

Abstract
Cancer cells have an abnormally high mitochondrial membrane potential (ΔΨm), which is associated with enhanced invasive properties in vitro and increased metastases in vivo. The mechanisms underlying the abnormal ΔΨm in cancer cells remain unclear. Research on different cell types has shown that ΔΨm is regulated by various intracellular mechanisms such as by mitochondrial inner and outer membrane ion transporters, cytoskeletal elements and biochemical signaling pathways. On the other hand, the role of extrinsic, tumor microenvironment (TME) derived cues in regulating ΔΨm is not well defined. In this review, we first summarize the existing literature on intercellular mechanisms of ΔΨm regulation, with a focus on cancer cells. We then offer our perspective on the different ways through which the microenvironmental cues such as hypoxia and mechanical stresses may regulate cancer cell ΔΨm.
Keywords: Mitochondrial membrane potential (ΔΨm), intracellular signaling, tumor microenvironment, metastasis
Graphical Abstract
1. Introduction
Nearly a century ago, Otto Warburg discovered that cancer cells have enhanced levels of glycolysis, and therefore abnormally high glucose uptake and lactate production even in the absence of hypoxic conditions (a phenomenon he called ‘aerobic glycolysis’) (Koppenol et al., 2011). It is now known that this phenomenon, i.e., the Warburg effect, is not a universal characteristic of all cancer cell types. Rather, tumors are highly heterogenous comprising of subpopulations of cancer cells displaying ‘Warburg-like’ metabolic characteristics (Hanahan & Weinberg, 2011). Nevertheless, this increased propensity for glucose consumption by some cancer cell subpopulations is routinely exploited for diagnostic fluorodeoxyglucose-positron emission tomography (FDG-PET) scans in many cancer types including breast and lung cancers (Zhu et al., 2011). Furthermore, such metabolic heterogeneity has emerged as an important cancer cell hallmark and a potential therapeutic target (Ashton et al., 2018; Ward & Thompson, 2012). Interestingly, studies investigating the mitochondrial activity of these metabolically heterogenous cancer cells have found an association between dysregulated mitochondrial activity and their invasiveness and metastatic potential (Dupuy et al., 2015; Porporato et al., 2014; Simoes et al., 2015; Whitaker-Menezes et al., 2011). A mechanistic understanding of the factors that lead to altered cancer cell mitochondrial activity is thus crucial to develop novel therapeutics, for instance, by targeting the mitochondrial support of metastatic behaviors.
A unique observation of cancer cell mitochondrial function is that many epithelial cancer cell types have an unusually high mitochondrial membrane potential (ΔΨm), compared to their normal counterparts (Chen, 1988; Summerhayes et al., 1982). Further studies showed an association of cancer cell ΔΨm with a range of cell behaviors including decreased susceptibility to apoptosis (Heerdt et al., 2003) and the acquisition of an invasive phenotype (Heerdt et al., 2005). In an in vivo mouse model of metastatic breast cancer, we found that mice injected with cancer cells of high ΔΨm have a greater lung metastatic burden than those with low ΔΨm cells, four weeks post tail-vein injection (Begum et al., 2019). The ΔΨm is thus associated with the metastatic development, and a greater understanding of the mechanisms by which it affects cell phenotypes will help in the discovery of novel therapeutic targets. However, whether the high ΔΨm is related to the ‘Warburg-phenotype’, and how it is differentially regulated in cancer cells, remains unclear.
One possibility is that the abnormally high ΔΨm in cancer cells is a result of genetic mutations that affect their intrinsic mitochondrial activity. Indeed, mutations in mitochondrial DNA (mtDNA) and variations in its copy number have been reported in several types of cancers (Ashton et al., 2018; Beadnell et al., 2018). Furthermore, it is possible that the altered ΔΨm results from the activity of oncogenes such as MYC, AKT, RAS, BRAF and reduction of tumor suppressors like p53, which were found to affect various metabolic pathways including those involving mitochondrial metabolism (Levine & Puzio-Kuter, 2010; Nagarajan et al., 2016; Ward & Thompson, 2012). While it is possible that the altered cancer cell ΔΨm is primarily a result of these cancer-specific mutations, there is also growing evidence supporting alternative mechanisms of physical interaction and biochemical signaling taking place within the tumor microenvironment (TME) (Pavlova & Thompson, 2016). In this review, we will briefly summarize how the ΔΨm is established and regulated, with a focus on TME-derived cues that have the capacity to regulate cancer cell ΔΨm. We will then discuss the current in vitro and in vivo techniques used to probe ΔΨm, and their potential applications in investigating ΔΨm-driven tumor progression.
2. How is the ΔΨm established?
Mitochondria are double membrane bound organelles. Transport of metabolites and ions across the outer mitochondrial membrane is regulated by large porin protein channels such as Voltage-Dependent Anion Channels (VDACs) that are permeable to solutes up to 5 kDa (Alberts et al., 2002). On the other hand, the mitochondrial inner membrane is largely impermeable, and transport of molecules across this membrane requires specialized membrane transport proteins (Alberts et al., 2002; Kühlbrandt, 2015). The inner membrane also forms deep invaginations into the mitochondrial matrix, called cristae (Fig. 1). The presence of cristae on the inner mitochondrial membrane greatly increases its effective surface area. It is also lined with respiratory complexes of the Electron Transport Chain (ETC) and is the site of mitochondrial adenosine triphosphate (ATP) synthesis. The mitochondrial matrix contains various enzymes that support the replication and transcription of mtDNA (Kühlbrandt, 2015). In addition, it the site of the tricarboxylic acid (TCA) cycle and contains TCA cycle associated enzymes.
Figure 1:

Schematic showing the tricarboxylic acid (TCA) cycle and electron transport chain (ETC) within the mitochondria.
Tricarboxylic acid (TCA) cycle:
Pyruvate from the cytosol (one of the end products of glycolysis) is transported into the mitochondrial matrix through a pyruvate carrier present on the inner mitochondrial membrane and converted to acetyl coenzyme A (acetyl CoA) by the enzyme pyruvate dehydrogenase. Acetyl CoA enters the TCA cycle, where it is oxidized to produce carbon dioxide and water (Pelley, 2012). During this process, electron carrier molecules nicotinamide adenine dinucleotide (NAD) and flavine adenine dinucleotide (FAD) are reduced, forming NADH and FADH2 from NAD+ and FAD respectively. These reduced electron carriers then take part in the ETC in the inner mitochondrial membrane (Pelley, 2012) (Fig. 1).
Electron transport chain (ETC):
The ETC takes place in the inner mitochondrial membrane and oxidizes electron carriers from the TCA cycle. Briefly, electrons from NADH and FADH2 are transferred to coenzyme Q by complexes I and II of the ETC respectively. Coenzyme Q carries these electrons to complex III, which donates them to cytochrome c (cyt c). Cyt c then diffuses along the membrane and transfers the electrons to complex IV, which then transfers these electrons to oxygen, eventually producing water (Pelley, 2012). In complexes I, III and IV, the activity of electron transfer is coupled to the pumping of protons into the mitochondrial intermembrane space (Pelley, 2012) (Fig. 1). This generates an electrochemical proton gradient across the mitochondrial inner membrane, which gives rise to the mitochondrial membrane potential (ΔΨm).
3. Functional significance of the ΔΨm
ATP synthesis:
Complex V of the ETC is an ATP synthase. Driven by the proton gradient generated by the ETC, it pumps protons back into the mitochondrial matrix and uses this energy to combine a molecule of adenosine diphosphate (ADP) with an inorganic phosphate to produce ATP (Pelley, 2012). This process of ATP production is termed oxidative phosphorylation (OXPHOS) since it requires the presence of oxygen. The ATP produced by the mitochondrial ATP synthase is transported out of the mitochondrial matrix in exchange for a molecule of cytosolic ADP by an adenine nucleotide transporter (ANT) (Fig. 1). Under some conditions of stress (such as ischemia), it has been reported that the ATP synthase and ANT reverse their activity – the ANT now transports ATP into the mitochondrial matrix in exchange for ADP, and the ATP synthase utilizes this ATP to form ADP and pump out protons into the mitochondrial intermembrane space, in an attempt to maintain the ΔΨm (Belous et al., 2003). In this state, mitochondria become major consumers of leftover cellular ATP, and try to maintain the ΔΨm in order to delay the onset of autophagic or apoptotic processes (which are associated with sustained ΔΨm depolarization). Zorova et al. suggest that the maintenance of ΔΨm at the expense of cellular ATP levels demonstrates how important it is for the cells to maintain stable ΔΨm levels (Zorova, Popkov, Plotnikov, Silachev, Pevzner, Jankauskas, Zorov, et al., 2018; Zorova, Popkov, Plotnikov, Silachev, Pevzner, Jankauskas, Babenko, et al., 2018).
Reactive oxygen species (ROS) production:
As explained in the previous section, there is a transport or shuttling of electrons across the complexes of the ETC in the inner mitochondrial membrane. However, this electron transfer is not perfect, and there is electron leakage at Complex I (in the direction of the mitochondrial matrix) and at Complex III (in both directions of the mitochondrial matrix and the intermembrane space) (Li et al., 2013). This leads to the partial reduction of molecular oxygen (O2) forming highly reactive free radicals called Reactive Oxygen Species (ROS) and include the superoxide anion, hydrogen peroxide and hydroxyl ions (Thannickal & Fanburg, 2000). In isolated rat heart mitochondria, ROS levels are low and unaffected by values of ΔΨm below 140 mV (Korshunov et al., 1997; Lee et al., 2001). Any increases in the ΔΨm beyond that threshold value leads to very high increases in ROS levels (Korshunov et al., 1997). Inhibiting the ATP synthase using oligomycin in cancer cells led to ΔΨm hyperpolarization along with increased ROS levels (Suski et al., 2012). These results suggest that a higher ΔΨm is associated with high ROS levels. However, in pathological conditions that affect the ETC complexes, low-ΔΨm levels have also been associated with high ROS levels (Lebiedzinska et al., 2010). Similarly, antimycin A (ETC Complex III inhibitor) treatment leads to a decrease in ΔΨm levels and increased mitochondrial ROS production (Suski et al., 2012). In summary, in mitochondrial disorders related to mitochondrial ATP synthase, increased ΔΨm is associated with high ROS levels; in disorders related to ETC subunits, decreased ΔΨm is associated with high ROS levels (Suski et al., 2012). To explain this apparent paradox, Li et al. propose an “ROS balance” hypothesis, suggesting that there is an optimal level of ΔΨm that is associated with physiological (low) levels of ROS (Li et al., 2013). Any extreme fluctuations in the ΔΨm levels (too low or too high) would lead to high ROS levels either by affecting the rate of electron transfer through the ETC or disrupting the balance of antioxidant enzymes (Li et al., 2013).
