

Abstract
Anti-PD-1/PD-L1 agents have transformed the treatment landscape of advanced non-small cell lung cancer (NSCLC). To expand our understanding of the molecular features underlying response to checkpoint inhibitors in NSCLC, we describe here the first joint analysis of the Stand Up To Cancer-Mark Foundation cohort, a resource of whole exome and/or RNA sequencing from 393 patients with NSCLC treated with anti-PD-(L)1 therapy, along with matched clinical response annotation. We identify a number of associations between molecular features and outcome, including (1) favorable (for example, ATM altered) and unfavorable (for example, TERT amplified) genomic subgroups, (2) a prominent association between expression of inducible components of the immunoproteasome and response and (3) a dedifferentiated tumor-intrinsic subtype with enhanced response to checkpoint blockade. Taken together, results from this cohort demonstrate the complexity of biological determinants underlying immunotherapy outcomes and reinforce the discovery potential of integrative analysis within large, well-curated, cancer-specific cohorts.
Subject terms: Non-small-cell lung cancer, Genome informatics, Cancer therapy
Genomic and transcriptomic analysis of 393 non-small cell lung cancer patients treated with checkpoint inhibitors identifies molecular features associated with response.
Main
The introduction of PD-1/PD-L1 inhibitors in the management of advanced non-small cell lung cancer (NSCLC) has led to a major paradigm shift in the treatment of the disease. Following multiple studies demonstrating improved overall survival, these agents have garnered approval either alone1–4 or in combination with chemotherapy5,6 or CTLA4 blockade7. However, with responses observed in only one in five unselected patients1–3, improved predictors of response are needed to identify patients most likely to benefit.
Given that the potential for long-term disease control is only realized in a minority of patients, extensive effort has been dedicated to identifying biomarkers of response and resistance. The dominant biomarkers to date are PD-L1 protein expression on tumor cell membranes4 and tumor mutational burden (TMB)8–10, which may underlie the generation of neoantigens that can serve as targets for immune recognition and targeting.
While additional features have begun to emerge including potential roles for mutation clonality11, an inflamed microenvironment12,13 and alterations in individual genes such as EGFR14,15 and STK11 (ref. 16), further identification and integration of relevant predictors have been hindered by the absence of large, multi-omic, NSCLC-specific patient cohorts.
Here we describe findings from the first integrative analysis of the Stand Up To Cancer-Mark Foundation (SU2C-MARK) NSCLC cohort, a dataset of 393 patients treated with checkpoint inhibitors in the advanced-stage setting. We performed whole exome sequencing (WES) and RNA sequencing (RNA-seq) along with detailed clinical response assessments, enabling the composite assessment of genomic and transcriptomic biomarkers of response and resistance. Collectively, these richly annotated data will be a resource to the field in furthering both the basic and applied investigation into the role of PD-1/PD-L1 agents in advanced NSCLC.
Results
Cohort description and mutation summary
We analyzed formalin-fixed paraffin-embedded (FFPE) tumor samples collected before receipt of checkpoint blockade (defined as the first line of therapy in which a patient received a PD-1/PD-L1 agent) from a total of 393 patients with advanced NSCLC across nine cancer centers (Table 1 and Fig. 1a). The majority of these patients were treated with single-agent therapy (81%), with additional subsets receiving combination therapy including either CTLA4 blockade (17%) or chemotherapy (1%). Both tumor and matched normal specimens (from blood, or in rare cases, adjacent normal tissue) underwent WES; for a subset of patients, tumor tissue was additionally profiled by whole transcriptome RNA-seq. After stringent quality control (Methods), a total of 309 WES and 152 RNA-seq specimens were included for analysis. The primary outcome was best overall response (BOR) determined by a dedicated review of clinical imaging and quantified using RECIST v1.1 criteria.
Table 1.
Baseline clinical characteristics of the SU2C-MARK cohort
| Patient characteristics (n = 393) | All patients, no. (%) |
|---|---|
| Age (years), median (range) | 64 (29–90) |
| Sex | |
| Male | 182 (46) |
| Female | 207 (53) |
| Smoking status | |
| Never | 46 (12) |
| Former | 283 (72) |
| Current | 60 (15) |
| Smoking pack-years | |
| 0 | 47 (12) |
| 1–10 | 46 (12) |
| 11–20 | 50 (13) |
| 21–40 | 125 (32) |
| >40 | 113 (29) |
| Histology | |
| Adenocarcinoma | 286 (73) |
| Squamous | 77 (20) |
| LCNE | 9 (2) |
| Other | 17 (4) |
| PD-L1 expression | |
| 0% | 56 (14) |
| ≥1% | 168 (43) |
| Prior lines of therapy | |
| 0 | 143 (36) |
| 1 | 150 (38) |
| ≥2 | 96 (24) |
| Therapy | |
| PD-(L)1 only | 317 (81) |
| PD-(L)1 + CTLA4 | 65 (17) |
| PD-(L)1 + chemotherapy | 2 (1) |
| BOR | |
| CR/PR | 142 (36) |
| SD | 110 (28) |
| PD | 132 (33) |
The SU2C-MARK cohort consists of 393 patients with NSCLC treated with immune checkpoint blockade therapy in the advanced setting. BOR to the first line containing a PD-(L)1 agent was recorded.
Fig. 1. Overview of the SU2C-MARK cohort and initial genomic characterization.

a, Overview of clinical and genomic data collected across the SU2C-MARK cohort (n = 393 patients). b, CoMut plot of SU2C-MARK cohort organized by response category. c, Log10 of the TMB as a function of response category. Significance was assessed via a two-sided Mann–Whitney U test. d, Volcano plot of logistic regression results for oncogenic mutations in known lung cancer drivers and binned BOR category comparing patients with a PR or CR to patients with SD or PD. ATM alterations reached significance (q < 0.1, Benjamini–Hochberg), while EGFR, RBM10, ARID1A, KEAP1 and SMARCA4 were all near significance (q < 0.25). e, Volcano plot of logistic regression results for gene-level copy number. Focal amplifications of TERT as well as the cytoband it is located on, 5p15.33 (Extended Data Fig. 3b), are associated with resistance to checkpoint blockade. f, Summary of exome-derived genomic features and logistic regression with response. Neoantigens were estimated using NetMHCpan-4.0 (ref. 60) following HLA allele identification with POLYSOLVER61. Subclone count was assessed via PhylogicNDT62. Aging, smoking and APOBEC burdens were calculated based on the mutation burden attributable to these processes (SBS5, SBS4 and SBS13, respectively) following mutational signature analysis (Extended Data Fig. 4 and Methods). HLA was estimated via LOHHLA24. B- and T-cell rearranged receptor abundance was estimated via MiXCR27. LOH, loss of heterozygosity; TMB, tumor mutation burden; PR, partial response; CR, complete response; SD, stable disease; PD, progressive disease.
As is typical for patients with NSCLC, the SU2C-MARK cohort consisted predominantly of adenocarcinoma (73%) and squamous cell carcinoma (20%), with smaller contributions from large cell neuroendocrine (LCNE) carcinoma (2%) and other histologies (4%; Extended Data Fig. 1a). Among patients with annotated PD-L1 staining assessments (224/393 available, 43% missing), 25% had a tumor proportion score (TPS) of less than 1%, 33% had PD-L1 TPS 1–49%, and 42% had PD-L1 TPS ≥ 50%. As expected, higher PD-L1 TPS was associated with an increased response rate to checkpoint blockade (Extended Data Fig. 1b). Thus, our dataset reflected the histologic and biomarker compositions typically observed in unselected, real-world NSCLC cohorts17,18.
Extended Data Fig. 1. Extended SU2C-MARK cohort characterization and genomic predictor evaluation.

(a) Distributions of clinical characteristics in the Stand Up To Cancer - Mark Foundation (SU2C-MARK) cohort. (b) Best overall response (BOR) distribution by PDL1 tumor proportion score (PDL1 TPS) category (significance assessed by two-sided Fisher’s exact test). CR = Complete Response, PR = Partial Response, SD = Stable Disease, PD = Progressive Disease, NE = Not Evaluable. (c,d) Kaplan-Meier curves for Progression-Free Survival (PFS) in EGFR mutated vs. unmutated patients (c) and KRAS/STK11 comutated patients vs. KRAS mutant STK11 umutated patients (d). Both EGFR mutated patients and KRAS/STK11 comutated patients demonstrated decreased progression-free survival relative to their counterparts (p = 0.03 and p = 0.001, respectively, logrank test).
Somatic alterations and PD-(L)1 blockade response in NSCLC
To better understand the relationship between mutational drivers and response, we assessed the prevalence of known drivers in lung cancer across our three response categories: partial or complete response (PR/CR), stable disease (SD) and progressive disease (PD; Fig. 1b and Extended Data Fig. 2a). Consistent with prior reports8–10, nonsynonymous TMB associated with response category (P = 6 × 10−9), with median TMB 14.0 mut/MB among those with PR/CR, compared to 9.0 mut/MB for SD, and 7.4 mut/MB for PD (Fig. 1c). Initial examination of the cohort was also consistent with previously observed driver associations15,16,19, such as EGFR alteration or KRAS/STK11 comutation being a negative predictor of checkpoint blockade response (Extended Data Fig. 1c,d).
Extended Data Fig. 2. Extended analysis of mutated genes in the SU2C-MARK cohort and comparison to external cohorts.

(a) Significant drivers identified independently in the SU2C-MARK cohort (left) as compared to TCGA Lung Adenocarcinoma (LUAD; right upper) and TCGA Lung Squamous Cell Carcinoma (LUSC; right lower). Of note, the SU2C-MARK cohort includes a mixture of frequent drivers observed in LUAD and LUSC, consistent with it representing pan-NSCLC histologies. (b) Kaplan-Meier curves comparing checkpoint blockade treated ATM mutant patients and ATM wildtype patients in the Memorial Sloan Kettering Cancer Center (MSKCC) Impact cohort. ATM mutated patients demonstrated improved survival compared to unmutated patients (p = 0.03, logrank test).
To facilitate a more comprehensive analysis, we performed logistic regression, testing the relationship between 49 known lung cancer drivers20,21 and response (that is, CR/PR versus SD/PD; Methods). In all, six genes achieved significance or near significance, defined as a false discovery rate (FDR) threshold of 10% or 25%, respectively (Fig. 1d). In this analysis, mutations in ATM appeared to be most favorable with respect to checkpoint blockade response (logistic regression FDR q = 0.04, OR = 3.5, CI95% (1.5, 8.0)), while EGFR alterations were least favorable (q = 0.12, OR = 0.29, CI95% (0.11, 0.79)). Given the strong association between ATM and response in our cohort, we tested this association in an independent cohort of patients with NSCLC treated with PD-(L)1 blockade and profiled by MSK-IMPACT22. In this external cohort, ATM alterations were associated with improved overall survival following checkpoint blockade (P = 0.03; Extended Data Fig. 2b). As this association was not seen at the cohort-wide level (P = 0.45), these results suggest a predictive rather than simply prognostic role for ATM alteration.
We next explored relationships between copy number alterations and response in the cohort (Extended Data Fig. 3a). Among focal events, only focal amplification of 5p15.33, the cytoband containing TERT, achieved significance, and was associated with reduced response to immunotherapy (q = 0.07, OR = 0.59, CI95% (0.40, 0.87); Fig. 1e and Extended Data Fig. 3b). Of note, this association was not reproduced in the MSK-IMPACT cohort, which may be a function of the more limited sensitivity of amplifications in panel data (Extended Data Fig. 3c). Taken together, these results suggest that in addition to the aggregate metric of TMB, individual driver events may also define favorable and unfavorable NSCLC subsets for checkpoint blockade.
Extended Data Fig. 3. Extended analysis of somatic copy number alterations within the SU2C-MARK Cohort.

