

Abstract
Isotope-assisted metabolic flux analysis (iMFA) is a powerful method to mathematically determine the metabolic fluxome from experimental isotope labeling data and a metabolic network model. While iMFA was originally developed for industrial biotechnological applications, it is increasingly used to analyze eukaryotic cell metabolism in physiological and pathological states. In this review, we explain how iMFA estimates the intracellular fluxome, including data and network model (inputs), the optimization-based data fitting (process), and the flux map (output). We then describe how iMFA enables analysis of metabolic complexities and discovery of metabolic pathways. Our goal is to expand the use of iMFA in metabolism research, which is essential to maximizing the impact of metabolic experiments and continuing to advance iMFA and biocomputational techniques.
Keywords: metabolic modeling, isotope labeling, mass spectrometry, fluxomics
The metabolic fluxome: a dynamic description of cellular phenotype
A detailed analysis of cellular metabolism is essential for a complete understanding of cellular function in health and disease, and metabolism is also an important parameter for biotechnology applications. Most cell metabolism studies focus on experimentally measured metabolite concentrations. However, metabolite concentrations can change for varied reasons (e.g., increased upstream vs. decreased downstream reaction rates) [1]. Alternatively, metabolite concentrations could remain unchanged despite changes in flux (e.g., if upstream and downstream fluxes change by exactly the same amount). Therefore, we gain limited information from metabolite concentrations alone.
Metabolic fluxes, defined as the metabolic reaction rate (moles/time), provide much more information than metabolite concentration alone. The fluxome indicates the traffic of carbon and other elements among metabolites and therefore provides information on both metabolite concentrations and reaction rates. Since the reaction rates depend on upstream and downstream enzyme expression and activation, the fluxome integrates the cellular metabolome with the transcriptome, proteome, and regulome to offer a dynamic, comprehensive representation of the cell metabolic state [2,3].
Metabolic fluxes include extracellular fluxes (transport reactions that cross the cell membrane) and intracellular fluxes. While extracellular fluxes can be directly measured by tracking metabolite concentrations in the culture medium, intracellular fluxes cannot be directly measured. They therefore must be inferred from metabolite labeling patterns of metabolites [4]. In simple systems, metabolic fluxes can be determined by simply measuring how an isotopically labeled nutrient is converted to intermediate metabolites and secreted products. However, manual analysis is usually not possible in more complex metabolic systems.
Isotope-assisted metabolic flux analysis (iMFA) is a mathematical technique based on optimization, which determines the metabolic fluxome by fitting metabolic labeling data onto a metabolic reaction network model [5,6]. iMFA was originally developed to engineer microbial strains (metabolic engineering); however, it has more recently been used to analyze a variety of eukaryotic systems. iMFA improves our ability to understand metabolic complexity, and by doing so, can advance metabolomic and fluxomic analysis beyond what is currently possible.
The goal of this review paper is to inspire more researchers to use iMFA to maximize analytical insight from their metabolomic data. We first describe how iMFA works, including the inputs, mathematical optimization, and output. We then describe how iMFA enables us to analyze various aspects of metabolic complexity, including iMFA advances that are currently being developed that will enable us to apply iMFA to even more diverse, complex systems.
iMFA components
Input: Network model
iMFA requires a metabolic network model (Figure 1), which describes the metabolic activities of the biological system. Metabolic reactions are categorized as source, sink, or internal (see Glossary) reactions. These reactions are then organized into compartments (e.g., cytosol, mitochondria) where they take place. Finally, atom transitions are explicitly defined for each biochemical reaction. For example, glucose-6-phosphate loses the first carbon atom when entering the pentose phosphate pathway (PPP). This detail must be included in the metabolic network model for the simulation to fit the experimental isotope labeling patterns. The final network model is a curated table of metabolites and reactions, with their corresponding stoichiometry, compartment, and atom transitions.
Figure 1. iMFA workflow.
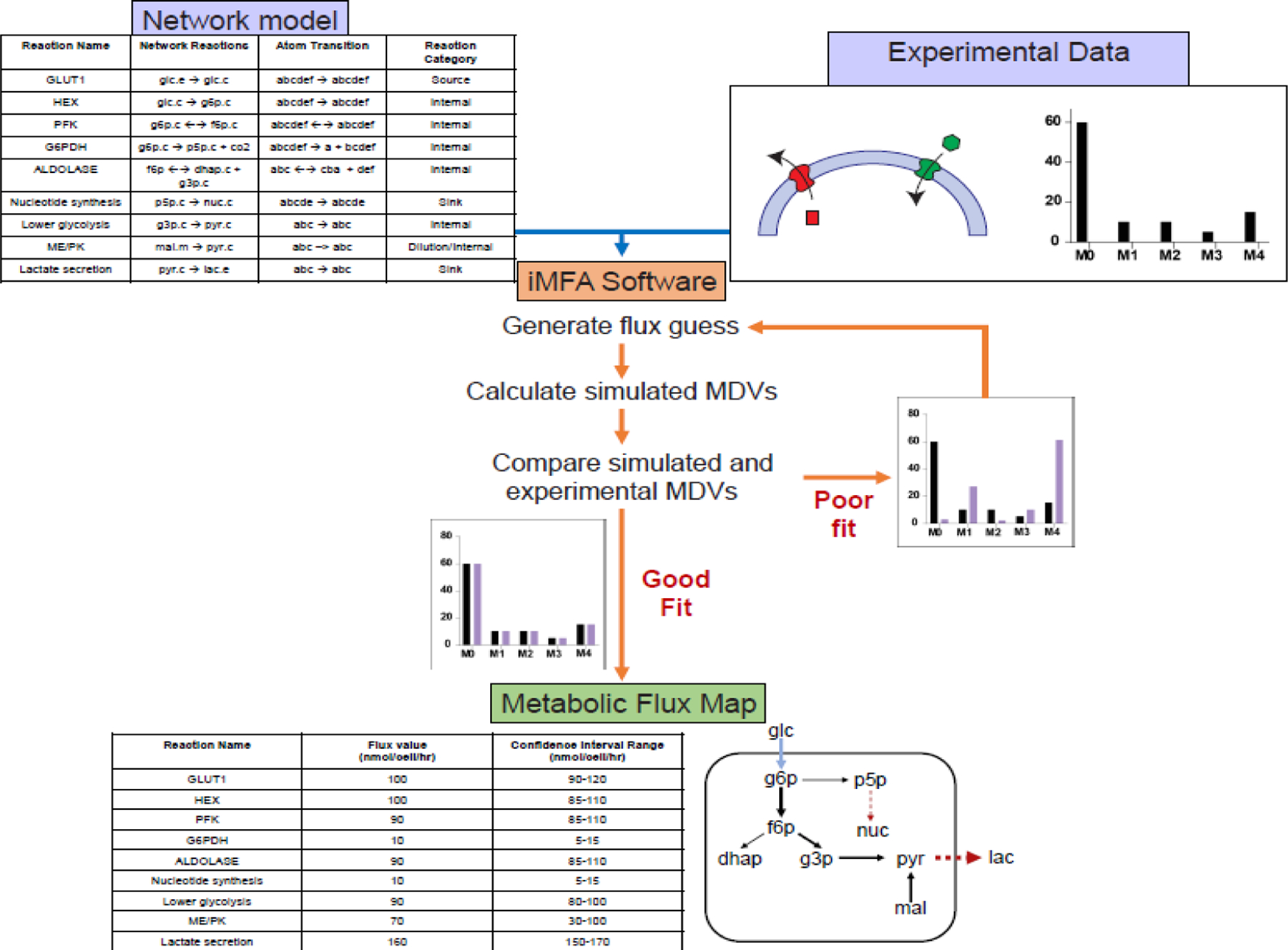
Experimental data, including MDVs and extracellular fluxes, and a metabolic network model are input into the iMFA software. Each reaction in the network model contains detailed information on the compartments of source and product metabolites, as well as the specific carbon atom transitions that occur in the reaction. In some software, such as INCA and eiFlux, compartments can be designated by adding a suffix after the metabolite name (e.g., cytoplasmic as “.c”, extracellular as “.e”, and mitochondrial as “.m”). The iMFA software then iteratively finds a set of fluxes that minimize the error between the simulated and experimental MDVs. When a good fit is achieved, the iMFA software outputs flux values and associated confidence intervals for each reaction flux. This information can be visualized as a flux map, in which arrow thickness represents the relative flux magnitude. The flux map shown here also indicates source reactions (light blue), sink reactions (dark red, dashed), and internal reactions (black).
Network models are typically curated by the end user via publicly available databases such as Kyoto Encyclopedia of Genes and Genomes (KEGG) [7,8], Virtual Metabolic Human [9], and MetaCyc [10,11]. To balance computational efficiency with metabolic accuracy, metabolic network models should be tailored to each biological system. For example, gluconeogenic pathways should be included in hepatocyte network models [12], while the TCA cycle can be pruned from red blood cell network models since these cells lack mitochondria [13,14]. Network construction is often iterative, since the initially curated network model may not include all necessary metabolites, reactions, or compartmentalization. The iterative process is described in detail in the iMFA output section below.
Challenges remain in developing comprehensive metabolic network models. While atom transition information can be attained from the literature as well as the aforementioned databases, the available atom mapping information may be limited for specific reactions. Standardizing atom mapping information across different databases, along with developing algorithms to map atom transitions in complex organic reactions, will improve the accuracy of metabolic network models.