Mitochondrial quality control:
The mitochondrial network within cells consists of motile mitochondria (Hyde et al., 2010) that can fuse together when they encounter each other (also called mitochondrial fusion) or split into daughter mitochondria (a process called mitochondrial fission). Together with mitophagy (selective elimination of damaged mitochondria), these processes form the cells’ quality control strategy for maintaining the bioenergetic efficiency and integrity of their mitochondrial networks (Twig, Hyde, et al., 2008). The role of ΔΨm in regulating the processes of mitochondrial fusion, fission and mitophagy is examined below.
Mitochondrial fusion:
The process of mitochondrial fusion facilitates rapid diffusion of mitochondrial content, allowing exchange of nutrients and metabolites within the mitochondrial network (Chen & Chan, 2004). Long-term tracking of fusion events in individual mitochondria showed that fusion occurs as frequently as one event every 5–20 minutes per mitochondrion in cell lines derived from rodent kidney and pancreatic beta-cells (Twig, Elorza, et al., 2008; Twig, Hyde, et al., 2008). Mitofusins Mfn1 and Mfn2 mediate mitochondrial outer membrane fusion whereas Opa1 mediates fusion of inner mitochondrial membranes (van der Bliek et al., 2013). Fusion appears to be a selective event based on ΔΨm levels: mitochondria with high-ΔΨm have a greater probability to undergo fusion when compared to low-ΔΨm mitochondria (Twig, Elorza, et al., 2008); reduction of ΔΨm by uncoupling drugs hinders fusion and enhances fragmentation of the mitochondrial network (Legros et al., 2002; Meeusen et al., 2004; Twig, Elorza, et al., 2008). Depolarizing, i.e., a reduction in the ΔΨm is associated with proteolytic processing and therefore inactivation of OPA-1 (Duvezin-Caubet et al., 2006; Liu et al., 2009), and this could be a potential mechanism by which the ΔΨm regulates fusion. ΔΨm repolarization was found to restore fusion in breast cancer cells (Legros et al., 2002). Together, these studies suggest that the process of mitochondrial fusion requires an intact ΔΨm, and controlling the ΔΨm can regulate mitochondrial fusion events. Conversely, inhibiting the mitofusin proteins leads to a decrease in the overall ΔΨm of the cells, suggesting that the overall ΔΨm of the cells is maintained through fusion by allowing intermixing of mitochondrial matrix as well as membrane components (Chen & Chan, 2004).
Mitochondrial fission:
Mitochondrial fission is the process by which an individual mitochondrion splits into two daughter mitochondria. Interestingly, this form of mitochondrial division appears to be asymmetrical – more than four-fifths of all tracked fission events led to the formation of a depolarized and a hyperpolarized daughter mitochondrion (Twig, Elorza, et al., 2008). The depolarized daughter mitochondrion is approximately six times less likely than the hyperpolarized daughter mitochondrion to undergo a subsequent fusion event (Twig, Elorza, et al., 2008). In this manner, fission generates a distinct pool of low-ΔΨm mitochondria that are now segregated from the overall mitochondrial network of the cell. Fission is thus a mechanism by which defective mitochondria can be compartmentalized and subsequently removed from the mitochondrial network (Chen & Chan, 2004). The process of mitochondrial fission is mediated by the protein Drp1. Under normal conditions, Drp1 is localized to the cytoplasm. During fission, it is recruited to the outer mitochondrial membrane, where it forms complexes that join and then separate the outer and inner membranes (Shirihai et al., 2015). In myoblast cultures, the presence of excess fatty acids like palmitate in the medium leads to enhanced mitochondrial fragmentation mediated by Drp1, and this is associated with a reduction in ΔΨm (Jheng et al., 2012). Inhibiting Drp1 in this model prevents the fission-associated loss of ΔΨm (Jheng et al., 2012). This suggests that the recruitment of Drp1 to the mitochondrial membranes could play a role in reducing ΔΨm.
Strikingly, Drp1 is markedly upregulated in invasive breast carcinoma and lymph node metastases when compared to normal breast tissue (Zhao et al., 2013). Inhibiting Drp1 in breast cancer cells is associated with a decrease in their in vitro migration and invasiveness (Zhao et al., 2013). Similar results are also obtained with hepatocellular carcinoma cells, where mitochondrial fission is associated with cell migration (Sun et al., 2018). These findings suggest that fission may play an important regulatory role in the development of metastases.
Mitophagy:
In instances of premature electron leak from the ETC or dysregulation of certain mitochondrial enzymes, there is an accumulation of ROS (Shirihai et al., 2015), which are known to oxidize and damage mitochondrial DNA as well as mitochondrial proteins including the respiratory proteins themselves (Marchi et al., 2012). From a quality control perspective, it is important for cells to eliminate these mitochondria from the larger mitochondrial network to prevent propagation of harmful mitochondrial DNA mutations or damaged mitochondrial proteins. This process of selectively degrading defective mitochondria is known as mitophagy (Mijaljica et al., 2014).
Depolarized mitochondria (mostly arising from fission with asymmetric distribution of ΔΨm) are targeted for removal by mitophagy. Interestingly, depolarization of ΔΨm occurs long before these mitochondria are eliminated by mitophagy, and inhibiting mitophagy does not recover their ΔΨm (Twig, Elorza, et al., 2008). Instead, it leads to an accumulation of low-ΔΨm mitochondria (Twig, Elorza, et al., 2008). Mitophagy is thus important to clear away the pool of low-ΔΨm mitochondria and maintain the overall cellular ΔΨm. Studies have shown that the mechanism mediating mitophagy of low-ΔΨm specific mitochondria involves the ΔΨm-dependent activation of PINK1-Parkin proteins (Twig & Shirihai, 2011). Briefly, low-ΔΨm stabilizes PINK1 which is present in the outer mitochondrial membrane (Vives-Bauza et al., 2010). Activated PINK1 then recruits cytosolic Parkin (Twig & Shirihai, 2011; Vives-Bauza et al., 2010). The recruited Parkin then ubiquitinates and inactivates the mitofusin proteins, thus isolating the low-ΔΨm mitochondria by preventing the mitochondria from undergoing fusion and rejoining the overall mitochondrial network (Tanaka et al., 2010; Twig & Shirihai, 2011). Parkin activation also leads to recruitment of pro-mitophagy proteins such as p97 (Tanaka et al., 2010). Mitophagy is thus an important mitochondrial quality control mechanism, and inhibition of mitophagy leading to accumulation of damaged mitochondria has been implicated in promoting tumorigenesis (Chourasia et al., 2015; Mijaljica et al., 2014).
Mitochondrial and cellular motility:
For cells to be motile and migrate in the direction of certain cues, they need increased ATP production especially at the leading edges of the migrating cell layer. It has been reported that the cells at the leading edge of an epithelial migrating sheet have higher ΔΨm (Chen, 1988). A higher ΔΨm has also been associated with increased migration in cancer cells (Heerdt et al., 2005). Thus, the upregulation of ΔΨm may play an important role in mediating cell migration. Additionally, even within individual cells, high-ΔΨm mitochondria are transported selectively towards local regions of increased energy demand (Hyde et al., 2010). In neurons for example, one study reported that mitochondria that travel from the cell body (soma) to the synapse regions have high ΔΨm compared to the mitochondria that are transported from the synapse back to the cell body (Saxton & Hollenbeck, 2012). Together, these results suggest that the ΔΨm is an important regulator of both overall cell migration as well as individual mitochondrial motility within the cells.
4. Intrinsic regulation of the ΔΨm
ΔΨm regulates important mitochondrial as well as cellular processes including production of ROS (which can serve as important cell signaling molecules when at physiological levels) (Thannickal & Fanburg, 2000), mitochondrial dynamics and quality control mechanisms, as well as mitochondrial and cell motility. But what regulates the ΔΨm? In the following section we will examine different intracellular mechanisms controlling the ΔΨm, from ion channels on the inner mitochondrial membrane to regulation by the cytoskeleton and cell signaling pathways (Fig. 2A).
Figure 2:

Schematic showing intrinsic (A) and extrinsic (B) factors that may regulate cancer cell ΔΨm.
Regulation of ΔΨm by inner membrane ion transporters:
To ensure that the electrochemical energy created by the proton gradient generated by the ETC is most effectively used for ATP production, the inner mitochondrial membrane (IMM) must be mostly impermeable to ions except through the ATP synthase. This would ensure that any ΔΨm dissipation is tightly coupled with ATP production. Conversely, the flux of various ions across the IMM through their respective ion channels (such as K+, Ca2+) can regulate ΔΨm and uncouple it from ATP production (O’Rourke, 2007).
Mitochondrial calcium uniporter (MCU):
These channels are the major source of Ca2+ influx into the mitochondrial matrix (Giorgi et al., 2018). ΔΨm provides the driving force for Ca2+ flux through MCUs; dissipating the ΔΨm with chemical uncouplers such as FCCP reduces Ca2+ flux through these channels (Friel & Tsien, 1994; Giorgi et al., 2018; Jean-Quartier et al., 2012). Furthermore, ΔΨm itself is regulated by the MCU activity. Influx of Ca2+ ions leads to short-term ΔΨm depolarization (Gunter & Pfeiffer, 1990; Kamer et al., 2018). Sustained high levels of Ca2+ in the mitochondrial matrix could induce the mitochondrial permeability transition pore (MPTP) and permanent loss of ΔΨm (Granatiero et al., 2019). Thus, MCUs can regulate ΔΨm by determining matrix Ca2+ levels. Indeed, several studies have shown that manipulating MCU levels can affect ΔΨm. In Drosophila S2 cells, knockdown (k/d) of MCU rescued hydrogen peroxide induced ΔΨm-loss (Choi et al., 2017). Interestingly, in liver cancer cells it was found that k/d of MCUR1, a regulator of MCU that promotes mitochondrial Ca2+ uptake, is associated with a decrease in ΔΨm (Ren et al., 2018). One possible explanation could be MCUR1 k/d leads to an overall decrease in matrix Ca2+, which could reduce the activity of Ca2+-sensitive ETC enzymes (O’Rourke et al., 2005) hence negatively affecting the H+ pump that gives rise to the ΔΨm.