(a) Somatic copy number alterations were analyzed using GISTIC2.0 (ref. 87) to identify significantly recurrent focal amplifications and deletions. Strong overlap between the events identified in the SU2C-MARK cohort and those previously described in TCGA LUAD and LUSC was observed. A subset of validated lung cancer drivers within regions of focal copy number alteration are annotated. (b) Volcano plot of logistic regression results for focal amplifications. Focal amplification of cytoband 5p15.33 (which contains TERT) is associated with resistance to checkpoint blockade. CR = Complete Response, PR = Partial Response, SD = Stable Disease, PD = Progressive Disease. (c) Kaplan-Meier curves comparing checkpoint blockade treated patients with and without TERT amplifications (AMP) in the Memorial Sloan Kettering Cancer Center (MSKCC) Impact cohort (p = 0.7, logrank test).
Predicted neoantigens, antigen presentation and response
To better understand how the determinants of immune recognition in our cohort related to response, we calculated the neoantigen burden for each exome in the SU2C-MARK cohort (Methods). Total neoantigen burden was significantly associated with response (q = 4 × 10−5, OR = 8.8, CI95% (4.2,19); Fig. 1f). As clonal neoantigens have been suggested to be more effective targets of immune recognition11, we additionally examined the role of clonal and subclonal neoantigen burden, along with total subclone count (Methods). Indeed, clonal neoantigen burden was also significantly associated with response (q = 2 × 10−4, OR = 5.4, CI95% (2.7,11)), whereas neither subclonal neoantigen burden nor total subclone count was significant (q = 0.7 and q = 0.6, respectively; Fig. 1f).
As different mutational processes may have different propensities for neoantigen generation, we also evaluated the mutation burden attributable to distinct mutational signatures (Extended Data Fig. 4a,b; Methods). Of the three dominant signatures, smoking was most strongly associated with response (q = 5 × 10−5), consistent with its association with clonal neoantigens, while aging (q = 0.05) and APOBEC (q = 0.01) were more weakly associated with response (Fig. 1f). We additionally observed a significant response association for indels (q = 2 × 10−5), which are suspected to be particularly immunogenic given their potential to generate new reading frames11,23.
Extended Data Fig. 4. Mutation signature analysis in the SU2C-MARK and TCGA-LCNE cohorts.

(a) Unsupervised mutational signature identification was performed using automatic relevance determination non-negative matrix factorization (ARD-NMF) on the combined SU2C-MARK and TCGA-LCNE cohorts. The TCGA-LCNE cohort comprises TCGA lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and published large cell neuroendocrine (LCNE)63 cohorts. Of the 7 signatures identified, the predominant signatures corresponded to COSMIC signatures for Aging (SBS5), Smoking (SBS4), and APOBEC (SBS13). Plots display mutational signatures identified in each sample based on mutation counts (left) as well as fraction of signature attributable mutations (right) with a shared color key for both plots. (b) Barplot of signature profiles demonstrating relative contribution from each 96-base context. Signatures were subsequently assigned to previously described COSMIC signatures based on cosine similarity84.
Previous studies have suggested that compromised antigen presentation, due to loss of heterozygosity (LOH) at HLA loci24, decreased total unique HLA alleles25, or loss of B2M26 may enable immune evasion in certain cancer types. We did not observe an association of any of these factors measured before therapy and response in this cohort (Fig. 1f), potentially suggesting disease-specific variation in mechanisms of resistance.
To further assess for variation in immune infiltrate, we used MiXCR27 to identify B- and T-cell clonotypes from rearranged VDJ reads in our WES data (Methods). Of these subsets, T-cell receptor (TCR) burden was associated with response but did not reach statistical significance (q = 0.3). Thus, among our expanded set of exome-derived features, tumor-intrinsic markers reflective of TMB as well as clonal mutation burden emerged as top predictors of response.
Transcriptional correlates of response
We next focused on the identification of transcriptional predictors of response. Using limma voom28, we performed a genome-wide analysis of differentially expressed genes between responders (PR/CR) and nonresponders (SD/PD; Fig. 2a and Methods). Initial assessment of these results identified three related genes that achieved cohort-wide significance (padj < 0.05; Methods): PSME1, PSME2 and PSMB9. These genes are notable for their prominent role in the function of the immunoproteasome (further described below), a noncanonical peptide processing complex thought to promote differential and enhanced antigen presentation in the setting of proinflammatory cytokines29. Examination of the broader collection of genes achieving nominal significance (nominal P < 0.05) revealed additional interferon-gamma (IFN-γ)-induced transcripts including TAP1 (a cytosolic peptide transporter in the antigen presentation pathway) and CD274 (which encodes PD-L1), inflammatory chemokines such as CXCL9, CXCL10 and CXCL11, and lymphocyte receptor genes (for example, CD3D and CD7), potentially surrogates for immune infiltration (Fig. 2a and Extended Data Fig. 5a). Top genes associated with nonresponse appear to span both developmental and immune-related pathways. AUTS2 and TCF7L1, interacting transcription factors within the Wnt/B-catenin signaling axis, are postulated to have roles in both stem cell30 and immune signaling31. Another nonresponse-associated gene, PDLIM3, is a member of a protein family thought to negatively regulate NF-κB-mediated inflammatory responses32. KALRN, a guanine nucleotide exchange factor expressed in stromal and myeloid cells, has been associated with inflammation in the context of atherogenesis (Extended Data Fig. 5a).
Fig. 2. Transcriptomic features associated with response and resistance in the SU2C-MARK cohort.

a, Volcano plot of limma voom results for top response-associated genes from RNA-seq samples in the SU2C-MARK cohort (n = 152 RNA samples). Nominal P values from two-sided significance testing are shown. Cutoffs of absolute log2(fold change) > 0.5 and P < 0.05 were used to identify significantly differentially expressed genes (red). b, Hallmark GSEA of response and resistance-associated pathways from limma voom. c, Dot plot of significance values for interferon-gamma (IFN-γ) targets (n = 198 genes), proteasome subunits (n = 56 genes) and immunoproteasome subunits (n = 5 genes). Boxplot overlay depicts the 25th percentile (minima), 50th percentile (center) and 75th percentile (maxima) of distribution with whiskers bounding points within 1.5× interquartile range (Q3–Q1) from each minimum and maximum. Immunoproteasome subunits as a set showed a greater association with response than IFN-γ targets and proteasome targets (P = 7 × 10−9 and P = 4 × 10−6, respectively, two-sided Mann–Whitney U test). d, Contour plot of a linear, 2D model predicting expression of representative immunoproteasome subunit PSMB8 as a function of the inflammatory cytokines IFNG and TNF. Contour levels correspond to roughly 1.2-fold TPM increments in PSMB8 expression. Patients with high expression of both IFNG and TNF demonstrated the highest PSMB8 expression (R2 = 0.31). e, Logistic regression summary results for tumor-associated immune cell signatures derived from single-cell sequencing37.
Extended Data Fig. 5. Extended response and resistance associated genes and signatures in the SU2C-MARK Cohort.

(a) Expression of top 10 significant protein-coding transcripts associated with response (PR/CR, left; N = 52 RNA samples) and nonresponse (SD/PD, right; N = 84 RNA samples). Boxplot overlay depicts 25th percentile (minima), 50th percentile (center), and 75th percentile (maxima) of distribution with whiskers bounding points within 1.5 X interquartile range (Q3–Q1) from each minimum and maximum. PR = Partial Response, CR = Complete Response, SD = Stable Disease, PD = Progressive Disease, TPM = transcripts per million (b) Volcano plot for Limma results for cohort wide analysis subsetted to Interferon Targets, Proteasome Subunits, and Immunoproteasome Subunits. (c) Scatterplots comparing 5 inducible components of the immunoproteasome against each other as well as IFNG. Regression line and bootstrapped 95% confidence interval are displayed.
To systematically identify differentially expressed pathways, we performed gene set enrichment analysis (GSEA) using the Hallmark Gene Sets33 (Fig. 2b). Top response-associated pathways included ALLOGRAFT_REJECTION, INTERFERON_GAMMA_RESPONSE and DNA_REPAIR, which has previously been observed as a predictor of checkpoint blockade response in urothelial carcinoma34,35. Pathways associated with resistance were diverse, with EPITHELIAL_MESENCHYMAL_TRANSITION, WNT_BETA_CATENIN_SIGNALING and TGF_BETA_SIGNALING gene sets all significantly associated with nonresponse (Fig. 2b). Taken together, these top genes and gene sets from bulk RNA-seq suggest the relevance of both immune and nonimmune components to the biology of checkpoint blockade.
Immunoproteasome expression and response
Given the remarkable convergence of all three genes (PSME1, PSME2 and PSMB9) on components of the proteasome/immunoproteasome system responsible for peptide generation, we expanded our exploration of genes specific to this antigen presentation pathway. Notably, PSME1 and PSME2 encode for the IFN-γ inducible PA28ɑβ complex that binds and enhances peptide processivity of both the constitutive and immunoproteasome. PSMB9 (LMP2) encodes the β1i IFN-γ inducible subunit that together with β2i (PSMB10) and β5i (PSMB8) represent the three inducible subunits whose incorporation transforms the constitutive proteasome into a specialized immunoproteasome with distinct peptide cleavage patterns29. Hence, as all the inducible components of this complex (PSMB8, PSMB9, PSMB10, PSME1 and PSME2) are known to be downstream of IFN-γ, which itself was nominally associated with response in our analysis (IFNG P = 0.001; log2 fold change 1.1), we evaluated the response association of these components alongside canonical IFN-γ targets (HALLMARK_INTERFERON_GAMMA_RESPONSE) as well as a comprehensive list of proteasome components (GOCC_PROTEASOME_COMPLEX; Fig. 2c). Notably, immunoproteasome components were enriched in terms of the significance of association with response relative to both IFN-γ targets more broadly, as well as all proteasome components (P = 9 × 10−9 and P = 2 × 10−5, respectively; Fig. 2c and Extended Data Fig. 5b).
Although the inducible subunits of the immunoproteasome were highly correlated with one another, increases in their expression could only partly be explained by elevated levels of IFN-γ (Extended Data Fig. 5c). Given that experimental evidence suggests they may also be induced by tumor necrosis factor-α (TNF-α)36, we evaluated whether higher levels of TNF may also contribute to upregulation of these components. Indeed, a linear combination of IFNG and TNF demonstrated improved model fit for immunoproteasome subunit expression (R2 = 0.31 for the combined model compared to 0.19 for the univariate model; Fig. 2d). Thus, immunoproteasome subunits appear to be singularly important predictors of response—even among the broader class of IFN-γ-induced transcripts—perhaps owing to their role as integrators of multiple cytokine cascades, enabling more efficient generation of peptide epitopes for HLA-I presentation.
Immune subset signatures
Given that both individual gene and pathway level analysis highlighted key roles for immune signaling, we aimed to better delineate discrete immune cell subsets in our bulk transcriptome data using previously identified signatures derived from single-cell RNA data37 (Methods). Of the 11 signatures we evaluated, exhausted CD8+ T-cells showed the strongest positive association with response, while the monocyte/macrophage and dendritic cell signatures were most strongly associated with resistance (Fig. 2e).
As a growing body of work suggests that distinct myeloid subsets may have differing roles in antitumor immunity38,39, we investigated more specific subsignatures related to these cell types. Using a marker list derived from a comprehensive single-cell RNA-seq study of infiltrating myeloid cells in human and mouse lung cancers40, we identified the hMono3 and hN3 subtypes as being particularly associated with resistance to checkpoint blockade (Extended Data Fig. 6). Notably, the hMono3 subtype is characterized by high expression of S100A8, a cytokine-like protein that can drive the accumulation of myeloid-derived suppressor cells41. The neutrophil hN3 subtype is defined by high expression of CXCR2, which has been shown to inhibit CD8 T-cell activation within the lung cancer microenvironment42. Thus, our focused analysis of immune subsets identified plausible mechanistic connections between myeloid infiltration and decreased response to checkpoint blockade.
Extended Data Fig. 6. Significance testing of single cell profiling derived myeloid subsets and checkpoint blockade response in the SU2C-MARK Cohort.
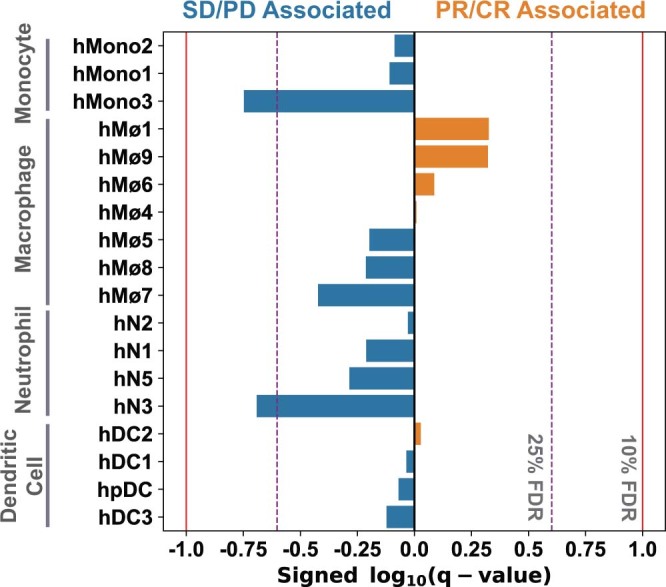
Logistic regression significance values for myeloid cell signatures derived from single cell profiling40 (Benjamini–Hochberg q-value). hMono3 and hN3 were classified as near-significant (q < 0.25) in their association with nonresponse. PR = Partial Response, CR = Complete Response, SD = Stable Disease, PD = Progressive Disease.
Microenvironmental (M) expression signatures
To identify M signatures relevant to immunotherapy response beyond individual cell types, we applied Bayesian non-negative matrix factorization (B-NMF) to our top 770 differentially expressed genes, yielding three distinct M signatures as follows: M-1, M-2 and M-3 (Fig. 3a,b; Methods). Because these signatures were derived from bulk sequencing, they are expected to reflect the complete microenvironmental signature, inclusive of both tumor and nontumor (that is, immune and stromal) sources. GSEA of these signatures revealed M-1 to be associated with epithelial–mesenchymal transition (a gene set that includes wound healing and fibrosis) and M-2 to be associated with allograft rejection/IFN-γ response, consistent with an inflamed immune environment (Fig. 3c). M-3 had a weak association with cell cycle-related E2F targets, potentially reflecting a proliferative tumor signature, which in conjunction with the relative depletion of infiltrating myeloid and lymphoid cells, most resembles the previously reported immune desert phenotype43 (Fig. 3d and Extended Data Fig. 7). Notably, the response rate to checkpoint blockade varied across these subtypes, with increased response rates observed in M-2 relative to M-1 and M-3 (P = 0.06; Fig. 3e). Overall, these results suggest that there may be at least two distinct transcriptional states associated with checkpoint blockade resistance in NSCLC.
Fig. 3. Derivation of M subtypes and association with checkpoint blockade response.