Input: Experimental data
The crucial experimental inputs to iMFA are the isotope labeling states (isotope labeling patterns or isotopomer abundances) that result when cells or tissues process isotopically labeled tracers. 13C-labeled tracers such as [U-13C6]glucose (uniformly labeled glucose), [1,2-13C2]glucose (glucose with 13C in carbon atoms C-1 and C-2), or [U-13C5]glutamine are frequently used to trace carbons. Isotopes of hydrogen (2H) and nitrogen (15N) can also be used to trace the respective elements [15]. Tracer selection requires thoughtful consideration as each tracer provides distinct metabolic information. For example, [U-13C6]glucose elucidates overall glycolytic rate and glucose contribution to the TCA cycle but provides little information on carbon partitioning in upper glycolysis. [1,2-13C2]glucose provides superior information on upper glycolysis and the PPP, especially at the glucose-6-phosphate dehydrogenase (G6PD) branchpoint or in distinguishing the oxidative from the non-oxidative PPP. [U-13C5]glutamine quantifies reductive carboxylation, gluconeogenesis, and glutamine contributions to the TCA cycle [1,16]. The key to a successful labeling experiment is to add physiological concentrations of labels to the culture medium to maximize experimental information. The tracers can be metabolically rearranged in the cells into unique patterns that reflect the underlying metabolic pathways and fluxes. Mathematical tools have been developed to enable researchers to decide the best tracer or best combination of tracers to investigate a metabolic scenario [17,18].
Ideally, cells or tissues are incubated in this labeled medium until the attainment of isotopic steady state, after which a single labeling pattern measurement is made. However, if attainment of isotopic steady state is impractical or does not give flux results, the labeling pattern has to be measured transiently en route to isotopic steady state and analyzed using instationary MFA (INST-MFA). INST-MFA incorporates intracellular metabolite pool size to estimate isotopomer distributions over time. INST-MFA is a more computationally demanding approach since the isotopomer balances are ordinary differential equations. However, many of current software options support INST-MFA and allow users to input labeling data from different time points.
At the end of a labeling experiment, cells or tissue are quenched to arrest metabolism, typically by submerging in liquid nitrogen or a cold organic solvent-water mixture, after which metabolites are extracted for analysis. The labeling state is then quantified by mass spectrometry (MS) or nuclear magnetic resonance imaging (NMR). If the isotope labeling is measured by mass spectrometry, the fractional enrichment of each mass isotopomer is then assembled into a mass distribution vector (MDV; Box 1) for each metabolite of interest.
Box 1. Isotopomers are arranged into mass distribution vectors (MDVs) for each metabolite.
Stable isotope labeling experiments can be quantified by measuring the incorporation of labeled atoms into metabolites. These isotope labeling patterns are known as isotopomers. For each metabolite, there are 2n possible unique isotopomers. Here we provide an example of isotopomers for pyruvate, which has 3 carbons and can therefore have 23 = 8 possible isotopomers (Figure I). Some isotopomers have the same number of labeled atoms but differ in the position of the labeled atoms. Isotopomers with the same total number of labeled atoms are known as isotopologues. These are designated by the mass shift or number of labeled atoms (M + n, where n is the number of total labeled atoms) (Figure I). The fractional or percent composition of isotopologues is referred to as a mass distribution vector (MDV). Traditional mass spectrometry tools such as gas chromatography-mass spectrometry (GC-MS) or liquid chromatography-mass spectrometry (LC-MS) can only discern between different isotopologues, while tandem MS/MS and nuclear magnetic resonance can distinguish isotopomers.
During LC-MS/GC-MS quantification, the ion count of each isotopologue is quantified. In the case of pyruvate, the unlabeled isotopologue (M+0) has a mass-to-charge ratio (m/z) of 88. Successive isotopologues each have one additional mass unit, with the heaviest isotopologue (M+3) having an m/z 91. By employing chromatographic separation and mass spectrometry, each isotopologue for each detectable metabolite can be quantified (Figure II). The ion count for each isotopologue is then normalized to the total ion count of each metabolite (the sum of ion counts for all isotopologues for the metabolite). This yields a fractional isotopologue composition, known as the mass distribution vector (MDV). Typically, the MDV must then be corrected for naturally occurring isotopes that can skew the calculated fractional isotopologue compositions. The final corrected MDV then serves as an input for iMFA software.
Figure I.
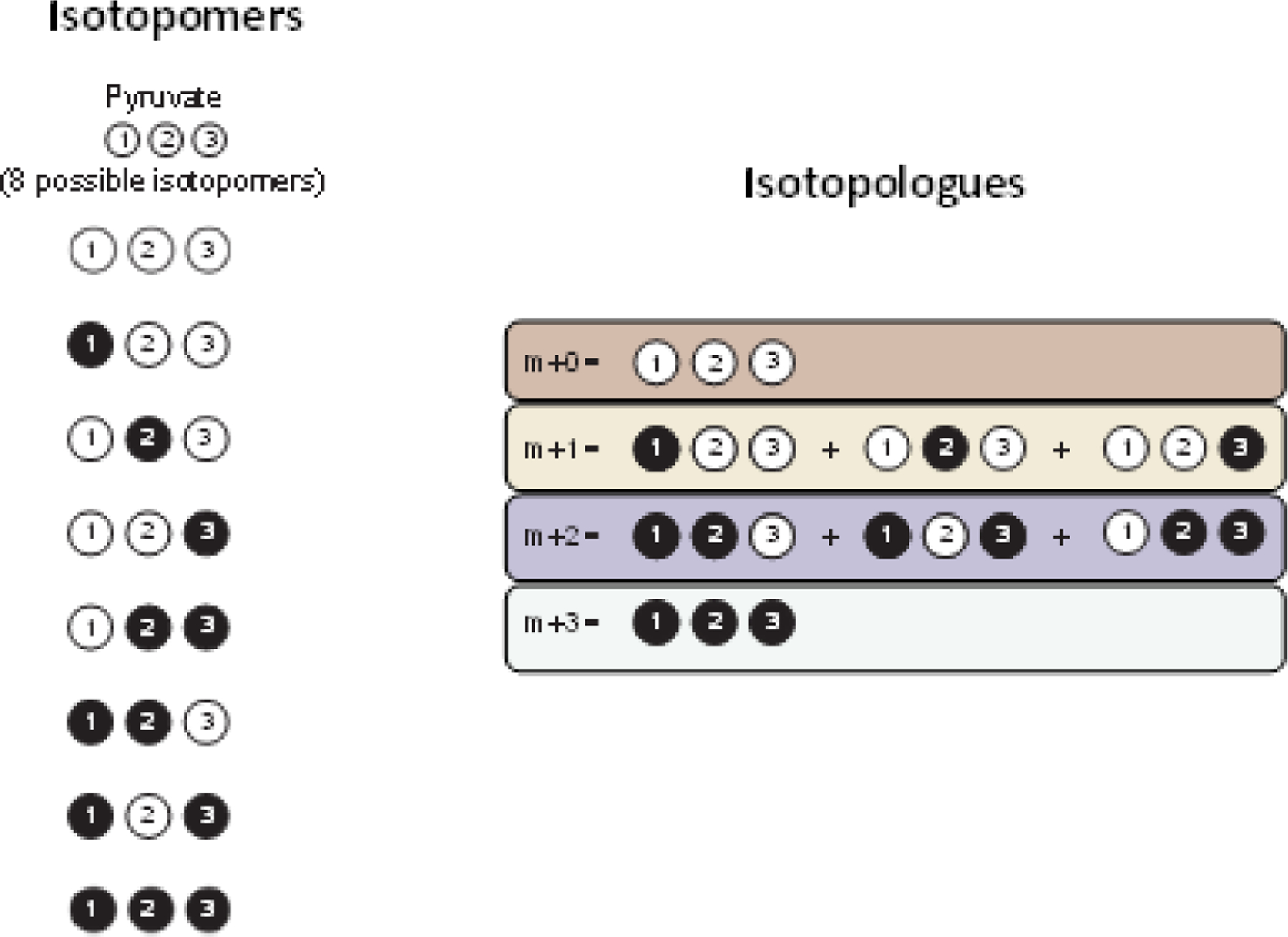
Isotopomers vs. isotopologues for pyruvate.
Figure II.
