

Abstract
Infections with Shiga toxin (Stx)-producing bacteria cause bloody diarrhea which may progress to life-threatening complications, including acute renal failure and neurological abnormalities. The precise mechanism of disease progression is unclear, although evidence suggests that the localized production of the host proinflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin-1 may exacerbate toxin-mediated vascular damage. Purified Stxs have been demonstrated to elicit proinflammatory cytokine synthesis from human peripheral blood mononuclear cells and monocytic cell lines in vitro. To understand toxin-monocyte interactions required for cytokine synthesis, we have treated differentiated THP-1 cells with purified wild-type toxins, enzymatic mutants, or B subunits and measured TNF-α production. Our data suggest that A subunit enzymatic activity is essential for cytokine production. THP-1 cells were treated with a series of protein kinase C (PKC), PKA, and protein tyrosine kinase inhibitors to examine the role of intracellular signaling molecules in Stx-mediated cytokine production. Treatment of cells with PKC and tyrosine kinase inhibitors blocked TNF-α secretion by Stx-stimulated THP-1 cells. Stx treatment directly activated PKC, which occurred at a point upstream of transcriptional activation of the gene encoding TNF-α.
Shiga toxins (Stxs) are a group of antigenically and functionally related protein toxins produced by the enteric pathogens Shigella dysenteriae serotype 1 and Escherichia coli serotypes categorized as enterohemorrhagic E. coli. All members of the Stx family are AB5 holotoxins composed of an approximately 32-kDa A subunit protein in noncovalent association with a pentameric ring of identical B subunit proteins, each with a molecular mass of ∼7.7 kDa. The A subunit is the enzymatic component of the toxins and acts as a highly specific N-glycosidase enzyme hydrolyzing the bond between ribose and a single adenine residue found on a prominent loop structure in the 28S rRNA component of eukaryotic ribosomes (6, 30). The depurination reaction results in the rapid inhibition of protein synthesis. Toxin binding to cells is mediated by B subunits which primarily associate with the membrane neutral glycolipids globotriaosylceramide (Gb3; Galα1→4Galβ1→4GlcCer) (16, 18) and globotetraosylceramide (Gb4; GalNAcβ1→3Galα1→4Galβ1→4GlcCer) (4, 29). S. dysenteriae serotype 1 produces the prototypical Stx, whereas E. coli has been shown to elaborate one or more toxins closely related to Stx, designated Stx1, Stx2, Stx2c, Stx2d, and Stx2e (reviewed in reference 22).
Although infections with Stx-producing organisms generally cause self-limited bloody diarrhea, patients are at increased risk for the development of life-threatening complications. Chief among these complications are progression to acute renal failure (hemolytic uremic syndrome) and neurological abnormalities, such as seizures, stroke, and coma (17, 34). A frequent pathologic finding in these sequelae is damage to capillaries in the colon, kidneys, and central nervous system (12). Thus, Stxs appear to selectively damage microvascular endothelial cells. This concept was supported by work demonstrating that human vascular endothelial cells were killed when exposed to purified Stx in vitro (23). It was subsequently shown that human endothelial cells were sensitized to the cytotoxic action of Stxs by treatment of the cells with the proinflammatory cytokines tumor necrosis factor alpha (TNF-α) or interleukin 1 (IL-1) (19, 38, 42). The mechanism of sensitization involved increased endothelial cell Gb3 biosynthesis through TNF receptor p55 signaling and protein kinase C (PKC) activation (20, 41). Thus, the proinflammatory cytokine response to Stxs may contribute to pathogenesis by sensitizing endothelial cells to the direct toxic effects of Stxs.
Cellular sources of proinflammatory cytokines produced in vivo in response to Stxs remain to be characterized. However, it has been shown that human monocytes and monocytic cell lines respond to the toxins by synthesizing and secreting TNF-α, IL-1β, and IL-6 (26, 43). Using Northern blot analyses, we showed that Stxs affect cytokine expression, at least in part, at the transcriptional level via mechanisms that may involve activation of the DNA-binding proteins nuclear factor κB (NF-κB) and activator protein-1 (28). The precise mechanisms by which Stxs, potent protein synthesis inhibitors that interact with membrane glycolipids, transduce signals in monocytes necessary to activate cytokine gene expression are not known. The experiments reported here are designed to elucidate the toxin components necessary for proinflammatory cytokine induction and to initially characterize host cell signaling pathways involved in cytokine expression.
MATERIALS AND METHODS
Toxins and cytokine inducers.
