

Abstract
Chronic pain involves sensitization of nociceptors and synaptic transmission of painful signals in nociceptive circuits in the dorsal horn of the spinal cord. We investigated the contribution of clathrin-dependent endocytosis to sensitization of nociceptors by G protein-coupled receptors (GPCRs) and to synaptic transmission in spinal nociceptive circuits. We determined whether therapeutic targeting of endocytosis could ameliorate pain. mRNA encoding dynamin (Dnm) 1–3 and adaptor-associated protein kinase 1 (AAK1), which mediate clathrin-dependent endocytosis, were localized to primary sensory neurons of dorsal root ganglia of mouse and human and to spinal neurons in the dorsal horn of the mouse spinal cord by RNAScope®. When injected intrathecally to mice, Dnm and AAK1 siRNA or shRNA knocked-down Dnm and AAK1 mRNA in dorsal root ganglia neurons, reversed mechanical and thermal allodynia and hyperalgesia, and normalized non-evoked behavior in preclinical models of inflammatory and neuropathic pain. Intrathecally administered inhibiters of clathrin, Dnm and AAK1 also reversed allodynia and hyperalgesia. Disruption of clathrin, Dnm and AAK1 did not affect normal motor functions of behaviors. Patch clamp recordings of dorsal horn neurons revealed that Dnm1 and AAK1 disruption inhibited synaptic transmission between primary sensory neurons and neurons in lamina I/II of the spinal cord dorsal horn by suppressing release of synaptic vesicles from presynaptic primary afferent neurons. Patch clamp recordings from dorsal root ganglion nociceptors indicated that Dnm siRNA prevented sustained GPCR-mediated sensitization of nociceptors. By disrupting synaptic transmission in the spinal cord and blunting sensitization of nociceptors, endocytosis inhibitors offer a therapeutic approach for pain treatment.
INTRODUCTION
Chronic pain is common, poorly understood and difficult to treat. The analgesic properties of µ-opioid agonists, a common treatment, dwindle with time and their usefulness is limited by life-threatening side effects [36]. The redundancy of pain signaling, where multiple receptors and channels activate the same neurons [2], may limit the effectiveness of selective ligands for the treatment of multi-modal forms of pain.
We investigated the contributions of clathrin-dependent endocytosis to sensitization of nociceptors and synaptic transmission in nociceptive circuits in the dorsal horn of the spinal cord. We hypothesized that disruption of endocytosis would reverse multiple modalities of pain. Mediators from damaged tissues activate G protein-coupled receptors (GPCRs), receptor tyrosine kinases and ligand-gated ion channels at the peripheral endings of nociceptors to initiate pain [2]. The central terminals of nociceptors in the dorsal horn of the spinal cord release substance P, calcitonin gene-related peptide (CGRP) and glutamate, which activate GPCRs on spinal neurons that transmit signals centrally. Synaptic vesicle (SV) cycling is required for synaptic transmission [6; 37]. SV exocytosis at presynaptic terminals releases neurotransmitters into the synapse. Clathrin-dependent endocytosis retrieves SVs from the plasma membrane of presynaptic neurons, which replenishes the releasable SV pool and is necessary for sustained neurotransmission. Dynamin (Dnm) GTPase mediates fission of clathrin-coated SVs and is required for endocytosis. Of the three Dnm isoforms (Dnm1, Dnm2, Dnm3), Dnm1 and Dnm3 are expressed in the nervous system [15]. Dnm1 deletion in mice impairs activity-dependent endocytosis of SVs at nerve terminals and disrupts neurotransmission during intense stimulation [29]. Although Dnm3 deletion alone does not severely disrupt synaptic transmission, deletion of Dnm3 and Dnm1 depletes SVs and causes accumulation of clathrin-coated pits (CCPs) in presynaptic terminals [29]. Dnm inhibitors recapitulate these effects [24]. Together with clathrin, adaptor protein 2 (AP2) constitutes the major coated protein of the endocytic vesicles. Adaptor associated kinase 1 (AAK1) recruits clathrin and AP2 to the plasma membrane and phosphorylates the µ2 subunit of AP2 (AP2M1), thereby stimulating cargo binding and recruitment, vesicle assembly and internalization [9]. Clathrin assembly promotes AAK1-dependent phosphorylation of AP2M1, which constitutes a feedforward loop for pit maturation [32]. Clathrin and Dnm also mediate endocytosis and sustained endosomal signaling of GPCRs, which underlies neuronal sensitization and nociception [18–20; 30; 40].
The contribution of clathrin, Dnm and AAK1 to nociception is not fully understood. A screen of knockout mice identified AAK1 as a mediator and target for neuropathic pain [21]. Aak1 deletion and inhibition attenuated neuropathic pain in mice and rats. The α subunit of the AP2 complex (AP2α2) is expressed in CGRP+ve nociceptors and AP2 shRNA suppressed nociception in mice [28]. However, the distribution of Dnm and AAK1 isoforms in nociceptive circuits is unknown and their role in nociception is unexplored. We used genetic and pharmacological approaches to investigate the contribution of endocytosis to nociception. Anatomical, electrophysiological and behavioral studies support the hypothesis that Dnm and AAK1 are necessary for sensitization of nociceptors and for nociceptive transmission in the spinal cord, and that disruption of endocytosis reverses nociception.
METHODS
Animals.
All experiments and procedures were in accordance with the guidelines recommended by the National Institute of Health, the International Association for the study of Pain, the National Centre for the Replacement, Refinement, and Reduction of Animals in Research ARRIVE guidelines, and were approved by the New York University Institutional Animal Care and Use Committee and the Monash University Animal Ethics Committee. Male and female C57BL/6 mice (8–10 weeks, Charles River) were housed four per cage at 22 ± 0.5°C under a controlled 14/10 h light/dark cycle with free access to food and water. Mice were randomly assigned to experimental groups, and the group size was based on our previous similar studies. Investigators were blind to treatments.
Collection of human tissues.
The collection of dorsal root ganglia (DRG) from deidentified organ donors was reviewed by the Institutional Review Board of the University of Cincinnati (#00003152, Study ID 2015–5302) and was deemed to be exempt. Donor information is provided in Table S1. DRG (L4, L5) were collected in the operating room within 90 min of aortic cross clamp and after removal of vital organs. DRG were placed in N-Methyl-D-glucamine-artificial cerebrospinal fluid (ACSF) solution at 4°C and were dissected as described [39]. For RNAScope, DRG were immersion fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) overnight at 4°C, cryoprotected in 30% sucrose for 24 h at 4°C, and embedded in Optimal Cutting Temperature (OCT) compound (Tissue Tek). Frozen sections (14 µm sections) were mounted onto Superfrost Plus slides (Fisher), dried (15 min) and stored at −20°C.
Collection of mouse tissues.
Mice were anesthetized (5% isoflurane) and perfused through the ascending aorta with PBS and then 4% paraformaldehyde in PBS. DRG, trigeminal ganglia (TG) and spinal cord were removed, post-fixed in 4% paraformaldehyde in PBS at 4°C for 1 h or overnight, respectively, placed in 30% sucrose solution for 24 h at 4°C, and embedded in OCT. Frozen sections (10–12 µm) were mounted onto Superfrost Plus slides (Fisher), dried (15 min) and stored at −20°C.
RNAScope® in situ hybridization and immunofluorescence.
The RNAScope® system (Advanced Cell Diagnostics) was used per manufacturer’s directions for fresh-frozen tissue except for omission of the initial on-slide fixation step. Probe hybridization and detection using the Multiplex Fluorescent Kit v2 followed the manufacturer’s directions. Probes to Mm-Dnm1 (#446931-C3), Mm-Dnm2 (#451831-C1), Mm-Dnm3 (#451841-C2), Mm-Aak1 (#1097711-C1) (mouse), Hs-Dnm1 (#1099091-C1), Hs-Dnm2 (#821511-C1), Hs-Dnm3 (#1105961-C2) and Hs-AAK1 (#531971-C1) (human) were used. Sections were incubated with Opal 620 reagent (1:1000, cat#FP1495001KT, Akoya Biosciences) for detection. To detect neurons, hybridized slides were incubated with NeuroTrace™ 500/525 Green Fluorescent Nissl Stain (1:500, cat#N21480, Invitrogen) (10 min, room temperature, RT). To detect satellite glial cells and peptidergic neurons, hybridized slides were incubated with rabbit anti-glutamine synthetase antibody (GS, 1:1000, cat#ab49873, Abcam, Cambridge, MA) or rabbit anti-CGRP, 1:1000, cat#C8198, Sigma), respectively (overnight, 4°C). Slides were washed and incubated with goat anti-rabbit Alexa Fluor® 488 (1:1000; cat#A21206, Invitrogen) (1 h, RT). Slides were washed and incubated with DAPI (1 µg/ml, 5 min) and mounted in ProLong® Gold Antifade (Thermo Fisher). Sections were observed using a Leica SP8 confocal microscope with HCX PL APO 40x (NA 1.30) oil objective.
RNAScope® quantification.
Dnm 1, 2 or 3 and Aak1 were localized by RNAScope®. Confocal images were analyzed using Fiji ImageJ (NIH) according to ACD Bio-Techne Technical Note. Regions of interest were defined by applying a threshold with the moments setting (Min & Max) and analyzing particles with size range from 0 to infinity. Regions of interest were overlaid on the original micrograph and the number of dots per area were quantified. Results are expressed as dots/mm2 tissue. A total of 3 images (20X magnification) were analyzed for each mouse (N=4 mice for control and treatment groups; 12 images analyzed per experimental group).
qRT-PCR.
RNA was isolated from snap frozen mouse tissues using Direct-zol RNA MiniPrep kit (cat#R2051 Zymo Research). cDNA was prepared using MultiScribe Reverse Transcriptase (cat#4311235 Thermo Fisher). cDNA (50 ng) was amplified for 40 cycles by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) using Dnm1 (DNM1, Mn00802468_m1), Dnm2 (DNM2, Mn00514582_m1), Dnm3 (DNM3, Mn00554098_m1), AAK1 (Mn01183675_m1) and GAPDH (Mn99999915_g1) primers, QuantStudio 3 Real-Time PCR System, and TaqMan Fast Advanced PCR Mastermix (cat#4444556 Thermo Fisher). All samples were analyzed at least in duplicate and normalized by GAPDH expression. The relative expression ratio per condition was calculated as described [27].
Intrathecal administration of siRNA to mice.
Cationic liposome and adjuvant anionic polymer (polyglutamate) were used to deliver siRNA [35]. The following siRNAs were used: ON-TARGETplus siRNA mouse Dnm1 siRNA (cat#L-043277–01-0005), mouse Dnm2 (cat#L-044919–02-0005), mouse Dnm3 (cat#L-059061–01-0005), mouse AAK1 (cat#L-065639–00-0005) or non-targeting control (CTR) siRNA (cat#D-001810–10-05) (Dharmacon) (Table S2). siRNAs (50 ng, 0.5 μl of 100 ng/μl stock) were mixed with 0.5 μl of adjuvant polyglutamate (0.1 μg/μl stock) and 1.5 μl sterile 0.15 M NaCl. Liposome solution, cationic lipid 2–3-[bis-(3-amino-propyl)-amino]-propylamino-N-ditetradecylcarbamoylmethyl-acetamide (DMAPAP) and L-α-dioleoyl phosphatidylethanolamine (DOPE) (DMAPAP/DOPE, 1/1 M:M) (2.5 μl of 200 μM) was added to siRNA/adjuvant, vortexed for 1 min, and incubated (30 min, RT). The siRNA lipoplexes were administered to conscious mice by intrathecal (i.t.) injection (L4-L5, 5 μl), 24 h after CFA intraplantar (i.pl.) injection or 10 d after SNI surgery. Dnm1, Dnm2, Dnm3 and Aak1 expression in DRG and spinal cord (L4-L5) were analyzed by RNAScope® in situ hybridization, 24 or 48 h after siRNA injection. For electrophysiology experiments, mouse Dnm1 siRNA, mouse AAK1 siRNA or CTR siRNA were mixed with PEI (cat#765090, Sigma) and incubated for 10 min at RT. The siRNA-PEI complex was diluted in saline and 5 µl was injected i.t. into mice 48 h before laminectomy for spinal slice preparation.
Intrathecal administration of shRNA to mice.
Mouse Dnm1 (cat# TL500548), AAK1 (cat# TL508098) shRNA plasmids and noneffective 29-mer scrambled shRNA cassette in pGFP-C-shLenti Vector were from OriGene (Table S2). The Dnm1 shRNA or AAK1 shRNA (1.25 µg) was mixed with polyethyleneimine-based transfection reagent (in vivo-jetPEI®, 201–50G; Polyplus) in an 8:1 N:P ratio (polyethyleneimine nitrogen to DNA phosphate ratio) [8]. The shRNAs in vivo-jetPEI® mixture were administered to conscious mice by i.t. injection (L4-L5, 5 μl), 24 h after CFA injection (i.pl.) or 10 d after SNI surgery. Dnm1 and Aak1 expression in DRG (L4-L5) were analyzed by RNAScope® in situ hybridization, 72 h after shRNA injection.
Intrathecal administration of endocytosis inhibitors to mice.
Dyngo4a (Dnm inhibitor, 50 nM), PitStop2 (clathrin inhibitor, 50 nM), inactive analogs (trademarks of Children’s Medical Research Institute, Newcastle Innovation and Freie Universitat Berlin), LP935509 (AAK1 inhibitor, cat#HY-117626, MedChemExpress; 1 – 10 µg/5µl), SGC-AAK1–1 (AAK1 inhibitor, cat#6528, Tocris; 1 – 10 µg/5µl) or vehicle (PBS, 5%DMSO/PBS) was injected i.t. (5 μl, L4/L5) into conscious mice. The inhibitors were injected 48 h after CFA injection (i.pl.) or 10 d after SNI surgery.
Inflammatory pain.
Complete Freund’s Adjuvant (CFA) (1 mg/ml) or vehicle (0.9% NaCl) was administered by i.pl. injection (10 µl) into the right hindpaw of sedated mice (2% isoflurane). siRNA, shRNA or inhibitors were injected i.t. 24 h or 48 h after CFA. Mechanical allodynia and thermal hyperalgesia were assessed.
Neuropathic pain.
Spared Nerve Injury (SNI) and sham surgeries were made as described [26]. Briefly, mice were anesthetized with isoflurane. A skin and muscle incision were made in the thigh to expose the sciatic nerve innervating the left hindpaw. The tibial and common peroneal nerves were ligated and transected distal to the ligature. The third branch, the sural nerve, was left intact. For sham controls, the nerves were exposed but not ligated or transected. siRNA, shRNA or inhibitors were injected i.t. at 10 d after surgery. Mechanical and cold allodynia were assessed.
Mechanical allodynia.