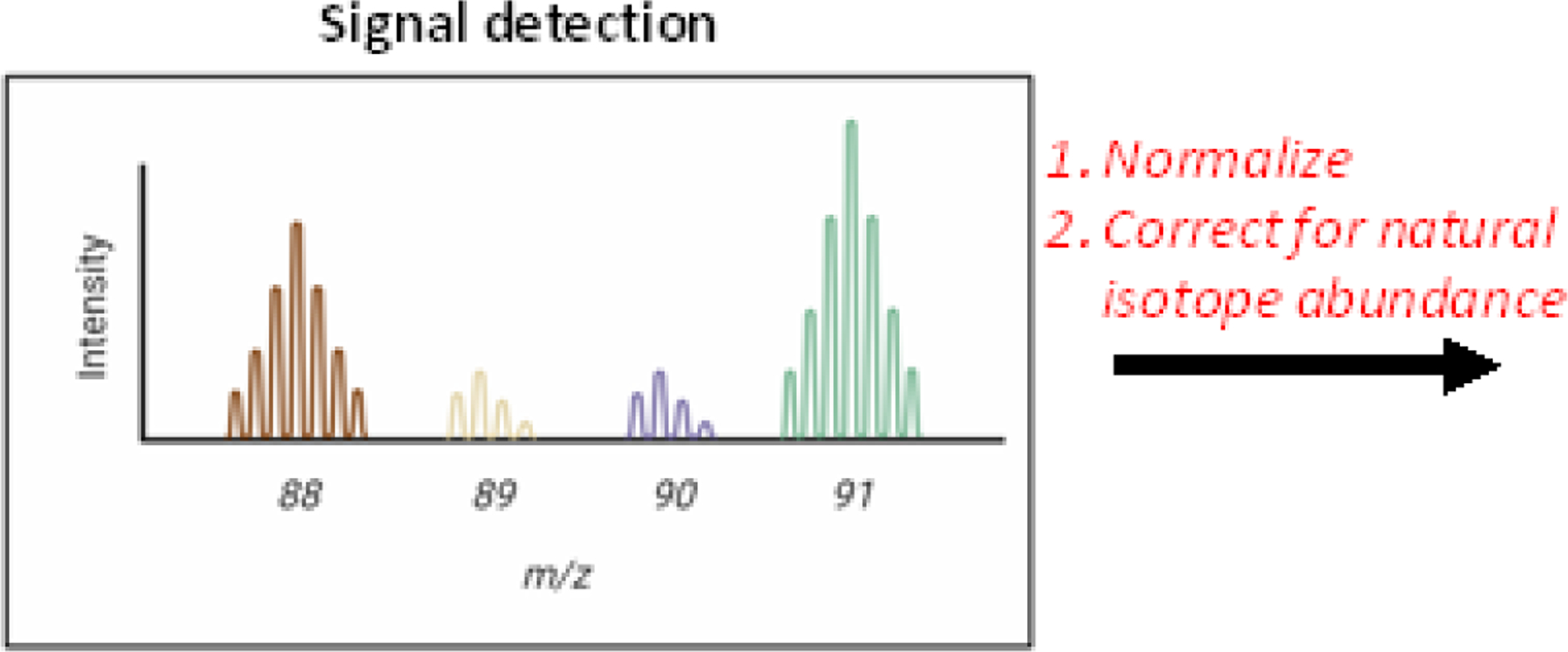
Fractional isotopologue composition is normalized and corrected for natural isotope abundance to determine the MDV.
Different isotope tracers provide complementary information that can be integrated into one holistic iMFA model. For example, [1,2-13C2]glucose could be used to provide information at the PPP branchpoint while [U-13C5]glutamine could be used to analyze reductive carboxylation in the TCA cycle. However, when two or more tracers with the same labeled carbon source are fed simultaneously, they can mask each other’s labeling patterns and cloud our ability to analyze metabolism. Instead, parallel labeling experiments, in which experiments are repeated with different labeled tracers, can also be used to target multiple metabolic pathways (e.g., labeled glucose for glycolysis and labeled glutamine for the TCA cycle). MDVs generated from parallel labeling experiments are challenging to interpret without a computational framework. The software tools discussed in this article enable the design and analysis of parallel labeling experiments.
The other essential experimental iMFA inputs are extracellular flux measurements, which constrain the intracellular fluxes by defining the absolute metabolite quantities that enter and leave the intracellular metabolic network. To determine extracellular fluxes, nutrients (e.g., glucose) and metabolic byproducts (e.g., lactate) in the cell culture medium are measured by high-performance liquid chromatography (HPLC) [19–22], gas/liquid chromatography (GC/LC)-mass spectrometry (MS) [23,24], enzymatic assays, or YSI biochemistry analyzers [25]. External biomass flux can also be determined by measuring change in cell biomass (e.g., dry weight) over time. The extracellular fluxes are then calculated by dividing the change in concentration by the time over which the change occurred. Finally, the fluxes are normalized to cell number and growth rate. Confounding factors such as metabolite degradation and liquid evaporation should also be included.
Current limitations in experimental metabolite and mass isotopomer measurements decrease the power of iMFA. Intracellular sinks and sources (e.g., glycogen) cannot be either measured or experimentally constrained, thus giving cells unknown intracellular metabolite inputs and/or outputs. Experimental mass isotopomer data are largely restricted to a subset of metabolites, primarily those in “core” canonical metabolic pathways such as glycolysis and the TCA cycle. Experiments also mix metabolites from different organelle pools during quenching and extraction, making it impossible to measure metabolite compartmentalization inside the cell. Organelle isolation can directly quantify compartmentalized metabolites; however, organelle isolation is a lengthy process that can itself perturb metabolism [26].
iMFA process: data fitting by mathematical optimization via iMFA software
iMFA is a mathematical optimization process that determines the intracellular metabolic fluxes that best account for the experimental measurements, given the user-defined network model. iMFA uses metabolite isotopomer mass balance equations to relate the amount of each isotopomer to its precursors and products using the metabolic reaction rates. The biological system is assumed to be at metabolic and often at isotopic steady state, which simplifies the iMFA mathematical problem from ordinary differential equations into algebraic equations.
Practically, users enter the experimental data and network model into iMFA software such as eiFlux [27], INCA [28,29], OpenMebius [30], METRAN, and 13CFLUX2 [31]. The iMFA software generally begins the optimization process with an initial guess for each metabolic flux in the system. The initial fluxes are then used to calculate a simulated MDV for each model metabolite. The simulated MDVs are compared to the experimental MDVs. The difference between the simulated and experimental MDVs is then represented as an error term, typically the standard sum of residuals (SSR). This process is repeated numerous times, and the flux values that result in the lowest SSR are returned. When the software does not achieve a satisfactory fit (SSR not low enough), the user must reformulate the network model (described in outputs). Once the model attains a satisfactory fit, techniques such as bootstrap Monte-Carlo [27,32] or parameter continuation [28,33] can be used to estimate confidence intervals for each flux in the network (described in detail in the next section).
Current iMFA software is being improved to make it more efficient and offer expanded features for biologically complex models. In most iMFA software, the state variables (isotopomers) and model parameters (fluxes) are kept distinct from each other, which requires the isotopomer abundances to be computed from scratch in each iteration. In contrast, eiFlux performs the optimization with no distinction between state variables and model parameters. Thus, during each iteration of the optimization, the system state is updated instead of being calculated from scratch. eiFlux also uses state-of-the-art optimization solvers such as the General Algebraic Modeling System (GAMS), thus facilitating efficient, robust parameter estimation and superior scalability to large metabolic networks [27].
New iMFA techniques are also being developed to analyze biological systems when metabolic steady state cannot be reached. Indeed, it is essential to study changes in cell metabolism during dynamic processes such as differentiation, activation, and proliferation. Dynamic 13C-MFA (DMFA) is an emerging non-steady state approach; however, DMFA has experimental and computational challenges, since varying MDVs and fluxes complicates measurements and flux estimation. In a recent study, curve fitting was used to estimate non-steady state glycolytic fluxes from insulin-treated adipocytes that were metabolically sampled over time. While the DMFA model replicated the experimental metabolic trends, the model did not achieve a statistically acceptable fit [34]. Additional progress in DMFA is essential to estimate non-steady state fluxes.
iMFA is also being adapted to incorporate tandem MS, an emerging technique potentially with the benefits of both MS and NMR. Tandem MS breaks precursor ions into product ion fragments, which enables position specific atom information to be acquired. Tandem MS has higher accuracy than GC-MS or LC-MS and improved detection of low abundance isotopomers compared to NMR. To incorporate tandem MS data, the mathematical iMFA framework must be expanded to integrate the fragmented isotopomer information. To date, only eiFlux, enables users to input tandem MS data [27].
Finally, iMFA software is progressing to make computationally feasible genome-scale models, which can improve the simulation fit to the experimental data. Parallelized algorithms have been used to perform genome-scale INST-MFA with improved speed [35]. Two-scale MFA, which uses iMFA data to constrain flux predictions in a genome-scale model, has also been implemented [36]. However, genome-scale models with large metabolic networks and limited experimental data can produce large flux variabilities. Parsimonious iMFA, which posits that evolutionary pressure selects for minimal energy expenditure, provides one solution. Parsimonious iMFA runs a second optimization after standard iMFA and selects the flux solution that minimizes the weighted sum of all fluxes [37]. Parsimonious iMFA can integrate transcriptomic data to minimize fluxes associated with low gene expression [38]. Recently, a proof-of-concept study demonstrated that when compared to traditional iMFA, parsimonious iMFA results in improved flux predictions compared to traditional iMFA. However, the computational complexity of this approach limits its applicability to genome-scale models [37]
Output: Metabolic Flux Map and Statistical Analysis
The statistical analysis of quantitative flux outputs is a key advantage of iMFA over manual tracer analysis. After an acceptable fit is achieved, iMFA generates a quantitative metabolic flux map of reaction flux estimates for all fluxes in the network, along with their respective confidence intervals. The confidence intervals for a given flux are then compared among experimental conditions, and if the confidence intervals do not overlap, then the fluxes are statistically significantly different. Statistical tests could be used to further test significance. Wide confidence intervals indicate high reaction flux uncertainty and suggest that the current experimental strategy is not suitable for flux resolution. In this case, the experimental design should be improved, for example by selecting different tracers or by integrating data from parallel labeling experiments [39,40].
