

Abstract
Natural Killer (NK) cells are best known for their killing of virus-infected cells and tumor cells via release of cytotoxic factors. However, NK cells can also produce growth factors and cytokines, and thus have potential to influence physiological processes such as wound healing. In this study we test the hypothesis that NK cells play a physiological role in skin wound healing of C57Bl/6J mice. Immunohistochemical and flow cytometry assays showed that NK cells accumulate in excisional skin wounds, peaking on day 5 post-injury. We also found that NK cells proliferate locally in wounds, and blocking IL-15 activity locally reduces NK cell proliferation and accumulation in wounds. Wound NK cells exhibit primarily a mature CD11b+CD27− and NKG2A+NKG2D− phenotype, and express LY49I and pro-inflammatory cytokines such as IFN-γ, Tnf-a and Il-1β. Systemic depletion of NK cells resulted in enhanced re-epithelization and collagen deposition, suggesting a negative role for these cells in skin wound healing. Depletion of NK cells did not influence accumulation of neutrophils or monocytes/macrophages in wounds but did reduce expression of IFN-γ, Tnf-a and Il-1β, indicating that NK cells contribute to pro-inflammatory cytokine expression in wounds. In short, NK cells may impede physiological wound healing via production of pro-inflammatory cytokines.
INTRODUCTION
The skin is a complex organ that functions as a protective barrier against environmental and pathogenic threats, and such functions require coordination between different cell types, soluble factors and extracellular matrix components. After skin injury, diverse populations of immune cells infiltrate the damaged tissue and interact with keratinocytes, fibroblasts and endothelial cells to prevent infection and promote an efficient healing response, resulting in wound closure and restoration of barrier function (1)(2).
The healing process can be divided in three distinct but overlapping phases: inflammatory, proliferative and remodeling (3). During the inflammatory phase, pro-inflammatory cells, including neutrophils and monocytes/macrophages (Mo/Mp) are recruited to the injury site and release pro-inflammatory factors to prevent infections and clear wound debris (4). During the proliferative phase, Mo/Mp and T cells downregulate pro-inflammatory factors and release soluble factors that promote proliferation and differentiation of fibroblasts, endothelial cells and keratinocytes (5). During the remodeling phase, Mo/Mp play a role in reorganization of the provisional matrix and then immune cells are cleared from the injury site and tissue homeostasis is restored (3)(5)(6). While the roles of neutrophils, Mo/Mp, and T cells in wound healing have been reasonably well studied, there is only limited information available on the role of another major class of immune cells: natural killer (NK) cells, a member of the Innate Lymphoid Cell (ILC) family.
NK cells are defined by their “natural killing” function through release of cytotoxic factors. NK cells act in host defense against virus-infected cells and tumor cells, inducing apoptosis of target cells upon release of perforin and granzyme granules and through production of cytokines such as interferon (IFN)-γ and tumor necrosis factor (TNF)-α (7)(8). In addition to pro-inflammatory cytokines, NK cells can also produce growth factors such as granulocyte-macrophage colony stimulating factor (GM-CSF) and transforming growth factor (TGF)-ß, and thus can shape immune responses through crosstalk with other immune cells such as Mo/Mp, T cells, B cells and dendritic cells as well as other tissue resident cells (9). In addition, hyperactivation or dysfunction of NK cells is associated with the pathogenesis of multiple diseases, including lupus, type 1 diabetes, autoimmune liver disease. Thus, NK cells may play protective or pathogenic roles depending on their activation state/phenotype and tissue context (10)(11).
NK cell-derived cytokines and chemokines participate in the inflammatory response to tissue injury and thus have potential to influence wound healing (12). Although NK cells have been extensively investigated in cancer and viral infection (8, 11, 13, 14), few studies have focused on the role of NK cells in skin wound healing. Of the studies investigating the role of NK cells in wound healing, one reported that NK cells dampen inflammation and play a positive role in healing after corneal epithelial abrasion (15). However, a second reported that NK cell ablation accelerates skin wound healing, suggesting a negative role in healing (16). Therefore, much remains to be learned about the role of NK cells in wound healing.
In this study, we demonstrate that NK cells accumulate in skin wounds, and proliferate locally in response to IL-15. Skin wound NK cells exhibit a primarily mature CD11b+CD27− and NKG2A+NKG2D− phenotype, express Ly49I and contribute to the expression of pro-inflammatory cytokines in wounds. Importantly, NK cell depletion enhanced re-epithelization and collagen deposition, suggesting a negative role for these cells in skin wound healing.
MATERIALS AND METHODS
Animals
C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred at the AALAC approved animal facility at the University of Illinois at Chicago. Mice were maintained in controlled environment conditions with 12hr light/dark cycles, water and food (standard diet) ad libitum and housed in groups of 5 animals until experimentation. Each experiment was performed with at least two cohorts with a total of n=6 male mice per group (age 11 – 14 weeks). All animal studies were approved by the Animal Care Committee (ACC) of University of Illinois at Chicago and followed the “Guide for the care and use of laboratory animals”, in compliance with the U.S. Department of Health and Human Services.
Wound Model
Full-thickness excisional skin wounding was performed as previously described (17)(18). Eight-mm excisional wounds were made on the dorsum of mice with a sterile 8-mm dermal biopsy punch, covered with Tegaderm® (3M) held in place with Coban® (3M) self-adhesive bandage wrap, as previously described (17). External wound closure was measured in digital images of the dorsum of each animal obtained immediately after wounding procedure (day 0) and prior to euthanasia on days 3, 5 and 7 post-injury. Wound area was measured using Fiji Image J and expressed as percentage reduction of the area compared to day 0. Animals were euthanized by isoflurane overdose followed by cervical dislocation and samples were collected for various assays as described below.
NK cell depletion
NK cells were depleted systemically as previously described (16) (19), using intraperitoneal (i.p.) injections of 100 μg of NK1.1 antibody (clone PK136, #108702, Biolegend, San Diego, CA), immediately after wounding (day 0), day 2, 4 and 6 post-injury. Controls were injected with equivalent doses of IgG2a control antibody (clone MOPC-173, #400202, Biolegend, San Diego, CA). Antibody solutions were diluted in sterile PBS with a total volume of 200 μL/injection. Samples were collected at days 3, 5 and 7 post-injury.
IL-15 neutralization
IL-15 was neutralized locally in wounds with intradermal (i.d.) injection of 4 μg of anti-mIL-15 antibody (polyclonal, #AF447, R&D Systems - Minneapolis, MN) per wound, immediately after wounding (day 0), day 2 and 4 post-injury. Controls were injected with equivalent doses of IgG control antibody (#I-2000-1, Vector Labs – Burlingame, CA). For i.d. injections, the total dose was distributed equally over four sites around periphery of the wound, as previously described (20). Samples were collected on day 5 post-injury.
Cell isolation
Wound cells, spleen cells, femoral bone marrow cells and peripheral blood cells were collected for cell sorting, qPCR and flow cytometry analysis. For peripheral blood cells, 100 μL of blood per animal was collected after deep isoflurane anesthesia but before euthanasia by cardiac puncture in tubes containing EDTA. Blood samples were then subjected to red blood cell lysis, then were filtered, washed and re-suspended in FACS buffer in a single cell suspension. For wound cells, after blood collection, mice were perfused with PBS through the left ventricle, the pelt was removed from the dorsum of the mouse, and wounds were harvested with an 8mm biopsy punch and subjected to enzymatic digestion and mechanical disruption to obtain a single cell suspension in a pool of 4 wounds/animal, as previously described (20). For bone marrow cells, two femurs per animal were collected and flushed with DMEM. Cells were then filtered, centrifuged and re-suspended in FACS buffer in a single cell suspension. For spleen (whole organ) and liver (left lobe) cells, samples from each animal were homogenized in DMEM with mechanical disruption and the help of a syringe and a 21G needle. Cells were then filtered, centrifuged and re-suspended in FACS buffer in a single cell suspension.
NK cell sorting
NK cells were sorted from single cell suspensions of blood, bone marrow, wound or spleen samples using MojoSort™ magnetic beads system (#480020, Biolegend - San Diego, CA), MojoSort™ Mouse anti-APC Magnetic Nanobeads (#480071, Biolegend – San Diego, CA), and NK1.1-APC antibody (clone PK136, #108710, Biolegend – San Diego, CA), following manufacturer instructions. Purity and yield of sorting was confirmed by flow cytometry using CD3-PE antibody (clone 145-2C11, #12-0031082, Invitrogen – Waltham, MA), NK1.1-APC antibody (clone PK136, #108710, Biolegend – San Diego, CA) and NK1.1-FITC antibody (clone PK136, #11-594182, Invitrogen – Waltham, MA).
