

Key Clinical Message
Coincidences in medicine are not so common. We are presenting a case of a patient diagnosed with Moya‐Moya disease and antiphospholipid syndrome (APS) who presented with clinical and laboratory characteristics of catastrophic APS versus TTP. The diagnosis was a challenge because characteristics were overlapping. Nevertheless, a decision to treat the patient for TTP was made with afterward improvement. MMD has been associated with multiple immune disorders; however, only one case of acquired thrombotic thrombocytopenic purpura has been documented in association with this disease. None has been associated with catastrophic antiphospholipid syndrome. We are presenting a challenging case where all these three medical conditions were present at the same time.
Keywords: antiphospholipid syndrome (APS), Moya‐Moya disease, Moya‐Moya syndrome, thrombotic thrombocytopenic purpura (TTP), treatment
1. BACKGROUND
Moyamoya disease is a cerebrovascular condition that predisposes affected patients to stroke. It is characterized by chronic progressive stenosis of the terminal portion of the bilateral internal carotid arteries or the proximal portion of the anterior and/or middle cerebral arteries, which leads to the formation of an abnormal vascular network composed of collateral pathways at the base of the brain. 1 Moyamoya is the Japanese term for a “puff of smoke,” used to describe the appearance of these collateral vessels on cerebral angiograms. 2 Patients with moyamoya spectrum vasculopathy have been associated with multiple autoimmune diseases, such as systemic lupus erythematosus, autoimmune thyroid disease, antiphospholipid syndrome, and congenital TTP. 3 , 4 Nevertheless, antiphospholipid (APS) syndrome is characterized by a heterogeneous group of autoantibodies strongly associated with arterial and venous thrombosis. Therefore, the endothelial cell interaction in the MMS could trigger antibodies exposing the components of the endothelial cell membrane. 5
On the other hand, thrombotic thrombocytopenic purpura (TTP) is rare thrombotic microangiopathy characterized by microangiopathic hemolytic anemia, severe thrombocytopenia, and ischemic end‐organ injury due to microvascular platelet‐rich thrombi. TTP results from a severe deficiency of the specific von Willebrand factor (VWF)‐cleaving protease, ADAMTS13. In adults, deficiency is most commonly acquired due to anti‐ADAMTS13 autoantibodies. 6 , 7 Currently, only one case of Moyamoya disease in association with acquired TTP has been described and published in 1994. 7 MMD manifests as progressive stenosis, and the hallmark of APS is endothelial damage through thrombosis, meaning endothelial damage, which could trigger TTP. Therefore, they share a possible pathophysiology pathway, but having these three diseases simultaneously is unusual. Hence, makes a challenging diagnosis and management.
2. CASE PRESENTATION
A 52‐ year‐ old female presented with a past medical history of multiple Moya‐Moya diseases (post‐endovascular repair in 2020), cerebrovascular accidents (CVA) with residual left‐side weakness, left upper extremity contracture, and antiphospholipid syndrome on anticoagulation.
It is essential to highlight that both entities (Moya‐Moya Disease and APS) were diagnosed 2 years before this presentation when the patient was admitted with the diagnosis of a second acute CVA with an unclear origin, found to have severe right medium cerebral artery stenosis (Figures 1 and 2) and consequent work up compatible with Moya Moya disease with posterior extracranial–intracranial endovascular repair.
FIGURE 1.

Cerebral angiogram coronal view.
FIGURE 2.
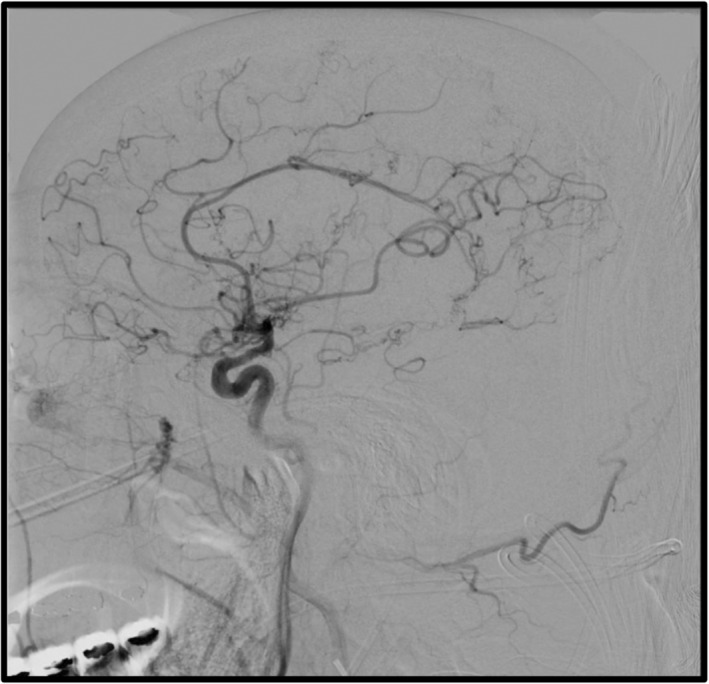
Cerebral angiogram sagittal view.