a, Overview of M signature generation using B-NMF. b, H-matrix of SU2C-MARK samples and normalized M signature activity from semisupervised B-NMF. c, Dot plot of hallmark GSEA results for B-NMF-derived M signatures. Nominal P values from the one-sided hypergeometric test are shown. d, Swarmplots of selected tumor-associated immune cell signatures by M clusters. Myeloid cells were generally enriched in the wound healing (M-1, n = 52 RNA samples) subtype, while most immune cell types were enriched in the immune-activated (M-2, n = 56 RNA samples) subtype and depleted in the immune desert (M-3, n = 44 RNA samples) subtype (P < 0.001 for all signatures, Kruskal–Wallis test). e, Response rate by M subtype. The immune-activated (M-2) subtype was enriched for responders compared to the wound healing (M-1) and immune desert (M-3) subtypes (P = 0.06, one-sided Fisher’s exact test).
Extended Data Fig. 7. Comparison of single-cell profiling derived myeloid subsets with Microenvironment (M) subtypes in the SU2C-MARK Cohort.

Swarmplot of myeloid cell signatures derived from single cell profiling40 across Microenvironmental subtypes. Significance of association was assessed by Kruskal–Wallis test (* p < 0.05, ** p < 0.01, *** p < 0.001).
Tumor intrinsic subtyping
Having explored aggregate microenvironmental states, we next turned our attention to tumor intrinsic expression factors that may have a relationship with response. To define relevant tumor intrinsic (TI) lung cancer subtypes, we assembled a large reference collection of over 1,000 transcriptomes (TCGA-LCNE) representing the three predominant NSCLC histologies, namely adenocarcinoma, squamous cell carcinoma and large cell neuroendocrine carcinoma (Fig. 4a and Methods). To define signatures of individual subtypes in this collection, we first performed B-NMF across this cohort, converging on a robust four-cluster solution (Fig. 4b and Extended Data Fig. 8a). Of these TI clusters, TI-1 and TI-2 contained predominantly adenocarcinomas, TI-3 was composed largely of squamous cell carcinomas, and TI-4 was primarily large cell neuroendocrine carcinomas (Extended Data Fig. 8b). Notably, unlike our M signatures above—which were derived solely from the subset of genes with significant response associations and were enriched for immune and stromal components—our TI signatures emerged from the unsupervised factorization of primary lung cohorts spanning three distinct histologies, explaining the high concordance between our TI subtypes and existing histologic categories.
Fig. 4. Derivation of TI NSCLC transcriptional subtypes.

a, Overview of B-NMF approach to the generation of TI subtype signatures. A total of 1,082 RNA-seq samples spanning the three dominant NSCLC histologies were used as input for signature identification. Specifically, the TCGA LUAD and LUSC cohorts were used in addition to a published LCNE Cohort by George et al.63 to generate the combined TCGA-LCNE cohort. b, H-matrix of TCGA-LCNE samples and normalized TI signature activity. c, Violin plots of cancer subtype immunohistochemistry markers based on membership in TI clusters TI-1 (n = 81 samples), TI-2 (n = 433 samples), TI-3 (n = 447) and TI-4 (n = 55). Dedifferentiated (TI-1) samples expressed lower levels of canonical adenocarcinoma and squamous markers, but notably high levels of markers associated with neighboring endodermal lineages (top row). Significance was assessed by the Kruskal–Wallis test (***P < 0.001).
Extended Data Fig. 8. Extended analysis of Tumor-Intrinsic (TI) subtypes.

(a) Alluvial plot of Tumor Intrinsic (TI) subtype downsampling analysis ranging from full TCGA-LCNE cohort (N = 1082) to under 50% downsample (N = 500). The TCGA-LCNE cohort comprises TCGA lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and published large cell neuroendocrine (LCNE)63 cohorts. Both overall distribution and individual sample membership were well preserved across downsamples. (b) Confusion matrix of TCGA-LCNE cohort comparing TI subtype assignment with study source. The novel de-differentiated (TI-1) subtype included predominantly TCGA LUAD samples, with a smaller contribution from TCGA LUSC. (c) Expression scatterplot of canonical adenocarcinoma and squamous cell carcinoma markers, NAPSA (Napsin A) and TP63 (encoding both p40 and p63), respectively, across the TCGA-LCNE Cohort. Samples are colored by TI cluster assignment, with neither de-differentiated (TI-1) nor LCNE (TI-4) samples showing strong canonical lineage marker expression. TPM = transcripts per million. (d) Tumor mutation burden (TMB) for Tumor Intrinsic subtypes TI-1 (N = 81 patients), TI-2 (N = 433 patients), TI-3 (N = 447 patients), and TI-4 (N = 55 patients) in the TCGA-LCNE Cohort. The De-differentiated (TI-1) subtype had an increased mutation burden compared to the Adeno (TI-2) and Squamous (TI-3) subtypes (p = 9 ×10−6 and p = 0.002, respectively, two-sided Mann–Whitney U test). (e) Violinplots of Tumor Intrinsic signatures by membership in Microenvironment clusters M-1 (N = 52 RNA samples), M-2 (N = 56 RNA samples), and M-3 (N = 44 RNA samples).
To understand these signatures in more detail, we explored the expression of canonical markers of adenocarcinoma and squamous differentiation, namely NAPSA (which encodes Napsin A) and TP63 (which encodes both p63 and p40), respectively (Extended Data Fig. 8c). While TI-2 and TI-3 showed the expected lineage marker preferences, TI-1 samples showed weak expression of both markers. Decreased expression of lung lineage markers has previously been described in a subtype of poorly differentiated adenocarcinomas in which markers for adjacent gut lineages (neighboring endodermal territories during development) can become activated44. Indeed, a comparison of these subtypes to immunohistochemical markers of various endodermal lineages revealed enrichment in these gut-specific marker genes in TI-1 samples, such as TFF1, FGA and CPS1 (Fig. 4c). TI-1 samples were also notable for an elevated TMB relative to the well-differentiated TI-2 adenocarcinoma subtype and the TI-3 squamous subtype (Extended Data Fig. 8d).
Having established a reference collection of TI expression signatures, we applied these signatures to RNA-seq data from the SU2C-MARK cohort and assessed their association with response to checkpoint inhibitors. Notably, the dedifferentiated TI-1 cluster was most closely associated with response (Fig. 5a), consistent with the elevated mutational burden in this subtype as well as its stronger association with the M-2 ‘immune-activated’ subtype (Fig. 5b and Extended Data Fig. 8e). Indeed, patients with both immune-activated (M-2) and dedifferentiation (TI-1) signatures had the highest response rates to checkpoint blockade (67% ORR; Fig. 5c). Thus, TI states and immune M signaling may independently and additively govern responses in NSCLC.
Fig. 5. Association between TI signatures, M signatures and response in the SU2C-MARK cohort.

a, Logistic regression analysis summary in the SU2C-MARK cohort between TI signatures and binned response category (PR/CR versus SD/PD). The dedifferentiated (TI-1) signature showed a significant association with response (q < 0.1, logistic regression with Benjamini–Hochberg adjustment). b, Kernel density estimate plot of the association between the activities of the dedifferentiated (TI-1) signature and the previously identified immune-activated (M-2) signature. c, Response rate in the SU2C-MARK cohort binned by expression of TI-1 and M-2 signatures. Patients with both high TI-1 and high M-2 show the highest response rate.
Integrative cohort analysis
Having evaluated a broad set of clinical, genomic and transcriptomic features relevant to checkpoint blockade response in NSCLC, we set out to better understand the relationships between these predictors. Combining the top predictive features from each analysis, we generated a cross-correlation matrix to better understand how they relate to each other as well as to previously published signatures relevant to tumor biology and immune response (Fig. 6 and Methods)35,45–50. Notably, three strong correlation blocks could be observed, with consistent response associations within each subset. The first correlation block (C1) appeared to reflect a canonical ‘wound healing’ microenvironment, including immunosuppressive myeloid and stromal signatures. The second correlation block (C2) reflected the more classic cytokine and immune milieu associated with ‘immune activation/exhaustion,’ including both infiltrating immune signatures and proteasome subunits. The third correlation block (C3) consisted of features related to mutational burden, presumably all proxies for neoantigen abundance and consequent enhanced immune recognition.
Fig. 6. Clinical, genomic and transcriptomic feature integration across the SU2C-MARK cohort.

