

Abstract
Heterochromatin is defined as chromosomal domains harboring repressive H3K9me2/3 or H3K27me3 histone modifications and relevant factors that physically compact the chromatin. Heterochromatin can restrict where transcription factors bind, providing a barrier to gene activation and cell identity changes. While heterochromatin thus helps maintain cell differentiation, it presents a barrier to overcome during efforts to reprogram cells for biomedical purposes. Recent findings reveal complexity in the composition and regulation of heterochromatin and that transiently disrupting the heterochromatin machinery can enhance reprogramming. Here, we discuss how heterochromatin is established and maintained in development and how our growing understanding of the mechanisms regulating H3K9me3-heterochromatin can be leveraged to improve our ability to direct changes in cell identity.
Keywords: heterochromatin, H3K9me3, H3K27me3, reprogramming, pioneer factors
Heterochromatin: Restricting Access to the Genome
Despite all cells containing the same genetic information, each cell type in multicellular organisms expresses a subset of genes corresponding to its distinct cellular function. The expression of cell type specific genes relies upon transcription factors acting in the context of chromatin. During development, the progressive expression of sets of transcription factors drives changes in cell identity and lineage commitment. Reprogramming involves the activation of a new cell identity, typically by the ectopic expression of a cocktail of transcription factors that activate alternative lineage genes. The ability to reprogram cells was originally discovered with the observation that nuclear transfer can change cell identity [1]. Reprogramming through the direct expression of transcription factors was first demonstrated by the ability of MyoD to convert fibroblasts to myoblasts [2] and later shown by reprogramming of B cells into macrophages through expression C/EBPα and C/EBPβ [3]. Finally, fibroblasts were converted to pluripotent stem cells following expression of the Oct4, Sox2, Klf4 and c-Myc (OSKM) transcription factors [4]. Reprogramming from one somatic cell lineage to another somatic cell lineage, also referred to as trans-differentiation, has been used to generate many cell types including hepatocytes [5], cardiomyocytes [6] and neurons [7].
Transcription factors can be restricted from binding to heterochromatic regions of the genome that are compact, inaccessible, and hence transcriptionally silent. By contrast, euchromatin is more open, accessible, and generally transcriptionally active. Transcription factors vary in their abilities to bind to free DNA, euchromatin, and silent, unmarked chromatin regions, but are largely blocked from activating target genes in heterochromatin regions during reprogramming [8,9]. Thus, learning how to overcome heterochromatin repression to enable transcription factor binding helps improve our ability to reprogram cells for basic science and therapeutic applications [10–12].
Here we review studies revealing an emerging view that heterochromatin is complex in composition. After reviewing such complexity, we will focus on the H3K9me3-heterochromatin subtype in mammalian cells, including how it is established and rearranged during early development, how it resists activation during reprogramming, and how it can be disrupted to enhance reprogramming (Figure 1). It appears that H3K9me3-heterochromatin achieves gene silencing through diverse mechanisms, resulting in structures and biochemical parameters that may interact differently with specific classes or families of transcription factors. Unraveling such specificity is a major goal for the future.
Figure 1.
