

Abstract
Purpose:
Resistance to endocrine therapy is the primary cause of treatment failure and death in patients with ER-positive (ER+)/luminal breast cancer. Expression and activation of the RET receptor tyrosine kinase may be driving poor outcomes. We aim to identify high-risk patients and druggable pathways for biomarker-based clinical trials.
Methods:
We obtained batch-normalized mRNA expression data from Breast Invasive Carcinoma - The Cancer Genome Atlas, PanCancer Atlas (BRCA-TCGA). To determine clinically significant cutoffs for RET expression, patients were grouped at different thresholds for Kaplan-Meier plotting. Differential gene expression (DGE) analysis and enrichment for gene sets was performed. transcriptomic dataset of antiestrogen-treated ER+ tumors stratified by clinical response was then analyzed.
Results:
High RET expression was associated with worse outcomes in patients with ER+ tumors, and stratification was enhanced by incorporating GDNF expression. High RET/GDNF patients had significantly lower overall survival (HR=2.04, p=0.012), progression-free survival (HR=2.87, p<0.001), disease-free survival (HR=2.67, p<0.001), and disease-specific survival (HR=3.53, p<0.001) than all other ER+ patients. High RET/GDNF tumors were enriched for estrogen-independent signaling and targetable pathways including NTRK, PI3K, and KRAS. Tumors with adaptive resistance to endocrine therapy were enriched for gene expression signatures of high RET/GDNF primary tumors.
Conclusion:
Expression and activation of the RET receptor tyrosine kinase may be driving poor outcomes in some patients with ER+ breast cancer. ER+ patients above the 75th percentile may benefit from clinical trials with tyrosine kinase inhibitors.
Keywords: Breast cancer, RET, GDNF, MAPK, estrogen receptor signaling
INTRODUCTION
Breast cancer is the most prevalent cancer and the second leading cause of cancer-related death in women globally[1,2]. Molecular subtypes have been identified with distinct transcriptomic profiles, treatment responses, and outcomes[3]. Approximately 80% of breast tumors are categorized within the luminal subtypes characterized by expression of the estrogen (ER) and progesterone receptors (PR)[3,4]. ER+/Luminal tumors have a relatively favorable prognosis compared to other subtypes[3,4]. However, roughly 30% of patients with ER+ tumors are innately resistant or develop resistance to endocrine therapy[5]. Endocrine resistance is a significant challenge in the treatment of ER+ breast cancer and various studies have been conducted to understand underlying mechanisms of resistance[4,6,7]. Multiple signaling pathways and modulators have been identified contributing to resistance including gain of function mutations in ESR1 encoding the estrogen receptor, dysregulation of MAPK signaling, and upregulation of receptor tyrosine kinases (RTKs)[1,6,8]. Nevertheless, specific drivers of endocrine resistance remain unidentified for the majority of ER+ breast cancer patients[6].
Several previous studies have established a connection between the RET RTK and response to endocrine therapy. RET activity results in downstream phosphorylation of ER when stimulated by its ligand glial cell derived neurotrophic factor (GDNF)[8,9] which leads to estrogen independent activation of estrogen response elements. Additionally, our prior work has demonstrated that RET is a significant modulator of ERK/MAPK activation and that genetic or pharmacologic inhibition of RET can enhance sensitivity to tamoxifen in ER+ breast cancer cell lines[9,10]. We hypothesize that a high expression of RET in human ER+ tumors is associated with poor outcomes and that RET activity may differentiate a distinct biological subset of ER+ tumors with specific gene expression patterns. Unique features of RET active tumors will identify biomarkers of patients at high risk for recurrence for future RET inhibitor trials.
METHODS
cBioPortal was used to access Breast Invasive Carcinoma The Cancer Genome Atlas, PanCancer Atlas (BRCA-TCGA) clinical and genomic data[11,12]. Gene expression data was obtained using batch-normalized mRNA expression RSEM data. Clinical and sample information was compiled from the clinical attributes and sample attributes files. Tumors were assigned PAM50 intrinsic subtype using a normalized centroid predictor[13]. 1,081 breast cancer patients with RSEM and clinical data were identified from BRCA-TCGA. Statistical analysis was performed using RStudio.
To analyze the distribution of gene expression across PAM50 subtypes and ER status, boxplots were generated using ggplot2, ggbreak, ggpubr, and scales R packages, and statistical significance was determined by student’s t-tests between groups as well as log2 fold changes (LogFC) in specific gene expression between groups of interest. Patients with missing data or indeterminate ER status were omitted from the analysis. GDNF expression was identified using established aliases[14,15].
To determine clinically significant cutoffs for differential gene expression analysis (DGE), patients were grouped and Kaplan-Meier (KM) plots were plotted for overall survival (OS), progression-free survival (PFS), disease-free survival (DFS) and disease-specific survival (DSS) using the survival[16] and survminer[17] packages. We systematically investigated various thresholds to determine an optimal RET expression threshold for high or low-expression groups.
For DGE, genes with less than 10 counts across all patients were omitted before analysis of RSEM data using the DESeq2[18] R package. Significant differentially expressed genes were grouped by regulation status using a LogFC cutoff of 1 for upregulation and −1 for downregulation, along with a false discovery rate (FDR) < 0.05. Genes with FDR < 0.05 but not meeting the upregulation or downregulation cutoff were indicated as increased or reduced in expression. Volcano plots of all genes were plotted using the EnhancedVolcano package and a heatmap plotted using the ComplexHeatmap[19] package. Enrichment of gene sets was then investigated through hypergeometric analysis of upregulated or downregulated elements using clusterProfiler[20]. Enrichment analysis was then performed using MSigDB[21] sets and gene ratios were computed as the number of genes enriched relative to the total number of genes from the specified set. Enriched sets were grouped by categories of interest and plotted using ggplot2[22]. Specific genes from enriched gene sets were then investigated through a heatmap using the ggplot2[22] package. Notable, enriched pathways were then analyzed through networks to map LogFC values of interacting genes using Cytoscape[23].
To determine changes in RET and associated gene expression with endocrine resistance, we obtained gene expression data from Xia et al.[7]. In that study, mRNA sequencing was performed from tumor samples while patients were taking endocrine therapy and annotated with clinical response to therapy (i.e. sensitivity and resistance). Gene counts were normalized to the upper quartile using NOISeq[24] and counts were log2 transformed. Scores for gene sets of interest were computed across each sample using singscore[25]. Boxplots were generated for RET and GDNF expression and gene set scores from TCGA analyses across resistant and sensitive samples. Scatter plots of RET and GDNF expression levels and bar plots of response type by RET and GDNF co-expression status were also generated using ggplot2[22].
AVAILABILITY OF DATA AND MATERIALS
The datasets and codes supporting the conclusions of this article are available in the GitHub repository, www.github.com/rashatk/HighRETGDNF.
RESULTS
RET Expression Across the Landscape of Human Breast Cancer
We used the TCGA transcriptional data to investigate expression of RET across intrinsic (PAM50) subtypes to determine how RET expression is associated with phenotypes RET expression was enriched in luminal and HER2-enriched subtypes compared to basal, claudin-low, and normal-like (p<0.001) (Figure 1a). Luminal B tumors had the highest median RET expression, (LumB vs Basal LogFC=4.20, p<0.001). Expression of RET in Luminal A tumors was significantly lower than Luminal B (LumA vs LumB LogFC=−0.44, p= 0.005) (Figure 1a). RET expression strongly correlated with clinical ER status, with the 794 ER+ patients having significantly higher RET expression compared to ER- (LogFC=2.90, p<0.001) (Figure 1b).
Fig. 1.