Cross-correlation heatmap of the top response and resistance-associated features in the SU2C-MARK cohort along with a selection of signatures previously described as relevant to tumor and immune biology35,45–50. The three strongest correlation blocks are outlined and roughly correspond to wound healing (C1), immune activation/exhaustion (C2) and neoantigens (C3). Of note, the direction of association (that is, positive or negative) with immune checkpoint blockade response was consistent for predictors within each of these highlighted correlation blocks.
The remaining nine features were somewhat loosely correlated as a fourth cluster (C4) enriched for single-gene alterations with potentially distinct immunobiologies. Notably, this cluster included EGFR mutations, which interestingly showed minimal association with the immune signatures but a moderate anticorrelation with mutational burden features, suggesting the intrinsic resistance of this subtype may predominantly be driven by insufficient neoantigens15 (Fig. 6 and Extended Data Fig. 9a).
Extended Data Fig. 9. Evaluation of correlation in TCGA data between top SU2C-MARK predictors and assessment of their ability to further stratify clinically relevant subgroups of the SU2C-MARK cohort.

(a) Cross-correlation heatmap of the top response and resistance associated features in the SU2C-MARK cohort as assessed in TCGA LUAD and LUSC combined datasets (N = 1018)35,45–50. Correlation cluster and response association colorbars based on designations in the SU2C-MARK cohort are plotted. Unsupervised hierarchical clustering re-identifies the previously recognized feature clusters corresponding to Wound Healing (C1), Immune Activation/Exhaustion (C2), and Neoantigens (C3). Nearly all features retain their original cluster designations (the relocation of the De-Differentiated TI-1 signature may relate to its association with high mutation burden as described earlier). (b) Contribution of SU2C-MARK predictors to clinically relevant biomarker subsets. The addition of features from the Wound Healing (C1) and Immune Activation/Exhaustion (C2) clusters meaningfully stratify traditionally favorable (for example, PDL1 high) and unfavorable (for example, PDL1 low) clinical subgroups (q = 0.06 and q = 0.16, respectively, Benjamini–Hochberg adjusted logrank test). TMB = Tumor Mutation Burden. (c) Association of top genomic predictors from SU2C-MARK cohort with Progression-Free Survival (PFS) for clinically relevant subgroups of NSCLC, namely high TMB ( > 10 mut/MB, top; favorable), high PD-L1 tumor proportion score (PDL1 TPS) corresponding to PDL1 TPS ≥ 50% (middle; favorable), and low PD-L1 expression (PDL1 TPS ≤ 1%, bottom; unfavorable). Signed FDR q-values based on Benjamini–Hochberg adjustment of logrank p-values are plotted for each feature (Methods).
To evaluate whether the additional genomic predictors identified in this study could augment existing biomarker-defined subsets of NSCLC, we selected the top two significant predictors from each cluster and evaluated their potential to further stratify progression-free survival (PFS) in three clinically relevant subgroups: TMB > 10 mut/MB (favorable; n = 27), PD-L1 TPS ≥ 50% (favorable; n = 34) and PD-L1 TPS ≤ 1% (unfavorable; n = 18). Following FDR correction, we identified multiple near-significant and significant associations (q < 0.25 and 0.1, respectively; Extended Data Fig. 9b,c and Methods), particularly when evaluating features from the immune activation/exhaustion and wound healing clusters (dedifferentiated TI-1 in PD-L1 TPS ≤ 1% q = 0.23; immune-activated M-2 in PD-L1 TPS ≤ 1% q = 0.16; macrophage/monocytes in PD-L1 TPS ≥ 50% q = 0.06; hMono3 in PD-L1 TPS ≥ 50% q = 0.11). Therefore, the presence of these factors may augment prediction based on standard clinical variables.
Feature analysis in single-cell data
Given that the predictors identified in this study were derived from bulk specimens, they likely reflect contributions from multiple distinct cell types within the tumor microenvironment. To gain additional insight into the specific cellular components that may be driving response and resistance, we explored these predictors in the context of published single-cell sequencing data from NSCLC within mixed tumor environments that may be contributing to these signals in bulk data51. Evaluation of the marker expression from the 13 cancer-related clusters revealed a straightforward mapping to several TI subtypes described earlier, including one cluster (cluster 12) which mapped to our dedifferentiated TI-1 subtype (Fig. 7a and Extended Data Fig. 10a; Methods).
Fig. 7. Exploration of top SU2C-MARK transcriptomic features in single-cell data.

a, Leiden clustering of single-cell RNA-seq data from NSCLC51 colored by cluster ID (upper) or cell-type label (lower). Exploration of tumor markers within the cancer-specific clusters enabled further resolution into NSCLC subtypes, including recapitulation of the dedifferentiated TI-1 subtype identified earlier from bulk RNA-seq data (Cluster 12; Extended Data Fig. 8a). b, Association between cell types identified in NSCLC single-cell data and selected genes and metagenes from the wound healing (C1) and immune activation/exhaustion (C2) feature clusters in the SU2C-MARK cohort or with previously described relationships to immunotherapy response35,45–50. Features within larger correlation blocks in bulk RNA-seq data did not always arise from the same single-cell sources (for example, TGF-β versus macrophages/monocytes in the wound healing cluster, and dedifferentiated TI-1 versus immune-activated M-2 in the immune activation/exhaustion cluster).
Extended Data Fig. 10.