At that time, there was also a high suspicion of an antiphospholipid syndrome as the patient had a history of one stillborn, one‐second trimester loss, and two early miscarriages with immunology testing positive for IgM anticardiolipin antibodies and for anti‐SS‐A antibodies also had an RPR positive result with no history of Syphilis. The patient was discharged on anticoagulation and has been found to be compliant during the outpatient follow‐up period. The serology was repeated 12 weeks apart, and the APS diagnosis was confirmed.
This time, the patient presented to the emergency department complaining of periumbilical abdominal pain for 1 day. The pain started at night and was described as achy, constant, radiating to the left lower quadrant, with mild relief after eating, and was associated with nausea.
Initial vital signs were normal. A physical exam was pertinent for epigastric abdominal tenderness to palpation. No signs of respiratory distress or poor mentation were observed.
Laboratory analysis showed severe thrombocytopenia 10, 000/uL, Hgb 6.7 g/dL, and haptoglobin levels of 10 mg/dL, below the minimum normal range parameter of 30 mg/dL, INR 2.5 due to anticoagulation with warfarin (Table 1). The peripheral blood smear showed anisopoikilocytosis, frequent schistocytes, moderate polychromasia, and severe thrombocytopenia and was compatible with microangiopathic hemolytic anemia (MAHA).
TABLE 1.
Pertinent laboratory results at admission.
| Exam | Result | Reference range |
|---|---|---|
| Hemoglobin | 6.7 | 11.2–15.7 gm/dL |
| Hct (%) | 20.7 | 34.1%–44.9% |
| MCV | 87.7 | 79.4–94.8 fL |
| Platelet count | 10 | 182–369 10*3/uL |
| Reticulocyte | 8.4 | 0.50%–1.70% |
| Absolute reticulocyte count | 0.1648 | 0.0164–0.0776 10*6/uL |
| Immature reticulocytes. | 35.7 | 3.0%–15.9% |
| Haptoglobin | 10 | 33–346 mg/dL |
| PT (s) | 23.7 | 9.1–11.7 s |
| INR (ratio) | 2.5 | 0.9–1.1 (ratio) |
| APTT | 39.6 | 23.2–31.6 s |
| Creatinine | 1.1 | 0.6–1.2 mg/dL |
| GFR | >60 | 60–999 mL/min/1.73 m2 |
| Fibrinogen | 407 | 200.0–450.0 mg/dL |
| ALT/SGPT | 24 | 4–36 IU/L |
| AST/SGOT | 39 | 8–33 IU/L |
| Bilirubin | 1.1 | 0.1–1.2 mg/dL |
| ADAMTS 13 antibody | 22 | <12 Units/mL |
| ADAMTS 13 activity | <2.0 | > 66.8% |
| Anticardiolipin Ab, IgG | 9 | 0–14 GLP U/mL |
| Anticardiolipin Ab, IgM | 22 | 0–12 MPL U/mL |
| B2 glycoprotein I Ab, IgG | <9 | 0–20 GPI IgG units |
| B2 glycoprotein I Ab, IgA | 14 | 0–25 GPI IgG units |
| B2 glycoprotein I Ab, IgM | <9 | 0–32 GPI IgG units |
| Antinuclear antibodies Screen | Negative | Negative |
| Complement C3, serum | 106 | 82–167 mg/dL |
| Complement C4, serum | 18 | 12–38 mg/dL |
The patient first received one unit of packed red blood cells with a minimal increase in hemoglobin level to 6.9 g/dL, then a second unit to achieve a hemoglobin level above 7.5 g/dL.