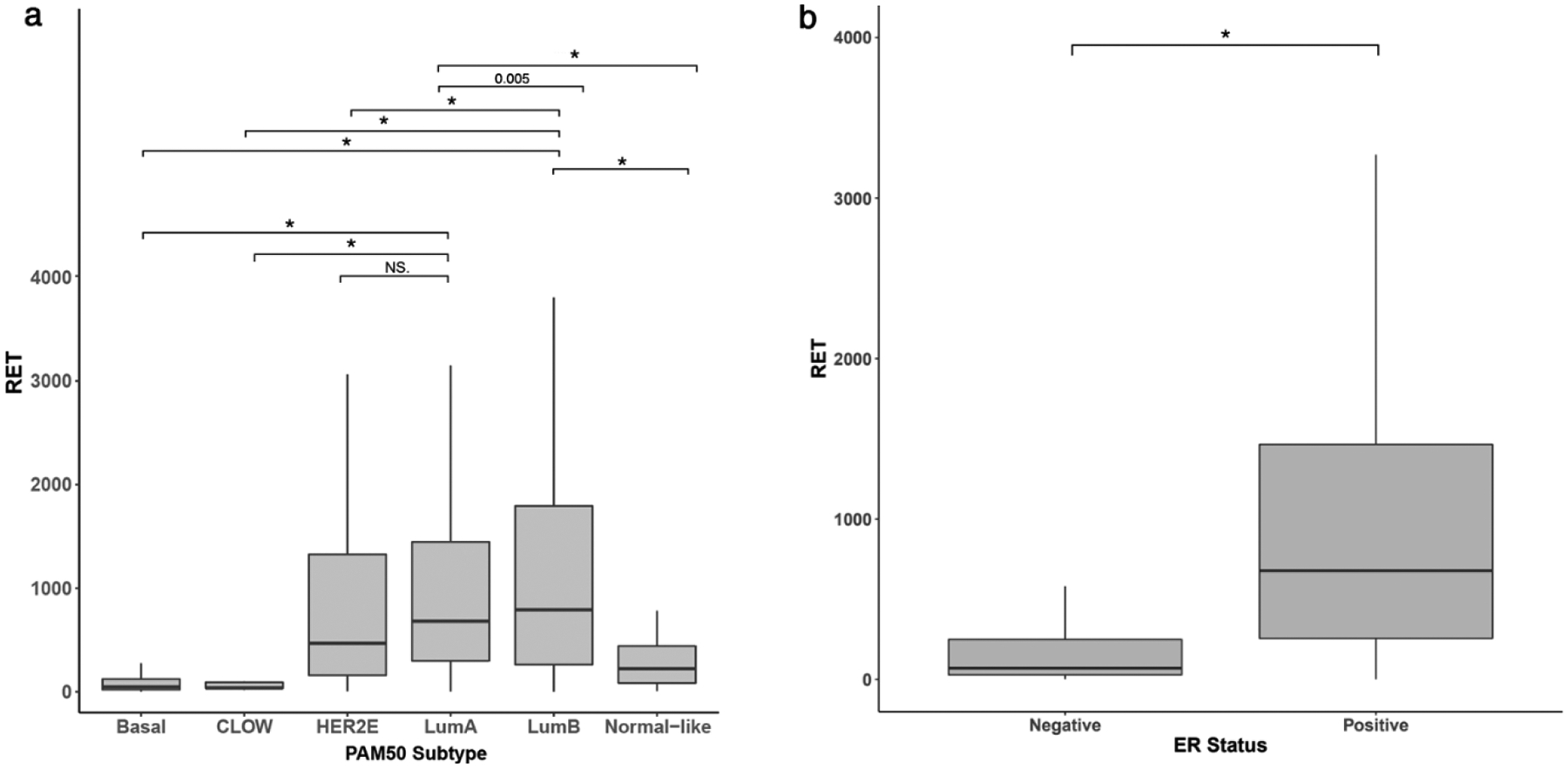
Distribution of RET gene counts in TCGA human breast tumors stratified by (A) PAM50 subtypes and (B) ER status (NS: Not significant, * p< 0.001).
RET Expression and Outcomes in ER+ Breast Tumors
ER positivity is widely clinically available and used for treatment decisions, so we analyzed the impact of RET expression on cancer outcomes in patients with ER+ breast cancer. We employed a systematic analysis based on the 10th, 25th, 50th, 75th and 90th percentiles to determine a biologically relevant threshold of RET expression that could function as a biomarker of worse outcome. RET expression above the 75th percentile (high RET) demonstrated the most robust stratification of outcomes. Kaplan-Meier (KM) plots demonstrated significantly lower progression-free survival (PFS) (HR=1.66, p=0.018), disease-free survival (DSS) (HR=1.77, p=0.049), and disease-specific survival (DSS) (HR=2.13, p=0.006) for high RET patients (Figure 2a). There was additionally a trend towards worse overall survival (OS) in high RET patients, (HR=1.38, p=0.13) (Figure 2a).
Fig. 2.
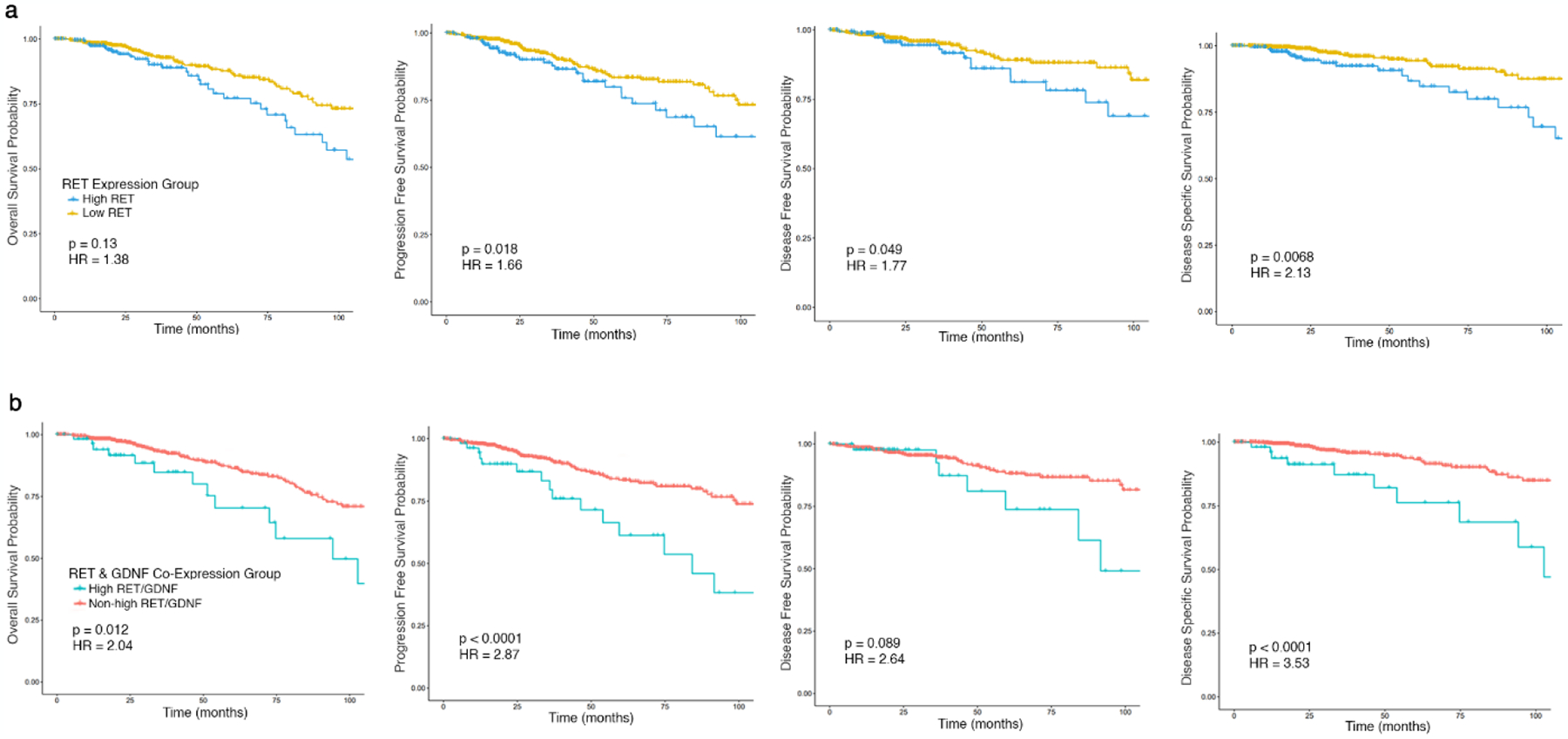
Kaplan-Meier survival plots comparing ER+ patients (A) above the upper quartile of RET expression (high RET) relative to those below (low RET) or (B) above the upper quartile of both RET and GDNF expression (high RET/GDNF) relative to all others (non-high RET/GDNF) by overall survival, progression-free survival, disease-free survival, and disease-specific survival.
Impact of RET-GDNF Co-expression on Survival in ER+ Patients
Because RET expression may not be sufficient for activity of the RET receptor and downstream signaling pathways, we investigated co-expression of RET with its activating ligand GDNF. We utilized the previously identified 75th percentile threshold for RET expression and stratified tumors based on both RET and GDNF expression above the 75th percentile (high RET/GDNF) compared to all other ER+ tumors. Fifty-six of 739 ER+ tumors (7.6%) were in the high RET/GDNF group. KM plots demonstrated that high RET/GDNF patients had significantly lower OS (HR=2.04, p=0.012), PFS (HR=2.87, p<0.001), DFS (HR=2.67, p<0.001), and DSS (HR=3.53, p<0.001) than all other ER+ patients (Figure 2b). Co-expression of GDNF and RET demonstrated more robust stratification of outcomes compared to RET expression alone.
We compared features of patients with high RET/GDNF to all other ER+ patients (Table 1). High RET/GDNF tumors were more common among white women (p=0.019) and tumors with ductal histology (p<0.001). Notably, there were no significant differences in age, stage, or PAM50 subtype, which imply that unique biology, rather than previously identified prognostic factors are driving poor outcomes.
Table 1.