Cell type identification and feature analysis from previously published single cell RNA-Seq data in NSCLC. (a) Analysis of top immunohistochemistry (IHC) markers for Tumor Intrinsic (TI) subtypes in single cell non-small cell lung cancer (NSCLC) data51. Leiden cluster 12 demonstrated moderate expression of all 3 IHC markers for the De-Differentiated TI-1 subtype identified from bulk RNA-Seq. Other cluster demonstrated Adeno, Squamous, or mixed Adeno/Squamous markers, with no predominantly Large Cell clusters. (b) Dotplots of the top 10 favorable (left) and unfavorable (right) single genes identified in limma voom analysis of the SU2C-MARK cohort, as expressed in single cell NSCLC data. As observed for features in the larger correlation blocks earlier (Fig. 7b), individual predictors with uncorrelated single cell profiles could be found within each category (for example, CXCL9 vs. CXCL11 among favorable predictors, and SIPA1L2 and PDLIM3 within unfavorable predictors).
Deconvolution of the unfavorable wound healing (C1) predictors suggested that the EMT and TGF-β signatures predominantly reflected fibroblasts and endothelial cells as opposed to a mesenchymal epigenetic state per se within the tumor cells; conversely, some of the dominant single-gene transcriptional predictors such as AUTS2 and TCF7L1 demonstrated substantial tumor intrinsic expression (Fig. 7b and Extended Data Fig. 10b). Similarly, analysis of the favorable predictors in the immune activation/exhaustion cluster (C2) revealed that while immunoproteasome subunits are expressed in most cell types, CXCL9 may be predominantly expressed by myeloid sources, and CXCL11 may be primarily derived from endothelial cells (Fig. 7b and Extended Data Fig. 10b). Finally, our favorable dedifferentiated (TI-1) and immune-activated (M-2) predictors, while correlated at the bulk level, did appear to identify distinct subpopulations (cancer cells and T-cells, respectively) at the single-cell level, consistent with our labeling of these signatures as tumor intrinsic versus microenvironmental (Fig. 7b). Taken together, these findings suggest the presence of rich, interacting ecosystems that may broadly underlie response and resistance to checkpoint blockade and provide a collection of specific signaling pathways and cell types that may be promising targets for future intervention.
Discussion
Comprehensive identification of predictors of checkpoint blockade response in patients with NSCLC has been limited by the availability of large, well-annotated patient cohorts with matched genomic data, particularly within individual cancer types. Here we present a joint analysis of the SU2C-MARK cohort, a collection of nearly 400 patients with NSCLC, enabling the identification of diverse molecular predictors of immunotherapy response.
Among the top genomic features identified were ATM mutation and TERT amplification. Given emerging literature associating ATM loss with the release of cytosolic DNA and activation of the cGAS/STING pathway in other cancer types52–54, it is conceivable that a similar mechanism underlies the association observed in our cohort between ATM loss and response. Although less well characterized in the context of immunotherapy, TERT amplification may serve a protective function against telomere crisis, thereby forestalling a parallel mechanism, which has been linked to cGAS/STING activation and subsequent sensitization to checkpoint blockade in mouse models55.
Transcriptomic analysis in the SU2C-MARK cohort was notable for the identification of immunoproteasome subunit genes as key predictors of response, with greater enrichment than general IFN-γ targets or proteasome subunits. These findings are consistent with those described in melanoma, where a supervised signature consisting specifically of PSMB8 and PSMB9 was found to be predictive of immune checkpoint blockade response56. We speculate that enhanced peptide supply to MHC-1 via increased expression of the PA28ɑβ complex and immunoproteasome may result in superior CD8+ T-cell responses. In addition, the altered cleavage specificity of the immunoproteasome relative to the constitutive proteasome—particularly in terms of preferences for branched-chain amino acids and chymotrypsin-like target sites29— may confer increased antigen quality in addition to quantity in immunoresponsive tumors.
Higher level organization of the strongest genes associated with response and resistance identified microenvironmental signatures previously associated with relevant immune states such as the immune-activated (M-2) signature and immune desert (M-3) signature. The wound healing (M-1) signature, although less well described in the context of lung cancer, does match the TGF-β transcriptional signature thought to drive T-cell exclusion in bladder cancer35. While the immune desert (M-3) signature was somewhat more enigmatic, the top-weighted genes appear to be largely tumor intrinsic, suggesting they may directly reflect a tumor state unfavorable to immune invasion. Consistent with this notion, one of the top-weighted genes in the signature, DSC3, is a component of intercellular desmosome junctions that can act as barriers to immune infiltration57.
In addition to features such as these global immune states that may have pan-cancer relevance, we also describe a dedifferentiated (TI-1) NSCLC-specific subtype identified independently in both bulk and single-cell data using unsupervised approaches. A similar subtype has been described in mouse lung cancer models featuring a decreased expression of classic lung lineage markers as well as enhanced expression of developmentally adjacent endodermal lineages44. The correlation between this tumor intrinsic state and our immune-activated (M-2) signature could represent an underlying differentiation state more susceptible to immune recognition (for example, via the presentation of oncofetal antigens)58, or conversely, a cell state change in response to an inflammatory cytokine milieu59. Establishing the direction of causality between these signatures may have important implications for further therapeutic intervention.
Finally, integrative analysis of our genomic features along with previously reported signatures relevant to immune and tumor biology supported the notion of a complex interplay between distinct signaling pathways (for example, CXCL9 versus TGF-β signaling) and distinct cell types (for example, myeloid cells versus fibroblasts), shedding light on some of the multifaceted interactions underlying checkpoint blockade responsiveness. Particularly noteworthy in this respect is the recognition that a number of features identified here may be truly tumor intrinsic predictors, which aside from a handful of specific driver events15,16 or defects in antigen presentation26 have been somewhat elusive in NSCLC. It is our hope that the SU2C-MARK cohort continues to serve as a rich resource for further unraveling the complex architecture of relevant genomic predictors, and for generating deeper insights into the biology of antitumor immunity.
Methods
Clinical cohort and assessment
All patients in the SU2C-MARK cohort consented through umbrella sequencing protocols approved under local institutional review board protocols at their respective cancer centers (Dana-Farber Cancer Institute 02-180, Massachusetts General Hospital 13-416, MD Anderson PA13-0589, Memorial Sloan Kettering 12-245, Columbia University IRB-AAA05706, University of California Davis LCRP-001, Yale 1411014879, Johns Hopkins IRB00100653). All samples in this study were from patients treated with anti-PD(L)1 therapy either as a single agent or in combination with other agents between 2009 and 2019. Although this cohort predominantly corresponds to standard-of-care therapy, a subset of patients from MSKCC treated with dual checkpoint blockade was derived from sequencing of specimens collected during the course of Checkmate 012 (NCT01454102; ref. 64).
Samples collected typically correspond to the first standard-of-care confirmation of metastatic disease, and therefore reflect a timepoint before receipt of any advanced therapy. Response data were assessed using RECIST v1.1 criteria through a dedicated radiologist review of standard-of-care clinical restaging studies (or in a subset of cases, imaging obtained while on a trial protocol). Confirmed BOR was determined using radiographic data following the first line of therapy involving a PD(L)1-based agent. PFS and overall survival were defined from the date of treatment start with a PD(L)1 agent until the first evidence of radiographic/clinical progression or date of death, respectively, and censoring was based on the date of last follow-up. To facilitate further analyses, WES and RNA-seq specimens were divided into two cohorts with cohort 1 corresponding to roughly the first 80% of available samples. Of note, a subset of these samples has been described previously in institution-specific collections65,66.
Informed consent was obtained under the institutional protocols listed above. Patients were not compensated for their participation. In all, the cohort consisted of 393 patients undergoing checkpoint blockade therapy. Patients in the cohort ranged in age from 29 to 90 years. In total 182 patients were male and 207 patients were female. Additional details on the cohort distribution are described in Extended Data Fig. 1a.
WES
WES of DNA was performed at the Genomics Platform of the Broad Institute of Harvard and MIT as described previously67,68, with the exception of samples previously sequenced at Johns Hopkins65 and Yale University69. In brief, DNA was extracted from FFPE tumor specimens and either matched normal whole blood, or in cases where this was unavailable, from adjacent normal FFPE specimens. Extraction was performed using the Qiagen AllPrep DNA/RNA Mini Kit (80204). A single aliquot of 150–500 ng input DNA in 100 μl TE buffer was used for library generation. Library preparation was performed using the Kapa HyperPrep kit, and quantification was performed using PicoGreen. Adapter ligation was performed using the TruSeq DNA exome kit from Illumina per manufacturer’s instructions. Sequencing of pooled libraries was performed using a HiSeq2500 with 76 bp paired-end reads. The mean target coverages for tumor and normal samples were 150× and 80×, respectively.
Somatic analysis of WES
Initial alignment of all samples to the hg19 genome was performed using the Broad Picard pipeline (v2.4.1), specifically with bwa 0.5.9 (ref. 70). The Broad Cancer Genome Analysis group somatic mutation pipeline was run in the cloud platform Firecloud/Terra. Specifically, the first-pass quality control was performed by assessing sample contamination using ContEst71 and identifying potential sample swaps using the Picard CrossCheckFingerprints tool (using software versions from the GATK 4.0.5.1 release). Somatic single nucleotide variants (SNVs) and indels were called using a combination of MuTect72, MuTect2 (ref. 73) and Strelka74. Recovery of somatic variants filtered due to tumor contamination in the matched normal was performed using DeTiN v1.7 (ref. 75) followed by annotation with Oncotator v1.9 (refs. 75,76). Adjacent SNV events were merged to di-nucleotide variants (DNVs), and filtering was performed using OxoG and FFPE Orientation Bias filters as well as removal of events observed in a panel of normals composed of TCGA and Illumina Capture Exome normals77. Finally, a BLAT realignment filter was implemented to eliminate potentially spurious variants resulting from mismapped reads78. To meet quality control criteria for inclusion in the exome cohort, samples were required to have mean and median target coverage >50×, contamination <5% and tumor purity >10% as assessed by ABSOLUTE (v1.5)79. Comparison of MutSig2CV80 driver analysis from the SU2C-MARK cohort agreed well with previously published results for TCGA lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) cohorts (Extended Data Fig. 2a).
TMB and mutation signature analysis
TMB was calculated as the natural log of nonsynonymous SNVs, DNVs and indels in a sample divided by the size of the Illumina exome capture territory in megabases. Signatures for the SU2C-MARK cohort were determined using the SignatureAnalyzer Bayesian NMF (v1.2) method81–83. In brief, we pooled TCGA LUAD21, TCGA LUSC20 and SU2C-MARK cohort samples to improve our power for detection of rare signatures and performed unsupervised signature extraction using 20 random initializations. Thirteen runs converged to a seven-signature solution, so the k = 7 solution with maximum posterior probability was selected for downstream analysis. Assessment of cosine similarity between the seven signatures identified and the previously described COSMIC signatures84 was used to assign labels to each, with the three dominant signatures representing aging, APOBEC and smoking. Signature attributable mutation burden was calculated as the relative projection strength for each signature in a given sample. Dominant signatures identified across the cohort are shown in Extended Data Fig. 4. Log values of the mutation, signature and clonal/subclonal burdens were calculated using a pseudocount of one event per MB.
Neoantigen analysis
Potential neoantigens were identified by first running POLYSOLVER (v1.0)61 to identify MHC Class I alleles from matched normal WES data. Predicted binding affinity for all possible 9mer and 10mer peptide sequences overlapping single and di-nucleotide somatic variants was assessed using NetMHCPan-4.0 (refs. 60,85,86). Neoantigens with percentile ranks of two or less for any Class I allele in the same patient were counted as predicted binders.
Somatic copy number alteration analysis and GISTIC evaluation
Somatic copy number alterations were assessed from WES using the GATK4 CNV pipeline on Firecloud/Terra (corresponding to GATK v4.0.8.0). A copy number panel of normals (n = 820 samples) was generated from a collection of FFPE as well as fresh frozen samples filtered to have less than 1% of tumor in normal contamination. GATK CNV bin length was set to zero, and read counts were processed using the hg19 Illumina Capture Exome (ICE) targets with padding of 250 bases. Intervals were filtered for having less than 1% of samples with zero coverage by setting—maximum-zeros-in-interval-percentage to 1. The minimum total allele count for informative heterozygous SNPs was set to 10. GISTIC2.0 was used to process the allelic somatic copy number data to identify recurrent copy number altered regions across the cohort87. The continuous copy number output values (rather than binned value) for focal and gene-specific events from GISTIC were used as inputs for downstream analysis. Comparison of significant recurrent alterations showed good consistency between the SU2C-MARK cohort and prior TCGA publications (Extended Data Fig. 3).