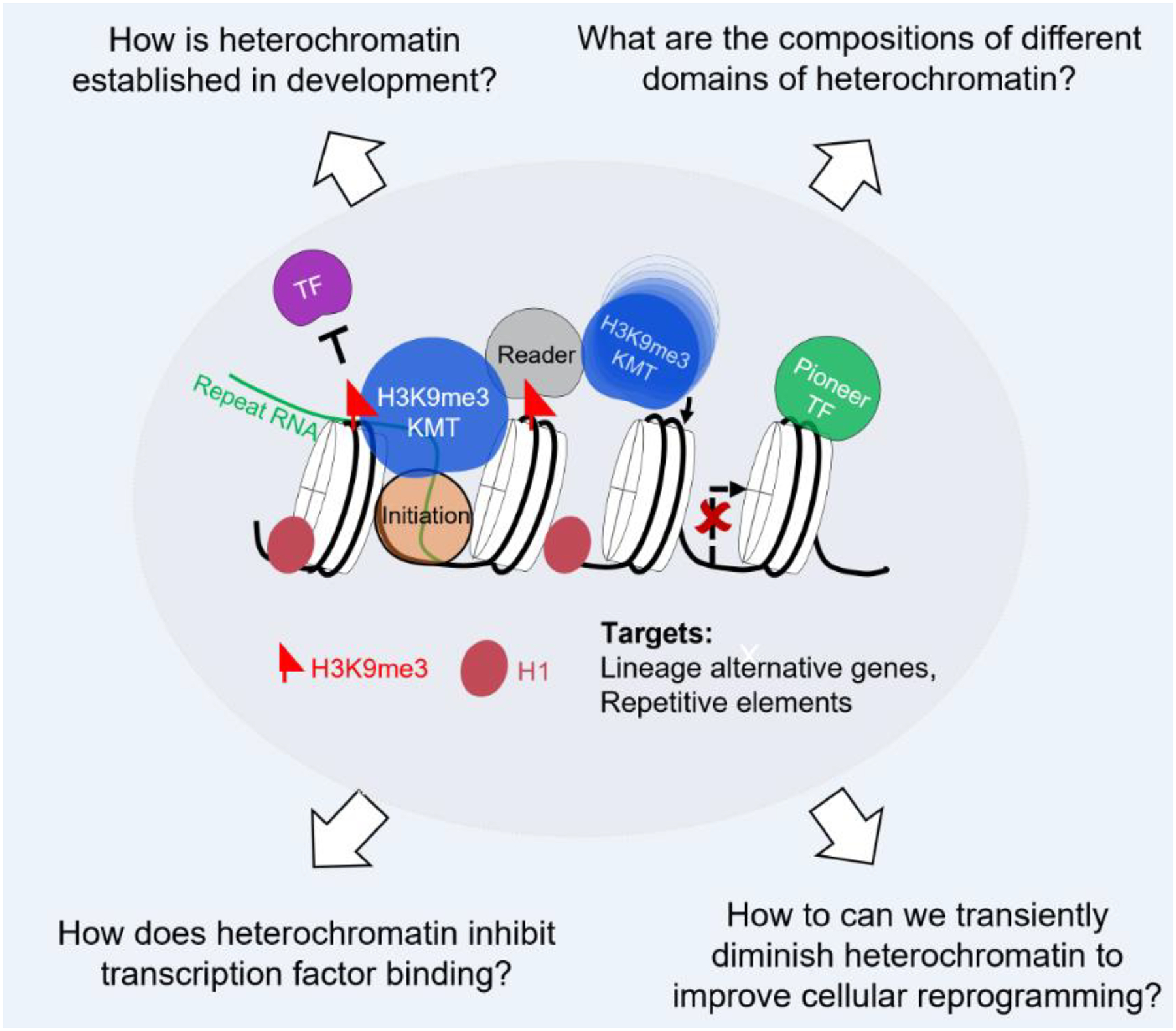
H3K9me3-heterochromatin as a barrier to cell fate change. Central to the functions of H3K9me3-heterochromatin is the “reader-writer” module, in which H3K9me3 mark deposited by H3K9me3 methyltransferases is recognized by reader proteins, including HP1α/β/γ, which further recruit methyltransferases to modify the neighboring nucleosomes. This leads to spreading of heterochromatin domains and stable maintenance of H3K9me3 domains over the cell cycle. Further enrichment of linker histone H1, HP1 proteins and other heterochromatin associated proteins lead to heterochromatin compaction and restricting the TFs from activating their targets. Building from this basic principle, we discussed how heterochromatin is established and maintained in development, different compositions of heterochromatin domains, how it molds the TF bindings and finally how to this knowledge to enhance cellular reprogramming.
Diverse Types of Heterochromatin
Functionally, heterochromatin silences alternative lineage genes during development [13–16], maintains repression of repeat elements, and promotes genome stability by suppressing recombination among different repeats across the genome [17]. The repressive function of heterochromatin is driven by its structure, biochemical modifications, and chromatin associated proteins and RNAs.