Flow Cytometry
Single cell suspensions from wounds, bone marrow and blood were Fc blocked with anti-CD16/32 antibody (clone 93, #101319, Biolegend – San Diego, CA) and labeled with CD3-PE (clone 145-2C11, #12-0031082, Invitrogen – Waltham, MA), NK1.1-FITC (clone PK136, #11-594182, Invitrogen – Waltham, MA), NK1.1-BV650 (clone PK136, #108735, Biolegend – San Diego, CA), CD11b-APC (clone M1/70, #101212, Biolegend – San Diego, CA), CD11b-FITC (M1/70, #101206, Biolegend – San Diego, CA), CD27-PE (clone LG.3A10, #124210, Biolegend - San Diego, CA), Ly6G-FITC (clone 1A8, #127606, Biolegend – San Diego, CA), Ly6C-PE (clone HK1.4, #128008, Biolegend – San Diego, CA), NKG2A-PeCy7 (clone 16A11, #142810, Biolegend – San Diego, CA), NKG2D-PE (clone A10, #115605, Biolegend - San Diego, CA). For wound samples, cells were also stained with Zombie Violet or Aqua (#423114 and #423102, Biolegend – San Diego, CA) to assess cell viability.
For proliferation/cell cycle analysis, cells were fixed and permeabilized using Cytofix/Cytoperm™ kit (#554722, BD Biosciences – Franklin Lakes, NJ), for intracellular labeling with anti-Ki67 antibody (polyclonal, #ab15580, Abcam – Cambridge, MA) followed by AF488 secondary antibody (polyclonal, #ab150077, Abcam – Cambridge, MA). Finally, cells were incubated with FxCycle™ Far Red (#F10348, Invitrogen – Waltham, MA), following manufacturer instructions.
All samples were read in a Cytoflex S (Beckman Coulter) cytometer and data analyzed with FlowJo software (FloyJo - Ashland, OR).
Wound Histology
Wound histology was assess essentially as described previously (18). Wounds were embedded in tissue freezing medium and snap frozen in isopentane cooled with liquid nitrogen, followed by sectioning at 10 μm thickness in a Leica CM1520 cryostat. Cryosections taken from the center of the wound (identified by sectioning from one edge of the wound to well past the middle of the wound) were used for H&E staining (re-epithelization), Trichrome staining (granulation tissue and collagen deposition) and immunohistochemistry (NK cell accumulation). For immunohistochemistry, M.O.M.® immunodetection kit (#BMK-2202, Vector Labs – Burlingame, CA) was used to minimize background staining and NK cells were labeled with NK1.1 primary antibody (clone PK136, #108702, Biolegend – San Diego, CA 1:50, Biolegend – San Diego, CA) following by staining with AEC (#SK-4200, Vector Labs – Burlingame, CA) and counterstained with Hematoxylin QS (#H3404-100, Vector Labs – Burlingame, CA). Digital images were obtained with Keyence BZ-X710 microscope with 2x (H&E and Trichrome) and 20x (Trichrome and immunohistochemistry) objectives and analyzed with Image J.
Percentage of re-epithelialization, length of epithelial tongues and granulation tissue area were measured in three sections per wound and averaged over sections to provide a representative value for each wound. Collagen deposition was quantified by automated counting of the number of clearly stained pixels based on a selected threshold intensity and normalized by total number of pixels in the image also in three sections per wound. Stained NK cells present in the wound bed were counted and expressed per mm2, also in three sections per wound.
Real Time Polymerase Chain Reaction (qPCR)
Total cells from 2 separated wounds/animal and sorted NK cells from a pool of 4 wounds/animal were used for qPCR analysis. Samples were collected in Trizol® reagent (#15596026, Invitrogen – Waltham, MA) and homogenized with a bead homogenizer for mRNA isolation and cDNA synthesis with SuperScript Vilo™ Kit (#11754-050, Invitrogen – Waltham, MA), following manufacturer instructions. PCR was performed in ABI 7500 Fast Real Time PCR using Fast Sybr Green® master mix (#4309155, Applied Biosystems – Foster City, CA). Expression of Il-10, Il-1b, Tnf-a and Ifn-γ genes were evaluated and Rpl-4 was used as housekeeping gene. Fold change was calculated using 2-delta delta Ct method and values are expressed as fold increase/decrease relative to control group (non-wounded skin).
Multiplex protein assay
The phenol phase from mRNA isolation with Trizol® reagent was saved for total protein isolation, following manufacturer instructions. Samples were then diluted and 10 μg of total protein was used for specific protein detection and quantification with LegendPlex™ Custom Multiplex (Biolegend – San Diego, CA) by flow cytometry, following manufacturer instructions. A custom multiplex assay was developed for detecting and quantifying IL-15, CX3CL1, CXCL10 and IFN-γ.
Statistics
Data are expressed as mean ± standard deviation (SD). Differences between groups were evaluated by t-test, one-way ANOVA + Tukey post hoc test and two-way ANOVA + Sidak post-hoc test as indicated. For NK cell depletion experiments, we used ANOVA models with main effects of treatment (2 levels: NK1.1 blocking antibody and isotype control antibody), and time point (3 levels: days 3, 5 and 7) and treatment-by-time interaction effects. Statistical significance (p<0.05) is shown in Figures using the symbol “#” for main effect of treatment from ANOVA results and symbol “*” for differences at specific time points from post-hoc testing.
RESULTS
NK cells accumulate in skin wounds of mice
We first established the time course of NK cell accumulation in excisional skin wounds of mice. Our immunohistochemistry analysis indicated that NK cells accumulated in the wound bed granulation tissue with a peak on day 5 post-injury in both male (Fig. 1 a/b), and female mice (Suppl. Fig. 1 a/b). Flow cytometry analysis corroborated this finding, demonstrating that CD3−NK1.1+ cells accumulate in wounds with a peak on day 5 post-injury (Fig. 1 d). In blood, CD3−NK1.1+ cell levels did not change from day 0 to day 3 post-injury, but significantly decreased on day 5 (Fig. 1 e). In bone marrow, there were no significant changes CD3−NK1.1+ cell levels, although a trend increase at late time points was observed (Fig. 1 f). In liver and spleen, CD3−NK1.1+ cell levels tended to decrease from day 0 to day 5 (Suppl. Fig. 1 c/d), (p=0.06 and p=0.09, respectively), indicating these tissues may be potential sources of wound NK cells. Interestingly, the ratio of CD3−NK1.1+ cells in blood and wounds was significantly lower at day 5 than the other time points, indicating a possible redistribution from blood to wounds (Fig. 1 g)
Figure 1. NK cells accumulate in skin wounds.