Mechanical allodynia was assessed by measuring hindpaw withdrawal response to von Frey filament stimulation using the up-and-down method [13]. Mice were acclimatized to the testing apparatus, which comprised individual clear Plexiglass boxes on an elevated wire mesh platform to facilitate access to the plantar surface of the hindpaws, for 1 h/d for 2 d. A series of von Frey filaments (0.02, 0.07, 0.16, 0.4, 1.0, and 2 g; Stoelting) were applied perpendicular to the plantar surface of hindpaw. The test began with an application of 0.4 g filament. A positive response was defined as a clear paw withdrawal or shaking. Whenever a positive response occurred, the next lower filament was applied, and whenever a negative response occurred, the next higher filament was applied. The testing consisted of 6 stimuli, and the pattern of response was converted to a 50% von Frey threshold [7].
Thermal hyperalgesia.
The Hargreaves apparatus was used to evaluate hypersensitivity to heat (Ugo Basile) [17]. Mice were acclimatized to the testing apparatus, which comprised individual clear Plexiglass chambers and a radiant heat source, for 1 h/d for 2 d. The infrared intensity was set at 50% and cut off time to a maximum of 30 s. The time between stimulus onset and paw withdrawal was measured automatically, giving an index of the thermal nociceptive threshold. Significant decreases in paw withdrawal latency were interpreted as evidence of thermal hyperalgesia. The latency, expressed in seconds, was evaluated before (basal) and at different time points after the treatment.
Cold allodynia.
Cold allodynia was assessed by measuring the acute nociceptive response to the acetone evoked evaporative cooling [38]. A droplet (50 μL) of acetone, formed on the flat-tip needle of a syringe, was gently touched to the plantar surface of the mouse hind paw. The time spent licking and lifting of the paw over a period of 60 s was evaluated before (basal) and at different time points after the treatment.
Non-evoked behavior.
Non-evoked behavior of mice was assessed using a behavioral spectrometer (Behavioral Instruments), a validated instrument for phenotyping rodents that avoids operator bias [3]. This approach has been used for studies of abhorrent behavior in mouse models of autism, restraint stress, and inflammatory, neuropathic and visceral pain [3; 5; 23]. The spectrometer comprised a 40 cm2 arena with a CCD camera mounted in the center of the ceiling and a door aperture in the front area of the arena. Movement was assessed by a floor mounted vibration sensor and 32 wall mounted infrared transmitter and receiver pairs. Mice were individually placed in the center of the behavioral spectrometer and their behavior was recorded for 20 min and analyzed using a combination of video tracking analysis (Viewer3, BiObserve) and vibration analysis. Total distance traveled in the open field (number of visits to a central area), average velocity of locomotion (cm/s), track length (cm), ambulation (% activity), time engaged in grooming (min), and wall distance (cm) were recorded and analyzed as described [3].
Spinal slice preparation.
Adult C57BL/6J mice were anesthetized (5% isoflurane), decapitated and the lumbar region of the spinal cord with the dorsal root exposed by laminectomy was removed. Parasagittal spinal cord slices with the dorsal root attached (300 μm) were sectioned on a vibratome (Leica VT 1200s) in ice cold (0–4°C) oxygenated sucrose‐based ACSF that contained (mM): 100 sucrose, 63 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 25 glucose, 25 NaHCO3 and 5 Na ascorbate. Slices were then incubated for 15 min at 34°C in NMDG‐based recovery ACSF composed of (mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na ascorbate, 2 thiourea, 3 Na pyruvate, 10 MgSO4 and 0.5 CaCl2 and adjusted to pH 7.4 with HCl. After the recovery incubation, slices were transferred to oxygenated ACSF with the following composition (mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 25 glucose and 25 NaHCO3 for 45 min at 36°C and then maintained at RT prior to transfer to the recording chamber. All ACSF solutions were equilibrated with 95% O2 and 5% CO2. Spinal cord slices were collected 48 h after administration of siRNA (i.t.). Some slices were preincubated with inhibitors of Dnm (Dyngo4a, 30 µM) or AAK1 (SGC-AAK1–1, 100 nM; LP-935509, 1 µM) for 10 min.
Spinal cord electrophysiology.
Slices were transferred to the recording chamber and continuously superfused with ACSF equilibrated with 95% O2/5% CO2 at a rate of 2ml/min at RT. Dodt-contrast optics were used to identify dorsal horn neurons in the translucent substantia gelatinosa layer of the superficial dorsal horn. Evoked and spontaneous excitatory post-synaptic currents (eEPSCs, sEPSCs respectively) were recorded in whole-cell voltage clamp using a CsCl-based internal solution composed of (mM): 140 CsCl, 10 EGTA, 5 HEPES, 2 CaCl2, 2 MgATP, 0.3 NaGTP, 5 QX-314.Cl and 0.1% biocytin (osmolarity 285–295 mosmol/l). Patch clamp electrodes had resistances between 3–5 MΩ and neurons were held at −65 mV (not corrected for the liquid junction potential of 4 mV). A bipolar stimulating electrode was placed in the dorsal root entry zone for electrical stimulation of evoked post-synaptic currents. For paired pulse experiments, evoked currents were elicited by two consecutive stimuli of identical strength separated by 40 ms. Paired pulse ratio (PPR) was calculated by dividing the second pulse by the first (PSC2/PSC1). All eEPSCs were recorded in gabazine (10 μM) and strychnine (0.5 μM). In experiments with endocytosis inhibitors, baseline recordings were made prior to superfusion of with inhibitors for 10 min, after which recordings were repeated in the presence of the inhibitor. In the inhibitor studies, the 1Hz protocol was reduced to 8 pulses to prevent plastic changes at the synapse occurring between the baseline recording and post-inhibitor recording.
SV imaging.
Lumbar spinal cord slices were incubated in Mg2+-free ACSF that contained (mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2.5 CaCl2, 10 glucose and 0.1 4-AP for 10 min to increase neuronal activity. Slices were transferred to Mg2+-free ACSF with 4-AP containing 8 μM FM1–43 (Abcam, Australia) for 3 min, washed with ACSF for 2 min, and then incubated in 1 mM ADVASEP-7 (Sigma) for 2 min. Slices were washed with ACSF for 2 min and the ADVASEP-7 incubation was repeated. Slices were placed in the recording chamber and washed for a further 15 min in ACSF prior to imaging. A bipolar stimulating electrode was placed in the dorsal root entry zone of the spinal cord and stimulated for 10 s at 1Hz, 4–6V to facilitate the release of vesicles from the presynaptic terminals. Optical recordings were completed on an upright fluorescence microscope (BX51W1, Olympus) under 40x magnification using a Cy3/TRITC filter set CCD camera (C11440 Orca Flash 4.0, Hamamatsu). Image sequences were analyzed using Fiji NIH image software.
Transmission electron microscopy.
Adult C57BL/6 mice were treated with Dnm1 or CTR siRNA (i.t.). After 48 h, mice were anaesthetized (5% isoflurane), decapitated and the lumbar region of the spinal cord exposed by laminectomy and removed. Spinal cord slices were prepared and a bipolar stimulating electrode was placed in the dorsal root entry zone for electrical stimulation of evoked post-synaptic currents. Spinal cord sections were fixed in 2% paraformaldehyde, 2.5% glutaraldehyde, 0.1M sucrose in 0.1M MBP (pH 7.4). Sections were post-fixed in 1% OsO4 and 1.5% K4Fe(CN)6 in ddH2O, dehydrated in graded series of ethanol and propylene oxide, and embedded in EMbed 812 (Electron Microscopy Sciences). Ultrathin sections (70 nm) were cut and stained with uranyl acetate and lead citrate. Stained grids were imaged with Talos120C transmission electron microscope (Thermo Fisher Scientific) and recorded using Gatan (4k x 4k) OneView Camera with software Digital Micrograph (Gatan Inc., Pleasanton, CA). Morphometry measurements were made using Image J by an investigator blinded to the experimental conditions. For analysis of SV numbers, 10331 vesicles in total from 212 Dnm1 siRNA and 182 CTR siRNA synapses from 2 separate preparations. To eliminate bias that could arise from choosing synapses based on their size or SV number, synapses were selected based on the presence of an active zone. All SVs within the adjacent SV cluster were then counted. In experiments designed to assess the effects of Dnm1 siRNA treatment on the abundance of CCPs, 13276 vesicles (CCP + SV) in total were counted; results were expressed as the percentage of CCPs relative to the total of SVs + CCPs/synapse (n=212 Dnm1 siRNA and 182 CTR siRNA synapses).
Trypsin-evoked nociception.
Dnm1+2+3 or CTR siRNA was administered by i.t. injection as described above. After 48 h, trypsin (10 µl, 80 nM) was injected (i.pl.) into the right hindpaw. Mechanical allodynia was assessed 1 h after trypsin injection [20].
Trypsin-evoked hyperexcitability of nociceptors.
Dnm1+2+3 or CTR siRNA was administered by i.t. injection as described above. After 48 h, DRG (L3-L5) were removed and dispersed by incubation in collagenase (4 mg/ml, Gibco) and dispase (4.7 mg/ml, Gibco) for 15 min at 37 °C and triturated with a 200 µL pipette tip. Neurons were plated onto coverslips coated with laminin (0.013 mg/ml) and poly-L-ornithine (0.1 mg/ml) in 12-well plates. Neurons were cultured in F12 medium (Sigma) containing 10% fetal bovine serum, penicillin and streptomycin and maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2 until retrieval (16 h) for patch clamp recordings. Small-diameter (<30 µm) neurons were selected for patch clamp recording. Changes in excitability were quantified by measuring rheobase. Whole-cell perforated patch-clamp recordings were made using Amphotericin B (240 μg/ml, Sigma Aldrich) in current clamp mode at RT. The recording chamber was perfused with external solution at 2 ml/min. Recordings were made using Multiclamp 700B amplifiers, digitized by Digidata 1440A, and processed using pClamp 10.7 software (Molecular Devices). Solutions had the following composition (mM): pipette - K-gluconate 110, KCl 30, HEPES 10, MgCl2 1, CaCl2 2; pH 7.25 with 1 M KOH; external - NaCl 140, KCl 5, HEPES 10, glucose 10, MgCl2 1, CaCl2 2; pH to 7.3–7.4 with 1 M NaOH. Neurons were preincubated with trypsin (100 nM) for 10 min and washed. Rheobase was measured at T=0 or T=30 min after washing.
Statistics.
Data are presented as mean ± standard error of the mean (SEM). Groups of n=6 to 10 mice were studied. Differences were assessed using Student’s two-tailed t test for two comparisons and 1- or 2-way ANOVA and Sídák, Tukey, Newman-Keuls or Dunnett’s post-hoc test for multiple comparisons. P<0.05 was considered significant at the 95% confidence level. Sample sizes and statistical tests are specified in figure legends.
RESULTS
Dnm1 and Dnm3 are expressed in neurons of sensory ganglia and the spinal cord dorsal horn
We used RNAScope® in situ hybridization to localize isoforms of Dnm1, Dnm2 and Dnm3 mRNAs in DRG, TG and spinal cord of mice. Neurons were identified by Nissl staining. Immunostaining for CGRP allowed identification of a subset of small diameter peptidergic nociceptors of sensory ganglia [2]. CGRP+ve nociceptors express AP2α2, a key endocytosis protein that contributes to nociception [28]. Satellite glial cells of sensory ganglia were identified by immunostaining for GS. Dnm1 and Dnm3 mRNAs were expressed in neurons of DRG and TG (Fig. 1A; Fig. S1A). Dnm2 mRNA was detected in DRG and TG neurons at lower levels (Fig. 1A; Fig. S1A). Although Dnm1, Dnm2 and Dnm3 mRNAs were detected in CGRP+ve nociceptors (Fig. S2), Dnm isoforms were expressed in most neurons regardless of CGRP expression and diameter. Dnm isoforms were also found in non-neuronal cells within sensory ganglia, including GS+ve satellite glial cells (Fig. 1A). Dnm1 and Dnm3 mRNAs were mainly detected in neurons of deeper laminae of the dorsal horn of the spinal cord, although some neurons with cell bodies in the superficial laminae (LI, LII, LIII) also expressed Dnm1 and Dnm3 mRNAs (Fig. 1B). Fewer neurons in the spinal cord expressed Dnm2 mRNA. Analysis of extracts of DRG, TG and spinal cord by qRT-PCR confirmed the higher levels of expression of Dnm1 and Dnm3 compared to Dnm2 mRNA (Fig. 1C, D; Fig. S1B). Dnm1 and Dnm3 mRNAs were also localized to neurons of human DRG (Fig. 1E), which adds translational relevance to these findings.
Fig 1. Localization of Dnm mRNA in DRG and spinal cord.
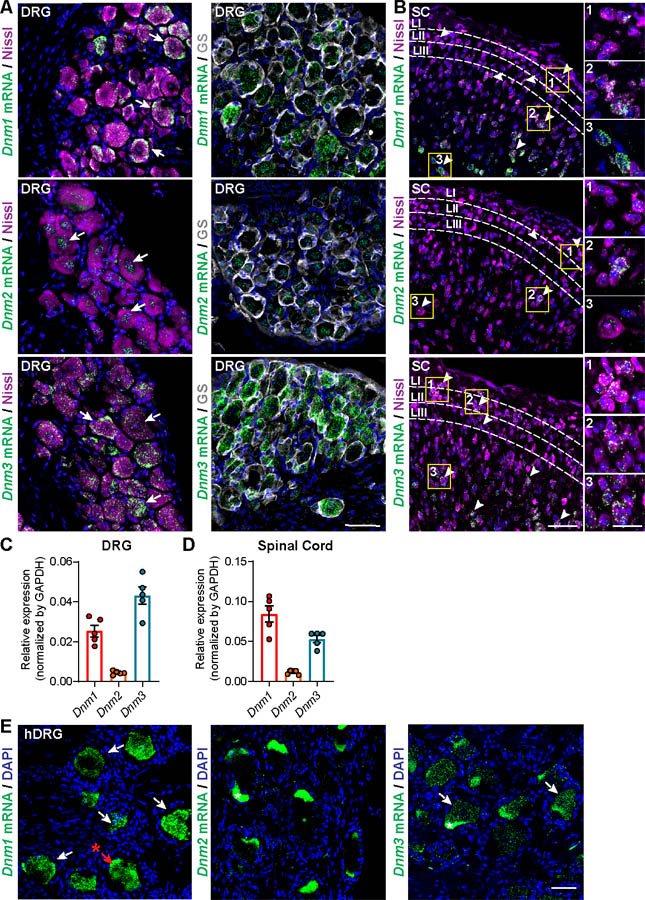
RNAScope® localization of Dnm1, Dnm2 and Dnm3 mRNA in DRG (A) and dorsal horn of the spinal cord (SC) (B) of mice. Arrows indicate mRNA expression within DRG and spinal cord neurons. Scale bar, 50 μm and in the detail box 20 μm. Representative images, n=5 mice per group. Quantification of expression of Dnm1, Dnm2 and Dnm3 mRNA in the DRG (C) and spinal cord (D) of mice determined by qRT-PCR, n=5 mice per group. RNAScope® localization of Dnm1, Dnm2 and Dnm3 mRNA in human DRG (E). Arrows indicate mRNA expression within DRG neurons. Scale bar, 50 μm. * Indicates fluorescent signal due to the presence of lipofuscin in human DRG neurons.