K+ channels:
There are several types of K+ channels on the IMM: mitoKATP, mitoBKCa and mitoKv1.3, which are regulated by ATP, Ca2+ and voltage respectively (Szewczyk et al., 2009). These mitochondrial K+ channels are thought to be involved in the regulation of ΔΨm, ROS generation as well as mitochondrial matrix volume (Laskowski et al., 2016). When there is ΔΨm hyperpolarization or an increase in ATP concentration, the opening of these channels leads to the dissipation of the ΔΨm (Bednarczyk, 2009; Szabò et al., 2012) and an amelioration of the oxidative stress associated with enhanced ROS levels at high ΔΨm (Laskowski et al., 2016). The ΔΨm of cervical cancer cells overexpressing the pore-forming subunit of the mitoKATP channel is significantly lower than that of the control cells, highlighting the ability of mitochondrial K+ flux in regulating ΔΨm (Paggio et al., 2019).
Uncoupling proteins (UCPs):
The ETC Complexes I, III and IV and the major producers of proton gradient; they pump protons into the mitochondrial intermembrane space. One of the major consumers of this proton gradient is the ATP synthase (ETC Complex V) when it is operating in its normal ATP-producing state. Over the past two decades, studies have found the presence of yet another class of consumers of the proton gradient on the inner mitochondrial membrane – the uncoupling proteins (UCPs) (Rousset et al., 2004). Originally found in brown adipose tissue (BAT), UCP1 is activated by free fatty acids and transports protons into the mitochondrial matrix, thus dissipating the ΔΨm without any resultant ATP production (Ricquier & Bouillaud, 2000). UCP1 is associated with energy dissipation as heat, which is essential in non-shivering thermogenesis in BAT (Ricquier & Bouillaud, 2000). Later, other types of UCPs were found to be expressed in several different cell types and associated with regulating cell functions such as proton leak, ΔΨm, ROS levels, insulin secretion and resting metabolic rate (Rousset et al., 2004). Notably, overexpression of UCP5 in neuroblastoma cells was found to decrease their ΔΨm (Kwok et al., 2010). In aortic endothelial cells, manipulations that decreased or increased UCP levels led to an increase or decrease in ΔΨm respectively (Shimasaki et al., 2013). These results provide compelling evidence that the ΔΨm can be regulated by the level and activity of UCPs in cells.
Regulation of ΔΨm by outer membrane ion transporters:
As mentioned earlier, VDACs form channels that selectively allow passage of solutes through the outer mitochondrial membrane based on their open/closed configuration. VDACs control mitochondrial function by regulating the transport of important charged metabolites through the mitochondria (such as succinate2- and ATP4-) (Colombini, 2004). Low ΔΨm lead to an ‘open’ configuration of VDAC which favors the selective transport of anions over cations through these channels. Higher magnitudes of ΔΨm (> 40 mV) lead to a ‘closed’ configuration which favors the transport of cations (Maldonado & Lemasters, 2012). Meanwhile, VDAC closure could regulate ΔΨm by reducing the OMM permeability to adenine nucleotides (like ANT) (Szabo & Zoratti, 2014). This would decrease substrate availability for the ΔΨm-consuming ATP synthase activity, thus increasing the overall ΔΨm. Evidence for VDAC regulation of ΔΨm has been reported in several studies. In neuroblastoma cells, dopamine stimulation leads to a transient decrease in ΔΨm, which can be prevented by modulating the open/close configuration of the VDACs (Premkumar & Simantov, 2002). Moreover, the ΔΨm-regulation by free cellular tubulin is also reported to be dependent on VDAC phosphorylation; inhibiting VDAC phosphorylation by protein kinase A (PKA) abrogates the inhibitory effect of enhanced free tubulin level on ΔΨm (Sheldon et al., 2011). Further investigation on VDAC isoforms shows that, while inhibition of different VDACs (using siRNA) all leads to a decrease in ΔΨm, VDAC3 k/d causes the greatest ΔΨm reduction (Maldonado & Lemasters, 2012; Maldonado et al., 2013). Additionally, VDAC activity can also be regulated by binding to hexokinase, a cytoplasmic enzyme involved in glycolysis (Pastorino & Hoek, 2008). K/d of hexokinase using shRNA decreases the ΔΨm of cancer cells, an effect that can be reversed by the presence of VDAC inhibitors (Dubey et al., 2016).
ΔΨm regulation by cytoskeletal elements:
The cytoskeleton regulates the spatial organization of subcellular organelles and couples them biochemically and physically to the extracellular cues (Fletcher & Mullins, 2010). It is mainly comprised of three types of polymers: microtubules, microfilaments (or actin filaments), and intermediate filaments (Fletcher & Mullins, 2010). Studies have shown that the cytoskeletal filaments can control mitochondrial motility, morphology and subcellular location (Kuznetsov et al., 2020). In this section, we will examine the role of these cytoskeletal filaments in regulating the ΔΨm.
Microtubules:
Microtubules are one of the main cytoskeletal elements associated with the transport of mitochondria within the cells (Kuznetsov et al., 2020). They are the stiffest cytoskeletal element (Fletcher & Mullins, 2010) and are made of tubulin heterodimers. Microtubule polymerization can be blocked by drugs that act as microtubule destabilizers such as nocodazole, rotenone and colchicine (Maldonado et al., 2010), which increase levels of free tubulin in cells (Maldonado et al., 2010). On the other hand, microtubule polymerization can be stabilized or enhanced by treatment with drugs such as paclitaxel, which decreases free tubulin levels as shown in hepatocellular carcinoma cells (Maldonado et al., 2010). Interestingly, microtubule depolymerization (increased free tubulin) decreases ΔΨm while its stabilization (decreased free tubulin) increases ΔΨm in cancer cells (Maldonado et al., 2010). This contrasts with non-cancer cells where microtubule stabilization does not affect ΔΨm (Maldonado et al., 2010). Other studies show that both depolymerizing and stabilizing microtubule decrease ΔΨm in myocytes (Kumazawa et al., 2014). In neurons, microtubule stabilization with paclitaxel leads to a significant decrease in the ΔΨm (Melli et al., 2008). In human dermal fibroblasts, depolymerizing microtubule with nocodazole does not change ΔΨm (Kandel et al., 2016). Together, these studies show that the microtubule control of ΔΨm is highly cell-type specific. A potential mechanism by which microtubule stability affects ΔΨm involves the binding of free tubulin to and closing of VDACs in the outer mitochondrial membrane (Rostovtseva et al., 2008), which reduces ADP transport into the mitochondria and overall mitochondrial activity (Maldonado et al., 2010; Rostovtseva et al., 2008).
Actin filaments (or microfilaments):
In contrast to microtubules which mediate long-distance transport of mitochondria within the cells, actin filaments have been associated with short-range mitochondrial transport as well as mediating the anchorage of the mitochondria in regions of high energy demand (Boldogh & Pon, 2006). Monomeric actin (G-actin) can polymerize and form diverse structures of filamentous actin (F-actin) (Fletcher & Mullins, 2010). The activity of several types of actin regulatory proteins can dictate the overall structure of the cell’s actin network (Fletcher & Mullins, 2010). For example, actin assembly in the presence of formins leads to the formation of liner actin filaments, whereas the activity of the Arp2/3 complex leads to branched actin networks (Davidson & Wood, 2016). In T-cells and neurons, gelsolin (an actin severing protein that caps actin filaments and prevents their elongation) is reported to prevent the loss of ΔΨm that is associated with the onset of apoptosis (Harms et al., 2004; Koya et al., 2000). Additionally, a small fraction of the overexpressed gelsolin in T-cells was found to colocalize with the mitochondria, suggesting that it may regulate ΔΨm by direct binding (Gourlay & Ayscough, 2005b; Koya et al., 2000). Interestingly, a more detailed study revealed that the mitochondrial fraction of the overexpressed gelsolin specifically bound to the VDACs, suggesting a regulatory mechanism of ΔΨm by gelsolin through VDACs (Kusano et al., 2000). Coronin, another actin binding protein that opposes the activity of Arp2/3 and thus inhibits F-actin formation is essential for maintaining ΔΨm in T-cells. Loss of Coronin-1 leads to a decrease in ΔΨm, which can be partially recovered by treatment with latrunculin A (an actin depolymerizing drug) (Foger et al., 2006). On the other hand, jasplakinolide treatment, which increases F-actin, leads to a decrease in ΔΨm (Dustin, 2006; Foger et al., 2006). These results suggest that the ratio of G-actin to F-actin in cells could potentially regulate their ΔΨm. A recent study in cancer cells reported that at any given time, about 20% of the mitochondria are associated with actin filaments (F-actin), and this association while regulating mitochondrial dynamics, had no effect on their ΔΨm (Moore et al., 2016). Regulation of ΔΨm by the actin cytoskeleton could therefore be highly context dependent and cell-type specific.
The relationship between actin dynamics and mitochondrial activity has also been explored in other model organisms such as yeast and plant cells. In yeast cells, mutations in actin regulatory proteins that promote the turnover of F-actin (Sla1p and End3p) lead to a loss of ΔΨm (Gourlay & Ayscough, 2005a). In plant root hair cells, both actin polymerizing and depolymerizing treatments (with jasplakinolide and latrunculin B respectively) reduce the ΔΨm (Wang et al., 2010). In agreement with these results, latrunculin treatment in mung bean plant cells also reduces the ΔΨm (Lo et al., 2011). Taken together, these studies provide compelling evidence that the dynamics of cellular actin cytoskeleton could regulate ΔΨm.
Intermediate Filaments (IFs):
IFs have the least stiffness out of the three types of cytoskeletal elements and are thus better suited for receiving mechanical stimuli of tensile rather than compressive nature (Fletcher & Mullins, 2010). Mutations in IF genes are associated with altered mitochondrial distribution and morphology in several pathological conditions including Charcot-Marie-Tooth disease and epidermolysis bullosa simplex (Matveeva et al., 2015). In mouse fibroblasts, a portion of vimentin IFs colocalizes with mitochondria (Nekrasova et al., 2011). Further, disrupting the vimentin IFs leads to a redistribution of the mitochondrial network as well as increased mitochondrial motility, suggesting that vimentin IFs are involved in regulating the spatial organization and motility of mitochondria within these cells (Nekrasova et al., 2011). Interestingly, motile mitochondria were found to have a lower ΔΨm than their stationary counterparts (Chernoivanenko et al., 2011); restoring vimentin expression in the vimentin-null fibroblasts decreased mitochondrial motility (Nekrasova et al., 2011) while increasing their ΔΨm (Chernoivanenko et al., 2015; Chernoivanenko et al., 2011). Conversely, silencing vimentin using shRNA in wild-type cells (which express normal levels of vimentin) leads to a decrease in ΔΨm (Chernoivanenko et al., 2015). A more detailed study revealed that the vimentin IF-mitochondria association could be disrupted by the phosphorylation of vimentin at its Ser-55 site by Rac-1, and activated Rac-1 increases mitochondrial motility and reduces the ΔΨm (Matveeva et al., 2015). Together, these results strongly suggest that the association of mitochondria with vimentin IFs helps anchor the mitochondria at specific intracellular locations (thus reducing their motility) and increases their ΔΨm.