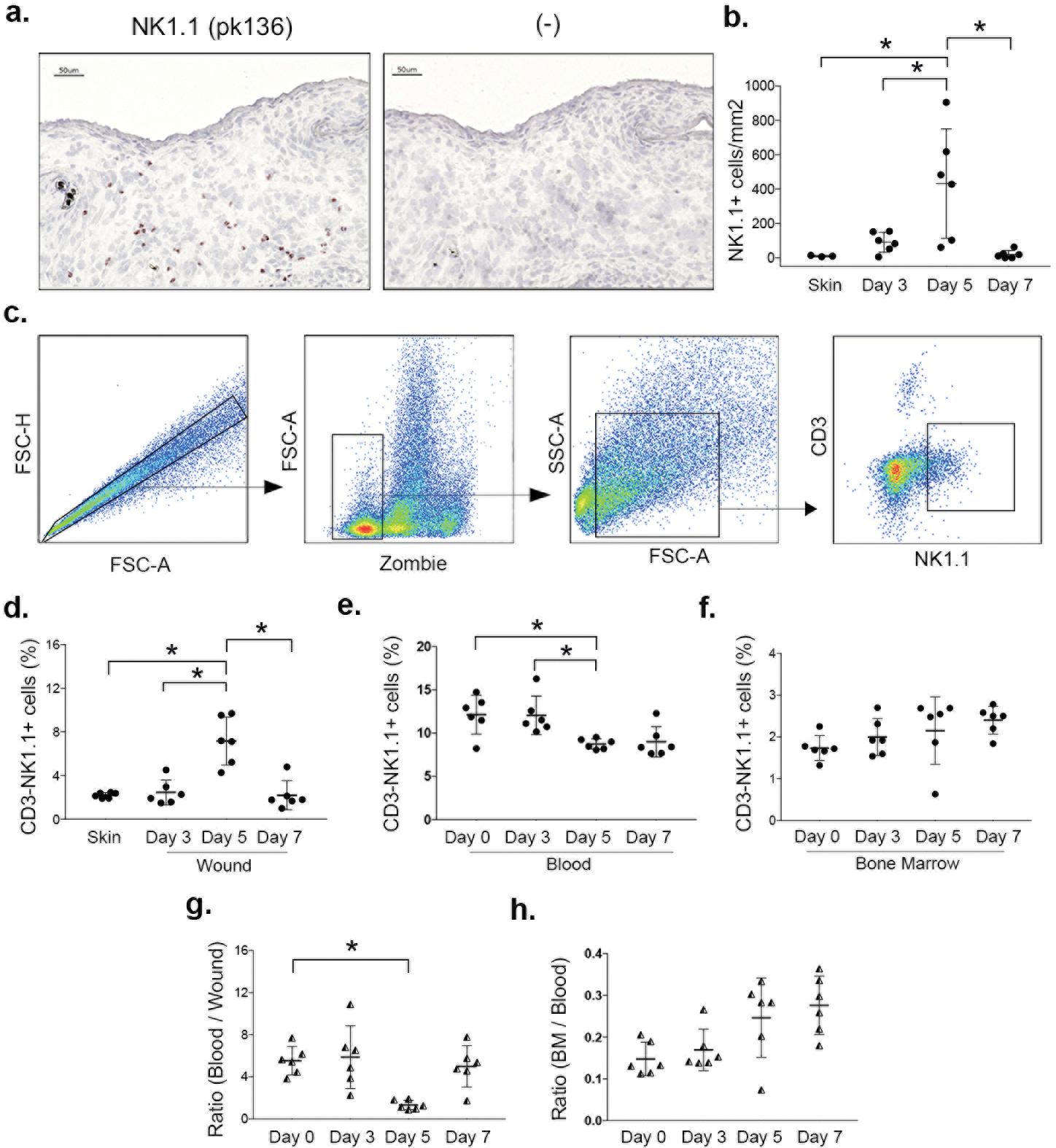
(a) NK cells identified in cryosections by immunohistochemical staining for NK1.1 (pk136) in male mice and (b) corresponding summary data for cell counts. (c) NK cells also assessed by flow cytometry: gating strategy shown for identifying NK cells (CD3−NK1.1+), Zombie dye used for gating viable cells from wounds. (d) Summary data for NK cells in wounds, (e) blood and (f) bone marrow. (g) Ratio of NK cells in blood/wound and (h) in bone marrow/blood. Data presented as mean ± SD, n = 6 mice, from two separate experiments in cohorts of 3 mice each. For each data set, one-way ANOVA and Tukey’s post hoc test were performed. *p < 0.05 between indicated time points.
As expected (21), CD3+NK1.1− T cells were also increased in wounds compared to uninjured skin, persisting from days 3 to 7 post-injury (Suppl. Fig. 1 e), while very few CD3+NK1.1+ NKT cells were observed in either uninjured skin or wounds, with no significant changes after wounding (Suppl. Fig. 1 f).
IL-15 promotes NK cell proliferation in wounds
Next, we determined whether NK cells proliferate in wounds and whether IL-15, a cytokine previously reported to mediate homeostatic proliferation of NK cells in different tissues (22)(23)(24), mediates NK cell proliferation in wounds. We first measured IL-15 protein levels in uninjured skin and in wounds and found that IL-15 was present, and demonstrated a non-significant trend of an increase after injury (Fig. 2 a).
Figure 2. NK cells proliferate in wounds in an IL-15-dependent manner.
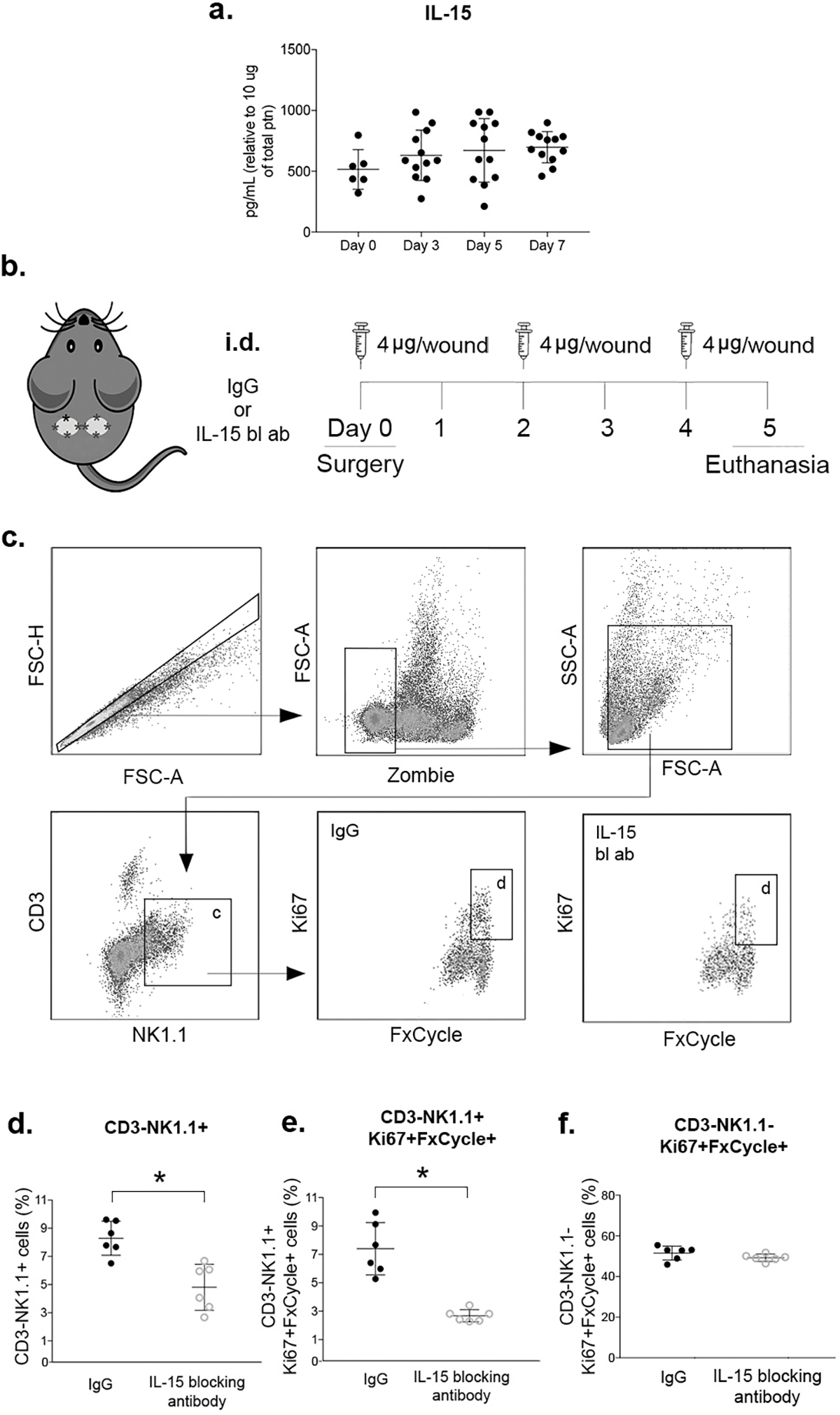
(a) Levels of IL-15 protein in wounds measured by flow cytometry multiplex assay. (b) Local IL-15 blocking strategy: animals were subjected to excisional wounding and then treated with intradermal (i.d.) injections of 4ug of anti-mIL-15 antibody or IgG isotype control antibody on days 0, 2 and 4. Wounds harvested on day 5. (c) Gating strategy for NK cells (CD3−NK1.1+) and proliferating NK cells (Ki67+FxCycle+) identification by flow cytometry, Zombie dye used for gating viable cells. (d) Summary data for NK cells (CD3−NK1.1+) in wounds. (e) Summary data for proliferating NK cells (CD3−NK1.1+Ki67+FxCycle+) in wounds. (f) Summary data for proliferating CD3−NK1.1− cells. Data presented as mean ± SD, n = 6 mice, from two separate experiments in cohorts of 3 mice. For IL-15 protein data, one-way ANOVA and Tukey’s post hoc test showed no significant differences. For IL-15 blocking antibody experiments, two-tailed t-tests showed significant differences between groups treated with anti-mIL-15 antibody and IgG isotype control;*p < 0.05.