ABSOLUTE analysis
Tumor purity and ploidy were estimated using ABSOLUTE (v1.5)79,88. Specifically, somatic mutation and copy number data were used as inputs, and purity/ploidy solutions were evaluated manually. In general, solutions were selected with a preference for describing the observed data well at modeled integer copy numbers, being parsimonious (for example, diploid as opposed to genome doubled), appropriately fitting full deletions and having an alpha/2 line centered within the highest somatic SNV allelic fraction peak (where alpha represents the model purity). A gene-specific integer copy number and LOH were inferred from integer copy number segmented output from total or allele-specific copy number analysis, respectively. Samples with less than 10% purity were excluded as a filtering step during WES quality control as above.
Subclone evaluation using PhylogicNDT
The subclonal architecture was inferred from ABSOLUTE input using PhylogicNDT (v1.0)62. Mutation clonality across single samples was modeled using a Dirichlet process, enabling the assignment of mutations to discrete subclones with imputed cancer cell fractions (CCFs). Variants assigned to clusters with CCF over 0.85 were classified as clonal, while the remainder were deemed subclonal. Subclone count was based on the total number of unique subclones identified by 1D Phylogic analysis.
T-cell and B-cell infiltrate analysis
Rearranged reads corresponding to T- and B-cell receptors were identified from WES data using MiXCR v3.0 (ref. 27). Primary BAM files were processed with the ‘analyze shotgun’ pipeline, and reads corresponding to TCR or Ig clonotypes with productive rearrangements (that is, those leading to in-frame rearrangements without stop codons) were summed to give a total TCR or Ig read count per sample. To infer relative T- or B-cell abundance, these read counts were normalized by calculating T-cell and B-cell burden89, defined as (rearranged receptor count reads + 1)/(aligned reads/106). Natural log of this burden metric was used during the significance assessment.
Response association testing
In total, 106 features derived from whole exome and transcriptome analysis were evaluated (Supplementary Table 30). Features reflecting mutation burden (for example, TMB, Neoantigens, etc.) were log-transformed before evaluation. Mutation and copy number features were filtered to include only those present in at least 5% of the cohort. Each feature was assessed in a univariate logistic regression model of BOR, binned as responders (PR/CR) versus nonresponders (SD/PD). FDR calculation was performed using the Benjamini–Hochberg method, with features categorized as significant (FDR < 0.1) or near-significant (FDR < 0.25).
Whole transcriptome sequencing
RNA-seq data were processed using the GTEx RNA-seq pipeline90 with the use of the GENCODE v19 reference transcriptome, followed by quality control evaluation using the RNA-SeQC2 (v1.0) pipeline90,91, generating both expression data as transcripts per million (TPM) as well as quality metrics. Specifically, this pipeline uses STAR (v1.0) alignment with the following settings: alignIntronMax = 1,000,000, alignIntronMin = 20, alignMatesGapMax = 1,000,000, alignSJDBoverhangMin = 1, alignSoftClipAtReferenceEnds = True, chimJunctionOverhangMin = 15, chimMainSegmentMultNmax = 1, chimSegmentMin = 15. Alignment is then followed by: (1) omission of reads that are unmapped, have secondary alignments (0 × 100 flag) or have the quality control fail flag (0 × 200), and (2) filtering for high-quality exonic reads that uniquely map as pairs (0 × 2 flag) and have fewer than six mismatches to ultimately generate gene-level expression data as well as associated quality metrics. Using the median exon TPM (CV), the number of genes detected, and other measures, we selected the highest quality samples (n = 152) for subsequent analysis.
RNA-seq differential expression analysis
To analyze differentially expressed genes, we restricted our search to protein-coding transcripts, and those minimally expressed at a log2TPM of 0.5 or higher in at least 30% of our samples. Using the BOR groupings of responders (PR/CR) versus nonresponders (SD/PD), we then used the R package limma voom to identify genes differentially expressed with respect to response.
Gene set enrichment analysis
Using the signed, log-transformed P values from the differential expression results, we performed enrichment analyses using the ‘fgsea’ package (v3.16)92 and the Hallmark Gene Sets from the Molecular Signatures Database (MSigDB)33.
RNA-seq supervised signature analysis
Using existing literature, we derived metagenes for clinically important features. Starting with groups of genes associated with a certain feature (for example, genes expressed according to B-cell abundance), we took the mean of the log2-transformed TPMs in our cohort, then compared samples to each other by z-scoring those averages. These analyses include metagenes for different groups of leukocytes93, which we use as a proxy for the level of immune infiltration indicated by RNA-seq. Additionally, we used previously published markers of developmental lineage44,94 and NSCLC subtypes95 to better understand the developmental identity of each sample. We also defined an additional gene set for neuroendocrine identity using markers from a published characterization of large-cell neuroendocrine lung cancer63. For cell type-specific characterization, we used metagenes from single-cell studies of lung cancer developmental subtypes and immune infiltrate37,40.
Non-negative matrix factorization-based expression subtyping
We applied the B-NMF algorithm81,89,96,97 to organize the significantly differentially expressed gene set from cohort 1 of our RNA-seq data (n = 123) into three distinct clusters, that is, our M subtypes. We first filtered our log2(TPM + 1) gene expression matrix to keep only genes with differential expression P value < 0.05 and absolute log-fold change > 0.5, thus limiting our analysis to genes potentially involved in response. We further filtered out genes with sparse or low expression, that is more than 10% NA or zero values, or in the bottom 10% of mean expression. We transformed the values to fold changes by subtracting the median for each gene, then obtained the Spearman correlation matrix of these fold changes, and performed hierarchical clustering while varying the number of clusters (K) from 2 to 10 and repeating 500 iterations for each K value. We then obtained consensus matrices for each K (calculating the number of times samples clustered together in the 500 iterations), summed these matrices across all K values, and normalized the resulting matrix by the number of iterations. Using B-NMF with a half-normal prior, this matrix was used to decide on the optimal number of clusters. Using this empirically determined value of K, we then applied the B-NMF algorithm to the original log2TPM gene expression matrix. In this case, the gene expression matrix is approximated by W*H, where H is the cluster membership matrix and W is the gene weight matrix. We used the W matrix to narrow down the genes most closely associated with each cluster, keeping only genes in the top 50% of normalized weights for each cluster, as well as those with the largest difference between within-cluster versus outside-cluster expression. Using this reduced marker gene list, we classified the remaining samples in cohort 2 into our three-cluster scheme. Of note, given re-annotation of the RNA sample from patient SU2CLC-DFC-DF0732 as having been post-treatment, this specimen was removed from our analysis (Supplementary Note and Supplementary Fig. 1). We used this same procedure to define the TI subtypes, with the exception of initially filtering to keep high-variance genes (instead of keeping genes of interest from the differential expression analysis, as in the M subtypes). We similarly used TI marker genes to classify additional samples.
Integrative predictor clustering
A collection of the top clinical, genomic and transcriptomic predictors identified in the SU2C-MARK cohort or published previously as relevant to antitumor immunity were first compared across samples in the SU2C-MARK cohort. Unsupervised hierarchical clustering was performed on this set, identifying four broad clusters that were ultimately designated wound healing, immune activation/exhaustion, neoantigens and others. As validation of these predictor classes, recalculation of these features was performed in TCGA data by combining publicly available mutation calling and RNA-seq data for the TCGA LUAD21 and TCGA LUSC20 cohorts (combined n = 1018). Unsupervised hierarchical clustering was again performed, and feature membership was compared to assignments made earlier from analysis within the SU2C-MARK dataset. As with other sections described here, integrative analysis was performed with Python (v3.7) and R (v3.4).
Single-cell analysis of predictor clusters
Using single-cell data from a previously published NSCLC cohort51, we performed preprocessing, integration and Leiden clustering in Scanpy (v1.9.1)98 to identify distinct cell types. For preprocessing, we filtered counts to cells with at least 200 genes, and then filtered out genes that were observed in fewer than 50 cells. Further filtering was performed on cells with between 1,000 and 8,000 genes, total counts between 3,000 and 100,000, percent of mitochondrial counts less than 15%, and percent of ribosome counts less than 20%. Cell cycle effects were regressed out using Scanpy, and samples were then integrated using Harmony (harmony2019)99. The cell type of the Leiden clusters was annotated based on gene markers described in Wu et al.51 as well as canonical IHC cancer subtype markers. Clusters were assigned one of the cell types alveolar, B cell, cancer, endothelial, epithelial, fibroblasts, myeloid or T cell based on these expression markers. Metagene expression level was calculated as the mean expression of the gene markers that comprised the metagene. For signatures M-2 and TI-1, the top ten genes by weight were selected. Of note, in some cases, single genes or individual genes in a signature did not pass filtering or were not detected, and therefore were not plotted/included in a given metagene.
Survival analysis
Single-feature survival analysis was performed using progression-free and overall survival data with censoring as described above. For the MSK impact cohort, patients with alterations in ATM found on panel sequencing who also had received checkpoint blockade therapy were included in the cohort. For integrative analysis across the feature list, the top two genomic features from each correlation cluster were selected for PFS analysis as follows: the monocyte/macrophage score, the hMono3 score, dedifferentiated signature TI-1, immune-activated signature M-2, TMB, TMB indel, ATM Mutation and TERT amplification. Participants were binned into high and low categories for each feature (using 0 as a cut point for z-score features, cluster identity for signatures, median for mutation burden features and the presence or absence of alteration/copy gain for single-gene features). FDR values were subsequently computed from the nominal P values obtained via the log-rank test using the Benjamini–Hochberg method. A complete list of the log-rank test results including median PFS for each subgroup is provided in Supplementary Table 31.
Statistics and reproducibility
This study was designed as a retrospective immunogenomic analysis of biospecimens from NSCLC patients receiving checkpoint blockade in the advanced setting. As such, no statistical method was used to predetermine the sample size. Patients who did not have at least one pretreatment whole exome or RNA-seq sample that passed QC following library construction or alignment were excluded from the analysis (as described above). There was no randomization or stratification performed during described analyses, and investigators were not blinded to participant outcomes during primary data analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41588-023-01355-5.
Supplementary information
Supplementary Note and Supplementary Fig. 1.
Supplementary Tables 1–31.
Acknowledgements
We express our deep gratitude to the patients and families whose participation enabled this study. We further thank the respective sequencing centers at Yale University, Johns Hopkins University and the Broad Institute of MIT and Harvard for processing the whole exome and RNA-seq data presented here. Funding for this study was provided by a Stand Up To Cancer—American Cancer Society Lung Cancer Dream Team Translational Research Grant (grant SU2C-AACR-DT17-15, Consortium Award). Stand Up to Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of Stand Up To Cancer. This work was additionally supported by The Mark Foundation for Cancer Research (grant 19-029-MIA, Consortium Award) Expanding Therapeutic Options for Lung Cancer (EXTOL) project. Additional funding was provided by a Conquer Cancer Foundation Young Investigator Award (A.R.), an NCI K99 Award for Outstanding Early-Stage Postdoctoral Investigators (A.R.) and Friends of Farber Award (A.R.). A.T.G. was supported, in part, by the Ruth L. Kirschstein National Research Service Award (NRSA) Institutional Research Training Grant 5T32GM007367-43, and in part by the NCI Ruth L. Kirschstein National Research Service Award (NRSA) Individual Fellowship F30CA257765. N.I.V. was supported by an SITC Genentech Fellowship Award, Conquer Cancer YIA and Damon Runyon Mark Foundation Physician-Scientist Training Award. B.R. was supported by a SITC-AstraZeneca Immunotherapy in Lung Cancer Clinical Fellowship Award. P.M.F. was supported by Bloomberg Philanthropies (BKI) and the LUNGevity Lung Cancer Foundation. J.W.R. was supported by a Cancer Center Support Grant (P30CA093373) and a Paul Calabresi Career Development Award for Clinical Oncology (K12CA138464). D.L.G. was supported by the generous philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shots Program (D.L.G. and J.V.H.). M.M. is partially supported by NIH R01 CA240317 (PathoGenetic Analysis of Invasive Mucinous Adenocarcinoma of the Lung). A.C. was supported by the NCI Cancer Target Discovery and Development Program (U01 CA217858), an NCI Outstanding Investigator Award (R35 CA197745) and NIH Shared Instrumentation Grants (S10 OD012351 and S1 0OD021764), as well as in part through the NCI Cancer Center Support Grant (P30 CA013696). B.D.G. was additionally supported in part through the NIH/NCI Cancer Center Support Grant P30 (CA008748); the Pershing Square Sohn Prize-Mark Foundation Fellowship; a collaboration by Stand Up To Cancer, a program of the Entertainment Industry Foundation, the Society for Immunotherapy of Cancer and the Lustgarten Foundation. J.W. was supported in part through the NIH/NCI Cancer Center Support Grant P30 (CA008748), the Ludwig Collaborative and Swim Across America Laboratory, the Parker Institute for Cancer Immunotherapy, Memorial Sloan Kettering Cancer Center, and Weill Cornell Medicine. N.H. is currently David P. Ryan, MD, Chair funded by a gift from Arther, Sandra and Sarah Irving, and is additionally supported by NIH/NCI (RO1CA208756). M.D.H. is a Damon Runyon Clinical Investigator supported in part by the Damon Runyon Cancer Research Foundation (CI-98-18). A.C. was supported by a Clinical Investigator Award from National Cancer Institute (K08 CA-248723).