Purified Stx1 and Stx2e were prepared from recombinant E. coli strains expressing toxin operons under control of thermoinducible promoters. Toxins were purified from bacterial sonicates by sequential DEAE-Sepharose and chromatofocusing column chromatography or MonoQ high-performance liquid chromatography as previously described (10, 29, 38). At each step, fractions were tested for Vero cell cytotoxicity by using the assay of Gentry and Dalrymple (8). A purified Stx1 enzymatic mutant, in which glutamate at position 167 and arginine at position 170 in the A subunit were replaced with glutamine and leucine, respectively (Stx1-E167Q-R170L), by oligonucleotide-directed site-specific mutagenesis, was the kind gift of Yoshifumi Takeda, National Institute of Infectious Diseases, Tokyo, Japan. The preparation and purification of Stx1-E167Q-R170L has been described previously (24). The mutant Stx1 holotoxin exhibited high-performance liquid chromatography and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) profiles similar to those of the wild-type toxin. Ouchterlony double gel diffusion with antisera raised against the mutant toxin showed a line of identity between Stx1-E167Q-R170L and wild-type Stx1, and both toxin preparations were shown to bind Gb3 by thin-layer chromatography (24, 45). A purified Stx2e enzymatic mutant in which glutamate at position 167 was replaced with glutamine (Stx2e-E167Q) was the kind gift of James E. Samuel, Texas A&M University Health Science Center, College Station, Tex. Stx2e-E167Q mutagenesis and purification procedures have been published (10). The E167Q-R170L and E167Q point mutations reduce verocytotoxicity approximately 105- to 106-fold compared to the verocytotoxicities of wild-type Stx1 and Stx2e, respectively (10, 24). Purified pentameric Stx1 B subunits were the kind gift of David Acheson, Tufts University School of Medicine (Boston, Mass.). Molecular biology and purification procedures for the Stx1 B subunit preparation have been reported (1). Endotoxin contamination of all toxin preparations was tested by Limulus amoebocyte lysate assay (Pyrotell, Associates of Cape Cod, Inc., Falmouth, Mass.) prior to use in cytokine induction experiments. Ricin and purified E. coli O111:B4 lipopolysaccharide (LPS) were purchased from Sigma Chemical Co., St. Louis, Mo. Murine monoclonal immunoglobulin M antibody pK002 directed against Gb3 was purchased from Accurate Chemical Corp. (Westbury, N.Y.).
Cells.
The human myelogenous leukemia cell line THP-1 (40) was purchased from American Type Culture Collection (Manassas, Va.) and maintained in RPMI 1640 (Gibco BRL, Grand Island, N.Y.) supplemented with penicillin (50 U/ml), streptomycin (50 μg/ml), and 10% fetal bovine serum (FBS; Hyclone Laboratories, Logan, Utah). Vero cells were grown in Minimum Essential Medium (Gibco BRL) with Earle's salts, 10 mM l-glutamine, and the supplements listed above. L929 cells were cultured in Iscove's modified Dulbecco's medium with 10% FBS. All cells were maintained at 37°C in humidified 5% CO2.
Macrophage differentiation.
THP-1 cells (106 cells/ml) grown in 60-mm-diameter culture dishes were induced to differentiate to a mature macrophage-like state by treatment with phorbol-12-myristate-13-acetate (PMA; Sigma) at 50 ng/ml for 48 h. Plastic-adherent, differentiated cells were then washed twice in cold Dulbecco's phosphate-buffered saline (PBS) and incubated with fresh medium lacking PMA for 72 h with daily medium changes prior to use in experiments. Unless otherwise noted, differentiated THP-1 cells were used in all experiments.
Analysis of protein synthesis.
Stx enzymatic activity was examined by measuring the inhibition of [3H]leucine incorporation into nascent polypeptides. Briefly, THP-1 or Vero cells grown in 24-well microtiter plates were incubated with purified Stxs (400 ng/ml) for various time points. At each time point, toxins were removed from the cells by three washes with cold sterile PBS. Forty microliters of labeling medium containing 10 μCi of [3H]leucine (DuPont New England Nuclear, Boston, Mass.) in leucine-free RPMI 1640 plus 10% FBS was added to each well, and the cells were incubated for 30 min at 37°C. The plates were then placed on ice, and excess radiolabel was removed by washing with cold PBS. Twenty microliters of RIPA buffer (0.15 M Tris-HCl [pH 7.4], 0.5% sodium deoxycholate, 0.5% NP-40, and 0.1% SDS) was added to each well. Lysates were transferred to microcentrifuge tubes. An additional 20 μl of RIPA buffer was added to the wells to collect any residual lysates. Lysates were centrifuged at 12,700 × g for 15 min at 4°C. Resultant supernatants were transferred to Whatman GF/C filter papers and allowed to dry. Filters were treated twice with 10% trichloroacetic acid and once with 100% ethanol and were allowed to air dry. Filters were placed in scintillant, and radioactivity was measured using a scintillation counter (Beckman LS8000; Beckman Instruments Inc., Fullerton, Calif.).
Analysis of TNF-α expression.
Amounts of immunoreactive TNF-α in culture supernatants were measured by enzyme-linked immunosorbent assay (Quantikine, R&D Systems, Minneapolis, Minn.) according to the manufacturer's instructions. The sensitivity of the assay is 4.4 pg/ml. TNF bioactivity in culture supernatants was measured by the lysis of actinomycin D-treated L929 cells as previously described (37). We have previously shown that purified toxins at 400 to 800 ng/ml induce optimal signals in these assays.
Kinase inhibitors.
PKC inhibitors H-7 and K252a, PKA inhibitor H-89, and protein tyrosine kinase inhibitor genistein were purchased from Calbiochem, La Jolla, Calif. H-7 and H-89 were dissolved in sterile water and stored at 4°C. K252a was dissolved in dimethyl sulfoxide (DMSO)-ethanol (50:50 [vol/vol]) at 500 μg/ml. Genistein was dissolved in DMSO at 500 μg/ml. K252a and genistein were stored at −20°C before use. Differentiated THP-1 cells were treated with the indicated concentrations of inhibitors for 2 h (genistein) or 3 h (H-7, H-89, and K252a). The cells were then washed twice in cold PBS and treated with the inhibitors in the presence or absence of Stx1, LPS, PMA, or vehicle control. Trypan blue exclusion studies showed no loss of cell viability in response to treatment of the cells with inhibitors alone. For all experiments, the percentage of organic solvents did not exceed 0.2% and had no effect on TNF-α expression.
Determination of PKC activity.