In some cases, iMFA does not find a set of metabolic fluxes that enable the simulation to fit the measured metabolite concentrations. When the SSR does not fall within the acceptable range, the user may need to change the metabolic network model (e.g., adding reaction reversibility, compartmentalization, or alternative routes). Visualization of the experimental and simulated MDVs with tools such as Escher-Trace [41] is helpful in determining which reactions should be altered. This iterative process can lead to valuable insights, especially in elucidating alternative or unique metabolic pathways.
iMFA resolves metabolic complexity
Large data sets
iMFA facilitates analysis of large data sets from parallel and instationary labeling experiments. These labeling strategies are well suited for mammalian cells, which can be prone to slow labeling due to high exchange rates between intracellular and extracellular metabolite pools [6,42]. Furthermore, the wealth of data can more accurately resolve metabolic fluxes in the context of the whole network, as well as buffer effects from measurement errors [43,44]. While these large data sets would be unwieldy to analyze manually, iMFA and INST-MFA are designed to easily incorporate extensive metabolic data.
In a recent INST-MFA study of resting and activated human platelets, the investigators used parallel labeling with mixed tracers to study both glucose and acetate metabolism. Platelets were incubated with either a mixture of [1,2–13C2]glucose, [U-13C6]glucose, and unlabeled glucose or [1–13C]acetate, [2–13C]acetate, and unlabeled acetate. Samples were then collected at 5 times for each labeling experiment. The MDVs and extracellular fluxes were integrated into an INST-MFA model using INCA. Through this approach, the authors determined that activated platelets redirect glucose flux from the PPP and TCA cycle towards lactate [45].
INST-MFA has also been used to analyze the metabolic impact of induced pluripotent stem cell (iPSC) differentiation into cardiomyocytes [46]; neural stem cell differentiation [47]; brown adipose tissue cold activation[48,49], paclitaxel treatment of cancer cells [50], and infection-induced metabolic changes [51,52]. Additional iMFA studies used parallel labeling, including combinations such as glucose/acetate [52], [1,2-13C2]glucose/[U-13C5]-glutamine [51,53–57], with and without palmitate [53]. The rigorous iMFA framework transforms complex data from these experiments into intuitive quantitative flux maps.
Cyclic pathways and reversible reactions
iMFA is especially useful in understanding labeling data from complex metabolic pathways with cyclic pathways, reversible reactions, and exchange fluxes. The TCA cycle is a classic example of metabolic complexity, since isotopomer labeling patterns change as metabolites pass through multiple TCA turns (Figure 2). Whereas manual MDV analysis can usually only account for one or two TCA cycle turns, iMFA can accurately model any number of TCA cycle turns (from one to infinity). Inclusion of infinite TCA cycle turns can clarify TCA cycle activity. For example, a recent study examined whether exogeneous citrate was first metabolized in the cytosol or the mitochondria in hypoxic hepatocellular carcinoma (HCC) cells [58]. When MDVs from HCC labeled with [2,4-13C2]citrate were manually interpreted assuming only one TCA cycle turn, TCA intermediate labeling patterns were indefinite and showed evidence of direct metabolism in both compartments. In contrast, iMFA with infinite TCA cycle turns unveiled significant cytosolic citrate metabolism, with exogeneous citrate primarily entering the mitochondria as α-ketoglutarate. Thus, by considering infinite TCA cycle turns, iMFA uncovered compartmentalization of exogenous citrate metabolism.
Figure 2. Isotope-assisted metabolic flux helps quantify multiple TCA cycle turns.
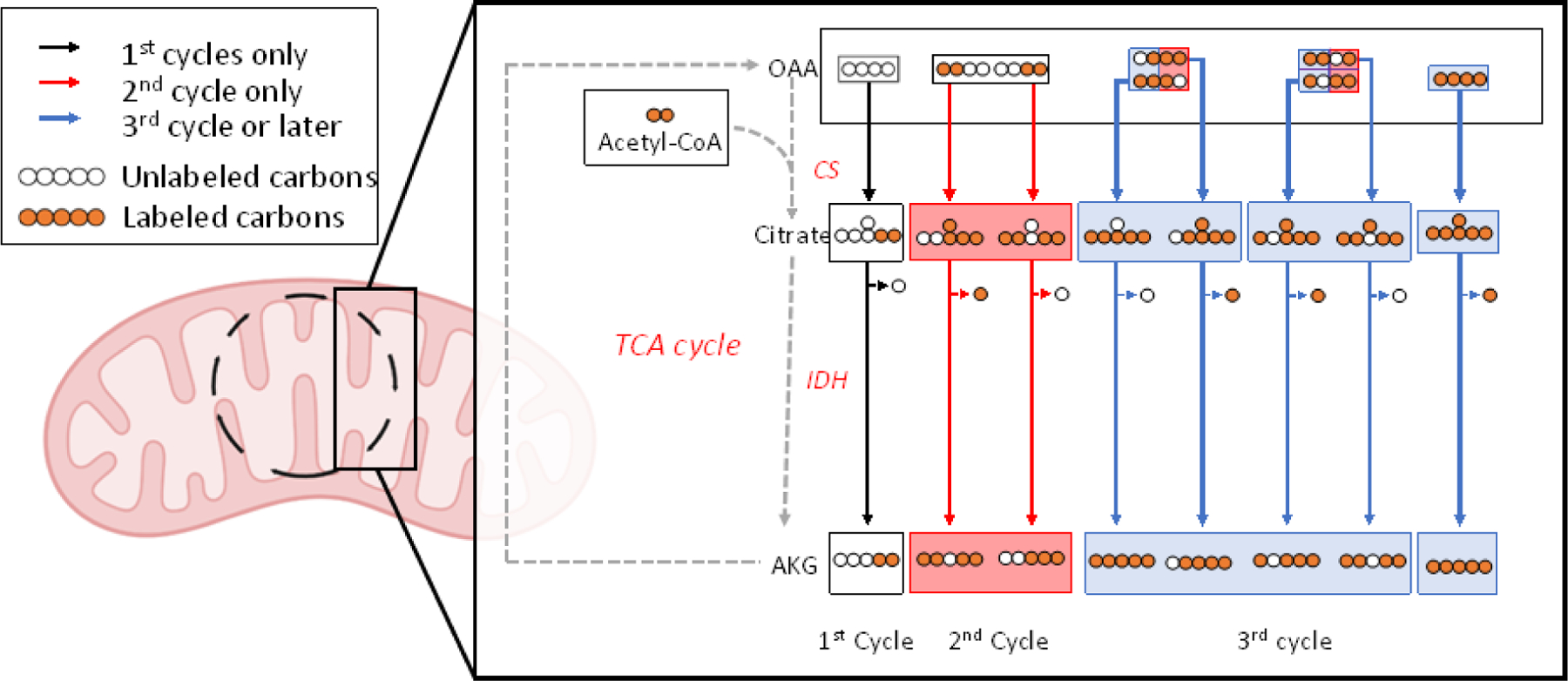
Cyclic metabolic networks, such as the TCA cycle, generate a different set of isotopomers at each turn. This simplified TCA cycle example assumes that uniformly labeled glucose and an unlabeled oxaloacetate (OAA) are the only carbon contributors. Each color represents a different TCA cycle turn. The initial unlabeled OAA reacts with glucose-derived, fully labeled acetyl-CoA to form [M+2] citrate via citrate synthase (CS). This citrate is then metabolized into α-ketoglutarate (AKG) via isocitrate dehydrogenase (IDH). As the AKG progresses through the TCA cycle (metabolites not shown), “flipped” isotopomers of OAA with either the first and second or the third and fourth carbons labeled can occur. When these two OAA isopomers react with fully labeled acetyl-CoA to form [M+4] citrate in the second TCA cycle turn, two unique citrate and AKG isotopomers are created. With each turn of the TCA cycle, more isotopomers are created in different concentrations until, with infinite TCA cycle turns, the metabolites reach metabolic steady state.
iMFA can also account for reversible TCA cycle reactions, in particular reductive carboxylation of α-ketoglutarate via reversed isocitrate dehydrogenase (IDH) flux. In human bronchial epithelial cells treated with cigarette smoke condensate, [U-13C5]glutamine labeling suggested increased glutamine enrichment of acetyl-CoA and [M+5]citrate. iMFA showed that cigarette smoke-treated cells completely reversed IDH flux, which was driven by glutamine consumption [59].
Similar to the TCA cycle, the PPP is complicated by both cyclic and reversible reactions, since fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphate (G3P) are both products and substrates of the PPP (Figure 3). In a recent study, iMFA revealed that oxidative PPP flux increased over 200% during oxidative burst in neutrophils. The change was fueled by a switch to a cyclic PPP phenotype, as well as reversal of the reaction from F6P into G6P. These insights were then confirmed by knockout of non-oxidative PPP enzymes[60].
Figure 3. iMFA helps analyze complexity from the cyclic and reversible PPP pathway.