Demographic and clinical features across ER+ patients with high RET/GDNF relative to all other ER+ patients.
| Characteristic | Overall ER+ N = 7941 |
High RET/GDNF N = 561 |
Non-high RET/GDNF N = 7381 |
p-value2 |
|---|---|---|---|---|
| Age | 59.29 (13.28) | 60.32 (14.17) | 59.21 (13.22) | 0.574 |
| Age Group | 0.8 | |||
| <30 | 7 (0.9%) | 1 (1.8%) | 6 (0.8%) | |
| 30–39 | 43 (5.4%) | 3 (5.4%) | 40 (5.4%) | |
| 40–49 | 148 (19%) | 8 (14%) | 140 (19%) | |
| 50–59 | 201 (25%) | 13 (23%) | 188 (25%) | |
| 60–69 | 215 (27%) | 16 (29%) | 199 (27%) | |
| Missing | 180 (23%) | 15 (27%) | 165 (22%) | |
| Sex | >0.9 | |||
| Female | 782 (98%) | 56 (100%) | 726 (98%) | |
| Male | 12 (1.5%) | 0 (0%) | 12 (1.6%) | |
| Race | 0.019* | |||
| Asian | 37 (4.7%) | 2 (3.6%) | 35 (4.7%) | |
| Black or African American | 109 (14%) | 1 (1.8%) | 108 (15%) | |
| White | 569 (72%) | 48 (86%) | 521 (71%) | |
| Missing | 79 (9.9%) | 5 (8.9%) | 74 (10%) | |
| Ethnicity | 0.5 | |||
| Hispanic or Latino | 29 (3.7%) | 2 (3.6%) | 27 (3.7%) | |
| Not Hispanic Or Latino | 621 (78%) | 47 (84%) | 574 (78%) | |
| Missing | 144 (18%) | 7 (12%) | 137 (19%) | |
| Histology | <0.001* | |||
| Infiltrating ductal carcinoma | 526 (66%) | 42 (75%) | 484 (66%) | |
| Infiltrating ductal carcinoma mixed with other types | 39 (4.9%) | 6 (11%) | 33 (4.5%) | |
| Lobular carcinoma | 184 (23%) | 3 (5.4%) | 181 (25%) | |
| Mucinous adenocarcinoma | 15 (1.9%) | 4 (7.1%) | 11 (1.5%) | |
| Other | 18 (2.3%) | 1 (1.8%) | 17 (2.3%) | |
| Missing | 12 (1.5%) | 0 (0%) | 12 (1.6%) | |
| T Stage | 0.093 | |||
| T1 | 209 (26%) | 10 (18%) | 199 (27%) | |
| T2 | 447 (56%) | 32 (57%) | 415 (56%) | |
| T3 | 111 (14%) | 9 (16%) | 102 (14%) | |
| T4 | 25 (3.1%) | 5 (8.9%) | 20 (2.7%) | |
| Missing | 2 (0.3%) | 0 (0%) | 2 (0.3%) | |
| N Stage | 0.2 | |||
| N0 | 351 (44%) | 18 (32%) | 333 (45%) | |
| N1 | 277 (35%) | 28 (50%) | 249 (34%) | |
| N2 | 89 (11%) | 6 (11%) | 83 (11%) | |
| N3 | 59 (7.4%) | 3 (5.4%) | 56 (7.6%) | |
| Missing | 18 (2.3%) | 1 (1.8%) | 17 (2.3%) | |
| M Stage | 0.3 | |||
| M0 | 643 (81%) | 50 (89%) | 593 (80%) | |
| M1 | 15 (1.9%) | 0 (0%) | 15 (2.0%) | |
| Missing | 136 (17%) | 6 (11%) | 130 (18%) | |
| AJCC Pathological Stage | 0.6 | |||
| Stage I | 139 (18%) | 7 (12%) | 132 (18%) | |
| Stage II | 431 (54%) | 30 (54%) | 401 (54%) | |
| Stage III | 191 (24%) | 18 (32%) | 173 (23%) | |
| Stage IV | 14 (1.8%) | 0 (0%) | 14 (1.9%) | |
| Missing | 19 (2.4%) | 1 (1.8%) | 18 (2.4%) | |
| PR Status | 0.2 | |||
| Positive | 668 (84%) | 48 (86%) | 620 (84%) | |
| Negative | 122 (15%) | 7 (12%) | 115 (16%) | |
| Indeterminate | 3 (0.4%) | 1 (1.8%) | 2 (0.3%) | |
| Missing | 1 | 0 | 1 | |
| HER2 Status | 0.4 | |||
| Positive | 120 (15%) | 9 (16%) | 111 (15%) | |
| Negative | 659 (83%) | 45 (80%) | 614 (83%) | |
| Indeterminate | 14 (1.8%) | 2 (3.6%) | 12 (1.6%) | |
| Missing | 1 | 0 | 1 | |
| PAM50 Subtype | 0.3 | |||
| Luminal A | 525 (66%) | 34 (61%) | 491 (67%) | |
| Luminal B | 196 (25%) | 20 (36%) | 176 (24%) | |
| HER2-Enriched | 29 (3.7%) | 2 (3.6%) | 27 (3.7%) | |
| Basal | 20 (2.5%) | 0 (0%) | 20 (2.7%) | |
| Claudin-Low | 1 (0.1%) | 0 (0%) | 1 (0.1%) | |
| Normal-Like | 23 (2.9%) | 0 (0%) | 23 (3.1%) |
n (%), mean (sd)
Fisher’s exact test; Pearson’s Chi-squared test
Statistically significant
Gene Expression Patterns of High RET & GDNF ER+ Breast Tumors
Differential gene expression (DGE) analysis comparing high RET/GDNF tumors to the remaining ER+ tumors revealed 195 significantly upregulated and 945 downregulated genes (Figure 3a). The top 40 upregulated and top 40 downregulated genes were visualized using a heatmap (Figure 3b).
Fig. 3.
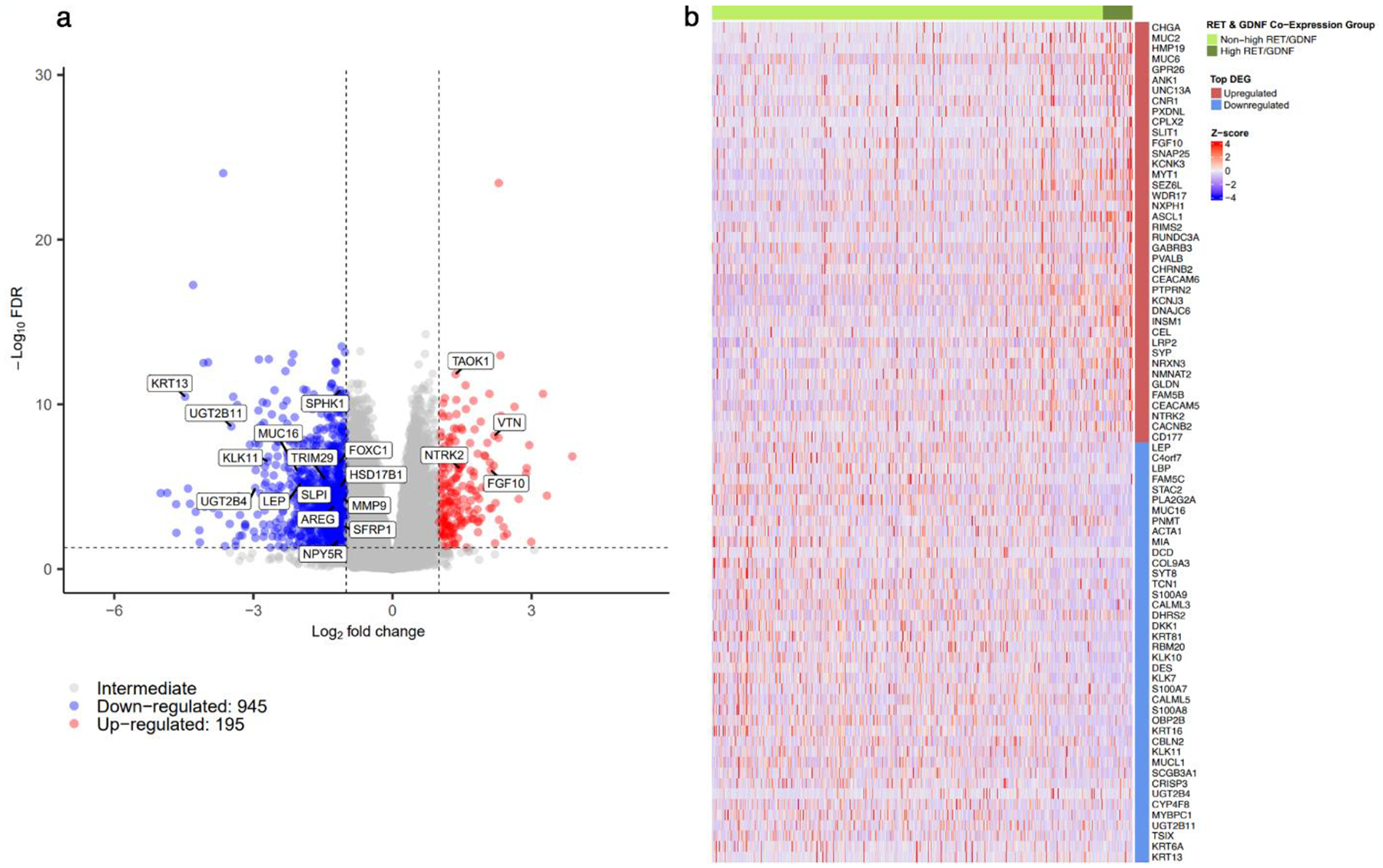
Differential expression analysis of ER+ patients above the 75th percentile of RET and GDNF expression (high RET/GDNF) relative to all other ER+ patients demonstrated by a (A) volcano plot of all genes with significantly upregulated and downregulated genes having L2FC > 1 and < −1 respectively along with FDR < 0.05 and (B) heatmap of top 40 significant upregulated and downregulated differentially expressed genes defining high RET/GDNF ER+ tumors.
Multiple genes with known roles in estrogen signaling were altered in high RET/GDNF tumors. These include FGF10 (LogFC=2.124, FDR<0.001), an FGFR2 ligand associated with reduced estrogen dependence, resistance to endocrine therapy, and poor prognosis in ER+ breast cancer.[26]. KCNK5 which promotes estrogen-dependent proliferation, is reduced (LogFC=−0.978, FDR<0.001)[27,28]. CCND1, an indirect transcriptional target of ER driving proliferation and tumor aggression, is increased (LogFC=0.559, FDR=0.004) in high RET/GDNF tumors[29].