Dnm siRNA knockdown in DRG reverses inflammatory and neuropathic pain
To study the contribution of Dnm isoforms to nociception, we administered (i.t. injection) Dnm1, Dnm2, Dnm3 or CTR siRNAs to mice. RNAScope® revealed that Dnm siRNA inhibited expression of Dnm1, Dnm2 and Dnm3 mRNA in DRG neurons after 48 h compared to CTR siRNA (Fig. 2A, B), while expression in the spinal cord was unaffected (Fig. S3A, B).
Fig 2. Dnm siRNA knockdown and inflammatory pain.
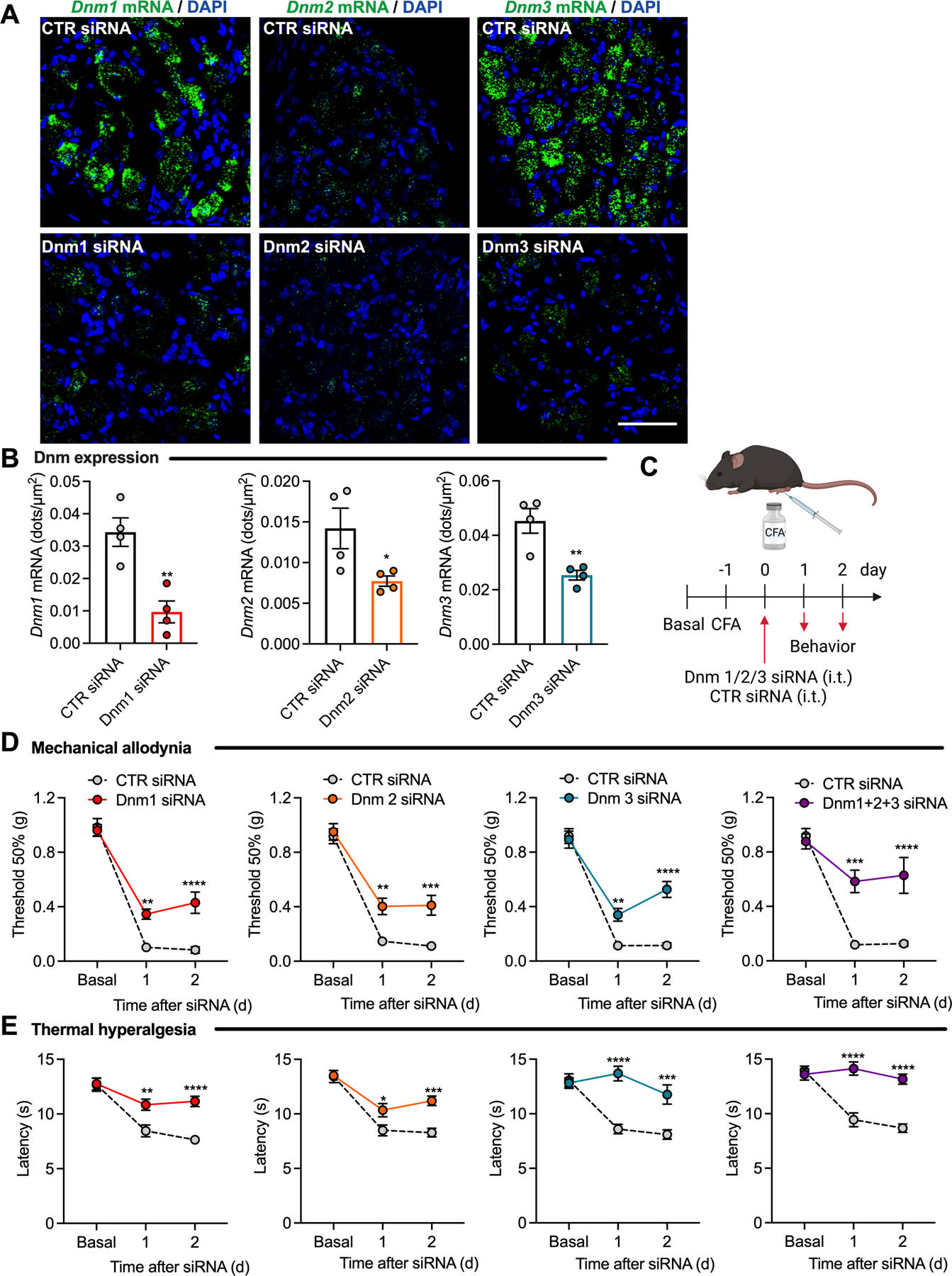
RNAScope® localization (A) and quantification (number of dots per area) (B) of Dnm1, Dnm2 and Dnm3 mRNA expression in mouse DRG at 2 d after administration of Dnm1, Dnm2, Dnm3 or control (CTR) siRNA, n=4 mice per group. Experimental timeline (C). Mechanical allodynia (D) and thermal hyperalgesia (E) induced by CFA measured 1 or 2 d after administration of Dnm1, Dnm2, Dnm3 or CTR siRNA, n=8 mice per group. Mean±SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. CTR siRNA. Parametric unpaired two-tailed t test (B) or 2-way ANOVA, Sídák multiple comparisons test (D, E).
We investigated the effects of Dnm knockdown in a preclinical model of inflammatory pain in mice induced by injection of CFA or vehicle (control) into the hindpaw (Fig. 2C). Dnm1, Dnm2, Dnm3 or CTR siRNA was administered 24 h after CFA. Withdrawal responses of the CFA-injected (left, ipsilateral) and non-injected (right, contralateral) hindpaws to stimulation with von Frey filaments and radiant heat were assessed daily to evaluate mechanical allodynia and thermal hyperalgesia, respectively. CFA-induced inflammation reduced both the withdrawal threshold to von Frey filaments and the withdrawal latency to heat in the ipsilateral paw for at least 4 d, consistent with mechanical allodynia and thermal hyperalgesia (Fig. 2D, E; Fig. S4A, B). Dnm1, Dnm2 or Dnm3 siRNAs partially reversed mechanical allodynia to a similar degree after 24 and 48 h, with a larger effect at 48 h when compared to CTR siRNA (Fig. 2D). When administered together, Dnm1+2+3 siRNAs reversed mechanical allodynia to 72±15% of baseline at 48 h (P<0.0001 compared to CTR siRNA). Dnm1, Dnm2 or Dnm3 siRNAs reversed thermal hyperalgesia after 24 and 48 h, with a larger effect at 24 h (Fig. 2E). Dnm1+2+3 siRNAs reversed thermal hyperalgesia by 100±4% at 24 h (P<0.0001). The anti-nociceptive actions of Dnm siRNA were lost after 72 h (Fig. S4A, B). None of the treatments (i.pl. CFA, i.t. siRNAs) affected withdrawal responses of the contralateral (non-injected) paw to mechanical stimuli (Fig. S4C–F).
Neuropathic pain was induced by SNI surgery of the hindpaw, which induces allodynia; control mice underwent sham surgery. Dnm1, Dnm2, Dnm3 or CTR siRNA was administrated (i.t. injection) 10 d after surgery and withdrawal responses of the operated (ipsilateral) and non-operated (contralateral) hindpaws to von Frey filaments and cold were assessed daily to evaluate mechanical and cold allodynia, respectively (Fig. 3A). Dnm1 siRNA reversed mechanical and cold allodynia after 24 and 48 h when compared to CTR siRNA, although the inhibitory effects were lost after 72 h (Fig. 3B). Dnm1+2+3 siRNAs strongly inhibited mechanical and cold allodynia at 24, 48 and 72 h (Fig. 3C). The largest effect on mechanical allodynia was after 48 h, when the response was 42±5% of baseline (P<0.0001). The largest effect on cold allodynia was after 24 h, when the response was 70±6% of baseline (P<0.0001). None of the treatments (SNI surgery, i.t. siRNA) affected withdrawal responses of the contralateral (non-operated) paw to mechanical stimuli (Fig. S5A).
Fig 3. Dnm siRNA knockdown and neuropathic pain.
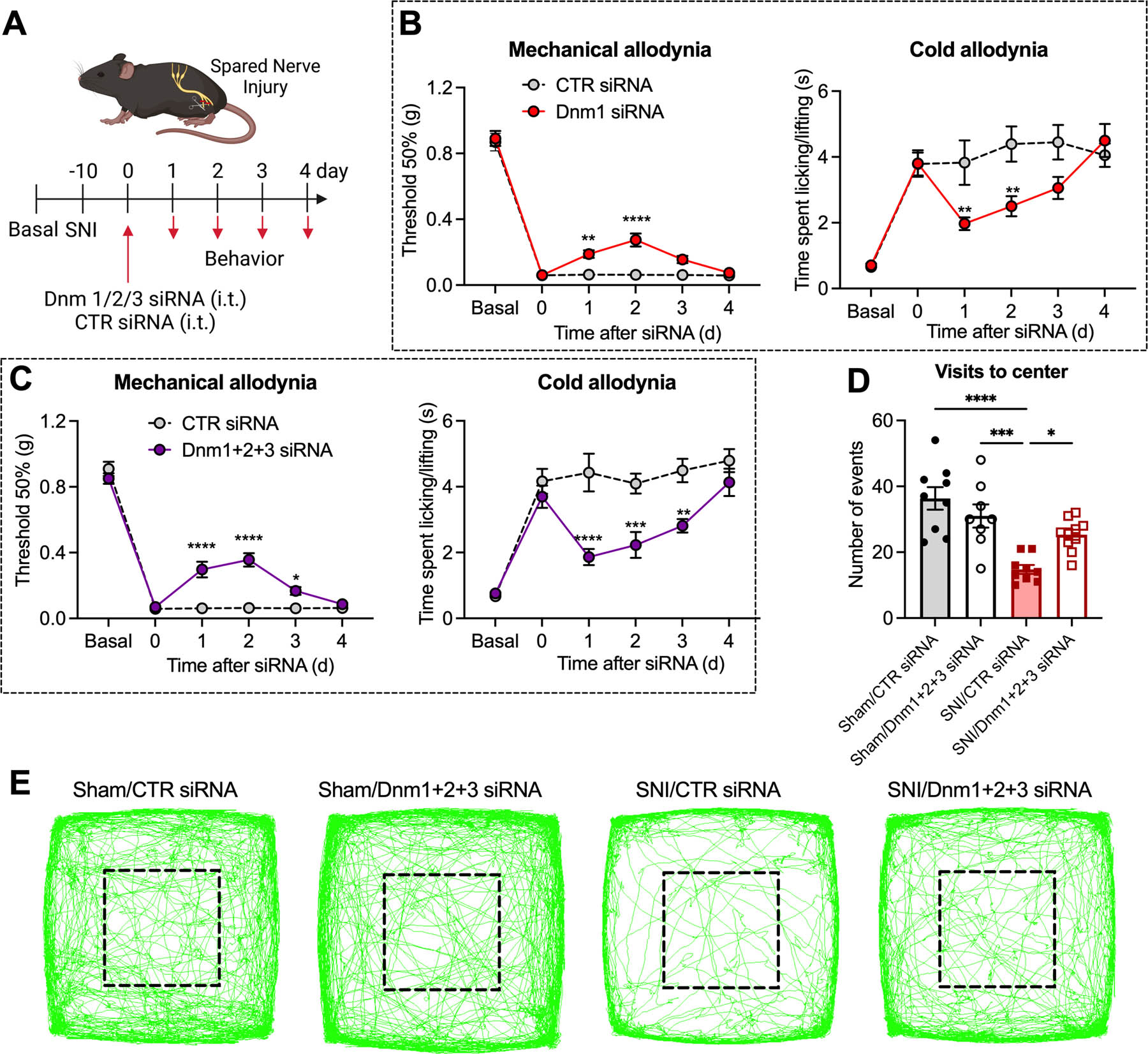
Experimental timeline (A). Mechanical and cold allodynia in SNI mice measured 1–4 d after administration of Dnm1 or CTR siRNA (B) or Dnm1+2+3 or CTR siRNA (C), n=10 mice per group. Non-evoked nociceptive behavior in SNI and sham control mice recorded for 20 min at 2 d after administration of Dnm1+2+3 or CTR siRNA. Visits to center area marked by black square (D) and representative images of the track records (E) are shown, n=8 mice per group for Sham/Dnm1+2+3 siRNA, n=9 mice per group for Sham/CTR siRNA and SNI/CTR siRNA and n=10 mice per group for SNI/Dnm1+2+3 siRNA. Mean±SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. CTR siRNA; Sham vs SNI; SNI/CTR siRNA vs SNI/Dnm1+2+3 siRNA. 2-way ANOVA, Sídák multiple comparisons test (B, C) or 1-way ANOVA, Tukey multiple comparisons test (D).
Non-evoked behavior was assessed using a behavioral spectrometer. Behavior was monitored for 20 min at 48 h after administration (i.t. injection) of Dnm or CTR siRNA into SNI and sham mice. In mice receiving CTR siRNA, the number of visits to the center area was reduced in SNI compared to sham mice (Fig. 3D, E). Dnm1+2+3 siRNAs normalized the number of visits to the center area in SNI mice but had no effects in sham mice. In sham mice, Dnm1+2+3 siRNAs did not affect average velocity, track length, total activity, ambulation, grooming or wall distance when compared with CTR siRNA (Fig. S5B).
AAK1 siRNA knockdown in DRG reverses inflammatory and neuropathic pain
RNAScope® revealed Aak1 mRNA in neurons of DRG, TG and spinal cord dorsal horn (Fig. 4A; Fig. S1A). Although Aak1 mRNA was detected in CGRP+ve peptidergic nociceptors of DRG, most neurons expressed Aak1 mRNA regardless of CGRP content or diameter (Fig. 4B). Aak1 mRNA was also detected in GS+ve satellite glial cells of sensory ganglia. qRT-PCR analyses confirmed expression of Aak1 mRNA in DRG, TG and spinal cord (Fig. 4C). AAK1 mRNA was also localized to neurons of human DRG (Fig. 4D).
Fig 4. Localization of Aak1 mRNA in DRG ganglia and spinal cord.

RNAScope® localization of Aak1 mRNA in DRG and dorsal horn of the spinal cord of mice (A). Arrows indicate mRNA expression within DRG and spinal cord neurons. Scale bar, 50 μm and in the detail box 20 μm. Representative images, n=5 mice per group. Localization of Aak1 mRNA in CGRP+ve DRG neurons (B). Scale bar, 50 μm. Representative images, n=3 mice per group. Quantification of expression of Aak1 mRNA in the DRG, spinal cord and TG of mice determined by qRT-PCR, n=4 mice per group (C). RNAScope® localization of Aak1 mRNA in human DRG (D). Arrows indicate mRNA expression within DRG neurons. Scale bar, 50 μm. * Indicates fluorescent signal due to the presence of lipofuscin in human DRG neurons.