Extended data
Author contributions
A.R., J.F.G., C.S., P.A.J., A.S., J.W., N.H., G.G. and M.D.H designed the study. J.F.G., A.T.G., N.I.V., M.S., V.K., H.R., P.M.F., V.A., J.W.R., D.L.G., N.A.P., V.V., J.V.H., R.S.H., J.R.B., K.A.S., V.E.V., B.S.H., N.R., P.A.J., M.M.A., A.T.S., J.W. and M.D.H. contributed patient materials and clinical annotations. J.F.G. and M.D.H. supervised clinical data collection. A.R., M.B.A., M.H., S.S.F., C.S., I.L., J.K., S.D., M.M., A.Chow, A. Califano, Y.A., V.N., N.I.V., A.T.G., B.R., B.D.G. and M.L. participated in primary data analysis, discussion and method development. N.H., G.G., J.F.G. and M.D.H. supervised the study. A.R., J.F.G., M.B.A., M.H., N.H., G.G. and M.D.H wrote the manuscript. All authors participated in the final assembly and revision of the manuscript.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
Raw sequencing data for WES and RNA-seq specimens in the SU2C-MARK cohort are available in dbGaP (phs002822.v1.p1), except for samples from Cleveland Clinic and UC Davis, as these sites did not explicitly include language around deposition of identifiable data in a controlled access repository. Further information about these collections can be obtained from the respective IRB teams (irb@ccf.org and hs-irbeducation@ucdavis.edu) and/or the PIs at each institution (UC Davis; PI: Riess – jwriess@ucdavis.edu; Cleveland Clinic; PI: Pennell – penneln@ccf.org). Data use restrictions specific to each site are also enumerated in the dbGaP accession and include Disease-Specific use (Dana-Farber Cancer Institute), Health/Medical/Biomedical use (MDA Anderson, Memorial Sloan Kettering), and General Research Use (Massachusetts General Hospital). Data from institution-specific cohorts is currently available in dbGaP under accession codes phs001618.v1.p1 (ref. 66) and phs001940.v2.p1 (ref. 65) as well as European Genome-phenome Archive EGAS00001003892 (ref. 65).
Code availability
Code generated for this study has been deposited in the linked Zenodo repository: 10.5281/zenodo.7625517 (ref. 100).
Competing interests
A.R. is a founder, equity owner, and consultant at Halo Solutions and has served as a consultant at Tyra Biosciences. J.F.G. has served as a compensated consultant or received honoraria from Bristol Myers Squibb, Genentech/Roche, Ariad/Takeda, Loxo/Lilly, Blueprint, Oncorus, Regeneron, Gilead, Moderna, Mirati, AstraZeneca, Pfizer, Novartis, iTeos, Nuvalent, Karyopharm, Beigene, Silverback Therapeutics, Merck and GlydeBio; research support from Novartis, Genentech/Roche and Ariad/Takeda; institutional research support from Bristol Myers Squibb, Tesaro, Moderna, Blueprint, Jounce, Array Biopharma, Merck, Adaptimmune, Novartis and Alexo; and has an immediate family member who is an employee with equity at Ironwood Pharmaceuticals. S.S.F is an inventor on provisional patent application No. 62/866,261 related to methods for predicting outcomes of checkpoint inhibition in melanoma and his salary was partially supported by research funding from IBM. I.L. owns equity and consults for ennov1, LLC, and additionally consults for PACT Pharma. J.K. is a current employee and equity owner of GlaxoSmithKline. N.I.V. is a consultant for Sanofi/Regeneron, Oncocyte, and Lilly. P.M.F. has served as a consultant for Amgen, AstraZeneca, BMS, Daichii, F-Star, G1, Genentech, Iteos, Janssen, Novartis, Sanofi, and Surface and has received research support from AstraZeneca, Biontech, BMS, and Novartis. V.A. has received research support to Johns Hopkins from Bristol Myers Squibb and AstraZeneca. J.W.R. has served as a consultant for Boehringer Ingelheim, Novartis, Blueprint, Daiichi Sankyo, EMD Serano, Jazz Pharmaceuticals, Bristol Myers Squibb, Janssen Oncology, Beigene, Turning Point Therapeutics, Genentech and receives research funding from AstraZeneca, Spectrum, Merck, Boehringer Ingelheim, Novartis, Revolution Medicines, GlaxoSmithKline. D.L.G. is an equity owner in Exact Sciences and Nektar; consults for Sanofi, GlaxoSmithKline, Alethia Biotherapeutics, Janssen Research & Development, Eli Lilly, Menarini Ricerche, and 4D Pharma; and receives research support from Janssen Research & Development, Takeda, AstraZeneca, Astellas, Ribon Therapeutics, and NGM Biopharmaceuticals. N.A.P. is a consultant for Astrazeneca, Merck, Pfizer, Eli Lilly/LOXO, Genentech, BMS, Amgen, Mirati, Inivata, G1 Therapeutics, Viosera, Xencor, Janssen, and Boehringer Ingelheim and receives research funding from LOXO, BMS, Merck, Heat Bio, WindMIL, Genentech, Astrazeneca, Spectrum, Mirati, Altor, Jounce, and Sanofi. V.V. is a consultant for BMS, Merck, AstraZeneca, Foundation Medicine, Novartis, Iteos Therapeutics, EMD Serono and receives research funding from AstraZeneca. S.R.D. provides independent image analysis for hospital-contracted clinical research trials programs for Merck, Pfizer, Bristol Myers Squibb, Novartis, Roche, Polaris, Cascadian, Abbvie, Gradalis, Bayer, Zai laboratories, Biengen, Resonance, and Analise and receives research support from Lunit Inc, GE, Vuno and Qure AI. M.M. is a consultant for AstraZeneca, H3 Biomedicine, BMS, Sanofi, Janssen Oncology; receives research funding from Novartis; and owns intellectual property in Elsevier. A.C. is a founder, equity holder, and consultant of Darwin Health Inc. (Columbia University is also an equity holder); holds intellectual property in US patent number 10,790,040 has been awarded related to this work, assigned to Columbia University with A.C. as an inventor, and US patent application number 20210327537 has been filed, also for assignment to Columbia University with A.C. as an inventor. J.V.H. is a consultant for AstraZeneca, BioCurity Pharmaceuticals, Boehringer Ingelheim Pharma, Bristol Myers Squibb, Chugai Biopharmaceuticals, Eli Lilly & Co, EMD Serono, Inc., Genentech, Janssen, Mirati Therapeutics, OncoCyte, Reflexion, Regeneron Pharmaceuticals, Sandoz Pharmaceuticals, Sanofi US Services, Takeda, uniQure, DAVA Oncology, BrightPath Biotherapeutics, Pneuma Respiratory, Eisai, Kairos Venture Investments, GlaxoSmithKline, Gritstone Oncology, Targeted Oncology, Intellisphere, LLC, Millennium Pharmaceuticals, Inc., Catalyst Pharmaceuticals, Guardant Health, Inc., Hengrui Therapeutics, Inc., and Leads Biolabs; receives research funding from AstraZeneca, GlaxoSmithKline, Spectrum; and has intellectual property in Spectrum. R.S.H. has equity in Immunocore, and Bolt, Checkpoint Therapeutics; consults for Immunocore, Junshi Pharmaceuticals, Abbvie, ARMO, AstraZeneca, Bayer, Bolt, Bristol Myers Squibb, Candel Therapeutics, Cybrexa Therapeutics, DynamiCure Biotechnology, eFFECTOR Therapeutics, Eli Lilly, EMD Serono, Foundation Medicine, Genentech/Roche, Genmab, Gliead, Halozyme, Heat Biologics, HiberCell, I-Mab Biopharma, Immune-Onc Therapeutics, Immunocore, Infinity Pharmaceuticals, Johnson and Johnson, Loxo Oncology, Merck, Mirati Therapeutics, Nektar, Neon Therapeutics, NextCure, Novartis, Ocean Biomedical, Oncocyte Corp, Oncternal Therapeutics, Pfizer, Refactor Health, Ribbon Therapeutics, Sanofi, Seattle Genetics, Shire PLC, Spectrum, STCube, Symphogen, Takeda, Tesaro, Tocagen, Ventana Medical Systems, WindMIL Therapeutics, and Xencor; and receives research support from AstraZeneca, Eli Lilly, Genentech/Roche, and Merck. J.R.B. is a consultant for Amgen, Johnson & Johnson, Merck, Bristol Myers Squibb, Sanofi, GlaxoSmithKline, Janssen, Bluprint, AstraZeneca, Regeneron, and Eli Lilly and receives research funding from Bristol Myers Squibb. K.A.S. is a consultant for Shattuck Labs, Pierre-Fabre, EMD Serono, Clinica Alemana de Santiago, Genmab, Takeda, Merck Sharpe & Dohme, Bristol Myers Squibb, AstraZeneca, Agenus and Torque Therapeutics and receives research funding from Navigate Biopharma, Tesaro/GSK, Moderna Inc., Takeda, Surface Oncology, Pierre-Fabre Research Institute, Merck Sharpe & Dohme, Bristol Myers Squibb, AstraZeneca, Ribon Therapeutics, Akoya Biosciences, Boehringer Ingelheim and Eli Lilly. V.E.V. is a founder of Delfi Diagnostics, serves as on the Board of Directors and as a consultant for this organization, and owns Delfi Diagnostics stock, which is subject to certain restrictions under university policy. Additionally, Johns Hopkins University owns equity in Delfi Diagnostics. V.E.V. divested his equity in Personal Genome Diagnostics (PGDx) to LabCorp in February 2022. V.E.V. is an inventor on patent applications submitted by Johns Hopkins University related to cancer genomic analyses and cell-free DNA for cancer detection that have been licensed to one or more entities, including Delfi Diagnostics, LabCorp, Qiagen, Sysmex, Agios, Genzyme, Esoterix, Ventana and ManaT Bio. Under the terms of these license agreements, the University and inventors are entitled to fees and royalty distributions. V.E.V. is an advisor to Viron Therapeutics and Epitope. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict-of-interest policies. N.A.R. is an equity owner in Synthekine and Gritstone; holds positions as CMO of Synthekine and member of Board of Directors and Scientific Advisory Board of Gristone; and holds intellectual property related to Determinants of cancer response to immunotherapy (PCT/US2015/062208) licensed to Personal Genome Diagnostics. P.A.J. is an equity owner in Gatekeeper Pharmaceuticals; consults for AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Chugai Pharmaceuticals, Eli Lilly Pharmaceuticals, Araxes Pharmaceuticals, SFJ Pharmaceuticals, Voronoi, Daiichi Sankyo, Biocartis, Novartis, Sanofi, Takeda Oncology, Mirati Therapeutics, Transcenta, Silicon Therapeutics, Syndax, Nuvalent, Bayer, Esai, Allorion Therapeutics, Accutar Biotech, and Abbvie; receives research support from AstraZeneca, Daiichi Sankyo, PUMA, Eli Lilly, Boehringer Ingelheim, Revolution Medicines, and Takeda Oncology and is a co-inventor and receives postmarketing royalties on a DFCI owned patent on EGFR mutations licensed to LabCorp. M.M.A. is a consultant for Genentech, Bristol Myers Squibb, Merck, AstraZeneca, AbbVie, Neon, Achilles, Maverick, Blueprint Medicine, Hengrui, Syndax, Ariad, Nektar, Gritstone, ArcherDX, Mirati, NextCure, Novartis, EMD Serono, Panvaxal/NovaRx, and Foundation Medicine; and is supported by research grants from Genentech, Lilly, Bristol Myers Squibb, and AstraZeneca. B.D.G. is an equity owner in Rome Therapeutics; consults for Darwin Health, Merck, PMV Pharma and Rome Therapeutics; has received research funding for Bristol Myers Squibb and Merck; and is an owner of intellectual property related to: Rna containing compositions and methods of their use (WO2016131048A1), Neoantigens and uses thereof for treating cancer (US20200232040A1), and Compositions and methods for inhibiting cancers and viruses (WO2020023776A2). M.L. is an owner of intellectual property related to: Neoantigens and uses thereof for treating cancer (WO2018136664A1). A.T.S. is an equity owner and current employee of Novartis. J.W. has equity in Tizona Pharmaceuticals, Imvaq, Beigene, Linneaus, Apricity, Arsenal IO, Georgiamune, Trieza, Maverick, Ascentage; consults for Amgen, Apricity, Ascentage Pharma, Arsenal IO, Astellas, AstraZeneca, Bayer, Bicara Therapeutics, Boehringer Ingelheim, Bristol Myers Squibb, Daiichi Sankyo, Dragonfly, Eli Lilly, F Star, Georgiamune, Idera, Imvaq, Maverick Therapeutics, Merck, Psioxus, Recepta, Tizona, Trieza, Truvax, Trishula, Sellas, Surface Oncology, Syndax, Syntalogic, Werewolf Therapeutics; receives research support from Bristol Myers Squibb and Sephora; and owns intellectual property related to: Xenogeneic DNA Vaccines, Alphavirus replicon particles expressing TRP2, Myeloid-derived suppressor cell (MDSC) assay, Newcastle Disease viruses for Cancer Therapy, Vaccinia virus mutants useful for cancer immunotherapy, Anti-PD-1 Antibody, Anti-CTLA4 antibodies, Anti-GITR antibodies and methods of use thereof, Identifying And Treating Subjects At Risk For Checkpoint Blockade Therapy Associated Colitis, Immunosuppressive follicular helper-like T cells modulated by immune checkpoint blockade, CD40 binding molecules and uses thereof, Phosphatidylserine Targeting Agents and uses thereof for adoptive T-cell therapies, Anti-CD40 agonist mAb fused to Monophosphoryl Lipid A (MPL) for cancer therapy, CAR+T cells targeting differentiation antigens as means to treat cancer. N.H. is an equity owner in BioNtech, Related Sciences/Danger Bio, and consults for Related Sciences/Danger Bio. G.G. is an equity holder in Scorpion Therapeutics; consults for Scorpion Therapeutics; receives research funding from IBM and Pharmacyclics; and is an inventor on patent applications related to MSMuTect, MSMutSig, MSIDetect, POLYSOLVER, SignatureAnalyzer-GPU and TensorQTL. M.D.H. has equity in Factorial, Immunai, Shattuck Labs, Arcus, and Avail Bio, and began as an employee and equity holder at AstraZeneca subsequent to the completion of this work; has consulted for Achilles, Adagene, Adicet, Arcus, AstraZeneca, Blueprint, BMS, DaVolterra, Eli Lilly, Genentech/Roche, Genzyme/Sanofi, Janssen, Immunai, Instil Bio, Mana Therapeutics, Merck, Mirati, Natera, Pact Pharma, Shattuck Labs, and Regeneron; has received research funding from Bristol Myers Squibb; and has intellectual property regarding a patent filed by Memorial Sloan Kettering related to the use of TMB to predict response to immunotherapy (PCT/US2015/062208), which is pending and licensed by PGDx. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Arvind Ravi, Matthew D. Hellmann, Monica B. Arniella.
These authors jointly supervised this work: Nir Hacohen, Gad Getz, Justin F. Gainor.
Contributor Information