CT scan of the brain was obtained. It showed residual findings from the previous right craniotomy, extensive ischemic encephalomalacia in the right parietal and temporal lobes, and bilateral caudate lacunar infarctions without acute intracranial pathology.
CTA of the abdomen revealed normal vascular anatomy, a spleen with two rounded low‐density lesions of 2.2 cm on the subcapsular upper pole and 1.2 cm in the medial portion of the spleen, calcified uterus fibroid, and a 2.8 cm left ovarian cyst.
The patient was admitted to the intensive care unit under the impression of microangiopathic hemolytic anemia. At that point, it was impossible to differentiate between catastrophic antiphospholipid syndrome versus thrombotic thrombocytopenic purpura (TTP); however, as both entities require rapid and aggressive management, the patient was started on daily plasma exchange with one plasma volume, prednisone 1 mg/kg and anticoagulation with enoxaparin. Platelet count normalized after two sessions of plasma exchange therapy and decreased to 109,000/ul on the third day. The patient received a total of 5 days of plasma exchange therapy. Hemoglobin levels remained stable above 8 g/dL and trended up through the therapy.
The liver function enzymes normalized on day #2 of admission, and bilirubin levels remained within normal limits throughout, with a resolution of laboratory evidence of hemolysis. Upon discharge, the patient continued with anticoagulation, resumed warfarin, and tapering doses of prednisone. Currently, the patient is following in the outpatient hematology clinic.
3. DISCUSSION
Moya moya disease (MMD) is a condition first described in 1957, which predisposes affected individuals to suffer from stroke due to progressive stenosis, most commonly of branches of the carotid arteries. 4 This disease has a higher prevalence in females than in males, and the highest prevalence was found in Japan, where moya‐moya translates to “hazy puff of smoke”. 8 This translation refers to the characteristic appearance of the vessels seen in angiography. The stenosis of larger vascular branches caused by this disease triggers neovascularization, and these thin new vessels cause a cloudy appearance.
Incidence rates are highest in eastern Asia; Japan reported 0.35 cases per 100,000 individuals.
On the other hand incidence rate in the United States is 0.09 per 10,000 individuals, and the highest incidence is among Asian Americans (0.28/100,000), followed by African Americans (0.13/100,000), Caucasian Americans (0.06/100,000), and Hispanics (0.03/100,000). Furthermore, the incidence reported between Asian and Asian American descendants was similar. 9 , 10 meaning that genetic factors have to be implicated in disease development. 11
The pathophysiology of MMD is still unknown. Since discovering this disease, multiple studies have shown that the RNF213 gene is implicated. Further studies have demonstrated that different genetic variants have more potential than others to trigger the disease. 12 , 13
The RNF213 was the first identifiable gene linked to an increased risk of MMD, but the exact mechanism is still poorly understood. 14 , 15 Several studies exist. The first one published in 2012 was a case–control looking at RNF213 variant c.14576G > A in patients with non‐MMD intracranial major artery stenosis/occlusion (ICASO); authors found an association between c.14576G > A and non‐MMD ICASO.
In 2013 the same group investigated the occurrence rate of the c.14576G > A variant in 323 ICASO patients and noted that some patients with distinct ICASO phenotypes might have a common genetic variant, RNF213 c.14576G > A, suggesting that the RNF213 c.14576G > A variant may be a high‐risk allele for ICASO. Two years later, another study was done on 78 MMD patients; nine had non‐atherosclerotic MMS showing the absence of the RNF213 c.14576G, a genetic variant in nonatherosclerotic MMS patients. In the absence of this particular genetic variant, patients with MMD did not present atherosclerosis, but RNF213 was present. 15 , 16 , 17
Furthermore, even in patients with RNF213 mutations, its presence does mean predict the development of this disease, which is why some investigators believe that this genetic variant might be a trigger and a second “hit” is necessary for the disease to develop. 18 How exactly MMD develops is still a mystery. The association between this disease and prothrombotic states is poorly understood and documented.
Autoimmunity has been documented in MMD and MMS; however, the specifics associations with immune disorders such as SLE, APLS, and further have not been documented. (Sigdel et al., 2013) (Miller et al., 2021).