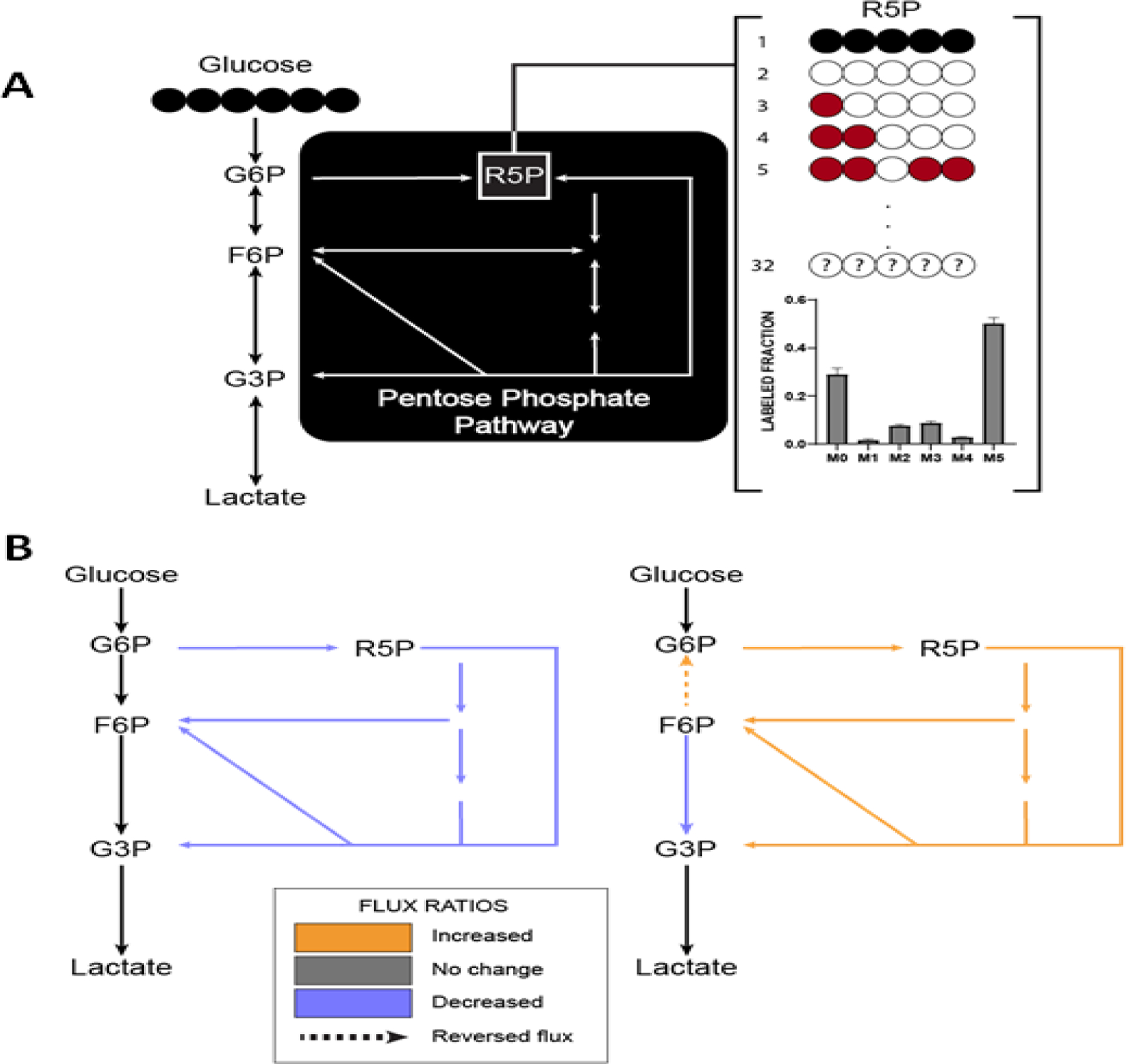
(A) In glycolysis, glucose is metabolized into glucose-6-phosphate (G6P), which can continue down glycolysis or get shuttled into the PPP. The PPP is a complex pathway due to multiple reversible reactions and cyclic interactions with the glycolytic intermediates F6P and G3P. If cells are labeled with uniformly labeled glucose, forward PPP flux will generate uniformly labeled ribose-5-phosphate (R5P). However, reaction reversibility and complex atom rearrangements can generate all 32 isotopomers possible R5P isotopomers (red). Even when simplified into MDVs, the data can still be too complex for manual interpretation. (B) Using iMFA, we showed that endothelial cells treated with dehydroandrosterone to inhibit the first and rate-limiting PPP reaction show decreased PPP fluxes and net fluxes in the forward direction [21] (left). iMFA also showed that neutrophils undergoing oxidative burst have forward PPP flux reversal of glucose-6-phosphate isomerase activity. This causes F6P to be metabolized into G6P, fueling cyclic PPP flux [53] (right).
iMFA can also quantify bidirectional metabolite fluxes in reversible reactions. Most biochemical reactions are reversible or bidirectional and are defined in terms of the net flux (difference between forward and reverse fluxes) and the exchange flux, (minimum of the forward and reverse fluxes). While the net flux indicates the magnitude and direction of the reaction, the exchange flux quantifies the amount by which the label back-mixes through the reaction [61]. Similar labeling patterns between a product and substrate metabolite (e.g., alanine and pyruvate or succinate and malate) often indicate high exchange fluxes. If a product and substrate metabolite are labeled differently, this can suggest low exchange, which can then be quantified using iMFA. Alternatively, labeling dilution can occur if there is high exchange flux with an unlabeled source [28].
Unlabeled sources and anaplerosis
iMFA can elucidate how different sources contribute to intracellular metabolic fluxes due to its ability to integrate many input and output metabolites into a single analysis. We used iMFA to study how inhibition of glycolytic side branch pathways (hexosamine biosynthetic pathway, PPP, polyol pathway) impacted holistic human endothelial cell glucose metabolism. Manual MDV analysis showed that inhibitors decreased labeling in each pathway as well as in the TCA cycle, which we initially interpreted to indicate decreased metabolic activity. However, by integrating extracellular fluxes and isotopomer data into an iMFA model, we discovered that decreased TCA cycle labeling was explained by influx of unlabeled glutamine, which diluted TCA cycle labeling and actually fueled increased TCA cycle activity [25].
The role of other anaplerotic substrates in metabolism can also be clarified using iMFA. Pyruvate is particularly important for the TCA cycle, as it can generate both oxaloacetate (via pyruvate carboxylase) and acetyl-CoA (via pyruvate dehydrogenase). Each route generates a unique set of isotopomers, complicating reaction flux determination from MDVs [62]. A modified iMFA model of whole-body glucose production and gluconeogenesis was used to understand how loss of pyruvate carboxylase impacts metabolic homeostasis in liver-specific pyruvate carboxylase knockout mice. iMFA showed that liver gluconeogenesis from TCA anaplerosis in the pyruvate carboxylase knockout mice was less than 15% of wild type liver. However, loss of pyruvate carboxylase depleted NADPH and glutathione to increase liver oxidative stress and inflammation [63]. In our study of human endothelial cell metabolism, iMFA showed that more pyruvate was routed through pyruvate dehydrogenase than pyruvate carboxylase, and that pyruvate dehydrogenase fluxes changed under metabolic inhibition while pyruvate carboxylase fluxes remained relatively stable [25].
Pathway discovery
iMFA also enables the discovery of new metabolic pathways. When simulated MDVs cannot fit the experimental MDVs, the metabolic network model must be modified. This process often suggests alternative metabolic pathways to then be tested experimentally. In lung cancer cells grown as spheroids or monolayers, iMFA initially could not fit simulated fluxes to the spheroid isotopomer labeling data because citrate showed reductive carboxylation but palmitate, which is produced from citrate, did not. The metabolic network model was then modified so that citrate produced by reductive carboxylation in the cytosol entered the mitochondria, where it mixed with mitochondrial citrate prior to being exported back to the cytosol for palmitate synthesis. This alteration significantly improved the simulation fit. Cytosolic citrate transport into the mitochondria was then validated experimentally using citrate transporter knockout cells [64]. iMFA was also used to uncover evidence that transketolase-like protein 1 (TKTL1) cleaves xylulose-5-phosphate to yield glyceraldehyde-3-phosphate and acetyl-CoA in mammalian cells, since including the TKTL1 reaction was the only way to achieve an acceptable fit [65]. In our study on endothelial metabolism, we initially could not fit the simulation to the experimental data due to excess unlabeled pyruvate. By iteratively modifying the model, we identified a novel four-carbon source that fed into the malate-pyruvate shuttle as a potential source for these unlabeled carbons [25].
Compartmentalization and cell-cell interactions
iMFA can analytically parse data that was mixed experimentally back into separate model compartments. In a study of Chinese hamster ovary cells, the initial iMFA model produced a poor fit due to low labeled pyruvate, alanine, lactate, and glutamate levels. A second model, which included a mitochondrial compartment separate from the cytosol, successfully fit the data. This model further predicted that malic enzyme, which catalyzes conversion of malate to pyruvate, was only active in mitochondria [42].
Cell co-culture systems, which are vital to understand physiological interactions among cells, are even more complex to analyze metabolically. Co-cultures have multiple cell types each with their own extracellular and intracellular fluxes. Experimentally, co-cultures can be created with varying degrees of interaction (Figure 4). Conditioned media experiments keep the cells distinct, enabling extracellular and intracellular metabolite separation; Transwell experiments allow cells to interact through the media while separating cells via a permeable membrane, enabling intracellular metabolite separation only; and direct co-culture mixes the cells and their metabolites. A combination of these experimental techniques is likely needed to describe metabolic interactions in a co-culture.
