGAS-expressing cells were examined under the microscope and the percentile of nuclear, cytosolic, or mitochondrial cGAS-expressing cells was determined.
Mouse strains and In vivo B16OVA tumor transplantation
Female six- to eight-week-old C57BL/6 mice were purchased from Jackson Laboratories. Mice were allowed one week to acclimate to the Boston Children’s Hospital (BCH) animal housing facilities. The experimental procedures conducted herein were approved by the Institutional Animal Care and Use Committee (IACUC) at BCH (protocol #00001432). Individual mice were randomly assigned to experimental treatment groups (n=5). OVA-expressing B16.F10 melanoma cell lines were engineered with Dox-inducible Class 1 cGAS∆N transgenes as described above. Mice were subcutaneously (s.c.) injected with 3 × 105 B16OVA cells resuspended in PBS, and to induce the expression of transgenic cGAS, mice received water including 2 mg/mL Dox and 1% sucrose (MilliporeSigma) ad libitum on day 0 - day 3, then again on day 20 - day 23 post transplantation. The size of tumor was assessed every other day and recorded as tumor area (length × width) measured with calipers (Digi-Max). Mouse survival was documented daily. Mice were considered nonsurvivors when reaching an endpoint (tumor measurements near or exceeding >15mm, or upon ulceration) and sacrificed in accordance with the IACUC protocol. Survival analysis (Kaplan-Meier) statistics and figures were generated using Prism (GraphPad Software) and differences among survival curves were considered significant for P-values <0.05.
Quantification and statistical analysis
Statistical significance was determined by two-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. Survival analysis was performed with Kaplan-Meier statistics. P value and Chi square were calculated by the Gehan-Breslow-Wilcoxon method, and P < 0.05 was considered as statistically significant. All statistical analyses were performed using Prism data analysis software (GraphPad Software). All experiments except in vivo mouse transplantation were performed at least three times, and the graphed data with error bars indicate the means with the standard error of the mean (SEM) of repeated experiments.
Supplementary Material
Movie S1. Human cGAS∆N localizes to mitochondria.
Reproducibility Checklist
Fig. S1. Human cGAS∆N induces type I IFN responses.
Fig. S2. cGAS∆N activities vary across mammalian species.
Fig. S3. Recombinant mammalian cGAS proteins are activated by dsDNA in vitro.
Fig. S4. cGAS-DNA aggregates interfere with biochemical analysis of cGAS subcellular distribution.
Fig. S5. Responses of membrane-targeted hcGAS∆N to non-self-DNA.
Data file S1. Raw data file (Excel spreadsheet)
Acknowledgments:
We thank all members of the Kagan laboratory for helpful discussions.
Funding:
This work was supported by NIH grants AI133524, AI093589, AI116550 and P30DK34854 to J.C.K. and from the Pew Biomedical Scholars program, The Mark Foundation for Cancer Research, and the NIH (1DP2GM146250-01) to P.J.K. D.C.H. is funded by 1R35GM142689-01 and a Recruitment of First-Time, Tenure-Track Faculty Award from the Cancer Prevention & Research Institute of Texas (RR 170047). K.M.S. is supported as an NCI F99 Graduate Fellow (1F99CA274660), W.Z. was supported in part through a Charles A. King Trust Postdoctoral Fellowship, A.A.G. was supported by a National Science Foundation graduate research fellowship.
Footnotes
Competing interests: J.C.K. consults for and holds equity in Corner Therapeutics, Larkspur Biosciences, and Neumora Therapeutics. None of these relationships influenced the work performed in this study. The other authors declare no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Requests for resources or reagents should be directed to and will be fulfilled by the lead contact, J.C.K.