AAK1 siRNA (i.t. injection) depleted Aak1 mRNA in DRG neurons after 24 h compared to CTR siRNA, determined by RNAScope® (Fig. 5A, B); Aak1 expression in the spinal cord was unaffected (Fig. S3A, B). To evaluate effects on nociception, AAK1 or CTR siRNA was administered (i.t. injection) to mice 24 h after CFA or 10 d after SNI surgery (Fig. 5C, D). AAK1 siRNA partially reversed CFA-evoked mechanical allodynia and almost completely reversed thermal hyperalgesia when compared to CTR siRNA (Fig. 5C). AAK1 siRNA reversed mechanical allodynia to 52±9% of baseline (P<0.0001 compared to CTR siRNA) and thermal hyperalgesia to 92±6% of baseline (P<0.01) after 48 h. AAK1 siRNA partially reversed SNI-evoked mechanical and cold allodynia (Fig. 5D). AAK1 siRNA reversed mechanical allodynia to 28±4% of baseline at 48 h (P<0.01) and reversed cold allodynia to 54±8% of baseline at 24 h (P<0.01). AAK1 siRNA did not affect withdrawal responses of the contralateral paw (Fig. S6A, B). In mice receiving CTR siRNA, the number of visits to the center area of the field was reduced in SNI compared to sham mice. AAK1 siRNA restored the number of visits to the center area in SNI mice but had no effects in sham mice (Fig. 5E, F). In sham mice, AAK1 siRNA did not affect any measured behavior (Fig. S6C).
Fig 5. AAK1 siRNA knockdown and inflammatory and neuropathic pain.
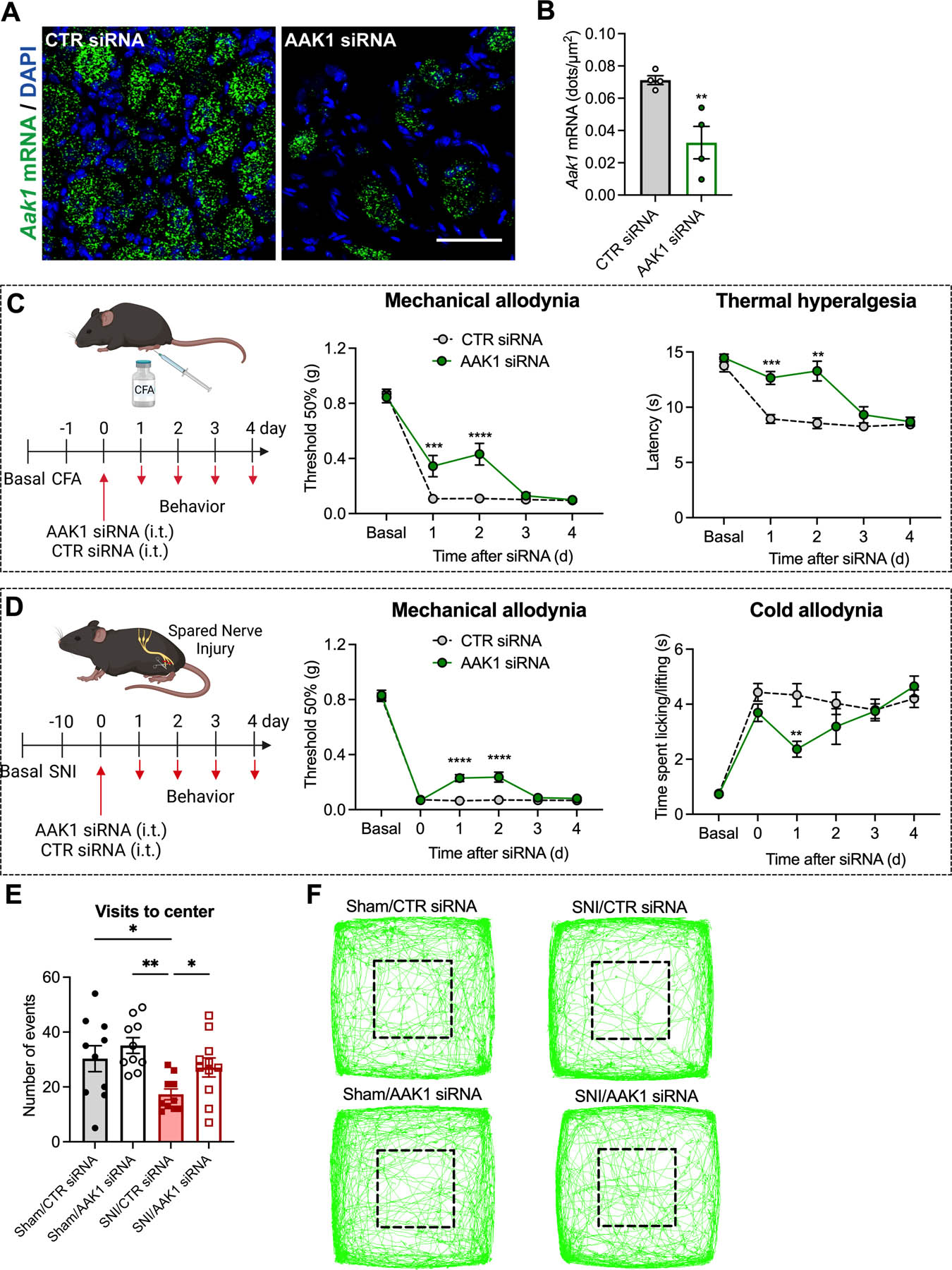
RNAScope® localization (A) and quantification (number of dots per area) (B) of Aak1 mRNA expression in mouse DRG at 1 d after administration of AAK1 or CTR siRNA, n=4 mice per group. Experimental timeline for CFA-evoked inflammatory pain, and mechanical allodynia and thermal hyperalgesia induced CFA measured 1–4 d after administration of AAK1 or CTR siRNA, n=8 mice per group (C). Experimental timeline for SNI-evoked neuropathic pain, and mechanical and cold allodynia in SNI mice measured 1–4 d after administration of AAK1 or CTR siRNA, n=10 mice per group (D). Non-evoked behavior in SNI and sham control mice recorded for 20 min at 2 d after administration of AAK1 or CTR siRNA. Visits to center area marked by black square (E) and representative images of the track records (F) are shown, n=10 mice per group for Sham/CTR siRNA, Sham/AAK1 siRNA and SNI/CTR siRNA and n=11 mice per group for SNI/AAK1 siRNA. Mean±SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. CTR siRNA. Parametric unpaired two-tailed t test (B), 2-way ANOVA, Sídák multiple comparisons test (C, D) or 1-way ANOVA, Newman-Keuls multiple comparisons test (E).
Dnm and Aak1 inhibitors reverse inflammatory and neuropathic pain
Inhibitors of endocytosis offer a pharmacological approach for pain management. To investigate this hypothesis, we examined the effects of inhibitors of Dnm (Dyngo4a) [33], clathrin (PitStop2) [34] and AAK1 (LP935509 [21], SGC-AAK1–1 [1]) on inflammatory and neuropathic pain.
Dyngo4a, PitStop2, inactive analogs (control) (5 µl, 50 µM), LP935509, SGC-AAK1–1 (5 µl, 1 – 10 µg) or vehicle (control) was administered (i.t. injection) 48 h after CFA (Fig. 6A). Dyngo4a partially reversed mechanical allodynia and thermal hyperalgesia when compared to vehicle, with a maximum inhibitory effect at 1 h of 80±11% for mechanical allodynia (P<0.0001 compared to vehicle) and 75±6% for thermal hyperalgesia (P<0.05) (Fig. 6B, C). PitStop2 reversed CFA-evoked mechanical allodynia and thermal hyperalgesia similarly to Dyngo4a (Fig. 6B, C). LP935509 dose-dependently inhibited mechanical allodynia, with a maximal effect for 10 µg at 2 h of 78±8% of baseline (P<0.0001) (Fig. 6D). LP935509 almost completely reversed thermal hyperalgesia, with a maximal effect for 10 µg at 2 h of 89±7% of baseline (P<0.0001) (Fig. 6E). Likewise, SGC-AAK1–1 reversed CFA-evoked mechanical allodynia and thermal hyperalgesia similarly to LP935509 (Fig. 6F, G).
Fig 6. Effects of Dnm, clathrin and AAK1 inhibitors on inflammatory and neuropathic pain.
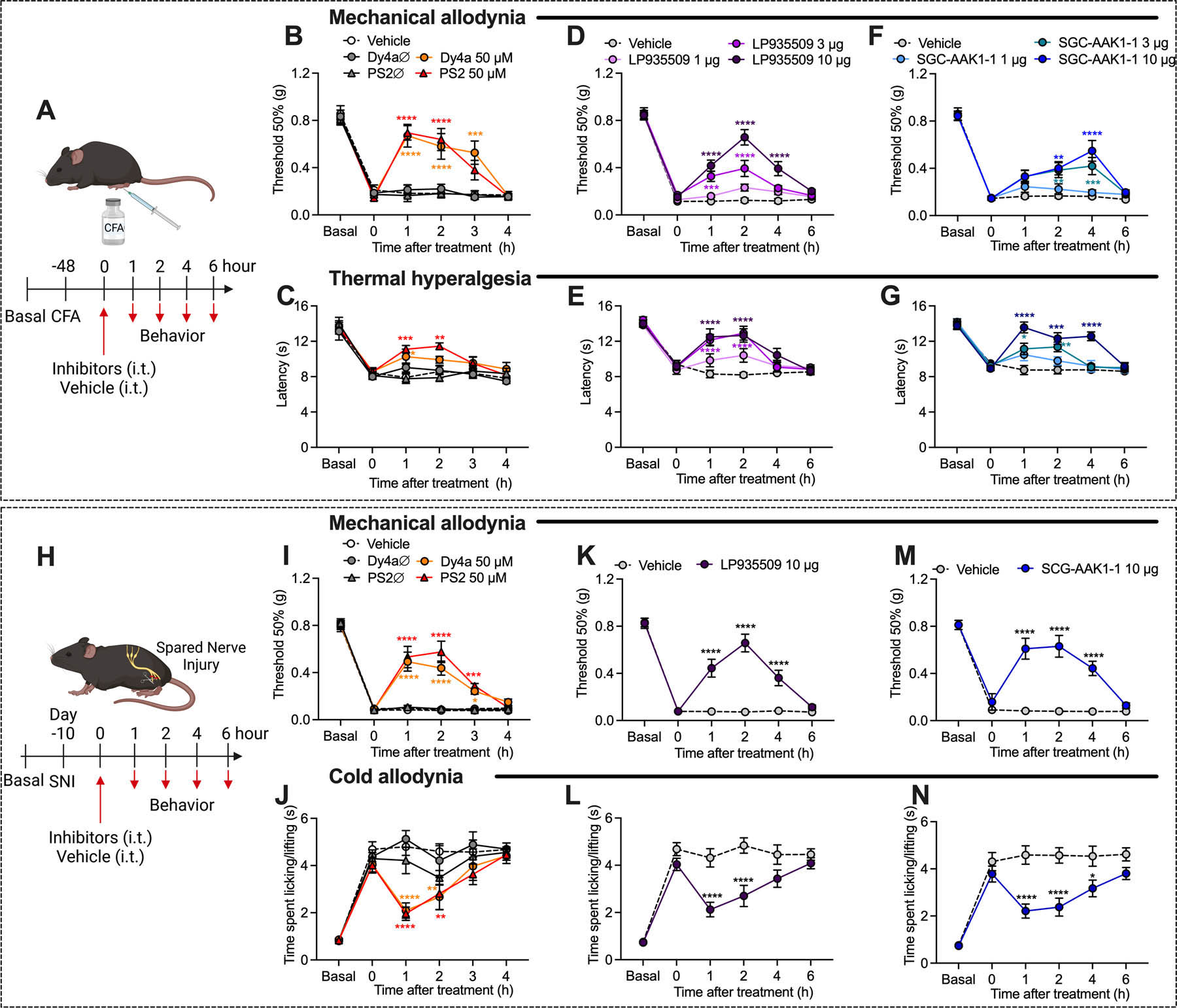
Experimental timeline for CFA-evoked inflammatory pain (A). Effects of Dyngo4a (Dy4a), PitStop2 (PS2), inactive analogs (Dy4aØ, PS2Ø) (all 5 µl, 50 μM, i.t.) or vehicle on CFA-evoked mechanical allodynia (B) and thermal hyperalgesia (C), n=6 mice per group for vehicle and inactive analogs and n=7 mice per group for Dyngo4a and PitStop2. Effects of LP935509 (5 µl, 1–10 µg, i.t.) or vehicle on CFA-evoked mechanical allodynia (D) and thermal hyperalgesia (E), n=8 mice per group. Effects of SGC-AAK1–1 (5 µl, 1–10 µg, i.t.) or vehicle on CFA-evoked mechanical allodynia (F) and thermal hyperalgesia (G), n=8 mice per group. Experimental timeline for SNI-evoked neuropathic pain (H). Effects of Dyngo4a, PitStop2, inactive analogs (5 µl, 50 μM, i.t.) or vehicle on SNI-evoked mechanical (I) and cold allodynia (J), n=7 mice per group for inactive analogs and n=8 mice per group for vehicle, Dyngo4a and PitStop2. Effects of LP935509 (5 µl, 10 µg, i.t.) or vehicle on SNI-evoked mechanical (K) and cold (L) allodynia, n=10 mice per group. Effects of SGC-AAK1–1 (5 µl, 10 µg, i.t.) or vehicle on SNI-evoked mechanical (M) and cold (N) allodynia, n=10 mice per group. Mean±SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. vehicle. 2-way ANOVA, Sídák multiple comparisons test.
Inhibitors of Dnm, clathrin or AAK1 were administered (i.t. injection) 10 d after SNI surgery (Fig. 6H). Dyngo4a reversed SNI-evoked mechanical and cold allodynia with a maximum inhibitory effect at 1 h of 61±10% of baseline for mechanical and 69±8% of baseline for cold allodynia (P<0.01) (Fig. 6I, J). PitStop2 similarly reversed SNI-induced mechanical and cold allodynia (Fig. 6I, J). LP935509 and SGC-AAK1–1 partially reversed SNI-evoked mechanical and cold allodynia from 1– 4 h (Fig. 6K–N). Maximal inhibitory effects of LP935509 (10 µg) were at 2 h for mechanical allodynia (79±9% of baseline, P<0.0001) and 1 h for cold allodynia (61±8% of baseline, P<0.0001).
Inactive analogs of Dyngo4a and PitStop2 or vehicle did not affect CFA- or SNI-evoked allodynia or hyperalgesia. None of the treatments affected withdrawal responses of the contralateral paw to mechanical stimuli (Fig. S7A–F).