To investigate whether IL-15 mediates NK cells proliferation in wounds, we neutralized IL-15 locally with i.d. injections of IL-15 neutralizing antibody. Neutralizing IL-15 significantly decreased CD3−NK1.1+ cells in wounds (Fig. 2 d) and decreased proliferating CD3−NK1.1+Ki67+FxCycle+ cells (Fig. 2 e) on day 5 post-injury, when compared to IgG isotype treated controls. These data indicate that IL-15 promotes NK cell proliferation, contributing to their accumulation in wounds. In contrast, the IL-15 blocking antibody did not have an effect on proliferation of CD3−NK1.1− cells (Fig. 2 f), indicating the effect was specific to NK cells.
In addition, chemokines, including those in the CXC and CX3C family, have been shown to direct NK cells to sites of inflammation in peripheral tissues (13)(25)(26)(27). Two such chemokines, CX3CL1 and CXCL10, were present in wounds (Suppl. Fig. 1 g/h). but only CX3CL1 was significantly increased over levels in uninjured skin (Suppl. Fig. 1 g).
NK cell phenotype in wounds
We then began to investigate the phenotypes of NK cells that accumulate in skin wounds. Our flow cytometry data show that the majority (~70%) of NK1.1+ cells sorted from wounds at all time points examined (days 3 to 7 post-injury), stained positively for CD11b but not CD27 on their surface, indicating a mature phenotype (28). Double positive CD11b+CD27+ staining was also observed for ~20% of NK1.1+ cells, which are considered intermediate maturity, and less than 3% of NK1.1+ cells were CD11b−CD27+, indicating only a minor contribution from this less mature subset (Fig. 3 b). Interestingly, the least mature subset CD11b−CD27− represented ~5–10% of NK1.1+ cells sorted from wounds (Fig. 3 b). The percentages of these subsets changed significantly at day 7 post-injury, at which point intermediate CD11b+CD27+ cells increased and mature CD11b+CD27− cells decreased, with no significant changes in CD11b−CD27− cells (Fig. 3 b).
Figure 3. NK cells express mature inflammatory phenotype in wounds.
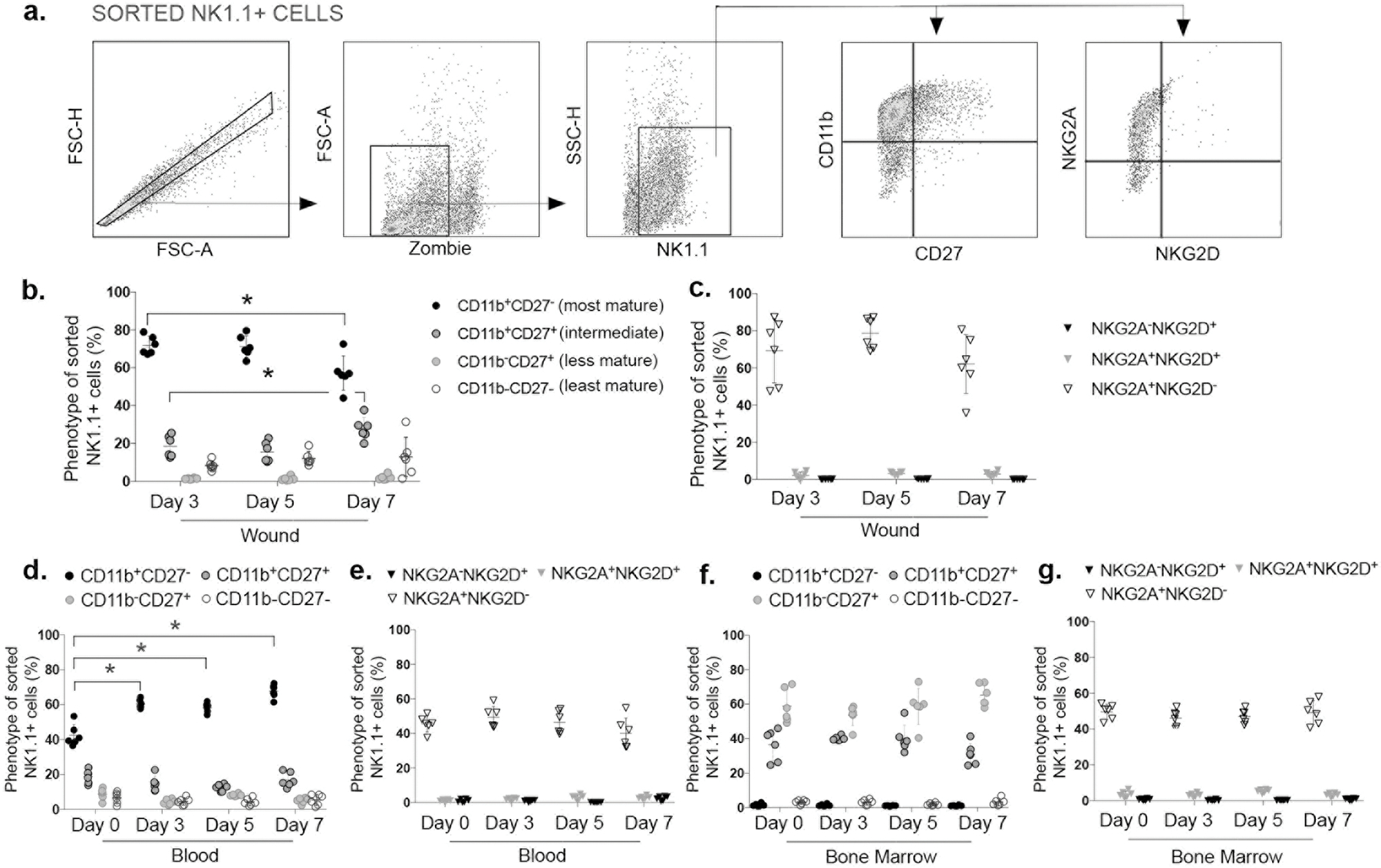
(a) Gating strategy for assessing phenotype of sorted NK cells (NK1.1+), based on maturation markers (CD11b, CD27), and activating/inhibitory receptors (NKG2A, NKG2D) by flow cytometry, Zombie dye used for gating viable cells in wound samples. Maturation stage of NK cells (NK1.1+) based on expression of CD11b and CD27 in (b) wounds, (d) blood and (f) bone marrow. activating/inhibitory receptors on NK cells (NK1.1+) based on expression of NKG2A and NKG2D in (c) wounds, (e) blood and (g) bone marrow. Data presented as mean ± SD, n = 6 mice, from two separate experiments in cohorts of 3 mice. For each data set, one-way ANOVA and Tukey’s multiple comparisons test were performed. * indicates significant differences between time points indicated; p < 0.05.
Next, we measured expression of activating NKG2D and inhibitory NKG2A receptors on NK1.1+ cells. Interestingly, ~75% of NK1.1+ cells from wounds present an NKG2A+NKG2D− phenotype and less than 5% present NKG2A+NKG2D+ or NKG2A-NKG2D+ phenotypes and there was little change in percentages of cells expressing these receptors from days 3 to 7 post-injury (Fig. 3 c). These data indicate that wound NK cells predominantly express the inhibitory receptor NKG2A but not the activating receptor NKG2D. Another inhibitory receptor, Ly49I was expressed in ~70% of CD3−NK1.1+ cells in uninjured skin and in ~90% of CD3−NK1.1+ cells in day 5 wounds (p=0.082) (Suppl. Fig. 2 f).