Our understanding of the structure of heterochromatin has undergone a dramatic shift, thanks to new insights provided by novel imaging, genomics, and biochemical advances. Compared to the uniform nucleosome compaction observed in vitro, recent experiments in vivo revealed a more complex picture of heterochromatin structures, with heterochromatin assuming multiple nucleosome configurations [18,19] and forming various higher-order structures [20]. Integrating how chromatin structural configurations correspond to specific histone modifications, protein and genomic compositions and their impact on transcription factor binding will provide key insights into heterochromatin regulation and function in development and reprogramming.
Heterochromatin is often characterized by the associated biochemical modifications that decorate the DNA and histones. The first heterochromatic mark discovered was DNA methylation, which is generally associated with transcriptional repression when occurring at CpG islands of gene promoters, but its function depends upon genomic context [21]. Covalent modification of the histone tails, including di- and tri-methylation of histone 3 lysine 9 (H3K9me2/3) [22], and tri-methylation of histone 3 lysine 27 (H3K27me3), are the most extensively studied histone modifications associated with heterochromatin. H3K27me3, catalyzed by Polycomb Repressive complex 2, has been associated with heterochromatin at developmental genes, including Hox clusters, which are dynamically regulated during development [23,24]. H3K9me2, catalyzed by the histone methyltransferases (HMTs) G9a/GLP, and H3K9me3, catalyzed by the HMTs SETDB1 and SUV39H1/H2 respectively [22] have long been known to repress repetitive elements. H3K9me2 and H3K9me3 are differentially distributed in the nucleus, with H3K9me2 signals mainly detected at the nuclear periphery and interacting with nuclear lamins through adaptor proteins [25,26], and H3K9me3 detected at both nuclear periphery and other more centrally located heterochromatin compartments, such as peri-nucleolar and pericentric heterochromatin [27]. Upon loss of H3K9me3 in C. elegans, H3K9me2 can maintain repression at some previously H3K9me3 repressed genes and repeats, but not all, indicating overlapping but not redundant repressive function [15]. Growing evidence has shown that H3K9me2/3 is dynamically regulated at genes and enhancers during development to enable lineage specifications and restrict alternative lineages [13,22,28,29]; H3K9me3 will be the major focus of this review. Additional repressive marks including H4K20me3 [30,31], H3K64me3, H2AK119ub1 [24,32], and histone variants [33], together contribute to the complex organization and regulation of heterochromatin.
H3K9me3-heterochromatin can be further decorated by associated proteins and RNAs, to enforce repression. Linker histone H1 associates with the “linker” DNA region between nucleosomes throughout most of the chromatin, i.e., both euchromatic and heterochromatin, but a higher density of H1 in heterochromatin domains contributes to chromatin compaction [34,35]. So-called histone modification-reader proteins include the heterochromatin binding proteins HP1α, HP1β, and HP1γ, which bind methylated lysines through their chromodomain and recruit SUV39H1/H2 and SETDB1 to spread H3K9me3 marks to neighboring nucleosomes, compacting the chromatin, and reinforce repression through the cell cycle [36]. Chromatin associated non-coding RNAs also play important roles in establishing and maintaining heterochromatin, such as the Xist RNA in X chromosome inactivation [37], satellite RNAs in the recruitment of SUV39H1 and SUV39H2 [38,39], pseudogene lncRNAs in the recruitment of SUV39H1 [40], and endogenous siRNAs which recruit HMTs through nuclear Argonaute [41]. Specific protein compositions of different heterochromatin compartments [8,37,42] may explain how heterochromatin can be uniquely deposited and rearranged in development and reprogramming.
Heterochromatin remodeling enables zygotic genome activation and totipotency
Mammalian embryos undergo extensive epigenetic reprogramming during pre-implantation development, erasing epigenetic information from the past generation and establishing new epigenetic programs to enable developmental progression [43]. Therefore, early development offers an important model to investigate molecular mechanisms of heterochromatin initiation, establishment, and maintenance, and its impact on cell potential (Figure 2).
Figure 2.