THP-1 cells were treated with Stx1 in the presence or absence of kinase inhibitors. The cells were washed twice in cold PBS and pelleted by centrifugation at 100 × g for 5 min at 4°C. Cells were resuspended in 50 mM Tris-HCl (pH 7.5) containing 0.3% (wt/vol) β-mercaptoethanol, 1.0 mM EDTA, 2.5 mM EGTA, 50 μg of phenylmethylsulfonyl fluoride per ml, and 10 mM benzamidine. The cells were disrupted by sonication, and the homogenates were cleared by centrifugation at 135,000 × g for 45 min. Total cellular PKC activity in sonicates was examined by measuring the phosphorylation of a synthetic PKC-specific substrate in the presence of a [γ-32P]ATP donor (Biotrak PKC System; Amersham Corp., Arlington Heights, Ill.) per the manufacturer's instructions. PKC activity (picomoles of phosphate transferred/minute/106 cells) is expressed as a percentage of basal (untreated cells) levels.
Northern blot analysis of TNF-α mRNA.
Total cellular RNA was extracted from THP-1 cells by the acid guanidinium isothiocyanate method (2) using Ultraspec II RNA isolation kits (Biotecx Laboratories, Houston, Tex.). RNA purity was assessed by optical density readings at 260 and 280 nm. Ten micrograms of RNA per lane was subjected to electrophoresis using 0.8% agarose–2.0 M formaldehyde gels in 1× MOPS (morpholinepropanesulfonic acid) running buffer at 50 V for 2 to 3 h. RNA was transferred to positively charged nylon membranes (GeneScreen Plus; NEN DuPont, Boston, Mass.) by using a Turboblotter Rapid Downward Transfer System (Schleicher and Schuell, Keene, N.H.) and was cross-linked by exposing the membrane to UV light (254 nm) for a total dose of 120 mJ/cm2 (UV Crosslinker; Bio-Rad, Hercules, Calif.). A human TNF-α probe, 5′-ATC TCT CAG CTC CAC GCC ATT GGC CAG GAG-3′ (Clontech, Palo Alto, Calif.), was 5′-end labeled with [γ-32P]ATP. An 18S RNA antisense control template (Ambion Inc., Austin, Tex.) was random prime labeled using MegaPrime DNA labeling kits (Amersham). After labeling, unincorporated nucleotides were removed using G-25 Sephadex columns. Membranes were treated with 7 to 10 ml of Rapid-Hyb hybridization buffer (Amersham) for 15 min at 42°C for the TNF-α probe or 65°C for the 18S RNA probe. Approximately 106 cpm of TNF-α probe was added per milliliter of hybridization buffer and hybridization was carried out at 42°C for 2 to 4 h. Membranes were washed in 2× SSC–0.1% SDS for 10 min at room temperature and were exposed to a phosphorimager screen overnight (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). The screen was analyzed by a phosphorimager (Molecular Dynamics, Sunnyvale, Calif.), and sums of counts above the background were calculated using ImageQuant software (Molecular Dynamics). Membranes were stripped by boiling in 0.1× SSC–0.1% SDS twice for 15 min and hybridized with 18S RNA probe at 65°C as an internal control for RNA loading. Ratios of counts of TNF-α transcripts to 18S RNA were calculated. To minimize interassay variability, relative levels of TNF-α mRNA from three separate experiments are shown by expressing the ratios as percentages above basal (unstimulated) levels by using the following formula:
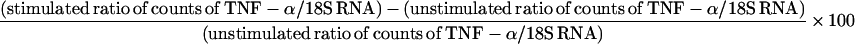 |
Phosphotyrosine analysis.
Eighteen hours prior to stimulation, differentiated THP-1 cells were serum starved in medium containing 0.5% FBS. The cells were then exposed to 1.0 μM genistein or an equivalent volume of DMSO vehicle control for 2 h prior to stimulation. The medium was replaced with medium containing purified Stx1 (400 ng/ml) with or without the kinase inhibitor. Following 1 h of incubation at 37°C, cells were washed twice with cold PBS and lysed with modified RIPA buffer (1.0% NP-40, 1.0% sodium deoxycholate, 150 nM NaCl, 10 mM Tris-HCl [pH 7.5], 5.0 mM sodium pyrophosphate, 1.0 mM NaVO4, 5.0 mM NaF, 1.0 μg of aprotinin/ml, 1.0 μg of leupeptin/ml, and 0.1 mM phenylmethylsulfonyl fluoride) for 15 min at 4°C. DNA was sheared by passing cell lysates through 22 1/2- and 26 3/8-gauge needles. Lysates were cleared by centrifugation at 14,900 × g for 15 min at 4°C. Protein content of cell lysates was determined using the Micro BCA assay kit (Pierce, Rockford, Ill.). Equivalent amounts of proteins were separated by SDS–12% PAGE and analyzed by Western blotting using an anti-phosphotyrosine monoclonal antibody (4G10; UBI, Lake Placid, N.Y.) and anti-mouse immunoglobulin G-horseradish peroxide (HRP) as the secondary antibody. Blots were developed using the ECL system (Amersham) by following the manufacturer's instructions.
Statistics.
Two sample-paired t tests were performed using Minitab Release 8 statistical software (Minitab, Inc., State College, Pa.).
RESULTS
Stx induction of TNF-α requires A subunit activity.