REFERENCES AND NOTES
- 1.Sun L, Wu J, Du F, Chen X, Chen ZJ, Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J et al. , Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Decout A, Katz JD, Venkatraman S, Ablasser A, The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol, (2021). [DOI] [PMC free article] [PubMed]
- 4.West AP et al. , Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackenzie KJ et al. , cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dou Z et al. , Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luecke S et al. , cGAS is activated by DNA in a length-dependent manner. EMBO Rep 18, 1707–1715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andreeva L et al. , cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Zhou W et al. , Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 174, 300–311 e311 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du M, Chen ZJ, DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnett KC et al. , Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 176, 1432–1446 e1411 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volkman HE, Cambier S, Gray EE, Stetson DB, Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tao J et al. , Nonspecific DNA Binding of cGAS N Terminus Promotes cGAS Activation. J Immunol 198, 3627–3636 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Kranzusch PJ, Lee AS, Berger JM, Doudna JA, Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep 3, 1362–1368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lahaye X et al. , NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 175, 488–501 e422 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Zierhut C et al. , The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 178, 302–315 e323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pathare GR et al. , Structural mechanism of cGAS inhibition by the nucleosome. Nature, (2020). [DOI] [PubMed]
- 18.Boyer JA et al. , Structural basis of nucleosome-dependent cGAS inhibition. Science, (2020). [DOI] [PMC free article] [PubMed]
- 19.Kujirai T et al. , Structural basis for the inhibition of cGAS by nucleosomes. Science, (2020). [DOI] [PMC free article] [PubMed]
- 20.Michalski S et al. , Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature, (2020). [DOI] [PubMed]
- 21.Zhao B et al. , The Molecular Basis of Tight Nuclear Tethering and Inactivation of cGAS. Nature, (2020). [DOI] [PMC free article] [PubMed]
- 22.Li T et al. , Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 371, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniels RW, Rossano AJ, Macleod GT, Ganetzky B, Expression of multiple transgenes from a single construct using viral 2A peptides in Drosophila. PLoS One 9, e100637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahier A, Jarriault S, Simultaneous expression of multiple proteins under a single promoter in Caenorhabditis elegans via a versatile 2A-based toolkit. Genetics 196, 605–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson KS et al. , Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 370, (2020). [DOI] [PubMed] [Google Scholar]
- 26.Chavarria-Smith J, Vance RE, Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9, e1003452 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hellmich KA et al. , Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS One 7, e49741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levinsohn JL et al. , Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8, e1002638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopes Fischer N, Naseer N, Shin S, Brodsky IE, Effector-triggered immunity and pathogen sensing in metazoans. Nat Microbiol 5, 14–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kufer TA, Creagh EM, Bryant CE, Guardians of the Cell: Effector-Triggered Immunity Steers Mammalian Immune Defense. Trends Immunol 40, 939–951 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ, Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 102, 17717–17722 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du JM et al. , cGAS and cancer therapy: a double-edged sword. Acta Pharmacol Sin 43, 2202–2211 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakhoum SF et al. , Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu H et al. , Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Woo SR et al. , STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H et al. , cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A 114, 1637–1642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng L et al. , STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demaria O et al. , STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A 112, 15408–15413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carozza JA et al. , Extracellular cGAMP is a cancer cell-produced immunotransmitter involved in radiation-induced anti-cancer immunity. Nat Cancer 1, 184–196 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcus A et al. , Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763 e754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hancks DC, Hartley MK, Hagan C, Clark NL, Elde NC, Overlapping Patterns of Rapid Evolution in the Nucleic Acid Sensors cGAS and OAS1 Suggest a Common Mechanism of Pathogen Antagonism and Escape. PLoS Genet 11, e1005203 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart SA et al. , Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campeau E et al. , A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kranzusch PJ et al. , Structure-guided reprogramming of human cGAS dinucleotide linkage specificity. Cell 158, 1011–1021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stetson DB, Medzhitov R, Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Human cGAS∆N localizes to mitochondria.
Reproducibility Checklist
Fig. S1. Human cGAS∆N induces type I IFN responses.
Fig. S2. cGAS∆N activities vary across mammalian species.
Fig. S3. Recombinant mammalian cGAS proteins are activated by dsDNA in vitro.
Fig. S4. cGAS-DNA aggregates interfere with biochemical analysis of cGAS subcellular distribution.
Fig. S5. Responses of membrane-targeted hcGAS∆N to non-self-DNA.
Data file S1. Raw data file (Excel spreadsheet)