Regulation of ΔΨm by signaling pathways:
The activities of several mitochondrial proteins, including the ETC complexes, can also regulate the ΔΨm. These activities can be expression of tissue-type specific isoforms of ETC subunits, their allosteric inhibition from interactions with small molecules (such as ADP), and their altered phosphorylation state by cell signaling molecules (Hüttemann et al., 2008). For example, both ETC Complexes I and IV, which contribute to ΔΨm through proton pumping, are regulated via PKA-mediated phosphorylation induced by c-AMP (Hüttemann et al., 2007). Akt was found to localize to the mitochondria following PI3K signaling, to phosphorylate ATP synthase (ETC Complex V) (Bijur & Jope, 2003). Whether the phosphorylation of ETC Complexes serves to activate or inhibit them seems to be dependent on the subunit type and site of phosphorylation. Notably, several other signaling molecules, including tyrosine kinases such as Abl, Src, EGFR and ErbB2 and serine threonine kinases like JNK, GSK3β, were also found to localize to the mitochondria and modulate ETC Complex activity (Lim et al., 2016). As such, various upstream signaling pathways can potentially regulate the ΔΨm.
To summarize, several studies suggest that the regulation of ΔΨm can be associated with mitochondrial ion channels, by cytoskeletal elements and cellular signaling pathways, and some of these results are derived from cancer cells. However, carefully designed experiments are needed to investigate specifically whether and how the ΔΨm is differentially regulated in normal vs. cancer cells by each of these factors.
5. ΔΨm of normal vs. cancer cells
More than thirty years ago, Summerhayes et al. first reported that most in vitro cultures of epithelial cancer cells have a much higher ΔΨm (as assessed by uptake and retention of ΔΨm-sensitive dye rhodamine 123) when compared to non-transformed cells (Summerhayes et al., 1982). Apart from these cancer cells, they found cardiac muscle cells and myotubes to possess such “unusually” high levels of ΔΨm (Summerhayes et al., 1982). Other cancer cell types screened from blood cancers, connective tissue derived cancers (osteosarcoma) and neuroblastomas do not have the high ΔΨm that is characteristic of epithelial carcinomas (Chen, 1988). To find out whether intrinsic differences in the ΔΨm of cancer cells can affect tumor growth and progression, Heerdt et al. subcloned isogenic colonic carcinoma cells with stable differences in their ΔΨm (Heerdt et al., 2003). They found that the intrinsic ΔΨm differences of these cells are associated with differences in mitochondrial mRNA levels, butyrate-induced cell cycle arrest and susceptibility to apoptosis (Heerdt et al., 2003). Further studies with these cells showed that the high-ΔΨm subclones have higher vascular endothelial growth factor (VEGF) and matrix metalloproteinase 7 (MMP7) secretion, as well as higher invasiveness (Heerdt et al., 2005, 2006). Similarly, high-ΔΨm breast cancer (MCF-7) subclones are also shown to have greater VEGF secretion than the low-ΔΨm subclones, suggesting a correlation of ΔΨm with invasive properties in these cells (Houston et al., 2011).
Bonnet et al. attempted to selectively target the high-ΔΨm of cancer cells. They found that treatment with dichloroacetate (DCA), a pharmacological inhibitor of mitochondrial pyruvate dehydrogenase kinase (PDK), selectively lowers the ΔΨm of cancer cells (breast and lung carcinoma cells, as well as glioma cells) while having no effect on the ΔΨm of non-cancerous epithelial cells (Bonnet et al., 2007). Several types of cancer cells have re-wired metabolic pathways, most notably, increased glycolysis relative to oxidative metabolism (Warburg effect). By inhibiting PDK (which phosphorylates and inactivates pyruvate dehydrogenase, thus reducing mitochondrial pyruvate influx), DCA increases mitochondrial pyruvate influx and shifts the balance from glycolysis to oxidative metabolism in cancer cells. Further, DCA treatment also causes release of cytochrome c from the mitochondria, thus inducing apoptosis (Bonnet et al., 2007). In in vivo studies, it was found that DCA treatment can reduce tumor cell proliferation and increase their apoptosis, thus reducing the overall tumor size (Bonnet et al., 2007).
Most studies that reported observations of high ΔΨm of cancer cells were performed using in vitro cultures. Recently, Momcilovic et al. used a positron emission tomography (PET) imaging technique to investigate the ΔΨm of lung cancer cells in vivo (Momcilovic et al., 2019). They found a tumor-subtype dependent ΔΨm heterogeneity in these mouse models – adenocarcinomatous tumors have a higher ΔΨm whereas small squamous cell carcinoma tumors have lower ΔΨm (Momcilovic et al., 2019). In summary, cancer cells are found to have highly dysregulated mitochondrial functions (Vyas et al., 2016), which can manifest as differences in their ΔΨm (Chen, 1988).
6. How does the TME regulate ΔΨm of cancer cells?
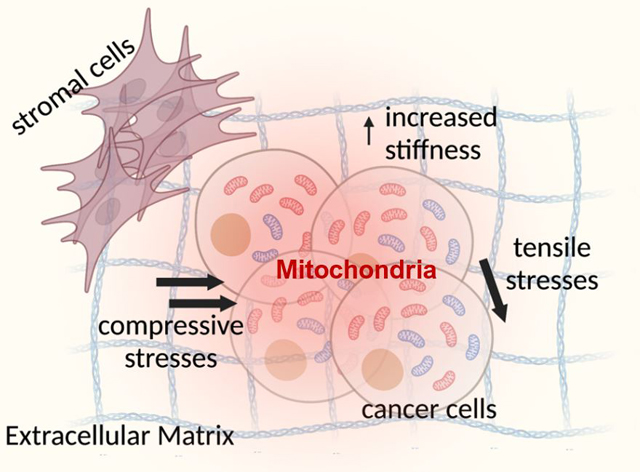
The immediate environment surrounding tumors in vivo, commonly referred to as the tumor microenvironment (TME), is a rich source of biophysical and biochemical cues that directly influence cancer cell behaviors. Several different cell types apart from the primary tumor cells are found in the TME: immune cells, fibroblasts, adipocytes, mesenchymal stem cells, endothelial cells and pericytes (Balkwill et al., 2012). Biochemical cues derived from cell-type specific signaling are important in directing tumor progression. For example, the interaction of cancer cells and dendritic cells in the TME is thought to regulate Treg expansion within the tumor and thereby mediate tumor escape from immune attacks (Whiteside, 2008). In addition, tumor progression in the TME has been associated with biophysical cues such as low oxygen concentrations (hypoxia) (Ando et al., 2017; Petrova et al., 2018), remodeling of the extracellular matrix (ECM) resulting in a change in both its composition and stiffness (Winkler et al., 2020), as well as compressive and shear stresses (Chaudhuri et al., 2018; Nia et al., 2020). These TME-derived cues are potential regulators of cancer cell mitochondrial activity and their roles in influencing the ΔΨm are explored below (Fig. 2B).
Hypoxia:
Exposure to hypoxia induces several changes in mitochondrial metabolism and the ΔΨm response to hypoxia appears to be highly cell-type specific and metabolic state-dependent. In myoblasts (Hawkins et al., 2010), infected macrophages (Wiese et al., 2012) and kidney proximal tubule cells (Weinberg et al., 2000), hypoxia has been reported to depolarize the ΔΨm (Khacho et al., 2014). However, in blastocysts (Ma et al., 2017) and hypoxia-sensitive glioblastoma cells (Turcotte et al., 2002), the ΔΨm increases in response to hypoxic stimuli. Furthermore, in hypoxia-resistant glioblastoma cells (Turcotte et al., 2002), uninfected macrophages (Wiese et al., 2012) and renal cell carcinoma cells (Papandreou et al., 2006), the ΔΨm remains unchanged under hypoxia. One potential explanation for these differences is in the distinct ways individual cell types adapt the efficiency and rate of mitochondrial respiration under hypoxia. In hypoxia-resistant glioblastoma cells, the ΔΨm remains constant under hypoxia, likely due to a compensatory decrease in overall oxygen consumption or stabilization of the ETC, leading to constant proton flux into the mitochondrial intermembrane space despite the reduced electron flux, which is not seen in the hypoxia-sensitive glioblastoma cells (Turcotte et al., 2002). Additionally, hypoxia has been associated with several other mechanisms that could determine the ΔΨm response. These include HIF-1α induced inhibition of pyruvate dehydrogenase, which decreases the overall mitochondrial respiration by reducing substrate availability, expression of hypoxia-specific isoforms of ETC complexes (cytochrome c oxidase), inhibition of ETC complex I, and the inhibition of ATP-synthase.
Substrate Stiffness and Composition:
Stromal secretion of lysyl oxidase (LOX) enzymes in the TME has been associated with increased ECM stiffness, promoting cancer cell migration and invasion (Winkler et al., 2020). Substrate stiffness has been shown to regulate the ΔΨm of several different cell types in vitro. Pulmonary arterial endothelial and smooth muscle cells (Bertero et al., 2016), and cardiomyocytes (Morishima et al., 2018) demonstrate enhanced ΔΨm when cultured on softer substrates. Another study on vascular smooth muscle cells found that their ΔΨm was the highest when cultured on a substrate with stiffness close to the in vivo stiffness of the tissue of origin (Bartolák-Suki et al., 2017), while both softer and stiffer substrates significantly reduced the ΔΨm of these cells. In addition to ECM stiffness, the overall ECM composition can also be modified in the TME which may in turn alter ΔΨm. In several tumor types, there is an increased deposition of fibrillar collagen, fibronectin and hyaluronan, leading to a desmoplastic phenotype that is linked to poor prognosis(Winkler et al., 2020). It was shown that pancreatic cancer cells cultured on substrates coated with fibronectin and laminin have a much higher ΔΨm than those on collagen coated or non-adherent substrates (Vaquero et al., 2003). Furthermore, altering the composition of anabolic vs. catabolic ECM proteins in nucleus pulposus cells was also found to change their ΔΨm (Wu et al., 2018). In vascular smooth muscle cells, silencing an ECM protein (cartilage oligomeric matrix protein) led to a decrease in their ΔΨm (Jia et al., 2018). Together, these studies suggest a possible role of ECM stiffness and composition in regulating cancer cell ΔΨm.