Earlier studies have shown that, in contrast to epithelial or endothelial cells, human peripheral blood monocytes and differentiated monocytic cell lines express low levels of membrane Gb3 and are relatively resistant to the cytotoxic action of purified Stxs, with 50% cytotoxic doses (CD50s) of >1.0 μg/ml (26). However, monocytic cells are not unresponsive to the toxins; they respond by internalizing toxins and synthesizing the proinflammatory cytokines TNF-α, IL-1β, and IL-6 (26, 43). Our preliminary studies showed that toxin receptor binding by purified Stx1 B subunits did not elicit monocyte cytokine production. We hypothesized, therefore, that enzymatically active Stx A subunits may be necessary for cytokine induction. We examined this hypothesis by treating differentiated THP-1 monocytic cells for 18 h with purified Stx holotoxins or Stx holotoxin molecules containing point mutations in the A subunit catalytic site (Stx1-E167Q-R170L, Stx2e-E167Q). The ability of Stxs to induce TNF-α production appeared to require A subunit enzymatic activity (Table 1). To further link TNF-α production with toxin enzymatic activity, THP-1 cells were treated with sublethal or lethal concentrations of ricin (CD50, ∼5.0 ng/ml), the Ricinus communis toxic lectin that shares N-glycosidase activity with Stx A subunits. Ricin treatment also elicited TNF-α production. In contrast, reagents which bind to Gb3 in the absence of enzymatic activity, i.e., purified Stx A subunit mutants, purified Stx1 B subunits, or anti-Gb3 monoclonal antibody, did not induce TNF-α synthesis.
TABLE 1.
TNF-α production by THP-1 cells requires Stx A subunit enzymatic activity
| Toxin treatment | Receptor specificity | TNF-α (pg/ml)a |
|---|---|---|
| Stx1 (800 ng/ml) | Gb3 | 149.2 ± 23.1 |
| Stx1A− (E167Q-R170L) (400 ng/ml) | Gb3 | 42.3 ± 1.0b |
| Stx1A− (E167Q-R170L) (800 ng/ml) | Gb3 | 24.2 ± 5.0b |
| Stx1A− (E167Q-R170L) (1 μg/ml) | Gb3 | 45.0 ± 1.1c |
| Stx1 B subunits | Gb3 | 12.9 ± 2.7 |
| Anti-Gb3 antibody | Gb3 | 17.3 ± 4.6 |
| Stx2e (800 ng/ml) | Gb4 | 275.2 ± 34.2 |
| Stx2eA− (E167Q) | Gb4 | 22.8 ± 4.9d |
| Ricin (0.33 ng/ml) | Terminal galactose residues | 66.2 ± 16.3 |
| Ricin (5.0 ng/ml) | Terminal galactose residues | 225.3 ± 26.1 |
| Ricin (10 ng/ml) | Terminal galactose residues | 222.4 ± 27.5 |
| LPS (200 ng/ml) | Multiple receptors | 1055 ± 55 |
| Media | 51.0 ± 10.8 |
Data are expressed as means ± standard errors of the mean. Results are from at least three independent experiments for each treatment.
P < 0.0001 for Stx1 versus Stx1A mutants.
P < 0.0002 for Stx1 versus Stx1A mutants at 1 μg/ml.
P < 0.0001 for Stx2e versus Stx2eA mutants.
To correlate ribosomal inactivation with cytokine production, we examined protein synthesis inhibition in cells treated with Stxs. When toxin-sensitive Vero cells (CD50 = 1.0 pg/ml) were treated with purified Stx1 or Stx2e (400 ng/ml), the incorporation of 3[H]leucine into nascent polypeptides was inhibited by approximately 90% within 1 h of toxin treatment (Fig. 1). We previously showed that undifferentiated THP-1 cells express relatively high levels of membrane Gb3, are sensitive to Stx1 (CD50 = 18 pg/ml), and fail to produce TNF-α when stimulated with Stxs (26). Within an hour of treatment with purified Stx1 (400 ng/ml), undifferentiated THP-1 cells manifested an approximate 75% reduction in protein synthesis. In contrast, when differentiated THP-1 cells were treated with an equivalent dose of Stx1, we noted transient increases in total protein synthesis 1 to 2 h after toxin treatment. Following treatment of cells with Stx2e, protein synthesis was inhibited by, at most, 20% over a 4-h time period. Thus, the sublethal amounts of Stxs used in these experiments to induce TNF-α expression from differentiated monocytic cells only marginally affected total protein synthesis.
FIG. 1.

Protein synthesis inhibition mediated by purified Stxs. Differentiated THP-1 cells were treated with 400 ng of purified Stx 1 (□) or Stx2e (○) per ml, and undifferentiated THP-1 cells (◊) and Vero cells (▵) were treated with 400 ng of Stx1/ml. At the indicated time points, cells were washed and incubated an additional 30 min in medium containing [3H]leucine. Following lysis of the cells and trichloroacetic acid precipitation of proteins, incorporation of radiolabeled leucine into nascent polypeptides was measured by scintillation counting. The data are expressed as percentages of basal protein synthesis (untreated cells) and are derived from at least three independent experiments.
Stx induction of TNF-α requires serine/threonine kinase activity.
Many studies have implicated serine/threonine kinases of the PKC family as monocyte/macrophage signal transduction molecules required for cytokine production (reviewed in reference 36). We hypothesized that PKC may also participate in Stx-mediated cell signaling. A series of PKC inhibitors was employed to monitor the effects of blocking kinase activity on TNF production by Stx1-treated THP-1 cells. Treatment of cells with the PKC inhibitor H-7 (Fig. 2A) or K252a (Fig. 2B) reduced TNF secretion in a dose-dependent manner following stimulation of cells with Stx1 for 12 h. Although H-7 and K252a are potent inhibitors of PKC, they can also inhibit cyclic nucleotide-dependent protein kinases (14). To exclude the involvement of PKA in Stx-induced TNF production, the PKA inhibitor H-89 was used. H-89 inhibits PKA activity at nanomolar concentrations, whereas the inhibitory concentration for PKC is several hundredfold higher. Treatment of THP-1 cells with concentrations of H-89 ranging from 0.1 to 1,000 nM did not result in significant decreases in TNF production in response to Stx1 (data not shown). These data suggest that members of the PKC family of serine/threonine kinases are involved in Stx signaling for cytokine production.