An extensive search was performed for associations between immune‐mediated diseases (SLE, APL, acquired and congenital TTP, etc.) and MMD, and we found only one case of acquired TTP in the setting of MMD. 19 However, in the case described by H Hiyama, et al. in 1994, the VWF‐cleaving protease was discovered in 1997 (Zheng, 2015); after this report, this case was reported today.
Congenital TTP associated with MMD has been more documented, but the association between ALP, TTP, and MMD has never been described.
Moya Moya terms refer to the characteristic pathological findings of stenosis and collateralization; MMD has often been associated with multiple other medical conditions, and in this case, it is described as a moya‐moya syndrome (MMS). 18
If asymptomatic, the usual treatment for MMD or MMS is aspirin. However, patients with proven decreased perfusion or once symptomatic should undergo a revascularization procedure. As seen in the case presented before, this patient underwent extracranial–intracranial bypass surgery after presenting with multiple strokes in the past.
MMD/MMS is associated with thrombophilic predispositions. H. Tsuda et al. conducted a study with 20 patients with either Moyamoya disease or quasi‐Moyamoya disease. Thrombophilia was assessed by measuring the activity and antigen levels of antithrombin III, protein C, protein S, fibrinogen, and plasminogen as well as detecting lupus anticoagulants. One‐third (four definite cases and three quasi‐cases) of the examined patients had either a congenital or acquired thrombophilic tendency. 19
The link between MMD/MMS and APS needs to be clarified. The formation of aPL antibodies could be associated with damage to the vascular endothelial or sub‐endothelial structures, secondary to the abnormal vasculature and/or altered flow. Furthermore, APS may play a role in promoting further thrombosis and recurrent ischemic events. 3 , 20
Most cases of APS and MMD/MMS cases coexist with other medical conditions; a substantial prevalence of up to 65% of other conditions like diabetes mellitus, hypothyroidism, Down syndrome, etc, was noted. 3 , 21
Hematologic disorders have been described in 16% of patients with MMD. We found 16 papers describing an association between moyamoya and hematologic disorders (spherocytosis, beta‐thalassemia, Fanconi anemia, etc.). 10
Congenital TTP associated with MMD has been described, but the pathophysiology mechanism is unclear. 22 , 23 Some of the theories suggest that increases in the proinflammatory and angiogenic cytokines further stimulate the synthesis of growth factor‐β and basic fibroblast growth factor, leading to angiogenesis and neovascularization. It is further hypothesized that microangiopathic changes are followed by macroangiopathic changes leading to the ischemic manifestations of Moya‐Moya syndrome. 22
Antiphospholipid syndrome (APS) is an autoimmune disorder characterized by the presence of several autoantibodies targeting phospholipids or phospholipid‐binding proteins. This syndrome is characterized by a procoagulant state, often leading to pregnancy loss and venous or arterial thrombosis. These antibodies are anticardiolipin, lupus anticoagulant, and beta2‐glycoprotein antibodies. 24 Diagnosis is usually made by detecting the presence of one or several of these antibodies and one or more episodes of arterial or venous thrombosis. In our patient, around the time she was diagnosed with MMD, she presented with elevated values of multiple anticardiolipin antibodies and a history of stillbirth, second‐trimester pregnancy loss, and multiple CVA; she was diagnosed with APS.
On this occasion, the presentation of a patient with MMS to the ED with abdominal pain was associated with nausea, autoimmune hemolytic anemia (positive direct Coombs test), decreased haptoglobin, and severe thrombocytopenia. The positivity of Coombs suggests autoimmune hemolysis; however, in this patient, this may be related to autoimmune para‐phenomena seen with APS. Furthermore, the presence of schistocytes made us believe this hemolysis was most likely microangiopathic, raising the urge to rule out any possibility of thrombotic microangiopathy (TMA).
Thrombotic thrombocytopenic purpura (TTP) is a disorder that can either be acquired or hereditary. It consists of a protein deficiency involved in cleaving Von Willebrand factor known as ADAMTS13. This causes an increase in the size of VWF, which leads to platelet aggregation and thrombus formation. It usually presents with nonspecific symptoms such as abdominal pain, nausea, vomiting, and weakness. A classic pentad of thrombocytopenia, MAHA, renal involvement, severe neurologic manifestations, and fever was described in TTP.
Nevertheless, TTP frequently presents without the full pentad. Fever and renal impairment are generally uncommon in the presentation, and in approximately 35% of patients with TTP, neurological signs are not seen. 2 Thrombocytopenia and MAHA are enough diagnostic criteria to raise concern for TTP, 2 although this presentation can be associated with any TMA.