Figure 4. A variety of experimental co-culture methods are essential to determining metabolic interactions among different cell types.
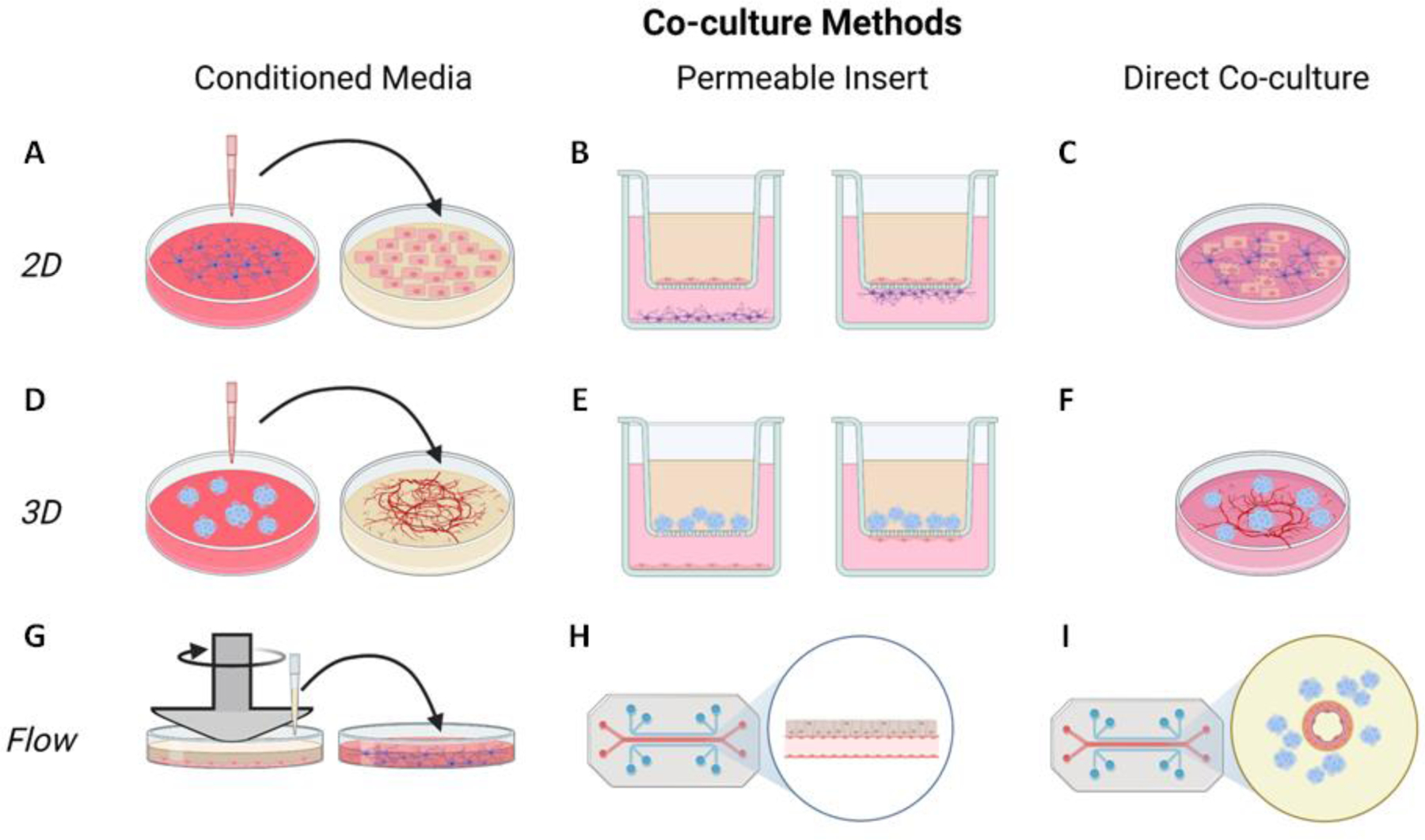
In 2D culture (top row), conditioned media from one cell type can be applied to a different cell type (A), cells can be co-cultured using a permeable insert so that they either share soluble factors through the media (left) or directly contact each other through the membrane (right, B), or cells can be directly co-cultured together in a dish (C). These same techniques can be used for 3D cell cultures (middle row), with cell spheroid media applied to an in vitro vessel-like network (D), cell spheroids co-cultured with a cell monolayer using a permeable membrane (E), or cell spheroids directly cultured on a vessel-like network (F). Finally, stimuli such as flow can be applied to co-cultures to examine metabolic interactions under more physiologically relevant conditions (bottom row). Flow can be applied to one set of cells using a cone and plate device, and conditioned media from the flow-adapted cells can be applied to another cell culture (G). Microfluidics can also be used to apply flow to cells in contact with another type of cells through a permeable membrane (H), or to 3D cell cultures like spheroids surrounding a perfused engineered vessel (I). Figure created with BioRender.
iMFA was used to analyze a co-culture of two Escherichia coli knockout strains without experimental separation. Each cell type was modeled in silico as a separate compartment with a complete metabolic reaction set. Each metabolite MDV was weighted by the fraction of each strain in the total cell population, and secreted metabolites were allowed to exchange between the two cell compartments. The co-culture two compartment network model achieved a satisfactory fit [66]. In another study, cell-cell interactions via extracellular vesicles (EV) were analyzed via a custom Exo-MFA model with terms for EV cargo packaging, secretion, and cargo release into recipient cells[67]. The study showed that TCA fluxes increased six hours after EV exposure, likely due to lactate, glutamine, and TCA intermediate cargo delivery. However, further work is required to improve co-culture analysis, including identifying novel labeling strategies and integrating multiple types of co-culture experiments.
In vivo flux determination
Over the past decade, iMFA has increasingly been used to interpret data from in vivo stable isotope labeling experiments in animals and humans [68]. Since in vivo iMFA is more complex (Box 2) and metabolite uptake and secretion cannot be measured in vivo, iMFA has primarily been used to study endogenous metabolite production. iMFA was used to investigate liver glucose production in mice that underwent short- and long-term fasting [41] and treadmill exercise [69]; renal glucose production in liver-specific pyruvate carboxylase knockout mice [70]; and liver ketogenesis [71]. Notably, the latter two examples required more complex, multicompartmental network models to integrate metabolites from multiple sources. In these studies, in vivo iMFA enabled the authors to both calculate metabolic fluxes in vivo, and quantify how diverse metabolic pathways (TCA cycle, gluconeogenesis, glycogen) contribute to endogenous metabolite production. For more detail, we refer to the reader to other reviews on this topic [72–74].
Box 2. iMFA limitations.
General iMFA limitations
Although iMFA provides a quantitative, systems-level perspective of metabolism, its value relies on the quality of acquired experimental data. When the selected tracer does not generate informative labeling patterns, the iMFA output fluxes will have low precision and large confidence intervals [16,78]. Measurement errors can also be problematic, as they skew flux estimates or impair model fit. These data-driven limitations can lead to erroneous conclusions, especially as the user may add or remove pathways and thereby generate an inaccurate metabolic model in an attempt to fit the data.
Current iMFA models only account for a limited subset of metabolic pathways (glycolysis, TCA, PPP, fatty acid metabolism). iMFA model sizes are limited by lack of information on atom transitions in some pathways, the inability to measure some metabolites, and computational power [79]. However, the exclusion of non-essential pathways can bias results.
Finally, most current iMFA approaches require systems to be at metabolic steady-state and can therefore not account for metabolic changes over time, which are often essential to fully understanding biological processes. Dynamic iMFA approaches that can estimate non-steady state metabolic fluxes approaches are still in their infancy [34,80].
In vivo specific iMFA limitations
iMFA can also be used to determine quantitative tissue-level and whole-body metabolic fluxes for in vivo studies. However, physiological complexity can greatly complicate iMFA. First, metabolite tracers must be carefully selected and validated to ensure they are metabolized by the tissue of interest and that the labeled tracers do not alter in vivo metabolic fluxes when added at concentrations sufficient to enrich downstream metabolites. Second, repeated sampling can be harmful to animals and therefore should be minimized for animal welfare. Thus, isotopic steady state may be difficult to measure for iMFA, and dynamic in vivo measurements are usually not possible for INST-MFA. Third, inter-organ metabolite exchange makes quantifying tissue-specific metabolite uptake and secretion difficult, which means that there are no constraining extracellular fluxes for iMFA.
Labeled substrates can be introduced to animals via oral gavage [75] or liquid diet [76]. These methods can lead to more biologically relevant metabolism but may not provide the necessary information (e.g., infusion rate) for iMFA. A key breakthrough for in vivo iMFA was development of a minimally invasive, dual arterial-venous catheterization technique to continuously infuse tracers [73,77]. This both minimizes stress due to repeat experimental sampling and provides a known infusion rate, which is important for flux value estimations. Dual arterial-venous catheterization, along with sensitive mass spectrometry tools, also enables sampling of microliter volumes, which make this approach practical in small animals [43]. Drawbacks to this method include technical complexity, cost, invasiveness, and reduced relevance for dietary studies [74].