FIG. 2.

Effect of PKC inhibitors on Stx1-mediated TNF production by THP-1 cells. PMA-differentiated THP-1 cells were pretreated with various doses of the PKC inhibitors H-7 (A) or K252a (B) for 3 h prior to stimulation with 800 ng of purified Stx1/ml. Twelve hours later, culture supernatants were collected and TNF bioactivity was determined by lysis of actinomycin D-treated L929 cells. Results shown are mean levels of TNF bioactivity (picograms/milliliter) ± standard errors of the means for three independent experiments.
The capacity of Stx1 to activate PKC was directly assessed. Total cellular PKC activity in lysates prepared from Stx1-treated monocytes was examined by measuring the transfer of the γ-phosphate group from [γ-32P]ATP to a PKC-specific substrate. Treatment of THP-1 cells with Stx1 increased total cellular PKC activity to ∼150% above basal levels in untreated cells (Fig. 3). Treatment of cells with H-7 or K252a prior to Stx1 exposure prevented elevated PKC activity, whereas H-89 treatment did not affect Stx1-mediated PKC activity. Short-term PMA treatment was used as a positive control and increased THP-1 PKC activity by ∼350%.
FIG. 3.

Effect of Stx1 on PKC activation in THP-1 cells. PMA-differentiated THP-1 cells were treated with purified Stx1 (800 ng/ml) for 3 h or PMA for 1 h. Some cells were pretreated with the PKC inhibitor H-7 (50 mM) or K252a (500 nM) or the PKA inhibitor H-89 (1000 nM) for 3 h prior to stimulation with 800 ng of purified Stx1/ml. PKC activation was assessed by measuring the transfer of 32P to a synthetic PKC substrate. Data shown are means ± standard errors of the means for three independent experiments.
PKC inhibitors block TNF-α mRNA synthesis in Stx-treated THP-1 cells.
Northern blot analyses of Stx-treated THP-1 cells demonstrated that the toxins act, at least in part, at the transcriptional level to increase quantities of TNF-α mRNA transcripts extracted from the cells (28). We used Northern blots to examine the role of PKC in Stx-induced TNF-α mRNA synthesis. Pretreatment of cells with the PKC inhibitors H-7 and K252a significantly blocked TNF-α transcript induction following stimulation with Stx1, while the PKA inhibitor H-89 did not affect toxin induction of TNF-α mRNA (Fig. 4). These data suggest PKC involvement in signaling events occurring upstream of TNF-α transcriptional activation. The positive control in these experiments, short-term treatment of THP-1 cells with TPA, triggered TNF-α mRNA synthesis.
FIG. 4.

Effect of PKC inhibitors on Stx1-mediated TNF-α mRNA synthesis by THP-1 cells. PMA-differentiated THP-1 cells were treated with purified Stx1 with or without protein kinase inhibitors as outlined in the legend to Fig. 3. Total RNA was isolated from the cells, and TNF-α mRNA was detected by Northern blot analysis. Ratios shown are the means ± standard errors of the means for three separate experiments.
Stx induction of TNF-α requires tyrosine kinase activity.
One of the earliest macrophage responses to LPS binding is the phosphorylation of multiple proteins at tyrosine residues (reviewed in reference 3). Treatment of THP-1 cells with purified Stxs also results in tyrosine phosphorylation (and dephosphorylation) of multiple proteins. Following toxin stimulation, major phosphoprotein bands at approximately 73, 70, 60, and 20 kDa appeared, while a 33-kDa band appeared dephosphorylated (Fig. 5). Toxin-mediated changes in phosphorylation appeared to be sensitive to the general tyrosine kinase inhibitor genistein, which reduced levels of phosphorylation to levels detected in untreated controls (Fig. 5, compare lanes 1 and 4). This finding led us to assess the role of protein tyrosine kinases in Stx-mediated TNF-α production. We treated monocytes with the tyrosine kinase inhibitor genistein prior to Stx1 exposure and monitored TNF-α secretion. Pretreatment of THP-1 cells with genistein for 2 h inhibited Stx1- and LPS-mediated TNF-α production (Fig. 6). Exposure of the cells to the carrier solvent DMSO or to genistein plus DMSO did not induce TNF-α production. Thus, Stx1 treatment of monocytic cells induced tyrosine phosphorylation, the inhibition of which was associated with the loss of cytokine expression.
FIG. 5.

Stx1-induced phosphorylation of tyrosine residues in THP-1 cells. Differentiated THP-1 cells were preincubated with 100 μM genistein or vehicle for 2 h followed by exposure to Stx1 (400 ng/ml) with or without the kinase inhibitor. Cells were also treated with the inhibitor or DMSO vehicle alone. Cells were lysed, and extracts were cleared by centrifugation. Equal amounts of protein were separated by SDS–12% PAGE and analyzed by Western blotting using an anti-phosphotyrosine monoclonal antibody, 4G10, as described in Materials and Methods. Closed arrows indicate phosphoproteins induced by Stx1 treatment that are sensitive to genistein. The open arrow indicates a phosphoprotein band of decreased intensity following Stx1 treatment that appears restored following inhibitor treatment.
FIG. 6.

Effect of the tyrosine kinase inhibitor genistein on Stx1-mediated TNF-α production by THP-1 cells. PMA-differentiated THP-1 cells were treated with purified Stx1 (400 ng/ml) or LPS (200 ng/ml) for 18 h. Some cells were pretreated with genistein (100 μM) for 2 h prior to stimulation with purified Stx1 or LPS. Culture supernatants were collected, and soluble TNF-α levels were determined by enzyme-linked immunosorbent assay. Data shown are the means ± standard errors of the means for three separate experiments.