TMA‐related disorders are characterized by the presence of microvascular thrombosis, which can be local or diffuse. Catastrophic antiphospholipid syndrome (CAPS) is a life‐threatening condition in patients with APS due to the rapid development of multiple thromboses. It often involves several vital organs, such as the kidneys, lungs, or heart. Most commonly, CAPS affects small vessels, resembling TMA, specifically very similar to the TTP presentation. This presentation, often challenging, has several life‐threatening differential diagnoses. Already mentioned above are TTP and CAPS, which were the main differentials in the case presented above. However, the differential also includes other entities like hemolytic uremic syndrome, disseminated intravascular coagulation, syndromes related to hypertension, pregnancy or drug‐induced, and even heparin‐induced thrombocytopenia. 25 , 26
The above case's main challenge was establishing a diagnosis and promptly identifying a potentially life‐threatening condition. In our patient with an established diagnosis of APS, we had to consider CAPS. Diagnosis of CAPS requires four criteria: evidence of involvement of three or more organs, the onset of manifestations are simultaneous or within a week of each other, histopathologic confirmation of small vessel occlusion of at least one organ, and laboratory confirmation of antiphospholipid antibodies. 26 Another severe condition to contemplate in this case was TTP. The PLASMIC score (Table 2) was developed to estimate the likelihood of severe ADAMTS13 deficiency, 27 and our patient had a PLASMIC score of 6, indicating a high likelihood of severe ADAMTS13 deficiency. The main concern initially was to differentiate between CAPS and TTP.
TABLE 2.
PLASMIC score.
| Points | |
|---|---|
| Platelet count <30,000/microL | 1 |
| Indicator of hemolysis: Reticulocyte count of >2.5%, or undetectable haptoglobin, or indirect bilirubin >2.0 mg/dL | 1 |
| No active cancer | 1 |
| No history of solid organ or hematopoietic stem cell transplant | 1 |
| MCV <90 fL | 1 |
| INR <1.5 | 1 |
| Creatinine <2.0 mg/dL | 1 |
Note: A score of 0–4 points denotes low risk; a score of 5 denotes intermediate risk; a score of 6–7 denotes high risk.
Clinically, differentiating between TTP and CAPS is impossible; therefore, we initiated treatment with plasma exchange and steroids which are effective in both conditions. In the case of CAPS, anticoagulation (AC) and high‐dose glucocorticoids (GC) are also known to be effective. The use of AC alone has demonstrated a significant positive effect on prognosis. In life‐threatening situations, the use of plasma exchange (PE) and or intravenous immunoglobulin (IVIG) should also be considered. Overall, evidence has shown superior survival rates when combining AC and GC with PE and/or IVIG. In patients with SLE and CAPS, the addition of cyclophosphamide is recommended based on evidence supporting a reduction in mortality. Several studies used Rituximab and eculizumab, but insufficient evidence supports either treatment. 25 In the case of TTP, the international society of Thrombosis and Hemostasis (ISTH) guidelines for treatment recommend using GC with PE when treating a first acute event. 28 This recommendation is made even in the context of relapse and despite the lack of evidence due to potential reductions in mortality with this treatment. Rituximab is a conditional recommendation due to the meager evidence of benefit in treatment. Nonetheless, in patients with known autoimmune disorders, more practitioners tend to consider the addition of Rituximab to PE and steroids. Caplacizumab is a monoclonal antibody against VWF, and studies support its use in cases of TTP refractory to plasma exchange therapy and glucocorticoids. Still, there are limited data regarding treating acute events of TTP. Due to a lack of availability and evidence, the guidelines do not provide specific recommendations. 29 In this case, therapy was initiated with PE and GC. Regarding anticoagulation, the patient was being treated with warfarin as an outpatient, but her compliance was unclear. While hospitalized, the patient began AC treatment with enoxaparin and was later transitioned to warfarin.
Our patient responded clinically and with laboratory improvement to plasma exchange, glucocorticoids, and anticoagulation treatment regimen. This improvement, along with evidence of decreased ADAMTS13 and elevated ADAMTS13 antibody levels, was consistent with the diagnosis of TTP.