We then examined genes associated with endocrine resistance. MUC2 has been implicated in poor outcomes with aromatase inhibitor therapy and is upregulated (LogFC=3.331, p<0.001)[30]. We also investigated genes established by Wang et al to have a role in tamoxifen resistance including MAPK1, ESR1 and RANBP2, which is implicated in JNK/c-JUN signaling[31]. Among high RET/GDNF patients, expression of MAPK1 (LogFC=0.377, p<0.001), ESR1 (LogFC=0.642, p=0.001), and RANBP2 (LogFC=0.46, p<0.001) was increased.
Transcription factors including NF-κB and JAK/STAT were also altered. NF-κB inhibitors such as IKBKB are implicated in anti-apoptotic signaling; IKBKB expression is increased (LogFC=0.304, FDR=0.012) and NF-κB levels are reduced (LFC=−0.626, FDR<0.001), suggestive of reduced NF-κB signaling[32]. Further, MUC16 is implicated in JAK/STAT signaling that leads to c-JUN overexpression[32]. MUC16 downregulation (LogFC=−2.054, FDR<0.001) and subsequent increase in c-JUN expression (LogFC=−0.532, FDR=0.001) also suggest reduced signaling. Downregulated NPY5R (LogFC=−1.171, FDR=0.020), which induces breast tumor apoptosis and reduces STAT3 activation, may indicate uncontrolled proliferation mediated by overactivation of STAT3 signaling[33]. SPHK1 expression is induced by TNFA and has been implicated in breast cancer proliferation and metastasis[34]. Reduced SPHK1 expression may thus result from reduced TNFA signaling.
Enriched Signaling Pathways Among ER+ Breast Tumors with High RET & GDNF
To define biologic differences in high RET/GDNF tumors, we analyzed enrichment of key pathway gene sets[21,35]. Gene sets consistent with increased ER signaling were enriched in high RET/GDNF tumors (Figure 4a), as were genes sets of reduced response to estrogen (Figure 4b) and increased antiestrogen resistance (Figure 4c). We compiled a heat plot of notable and significantly altered genes within these sets (Figure 4d), which identified several important genes. FOXC1 has been shown to repress expression of ESR1; and downregulation of FOXC1 (LogFC=−1.161, FDR<0.001) was seen in high RET/GDNF tumors[36]. HSD17B1 promotes conversion of estrone into estrogen in the final step of estrogen synthesis[37], so downregulated HSD17B1 (LogFC=−1.117, FDR<0.001) may indicate divergence towards estrogen-independent ER activity. Similarly, LEP is implicated in estrogen production and is thus implicated in estrogen-dependent breast cancer growth[38]. Downregulated LEP (LogFC=−1.974, FDR<0.001) suggests low estrogen synthesis and estrogen independence. Further, TRIM29 is a tumor suppressor in ER+ breast cancer that interacts with ESR1 to reduce binding to estrogen response elements (ERE) that typically results in ER-dependent gene expression[39]. TRIM29 downregulation (LogFC=−1.469, FDR<0.001) in high RET/GDNF tumors may indicate increased ESR1 binding to ERE. Additionally, MYC is an established direct target of ER and is associated with poor survival, tumor stemness, and aggressive phenotype[40], was enriched (Figure 4e).
Fig. 4.
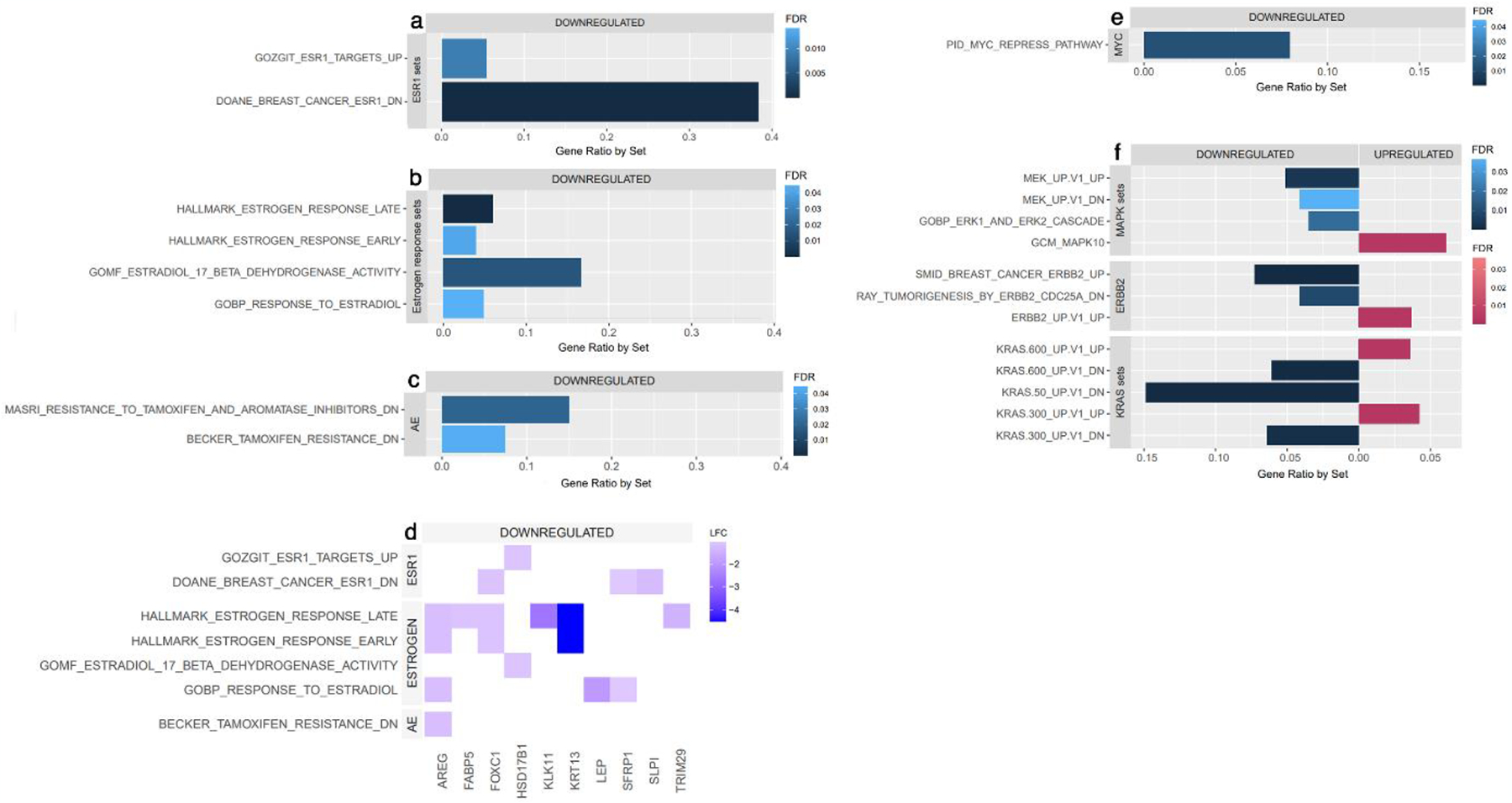
Enriched gene sets among high RET/GDNF tumors relative to non-high RET/GDNF tumors across (A) ESR1, (B) estrogen, and (C) antiestrogen (AE) signaling sets. (D) Heat plot of notable and significantly regulated genes across enriched estrogen, ESR1 and antiestrogen (AE) signaling sets. Enriched gene sets among high RET/GDNF tumors across (E) MYC and (F) MAPK, ERBB2, and KRAS signaling sets.
Because RET is upstream of ERK/MAPK signaling, we defined expression of genes associated with these pathways in high RET/GDNF ER+ tumors. KRAS and MAPK10 signaling genes were enriched (Figure 4f). DUSP proteins dephosphorylates MAPK proteins leading to reduced MAPK signaling activity[41–43]. Increased expression of DUSP7 (LogFC=−0.245, p=0.003) and DUSP4 (LogFC=0.964, FDR<0.001) in high RET/GDNF tumors may activate MAPK. Increased DCLK1 (LogFC=0.922, FDR<0.001) and EFR3B (LogFC=0.816, FDR<0.001) may contribute to increased RAS/ERK activity[44,45]. Expression of PDPK1 which activates AKT is increased (LogFC=0.441, FDR<0.001)[32]. Upstream targets of MAPK and PI3K/AKT signaling were also enriched, including ERBB2 pathway. TAOK1 (LogFC=1.360, FDR<0.001) and ERBB4 (LogFC=0.629, FXR=0.004) which activate MAPK were enriched[46,47]. Expression of AREG, an EGFR ligand, is significantly downregulated (LogFC=−1.254, FDR<0.001)[48]. Further, expression of VEGF and VEGFR, which are also upstream regulators of MAPK and AKT signaling, are downregulated[49]. These findings further indicate RET as the driver of KRAS, MAPK and PI3K/AKT signaling in these tumors.