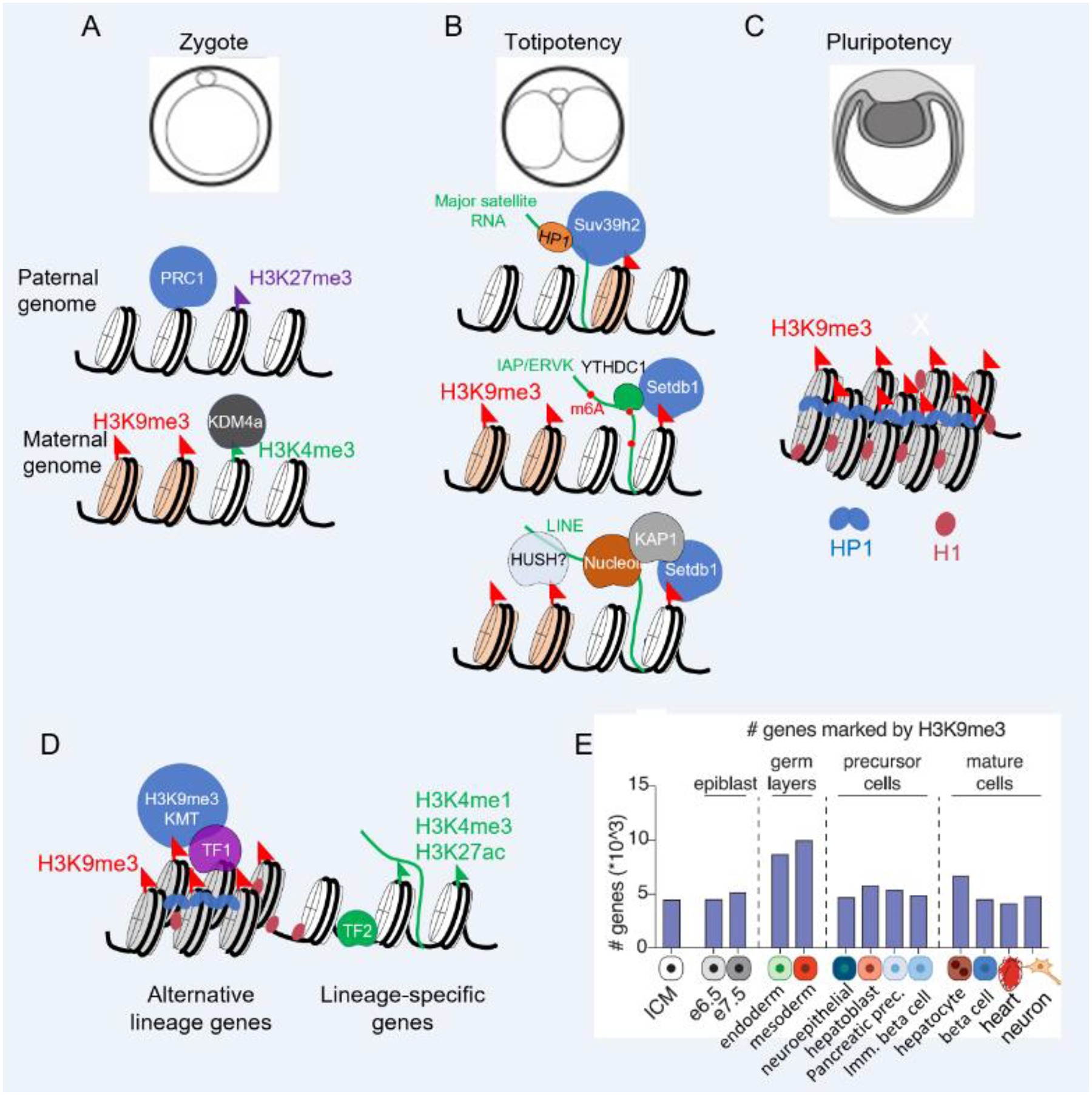
Heterochromatin is dynamic during development. (A) Heterochromatin remodeling accompanies developmental progression during early mouse development. In the zygote, the maternal genome possesses H3K9me3 heterochromatin marks at centromeric and pericentromeric regions, whereas paternal genome does not. (B) An ensuing heterochromatin remodeling creates an open chromatin environment, a hallmark of totipotent states and leads to activation of repeat regions, which recruit heterochromatin machinery to establish heterochromatin and promote the transition from totipotency to pluripotency. (C) Heterochromatin domains in pluripotent stem cells are decorated with H3K9me3 marks, compacted by linker histone H1 and recruit heterochromatin associated proteins, including HP1. (D) During lineage specifications in mouse development, transcription factors, including KRAB-ZNF proteins direct heterochromatin machinery to repress alternative lineage-specific genes to maintain the cell fate. (E) Genes are increasingly marked by H3K9me3 for repression during germ layer development, but this mark is removed from key functional genes upon lineage specification [13].