4. LEARNING POINTS
Describe the association between Moya‐Moya spectrum disease and acquired thrombotic thrombocytopenic purpura.
Discuss the approach to challenging diagnosis and further management of a patient with an autoimmune disease and thrombotic thrombocytopenic purpura.
Create awareness in the medical community about the multiple diseases associated with Moya‐Moya spectrum diseases, such as antiphospholipid syndrome and TTP.
5. CONCLUSION
MMD can often be associated with multiple other conditions, including hematologic conditions. This patient with MMS and APS posed a diagnostic challenge due to the presence of overlapping signs and symptoms of TTP and APS, both of which can be associated with MMD. However, the main approach in patients presenting with severe thrombocytopenia and MAHA is to treat a potentially life‐threatening condition, such as TTP. Treatment should start on clinical grounds without further delays.
DECLARATIONS
A preprint has previously been published. Nehemias Guevara‐Rodriguez, Gabriela Marmanillo‐Mendoza, Jorge Castelar et al. A challenging diagnosis and management of patient acquired thrombotic thrombocytopenic purpura (TTP) versus catastrophic antiphospholipid syndrome in a patient with Moya‐Moya disease, case report, and literature review, 23 September 2022, PREPRINT (Version 1) available at Research Square [https://doi.org/10.21203/rs.3.rs‐2062954/v1].
AUTHOR CONTRIBUTIONS
Nehemias Guevara‐Rodriguez: Conceptualization; investigation; supervision; writing – original draft; writing – review and editing. Gabriela Marmanillo‐Mendoza: Writing – original draft. Jorge Castelar: Writing – original draft. Camelia Ciobanu: Writing – original draft. Ilmana Fulger: Conceptualization; supervision; writing – review and editing.
FUNDING INFORMATION
This case and literature review have not to been funded by any entity.
CONFLICT OF INTEREST STATEMENT
Not applicable.
CONSENT
Written informed consent was obtained from the patient to publish this case report and is available upon request.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
Not applicable.
Guevara‐Rodriguez N, Marmanillo‐Mendoza G, Castelar J, Ciobanu C, Fulger I. Unusual presentation of acquired thrombotic thrombocytopenic purpura (TTP) versus catastrophic antiphospholipid syndrome in a patient with Moya‐Moya disease, case report, and literature review. Clin Case Rep. 2023;11:e7317. doi: 10.1002/ccr3.7317
DATA AVAILABILITY STATEMENT
Data are available upon request, following HIPAA regulations.
REFERENCES
- 1. Jr D, Teixeira BC, Koppe GL, et al. Moyamoya Disease and Syndrome a Review. Radiol Bras. 2022;55(1):31‐37. Available from: https://www.scielo.br/j/rb/a/RnSpk536gD9rhd3QX7HykHj/?lang=en&format=pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scully M, Hunt BJ, Benjamin S, et al. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol. 2012;158(3):323‐335. [DOI] [PubMed] [Google Scholar]
- 3. Wang Z, Fu Z, Wang J, Cui H, Zhang Z, Zhang B. Moyamoya syndrome with antiphospholipid antibodies: a case report and literature review. Lupus. 2014;23(11):1204‐1206. [DOI] [PubMed] [Google Scholar]
- 4. Scott RM, Smith ER. Moyamoya disease and Moyamoya syndrome. N Engl J Med. 2009;360(12):1226‐1237. [DOI] [PubMed] [Google Scholar]
- 5. Shuja‐Ud‐Din MA, Ahamed SA, Baidas G, Naeem M. Moyamoya syndrome with primary antiphospholipid syndrome. Med Princ Pract Int J Kuwait Univ Health Sci Cent. 2006;15(3):238‐241. [DOI] [PubMed] [Google Scholar]
- 6. Sukumar S, Lämmle B, Cataland SR. Thrombotic thrombocytopenic purpura: pathophysiology, diagnosis, and management. J Clin Med. 2021;10(3):536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hiyama H, Kusano R, Muragaki Y, Miura N. Moyamoya disease associated with thrombotic thrombocytopenic purpura (TTP). No Shinkei Geka. 1994;22(6):567‐572. [PubMed] [Google Scholar]