Compressive and tensile stresses:
As tumors grow, they push against the surrounding stromal tissue and accumulate solid stresses within the tumor. It was shown that there are compressive forces at the tumor core and tensile stresses at the tumor periphery (Jain et al., 2014). While the compressive stress has been reported to enhance the migration of breast cancer (Tse et al., 2012) and glioma (Kalli et al., 2019) cells, its effect on ΔΨm is still unclear and remains to be investigated. On the other hand, tensile stresses have found to regulate ΔΨm in various cell types in vitro. In human neuroblastoma cells, mild to moderate stretch was shown to depolarize the ΔΨm (Wang et al., 2014). Interestingly, sustained mechanical stretch reduces the ΔΨm of cardiomyocytes but has no effect on that of cardiac fibroblasts (Liao et al., 2004). Interestingly, bovine aortic smooth muscle cells have increased ΔΨm under both monotonous and variable stretching (Bartolak-Suki et al., 2015). These data demonstrate a potential role of static and dynamic tensile stresses in TME in altering the ΔΨm in cancer cells.
7. Techniques to investigate ΔΨm
In vivo techniques:
Determining the ΔΨm of cells in vivo is not very straightforward and remains challenging through mitochondrial dyes and direct imaging. Some recent studies have reported the use of novel approaches to measure the ΔΨm of live cells in vivo. Lipophilic cations such as triphenylphosphonium (TPP) can readily accumulate in the mitochondria (owing to the ΔΨm), which can be conjugated with compounds for mitochondrial targeting (Zielonka et al., 2017). A novel TPP-based approach was reported by Logan et al. where two TPP-based probes with complementary click chemistry moieties are injected in vivo (in mouse models) that accumulate in the mitochondria in a ΔΨm (and plasma membrane potential) dependent manner, where they form a stable “MitoClick” compound. The amount of “MitoClick” accumulated in different cell types or tissues can be quantified by using liquid chromatography-tandem mass spectrometry (LC-MS/MS), and this approach was shown to sensitively report small changes in ΔΨm in vivo (Logan et al., 2016). The second TPP-based approach to measure ΔΨm in vivo uses a radiolabeled TPP tracer that accumulates in tissues in a ΔΨm-dependent manner and can then be imaged using positron emission tomography (PET). Using this approach in a mouse model of lung tumor, Momcilovic et al. report that the lung tumors could be segregated into distinct subtypes (adenocarcinoma vs. squamous cell carcinoma) based on their estimated in vivo ΔΨm (Momcilovic et al., 2019). These TPP-based approaches are promising new techniques for in vivo estimation of ΔΨm.
In vitro techniques:
The most widely used techniques to monitor the ΔΨm in vitro are based on lipophilic cationic fluorescent dyes that accumulate in the mitochondria in a ΔΨm-dependent manner (Perry et al., 2011). The ΔΨm can then be assessed by quantifying the fluorescence of these mitochondria-accumulated dyes using either fluorescence microscopy, flow cytometry or microplate reader-based approaches. Some of the commonly used ΔΨm fluorescent probes include tetramethylrhodamine methyl ester (TMRM), 5,5′,6,6’-tetrachloro-1,1’,3,3′- tetraethylbenzimidazolylcarbocyanine iodide (JC-1), 1,1′,3,3,3′,3′-hexamethylindodicarbo - cyanine iodide (DiIC1(5)), and rhodamine 123 (Rho123) (Martin et al., 2011; Perry et al., 2011; Walsh et al., 2017). Out of these dyes, TMRM is known to have the least inhibitory effect on mitochondrial activity and hence is the most preferred dye to use for in vitro ΔΨm measurements (Perry et al., 2011). Dye-based imaging of ΔΨm has several advantages such as real-time measurement of ΔΨm in response to administered cues (for reversible dyes), and spatial resolution allowing visualization of spatially resolved ΔΨm heterogeneity within a cell population. However, with many of these dyes, there is a concentration dependence that would determine the interpretation of the observed fluorescence intensity. As Perry et al. have reported, there is a ‘non-quenching’ mode where the dyes are used between 0.5 – 30 nM concentration, where ΔΨm hyperpolarization would lead to increased fluorescence intensity whereas ΔΨm depolarization would decrease observed fluorescence intensity. However, when these dyes are used at higher concentrations between 50 – 100 nM, increased aggregation of the dye in the mitochondria in response to ΔΨm hyperpolarization would result in ‘quenching’ of the dye fluorescence. Furthermore, in our own experience, the concentration of the ΔΨm-dyes as well as the staining time needs to be optimized for each cell line, such that any experimental treatments performed to investigate changes in the ΔΨm must be performed in the phase of dye-loading where the observed dye fluorescence plateaus. Another challenge with reversible dye-based approaches is the fact that the dye needs to remain in the imaging buffer, or it would lead to a steady loss of fluorescence over time and the samples that are imaged at the beginning would seem to have a higher- ΔΨm than the ones imaged at the end. However, having the dye in the imaging buffer would also interfere with imaging quality and resolution. There is hence a need for novel approaches to probe ΔΨm within cells. Recently, a new technique to probe ΔΨm has been reported that utilizes photoinduced electron transfer (PeT)-based Rhodamine Voltage Reporter (RhoVR) instead of the traditional lipophilic dyes that accumulate in the mitochondria in a ΔΨm-dependent manner (Klier et al., 2021) and has been shown to be sensitive to FCCP-induced ΔΨm-depolarization as well as allowing simultaneous probing of cells for cytosolic Ca2+ and plasma membrane potential with other probes. Additionally, Okkelman et al. report using TMRM for fluorescence lifetime imaging microscopy (FLIM), and shows promise for simultaneous probing of mitochondrial bioenergetics and ΔΨm (Okkelman et al., 2020).
In vitro models to study the microenvironmental regulation of ΔΨm:
As summarized in the previous sections, ΔΨm can be differentially regulated by cues that remodel the cellular cytoskeleton. The TME is a rich source of biophysical cues that play a role in remodeling cancer cell cytoskeleton to facilitate their invasive and migratory properties (Li & Wang, 2020). We have previously developed an in vitro micropatterning approach to recapitulate the confinement of tumor islands by stromal cells (Begum et al., 2019; Shen et al., 2014). A spatial distribution of ΔΨm was found within the micropatterned tumor islands, where the MCF-7 breast cancer cells at the edges of the micropattern had higher ΔΨm compared to those at the micropattern centers (Begum et al., 2019). Interestingly, altering the density of stromal cells surrounding the micropatterned tumor island can also regulate the spatial distribution of ΔΨm – a high degree of stromal confinement led to a very narrow region of cancer cells with high ΔΨm (Begum et al., 2019). To ensure that these results were not confounded by biochemical signals derived from the stromal cells, we mechanically confined the cancer cells in a micropattern surrounded by a thin layer of polydimethylsiloxane (PDMS). This led to a uniformly low-ΔΨm throughout the micropatterned tumor island, indicating that physical confinement as a key mechanism by which the surrounding stromal cells regulate the spatial ΔΨm distribution in the micropatterned tumor model.
Interestingly, RNA-sequencing of cancer cells from the centers and edges of the tumor micropatterns revealed that pathways related to adherens junctions (E-cadherin mediated intercellular adhesions) are upregulated at the micropattern centers (Begum et al., 2021). Through inhibition, overexpression, and knockdown of E-cadherin, we confirmed that the confinement cues from the TME regulate cancer cell ΔΨm through their effects on E-cadherin mediated intercellular adhesions. Thus, in vitro models recapitulative of the TME can be important tools in discovering novel mechanisms that regulate the ΔΨm in cancer cells.
8. Conclusions
It has been known for decades that cancer cells have abnormally high ΔΨm. However, it remains unclear whether this phenotype is a result of cell-intrinsic mutations specific to and characteristic of cancer cells, or due to the activation of aberrant intracellular signaling pathways or from extrinsic cues in the TME. Given the crucial role of the TME in dictating cancer cell behavior, we propose that specific cues from the TME can alter the ΔΨm of cancer cells through intracellular physicochemical signaling. A better understanding of the source of altered cancer cell ΔΨm can lead to the discovery of new targets for attacking cancer cells, by reducing their metabolic plasticity and potentially their metastatic ability. Since the abnormally high ΔΨm has been observed in a wide variety of epithelial carcinoma cells, a detailed understanding of the underlying mechanisms giving rise to this phenotype will be beneficial for developing new therapeutic targets for a wide range of carcinomas.
Acknowledgments
This work was supported by an NIH National Cancer Institute grant (R01CA220012), an NIH National Institute of Biomedical Imaging and Bioengineering Trailblazer Award (R21EB024748), a STOP CANCER Marni Levine Memorial Research Career Development Award, the USC Viterbi School of Engineering, and the USC Provost’s PhD Fellowship. This research was also supported by shared resources from an NIH National Cancer Institute Award (P30CA014089). We would also like to thank Chelsea Mariano and Irene Li for their help with literature review.