In peripheral blood of uninjured mice, NK1.1+ cells showed a more mixed distribution of phenotypes with ~40% mature CD11b+CD27− cells, ~25% intermediate CD11b+CD27+ and ~10% less mature CD11b−CD27+ and CD11b−CD27− cells, similar to previous reports of NK cell phenotype distribution in blood (29). After skin wounding, the phenotype of peripheral blood NK1.1+ cells significantly changed towards that observed in wounds, with ~60% of NK1.1+ cells presenting a mature CD11b+CD27− phenotype, ~20% intermediate CD11b+CD27+ and ~10% immature CD11b−CD27+ or CD11b−CD27− cells (Fig. 3 d). Similar to wounds, blood NK1.1+ cells predominantly express the inhibitory NKG2A receptor (~50%) whereas less than 5% are either NKG2A+NKG2D+ or NKG2A−NKG2D+ cells, with little change over the time course analyzed (Fig. 3 c/e).
In bone marrow, the phenotype of NK1.1+ cells differed from that observed in wounds and peripheral blood. Bone marrow NK1.1+ cells expressed a less mature phenotype with ~60% CD11b−CD27+ cells, ~40% intermediate CD11b+CD27+ cells and less than 1% of mature CD11b+CD27− cells, which did not differ significantly from day 0 to day 7 post-injury (Fig. 3 f); these phenotypes are similar as those reported previously for bone marrow NK cells (29). Finally, bone marrow NK1.1+ cells exhibited a similar pattern of NKG2 receptor expression as in wounds and blood, with ~60% NK1.1+ cells expressing inhibitory NKG2A but not activating NKG2D receptor and less than 5% NKG2A+NKG2D+ or NKG2A−NKG2D+ cells from day 0 to day 7 post-injury (Fig. 3 c/e/g).
In liver and spleen, CD3−NK1.1+ cells, expressed a generally mature phenotype with ~50–60% CD11b+CD27− cells, while < 20% of cells expressed intermediate CD11b+CD27+ and less mature CD11b−CD27+/CD11b−CD27− phenotypes each (Suppl. Fig. 2 b/c). Additionally, the inhibitory Ly49I receptor was expressed in ~90% and ~50% of CD3−NK1.1+ cells of liver and spleen, respectively (Suppl. Fig. 2 d/e).
NK cell depletion results in enhanced wound re-epithelization and collagen deposition
Next, we sought to determine the role of NK cells in skin wound healing using a model of systemic NK cell depletion (19)(14). First, we confirmed that serial i.p. injections of the NK1.1 PK136 antibody significantly decreased CD3−NK1.1+ cells in peripheral blood at each time point post-injury, reducing their levels from ~5–7% of the blood cell population for mice treated with IgG2a isotype controls to ~1–2% for mice treated with NK1.1 antibody (Fig. 4 c). The blocking antibody also decreased CD3−NK1.1+ cells in skin wounds at each time point, including the time point of peak accumulation (day 5), at which point systemic depletion reduced wound NK cells from ~8% to ~2% of viable gated cells when compared to mice treated with control antibody (Fig. 4 d). Thus, the systemic NK cell depletion successfully reduced NK cell accumulation in skin wounds.
Figure 4. NK cell depletion results in accelerated wound closure.
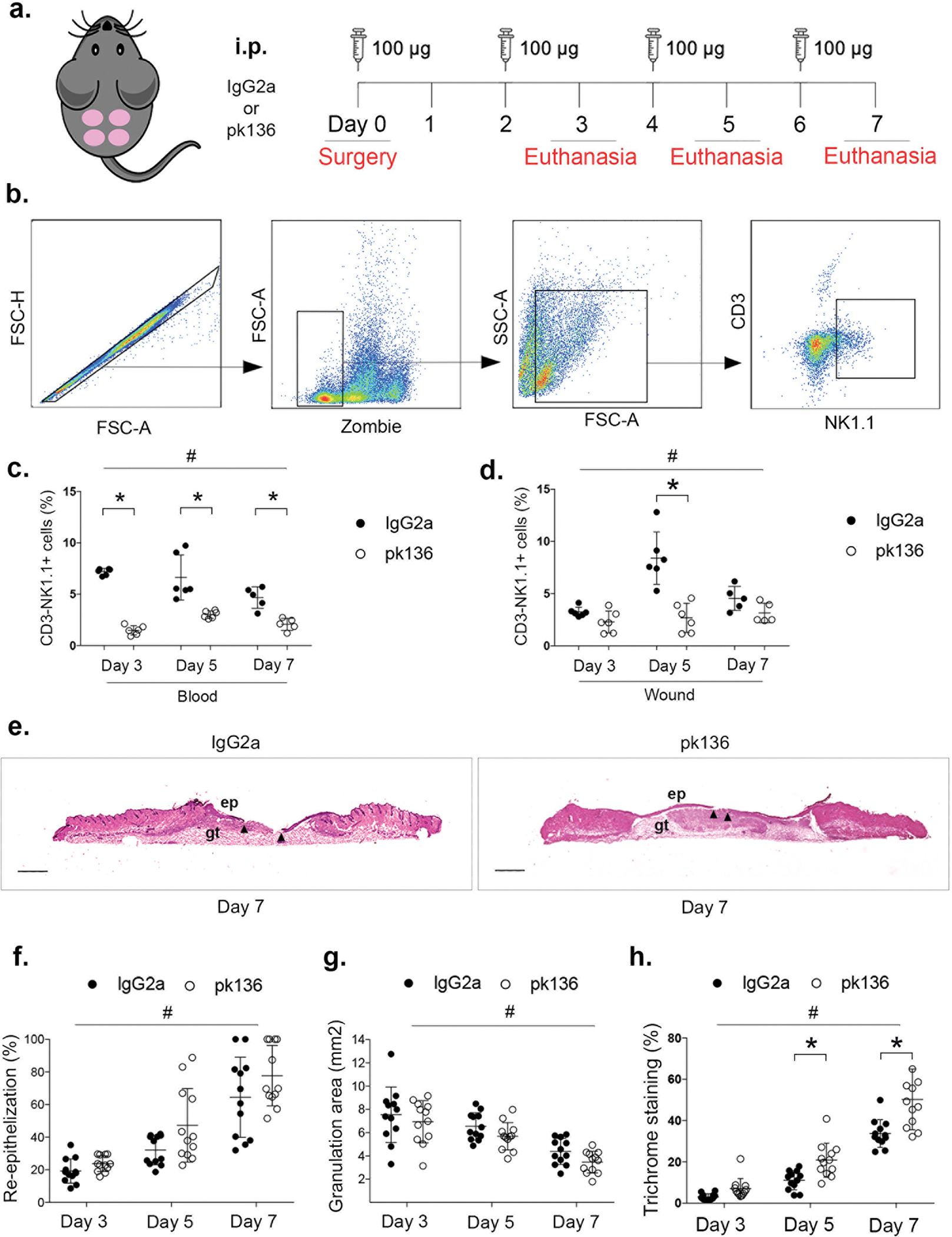
(a) Strategy for systemic NK cell depletion: mice were subjected to full-thickness wounding and 100ug of NK1.1(pk136) antibody or IgG2a isotype control antibody injected intraperitoneally (i.p.) on days 0, 2, 4 and 6 post-wounding. Wounds were harvested on days 3, 5 or 7. (b) Gating strategy for assessing wound NK cells (CD3−NK1.1+) by flow cytometry, Zombie dye used for gating viable cells in wound samples. Summary data for NK cells in (c) blood and (d) wounds. (e) Representative images of hematoxylin and eosin stained cryosections of center of day 7 wounds. ep: epithelium; gt: granulation tissue. Arrows indicate tips of epithelial tongues migrating into wound. Scale bar = 1mm. (f) Summary data for re-epithelization assessed in hematoxylin and eosin stained cryosections. (g) Summary data for granulation tissue area assessed in hematoxylin and eosin stained cryosections. (h) Summary data for collagen deposition assessed in Trichrome stained cryosections. Data presented as mean ± SD, n = 6 mice, from two separate experiments in cohorts of 3 mice. For each data set, two-way ANOVA and Sidak’s multiple comparisons test were performed. # indicates significant main effect of pk136 versus isotype control antibody over all time points, * indicates significant difference between groups at indicated time point (p < 0.05).