Dnm1 and AAK1 shRNA knockdown induces long-lasting reversal of inflammatory and neuropathic pain
To determine whether a more sustained knockdown of Dnm or AAK1 would have a larger and longer-lasting antinociceptive effect, we administered Dnm1, AAK1 or CTR shRNA to mice (i.t. injection). Dnm1 and AAK1 shRNA depleted Dnm1 mRNA by 42±6% and Aak1 mRNA by 28±4% (both P<0.01 to CTR), respectively, in DRG neurons after 72 h, determined by RNAScope®. Dnm1, AAK1 or CTR shRNA was administered to mice 24 h after CFA or 10 d after SNI surgery. Dnm1 and AAK1 shRNA caused a long-lasting reversal of CFA-evoked and SNI-evoked nociception that was fully sustained for at least 7 d (Fig. 7A, B). In CFA-treated mice, Dnm1 and AAK1 shRNA, respectively, reversed mechanical allodynia to 78±8% and 71±6% of baseline (P<0.0001 compared to CTR shRNA) and thermal hyperalgesia to 86±4% and 83±4% of baseline (P=0.0001; P=0.001) after 72 h. In SNI mice, Dnm1 and AAK1 shRNA respectively reversed mechanical allodynia to 69±11% and 41±5% of baseline (P<0.0001 compared to CTR shRNA) and cold allodynia to 79±4% and 97±3% of baseline (P<0.0001) after 72 h (Fig. 7D). Dnm1 or AAK1 shRNA did not affect withdrawal responses of the contralateral paw to mechanical stimulation, heat or cold (Fig. S8A, B).
Fig 7. Dnm1 and AAK1 shRNA knockdown and inflammatory and neuropathic pain.
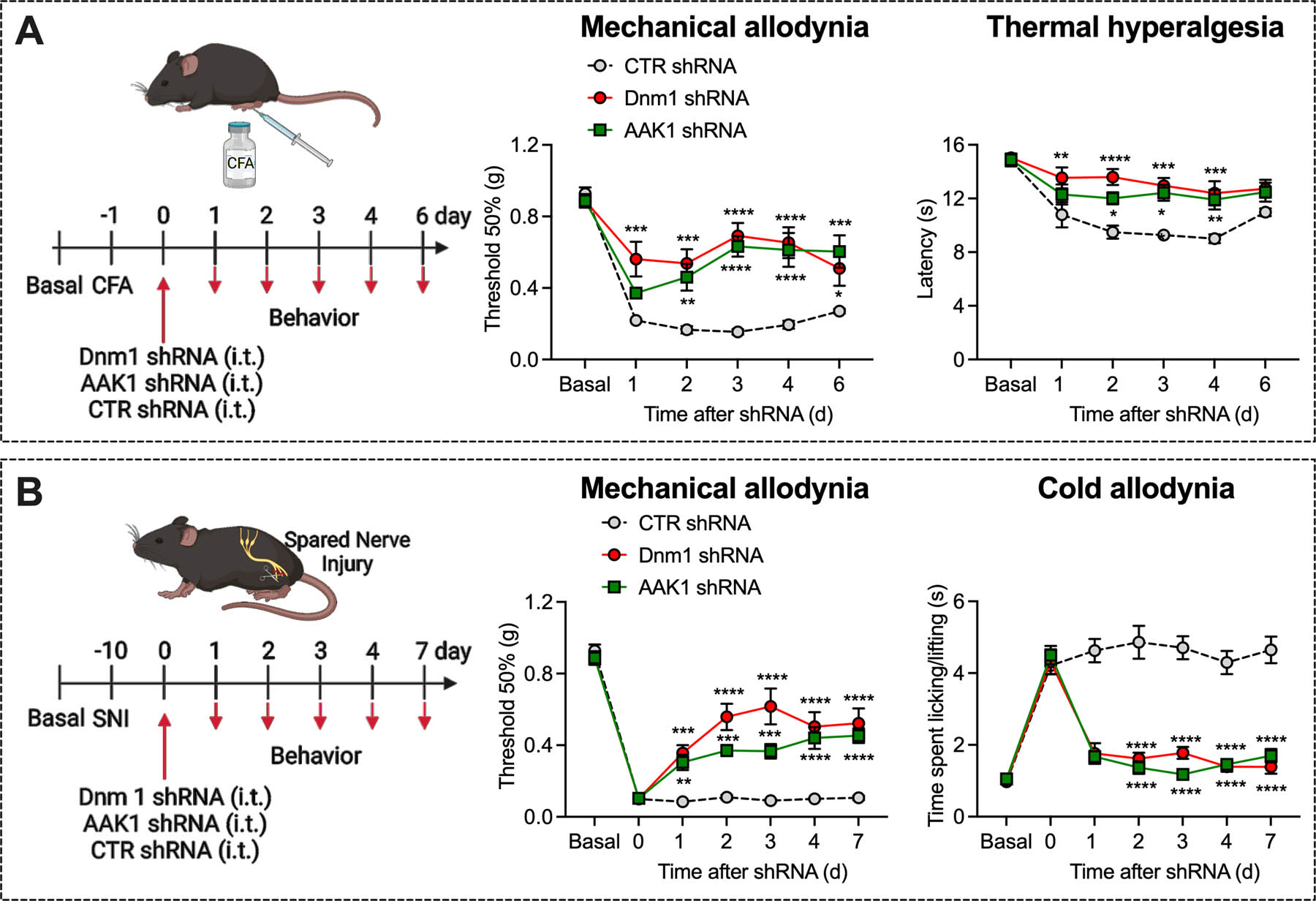
Experimental timeline for CFA-evoked inflammatory pain, and mechanical allodynia and thermal hyperalgesia measured 1–6 d after administration of Dnm1, AAK1 or CTR shRNA, n=8 mice per group (A). Experimental timeline for SNI-evoked neuropathic pain, and mechanical and cold allodynia measured 1–7 d after administration of Dnm1, AAK1 or CTR shRNA, n=8 mice per group (B). Mean±SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. CTR shRNA. 2-way ANOVA, Sídák multiple comparisons test.
Thus, Dnm and AAK1 siRNA, shRNA and inhibitors reverse nociception in preclinical models of inflammatory and neuropathic pain without discernable effects on normal motor functions or behavior.
Dnm1 siRNA increases the threshold required to elicit dorsal root evoked synaptic currents in spinal neurons
To investigate the contribution of Dnm to synaptic transmission in nociceptive circuits, dorsal root-evoked synaptic currents were recorded from spinal neurons in parasagittal slice preparations of mouse spinal cord 48 h after injection of Dnm1 or CTR siRNA (i.t.). Dorsal roots were stimulated and eEPSCs were recorded in whole cell voltage clamp in superficial dorsal horn (lamina I-II) neurons. The stimulus was incrementally increased from 1–10 V to assess the stimulus intensity required to elicit a synaptic response. The threshold voltage required to stimulate an eEPSC was higher in the Dnm1 siRNA group compared to the CTR siRNA group, and current amplitudes were higher in the control group at lower stimulus strength (Fig. 8A, B), which may be due to increased release probability at higher stimulus intensities and recruitment of high threshold fibers. In the control group, the 50% maximal eEPSC amplitude was 2.7 V and maximal amplitude was 5 V, above which the responses were not significantly different from one another. In the Dnm1 siRNA group, the 50% maximal eEPSC amplitude was 4.26 V and the maximal eEPSC amplitude was 6 V (repeated measures 1-way ANOVA, with Sidak’s multiple comparisons test).
Fig 8. Contributions of Dnm1-mediated endocytosis to synaptic transmission in the spinal cord dorsal horn.
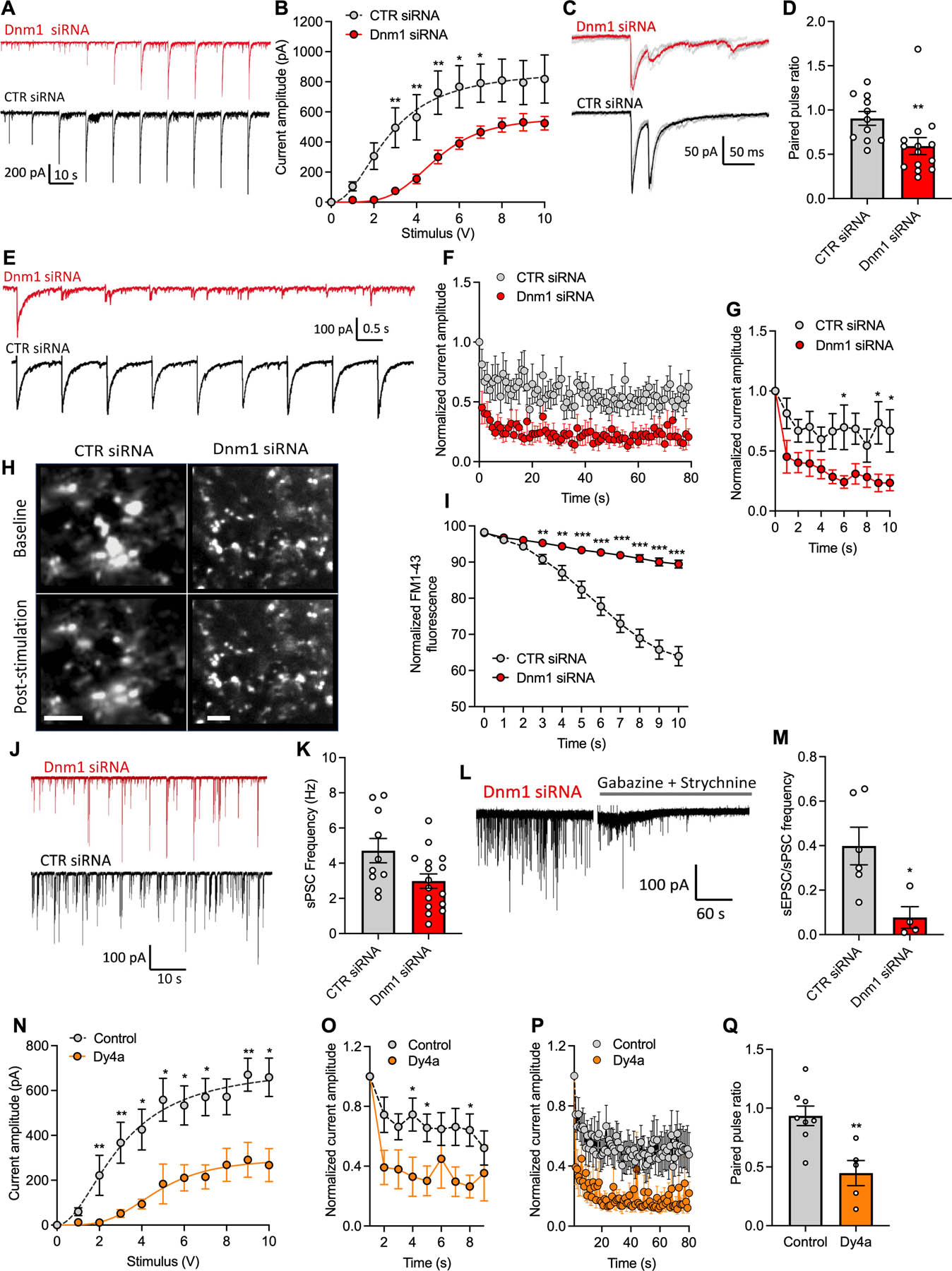
Input-output responses of electrically-evoked postsynaptic currents from spinal cord slices of mice at 2 d after intrathecal injection of Dnm1 or CTR siRNA. Representative traces show eEPSCs recorded in whole-cell voltage-clamp from dorsal horn neurons in response to increasing intensity of presynaptic dorsal root electrical stimulus (1–10 V) (A). Mean eEPSC amplitude from Dnm1 (n=14) and CTR (n=10) siRNA treated mice (B). Traces show averaged paired-pulse responses for neurons of Dnm1 and CTR siRNA treated mice (C). Paired-pulse ratio of Dnm1 (n=14) and CTR (n=11) siRNA groups (D). Traces showing eEPSC responses to 1Hz presynaptic stimulation in Dnm1 and CTR siRNA treated groups (E). Normalized amplitude of eEPSCs over (F) 80 s and the initial 10 s (G) of 1 Hz presynaptic stimulation in Dnm1 (n=11) and CTR (n=8) siRNA treated mice. Images of FM1–47 fluorescence detecting activity of individual synapses in the dorsal horn of Dnm1 and CTR siRNA mice pre- and post-1 Hz stimulation (H). Scale bar, 5 μm. Normalized FM1–47 fluorescence from the start of stimulation in Dnm1(n=36) and CTR (n=33) siRNA groups (I). Traces show spontaneous postsynaptic currents (PSPs) recorded in voltage-clamped chloride-loaded spinal neurons (J). Frequency of sPSC events in spinal neurons from Dnm1 and CTR siRNA groups (K). Trace showing baseline sPSC events, which are reduced by GABA and glycine receptor antagonists (L). The frequency of sEPSCs as a proportion of total sPSCs is reduced in neurons from Dnm1 siRNA treated mice (M). Input-output responses showing current amplitude in response to increasing stimulus intensity (1–10V) in Dyngo4a (30 µM, n=5) and vehicle (n=13) treated spinal cord slices (N). Normalized amplitude of eEPSCs over the initial 8 s (O) and over 80 s (P) of 1 Hz stimulation in Dyngo4a (n=5) and vehicle (n=7) treated spinal cord slices. Paired-pulse ratio of Dyngo4a (n=5) and vehicle (n=8) treated spinal cord (Q). Mean±SEM. *P<0.05, **P<0.01, ***P<0.001 vs. CTR. 2-way ANOVA, Sídák (B, G) or Dunnett’s (I) multiple comparisons test or Mann-Whitney test (D, M, N, O, Q).
Dnm1 siRNA reduces the probability of SV release and prevents sustained SV release from presynaptic primary afferent neurons
To investigate the effect of Dnm1 knockdown on the probability of SV release and to determine the contribution of pre- or post-synaptic sites, we measured paired pulse ratios of eEPSCs in superficial neurons using a paired dorsal root stimulation at 40 ms intervals. Compared to controls, the paired pulse ratio was depressed in the Dnm1 siRNA group (P=0.0042, Mann Whitney test), which indicates a presynaptic mechanism caused by a reduction in release probability from primary afferent neurons (Fig. 8C, D). The prominent expression of Dnm1 mRNA in DRG neurons and the preferential knockdown of Dnm1 mRNA in DRG neurons support a presynaptic mechanism.
Since a reduction in release probability is likely to affect the sustained responses to repetitive stimuli, we tested the eEPSC responses to 1 Hz stimulation of the dorsal roots. A 1 Hz frequency was chosen to allow repetitive activation of the slower latency C-fibers, which would be lost at higher frequencies, as well as A-fiber mediated signals. In spinal cord from mice treated with CTR siRNA, consistent eEPSC amplitudes were observed throughout the 80 s train of stimuli, whereas the eEPSC amplitudes rapidly decreased after repeated stimulation in the Dnm1 siRNA group (Fig. 8E, F). This decrease was most apparent within the first 10 stimuli, where normalized amplitudes were significantly different between the two groups by the 6th stimulus (Fig. 8G).