ER and MAPK Networks in High RET/GDNF ER+ Tumors
We used Cytoscape[23] to define interactions in key pathways, including RAS. In this pathway, GAB1 PTPN11 and RAB5A expression was increased in high RET/GDNF tumors. NTRK2 expression was significantly upregulated (LogFC=1.447, FDR<0.001). NTRK2 has been associated with poor prognosis of invasive breast cancer and represents a druggable target with histology agnostic FDA approval[50–52]. Within PI3K/AKT signaling,, VTN (LogFC=2.197, FDR<0.001) was upregulated in high RET/GDNF tumors[53]. Genes downstream of the classical estrogen-dependent ER transcriptional activity, such as TP53, FGF2, and E2F1, and the nonclassical estrogen-dependent ER transcriptional activity mediated by transcription factors including SP1 and c-JUN such as BCL2, IGF1, and MMP1, are not significantly altered (Figure 5)[54,55]. However, genes downstream of estrogen-independent ER transcriptional activity are significantly altered, with RBBP4, PTPRF, and RAB14 increased in expression[56].
Fig. 5.
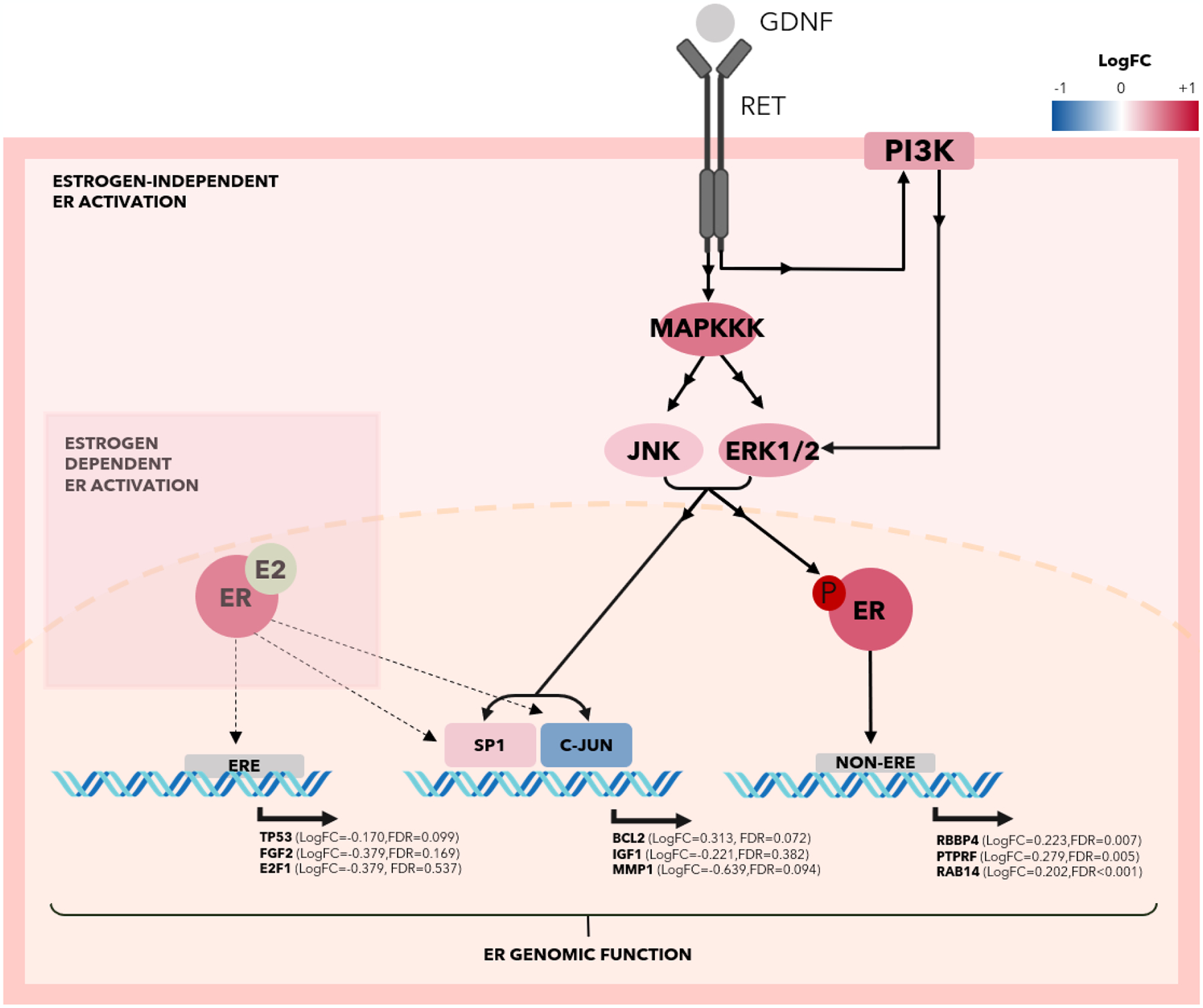
Graphical representation using Cytoscape of RET activation by GDNF driving activation of MAPK pathway and enriching estrogen-independent ER activity.
RET Expression in Endocrine Therapy Resistant ER+ Breast Cancer
Because RET reduces sensitivity to endocrine therapy in vitro, we hypothesized that RET expression would be enriched in ER+ tumors that are resistant to endocrine therapy. To test this hypothesis, we obtained a transcriptomic dataset from patients with unresectable or metastatic ER+ breast cancer treated with endocrine therapy. Serial biopsies were obtained for whole transcriptome profiling with clinical sensitivity annotation[7].
We compared RET and GDNF gene expression stratified by endocrine therapy sensitivity. Although GDNF levels did not significantly differ among endocrine-resistant tumors relative to sensitive tumors, RET gene expression was significantly higher (LogFC=1.025, FDR=0.01) (Figure 6a). RET expression was also significantly higher in adaptive resistance samples relative to all sensitive samples (LogFC=1.113, FDR=0.014) (Figure 6b). To directly assess if RET and GDNF are overexpressed in response to endocrine therapy with emerging resistance, we compared adaptive resistance samples to patient-matched sensitive samples (i.e. sensitive tumors that developed adaptive resistance), which were labeled “patient-matched pre-resistant” (PMP). No differences were observed in RET or GDNF expression between matched specimens (Figure 6c). Resistant samples demonstrated higher RET expression (Figure 6d). Samples were then divided into high RET/GDNF (75th percentile or non-high RET/GDNF. Most sensitive samples were non-high RET/GDNF, whereas the high RET/GDNF samples were enriched for PMP and adaptive resistance samples (Figure 6e).
Fig. 6.
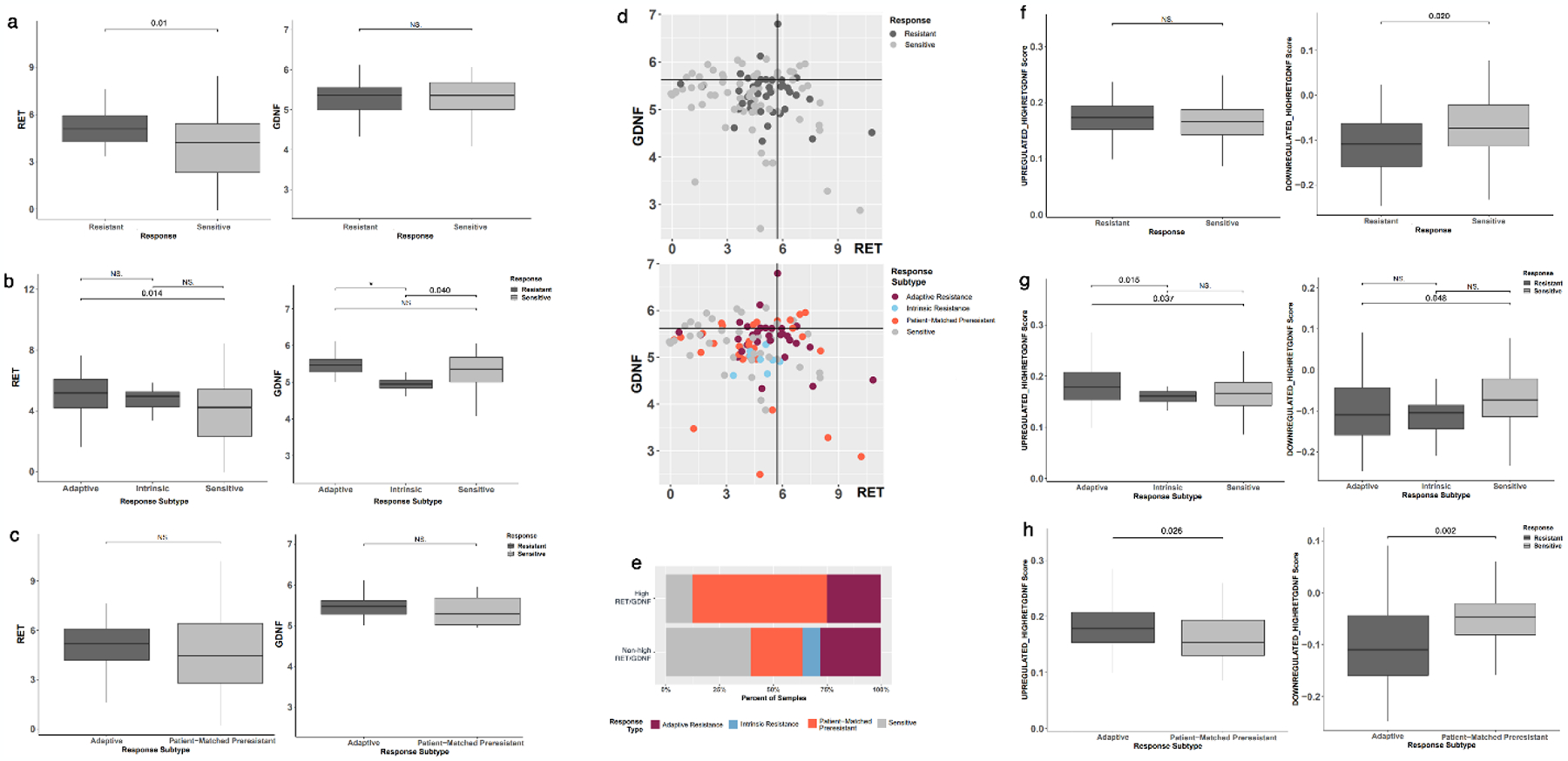
Expression of RET, GDNF, and signatures of high RET/GDNF profile in a gene expression dataset of endocrine therapy-resistant tumors[7]. (A) RET expression is increased in endocrine therapy-resistant tumors compared to sensitive ones with no change in GDNF expression. (B) Adaptive-resistance samples have increased RET expression compared to sensitive samples, with no statistically significant difference between intrinsic resistant and sensitive specimens. (C) No change in RET or GDNF expression is seen in adaptive-resistance samples and patient-matched resistant (PMP) samples. (D) Scatter plot of RET and GDNF expression across all samples by the response to endocrine therapy with cutoffs at the 75th percentile of gene expression. (E) Bar plot of response type distribution for samples with high RET/GDNF compared to non-high RET/GDNF samples across all samples. Distribution of enrichment scores for TCGA-derived top 40 upregulated or downregulated gene sets in high RET/GDNF tumors by (F) tumor response to endocrine therapy and (G) adaptive or intrinsic resistance. (H) a comparison between adaptive resistant samples and patient-matched resistant samples demonstrates significant enrichment of upregulated genes and depletion of downregulated genes associated with high RET/GDNF tumors. (NS: Not significant, * p < 0.001).