Importantly, NK cell depletion resulted in significantly accelerated wound closure (mean difference of 7.4% over all time points) in digital images of the wound surface (Suppl. Fig. 2 g,h) and significantly increased histological measurements of re-epithelialization (mean difference of 11.0% over all time points) (Fig. 4 e,f), as evidenced by a significant main effect of NK1.1 antibody treatment compared to control antibody in ANOVA analysis of each dataset; each dataset also showed a significant main effect of time point but no interaction effect. NK cell depletion also significantly reduced granulation tissue area (Fig. 4 g) as evidenced by a significant main effect of antibody treatment in ANOVA analysis of histological measurements, although the effect size was small (mean difference of 0.8 mm2). Finally, NK cell depletion significantly increased collagen deposition, assessed by Trichrome staining (Suppl. Fig. 2 i). The ANOVA analysis for trichrome staining showed a significant main effect of antibody treatment as well as and significant interaction between treatment and time point. Post-hoc testing showing significant differences on days 5 and 7 post-injury; blue collagen staining increased from ~30% to ~50% at day 7 post-injury (Fig. 4 h). NK cell depletion did not change levels of dead cells isolated from wounds (Suppl. Fig. 2 a), providing evidence that NK cells do not kill cells in wounds. In summary, these data indicate a small but significant positive effect of NK cell depletion on wound healing.
NK cell depletion does not alter accumulation of other leukocytes in wounds but contribute to pro-inflammatory response in physiological wound healing
Finally, to assess the role of NK cells role in the inflammatory response during wound healing, we determined whether NK cell depletion altered the accumulation of other leukocytes, including Np and Mo/Mp. Our data demonstrate that NK cells ablation did not alter the levels of neutrophils (Ly6G+CD11b+ cells) (Fig. 5 b), monocytes (Ly6G−Ly6ChiCD11b+) (Fig. 5 c) or macrophages (Ly6G−Ly6CloCD11b+ cells) in wounds (Fig. 5 d). Nonetheless, NK cell depletion significantly reduced mRNA expression of inflammatory cytokines in wounds, including Ifn-γ (Fig. 5 e), Tnf-α (Fig. 5 f) and Il-1β (Fig. 5 g), as evidenced by a significant main effect of NK1.1 PK136 treatment compared to control antibody treatment in ANOVA analysis. In addition, mRNA expression of Il-1β and Tnf-α showed significant interaction effects, and post-hoc analysis showed that Tnf-α was significantly lower at day 5 post-injury (Fig. 5 f) and Il-1β was significantly lower at day 3 post-injury (Fig. 5 g), compared with control antibody treatment. In contrast, NK cell ablation did not alter expression of Il-10 in wounds (Fig. 5 h). The mRNA data were corroborated by protein data showing reduced IFN-γ in wounds of NK depleted mice compared with controls (Suppl. Fig. 2 j). Consistent with the depletion data, NK1.1+ cells sorted from wounds expressed elevated levels of Tnf-α and Il-1β, particularly on day 3 post-injury compared to control NK1.1+ cells sorted from spleen (Suppl. Fig. 2 k/l). Moreover, wound NK cells expressed Ifn-γ but it did not change from days 3 to 7 post-injury (Suppl. Fig. 2 m). Finally, expression of Il-10 was not detectable in sorted NK1.1+ cells from spleen or wounds (data not shown). In summary, these data indicate that NK cells contribute to the expression of pro-inflammatory genes during wound healing.
Figure 5. NK cells depletion reduces cytokine expression in wounds.
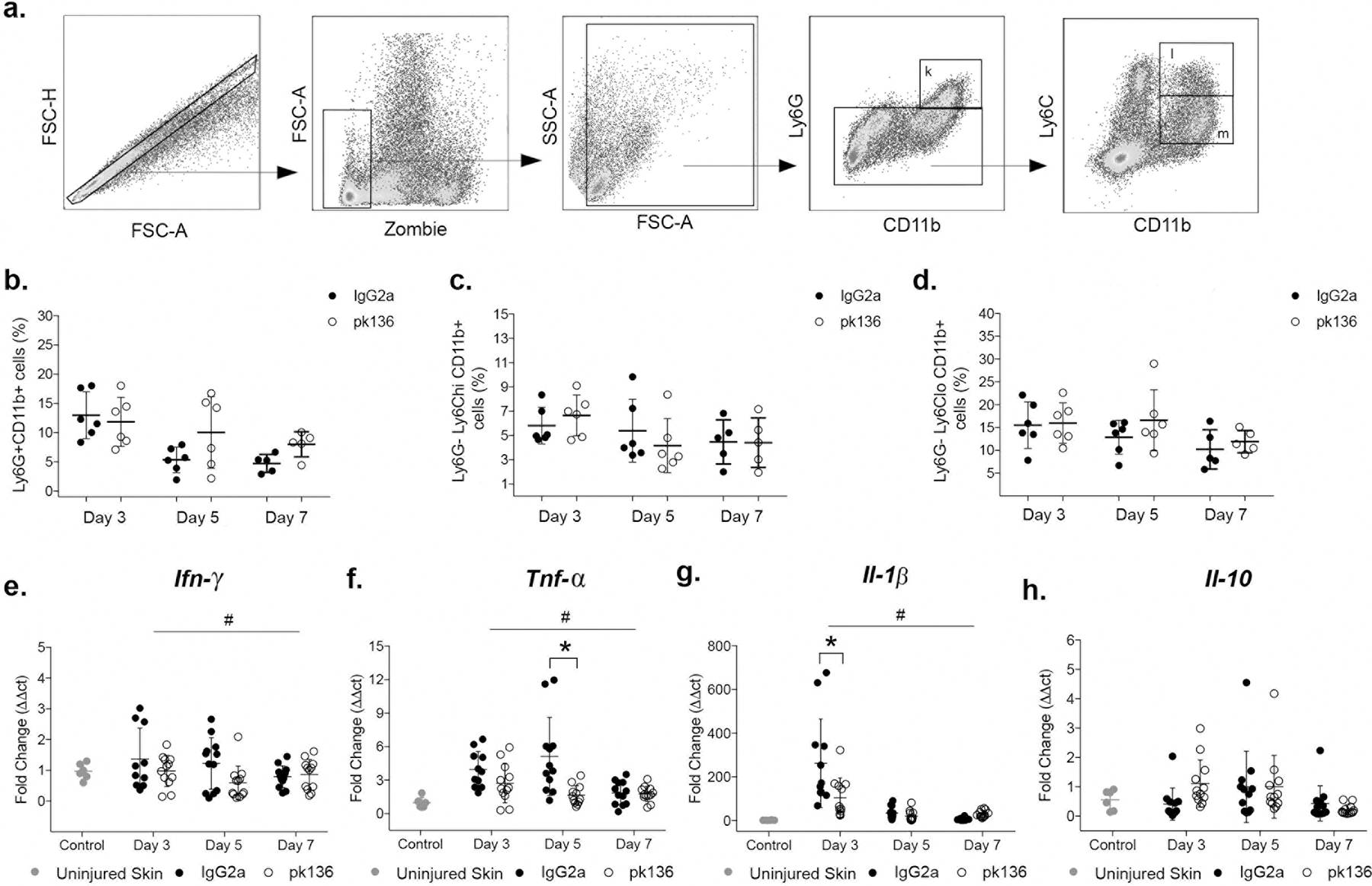
Mice were subjected to full-thickness wounding and 100ug of NK1.1(pk136) antibody or IgG2a isotype control antibody injected intraperitoneally (i.p.) on days 0, 2, 4 and 6 post-wounding. Wounds were harvested on days 3, 5 or 7. (a) Gating strategy for identification of neutrophils, monocytes and macrophages by flow cytometry, Zombie dye used for gating viable cells in wound samples. Summary data for (b) neutrophils (Ly6G+CD11b+), (c) monocytes (Ly6G−Ly6ChiCD11b+) and (d) macrophages (Ly6G−Ly6loCD11b+) in wounds. Summary data for gene expression (fold change) of (e) Ifn-γ, (f) Tnf-α, (g) Il-1β, (h) Il-10 and (i) Tgf-β. Data presented as mean ± SD, n = 6 mice, from two separated experiments in cohorts of 3 mice. For each data set, two-way ANOVA and Sidak’s multiple comparisons test were performed. # indicates significant main effect of pk136 versus isotype control antibody over all time points, * indicates significant difference between groups at indicated time point (p < 0.05).