Styryl dyes can be used to observe SV recycling [10]. Dye molecules reversibly partition into the plasma membrane outer leaflet but do not penetrate cells due to cationic charge. Upon stimulation and SV exocytosis, dye molecules internalize during compensatory endocytosis and label newly formed SVs. When labeled vesicles undergo exocytosis in dye-free medium, dye molecules dissociate from the plasma membrane and lose fluorescence. To further support findings from electrophysiological studies showing reduced SV release after Dnm1 knockdown, FM1–47 was used to track SV recycling. Spinal cord slices were incubated in Mg2+-free ACSF containing 4-aminopyridine (4-AP) to enhance calcium-dependent neurotransmitter release. Tissues were incubated with FM1–43 for 3 min, washed and incubated with ADVASEP-7 to scavenge unincorporated FM1–43. The dorsal roots were stimulated at 1 Hz 4–6V to facilitate SV release from presynaptic terminals, which were imaged by fluorescence microscopy. After 1 Hz dorsal root stimulation, fluorescence at individual synapses declined in the CTR siRNA group, representing exocytosis and consequent loss of labeled SVs. In contrast, synaptic fluorescence was maintained in the Dnm1 siRNA group (Fig. 8H, I).
Dnm1 siRNA inhibits spontaneous excitatory but not inhibitory events in the spinal cord
No significant difference in the frequency of spontaneous synaptic events (total sPSC, including sEPSCs and spontaneous inhibitory post-synaptic currents (sIPSCs) were observed between Dnm1 and CTR siRNA groups (Fig. 8J, K), which suggests the underlying network activity in the dorsal horn remains functional after Dnm1 knockdown. However, when the proportion of excitatory to inhibitory synaptic events was compared by measuring sEPSC frequency in the presence of antagonists of GABA (gabazine) and glycine (strychnine) receptors as a proportion of baseline sPSC frequency, there was a significant reduction in the excitatory component in the Dnm1 siRNA group (P=0.019, Mann-Whitney test) (Fig. 8L, M). This change is likely to be due to a reduction in spontaneous excitatory activity generated by primary afferent neurons, while the spontaneous glycinergic and GABAergic events that originate from inhibitory neurons in laminae II-III remain the same, leading to reduced nociceptive signaling.
Dnm inhibition reduces the probability of SV release and prevents sustained release from presynaptic primary afferent neurons
To corroborate the findings with Dnm1 siRNA, we investigated the effect of the Dnm inhibitor Dyngo4a on synaptic transmission. Dyngo4a (30 µM) reduced eEPSC amplitude in response to dorsal root stimulation at increasing intensities compared to vehicle (DMSO/ACSF) (Fig. 8N). Dyngo4a reduced the sustained neurotransmitter release when afferent inputs were stimulated with a 1 Hz train of impulses (Fig. 8O, P). There was a decrease in paired pulse ratio of eEPSCs in superficial dorsal horn neurons following incubation in Dyngo4a, which suggests that presynaptic release is driving these changes in synaptic activity (Fig. 8Q).
AAK1 siRNA reduces the probability of SV release and prevents sustained release from presynaptic primary afferent neurons
We administered AAK1 or CTR siRNA to mice (i.t.) and recorded eEPSCs in response to stimulation of the dorsal roots. The threshold voltage required to stimulate an eEPSC was higher in the AAK1 siRNA group and current amplitudes were higher in the control group at lower stimulus strength (Fig. 9A). AAK-1 siRNA also suppressed sustained neurotransmitter release when afferent inputs were stimulated with a 1 Hz train (Fig. 9B, C).
Fig 9. Contributions of AAK1-mediated endocytosis to synaptic transmission in the spinal cord dorsal horn.

Input-output responses showing mean eEPSC amplitude of electrically evoked postsynaptic currents recorded from neurons from spinal cord slices of AAK-1 (n=16) and CTR (n=10) siRNA treated mice (A). Normalized current amplitude of eEPSCs over 80 s (B) or the first 10 s (C) of 1 Hz dorsal root stimulation from lamina II neurons from AAK1 (n=16) and CTR (n=8) siRNA treated mice. Effect of LP-935509 (1 µM, n=9) (D) and SGC-AAK1–1 (100 nM, n=4) (E) on eEPSC amplitude in response to increasing electrical stimulus of the dorsal root (1–10 V). Baseline recordings are shown in grey and responses following superfusion AAK1 inhibitor in red. Normalized current amplitude of eEPSCs over 8 s of 1 Hz dorsal root stimulation for baseline and after LP-935509 (F) and SGC-AAK1–1 (G) incubation. Paired pulse ratios of LP-935509 (H) and SGC-AAK1–1 (I) treated slices compared to baseline. Mean±SEM. *P<0.05, **P<0.01, ***P<0.001 vs. CTR siRNA or baseline. 2-way ANOVA, Sídák multiple comparisons test (A) or Paired Wilcoxin tests (D, G).
AAK1 inhibitors reduce the probability of SV release and prevent sustained release from presynaptic primary afferent neurons
The AAK1 inhibitors LP-935509 (1 µM) or SCG-AAK1–1 (100 nM) were bath-applied following baseline recordings in the presence of the vehicle. Both compounds reduced eEPSC amplitude in response to dorsal root stimulation at increasing intensities compared to baseline control (Fig. 9D, E), but the effects were less significant than data from AAK-1 siRNA treated animals (Fig. 9A). When afferent inputs were stimulated with a 1Hz train in the presence of these compounds, there was a significant reduction in sustained release in the LP-935509 treated group compared to baseline control (Fig. 9F) but not in the SGC-AAK1–1 group (Fig. 9G). The train of stimuli were limited to 8 pulses in these experiments to avoid synaptic potentiation between the baseline control and drug treatment recordings. Paired pulse ratios were not significantly different with either treatment (Fig. 9H, I).
Thus, Dnm1 and AAK1 siRNA and inhibitors deplete the readily releasable SV pool and suppress SV release from presynaptic terminals, which disrupts neurotransmission in nociceptive circuits in the spinal cord.
Dnm1 siRNA reduces the number of SVs in spinal cord synapses
Transmission electron microscopy was used to examine the effects of knockdown of Dnm mRNA on SV recycling in the dorsal horn. A characteristic feature of synapses in mice treated with Dnm1 siRNA was a reduction in the number of SVs and an increase in the proportion of clathrin-coated vesicular profiles when compared to synapses in mice treated with CTR siRNA (Fig. 10A–H). Many of the coated profiles in Dnm1 siRNA-treated mice appeared to be interconnected clathrin-coated buds (Fig. 10E, F). These results are in line with the effects of Dnm1 deletion on the morphological appearance of synapses [29].
Fig 10. Ultrastructural analysis of spinal cord slices after Dnm1 knockdown.
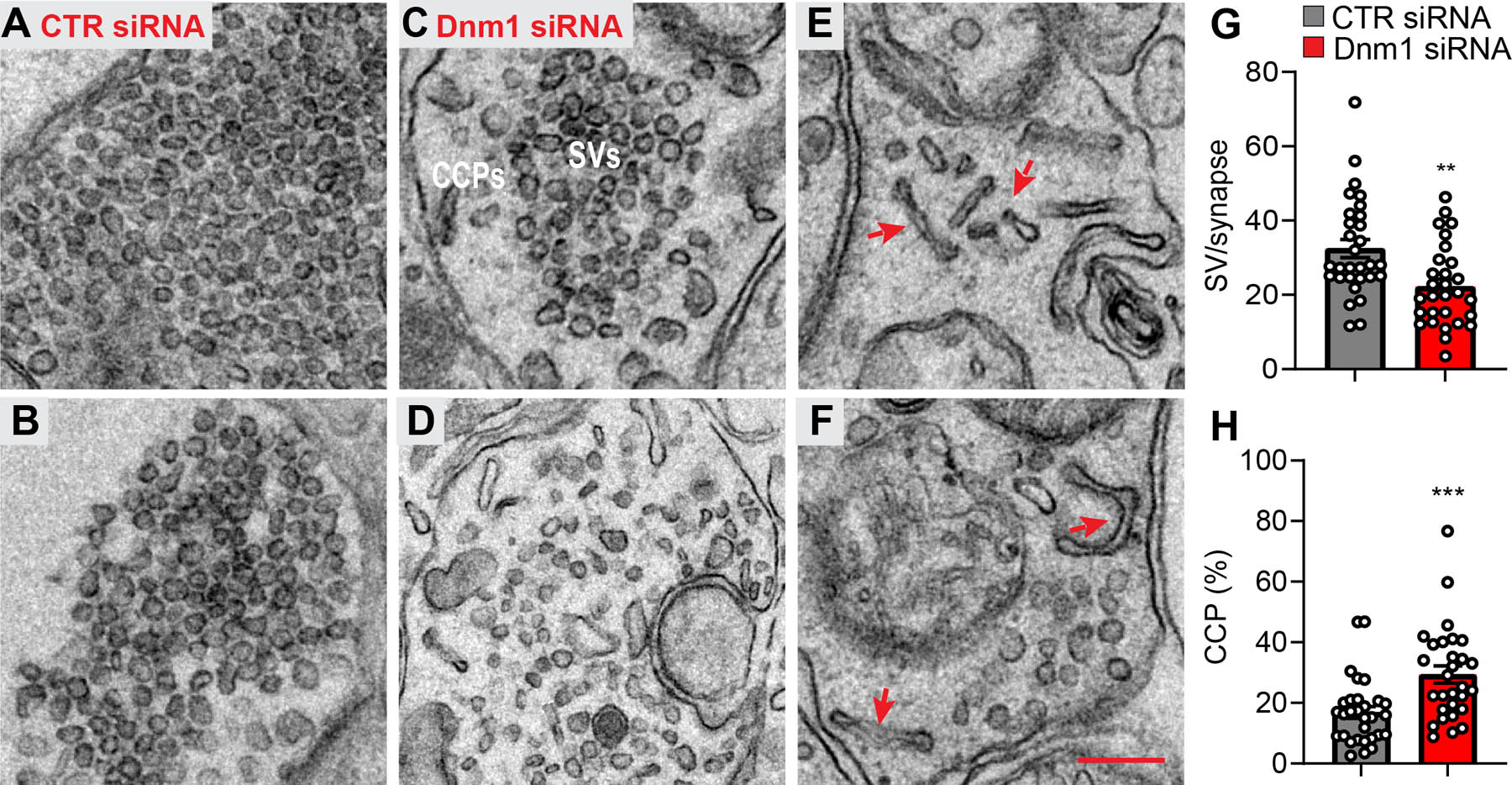
Duplicate electron micrographs of spinal cord from mice at 2 d after administration of CTR siRNA showing presynaptic terminals filled with abundant SVs (A, B). Duplicate low and high power electron micrographs of spinal cord from mice at 2 d after administration of Dnm1 siRNA (C-F). Images of presynaptic terminals reflect the range in the severity of the phenotype from nearly normal (C) to a major accumulation of CCPs with few remaining SVs (D). Red arrows show interconnected CCPs in synapses from Dnm1 siRNA treated mice. Scale bars, 200 nm. Quantification of SVs in Dnm1 and CTR siRNA treated neurons expressed as the number per synaptic profile (G). Quantification of CCPs expressed as the percentage of CCPs relative to the total of SVs + CCPs/synapse (Dnm1, n=212; CTR n=182 synapses) (H). Mean±SEM. **P<0.01, ***P<0.001 vs. CTR siRNA. Parametric unpaired two-tailed t test.
Dnm siRNA attenuates activation of nociceptors
In addition to inhibiting synaptic transmission, endocytosis inhibitors curtail signaling of GPCRs, including protease-activated receptor-2 (PAR2), in nociceptors and thereby blunt persistent nociception [20]. To examine the role of Dnm in GPCR-evoked nociception, Dnm or CTR siRNA was administered (i.t.) 48 h before injection of trypsin (10 μL, 80 nM i.pl.; PAR2 agonist). In mice receiving CTR siRNA, trypsin induced mechanical allodynia after 60 min (Fig. 11A). Dnm1+2+3 siRNA inhibited trypsin-evoked allodynia. To ascertain whether these effects depend on disruption of trypsin-evoked sensitization of nociceptors, we measured the rheobase (minimal input current to fire action potential) of small diameter DRG neurons by patch-clamp recording. DRG were preincubated with trypsin (100 nM, 10 min), washed and rheobase was measured 0 or 30 min later (Fig. 11B). In DRG from CTR siRNA mice, trypsin decreased the rheobase at 0 and 30 min (control 79±15 pA; 0 min 30±4 pA, P=0.002 vs. control; 30 min 26±4 pA, P=0.001 vs. control, 1-way ANOVA, Dunnett’s test, Fig. 11C). Dnm1+2+3 siRNA did not block the initial effects of trypsin (control 72±12 pA, 0 min 34±5 pA, P=0.003 vs. control) but prevented the sustained effects of trypsin (30 min, 68±8 pA, P=0.7 vs. control, Fig. 11D). Thus, trypsin causes an immediate and a sustained hyperexcitability of nociceptors; the sustained effect requires PAR2 endosomal signaling.
Fig 11. Dnm-mediated sensitization of nociceptors.