Gene signatures can be more representative of underlying biology than individual genes, so we characterized the enrichment and depletion of high RET/GDNF gene expression signatures derived from the TCGA analysis. The top 40 upregulated genes and 40 downregulated genes were compiled into gene sets. The upregulated high RET/GDNF signature was not enriched in resistant tumors, but resistant samples were depleted for the downregulated signature (LogFC=−0.037, FDR=0.020) (Figure 6f). Adaptive resistance samples, however, were enriched for the upregulated signature compared to both sensitive (LogFC=0.024, FDR=0.003) and intrinsic resistance (LogFC=0.029, FDR=0.015) samples, as well as significantly lower gene expression for the downregulated gene set relative to sensitive samples (LogFC=−0.035, FDR=0.048) (Figure 6g). Further patient-matched comparison demonstrated significantly higher expression scores of upregulated genes in adaptive resistant samples relative to PMP samples (LogFC=0.032, FDR=0.002), and lower expression of downregulated genes (LogFC=−0.058, FDR=0.002) (Figure 6h). These findings suggest that the development of resistance to endocrine therapy may involve an adaptive response resulting in a gene expression profile characteristic of high RET/GDNF tumors.
DISCUSSION
In this study, we demonstrate a distinct, high-risk subpopulation of ER+ breast cancer characterized by high expression of genes encoding the RET receptor and its activating ligand GDNF with poor survival. High RET/GDNF ER+ tumors have a distinct transcriptional profile that underlies important biologic distinctions with other ER+ breast tumors, and could inform treatment strategies for these patients.
We have previously shown that RET inhibition reduces proliferation in ER+ breast cancer cell lines and xenografts[10,57]. However, clinical trials of tyrosine kinase inhibitors in non-HER2 amplified breast cancer have had disappointing results[58–61] which may be due to lack of biomarkers of patients likely to respond. Supporting this, two recent trials of TKI with anti-RET activity were positive by objective response rate in ER+/HER2-patients[62,63]. Biomarker-based selection of patients will be key to identifying patients most likely to benefit in future clinical trials. The data in this manuscript support the selection of patients above the 75th percentile of both RET and GDNF expression which identifies patients with poor outcomes and with biological evidence of downstream kinase and estrogen signaling pathways, making this an ideal group for future TKI investigation.
We also identify several key enriched features of high RET/GDNF tumors that highlight differences in tumor biology and suggest actionable targets in these high-risk patients. High RET/GDNF drives downstream upregulation of both estrogen-dependent and estrogen-independent ER activity. Interestingly, this appears to be driven by a shift towards estrogen independent ER activity which is associated with increased proliferation, invasion, and metastasis. Because high RET/GDNF tumors have a shift in ER activity towards estrogen-independent ER activity, ER degraders such as fulvestrant may be preferred over agents such as tamoxifen or aromatase inhibitors that act on estrogen-dependent activation of ER. We have identified multiple druggable targets, including NTRK2 and TAOK1 which may demonstrate potential through further investigation to improve outcomes in this population through TKI treatment.
The shift towards ER-independent ER activity may be due to the activation of MAPK pathways downstream of RET. MAPK activation has been shown to drive estrogen-independent activation of estrogen response elements in breast cancer cell lines[64], and activating mutations in MPAK pathway can drive endocrine resistance[6,65]. We found enrichment of MAPK activity signatures in high RET/GDNF ER+ tumors, demonstrating that these pathways are maintained in human breast tumors and may enhance estrogen-independent ER activity (Figure 7). Supporting this, our preoperative window trial using a short treatment of the tyrosine kinase inhibitor vandetanib in primary ER+ tumors results in a reduction in ERK activity[66]. Several inhibitors of ERK/MAPK are currently in clinical trials for breast cancer, and patients with high RET/GDNF ER+ tumors may be the patient population most likely to benefit from these therapies. Further study is needed to determine the biological mechanisms of RET and GDNF dependent reprogramming kinase signaling pathways, estrogen response elements, and effects on emerging endocrine resistance.
Fig. 7.
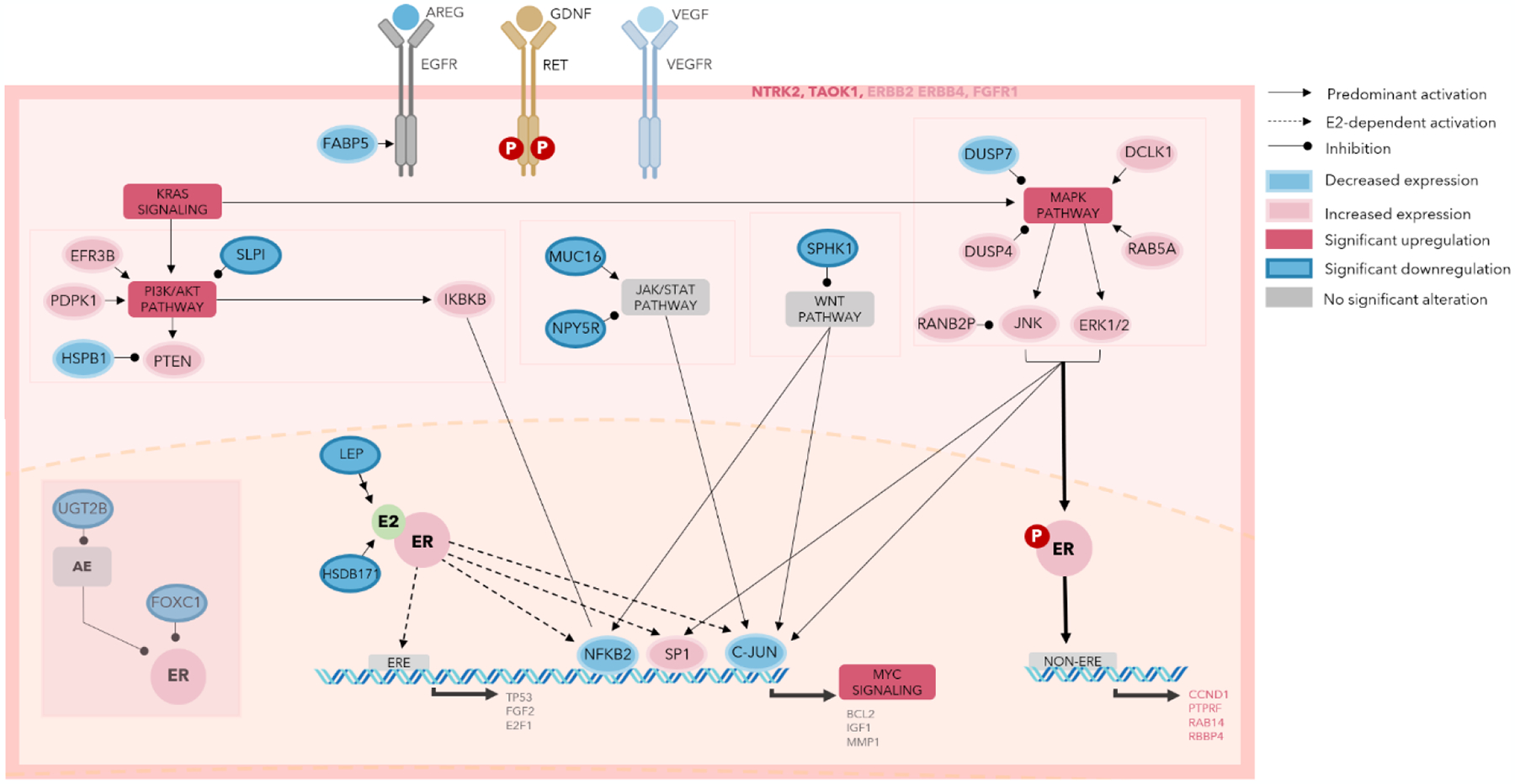
Summary schematic of key receptors, pathways, and interactions in high RET/GDNF tumors.