DISCUSSION
Data presented in this study and elsewhere (26, 43) suggest that sublethal concentrations of Stxs induce TNF-α and IL-1β expression by human monocytes and monocytic cell lines. We demonstrate that cytokine production in response to Stx treatment correlates with toxin A subunit enzymatic activity, as mutants with markedly reduced N-glycosidase activity, but retaining the ability to form AB5 holotoxins and bind to the cells via Gb3 or Gb4 receptors, fail to elicit TNF-α production. Furthermore, treatment of monocytes with ricin, a toxin possessing an identical mechanism of action as Stx A subunits, elicits TNF-α production. Binding of Gb3 by Stx1 B subunits or antibody against Gb3 was not sufficient to trigger cytokine production.
The precise mechanisms by which Stxs trigger increased cytokine synthesis and secretion by monocytes is unknown. A number of well-characterized transmembrane signaling pathways are triggered by growth factor-growth factor receptor interactions (31). The lipid A component of LPS has been shown to bind to a number of monocyte/macrophage membrane proteins, including CD14, β2 leukocyte integrins (CD11a,b,c/CD18), and the scavenger receptor (reviewed in reference 7). Following LPS binding to membrane receptors, transmembrane signaling may be initiated by toll-like receptors, a series of signal transfer proteins. In contrast to growth factors or LPS, Stxs and ricin do not appear to stimulate host cell signaling cascades solely through the simple interaction of toxins with membrane receptors. Rather, it appears that wild-type toxins must enter cells, undergo retrograde intracellular routing, and initiate cell signaling cascades from within the cell. A likely starting point for cell signal initiation is the ribosome (15), and studies are under way to link Stx-mediated ribosome inactivation with cytokine production by monocytic cells.
Stx-mediated cytokine induction by monocytes was dependent on cell differentiation and toxin dose. Treatment of toxin-sensitive undifferentiated THP-1 cells with Stxs (400 ng/ml) resulted in cell death without cytokine release (26). In contrast, cytokine production manifested in differentiated THP-1 cells following treatment with an identical dose of Stx1 which was sublethal and did not drastically alter protein synthesis. The cytokine induction ability of sublethal doses of Stxs may not be limited to proinflammatory cytokine production by myeloid cells. Recently, it was shown that human intestinal epithelial cells, although relatively sensitive to Stxs, secreted the chemokine IL-8 when treated with doses of purified Stxs that mediated only ∼10% of the cell killing (39, 45). Interestingly, IL-8 production by the human colonic epithelial cell line Caco2 also required A subunit activity (45). These data suggest that cytokine and chemokine induction within localized tissues may occur in the presence of concentrations of Stxs that are sublethal for cells in vitro.
To determine signaling components involved in Stx-mediated TNF-α production, we examined the role of PKC, PKA, and protein tyrosine kinases in induction. The PKC family of serine/threonine kinases contains at least 12 isoforms that are involved in signal transduction, regulation of gene expression, and myeloid differentiation (21). The PKC family is subdivided into three groups: (i) the conventional PKC group containing isoforms requiring both Ca2+ and diacylglycerol (DAG), phosphoserine, or phorbol esters for activation, (ii) the Ca2+-independent PKC group, requiring only DAG, phosphoserine, or phorbol esters for activation, and (iii) the atypical PKC isoforms, which require neither Ca2+ nor lipids for activation and are insensitive to phorbol esters. Various PKC isoforms have been implicated in LPS-mediated signaling, but their role in induction of TNF-α remains controversial (25, 32, 33). Shapira et al. (33) showed that LPS-stimulated human monocyte production of TNF-α and IL-1 was blocked by both tyrosine kinase and PKC inhibitors. However, a DAG-dependent kinase inhibitor did not affect LPS-induced TNF-α production, suggesting that atypical PKC isoforms may be involved in LPS signaling. Herrera-Velit et al. (13) showed that LPS treatment of human monocytes resulted in the selective activation of the atypical PKC-ζ isoform. Atypical PKC isoforms may, in turn, be linked to activation of downstream kinases, such as the extracellular signal-regulated kinase cascade, involved in LPS-mediated signaling (9, 27, 44). The results presented here suggest that PKC activation in differentiated THP-1 cells is essential in Stx-mediated signaling for TNF-α production. The position of PKC in the signaling pathway and the identity of the PKC isoform(s) involved in Stx signaling remain to be determined.
Using the tyrosine kinase inhibitor genistein, TNF-α production by THP-1 cells stimulated by Stx1 was decreased. Thus, protein tyrosine kinases are also involved in Stx-mediated signaling. Several possible candidate kinase pathways that contain tyrosine kinase activity have been characterized in LPS-treated monocytes, including the extracellular signal-regulated kinase, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase cascades (5, 12, 35, 44). It will be necessary to screen specific components of these pathways to establish their role in Stx signaling since the parallels between LPS- and Stx-induced signaling are currently unknown.
ACKNOWLEDGMENTS
We thank Anastasia Green and Belakere Ramegowda for excellent technical assistance. We thank David Acheson, Jim Samuel, and Yoshifumi Takeda for kindly sharing reagents essential for the completion of this study.