DISCUSSION
In this study we showed that NK cells accumulate in excisional skin wounds, began to characterize their phenotype in wounds and assessed their role in wound healing using antibody depletion experiments. Our findings showed that NK cell accumulation peaked on day 5 after wounding and that IL-15-dependent proliferation contributes to their accumulation in wounds. In addition, wound NK cells presented a mature phenotype (primarily CD11b+CD27−) and contributed to the expression of pro-inflammatory cytokines in wounds. Finally, NK cell depletion resulted in enhanced re-epithelization and collagen deposition suggesting a negative role for NK cells in wound healing.
IL-15 is crucial for NK cell development, NK cell proliferation and survival, not only in bone marrow but also in secondary lymphoid tissues such as spleen (22)(24)(30). Thus, we investigated the role of IL-15 in the accumulation of NK cells in skin wounds. Importantly, we show that IL-15 inhibition in wounds resulted in significant decrease in both total and proliferating NK cells, indicating that IL-15 contributes to NK cell accumulation in wounds at least partly by inducing their proliferation.
Interestingly, peripheral blood NK cell levels decreased at the time of peak wound NK cell accumulation, suggesting a redistribution from blood to wound sites, and that chemokines likely also play a role in wound NK cell accumulation. In addition, there were trends of decreased NK cell levels in liver and spleen after wounding, suggesting these tissues may be sources of wound NK cells, while no differences were observed in in bone marrow NK cells. Further investigation is warranted to determine the source of NK cells in skin wounds and mechanisms involved in their trafficking.
Factors that might contribute to NK cell infiltration into the wound includes the chemokines CX3CL1 and CXCL10, which are produced by Mo/Mp, fibroblast, endothelial and dendritic cells (13)(27)(31). We found that CX3CL1 and CXCL10 were expressed in wounds, and CX3CL1 levels increased after wounding, but the time course of this increase did not match that of wound NK cells. Thus, further study is needed to identify the chemokines responsible for NK cell accumulation in skin wounds.
NK cells progress from the least mature CD11b−CD27− phenotype, through CD11b−CD27+ and CD11b+CD27+ phenotypes, finally to the most mature CD11b+CD27− phenotype (28) (32). As expected, in uninjured mice, bone marrow NK cells presented with phenotypes that were less mature, while in blood these cells exhibit primarily a mature phenotype; these results are consistent with previous studies (29)(33)(34). After injury, whereas bone marrow NK cells remained primarily less mature, levels of mature NK cells increased in blood, suggesting that factors produced after skin injury might contribute to NK cells maturation systemically. In wounds, the majority of NK cells present the mature CD11b+CD27− phenotype at all time points examined post-injury, with a smaller contribution from intermediate CD11b+CD27+ cells. As has been found in other studies, the mature CD11b+CD27− phenotype of wound NK cells, was associated with expression of the inhibitory receptor NKG2A, but not the activating receptor NKG2D (32). However, NK cells can express numerous activating and inhibitor receptors, including NKG2A, NKG2D and Ly49I, and we have only begun characterizing wound NK cells. For example, a study utilizing used mass cytometry to simultaneously analyze 35 cell surface antigens, including 28 NK cell receptors, indicated that there can be up to 30,000 distinct NK cell phenotypes in an individual (35).
Two prominent changes in the function of NK cells as they mature are loss of proliferative ability and gain of effector function (32)(34). The proliferative ability of CD11b+CD27− and CD11b+CD27+ NK cells has been reported to be lower than that of CD11b−CD27+ and CD11b−CD27− NK cells; however, the more mature cells retained some proliferative capacity, especially during NK cell replenishment after their depletion (32). These latter data are consistent with results from our study suggesting proliferation of mature NK cells in skin wounds. In addition, NK cell effector functions, including cytokine production, are reported to be enhanced as the cells mature and decrease expression of CD27 receptor (28)(32)(34). Our data demonstrates that NK cells from wounds, which exhibit a more mature phenotype, expressed Tnf-α, Il-1β and IFN-γ, and that depletion of NK cells reduced expression of these cytokines in wound homogenates, suggesting that NK cells contribute to pro-inflammatory cytokine expression during the inflammatory stage of healing. Additionally, wound NK cells did not appear to contribute to IL-10 expression in wounds arguing against a regulatory function of these cells, previously described in infection models (36)(37).
Importantly, in our study, depletion of NK cells resulted in enhanced re-epithelization, decreased granulation area, increased collagen deposition and did not change infiltration of other leukocytes such as Np and Mo/Mp. These findings are consistent with those of a previous study showing that NK cell depletion resulted in accelerated wound closure assessed by external measurements (16). This previous study focused on the role of Hypoxia-Inducible Transcription Factor (HIF)-1α in NK cells in regulating a trade-off between wound healing and bacterial defense (16). In this previous study, the lack of HIF-1α in NK cells enhanced neovascularization and accelerated wound closure, but compromised antibacterial defense. Lack of HIF-1α in NK cells also decreased expression of IFN-γ and TNF-α in wounds, similar to our findings with NK cell depletion. Thus, HIF-1α may promote the pro-inflammatory effects of NK cells observed in both the current and previous studies.
In contrast, a study investigating the role of NK cells in a model of corneal epithelial abrasion suggested that NK cells play a positive role in corneal healing, limiting innate acute inflammation upon wounding (15). This study indicated that NK cell ablation resulted in increased neutrophil influx, exacerbated inflammation, inhibited corneal nerve regeneration and inhibited epithelial healing (15). Importantly, the cornea is associated with so-called immune privilege, which involves tissue-specific tolerance and/or suppression of immune responses. Thus, such tissue-specific differences may contribute to differences observed between skin and cornea (38).
Two additional studies focusing on a different subset of NK cells, namely NKT cells, reported that mice deficient in NKT cells exhibited accelerated wound closure, enhanced collagen deposition but no alterations in infiltration or proliferation of other leukocytes, including Mo/Mp and T cells. For identifying NKT cells, the group considered a CD1d dimer+Ly49c+ population. (39)(40). In our study, NKT cells identified as NK1.1+CD3+ cells were extremely rare; the differences between studies may reflect the different sets of markers used. In contrast, another study reported that mice lacking invariant NKT cells exhibited prolonged neutrophilic inflammation and delayed wound healing, suggesting that invariant NKT cells play a positive role in wound healing (41)(42). The different results of these studies indicate that more research is needed to elucidate the role of different NK cell subsets in wound healing.
Limitations of this study included the use of NK cells from spleen of uninjured mice as a control when evaluating gene expression of NK cells; NK cells in uninjured skin are rare, sometimes undetectable, limiting the assessment of NK cells in such samples. Although like wound NK cells, spleen cells express predominantly a mature CD11b+CD27− phenotype, tissue specific gene expression differences could exist. We also acknowledge that sample size (n=6) may not have provided sufficient power to detect differences at specific time points for some assays. We performed a power analysis for each assay performed, which indicated power of > 80% for detecting main effects of time and treatment, as well as interaction between time and treatment for most assays. A notable exception was the re-epithelialization data in Fig 4f, which showed > 99% power to detect main effect of time, 81% power to detect main effect of antibody treatment but only 17% power to detect time by treatment interactions, thus limiting our ability to detect differences at specific time points. In addition, the IFN-γ expression data in Fig 5e showed low power for each effect, 36% for main effect of time, 49% for main effect of antibody treatment, and 38% for the interaction effect between time and treatment, again indicating lack power to detect differences for this assay.
In conclusion, we demonstrate here that NK cells accumulate and proliferate in wounds in an IL-15-dependent manner. We also show that NK cells contribute to the expression of pro-inflammatory cytokines in wounds, including IFN-γ and TNF-α. Finally, NK cell depletion resulted in enhanced re-epithelization and increased collagen deposition, indicating a negative role of NK cells in skin wound healing. Future studies should further investigate the factors that contribute to NK cell accumulation in wounds and their interaction with other wound cells that influence the healing response.
Supplementary Material
KEY POINTS:
Natural killer cells accumulate and proliferate in skin wounds
Natural killer cells express pro-inflammatory factors in skin wounds
Depletion of natural killer cells enhances re-epithelization and collagen deposition
ACKNOWLEDGEMENTS
The authors thank Dr. Giamila Fantuzzi, University of Illinois at Chicago, for critical comments on a previous draft of the manuscript and Rachel Lane, MS from Biostatistics Core in the Center for Clinical and Translational Science at University of Illinois at Chicago, for advice on statistical analysis.
Funding statement
This study was supported by NIGMS through grant R35GM136228 to TJK.