CONCLUSION
A sub-population of ER+ breast tumors with high RET/GDNF expression confers poor survival which is associated with MAPK activation and estrogen-independent ER activation. Our analysis identifies the top quartile of GDNF and RET co-expression as a candidate biomarker for trials of anti-RET therapeutics. Additionally, we identify several targetable pathways including NTRK, PI3K, and KRAS that could improve outcomes for these high-risk patients. Further study is needed to understand how RET is driving oncogenic pathways in human ER+ tumors and treatment strategies to improve outcomes for patients with RET-driven tumors.
Acknowledgements:
This work was presented, in part, as an oral presentation at the 18th Annual Academic Surgical Congress, February 8, 2023 in Houston, TX.
Funding:
Dr Philip Spanheimer is supported by the National Institutes of Health grant P50CA058223. This work was supported by a UNC Lineberger Comprehensive Cancer Center Developmental Award which is supported in part by P30 CA016086 Cancer Center Core Support Grant.
Footnotes
Competing interests: The authors declare no potential conflicts of interest.
Data availability:
The datasets used and code generated during the current study are available on a public GitHub repository (www.github.com/rashatk/HighRETGDNF). The dataset obtained from Xia et al. can be accessed at doi:10.1158/1078-0432.CCR-21-3189.
REFERENCES
- 1.Rondón-Lagos M, Villegas V, Rangel N, Sánchez M, Zaphiropoulos P. Tamoxifen Resistance: Emerging Molecular Targets. IJMS. 2016. Aug 19;17(8):1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lei S, Zheng R, Zhang S, Wang S, Chen R, Sun K, Zeng H, Zhou J, Wei W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun (Lond). 2021. Nov;41(11):1183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lønning PE, Børresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001. Sep 11;98(19):10869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanker AB, Sudhan DR, Arteaga CL. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell. 2020. Apr;37(4):496–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X ran, Zhang R yan, Gong H, Rugo HS, Chen L bo, Fu Y, Che J wei, Tie J, Shao B, Wan F ling, Kong W yao, Song G hong, Jiang H fang, Xu G bing, Li H ping. Methylome Variation Predicts Exemestane Resistance in Advanced ER + Breast Cancer. Technol Cancer Res Treat. 2020. Jan 1;19:153303381989633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, Cai Y, Bielski CM, Donoghue MTA, Jonsson P, Penson A, Shen R, Pareja F, Kundra R, Middha S, Cheng ML, Zehir A, Kandoth C, Patel R, Huberman K, Smyth LM, Jhaveri K, Modi S, Traina TA, Dang C, Zhang W, Weigelt B, Li BT, Ladanyi M, Hyman DM, Schultz N, Robson ME, Hudis C, Brogi E, Viale A, Norton L, Dickler MN, Berger MF, Iacobuzio-Donahue CA, Chandarlapaty S, Scaltriti M, Reis-Filho JS, Solit DB, Taylor BS, Baselga J. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell. 2018. Sep;34(3):427–438.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia Y, He X, Renshaw L, Martinez-Perez C, Kay C, Gray M, Meehan J, Parker JS, Perou CM, Carey LA, Dixon JM, Turnbull A. Integrated DNA and RNA Sequencing Reveals Drivers of Endocrine Resistance in Estrogen Receptor–Positive Breast Cancer. Clinical Cancer Research. 2022. Aug 15;28(16):3618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plaza-Menacho I, Morandi A, Robertson D, Pancholi S, Drury S, Dowsett M, Martin LA, Isacke CM. Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene. 2010. Aug 19;29(33):4648–57. [DOI] [PubMed] [Google Scholar]
- 9.Spanheimer PM, Cyr AR, Gillum MP, Woodfield GW, Askeland RW, Weigel RJ. Distinct pathways regulated by RET and estrogen receptor in luminal breast cancer demonstrate the biological basis for combination therapy. Ann Surg. 2014. Apr;259(4):793–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spanheimer PM, Park JM, Askeland RW, Kulak MV, Woodfield GW, De Andrade JP, Cyr AR, Sugg SL, Thomas A, Weigel RJ. Inhibition of RET Increases the Efficacy of Antiestrogen and Is a Novel Treatment Strategy for Luminal Breast Cancer. Clinical Cancer Research. 2014. Apr 15;20(8):2115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery. 2012. May 1;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal [Internet]. 2013. Apr 2 [cited 2022 Oct 22];6(269). Available from: https://www.science.org/doi/10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X, Rødland EA, Tibshirani R, Plevritis S. Molecular subtyping for clinically defined breast cancer subgroups. Breast Cancer Res. 2015. Dec;17(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.GDNF Protein Expression Summary [Internet]. The Human Protein Atlas. Available from: https://www.proteinatlas.org/ENSG00000168621-GDNF
- 15.GDNF [Internet]. NCBI Gene Testing Registry. Available from: https://www.ncbi.nlm.nih.gov/gtr/genes/2668/
- 16.Therneau T A Package for Survival Analysis in R [Internet] [Internet]. 2020. Available from: https://CRAN.R-project.org/package=survival [Google Scholar]
- 17.survminer: Survival Analysis and Visualization. [Internet]. Available from: https://rpkgs.datanovia.com/survminer/index.html.
- 18.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014. Dec;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016. Sep 15;32(18):2847–9. [DOI] [PubMed] [Google Scholar]
- 20.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, Fu X, Liu S, Bo X, Yu G. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation. 2021. Aug;2(3):100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005. Oct 25;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wickham H ggplot2: Elegant Graphics for Data Analysis. 2nd ed. 2016. Cham: Springer International Publishing : Imprint: Springer; 2016. 1 p. (Use R!). [Google Scholar]
- 23.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003. Nov;13(11):2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarazona S, Furió-Tarí P, Turrà D, Pietro AD, Nueda MJ, Ferrer A, Conesa A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015. Jul 16;gkv711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foroutan M, Bhuva DD, Lyu R, Horan K, Cursons J, Davis MJ. Single sample scoring of molecular phenotypes. BMC Bioinformatics. 2018. Dec;19(1):404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clayton NS, Grose RP. Emerging Roles of Fibroblast Growth Factor 10 in Cancer. Front Genet. 2018. Oct 24;9:499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou Y, Xie J, Tian W, Wu L, Xie Y, Huang S, Tang Y, Deng X, Wu H, Xie X. Integrative Analysis of KCNK Genes and Establishment of a Specific Prognostic Signature for Breast Cancer. Front Cell Dev Biol. 2022. May 17;10:839986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alvarez-Baron CP, Jonsson P, Thomas C, Dryer SE, Williams C. The Two-Pore Domain Potassium Channel KCNK5: Induction by Estrogen Receptor α and Role in Proliferation of Breast Cancer Cells. Molecular Endocrinology. 2011. Aug 1;25(8):1326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valla M, Klæstad E, Ytterhus B, Bofin AM. CCND1 Amplification in Breast Cancer -associations With Proliferation, Histopathological Grade, Molecular Subtype and Prognosis. J Mammary Gland Biol Neoplasia. 2022. Mar;27(1):67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Astashchanka A, Shroka TM, Jacobsen BM. Mucin 2 (MUC2) modulates the aggressiveness of breast cancer. Breast Cancer Res Treat. 2019. Jan;173(2):289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Wang S. Identification of key genes involved in tamoxifen-resistant breast cancer using bioinformatics analysis. Transl Cancer Res TCR. 2021. Dec;10(12):5246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lakshmanan I, Ponnusamy MP, Das S, Chakraborty S, Haridas D, Mukhopadhyay P, Lele SM, Batra SK. MUC16 induced rapid G2/M transition via interactions with JAK2 for increased proliferation and anti-apoptosis in breast cancer cells. Oncogene. 2012. Feb 16;31(7):805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Wang X, Sun J, Chen Y, Li J, Huang J, Du H, Gan L, Qiu Z, Li H, Ren G, Wei Y. The Novel Methylation Biomarker NPY5R Sensitizes Breast Cancer Cells to Chemotherapy. Front Cell Dev Biol. 2022. Jan 11;9:798221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng X, Zhang Y, Qi X, Wang M, Sun P, Zhang Y, Jiang J. Role of Sphk1 in the malignant transformation of breast epithelial cells and breast cancer progression. Indian J Cancer. 2014;51(4):524. [DOI] [PubMed] [Google Scholar]