This work was supported by Public Health Service grant AI-34530 from the National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Calderwood S B, Acheson D W K, Goldberg M B, Boyko S A, Donohue-Rolfe A. A system for production and rapid purification of large amounts of the Shiga toxin/Shiga-like toxin I B subunit. Infect Immun. 1990;58:2977–2982. doi: 10.1128/iai.58.9.2977-2982.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 3.DeFranco A L, Finn A J, Hambleton J, Crowley M T, MacKichan M L, Weinstein S L. The role of MAP kinases, phosphatidylinositol 3-kinase, and ceramide in LPS-induced signaling in macrophages. In: Brade H, Opal S M, Vogel S N, Morrison D C, editors. Endotoxin in health and disease. New York, N.Y: Marcel Dekker; 1999. pp. 473–482. [Google Scholar]
- 4.DeGrandis S, Law H, Brunton J, Gyles C, Lingwood C A. Globotetraosylceramide is recognized by the pig edema disease toxin. J Biol Chem. 1989;264:12520–12525. [PubMed] [Google Scholar]
- 5.Durando M M, Meier K E, Cook J A. Endotoxin activation of mitogen-activated protein kinase in THP-1 cells: diminished activation following endotoxin desensitization. J Leukoc Biol. 1998;64:259–264. doi: 10.1002/jlb.64.2.259. [DOI] [PubMed] [Google Scholar]
- 6.Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur J Biochem. 1988;171:45–50. doi: 10.1111/j.1432-1033.1988.tb13756.x. [DOI] [PubMed] [Google Scholar]
- 7.Fenton M J, Golenbock D T. LPS-binding proteins and receptors. J Leukoc Biol. 1998;64:25–32. doi: 10.1002/jlb.64.1.25. [DOI] [PubMed] [Google Scholar]
- 8.Gentry M K, Dalrymple J M. Quantitative microtiter assay for Shigella toxin. J Clin Microbiol. 1980;12:361–366. doi: 10.1128/jcm.12.3.361-366.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geppert T D, Whitehurst C E, Thompson P, Beutler B. Lipopolysaccharide signals activation of tumor necrosis factor biosynthesis through the Ras/Raf-1/MEK/MAPK pathway. Mol Med. 1994;1:93–103. [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon V M, Whipp S C, Moon H W, O'Brien A D, Samuel J E. An enzymatic mutant of Shiga-like toxin II variant is a vaccine candidate for edema disease of swine. Infect Immun. 1992;60:485–490. doi: 10.1128/iai.60.2.485-490.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Habib R. Pathology of the hemolytic uremic syndrome. In: Kaplan B S, Trompeter R S, Moake J L, editors. Hemolytic-uremic syndrome and thrombotic thrombocytopenic purpura. New York, N.Y: Marcel Dekker; 1992. pp. 315–353. [Google Scholar]
- 12.Hambleton J, Weinstein S L, Lem L, DeFranco A L. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci USA. 1996;93:2774–2778. doi: 10.1073/pnas.93.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrera-Velit P, Knutson K L, Reiner N E. Phosphatidylinositol 3-kinase-dependent activation of protein kinase C-ζ in bacterial lipopolysaccharide-treated human monocytes. J Biol Chem. 1997;272:16445–16452. doi: 10.1074/jbc.272.26.16445. [DOI] [PubMed] [Google Scholar]
- 14.Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulphomides: novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- 15.Iordanov M S, Pribnow D, Magun J L, Dinh T-H, Pearson J A, Chen S L-E, Magun B E. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyltransferase reaction and by sequence-specific RNA damage to the α-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997;17:3373–3381. doi: 10.1128/mcb.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacewicz M, Clausen H, Nudelman E, Donohue-Rolfe A, Keusch G T. Pathogenesis of Shigella diarrhea. XI. Isolation of a Shigella toxin-binding glycolipid from rabbit jejunum and HeLa cells and its identification as globotriaosylceramide. J Exp Med. 1986;163:1391–1404. doi: 10.1084/jem.163.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karmali M A, Petric M, Lim C, Fleming P C, Arbus G S, Lior H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis. 1985;151:775–782. doi: 10.1093/infdis/151.5.775. [DOI] [PubMed] [Google Scholar]
- 18.Lindberg A A, Schultz J E, Westling M, Brown J E, Rothman S W, Karlsson K-A, Strömberg N. Identification of the receptor glycolipid for Shiga toxin produced by Shigella dysenteriae type 1. In: Lark D L, editor. Protein-carbohydrate interactions. London, England: Academic Press, Ltd.; 1986. pp. 439–446. [Google Scholar]
- 19.Louise C B, Obrig T G. Shiga toxin-associated hemolytic uremic syndrome: combined cytotoxic effects of Shiga toxin, interleukin-1β, and tumor necrosis factor alpha on human vascular endothelial cells in vitro. Infect Immun. 1991;59:4173–4179. doi: 10.1128/iai.59.11.4173-4179.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louise C B, Tran M C, Obrig T G. Sensitization of human umbilical vein endothelial cells to Shiga toxin: involvement of protein kinase C and NF-κB. Infect Immun. 1997;65:3337–3344. doi: 10.1128/iai.65.8.3337-3344.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mellor H, Parker P J. The extended protein kinase C superfamily. Biochem J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melton-Celsa A R, O'Brien A D. Structure, biology and relative toxicity of Shiga toxin family members for cells and animals. In: Kaper J B, O'Brien A D, editors. Escherichia coli O157:H7 and other Shiga toxin-producing strains of E. coli. Washington, D.C.: ASM Press; 1998. pp. 121–128. [Google Scholar]