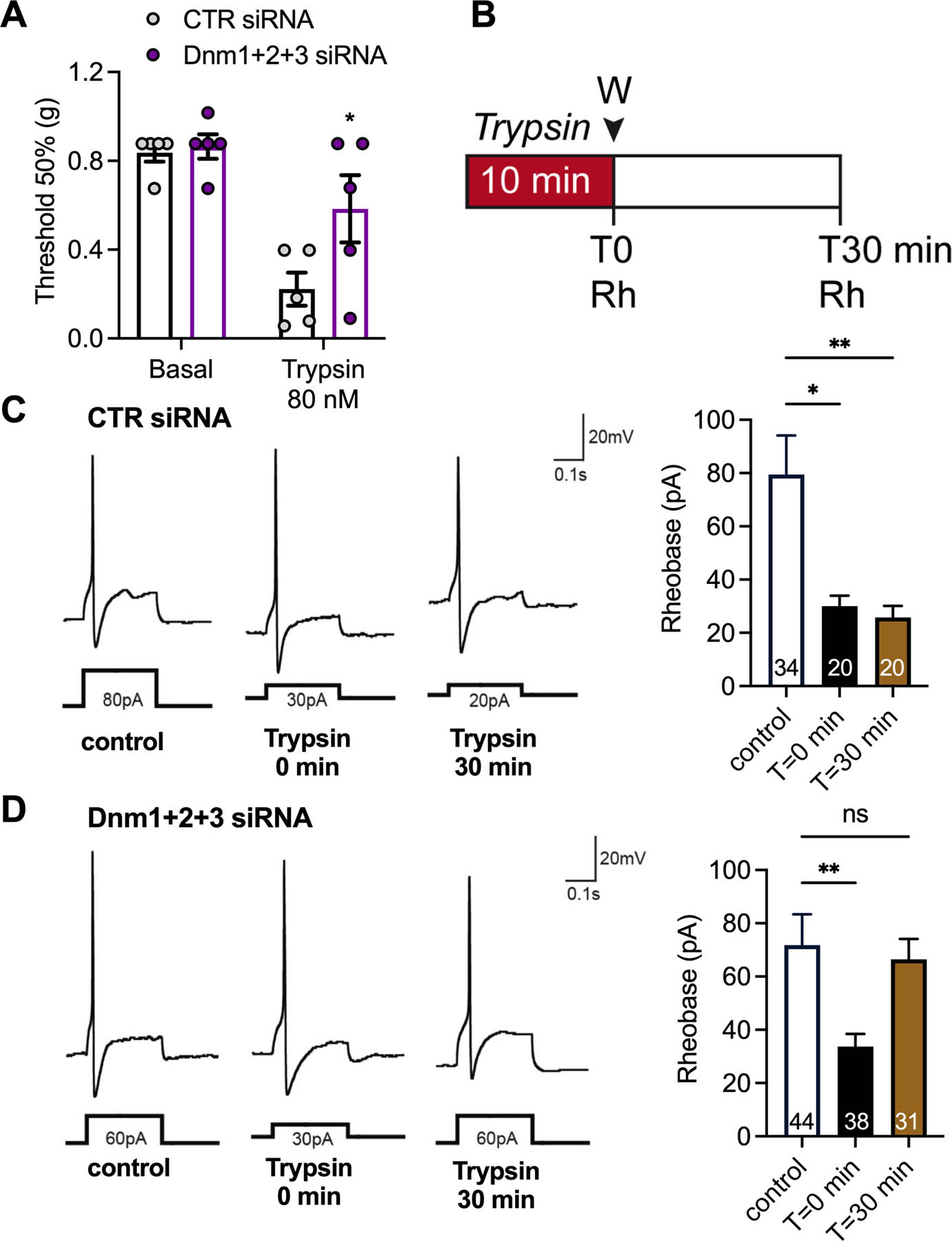
Effects of trypsin (80 nM, 10 µl, i.pl.) on mechanical allodynia in mice at 2 d after intrathecal injection of Dnm1+2+3 or CTR siRNA (A). Mechanical allodynia was measured at 60 min after trypsin, n=5 mice per group. Experimental timeline for electrophysiology experiments (B). Representative traces of rheobase (Rh) and mean rheobase response of DRG neurons at 2 d after administration of CTR siRNA (C) or Dnm1+2+3 siRNA (D). DRG neurons were challenged with trypsin and washed. Rheobase was measured at T=0 and T=30 min following trypsin challenge. Numbers in bars denote neurons measured. Mean±SEM. *P<0.05, **P<0.01 vs. control or CTR siRNA. 2-way ANOVA, Sídák multiple comparisons test (B) or 1-way ANOVA, Dunnett’s multiple comparisons test (C, D).
DISCUSSION
We report a major role for Dnm and AAK1 in synaptic transmission within nociceptive circuits in the dorsal horn of the spinal cord. Dnm1, Dnm3 and Aak1 mRNA were expressed in mouse and human DRG, including in peptidergic nociceptors. Intrathecal siRNA or shRNA down-regulated Dnm and Aak1 mRNA in DRG neurons and reversed nociception in preclinical models of inflammatory and neuropathic pain. Dnm, clathrin and AAK1 inhibitors replicated the effects of siRNA, supporting selectivity. Dnm1 and AAK1 knockdown and inhibition blunted electrically-evoked synaptic transmission between primary sensory neurons and dorsal horn neurons. These changes were coincident with the accumulation of SVs within presynaptic nerve terminals. The results support the hypothesis that Dnm and AAK1 mediate the endocytosis of SVs in nociceptive circuits and thereby sustain nociceptive transmission (Fig. 12). Disruption of this process ameliorates pain.
Fig. 12. Hypothesized contributions of endocytosis in nociceptors to pain transmission.
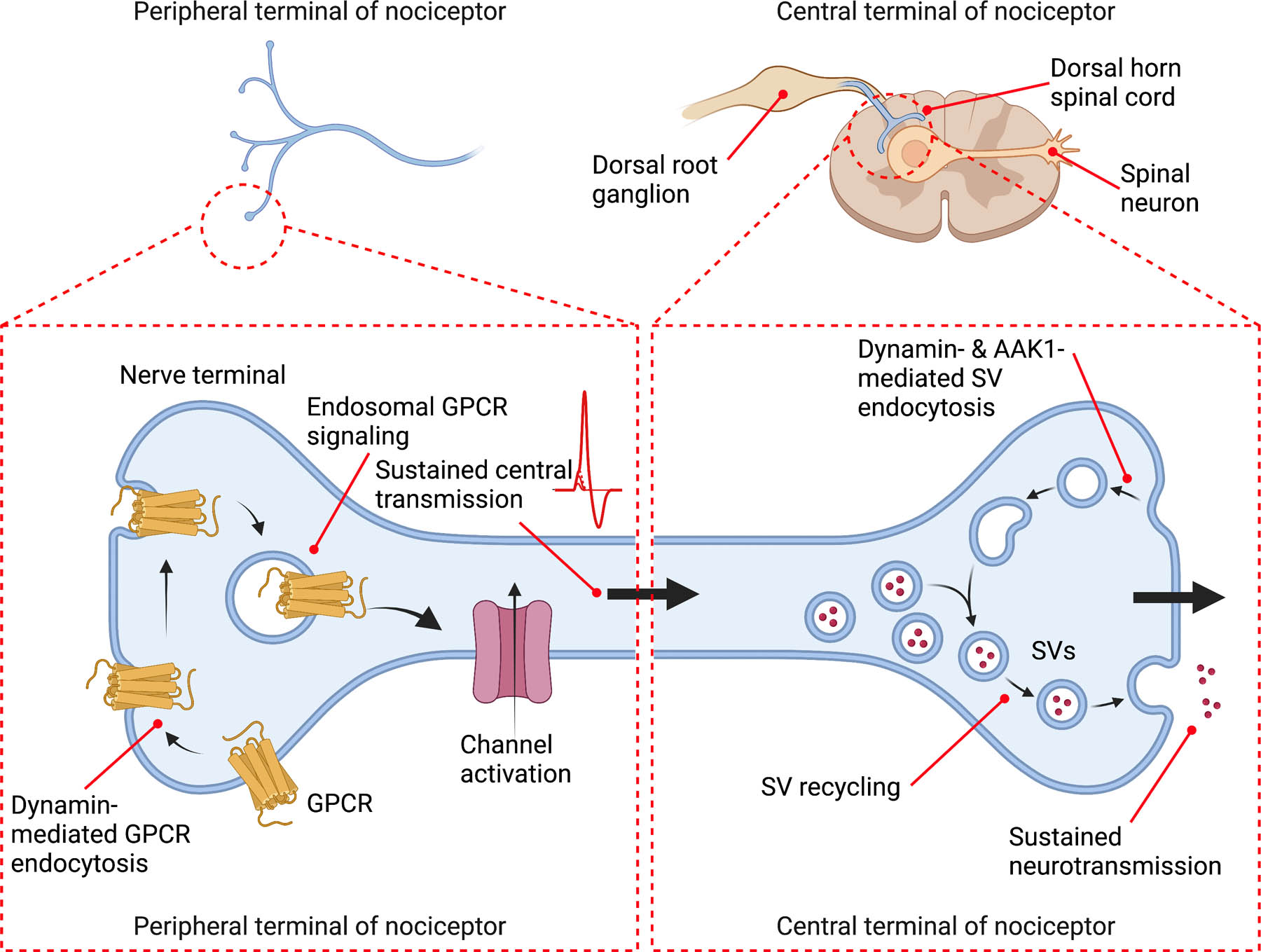
The role of endocytosis for GPCR signaling in the periphery and SV recycling centrally are depicted. At the peripheral terminals of nociceptors, clathrin- and Dnm-mediated endocytosis of GPCRs, such as PAR2, enables sustained endosomal signaling that is necessary for activation of ion channels and ongoing central transmission. At the central terminals of nociceptors, Dnm- and AAK1-mediated endocytosis of SVs replenishes the releasable pool of SVs that is necessary for sustained synaptic transmission.
The conclusion that Dnm mediates synaptic transmission in nociceptive spinal circuits is supported by the expression of Dnm1 and Dnm3 mRNA in DRG and spinal cord neurons. Dnm1 and Dnm3 mRNA were also expressed in human DRG neurons, providing translational relevance. Single-nucleus RNA sequencing in mouse and human DRG tissue identified Dnm1 and Dnm3 in all sensory neuron subtypes [25; 31]. Dnm1 and Dnm3 mRNA were coexpressed with CGRP in peptidergic nociceptors. Peptidergic nociceptors are sensitized during pain and release neuropeptides that mediate pain transmission in the spinal cord [2; 16]. However, Dnm isoforms were expressed by most primary sensory neurons as well as by satellite glial cells, and thus participate in multiple processes. Intrathecal Dnm siRNA or shRNA down-regulated Dnm mRNA in DRG neurons and reversed nociception in mice with persistent inflammatory and neuropathic pain. Isoform-selective knockdown had anti-nociceptive actions, suggesting redundancy. Differences in the magnitude of the antinociceptive actions of Dnm siRNA in models of inflammatory and neuropathic pain could be attributable to different mechanisms of nociceptive transmission; further studies are required to investigate this possibility. Simultaneous knockdown of all Dnm isoforms was more efficacious and sustained than knockdown of individual isoforms, and also normalized behavioral changes in a model of neuropathic pain. Dnm1 shRNA had large antinociceptive actions that were fully sustained for at least 7 d in mouse models of inflammatory and neuropathic pain. A limitation of this study is that knockdown was assessed at the level of mRNA rather than protein. Future studies will investigate the correlation between the antinociceptive actions of shRNA and the degree of protein knockdown over time.
Electrophysiological studies support the conclusion that clathrin and Dnm sustain synaptic transmission in nociceptive circuits by mediating the endocytosis and replenishing the releasable pool of SVs of presynaptic terminals. Dnm1 knockdown and inhibition suppressed eEPSCs in superficial dorsal horn neurons and depressed the paired pulse ratio of eEPSCs, which implicates a presynaptic mechanism caused by a reduction in the probability of neurotransmitter release from primary afferent neurons. The finding that Dnm1 siRNA disrupted the maintenance of eEPSCs during repetitive stimulation of dorsal roots implicates Dnm in sustained synaptic transmission in nociceptive circuits. The imaging of SV recycling with a styryl dye revealed that Dnm1 siRNA disrupts SV recycling, which would be secondary to disruption of SV endocytosis. Ultrastructural studies showed that Dnm1 mRNA knockdown reduced the number of SVs and caused an accumulation of CCPs in presynaptic nerve terminals of dorsal horn synapses. These morphological changes are consistent with the role of Dnm in SV endocytosis and are in accordance with reported changes in SVs and CCPs in neurons cultured from Dnm1 and Dnm1+3 knockout mice [14; 29]. Thus, behavioral, electrophysiological and morphological studies support the conclusion that Dnm is necessary for endocytosis of SVs in presynaptic nerve terminals in nociceptive circuits in the dorsal horn. This process enables SV recycling, which is required for ongoing neurotransmission that underlies nociceptive signaling within the spinal cord.
AAK1 participates in clathrin-mediated endocytosis [9; 32] and is a target for treatment of neuropathic pain [21]. Anatomical, behavioral and electrophysiological studies support the hypothesis that AAK1, like Dnm, mediates endocytosis of SVs in presynaptic neurons of nociceptive circuits in the dorsal horn and is thus necessary for ongoing synaptic transmission of nociceptive signals. Aak1 mRNA was prominently expressed by primary sensory neurons of DRG, including peptidergic nociceptors, and in neurons of the spinal cord. In line with these results, the α-subunit isoform of the AP2 complex, which it is activated by the AAK1 phosphorylation, is preferentially expressed within CGRP+ve nociceptors [28]. Single-nucleus RNA sequencing studies demonstrated expression of Aak1 in all DRG sensory neuron subtypes of mouse and human [25; 31]. Intrathecal AAK1 siRNA downregulated the Aak1 mRNA expression in DRG neurons, reversed mechanical and thermal nociception in mice with inflammatory and neuropathic pain, and suppressed electrically-evoked synaptic currents and neurotransmitter release from presynaptic afferent neurons in the spinal cord. AAK1 shRNA had large and long-lasting antinociceptive actions. The observations that two distinct inhibitors of AAK1 replicated the effects of AAK1 siRNA by reversing nociceptive behavior and reducing the probability of SV release supports selectivity. These results accord with genetic and pharmacological studies that revealed a major role of AAK1 in neuropathic pain through global deletion or systemic antagonism of AAK1 [21]. The current research identifies an anatomical site and mechanism of the pronociceptive actions of AAK1. The results support the hypothesis that AAK1 mediates SV endocytosis in presynaptic terminals of nociceptors in the dorsal horn of the spinal cord, which underlies SV recycling and sustained synaptic transmission of pain.
The observation that Dnm and AAK1 siRNA preferentially knockdown Dnm and Aak1 mRNA in DRG, rather than the spinal cord, supports the hypothesis that Dnm and AAk1 mediate pain transmission by regulating presynaptic neurotransmission in the dorsal horn. The lack of discernable knockdown in the spinal cord may be attributable to poor tissue penetration and targeting of neurons in deeper laminae that are the main site of Dnm and Aak1 mRNA expression. The current studies do not preclude an important role for endocytosis in spinal neurons, which deserves further investigation.
In addition to mediating endocytosis of SVs in presynaptic nerve terminals, Dnm and AAK1 may contribute to nociception by mediating endosomal signaling of pronociceptive GPCRs in primary afferent neurons and spinal neurons (Fig. 12). Clathrin and Dnm mediate endocytosis of PAR2 by nociceptors and endocytosis of substance P and CGRP receptors by spinal neurons [18; 20; 40]. Endosomal signaling of these GPCRs activates kinases that mediate sustained activation of neurons, which is necessary for nociception. Accordingly, inhibitors of Dnm and lipid-conjugated or nanoparticle-encapsulated antagonists that target GPCRs in endosomes have sustained anti-nociceptive effects [11; 18; 20; 22; 23; 40]. In the current study, we observed that trypsin evokes mechanical allodynia in mice, which is known to be mediated by PAR2 [20]. Trypsin caused an immediate and a sustained increase in excitability of nociceptors. Intrathecal Dnm1+2+3 siRNA inhibited mechanical allodynia and sustained hyperexcitability of nociceptors. These results are in agreement with the effects of Dyngo4a on trypsin-evoked nociception [20]. These findings support the hypothesis that Dnm also contributes to nociception by mediating the endosomal signaling of PAR2 (and other GPCRs) that underlies neuronal hyperexcitability and pain.
Is endocytosis in nociceptors a potential target for the treatment of pain?