- 35.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003. Jul;34(3):267–73. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Xu Y, Li L, Wang L, Yao R, Sun Q, Du G. FOXC1 is associated with estrogen receptor alpha and affects sensitivity of tamoxifen treatment in breast cancer. Cancer Med. 2017. Jan;6(1):275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaudet MM, Chanock S, Dunning A, Driver K, Brinton LA, Lissowska J, Peplonska B, Pharoah P, Garcia-Closas M. HSD17B1 Genetic Variants and Hormone Receptor–Defined Breast Cancer. Cancer Epidemiology, Biomarkers & Prevention. 2008. Oct 1;17(10):2766–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin TY, Saindane M, Park KS, Kim S, Nam S, Yoo Y, Yang JH, Yun I. LEP as a potential biomarker in prognosis of breast cancer: Systemic review and meta analyses (PRISMA). Medicine. 2021. Aug 20;100(33):e26896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Welm B, Boucher KM, Ebbert MTW, Bernard PS. TRIM29 Functions as a Tumor Suppressor in Nontumorigenic Breast Cells and Invasive ER+ Breast Cancer. The American Journal of Pathology. 2012. Feb;180(2):839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin L, Li Q, Mrdenovic S, Chu GCY, Wu BJ, Bu H, Duan P, Kim J, You S, Lewis MS, Liang G, Wang R, Zhau HE, Chung LWK. KRT13 promotes stemness and drives metastasis in breast cancer through a plakoglobin/c-Myc signaling pathway. Breast Cancer Res. 2022. Dec;24(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.National Library of Medicine Gene Database. [Internet]. Available from: https://www.ncbi.nlm.nih.gov/gene/?term=
- 42.Guo X, Ramirez I, Garcia YA, Velasquez EF, Gholkar AA, Cohn W, Whitelegge JP, Tofig B, Damoiseaux R, Torres JZ. DUSP7 regulates the activity of ERK2 to promote proper chromosome alignment during cell division. J Biol Chem. 2021;296:100676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Wang N, Xiong Y, Guo G, Zhu M, Gu Y. Transcription Factor FOSL1 Enhances Drug Resistance of Breast Cancer through DUSP7-Mediated Dephosphorylation of PEA15. Mol Cancer Res. 2022. Apr 1;20(4):515–26. [DOI] [PubMed] [Google Scholar]
- 44.Adhikari H, Kattan WE, Kumar S, Zhou P, Hancock JF, Counter CM. Oncogenic KRAS is dependent upon an EFR3A-PI4KA signaling axis for potent tumorigenic activity. Nat Commun. 2021. Sep 9;12(1):5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu H, Wen T, Zhou Y, Fan X, Du T, Gao T, Li L, Liu J, Yang L, Yao J, Ge Y, An G. DCLK1 Plays a Metastatic-Promoting Role in Human Breast Cancer Cells. BioMed Research International. 2019. May 15;2019:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sundvall M, Iljin K, Kilpinen S, Sara H, Kallioniemi OP, Elenius K. Role of ErbB4 in Breast Cancer. J Mammary Gland Biol Neoplasia. 2008. Jun;13(2):259–68. [DOI] [PubMed] [Google Scholar]
- 47.Chao MW, Lin TE, HuangFu WC, Chang CD, Tu HJ, Chen LC, Yen SC, Sung TY, Huang WJ, Yang CR, Pan SL, Hsu KC. Identification of a dual TAOK1 and MAP4K5 inhibitor using a structure-based virtual screening approach. Journal of Enzyme Inhibition and Medicinal Chemistry. 2021. Jan 1;36(1):98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lo HW, Hsu SC, Hung MC. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res Treat. 2006. Feb;95(3):211–8. [DOI] [PubMed] [Google Scholar]
- 49.Bose D, Banerjee S, Singh RK, Wise LM, Robertson ES. Vascular endothelial growth factor encoded by Parapoxviruses can regulate metabolism and survival of triple negative breast cancer cells. Cell Death Dis. 2020. Nov 20;11(11):996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao C, Li H, Zhuang J, Zhang H, Wang K, Yang J, Liu C, Liu L, Zhou C, Sun C. The construction and analysis of ceRNA networks in invasive breast cancer: a study based on The Cancer Genome Atlas. CMAR. 2018. Dec;Volume 11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, Nathenson M, Doebele RC, Farago AF, Pappo AS, Turpin B, Dowlati A, Brose MS, Mascarenhas L, Federman N, Berlin J, El-Deiry WS, Baik C, Deeken J, Boni V, Nagasubramanian R, Taylor M, Rudzinski ER, Meric-Bernstam F, Sohal DPS, Ma PC, Raez LE, Hechtman JF, Benayed R, Ladanyi M, Tuch BB, Ebata K, Cruickshank S, Ku NC, Cox MC, Hawkins DS, Hong DS, Hyman DM. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med. 2018. Feb 22;378(8):731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joshi SK, Qian K, Bisson WH, Watanabe-Smith K, Huang A, Bottomly D, Traer E, Tyner JW, McWeeney SK, Davare MA, Druker BJ, Tognon CE. Discovery and characterization of targetable NTRK point mutations in hematologic neoplasms. Blood. 2020. Jun 11;135(24):2159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bera A, Subramanian M, Karaian J, Eklund M, Radhakrishnan S, Gana N, Rothwell S, Pollard H, Hu H, Shriver CD, Srivastava M. Functional role of vitronectin in breast cancer. Ramos JW, editor. PLoS ONE. 2020. Nov 19;15(11):e0242141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL. Estrogen Receptor Binding to DNA Is Not Required for Its Activity through the Nonclassical AP1 Pathway. Journal of Biological Chemistry. 2001. Apr;276(17):13615–21. [DOI] [PubMed] [Google Scholar]
- 55.Bourdeau V, Deschênes J, Métivier R, Nagai Y, Nguyen D, Bretschneider N, Gannon F, White JH, Mader S. Genome-Wide Identification of High-Affinity Estrogen Response Elements in Human and Mouse. Molecular Endocrinology. 2004. Jun;18(6):1411–27. [DOI] [PubMed] [Google Scholar]
- 56.Hallett RM, Hassell JA. Estrogen independent gene expression defines clinically relevant subgroups of estrogen receptor positive breast cancer. BMC Cancer. 2014. Dec;14(1):871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spanheimer PM, Woodfield GW, Cyr AR, Kulak MV, White-Baer LS, Bair TB, Weigel RJ. Expression of the RET Proto-oncogene Is Regulated by TFAP2C in Breast Cancer Independent of the Estrogen Receptor. Ann Surg Oncol. 2013. Jul;20(7):2204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller KD, Trigo JM, Wheeler C, Barge A, Rowbottom J, Sledge G, Baselga J. A multicenter phase II trial of ZD6474, a vascular endothelial growth factor receptor-2 and epidermal growth factor receptor tyrosine kinase inhibitor, in patients with previously treated metastatic breast cancer. Clin Cancer Res. 2005. May 1;11(9):3369–76. [DOI] [PubMed] [Google Scholar]
- 59.Yardley DA, Dees EC, Myers SD, Li S, Healey P, Wang Z, Brickman MJ, Paolini J, Kern KA, Citrin DL. Phase II open-label study of sunitinib in patients with advanced breast cancer. Breast Cancer Res Treat. 2012. Dec;136(3):759–67. [DOI] [PubMed] [Google Scholar]
- 60.Mayer EL, Isakoff SJ, Klement G, Downing SR, Chen WY, Hannagan K, Gelman R, Winer EP, Burstein HJ. Combination antiangiogenic therapy in advanced breast cancer: a phase 1 trial of vandetanib, a VEGFR inhibitor, and metronomic chemotherapy, with correlative platelet proteomics. Breast Cancer Res Treat. 2012. Nov;136(1):169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boér K, Láng I, Llombart-Cussac A, Andreasson I, Vivanco GL, Sanders N, Pover GM, Murray E. Vandetanib with docetaxel as second-line treatment for advanced breast cancer: a double-blind, placebo-controlled, randomized Phase II study. Invest New Drugs. 2012. Apr;30(2):681–7. [DOI] [PubMed] [Google Scholar]
- 62.Matias-Guiu X, Calabuig R, Badia F, Serra J, La Calle JP. Spontaneous infarcts in fibroadenomas of the breast. Curr Surg. 1988;45(4):277–9. [PubMed] [Google Scholar]
- 63.Lim JSJ, Wong ALA, Ow SGW, Ngoi NYL, Chan GHJ, Ang YLE, Chong WQ, Lim SE, Lim YW, Lee M, Choo JRE, Tan HL, Yong WP, Soo RA, Tan DSP, Chee CE, Sundar R, Yadav K, Jain S, Wang L, Tai BC, Goh BC, Lee SC. Phase Ib/II Dose Expansion Study of Lenvatinib Combined with Letrozole in Postmenopausal Women with Hormone Receptor-Positive Breast Cancer. Clin Cancer Res. 2022. Jun 1;28(11):2248–56. [DOI] [PubMed] [Google Scholar]
- 64.Peng W xin, Huang J guo, Yang L, Gong A hua, Mo YY. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol Cancer. 2017. Dec;16(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferrando L, Vingiani A, Garuti A, Vernieri C, Belfiore A, Agnelli L, Dagrada G, Ivanoiu D, Bonizzi G, Munzone E, Lippolis L, Dameri M, Ravera F, Colleoni M, Viale G, Magnani L, Ballestrero A, Zoppoli G, Pruneri G. ESR1 gene amplification and MAP3K mutations are selected during adjuvant endocrine therapies in relapsing Hormone Receptor-positive, HER2-negative breast cancer (HR+ HER2- BC). PLoS Genet. 2023. Jan;19(1):e1010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spanheimer PM, Bashir A, Lorenzen AW, Beck AC, Liao J, Lizarraga IM, Erdahl LM, Sugg SL, Karwal MW, Weigel RJ. A Pilot Study of Preoperative Vandetanib on Markers of Proliferation and Apoptosis in Breast Cancer. Am J Clin Oncol. 2021. Sep 1;44(9):456–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets and codes supporting the conclusions of this article are available in the GitHub repository, www.github.com/rashatk/HighRETGDNF.
The datasets used and code generated during the current study are available on a public GitHub repository (www.github.com/rashatk/HighRETGDNF). The dataset obtained from Xia et al. can be accessed at doi:10.1158/1078-0432.CCR-21-3189.