Footnotes
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Alberts B, Johnson A, & Lewis J. (2002). The Mitochondrion. In (4th editio ed.). [Google Scholar]
- Ando Y, Ta HP, Yen DP, Lee S. s., Raola S, & Shen K. (2017). A Microdevice Platform Recapitulating Hypoxic Tumor Microenvironments. Scientific Reports, 7: 15233, 1–12. 10.1038/s41598-017-15583-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton TM, McKenna WG, Kunz-Schughart LA, & Higgins GS (2018). Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res, 24(11), 2482–2490. 10.1158/1078-0432.CCR-17-3070 [DOI] [PubMed] [Google Scholar]
- Balkwill FR, Capasso M, & Hagemann T. (2012). The tumor microenvironment at a glance. J Cell Sci, 125(Pt 23), 5591–5596. 10.1242/jcs.116392 [DOI] [PubMed] [Google Scholar]
- Bartolak-Suki E, Imsirovic J, Parameswaran H, Wellman TJ, Martinez N, Allen PG, . . . Suki B. (2015). Fluctuation-driven mechanotransduction regulates mitochondrial-network structure and function. Nat Mater, 14(10), 1049–1057. 10.1038/nmat4358 [DOI] [PubMed] [Google Scholar]
- Bartolák-Suki E, Imsirovic J, Nishibori Y, Krishnan R, & Suki B. (2017). Regulation of mitochondrial structure and dynamics by the cytoskeleton and mechanical factors. International Journal of Molecular Sciences, 18(8), 7–11. 10.3390/ijms18081812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadnell TC, Scheid AD, Vivian CJ, & Welch DR (2018). Roles of the mitochondrial genetics in cancer metastasis: not to be ignored any longer. Cancer Metastasis Rev, 37(4), 615–632. 10.1007/s10555-018-9772-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarczyk P. (2009). Potassium channels in brain mitochondria. Acta Biochimica Polonica, 56(3), 385–392. 10.18388/abp.2009_2471 [DOI] [PubMed] [Google Scholar]
- Begum HM, Mariano C, Zhou H, & Shen K. (2021). E-Cadherin Regulates Mitochondrial Membrane Potential in Cancer Cells. Cancers, 13(20). 10.3390/cancers13205054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum HM, Ta HP, Zhou H, Ando Y, Kang D, Nemes K, . . . Shen K. (2019). Spatial Regulation of Mitochondrial Heterogeneity by Stromal Confinement in Micropatterned Tumor Models. Sci Rep, 9(1), 11187. 10.1038/s41598-019-47593-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belous A, Knox C, Nicoud IB, Pierce J, Anderson C, Pinson CW, & Chari RS (2003). Reversed activity of mitochondrial adenine nucleotide translocator in ischemia-reperfusion. Transplantation, 75(10), 1717–1723. 10.1097/01.TP.0000063829.35871.CE [DOI] [PubMed] [Google Scholar]
- Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, . . . Chan SY (2016). Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest, 126(9), 3313–3335. 10.1172/JCI86387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, & Jope RS (2003). Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem, 87(6), 1427–1435. 10.1046/j.1471-4159.2003.02113.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldogh IR, & Pon LA (2006). Interactions of mitochondria with the actin cytoskeleton. Biochim Biophys Acta, 1763(5–6), 450–462. 10.1016/j.bbamcr.2006.02.014 [DOI] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, . . . Michelakis ED (2007). A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell, 11(1), 37–51. 10.1016/j.ccr.2006.10.020 [DOI] [PubMed] [Google Scholar]
- Chaudhuri PK, Low BC, & Lim CT (2018). Mechanobiology of Tumor Growth. Chem Rev, 118(14), 6499–6515. 10.1021/acs.chemrev.8b00042 [DOI] [PubMed] [Google Scholar]
- Chen H, & Chan DC (2004). Mitochondrial Dynamics in Mammals. In (Vol. 59, pp. 119–144). [DOI] [PubMed] [Google Scholar]
- Chen LB (1988). Mitochondrial Membrane Potential in Living Cells. Annual Review of Cell Biology, 4(1), 155–181. 10.1146/annurev.cb.04.110188.001103 [DOI] [PubMed] [Google Scholar]
- Chernoivanenko IS, Matveeva EA, Gelfand VI, Goldman RD, & Minin AA (2015). Mitochondrial membrane potential is regulated by vimentin intermediate filaments. FASEB Journal, 29(3), 820–827. 10.1096/fj.14-259903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoivanenko IS, Matveeva EA, & Minin AA (2011). Vimentin intermediate filaments increase mitochondrial membrane potential. Biochemistry (Moscow) Supplement Series A: Membrane and Cell Biology, 5(1), 21–28. 10.1134/S1990747811010041 [DOI] [Google Scholar]
- Choi S, Quan X, Bang S, Yoo H, Kim J, Park J, . . . Chung J. (2017). Mitochondrial calcium uniporter in Drosophila transfers calcium between the endoplasmic reticulum and mitochondria in oxidative stress-induced cell death. Journal of Biological Chemistry, 292(35), 14473–14485. 10.1074/jbc.M116.765578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Boland ML, & Macleod KF (2015). Mitophagy and cancer. Cancer and Metabolism, 3(1), 1–11. 10.1186/s40170-015-0130-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombini M. (2004). VDAC: The channel at the interface between mitochondria and the cytosol. Molecular and Cellular Biochemistry, 107–115. [DOI] [PubMed] [Google Scholar]
- Davidson AJ, & Wood W. (2016). Unravelling the Actin Cytoskeleton: A New Competitive Edge? Trends in Cell Biology, 26(8), 569–576. 10.1016/j.tcb.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey AK, Godbole A, & Mathew MK (2016). Regulation of VDAC trafficking modulates cell death. Cell Death Discovery, 2(1). 10.1038/cddiscovery.2016.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, . . . Siegel PM (2015). PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab, 22(4), 577–589. 10.1016/j.cmet.2015.08.007 [DOI] [PubMed] [Google Scholar]
- Dustin ML (2006). When F-actin becomes too much of a good thing. Science, 313(5788), 767–768. 10.1126/science.1131714 [DOI] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, . . . Reichert AS (2006). Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. Journal of Biological Chemistry, 281(49), 37972–37979. 10.1074/jbc.M606059200 [DOI] [PubMed] [Google Scholar]
- Fletcher DA, & Mullins RD (2010). Cell mechanics and the cytoskeleton. Nature, 463(7280), 485–492. 10.1038/nature08908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foger N, Rangell L, Danilenko DM, & Chan AC (2006). Requirement for Coronin 1 in T Lymphocyte Trafficking and Cellular Homeostasis. Science, 313(August), 839–842. 10.1016/b978-1-4832-3292-8.50018-7 [DOI] [PubMed] [Google Scholar]
- Friel DD, & Tsien RW (1994). An FCCP-sensitive Ca2+store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. Journal of Neuroscience, 14(7), 4007–4024. 10.1523/jneurosci.14-07-04007.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Marchi S, & Pinton P. (2018). The machineries, regulation and cellular functions of mitochondrial calcium. Nature Reviews Molecular Cell Biology, 19(11), 713–730. 10.1038/s41580-018-0052-8 [DOI] [PubMed] [Google Scholar]
- Gourlay CW, & Ayscough KR (2005a). Identification of an upstream regulatory pathway controlling actin-mediated apoptosis in yeast. Journal of Cell Science, 118(10), 2119–2132. 10.1242/jcs.02337 [DOI] [PubMed] [Google Scholar]
- Gourlay CW, & Ayscough KR (2005b). The actin cytoskeleton: A key regulator of apoptosis and ageing? Nature Reviews Molecular Cell Biology, 6(7), 583–589. 10.1038/nrm1682 [DOI] [PubMed] [Google Scholar]
- Granatiero V, Pacifici M, Raffaello A, De Stefani D, & Rizzuto R. (2019). Overexpression of Mitochondrial Calcium Uniporter Causes Neuronal Death. Oxidative Medicine and Cellular Longevity, 2019. 10.1155/2019/1681254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, & Pfeiffer DR (1990). Mechanisms by which mitochondria transport calcium. American Journal of Physiology - Cell Physiology, 258(5 27–5), C755-C786. 10.1152/ajpcell.1990.258.5.c755 [DOI] [PubMed] [Google Scholar]
- Hanahan D, & Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 646–674. https://doi.org/S0092-8674(11)00127-9[pii] 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Harms C, Bösel J, Lautenschlager M, Harms U, Braun JS, Hörtnagl H, . . . Endres M. (2004). Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Molecular and Cellular Neuroscience, 25(1), 69–82. 10.1016/j.mcn.2003.09.012 [DOI] [PubMed] [Google Scholar]
- Hawkins BJ, Levin MD, Doonan PJ, Petrenko NB, Davis CW, Patel VV, & Madesh M. (2010). Mitochondrial complex II prevents hypoxic but not calcium- and proapoptotic Bcl-2 protein-induced mitochondrial membrane potential loss. J Biol Chem, 285(34), 26494–26505. 10.1074/jbc.M110.143164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerdt BG, Houston MA, & Augenlicht LH (2005). The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res, 65(21), 9861–9867. 10.1158/0008-5472.CAN-05-2444 [DOI] [PubMed] [Google Scholar]
- Heerdt BG, Houston MA, & Augenlicht LH (2006). Growth properties of colonic tumor cells are a function of the intrinsic mitochondrial membrane potential. Cancer Res, 66(3), 1591–1596. 10.1158/0008-5472.CAN-05-2717 [DOI] [PubMed] [Google Scholar]
- Heerdt BG, Houston MA, Wilson AJ, & Augenlicht LH (2003). The intrinsic mitochondrial membrane potential (δψm) is associated with steady-state mitochondrial activity and the extent to which colonic epithelial cells undergo butyrate-mediated growth arrest and apoptosis. Cancer Research, 63(19), 6311–6319. [PubMed] [Google Scholar]
- Houston MA, Augenlicht LH, & Heerdt BG (2011). Stable differences in intrinsic mitochondrial membrane potential of tumor cell subpopulations reflect phenotypic heterogeneity. Int J Cell Biol, 2011, 978583. 10.1155/2011/978583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde BB, Twig G, & Shirihai OS (2010). Organellar vs cellular control of mitochondrial dynamics. Seminars in Cell and Developmental Biology, 21(6), 575–581. 10.1016/j.semcdb.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Lee I, Pecinova A, Pecina P, Przyklenk K, & Doan JW (2008). Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. Journal of Bioenergetics and Biomembranes, 40(5), 445–456. 10.1007/s10863-008-9169-3 [DOI] [PubMed] [Google Scholar]
- Hüttemann M, Lee I, Samavati L, Yu H, & Doan JW (2007). Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochimica et Biophysica Acta - Molecular Cell Research, 1773(12), 1701–1720. 10.1016/j.bbamcr.2007.10.001 [DOI] [PubMed] [Google Scholar]