In the zygote, the paternal genome in sperm is largely packaged with protamines, while the remaining canonical histones are largely devoid of H3K9me3 [28,44]. The zygotic maternal genome possesses canonical H3K9me3-, H3K40me3-, and H3K64me3-marked heterochromatin at centromeric, pericentromeric, and telomeric regions [44,45]. De novo H3K9me3 on paternal genomes by SUV39H2 starts as early as the late zygote stage [46], although the association of the SUV39H2 RNA-binding domain with the pericentromeric RNA transcribed from the paternal genome limits its methyltransferase activities [46,47]. SUV39H1 lacks RNA binding domains [39] and overexpression of SUV39H1 induces precocious H3K9me3 heterochromatin in zygotes, causing a developmental arrest at the 2-cell stage and reducing nuclear transfer efficiency by the oocyte [46,48]. Similarly, depleting KDM4a in oocyte, the major H3K9me3 demethylase expressed in mouse and human oocytes, leads to invasion of H3K9me3 domains into euchromatin and disrupts zygotic gene activation [49]. We can conclude that precisely coordinated heterochromatin resetting is crucial for establishing a permissive chromatin environment for zygotic genome activation and establishing totipotency (Figure 2).
Heterochromatin re-establishment in early embryo drives the transition from totipotency to pluripotency
Heterochromatin maintains genome integrity by preventing the recombination between repeat sequences and silencing transcription from repetitive elements to prevent the formation of RNA:DNA hybrids (reviewed in [17]). However, the newly established heterochromatin domains before the 8-cell stage lack HP1α [30,50] and most of the linker histone H1 variants [51], which normally are molecular hallmarks of compact heterochromatin domains [34], consistent with the notion that heterochromatin domains prior to the 8 cell stage harbor a noncanonical, non-repressive structure [52]. Consequently, the resetting of H3K9me3, along with erasure of other heterochromatin marks, H3K64me3 and H4K20me3, and DNA methylation (reviewed in [53]) from the 2-cell to blastocyst stage leads to transient activation of satellite repeats and many retrotransposons and during pre-implantation development [54].
Interestingly, a pulse of major satellite RNA transcribed from the paternal genome during the zygote stage recruits SUV39H2 to pericentromeric regions [39] and the transcription from both strands may lead to dsRNA formation, reminiscent of RNAi mechanisms in S. pombe and C.elegans (reviewed in [22]). Retrotransposons constitute a large proportion of the mammalian genome, and mounting evidence suggests that RNAs transcribed from diverse classes of retrotransposons can direct different heterochromatin machineries to silence the repetitive DNA and target genes [55–58] (Figure 2B). The retrotransposons and can be broadly divided into non-LTR elements, including LINE and SINE elements, and LTR elements, including ERV1, ERV2, ERV3 and MaLR (reviewed in [59]). LINE elements constitutes 10%−30% of eutherian genomes [60] and its transcripts, abundant in 2-cell embryos, recruit Nucleolin and KAP1 (TRIM28) to repress Dux, master regulator of 2-cell totipotency genes, and therefore drive the exit from totipotency [56].
In addition, the HUSH complex recognizes L1 (LINE) RNAs and recruits SETDB1 to silence L1 retrotransposons, although direct evidence for a function of the HUSH complex in early development is still lacking. LTR elements represent around 25% of retrotransposons in the mammalian genome, and their transcripts are detected from zygote to morula stages, some of which show remarkable stage-specificities (reviewed in [60]). ERV2 families, including IAP and ERVK are among the most abundant ERV elements in the mouse genome [59,61]. RNA m6A modifications by Mttl3/4 on IAPs RNAs can mark the RNAs for degradation [58] and recruit YTHDC1, which further recruits Setdb1 to initiate heterochromatin formation at IAP elements [55]. It is currently unknown if the RNA-directed mechanism also plays a role in silencing other LTR families.