- 23.Obrig T G, DelVecchio P J, Brown J E, Moran T P, Rowland B M, Judge T K, Rothman S W. Direct cytotoxic action of Shiga toxin on human vascular endothelial cells. Infect Immun. 1988;56:2373–2378. doi: 10.1128/iai.56.9.2373-2378.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohmura M, Yamasaki S, Kurazono H, Kashiwagi K, Igarashi K, Takeda Y. Characterization of non-toxic mutants of Verotoxin 1 that were constructed by replacing amino acids in the A subunit. Microb Pathog. 1993;15:169–176. doi: 10.1006/mpat.1993.1067. [DOI] [PubMed] [Google Scholar]
- 25.Prabhakar U, Lipschutz D, Pullen M, Turchin H, Kassis S, Nambi P. Protein kinase C regulates TNF-α production by human monocytes. Eur Cytokine Netw. 1993;4:31–37. [PubMed] [Google Scholar]
- 26.Ramegowda B, Tesh V L. Differentiation-associated toxin receptor modulation, cytokine production and sensitivity to Shiga-like toxins in human monocytes and monocytic cell lines. Infect Immun. 1996;64:1173–1180. doi: 10.1128/iai.64.4.1173-1180.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reimann T, Büscher D, Hipskind R A, Krautwald S, Lohmann-Matthew M-L, Baccarini M. Lipopolysaccharide induces activation of the Raf-1/MAP kinase pathways. J Immunol. 1994;153:5740–5749. [PubMed] [Google Scholar]
- 28.Sakiri R, Ramegowda B, Tesh V L. Shiga toxin type 1 activates tumor necrosis factor-α gene transcription and nuclear translocation of transcriptional activators nuclear factor-κB and activator protein-1. Blood. 1998;92:558–566. [PubMed] [Google Scholar]
- 29.Samuel J E, Perera L P, Ward S, O'Brien A D, Ginsberg V, Krivan H C. Comparison of the glycolipid receptor specificities of Shiga-like toxin type II and Shiga-like toxin type II variants. Infect Immun. 1990;58:611–618. doi: 10.1128/iai.58.3.611-618.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saxena S K, O'Brien A D, Ackerman E J. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28 S RNA when microinjected into Xenopus oocytes. J Biol Chem. 1989;264:596–601. [PubMed] [Google Scholar]
- 31.Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- 32.Shames B D, Selzman C H, Pulido E J, Meng X, Meldrum D R, McIntyre R C, Jr, Harken A H, Banerjee A. LPS-induced NF-κB activation and TNF-α release in human monocytes are protein tyrosine kinase dependent and protein kinase C independent. J Surg Res. 1999;83:69–74. doi: 10.1006/jsre.1998.5564. [DOI] [PubMed] [Google Scholar]
- 33.Shapira L, Takashiba S, Champagne C, Amar S, Van Dyke T E. Involvement of protein kinase C and tyrosine kinase in lipopolysaccharide-induced TNF-α and IL-1β production by human monocytes. J Immunol. 1994;153:1818–1824. [PubMed] [Google Scholar]
- 34.Siegler R L. The hemolytic uremic syndrome. Pediatr Clin N Am. 1995;42:1505–1529. doi: 10.1016/s0031-3955(16)40096-9. [DOI] [PubMed] [Google Scholar]
- 35.Swantek J, Cobb M H, Geppert T D. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor-α (TNF-α) translation: glucocorticoids inhibit TNF-α translation by blocking JNK/SAPK. Mol Cell Biol. 1997;17:6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sweet M J, Hume D A. Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996;60:8–26. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 37.Tesh V L, Ramegowda B, Samuel J E. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect Immun. 1994;62:5085–5094. doi: 10.1128/iai.62.11.5085-5094.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tesh V L, Samuel J E, Perera L P, Sharefkin J B, O'Brien A D. Evaluation of the role of Shiga toxin and Shiga-like toxins in mediating direct damage to human vascular endothelial cells. J Infect Dis. 1991;164:344–352. doi: 10.1093/infdis/164.2.344. [DOI] [PubMed] [Google Scholar]
- 39.Thorpe C M, Hurley B P, Lincicome L L, Jacewicz M S, Keusch G T, Acheson D W K. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect Immun. 1999;67:5985–5993. doi: 10.1128/iai.67.11.5985-5993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1) Int J Cancer. 1980;26:171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 41.Van de Kar N C A J, Kooistra T, Vermeer M, Lesslauer W, Monnens L A H, van Hinsbergh V W M. Tumor necrosis factor-α induces endothelial cell galactosyl transferase activity and verocytotoxin receptors: role of specific tumor necrosis factor receptors and protein kinase C. Blood. 1995;85:734–743. [PubMed] [Google Scholar]
- 42.Van de Kar N C A J, Monnens L A H, Karmali M A, van Hinsbergh V W M. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: implications for the pathogenesis of the hemolytic uremic syndrome. Blood. 1992;80:2755–2764. [PubMed] [Google Scholar]
- 43.Van Setten P A, Monnens L A H, Verstraten R G G, van den Heuvel L P W J, van Hinsbergh V W M. Effects of verocytotoxin-1 on nonadherent human monocytes: binding characteristics, protein synthesis and cytokine release. Blood. 1996;88:174–183. [PubMed] [Google Scholar]
- 44.Weinstein S L, Sanghera J S, Lemke K, DeFranco A L, Pelech S L. Bacterial lipopolysaccharide induces tyrosine phosphorylation and activation of mitogen-activated protein kinases in macrophages. J Biol Chem. 1992;267:14955–14962. [PubMed] [Google Scholar]
- 45.Yamasaki C, Natori Y, Zeng X-T, Ohmura M, Yamasaki S, Takeda Y, Natori Y. Induction of cytokines in a human colon epithelial cell line by Shiga toxin 1 (Stx1) and Stx2 but not by non-toxic mutant Stx1 which lacks N-glycosidase activity. FEBS Lett. 1999;442:231–234. doi: 10.1016/s0014-5793(98)01667-6. [DOI] [PubMed] [Google Scholar]