- Jain RK, Martin JD, & Stylianopoulos T. (2014). The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng, 16, 321–346. 10.1146/annurev-bioeng-071813-105259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Quartier C, Bondarenko AI, Alam MR, Trenker M, Waldeck-Weiermair M, Malli R, & Graier WF (2012). Studying mitochondrial Ca 2+ uptake - A revisit. Molecular and Cellular Endocrinology, 353(1–2), 114–127. 10.1016/j.mce.2011.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, . . . Tsai YS (2012). Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Molecular and Cellular Biology, 32(2), 309–319. 10.1128/mcb.05603-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Wang M, Mao C, Yu F, Wang Y, Xiao R, . . . Kong W. (2018). COMP-prohibitin 2 interaction maintains mitochondrial homeostasis and controls smooth muscle cell identity. Cell Death Dis, 9(6), 676. 10.1038/s41419-018-0703-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli M, Voutouri C, Minia A, Pliaka V, Fotis C, Alexopoulos LG, & Stylianopoulos T. (2019). Mechanical Compression Regulates Brain Cancer Cell Migration Through MEK1/Erk1 Pathway Activation and GDF15 Expression. Front Oncol, 9, 992. 10.3389/fonc.2019.00992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, Sancak Y, Fomina Y, Meisel JD, Chaudhuri D, Grabarek Z, & Mootha VK (2018). MICU1 imparts the mitochondrial uniporter with the ability to discriminate between Ca2+ and Mn2+. Proceedings of the National Academy of Sciences of the United States of America, 115(34), E7960–E7969. 10.1073/pnas.1807811115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel J, Angelin AA, Wallace DC, & Eckmann DM (2016). Mitochondrial respiration is sensitive to cytoarchitectural breakdown. Integrative Biology (United Kingdom), 8(11), 1170–1182. 10.1039/c6ib00192k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, . . . Slack RS (2014). Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun, 5, 3550. 10.1038/ncomms4550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klier P, Martin J, & Miller E. (2021). Imaging reversible mitochondrial membrane potential dynamics with a masked rhodamine voltage reporter. In. J Am Chem Soc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, & Dang CV (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer, 11(5), 325–337. 10.1038/nrc3038 [DOI] [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, & Starkov AA (1997). High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Letters, 416(1), 15–18. 10.1016/S0014-5793(97)01159-9 [DOI] [PubMed] [Google Scholar]
- Koya RC, Fujita H, Shimizu S, Ohtsu M, Takimoto M, Tsujimoto Y, & Kuzumaki N. (2000). Gelsolin inhibits apoptosis by blocking mitochondrial membrane potential loss and cytochrome c release. Journal of Biological Chemistry, 275(20), 15343–15349. 10.1074/jbc.275.20.15343 [DOI] [PubMed] [Google Scholar]
- Kumazawa A, Katoh H, Nonaka D, Watanabe T, Saotome M, Urushida T, . . . Hayashi H. (2014). Microtubule disorganization affects the mitochondrial permeability transition pore in cardiac myocytes. Circulation Journal, 78(5), 1206–1215. 10.1253/circj.CJ-13-1298 [DOI] [PubMed] [Google Scholar]
- Kusano H, Shimizu S, Koya RC, Fujita H, Kamada S, Kuzumaki N, & Tsujimoto Y. (2000). Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene, 19, 4807–4814. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Javadov S, Grimm M, Margreiter R, Ausserlechner MJ, & Hagenbuchner J. (2020). Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells. Cells, 9, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok KHH, Ho PWL, Chu ACY, Ho JWM, Liu HF, Yiu DCW, . . . Ho SL (2010). Mitochondrial UCP5 is neuroprotective by preserving mitochondrial membrane potential, ATP levels, and reducing oxidative stress in MPP+ and dopamine toxicity. Free Radical Biology and Medicine, 49(6), 1023–1035. 10.1016/j.freeradbiomed.2010.06.017 [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W. (2015). Structure and function of mitochondrial membrane protein complexes. BMC Biology, 13(1), 1–11. 10.1186/s12915-015-0201-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski M, Augustynek B, Kulawiak B, Koprowski P, Bednarczyk P, Jarmuszkiewicz W, & Szewczyk A. (2016). What do we not know about mitochondrial potassium channels? Biochimica et Biophysica Acta - Bioenergetics, 1857(8), 1247–1257. 10.1016/j.bbabio.2016.03.007 [DOI] [PubMed] [Google Scholar]
- Lebiedzinska M, Karkucinska-Wieckowska A, Giorgi C, Karczmarewicz E, Pronicka E, Pinton P, . . . Wieckowski MR (2010). Oxidative stress-dependent p66Shc phosphorylation in skin fibroblasts of children with mitochondrial disorders. Biochimica et Biophysica Acta - Bioenergetics, 1797(6–7), 952–960. 10.1016/j.bbabio.2010.03.005 [DOI] [PubMed] [Google Scholar]
- Lee I, Bender E, Arnold S, & Kadenbach B. (2001). New control of mitochondrial membrane potential and ROS formation - A hypothesis. Biological Chemistry, 382(12), 1629–1636. 10.1515/BC.2001.198 [DOI] [PubMed] [Google Scholar]
- Legros F. d. r., Lombe A, Frachon P, & Rojo M. (2002). Mitochondrial Fusion in Human Cells is Efficient, Requires the Inner Membrane Potential, and Is Mediated by Mitofusins. Molecular Biology of the Cell, 13(December), 4343–4354. 10.1091/mbc.E02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, & Puzio-Kuter AM (2010). The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science, 330. [DOI] [PubMed] [Google Scholar]
- Li X, Fang P, Mai J, Choi ET, Wang H, & Yang XF (2013). Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. Journal of Hematology and Oncology, 6(1), 1–19. 10.1186/1756-8722-6-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, & Wang J. (2020). Mechanical tumor microenvironment and transduction: cytoskeleton mediates cancer cell invasion and metastasis. Int J Biol Sci, 16(12), 2014–2028. 10.7150/ijbs.44943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao XD, Wang XH, Jin HJ, C. L. Y, & Chen Q. (2004). Mechanical stretch induces mitochondria-dependent apoptosis in neonatal rat cardiomyocytes and G2/M accumulation in cardiac fibroblasts. Cell Research, 14. [DOI] [PubMed] [Google Scholar]
- Lim S, Smith KR, Lim STS, Tian R, Lu J, & Tan M. (2016). Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell and Bioscience, 6(1), 1–15. 10.1186/s13578-016-0089-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weaver D, Shirihai O, & Hajnóczky G. (2009). Mitochondrial kiss-and-run: Interplay between mitochondrial motility and fusion-fission dynamics. EMBO Journal, 28(20), 3074–3089. 10.1038/emboj.2009.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YS, Cheng N, Hsiao LJ, Annamalai A, Jauh GY, Wen TN, . . . Chiang KS (2011). Actin in mung bean mitochondria and implications for its function. Plant Cell, 23(10), 3727–3744. 10.1105/tpc.111.087403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan A, Pell VR, Shaffer KJ, Evans C, Stanley NJ, Robb EL, . . . Murphy MP (2016). Assessing the mitochondrial membrane potential in cells and in vivo using targeted click chemistry and mass spectrometry. Cell Metabolism, 23(2), 379–385. 10.1016/j.cmet.2015.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YY, Chen HW, & Tzeng CR (2017). Low oxygen tension increases mitochondrial membrane potential and enhances expression of antioxidant genes and implantation protein of mouse blastocyst cultured in vitro. J Ovarian Res, 10(1), 47. 10.1186/s13048-017-0344-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado EN, & Lemasters JJ (2012). Warburg revisited: Regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. Journal of Pharmacology and Experimental Therapeutics, 342(3), 637–641. 10.1124/jpet.112.192153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado EN, Patnaik J, Mullins MR, & Lemasters JJ (2010). Free Tubulin Modulates Mitochondrial Membrane Potential in Cancer Cells. Cancer Research, 70(24), 10192–10201. 10.1158/0008-5472.can-10-2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM, . . . Lemasters JJ (2013). Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem, 288(17), 11920–11929. 10.1074/jbc.M112.433847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, . . . Pinton P. (2012). Mitochondria-Ros Crosstalk in the Control of Cell Death and Aging. Journal of Signal Transduction, 2012, 1–17. 10.1155/2012/329635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SA, Hewish M, Sims D, Lord CJ, & Ashworth A. (2011). Parallel high-throughput RNA interference screens identify PINK1 as a potential therapeutic target for the treatment of DNA mismatch repair-deficient cancers. Cancer Res, 71(5), 1836–1848. 10.1158/0008-5472.CAN-10-2836 [DOI] [PubMed] [Google Scholar]
- Matveeva EA, Venkova LS, Chernoivanenko IS, & Minin AA (2015). Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol Open, 4(10), 1290–1297. 10.1242/bio.011874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S, McCaffery JM, & Nunnari J. (2004). Mitochondrial fusion intermediates revealed in vitro. Science, 305(5691), 1747–1752. 10.1126/science.1100612 [DOI] [PubMed] [Google Scholar]
- Melli G, Taiana M, Camozzi F, Triolo D, Podini P, Quattrini A, . . . Lauria G. (2008). Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Experimental Neurology, 214(2), 276–284. 10.1016/j.expneurol.2008.08.013 [DOI] [PubMed] [Google Scholar]
- Mijaljica D, Prescott M, & Devenish RJ (2014). Mitophagy: An Overview (Fourth Edi ed., Vol. 4). Elsevier Inc. 10.1016/B978-0-12-405528-5.00005-5 [DOI] [Google Scholar]
- Momcilovic M, Jones A, Bailey ST, Waldmann CM, Li R, Lee JT, . . . Shackelford DB (2019). In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature, 575(7782), 380–384. 10.1038/s41586-019-1715-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Wong YC, Simpson CL, & Holzbaur EL (2016). Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat Commun, 7, 12886. 10.1038/ncomms12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima M, Horikawa K, & Funaki M. (2018). Cardiomyocytes cultured on mechanically compliant substrates, but not on conventional culture devices, exhibit prominent mitochondrial dysfunction due to reactive oxygen species and insulin resistance under high glucose. PLoS One, 13(8), e0201891. 10.1371/journal.pone.0201891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan A, Malvi P, & Wajapeyee N. (2016). Oncogene-directed alterations in cancer cell metabolism. Trends Cancer, 2(7), 365–377. 10.1016/j.trecan.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasova OE, Mendez MG, Chernoivanenko IS, Tyurin-Kuzmin PA, Kuczmarski ER, Gelfand VI, . . . Minin AA (2011). Vimentin intermediate filaments modulate the motility of mitochondria. Molecular Biology of the Cell, 22(13), 2282–2289. 10.1091/mbc.E10-09-0766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nia HT, Munn LL, & Jain RK (2020). Physical traits of cancer. Science, 370(6516). 10.1126/science.aaz0868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B. (2007). Mitochondrial ion channels. Annual Review of Physiology, 69, 19–49. 10.1146/annurev.physiol.69.031905.163804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B, Cortassa S, & Aon MA (2005). Mitochondrial ion channels: Gatekeepers of life and death. Physiology, 20(5), 303–315. 10.1152/physiol.00020.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkelman I, Papkovsky D, & Dmitriev R. (2020). Estimation of the Mitochondrial Membrane PotentialUsing Fluorescence Lifetime Imaging Microscopy. In. Journal of Quantitative Cell Science. [DOI] [PubMed] [Google Scholar]
- Paggio A, Checchetto V, Campo A, Menabò R, Di Marco G, Di Lisa F, . . . De Stefani D. (2019). Identification of an ATP-sensitive potassium channel in mitochondria. Nature, 572(7771), 609–613. 10.1038/s41586-019-1498-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, & Denko NC (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab, 3(3), 187–197. 10.1016/j.cmet.2006.01.012 [DOI] [PubMed] [Google Scholar]