It is possible that RNA transcribed at heterochromatin domains may directly recruit HP1 proteins through interactions with the HP1 hinge domains [62]. Taken together, the extant studies indicate that RNA is at the core of heterochromatin initiation and maintenance and provides targeting specificity for heterochromatin machineries to silence diverse repeat families. Recently, of 172 proteins found to be associated with H3K9me3-heterochromatin in human fibroblast cells, many are strongly enriched for RGG RNA-binding motif [8], hinting that the RNA binding is a common mechanism for heterochromatin formation and maintenance.
In addition to the RNA-directed heterochromatin initiation mechanisms mentioned above, many transcription factors directly interact with SUV39H1/H2, SETDB1, and HP1 to recruit heterochromatin machinery to repress diverse retrotransposon families and lineage specific genes (Figure 2D) [63–65]. KRAB-ZFP proteins represent a repertoire of constantly evolving transcription factors that recruit SETDB1 through the bridging factor KAP1 to silence invading retrotransposons (reviewed in [66]). Some zinc-finger proteins, including ZFP809 [67], KLF4, KLF17 [68], and ZFP93 [69], are highly expressed in early embryos and bind to specific families of retrotransposons, indicating that ZFPs can recruit H3K9me3 machineries to establish H3K9me3 heterochromatin at specific retrotransposons in early development. Interestingly, the maturation of heterochromatin domains requires additional heterochromatin associated proteins, including CAF-1, linker histone H1 and SUMOylation pathway (Figure 2C) [46,56,70]. Depleting SETDB1 and the aforementioned heterochromatin associated proteins in early embryos causes a developmental arrest at the 2-cell stage and de-represses totipotent genes Dux and Zscan4 in pluripotent ES cells, causing reversion to 2 cell-like totipotent state. Therefore, the re-establishment of H3K9me3 heterochromatin directed by RNA and TFs plays important roles in repressing the 2C totipotency program and driving the transition to pluripotency at the blastocyst stage.
Dynamic heterochromatin changes enable lineage specification in development
In addition to repressing retrotransposons, H3K9me3-marked heterochromatin plays important roles in delineating lineage specification during and after gastrulation (Figure 2D–E) [13]. Mapping H3K9me3 changes at protein-coding genes, from the germ-layer stage to endoderm progenitors, and then to differentiated hepatic and pancreatic cells, reveals that, in addition to the expected acquisition of H3K9me3 at genes that become silent in terminal differentiation, surprisingly many genes are marked by H3K9me3 heterochromatin at the germ layer stage and gradually lose the mark during lineage progression (Figure 2E). Further genetic studies with Suv39h1/Suv39h2 and Setdb1 triple KO or Setdb1 knockdown show that H3K9me3 heterochromatin functions to restrict late developmental genes and repress alternative lineages [13,28] (comprehensively reviewed in [22]). Thus, H3K9me3 dynamics at protein coding genes are critical for embryologic differentiation to progress properly. Interestingly, in addition to the roles of ZFPs in heterochromatin initiation mentioned above, some of the KRAB-ZNF proteins also show lineage specific expression and functions, such as ZNF417/ZNF587 in human neurons [71], ZNF558 in human neural progenitors [72], ZNF589 in human hematopoietic system [73], and ZNF808 in human pancreatic development [74]. Thus, that KRAB-ZNF proteins and the transposable elements that they target can be co-opted by the host genome to expand the lineage and species-specific regulatory network [75,76].
In summary, H3K9me3-heterochromatin dynamics are critical for early development. Interestingly, the drastic heterochromatin remodeling in early development does not necessarily lead to genome instability, and similarly no genome-wide genome instabilities in liver were observed after global loss of H3K9me3 caused by compound SUV39H1/2 and SETDB1 deletions [13], suggesting that the roles of H3K9me3 heterochromatin in safeguarding genome stabilities are cell context dependent. Understanding how different heterochromatin associated proteins direct diverse heterochromatin patterns during development has inspired novel screens to perturb heterochromatin machineries to help reprogram cells [10].