An advantage is that targeting endocytosis may surmount the redundancy of pain mechanisms. A plethora of mediators initiate and maintain pain, which might explain the lack of efficacy of some highly selective inhibitors. A disadvantage of targeting Dnm and AAK1 is their widespread distribution and multiple roles beyond pain. We enhanced selectivity by administering siRNA, shRNA and antagonists into the intrathecal space, which preferentially targeted DRG neurons. Notably, Dnm and AAk1 siRNA, shRNA and inhibitors did not affect normal motor functions in mice with inflammatory or neuropathic pain, assessed by measuring withdrawal responses of the contralateral paw. These treatments also did not affect multiple behaviors in sham-operated mice, monitored using a behavioral spectrometer. Moreover, these treatments preferentially blocked electrically evoked synaptic transmission, with no effects on basal synaptic transmission. The systemic administration of Dnm inhibitors could disrupt essential cellular processes that depend upon endocytosis. Indeed, deletion of Dnm 1 and Dnm3 is lethal in mice [14; 29]. However, global deletion of Aak1 and systemic administration of the AAK1 antagonist LP935509 inhibits nociception in mice and rats without effects on normal motor function [21]. The AAK1 antagonist LX9211 was safe and well tolerated in healthy subjects in phase 1 clinical trials [4]. The AAK1 inhibitor SGC-AAK1–1 shows improved selectivity over the LX9211 [1]; to our knowledge its therapeutic potential has not been evaluated beyond the current study. Another limitation is that we assessed nociception and did not evaluate the unpleasant emotional aspects of pain, which requires assessment of affective responses to painful stimuli [12].
This study defined the contribution of Dnm and AAK1 to SV endocytosis in presynaptic terminals of nociceptive circuits in the spinal cord, which is necessary for sustained transmission of nociceptive signals. It also confirmed the contribution of endocytosis to GPCR-mediated sensitization of nociceptors. Disruption of endocytosis and consequent inhibition of nociceptor sensitization and nociceptive neurotransmission offers a non-opioid approach to treat pain.
Supplementary Material
ACKNOWLEDGMENTS:
(1) We thank Daniel P. Poole, Nicholas A. Veldhuis and Phillip Robinson for valuable discussion. (2) We thank the NYU DART Microscopy Laboratory for consultation and assistance with electron microscopy; this Core is partially supported by NYU Cancer Center Support Grant NIH/NCI P30CA016087. Fig. 10E was made using BioRender. (3) Supported by grants from: National Institutes of Health (NS102722, DE026806, DK118971, DE029951, NWB, BLS; RF1 NS113881, SD), Department of Defense (W81XWH1810431, W81XWH-22–1-0239, Expansion Award, NWB, BLS), National Health and Medical Research Council (NHMRC APP1125877, APP1139586, WLI) and Australian Research Council (ARC DP190102854, WLI). (4) N.W.B. is a founding scientist of Endosome Therapeutics Inc. Research in N.W.B.’s laboratory is partly supported by Takeda Pharmaceuticals Inc.
Data Availability.
Contact the corresponding author (Nigel W. Bunnett at nwb2@nyu.edu) to obtain original data.
REFERENCES
- [1].Agajanian MJ, Walker MP, Axtman AD, Ruela-de-Sousa RR, Serafin DS, Rabinowitz AD, Graham DM, Ryan MB, Tamir T, Nakamichi Y, Gammons MV, Bennett JM, Counago RM, Drewry DH, Elkins JM, Gileadi C, Gileadi O, Godoi PH, Kapadia N, Muller S, Santiago AS, Sorrell FJ, Wells CI, Fedorov O, Willson TM, Zuercher WJ, Major MB. WNT Activates the AAK1 Kinase to Promote Clathrin-Mediated Endocytosis of LRP6 and Establish a Negative Feedback Loop. Cell Rep 2019;26(1):79–93 e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell 2009;139(2):267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brodkin J, Frank D, Grippo R, Hausfater M, Gulinello M, Achterholt N, Gutzen C. Validation and implementation of a novel high-throughput behavioral phenotyping instrument for mice. J Neurosci Methods 2014;224:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bundrant L, Hunt TL, Banks P, Gopinathan S, Boehm KA, Kassler-Taub K, Tyle P, Wilson A, Warner C, Wason S. Results of two Phase 1, Randomized, Double-blind, Placebo-controlled, Studies (Ascending Single-dose and Multiple-dose Studies) to Determine the Safety, Tolerability, and Pharmacokinetics of Orally Administered LX9211 in Healthy Participants. Clin Ther 2021;43(6):1029–1050. [DOI] [PubMed] [Google Scholar]
- [5].Castro J, Harrington AM, Lieu T, Garcia-Caraballo S, Maddern J, Schober G, O’Donnell T, Grundy L, Lumsden AL, Miller P, Ghetti A, Steinhoff MS, Poole DP, Dong X, Chang L, Bunnett NW, Brierley SM. Activation of pruritogenic TGR5, MrgprA3, and MrgprC11 on colon-innervating afferents induces visceral hypersensitivity. JCI Insight 2019;4(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chanaday NL, Cousin MA, Milosevic I, Watanabe S, Morgan JR. The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J Neurosci 2019;39(42):8209–8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53(1):55–63. [DOI] [PubMed] [Google Scholar]
- [8].Cheng CF, Cheng JK, Chen CY, Rau RH, Chang YC, Tsaur ML. Nerve growth factor-induced synapse-like structures in contralateral sensory ganglia contribute to chronic mirror-image pain. Pain 2015;156(11):2295–2309. [DOI] [PubMed] [Google Scholar]
- [9].Conner SD, Schmid SL. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J Cell Biol 2002;156(5):921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cousin MA, Robinson PJ. Mechanisms of synaptic vesicle recycling illuminated by fluorescent dyes. J Neurochem 1999;73(6):2227–2239. [DOI] [PubMed] [Google Scholar]
- [11].De Logu F, Nassini R, Hegron A, Landini L, Jensen DD, Latorre R, Ding J, Marini M, Souza Monteiro de Araujo D, Ramirez-Garcia P, Whittaker M, Retamal J, Titiz M, Innocenti A, Davis TP, Veldhuis N, Schmidt BL, Bunnett NW, Geppetti P. Schwann cell endosome CGRP signals elicit periorbital mechanical allodynia in mice. Nat Commun 2022;13(1):646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Deuis JR, Dvorakova LS, Vetter I. Methods Used to Evaluate Pain Behaviors in Rodents. Front Mol Neurosci 2017;10:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 1980;20(1):441–462. [DOI] [PubMed] [Google Scholar]
- [14].Ferguson SM, Brasnjo G, Hayashi M, Wolfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, O’Toole E, Flavell R, Cremona O, Miesenbock G, Ryan TA, De Camilli P. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science 2007;316(5824):570–574. [DOI] [PubMed] [Google Scholar]
- [15].Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 2012;13(2):75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Finnerup NB, Kuner R, Jensen TS. Neuropathic Pain: From Mechanisms to Treatment. Physiol Rev 2021;101(1):259–301. [DOI] [PubMed] [Google Scholar]
- [17].Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988;32(1):77–88. [DOI] [PubMed] [Google Scholar]
- [18].Jensen DD, Lieu T, Halls ML, Veldhuis NA, Imlach WL, Mai QN, Poole DP, Quach T, Aurelio L, Conner J, Herenbrink CK, Barlow N, Simpson JS, Scanlon MJ, Graham B, McCluskey A, Robinson PJ, Escriou V, Nassini R, Materazzi S, Geppetti P, Hicks GA, Christie MJ, Porter CJH, Canals M, Bunnett NW. Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci Transl Med 2017;9(392). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jimenez-Vargas NN, Gong J, Wisdom MJ, Jensen DD, Latorre R, Hegron A, Teng S, DiCello JJ, Rajasekhar P, Veldhuis NA, Carbone SE, Yu Y, Lopez-Lopez C, Jaramillo-Polanco J, Canals M, Reed DE, Lomax AE, Schmidt BL, Leong KW, Vanner SJ, Halls ML, Bunnett NW, Poole DP. Endosomal signaling of delta opioid receptors is an endogenous mechanism and therapeutic target for relief from inflammatory pain. Proc Natl Acad Sci U S A 2020;117(26):15281–15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jimenez-Vargas NN, Pattison LA, Zhao P, Lieu T, Latorre R, Jensen DD, Castro J, Aurelio L, Le GT, Flynn B, Herenbrink CK, Yeatman HR, Edgington-Mitchell L, Porter CJH, Halls ML, Canals M, Veldhuis NA, Poole DP, McLean P, Hicks GA, Scheff N, Chen E, Bhattacharya A, Schmidt BL, Brierley SM, Vanner SJ, Bunnett NW. Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc Natl Acad Sci U S A 2018;115(31):E7438–E7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kostich W, Hamman BD, Li YW, Naidu S, Dandapani K, Feng J, Easton A, Bourin C, Baker K, Allen J, Savelieva K, Louis JV, Dokania M, Elavazhagan S, Vattikundala P, Sharma V, Das ML, Shankar G, Kumar A, Holenarsipur VK, Gulianello M, Molski T, Brown JM, Lewis M, Huang Y, Lu Y, Pieschl R, O’Malley K, Lippy J, Nouraldeen A, Lanthorn TH, Ye G, Wilson A, Balakrishnan A, Denton R, Grace JE, Lentz KA, Santone KS, Bi Y, Main A, Swaffield J, Carson K, Mandlekar S, Vikramadithyan RK, Nara SJ, Dzierba C, Bronson J, Macor JE, Zaczek R, Westphal R, Kiss L, Bristow L, Conway CM, Zambrowicz B, Albright CF. Inhibition of AAK1 Kinase as a Novel Therapeutic Approach to Treat Neuropathic Pain. J Pharmacol Exp Ther 2016;358(3):371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Latorre R, Hegron A, Peach CJ, Teng S, Tonello R, Retamal JS, Klein-Cloud R, Bok D, Jensen DD, Gottesman-Katz L, Rientjes J, Veldhuis NA, Poole DP, Schmidt BL, Pothoulakis CH, Rankin C, Xie Y, Koon HW, Bunnett NW. Mice expressing fluorescent PAR2 reveal that endocytosis mediates colonic inflammation and pain. Proc Natl Acad Sci U S A 2022;119(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Latorre R, Ramirez-Garcia PD, Hegron A, Grace JL, Retamal JS, Shenoy P, Tran M, Aurelio L, Flynn B, Poole DP, Klein-Cloud R, Jensen DD, Davis TP, Schmidt BL, Quinn JF, Whittaker MR, Veldhuis NA, Bunnett NW. Sustained endosomal release of a neurokinin-1 receptor antagonist from nanostars provides long-lasting relief of chronic pain. Biomaterials 2022;285:121536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lu W, Ma H, Sheng ZH, Mochida S. Dynamin and activity regulate synaptic vesicle recycling in sympathetic neurons. J Biol Chem 2009;284(3):1930–1937. [DOI] [PubMed] [Google Scholar]
- [25].Nguyen MQ, von Buchholtz LJ, Reker AN, Ryba NJ, Davidson S. Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. Elife 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pertin M, Gosselin RD, Decosterd I. The spared nerve injury model of neuropathic pain. Methods Mol Biol 2012;851:205–212. [DOI] [PubMed] [Google Scholar]
- [27].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001;29(9):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Powell R, Young VA, Pryce KD, Sheehan GD, Bonsu K, Ahmed A, Bhattacharjee A. Inhibiting endocytosis in CGRP(+) nociceptors attenuates inflammatory pain-like behavior. Nat Commun 2021;12(1):5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Raimondi A, Ferguson SM, Lou X, Armbruster M, Paradise S, Giovedi S, Messa M, Kono N, Takasaki J, Cappello V, O’Toole E, Ryan TA, De Camilli P. Overlapping role of dynamin isoforms in synaptic vesicle endocytosis. Neuron 2011;70(6):1100–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ramirez-Garcia PD, Retamal JS, Shenoy P, Imlach W, Sykes M, Truong N, Constandil L, Pelissier T, Nowell CJ, Khor SY, Layani LM, Lumb C, Poole DP, Lieu T, Stewart GD, Mai QN, Jensen DD, Latorre R, Scheff NN, Schmidt BL, Quinn JF, Whittaker MR, Veldhuis NA, Davis TP, Bunnett NW. A pH-responsive nanoparticle targets the neurokinin 1 receptor in endosomes to prevent chronic pain. Nat Nanotechnol 2019;14(12):1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Renthal W, Tochitsky I, Yang L, Cheng YC, Li E, Kawaguchi R, Geschwind DH, Woolf CJ. Transcriptional Reprogramming of Distinct Peripheral Sensory Neuron Subtypes after Axonal Injury. Neuron 2020;108(1):128–144 e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ricotta D, Conner SD, Schmid SL, von Figura K, Honing S. Phosphorylation of the AP2 mu subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J Cell Biol 2002;156(5):791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Robertson MJ, Deane FM, Robinson PJ, McCluskey A. Synthesis of Dynole 34–2, Dynole 2–24 and Dyngo 4a for investigating dynamin GTPase. Nat Protoc 2014;9(4):851–870. [DOI] [PubMed] [Google Scholar]
- [34].Robertson MJ, Deane FM, Stahlschmidt W, von Kleist L, Haucke V, Robinson PJ, McCluskey A. Synthesis of the Pitstop family of clathrin inhibitors. Nat Protoc 2014;9(7):1592–1606. [DOI] [PubMed] [Google Scholar]
- [35].Schlegel A, Largeau C, Bigey P, Bessodes M, Lebozec K, Scherman D, Escriou V. Anionic polymers for decreased toxicity and enhanced in vivo delivery of siRNA complexed with cationic liposomes. J Control Release 2011;152(3):393–401. [DOI] [PubMed] [Google Scholar]
- [36].Shipton EA, Shipton EE, Shipton AJ. A Review of the Opioid Epidemic: What Do We Do About It? Pain Ther 2018;7(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci 2004;27(1):509–547. [DOI] [PubMed] [Google Scholar]
- [38].Tonello R, Fusi C, Materazzi S, Marone IM, De Logu F, Benemei S, Goncalves MC, Coppi E, Castro-Junior CJ, Gomez MV, Geppetti P, Ferreira J, Nassini R. The peptide Phalpha1beta, from spider venom, acts as a TRPA1 channel antagonist with antinociceptive effects in mice. Br J Pharmacol 2017;174(1):57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Valtcheva MV, Copits BA, Davidson S, Sheahan TD, Pullen MY, McCall JG, Dikranian K, Gereau RWt. Surgical extraction of human dorsal root ganglia from organ donors and preparation of primary sensory neuron cultures. Nat Protoc 2016;11(10):1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yarwood RE, Imlach WL, Lieu T, Veldhuis NA, Jensen DD, Klein Herenbrink C, Aurelio L, Cai Z, Christie MJ, Poole DP, Porter CJH, McLean P, Hicks GA, Geppetti P, Halls ML, Canals M, Bunnett NW. Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission. Proc Natl Acad Sci U S A 2017;114(46):12309–12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Contact the corresponding author (Nigel W. Bunnett at nwb2@nyu.edu) to obtain original data.