- Pastorino JG, & Hoek JB (2008). Regulation of hexokinase binding to VDAC. Journal of Bioenergetics and Biomembranes, 40(3), 171–182. 10.1007/s10863-008-9148-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, & Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab, 23(1), 27–47. 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelley JW (2012). Citric Acid Cycle, Electron Transport Chain, and Oxidative Phosphorylation. Elsevier’s Integrated Review Biochemistry, 57–65. 10.1016/b978-0-323-07446-9.00007-6 [DOI] [Google Scholar]
- Perry SW, Norman JP, Barbieri J, Brown EB, & Gelbard HA (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques, 50(2), 98–115. 10.2144/000113610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova V, Annicchiarico-Petruzzelli M, Melino G, & Amelio I. (2018). The hypoxic tumour microenvironment. Oncogenesis, 7(1). 10.1038/s41389-017-0011-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, . . . Sonveaux P. (2014). A mitochondrial switch promotes tumor metastasis. Cell Rep, 8(3), 754–766. 10.1016/j.celrep.2014.06.043 [DOI] [PubMed] [Google Scholar]
- Premkumar A, & Simantov R. (2002). Mitochondrial voltage-dependent anion channel is involved in dopamine-induced apoptosis. Journal of Neurochemistry, 82(2), 345–352. 10.1046/j.1471-4159.2002.00966.x [DOI] [PubMed] [Google Scholar]
- Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu Y, . . . Xing J. (2018). MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxidants and Redox Signaling, 28(12), 1120–1136. 10.1089/ars.2017.6990 [DOI] [PubMed] [Google Scholar]
- Ricquier D, & Bouillaud F. (2000). Mitochondrial uncoupling proteins: From mitochondria to the regulation of energy balance. Journal of Physiology, 529(1), 3–10. 10.1111/j.1469-7793.2000.00003.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, & Sackett DL (2008). Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proceedings of the National Academy of Sciences of the United States of America, 105(48), 18746–18751. 10.1073/pnas.0806303105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, & Ricquier D. (2004). The Biology of Mitochondrial Uncoupling Proteins. Diabetes, 53(SUPPL. 1). 10.2337/diabetes.53.2007.s130 [DOI] [PubMed] [Google Scholar]
- Saxton WM, & Hollenbeck PJ (2012). The axonal transport of mitochondria. Journal of Cell Science, 125(9), 2095–2104. 10.1242/jcs.053850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon KL, Maldonado EN, Lemasters JJ, Rostovtseva TK, & Bezrukov SM (2011). Phosphorylation of voltage-dependent anion channel by serine/threonine kinases governs its interaction with tubulin. PLoS One, 6(10), 1–10. 10.1371/journal.pone.0025539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Luk S, Hicks DF, Elman JS, Bohr S, Iwamoto Y, . . . Parekkadan B. (2014). Resolving cancer-stroma interfacial signalling and interventions with micropatterned tumour-stromal assays. Nat Commun, 5, 5662. 10.1038/ncomms6662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimasaki Y, Pan N, Messina LM, Li C, Chen K, Liu L, . . . Keaney JF (2013). Uncoupling Protein 2 Impacts Endothelial Phenotype via p53-Mediated Control of Mitochondrial Dynamics. Circulation Research, 113(7), 891–901. 10.1161/CIRCRESAHA.113.301319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirihai OS, Song M, & Dorn GW (2015). How mitochondrial dynamism orchestrates mitophagy. Circulation Research, 116(11), 1835–1849. 10.1161/CIRCRESAHA.116.306374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes RV, Serganova IS, Kruchevsky N, Leftin A, Shestov AA, Thaler HT, . . . Ackerstaff E. (2015). Metabolic plasticity of metastatic breast cancer cells: adaptation to changes in the microenvironment. Neoplasia, 17(8), 671–684. 10.1016/j.neo.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerhayes IC, Lampidis TJ, Bernal SD, Nadakavukaren JJ, Nadakavukaren KK, Shepherd EL, & Chen LB (1982). Unusual retention. of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proc Natl Acad Sci U S A, 79, 5292–5296,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Cao H, Zhan L, Yin C, Wang G, Liang P, . . . Xing, J. (2018). Mitochondrial fission promotes cell migration by Ca 2+ /CaMKII/ERK/FAK pathway in hepatocellular carcinoma. Liver International, 38(7), 1263–1272. 10.1111/liv.13660 [DOI] [PubMed] [Google Scholar]
- Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, & Wieckowski MR (2012). Relation Between Mitochondrial Membrane Potential and ROS formation. In (Vol. 810, pp. 103–117). 10.1007/978-1-61779-382-0 [DOI] [PubMed] [Google Scholar]
- Szabo I, & Zoratti M. (2014). Mitochondrial channels: Ion fluxes and more. Physiological Reviews, 94(2), 519–608. 10.1152/physrev.00021.2013 [DOI] [PubMed] [Google Scholar]
- Szabò I, Leanza L, Gulbins E, & Zoratti M. (2012). Physiology of potassium channels in the inner membrane of mitochondria. Pflugers Archiv European Journal of Physiology, 463(2), 231–246. 10.1007/s00424-011-1058-7 [DOI] [PubMed] [Google Scholar]
- Szewczyk A, Jarmuszkiewicz W, & Kunz WS (2009). Mitochondrial potassium channels. IUBMB Life, 61(2), 134–143. 10.1002/iub.155 [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, & Youle RJ (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. Journal of Cell Biology, 191(7), 1367–1380. 10.1083/jcb.201007013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ, & Fanburg BL (2000). Reactive oxygen species in cell signaling. American Journal of Physiology-Lung Cellular and Molecular Physiology, 279, L1005–L1028. 10.1002/9780470988565.ch9 [DOI] [PubMed] [Google Scholar]
- Tse JM, Cheng G, Tyrrell JA, Wilcox-Adelman SA, Boucher Y, Jain RK, & Munn LL (2012). Mechanical compression drives cancer cells toward invasive phenotype. Proc Natl Acad Sci U S A, 109(3), 911–916. 10.1073/pnas.1118910109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte ML, Parliament M, Franko A, & Allalunis-Turner J. (2002). Variation in mitochondrial function in hypoxia-sensitive and hypoxia-tolerant human glioma cells. British Journal of Cancer, 86, 619–624. 10.1038/sj/bjc/6600087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, . . . Shirihai OS (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO Journal, 27(2), 433–446. 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Hyde B, & Shirihai OS (2008). Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochimica et Biophysica Acta - Bioenergetics, 1777(9), 1092–1097. 10.1016/j.bbabio.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, & Shirihai OS (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxidants and Redox Signaling, 14(10), 1939–1951. 10.1089/ars.2010.3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek AM, Shen Q, & Kawajiri S. (2013). Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harbor Perspectives in Biology, 5(6). 10.1101/cshperspect.a011072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero EC, Edderkaoui M, Nam KJ, Gukovsky I, Pandol SJ, & Gukovskaya AS (2003). Extracellular matrix proteins protect pancreatic cancer cells from death via mitochondrial and nonmitochondrial pathways. Gastroenterology, 125(4), 1188–1202. 10.1016/s0016-5085(03)01203-4 [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, De Vries RLA, Tocilescu M, & Przedborski S. (2010). PINK1/Parkin direct mitochondria to autophagy. Autophagy, 6(2), 315–316. 10.4161/auto.6.2.11199 [DOI] [PubMed] [Google Scholar]
- Vyas S, Zaganjor E, & Haigis MC (2016). Mitochondria and Cancer. Cell, 166(3), 555–566. 10.1016/j.cell.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Siebenwirth C, Greubel C, Ilicic K, Reindl J, Girst S, . . . Dollinger G. (2017). Live cell imaging of mitochondria following targeted irradiation in situ reveals rapid and highly localized loss of membrane potential. In. Scientific Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Franco R, Skotak M, Hu G, & Chandra N. (2014). Mechanical stretch exacerbates the cell death in SH-SY5Y cells exposed to paraquat: mitochondrial dysfunction and oxidative stress. Neurotoxicology, 41, 54–63. 10.1016/j.neuro.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu Y, Ling Y, Zhang H, Liu P, Baluška F, . . . Wang Q. (2010). Disruption of actin filaments induces mitochondrial Ca2+release to the cytoplasm and [Ca2+]cchanges in Arabidopsis root hairs. BMC Plant Biology, 10. 10.1186/1471-2229-10-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, & Thompson CB (2012). Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell, 21(3), 297–308. 10.1016/j.ccr.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JM, Venkatachalam MA, Roeser NF, & Nissim I. (2000). Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci U S A, 97, 2826–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, . . . Sotgia F. (2011). Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle, 10(23), 4047–4064. 10.4161/cc.10.23.18151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside TL (2008). The tumor microenvironment and its role in promoting tumor growth. Oncogene, 27(45), 5904–5912. 10.1038/onc.2008.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese M, Gerlach RG, Popp I, Matuszak J, Mahapatro M, Castiglione K, . . . Jantsch J. (2012). Hypoxia-mediated impairment of the mitochondrial respiratory chain inhibits the bactericidal activity of macrophages. Infect Immun, 80(4), 1455–1466. 10.1128/IAI.05972-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, & Werb Z. (2020). Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nature Communications, 11(1), 1–19. 10.1038/s41467-020-18794-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Song Y, Li S, Liu X, Hua W, Wang K, . . . Yang C. (2018). Pramlintide regulation of extracellular matrix (ECM) and apoptosis through mitochondrial-dependent pathways in human nucleus pulposus cells. Int J Immunopathol Pharmacol, 31, 394632017747500. 10.1177/0394632017747500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, . . . Tu Y. (2013). Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene, 32(40), 4814–4824. 10.1038/onc.2012.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Lee D, & Shim H. (2011). Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin Oncol, 38(1), 55–69. 10.1053/j.seminoncol.2010.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, . . . Kalyanaraman B. (2017). Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem Rev, 117(15), 10043–10120. 10.1021/acs.chemrev.7b00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorova LD, Popkov VA, Plotnikov EJ, Silachev DN, Pevzner IB, Jankauskas SS, . . . Zorov DB (2018). Functional Significance of the Mitochondrial Membrane Potential. Biochemistry (Moscow) Supplement Series A: Membrane and Cell Biology, 12(1), 20–26. 10.1134/S1990747818010129 [DOI] [Google Scholar]
- Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, . . . Zorov DB (2018). Mitochondrial membrane potential. Anal Biochem, 552, 50–59. 10.1016/j.ab.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]