Nir Hacohen, Email: nhacohen@broadinstitute.org.
Gad Getz, Email: gadgetz@broadinstitute.org.
Justin F. Gainor, Email: jgainor@mgh.harvard.edu
Extended data
is available for this paper at 10.1038/s41588-023-01355-5.
Supplementary information
The online version contains supplementary material available at 10.1038/s41588-023-01355-5.
References
- 1.Brahmer J, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbst RS, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–1550. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 4.Reck M, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 5.Paz-Ares L, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 2018;379:2040–2051. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 6.Gandhi L, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 7.Hellmann MD, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N. Engl. J. Med. 2019;381:2020–2031. doi: 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 8.Rizvi, H. et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol.36, 633–641 (2018). [DOI] [PMC free article] [PubMed]
- 9.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kowanetz M, et al. Tumor mutation load assessed by FoundationOne (FM1) is associated with improved efficacy of atezolizumab (atezo) in patients with advanced NSCLC. Ann. Oncol. 2016;27:V123. [Google Scholar]
- 11.McGranahan N, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cristescu R, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362:eaar3593. doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Litchfield K, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184:596–614. doi: 10.1016/j.cell.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee, C. K. et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer—a meta-analysis. J. Thorac. Oncol.12, 403–407 (2017). [DOI] [PubMed]
- 15.Gainor JF, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin. Cancer Res. 2016;22:4585–4593. doi: 10.1158/1078-0432.CCR-15-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skoulidis F, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waterhouse D, et al. Real-world outcomes of immunotherapy-based regimens in first-line advanced non-small cell lung cancer. Lung Cancer. 2021;156:41–49. doi: 10.1016/j.lungcan.2021.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Khozin S, et al. Real-world progression, treatment, and survival outcomes during rapid adoption of immunotherapy for advanced non-small cell lung cancer. Cancer. 2019;125:4019–4032. doi: 10.1002/cncr.32383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hastings K, et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann. Oncol. 2019;30:1311–1320. doi: 10.1093/annonc/mdz141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature489, 519–525 (2012). [DOI] [PMC free article] [PubMed]
- 21.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature511, 543–550 (2014). [DOI] [PMC free article] [PubMed]
- 22.Cheng DT, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turajlic S, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18:1009–1021. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- 24.McGranahan N, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259–1271. doi: 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chowell D, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359:582–587. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sade-Feldman M, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017;8:1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolotin DA, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods. 2015;12:380–381. doi: 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- 28.Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol.15, R29 (2014). [DOI] [PMC free article] [PubMed]
- 29.Murata S, Takahama Y, Kasahara M, Tanaka K. The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nat. Immunol. 2018;19:923–931. doi: 10.1038/s41590-018-0186-z. [DOI] [PubMed] [Google Scholar]
- 30.Moreira S, et al. Endogenous BioID elucidates TCF7L1 interactome modulation upon GSK-3 inhibition in mouse ESCs. iScience. 2019 doi: 10.2139/ssrn.3348349. [DOI] [Google Scholar]
- 31.Oksenberg N, et al. Genome-wide distribution of Auts2 binding localizes with active neurodevelopmental genes. Transl. Psychiatry. 2014;4:e431. doi: 10.1038/tp.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ono R, Kaisho T, Tanaka T. PDLIM1 inhibits NF-κB-mediated inflammatory signaling by sequestering the p65 subunit of NF-κB in the cytoplasm. Sci. Rep. 2015;5:18327. doi: 10.1038/srep18327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liberzon A, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X, et al. R-spondin1/LGR5 activates TGFβ signaling and suppresses colon cancer metastasis. Cancer Res. 2017;77:6589–6602. doi: 10.1158/0008-5472.CAN-17-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mariathasan S, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altun M, et al. Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Res. 2005;65:7896–7901. doi: 10.1158/0008-5472.CAN-05-0506. [DOI] [PubMed] [Google Scholar]
- 37.Sade-Feldman M, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2019;176:404. doi: 10.1016/j.cell.2018.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 39.Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021;21:485–498. doi: 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zilionis R, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50:1317–1334. doi: 10.1016/j.immuni.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinha P, et al. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J. Immunol. 2008;181:4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Y, et al. Targeting CXCR2 inhibits the progression of lung cancer and promotes therapeutic effect of cisplatin. Mol. Cancer. 2021;20:62. doi: 10.1186/s12943-021-01355-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 44.Tata PR, et al. Developmental history provides a roadmap for the emergence of tumor plasticity. Dev. Cell. 2018;44:679–693. doi: 10.1016/j.devcel.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willingham SB, et al. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti-PD-(L)1 and anti-CTLA-4 in preclinical models. Cancer Immunol. Res. 2018;6:1136–1149. doi: 10.1158/2326-6066.CIR-18-0056. [DOI] [PubMed] [Google Scholar]
- 46.Hedegaard J, et al. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell. 2016;30:27–42. doi: 10.1016/j.ccell.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 47.Chen J, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567:530–534. doi: 10.1038/s41586-019-0985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao J, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397–404. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Şenbabaoğlu Y, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016;17:231. doi: 10.1186/s13059-016-1092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ayers M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu F, et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021;12:2540. doi: 10.1038/s41467-021-22801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu M, et al. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J. Clin. Invest. 2021;131:e139333. doi: 10.1172/JCI139333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J. Clin. Invest. 2020;130:5951–5966. doi: 10.1172/JCI130445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Q, et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 2019;79:3940–3951. doi: 10.1158/0008-5472.CAN-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mender I, et al. Telomere stress potentiates STING-dependent anti-tumor immunity. Cancer Cell. 2020;38:400–411. doi: 10.1016/j.ccell.2020.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kalaora S, et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat. Commun. 2020;11:896. doi: 10.1038/s41467-020-14639-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chae YK, et al. Overexpression of adhesion molecules and barrier molecules is associated with differential infiltration of immune cells in non-small cell lung cancer. Sci. Rep. 2018;8:1023. doi: 10.1038/s41598-018-19454-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan C, et al. Cancer/testis antigens: from serology to mRNA cancer vaccine. Semin. Cancer Biol. 2021;76:218–231. doi: 10.1016/j.semcancer.2021.04.016. [DOI] [PubMed] [Google Scholar]
- 59.Hölzel M, Bovier A, Tüting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer. 2013;13:365–376. doi: 10.1038/nrc3498. [DOI] [PubMed] [Google Scholar]
- 60.Jurtz V, et al. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shukla SA, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 2015;33:1152–1158. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leshchiner, I, et al. Comprehensive analysis of tumour initiation, spatial and temporal progression under multiple lines of treatment. Preprint at bioRxiv10.1101/508127 (2019).
- 63.George J, et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat. Commun. 2018;9:1048. doi: 10.1038/s41467-018-03099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hellmann MD, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18:31–41. doi: 10.1016/S1470-2045(16)30624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anagnostou V, et al. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat. Cancer. 2020;1:99–111. doi: 10.1038/s43018-019-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gettinger SN, et al. A dormant TIL phenotype defines non-small cell lung carcinomas sensitive to immune checkpoint blockers. Nat. Commun. 2018;9:3196. doi: 10.1038/s41467-018-05032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Allen EM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wagle N, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014;371:1426–1433. doi: 10.1056/NEJMoa1403352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kadara H, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann. Oncol. 2017;28:75–82. doi: 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cibulskis K, et al. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27:2601–2602. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benjamin, D. et al. Calling somatic SNVs and indels with Mutect2. Preprint at bioRxiv10.1101/861054 (2019).
- 74.Kim S, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 75.Taylor-Weiner A, et al. DeTiN: overcoming tumor-in-normal contamination. Nat. Methods. 2018;15:531–534. doi: 10.1038/s41592-018-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramos AH, et al. Oncotator: cancer variant annotation tool. Human Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ellrott K, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018;6:271–281. doi: 10.1016/j.cels.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res.12, 656–664 (2002). [DOI] [PMC free article] [PubMed]
- 79.Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan VYF, Févotte C. Automatic relevance determination in non-negative matrix factorization with the β-divergence. IEEE Trans. Pattern Anal. Mach. Intell. 2013;35:1592–1605. doi: 10.1109/TPAMI.2012.240. [DOI] [PubMed] [Google Scholar]
- 82.Kasar S, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun. 2015;6:8866. doi: 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim J, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016;48:600–606. doi: 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoof I, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mermel CH, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Landau DA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Freeman, S. S. et al. Combined tumor and immune signals from genomes or transcriptomes predict outcomes of checkpoint inhibition in melanoma. Cell Rep. Med.3, 100500 (2021). [DOI] [PMC free article] [PubMed]
- 90.GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Graubert A, Aguet F, Ravi A, Ardlie KG, Getz G. RNA-SeQC 2: efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics. 2021 doi: 10.1093/bioinformatics/btab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv10.1101/060012 (2021).
- 93.Danaher P, et al. Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer. 2017;5:18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Laughney AM, et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 2020;26:259–269. doi: 10.1038/s41591-019-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen F, et al. Multiplatform-based molecular subtypes of non-small-cell lung cancer. Oncogene. 2017;36:1384–1393. doi: 10.1038/onc.2016.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim J, et al. The cancer genome atlas expression subtypes stratify response to checkpoint inhibition in advanced urothelial cancer and identify a subset of patients with high survival probability. Eur. Urol. 2019;75:961–964. doi: 10.1016/j.eururo.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 97.Robertson AG, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2018;174:1033. doi: 10.1016/j.cell.2018.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Korsunsky I, et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Holton, M., Arniella, M., Ravi, A. & Getz, G. Genomic and transcriptomic analysis of checkpoint blockade response in advanced non-small cell lung cancer. Zenodo10.5281/ZENODO.7625517 (2023). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Note and Supplementary Fig. 1.
Supplementary Tables 1–31.
Data Availability Statement
Raw sequencing data for WES and RNA-seq specimens in the SU2C-MARK cohort are available in dbGaP (phs002822.v1.p1), except for samples from Cleveland Clinic and UC Davis, as these sites did not explicitly include language around deposition of identifiable data in a controlled access repository. Further information about these collections can be obtained from the respective IRB teams (irb@ccf.org and hs-irbeducation@ucdavis.edu) and/or the PIs at each institution (UC Davis; PI: Riess – jwriess@ucdavis.edu; Cleveland Clinic; PI: Pennell – penneln@ccf.org). Data use restrictions specific to each site are also enumerated in the dbGaP accession and include Disease-Specific use (Dana-Farber Cancer Institute), Health/Medical/Biomedical use (MDA Anderson, Memorial Sloan Kettering), and General Research Use (Massachusetts General Hospital). Data from institution-specific cohorts is currently available in dbGaP under accession codes phs001618.v1.p1 (ref. 66) and phs001940.v2.p1 (ref. 65) as well as European Genome-phenome Archive EGAS00001003892 (ref. 65).
Code generated for this study has been deposited in the linked Zenodo repository: 10.5281/zenodo.7625517 (ref. 100).