Concluding remarks and future perspectives
iMFA generates quantitative flux maps from complex metabolomic data, enabling the end users to discover metabolic differences among cell states and generate new hypotheses that then can be tested experimentally. Many iMFA discoveries could not have been intuited from manual analysis of isotope labeling. We envision that iMFA will continue to yield new insights into metabolism, especially as larger models and dynamic approaches are developed. iMFA has great potential in elucidating the importance of cell metabolism in stem cell differentiation, rare inherited metabolic disorders, and diseases related to metabolic dysfunction such as cardiovascular disease, diabetes, and obesity. However, to achieve this potential, iMFA must continue to improve in its ability to manage metabolic complexity (see Outstanding questions). As computational and experimental techniques advance, iMFA approaches should be both increasingly accessible and increasingly powerful for metabolism research.
Outstanding questions:
iMFA co-culture analysis is complicated by the experimental mixing of metabolite pools from multiple cell types. What experimental and computational strategies can be used to determine cell-specific fluxomics and cell-cell metabolic interactions in co-culture models?
Currently, iMFA requires systems to be at a metabolic steady state; however, cell metabolism plays a critical role in dynamic cell processes. How can we advance iMFA approaches to analyze dynamic metabolic systems?
Genome-scale iMFA is limited by both experimental and computational approaches. For experimental approaches, how can we improve MS and NMR tools to collect additional isotopologues? For computational approaches, how can we increase computational efficiency to scale and integrate genome-scale isotopologue data?
Highlights:
Isotope-assisted metabolic flux analysis (iMFA) is a mathematical technique that estimates intracellular metabolic fluxes for complex biological systems.
iMFA software uses experimental data (extracellular fluxes, isotope labeling patterns) and a curated network model as inputs and produces a quantitative metabolic flux map as the output.
iMFA determines a set of flux values that produce the best match between the simulated and experimental MDVs using an iterative optimization process.
iMFA is essential to metabolic analysis of complex systems and enables the discovery of new metabolic pathways.
Advances in iMFA are needed to enable analysis of dynamic metabolic states, multicellular co-cultures, and genome-scale networks. These new tools will significantly advance our understanding of metabolic complexity.
Acknowledgements
This research was funded by NSF CBET-2211966 to A.C. and G.S., NIH 1R01HL140239 to A.C, NSF DGE-1632976 to B.M, NSF MCB-1517671 to G.S., and a seed grant from the Maryland Brain and Behavior Institute to A.C and G.S.
Glossary
- Anaplerotic
Reactions that replenish intermediates of a cyclic metabolic pathway that are diverted toward biosynthesis, particularly in the case of TCA cycle intermediates. An example is the reaction pyruvate + CO2 → oxaloacetate, which replenishes the oxaloacetate diverted from the TCA cycle toward the synthesis of the aspartic family of amino acids
- Exchange flux
consider a pair of reversible reactions A ↔ B, where one reaction, say A → B, has flux v1 and the other reaction B → A has flux v2 with both v1 and v2 ≥ 0. In this case, the net flux of the reversible reaction pair is the absolute value of the difference between v1 and v2 (|v1 – v2|), while the exchange flux is the minimum of v1 and v2 (min[v1, v2])
- Fluxomics
An -omics method that measures intracellular and extracellular metabolic reaction rates
- Isotopic steady state
A state in which fractional isotope enrichment is constant over time. Isotopic steady state can be difficult to achieve in mammalian cell cultures because high metabolite exchange leads to slow labeling, and changes in the cell state may cause metabolic variations. It is therefore difficult to maintain metabolic steady state long enough to reach isotopic steady state
- Isotopologues
Metabolites that have different numbers of labeled atoms and therefore mass but are otherwise identical. Isotopologues can be differentiated by LC-MS or GC-MS which detect mass-to-charge ratios (m/z)
- Isotopomers
Isotopologues with the same number of labeled atoms and mass but different labeled atom positions. Distinguishing isotopomers requires position-specific information from tandem MS or NMR
- Instationary MFA (INST-MFA)
INST-MFA enables analysis of a system at metabolic but not isotopic steady state. INST-MFA requires the measurement of isotopomer distributions over time and preferably intracellular metabolite concentrations. INST-MFA is computationally more time-consuming than steady-state iMFA
- Internal reactions
Reactions that both consume and produce metabolites and link source and sink reactions
- Liquid chromatography mass spectrometry (LC-MS) and gas chromatography mass spectrometry (GC-MS)
LC-MS and GC-MS separate samples using liquid or gas chromatographic methods prior to detection by mass spectrometry. LC-MS has simpler sample preparation and detects a broader metabolite range. GC-MS better detects sterols, sugars, and very-short-chain fatty acids
- Mass distribution vector (MDV)
A distribution of isotopologues for a given metabolite. Each isotopologue’s distribution is determined by dividing its ion count by the total metabolite ion count, which is also the sum of all isotopologues
- Metabolic steady state
A state in which metabolic parameters (fluxes and metabolite concentrations) are constant over time, resulting in no net metabolite accumulation. Cell cultures are typically considered at metabolic steady state when they are not proliferating or proliferating at a constant rate. The metabolic steady state assumption eliminates kinetic parameters to simplify iMFA calculations
- Net flux
See Exchange flux above
- Sink reactions
Reactions that serve as an end output for the model. Examples include metabolite efflux (lactate secretion) or intracellular sinks (glycogen storage)
- Source reactions
Reactions that generate metabolites that are only consumed but not produced by other reactions in the model. Examples include glucose or amino acid extracellular uptake
- Sum of squared residuals (SSR)
SSR is the sum of the magnitude of difference between model estimates and data measurements. In iMFA, residuals (deviations between measured and simulated isotopologues) are calculated for each metabolite and then summed to obtain the SSR
- Tandem mass-spectrometry (tandem MS)
Tandem MS uses multiple mass detectors in tandem to distinguish isotopologues with similar m/z ratios. After molecules are fragmented in the first MS, they metabolites are further fragmented in a second MS. These “fragments of fragments” provide position specific labeling information, improved sensitivity, and broader coverage
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
None are declared by the authors.
References
- 1.Jang C et al. (2018) Metabolomics and Isotope Tracing. Cell 173, 822–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suthers PF et al. (2007) Metabolic flux elucidation for large-scale models using 13C labeled isotopes. Metab Eng 9, 387–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nielsen J (2003) It Is All about Metabolic Fluxes. J Bacteriol 185, 7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buescher JM et al. (2015) A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol 34, 189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antoniewicz MR (2015) Methods and advances in metabolic flux analysis: a mini-review. J Ind Microbiol Biotechnol 42, 317–325 [DOI] [PubMed] [Google Scholar]
- 6.Dai Z and Locasale JW (2017) Understanding metabolism with flux analysis: From theory to application. Metab Eng 43, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlsen E et al. (2018) Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinformatics 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanehisa M et al. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36, D480–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noronha A et al. (2018) The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res 47, D614–D624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caspi R et al. (2020) The MetaCyc database of metabolic pathways and enzymes-a 2019 update. Nucleic Acids Res 48, D455–D453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caspi R et al. (2017) The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res 46, D633–D639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orman MA et al. (2012) Stoichiometry based steady-state hepatic flux analysis: Computational and experimental aspects Metabolites, 2MDPI AG, 268–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiback SJ and Palsson BO (2002) Extreme pathway analysis of human red blood cell metabolism. Biophys J 83, 808–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang ZW et al. (2011) Red blood cell extrudes nucleus and mitochondria against oxidative stress. IUBMB Life 63, 560–565 [DOI] [PubMed] [Google Scholar]
- 15.Lewis CA et al. (2014) Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol Cell 55, 253–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metallo CM et al. (2009) Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J Biotechnol 144, 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nargund S and Sriram G (2013) Designer labels for plant metabolism: Statistical design of isotope labeling experiments for improved quantification of flux in complex plant metabolic networks. Mol Biosyst 9, 99–112 [DOI] [PubMed] [Google Scholar]
- 18.Nöh K et al. (2018) A Pareto approach to resolve the conflict between information gain and experimental costs: Multiple-criteria design of carbon labeling experiments. PLoS Comput Biol 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergman T et al. (1986) Amino Acid Analysis by High Performance Liquid Chromatography of Phenylthiocarbamyl Derivatives BT - Advanced Methods in Protein Microsequence Analysis. In (Wittmann-Liebold B et al., eds), pp. 45–55, Springer Berlin Heidelberg [Google Scholar]
- 20.Sherwood RA (1990) Amino acid measurement by high-performance liquid chromatography using electrochemical detection. J Neurosci Methods 34, 17–22 [DOI] [PubMed] [Google Scholar]
- 21.Ling Z et al. (2016) Sensitive determination of glucose in Dulbecco’s modified Eagle medium by high-performance liquid chromatography with 1-phenyl-3-methyl-5-pyrazolone derivatization: application to gluconeogenesis studies. Biomed Chromatogr 30, 601–605 [DOI] [PubMed] [Google Scholar]
- 22.Robitaille L and Hoffer LJ (1988) Measurement of branched chain amino acids in blood plasma by high performance liquid chromatography. Can J Physiol Pharmacol 66, 613–617 [DOI] [PubMed] [Google Scholar]
- 23.Zhu W et al. (2011) Quantitative profiling of tryptophan metabolites in serum, urine, and cell culture supernatants by liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 401, 3249–3261 [DOI] [PubMed] [Google Scholar]