REFERENCES
- 1.Kobayashi T, Ricardo-Gonzalez R, and Moro K. 2020. Skin-resident innate lymphoid cells – cutaneous innate guardians and regulators. Trends in Immunology 41: 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kabashima K 2019. The immunological anatomy of the skin. Nature Reviews in Immunology 19: 19–30. [DOI] [PubMed] [Google Scholar]
- 3.Singer A, and Clark R. 1999. Cutaneous wound healing. New England Journal of Medicine 341: 738–746. [DOI] [PubMed] [Google Scholar]
- 4.Mirza R, and Koh T. 2015. Contributions of cell subsets to cytokine production during normal and impaired wound healing. Cytokine 71: 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novak M, and Koh T. 2013. Phenotypic transitions of macrophages orchestrate tissue repair. American Journal of Pathology 183: 1352–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mirza R, DiPietro L, and Koh T. 2009. Selective and specific macrophage ablation is detrimental do wound healing in mice. American Journal of Pathology 175: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon S, Kim T, and Choi I. 2015. Understanding of molecular mechanisms in natural killer cell therapy. Experimental and Molecular Medicine 47: e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandstadter J, and Yang Y. 2011. Natural killer cell responses to viral infection. Journal of Innate Immunology 3: 274–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson M 2002. Role of chemokines in the biology of natural killer cells. Journal of Leukocyte Biology 71: 173–183. [PubMed] [Google Scholar]
- 10.Liu M, Liang S, and Zhang C. 2021. NK cells in autoimmune diseases: Protective or pathogenic? Frontiers in Immunology 12: 624687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zitti B, and Bryceson Y. 2018. Natural killer cells in inflammation and autoimmunity. Cytokine and Growth Factor Reviews 42: 37–46. [DOI] [PubMed] [Google Scholar]
- 12.Klose C, and Artis D. 2016. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nature Immunology 17: 765–774. [DOI] [PubMed] [Google Scholar]
- 13.Wendel M, Galani I, Suri-Payer E, and Cerwenka A. 2008. Natural Killer Cell Accumulation in Tumors Is Dependent on IFN-γ and CXCR3 Ligands. Cancer Research 68: 8437–8445. [DOI] [PubMed] [Google Scholar]
- 14.Waggoner S, Cornberg M, Selin L, and Welsh R. 2012. Natural killer cells act as rheostats modulating antiviral T cells. Nature 481: 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Q, Smith C, Zhang W, Burns A, and Li Z. 2012. NK cells modulate the inflammatory response to corneal epithelial abrasion and thereby support wound healing. American Journal of Pathology 181: 452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobecki M, Krzywinska E, Nagarajan S, Audige A, Huynh K, Zacharjasz J, Debbache J, Kerdiles Y, Gotthardt D, Takeda N, Fandrey J, Sommer L, Sexl V, and Stockmann C. 2021. NK cells in hypoxic skin mediate a trade-off between wound healing and antibacterial defence. Nature Communications 12: 4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva J, Pitta I, Pitta M, Koh T, and Abdalla D. 2019. New peroxisome proliferator-activated receptor agonist (GQ-11) improves wound healing in diabetic mice. Advances in Wound Healing 8: 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts R, Cavalcante-Silva J, Kineman R, and Koh T. 2021. Liver is a primary source of insulin-like growth factor-1 in skin wound healing. Journal of Endocrinology 252: 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Touw W, Burrell B, Lal G, and Bromberg J. 2012. NK cells are required for co-stimulatory blockade induced tolerance to vasculatory allografts. Transplantation 94: 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirza R, Fang M, Ennis W, and Koh T. 2013. Blocking Interleukin-1 induces a healing-associated wound macrophage phenotype and improves healing in type 2 diabetes. Diabetes 62: 2579–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Havran W, and Jameson J. 2010. Epiderminal T cells and wound healing. The Journal of Immunology 184: 5423–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawamura T, Koka R, Averil M, and Vinay K. 2003. Differential Roles for IL-15R α-Chain in NK Cell Development and Ly-49 Induction. The Journal of Immunology 171: 5085–5090. [DOI] [PubMed] [Google Scholar]
- 23.Prlic M, Blazar B, Farrar M, and Jameson S. 2003. In vivo survival and homeostatic proliferation of natural killer cells. Journal of Experimental Medicine 197: 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desbois M, Beal C, Charrier m., Besse B, Meurice G, Cagnard N, Jacques Y, Bechard d., Cassard L, and Chaput N. 2020. IL-15 superagonist RLI has potent immunostimulatory properties on NK cells: implications for antimetastatic treatment. Journal for immunostimulatory properties on NK cells: implications for antimetastatic treatment. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin-Fontecha A, Thomsen L, Brett S, Gerard C, Lipp M, Lanzavecchia A, and Sallusto F. 2004. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nature 5: 1260–1265. [DOI] [PubMed] [Google Scholar]
- 26.Fong A, Robinson L, and Steeber D. 1998. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. Journal of Experimental Medicine 188: 1413–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imai T, Hieshima K, and Haskell C. 1997. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91: 521–530. [DOI] [PubMed] [Google Scholar]
- 28.Abel A, Yang C, Thakar M, and Malarkannan S. 2018. Natural killer cells: development, maturation and clinical utilization. Frontiers in Immunology 9: 1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crinier A, Narni-Mancinelli E, Ugolini S, and Vivier E. 2020. Snapshot: natural killer cells. Cell 180. [DOI] [PubMed] [Google Scholar]
- 30.Oka N, Markova T, Tsuzuki K, Li W, El-Darawish Y, Pancheva-Demireva M, Yamanishi K, Yamanishi H, Sakagami M, Tanaka Y, and Okamura H. 2020. IL-12 regulates the expansion, phenotype, and function of murine NK cells activated by IL-15 and IL-18. Cancer Immunology and Immunotherapy 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu M, Guo S, and Stiles J. 2011. The emerging role of CXCL10 in cancer. Oncology Letters 2: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, and Walzer T. 2009. Maturation of mouse NK cells is a 4-stage developmental program. Blood 113: 5488–5946. [DOI] [PubMed] [Google Scholar]
- 33.Fu B, Wang F, Sun R, Ling B, Tian Z, and Wei H. 2011. CD11b and CD27 reflect distinct population and functional specialization in human natural killer cells. Immunology 133: 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayakawa Y, and Smyth M. 2006. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. The Journal of Immunology 176: 1517–1524. [DOI] [PubMed] [Google Scholar]
- 35.Horowits A, Strauss-Albee D, Leipold M, Kubo J, Nemat-Gorgani N, and Dogan O. 2013. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Science and Translational Medicine 5: 208ra145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vivier E, and Ugolini S. 2009. Regulatory natural killer cells: New players in the IL-10 anti-inflammatory response. Cell Host Microbe 6: 493–495. [DOI] [PubMed] [Google Scholar]
- 37.Martinez-Espinosa I, Serrato J, and Ortiz-Quintero B. 2022. Role of IL-10-producing natural killer cells in the regulatory mechanisms of inflammation during systemic infection. Biomolecules 12: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bukowiecki A, Hos D, Cursiefen C, and Eming S. 2017. Wound-Healing Studies in Cornea and Skin: Parallels, Differences and Opportunities. International Journal of Molecular Sciences 18: 1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider D, Palmer J, Tulley J, Speicher J, Kovacs E, Gamelli R, and Faunce D. 2011. A Novel Role for NKT Cells in Cutaneous Wound Repair. Journal of Surgical Research 168: 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider D, Palmer J, Tulley J, Kovacs E, Gamelli R, and Faunce D. 2011. Prevention of NKT Cell Activation Accelerates Cutaneous Wound Closure and Alters Local Inflammatory Signals. Journal of Surgical Research 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanno H, Kawakami K, Kanno E, Suzuki A, Takagi N, Yamamoto H, Ishii K, Yoshimichi I, Maruyama R, and Tachi M. 2017. Invariant NKT cells promote skin wound healing by preventing a prolonged neutrophilic inflammatory response. Wound Repair and Regeneration 25: 805–815. [DOI] [PubMed] [Google Scholar]
- 42.Tanno H, Kawakami K, Ritsu M, Imai Y, Maruyama R, and TAchi M. 2015. Contribution of Invariant Natural Killer T Cells to Skin Wound Healing. The American Journal of Pathology 185: 3248–3257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.