- 8. Burke GM, Burke AM, Sherma AK, Hurley MC, Batjer HH, Bendok BR. Moyamoya disease: a summary. Neurosurg Focus. 2009;26(4):E11. [DOI] [PubMed] [Google Scholar]
- 9. Uchino K, Johnston SC, Becker KJ, Tirschwell DL. Moyamoya disease in Washington state and California. Neurology. 2005;65(6):956‐958. [DOI] [PubMed] [Google Scholar]
- 10. Miller R, Unda SR, Holland R, Altschul DJ. Western Moyamoya phenotype: a scoping review. Cureus. 2021;13(11):e19812. Available from: https://www.cureus.com/articles/67075‐western‐moyamoya‐phenotype‐a‐scoping‐review [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamauchi T, Tada M, Houkin K, et al. Linkage of familial moyamoya disease (spontaneous occlusion of the circle of Willis) to chromosome 17q25. Stroke. 2000;31(4):930‐935. [DOI] [PubMed] [Google Scholar]
- 12. Kamada F, Aoki Y, Narisawa A, et al. A genome‐wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56(1):34‐40. [DOI] [PubMed] [Google Scholar]
- 13. Bang OY, Fujimura M, Kim SK. The pathophysiology of Moyamoya disease: an update. J Stroke. 2016;18(1):12‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One. 2011;6(7):e22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miyawaki S, Imai H, Shimizu M, et al. Genetic analysis of RNF213 c.14576G>a variant in nonatherosclerotic quasi‐Moyamoya disease. J Stroke Cerebrovasc Dis. 2015;24(5):1075‐1079. [DOI] [PubMed] [Google Scholar]
- 16. Miyawaki S, Imai H, Shimizu M, et al. Genetic variant RNF213 c.14576G>a in various phenotypes of intracranial major artery stenosis/occlusion. Stroke. 2013;44(10):2894‐2897. [DOI] [PubMed] [Google Scholar]
- 17. Miyawaki S, Imai H, Takayanagi S, Mukasa A, Nakatomi H, Saito N. Identification of a genetic variant common to moyamoya disease and intracranial major artery stenosis/occlusion. Stroke. 2012;43(12):3371‐3374. [DOI] [PubMed] [Google Scholar]
- 18. Fox BM, Dorschel KB, Lawton MT, Wanebo JE. Pathophysiology of vascular stenosis and remodeling in Moyamoya disease. Front Neurol. 2021;12. doi: 10.3389/fneur.2021.661578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsuda H, Hattori S, Tanabe S, et al. Thrombophilia found in patients with Moyamoya disease. Clin Neurol Neurosurg. 1997;99:S229‐S233. [DOI] [PubMed] [Google Scholar]
- 20. de Veber G. Vascular occlusion in Moyamoya: a multitude of mechanisms? Stroke. 2001;32:1791‐1792. [Google Scholar]
- 21. Chen JB, Liu Y, Zhou LX, Sun H, He M, You C. Increased prevalence of autoimmune disease in patients with unilateral compared with bilateral moyamoya disease. J Neurosurg. 2016;124(5):1215‐1220. [DOI] [PubMed] [Google Scholar]
- 22. Jain P, Yoganathan S, Sharma S, et al. Congenital thrombotic thrombocytopenic purpura associated with Moyamoya syndrome in a 3‐year‐old girl: a case report. J Child Neurol. 2012;27(10):1331‐1335. [DOI] [PubMed] [Google Scholar]
- 23. Park HW, Oh D, Kim N, et al. Congenital thrombotic thrombocytopenic purpura associated with unilateral moyamoya disease. Pediatr Nephrol. 2008;23(9):1555‐1558. [DOI] [PubMed] [Google Scholar]
- 24. Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002;346(10):752‐763. [DOI] [PubMed] [Google Scholar]
- 25. Cervera R, Rodríguez‐Pintó I, Espinosa G. The diagnosis and clinical management of the catastrophic antiphospholipid syndrome: a comprehensive review. J Autoimmun. 2018;92:1‐11. [DOI] [PubMed] [Google Scholar]
- 26. Cervera R. Antiphospholipid syndrome. Thromb Res. 2017;151:S43‐S47. [DOI] [PubMed] [Google Scholar]
- 27. Bendapudi PK, Hurwitz S, Fry A, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4(4):e157‐e164. [DOI] [PubMed] [Google Scholar]
- 28. Zheng XL. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med. 2015;66:211‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng XL, Vesely SK, Cataland SR, et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2496‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available upon request, following HIPAA regulations.