- 24.Lau SKP et al. (2015) Identification of specific metabolites in culture supernatant of Mycobacterium tuberculosis using metabolomics: exploration of potential biomarkers. Emerg Microbes Infect 4, e6–e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moiz B et al. (2021) (13)C Metabolic Flux Analysis Indicates Endothelial Cells Attenuate Metabolic Perturbations by Modulating TCA Activity. Metabolites 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee WD et al. (2019) Spatial-fluxomics provides a subcellular-compartmentalized view of reductive glutamine metabolism in cancer cells. Nat Commun 10, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lugar DJ and Sriram G (2022) Isotope-assisted metabolic flux analysis as an equality-constrained nonlinear program for improved scalability and robustness. PLoS Comput Biol 18, e1009831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Young JD (2014) INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30, 1333–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahim M et al. (2022) INCA 2.0: A tool for integrated, dynamic modeling of NMR- and MS-based isotopomer measurements and rigorous metabolic flux analysis. Metab Eng 69, 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kajihata S et al. (2014) OpenMebius: an open source software for isotopically nonstationary 13C-based metabolic flux analysis. Biomed Res Int 2014, 627014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weitzel M et al. (2013) 13CFLUX2--high-performance software suite for (13)C-metabolic flux analysis. Bioinformatics 29, 143–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nargund S and Sriram G (2014) Mathematical modeling of isotope labeling experiments for metabolic flux analysis. Methods in Molecular Biology 1083, 109–131 [DOI] [PubMed] [Google Scholar]
- 33.Antoniewicz MR et al. (2006) Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab Eng 8, 324–337 [DOI] [PubMed] [Google Scholar]
- 34.Quek LE et al. (2020) Dynamic 13C Flux Analysis Captures the Reorganization of Adipocyte Glucose Metabolism in Response to Insulin. iScience 23, 100855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z et al. (2020) Parallel isotope differential modeling for instationary 13C fluxomics at the genome scale. Biotechnol Biofuels 13, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ando D and Garcia MH (2018) Two-Scale 13C Metabolic Flux Analysis for Metabolic Engineering. Methods in Molecular Biology 1671, 170–179 [DOI] [PubMed] [Google Scholar]
- 37.Foguet C et al. (2019) p13CMFA: Parsimonious 13C metabolic flux analysis. PLoS Comput Biol 15, e1007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jenior ML et al. (2020) Transcriptome-guided parsimonious flux analysis improves predictions with metabolic networks in complex environments. PLoS Comput Biol 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y et al. (2020) Metabolic Flux Analysis—Linking Isotope Labeling and Metabolic Fluxes. Metabolites 10, 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long CP and Antoniewicz MR (2019) High-resolution C metabolic flux analysis. 14 [DOI] [PubMed] [Google Scholar]
- 41.Kumar A et al. (2020) Escher-Trace: A web application for pathway-based visualization of stable isotope tracing data. BMC Bioinformatics 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicolae A et al. (2014) Non-stationary 13C metabolic flux analysis of Chinese hamster ovary cells in batch culture using extracellular labeling highlights metabolic reversibility and compartmentation. BMC Syst Biol 8, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasenour CM et al. (2015) Mass spectrometry-based microassay of 2H and 13C plasma glucose labeling to quantify liver metabolic fluxes in vivo. American Journal of Physiology-Endocrinology and Metabolism 309, E191–E203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crown SB et al. (2016) Comprehensive metabolic modeling of multiple13C-isotopomer data sets to study metabolism in perfused working hearts. Am J Physiol Heart Circ Physiol 311, H881–H891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sake CL et al. (2022) Isotopically nonstationary 13C metabolic flux analysis in resting and activated human platelets. Metab Eng 69, 313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Correia C et al. (2017) 3D aggregate culture improves metabolic maturation of human pluripotent stem cell derived cardiomyocytes. Biotechnol Bioeng 115, 630–644 [DOI] [PubMed] [Google Scholar]
- 47.Sá JV et al. (2017) Quantification of Metabolic Rearrangements During Neural Stem Cells Differentiation into Astrocytes by Metabolic Flux Analysis. Neurochem Res 42, 244–253 [DOI] [PubMed] [Google Scholar]
- 48.Wang Z et al. (2020) Chronic cold exposure enhances glucose oxidation in brown adipose tissue. EMBO Rep 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Held NM et al. (2018) Pyruvate dehydrogenase complex plays a central role in brown adipocyte energy expenditure and fuel utilization during short-term beta-adrenergic activation. Sci Rep 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Araki C et al. (2018) Mass Spectrometry-Based Method to Study Inhibitor-Induced Metabolic Redirection in the Central Metabolism of Cancer Cells. Mass Spectrometry 7, A0067–A0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carinhas N et al. (2017) 13C-metabolic flux analysis of human adenovirus infection: Implications for viral vector production. Biotechnol Bioeng 114, 195–207 [DOI] [PubMed] [Google Scholar]
- 52.Mackenzie JS et al. (2020) Bedaquiline reprograms central metabolism to reveal glycolytic vulnerability in Mycobacterium tuberculosis. Nat Commun 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parker SJ et al. (2017) LKB1 promotes metabolic flexibility in response to energy stress. Metab Eng 43, 208–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Araki C et al. (2018) Mass Spectrometry-Based Method to Study Inhibitor-Induced Metabolic Redirection in the Central Metabolism of Cancer Cells. Mass Spectrom (Tokyo) 7, A0067–A0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noguchi S et al. (2020) Direct and quantitative analysis of altered metabolic flux distributions and cellular ATP production pathway in fumarate hydratase-diminished cells. Sci Rep 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lesner NP et al. (2020) α-ketobutyrate links alterations in cystine metabolism to glucose oxidation in mtDNA mutant cells. Metab Eng 60, 157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Odenwelder DC et al. (2021) Induced pluripotent stem cells can utilize lactate as a metabolic substrate to support proliferation. Biotechnol Prog 37, e3090. [DOI] [PubMed] [Google Scholar]
- 58.Kumar A et al. (2021) NaCT/SLC13A5 facilitates citrate import and metabolism under nutrient-limited conditions. Cell Rep 36, 109701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rahman SMJ et al. (2016) The airway epithelium undergoes metabolic reprogramming in individuals at high risk for lung cancer. JCI Insight 1, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Britt EC et al. (2022) Switching to the cyclic pentose phosphate pathway powers the oxidative burst in activated neutrophils. Nat Metab 4, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiechert W (2007) The Thermodynamic Meaning of Metabolic Exchange Fluxes. Biophys J 93, 2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alves TC et al. (2015) Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab 22, 936–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cappel DA et al. (2019) Pyruvate-Carboxylase-Mediated Anaplerosis Promotes Antioxidant Capacity by Sustaining TCA Cycle and Redox Metabolism in Liver. Cell Metab 29, 1291–1305.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang L et al. (2017) Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein. Metab Eng 43, 198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ahn WS et al. (2016) Evidence for transketolase-like TKTL1 flux in CHO cells based on parallel labeling experiments and 13C-metabolic flux analysis. Metab Eng 37, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gebreselassie NA and Antoniewicz MR (2015) 13C-metabolic flux analysis of co-cultures: A novel approach. Metab Eng 31, 132–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Achreja A et al. (2017) Exo-MFA – A 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab Eng 43, 156–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fernández-García J et al. (2020) Stable Isotopes for Tracing Mammalian-Cell Metabolism In Vivo. Trends Biochem Sci 45, 185–201 [DOI] [PubMed] [Google Scholar]
- 69.Hughey CC et al. (2017) Loss of hepatic AMP-activated protein kinase impedes the rate of glycogenolysis but not gluconeogenic fluxes in exercising mice. J Biol Chem 292, 20125–20140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rahim M et al. (2021) Multitissue 2H/13C flux analysis reveals reciprocal upregulation of renal gluconeogenesis in hepatic PEPCK-C–knockout mice. JCI Insight 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deja S et al. (2021) In vivo estimation of ketogenesis using metabolic flux analysis-Technical aspects and model interpretation. Metabolites 11, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moiz B et al. (2022) Isotope-Assisted Metabolic Flux Analysis: A Powerful Technique to Gain New Insights into the Human Metabolome in Health and Disease. Metabolites 12, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bednarski TK et al. (2021) In vivo 2H/13C flux analysis in metabolism research. Curr Opin Biotechnol 71, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fernández-García J et al. (2020) Stable Isotopes for Tracing Mammalian-Cell Metabolism In Vivo. Trends Biochem Sci 45, 185–201 [DOI] [PubMed] [Google Scholar]
- 75.Williams HC et al. (2020) Oral Gavage Delivery of Stable Isotope Tracer for In Vivo Metabolomics. Metabolites 10, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun RC et al. (2017) Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nature Communications 2017 8:1 8, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hasenour CM et al. (2015) Mass spectrometry-based microassay of 2H and 13C plasma glucose labeling to quantify liver metabolic fluxes in vivo. American Journal of Physiology-Endocrinology and Metabolism 309, E191–E203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Walther JL et al. (2012) Optimization of 13C isotopic tracers for metabolic flux analysis in mammalian cells. Metab Eng 14, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Basler G and Fernie AR (2018) Advances in metabolic flux analysis toward genome-scale profiling of higher organisms. 0, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martínez VS et al. (2015) Dynamic metabolic flux analysis using B-splines to study the effects of temperature shift on CHO cell metabolism. Metab Eng Commun 2, 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]