Heterochromatin blocks transcription factor binding and gene activation
To elicit cellular reprogramming, transcription factors are induced to bind and activate genes of a new cell identity. Many reprogramming protocols have been developed to enable conversion to diverse cell identities, including pluripotent stem cells [4], macrophages [3], hepatocytes [5], cardiomyocytes [6] and neurons [7]. However, in most cases, the reprogramming elicited by the ectopic expression of transcription factors is limited and does not reflect the desired, fully differentiated cell state [77]. Indeed, reprogramming transcription factors are often impeded from binding terminal differentiation genes of alternative fates because of repressive chromatin at important differentiation genes, particularly H3K9me3-heterochromatin [8] [9,15,78]. Transcription factors possess different capacities to bind and open closed chromatin.
Pioneer transcription factors have DNA binding domains that can bind a partial motif displayed on the surface of a nucleosome [79], leading to chromatin opening and enabling additional factors to bind [80]. Hence pioneer factors can scan closed chromatin regions, in contrast to transcription factors that primarily target open chromatin regions [81,82]. Analysis of heterochromatin compartments and diverse transcription factors by single molecule tracking demonstrated that the pioneer transcription factors’ nonspecific DNA and nucleosome binding ability enabled access to the most restricted heterochromatin [83]. Loss of OCT4 nucleosome binding ability, without compromising free DNA binding affinity, was sufficient to exclude OCT4 from binding closed chromatin and abolish its reprogramming capacities [84]. Therefore, pioneer factor binding initiates structural changes between the DNA and histones [85,86] and facilitates binding of other transcription factors and remodelers [87].
Despite their abilities to bind nucleosomes, the pioneer transcriptional factors Sox2 and Oct4 are largely excluded from the H3K9me3 marked heterochromatin during reprogramming [9,88]. For instance, in human pluripotent cells Oct4, Sox2 and Klf4 are bound to pluripotency genes such as Nanog and Prdm14, but these genes are buried in H3K9me3-marked heterochromatin domains in human fibroblasts. The activation of such pluripotent genes in H3K9me3 heterochromatin occurs at the final stage of iPSC reprogramming and is a rate-limiting step. Similarly, during fibroblast to hepatic cell reprogramming by pioneer factor FoxA3 with transcription factors HNF1α and HNF4α, hepatic genes repressed by H3K9me3-marked heterochromatin are more resistant to activation than the genes marked by H3K27me3 or silenced chromatin marked by neither H3K9me3 and H3K27me3 [8]. During pro-opiomelanocortin to melanotropes differentiation, binding of the pioneer factor PAX7 was also blocked from regions with high H3K9me3 [89]. H3K27me3 heterochromatin can also block MyoD in undifferentiated muscle cells [90] and multiple lineage specific transcription factors during early mouse and human embryonic stem cell differentiation [91]. Although heterochromatin has been shown to exclude transcription factor binding in many cell contexts, the ability to bind or being excluded from specific chromatin contexts varies among specific pioneer factors [83,92].
Heterochromatin can be de-repressed to enhance reprogramming gene activation
Loss of all H3K9 methylation through disruption and deletion of all H3K9 lysine methyltransferases leads to global chromatin decompaction, including loss of electron-dense heterochromatin and derepression of protein-coding genes and repeat elements [93]. Transiently depleting diverse non-enzymatic proteins important in maintaining H3K9me3 enhances activation of genes in heterochromatin and improves reprogramming [8–10,12,15,94] (Figure 3A). However, inhibition of H3K27me3 by knockdown of PRC2 components EED, EZH2, or SUZ12 decreased iPSC reprogramming, potentially due to a failure to silence fibroblast specific transcripts which gain H3K27me3 during successful iPSC reprogramming [11] (Figure 3A). Disruption of MBD3 or GATAD2A in the NuRD complex, which normally facilitates repression through histone de-acetylation and remodeling, enhanced iPSC reprogramming [95]. GATAD2A siRNA knockdown was also shown to improve the activation of genes located in H3K27me3 heterochromatin during fibroblast to hepatocyte reprogramming [10], potentially due to decreased H3K27 de-acetylation [95]. Gene de-repression alone is typically not sufficient for activation during reprogramming which requires both de-repression as well as the presence of an activating transcription factor [10,15].
Figure 3.
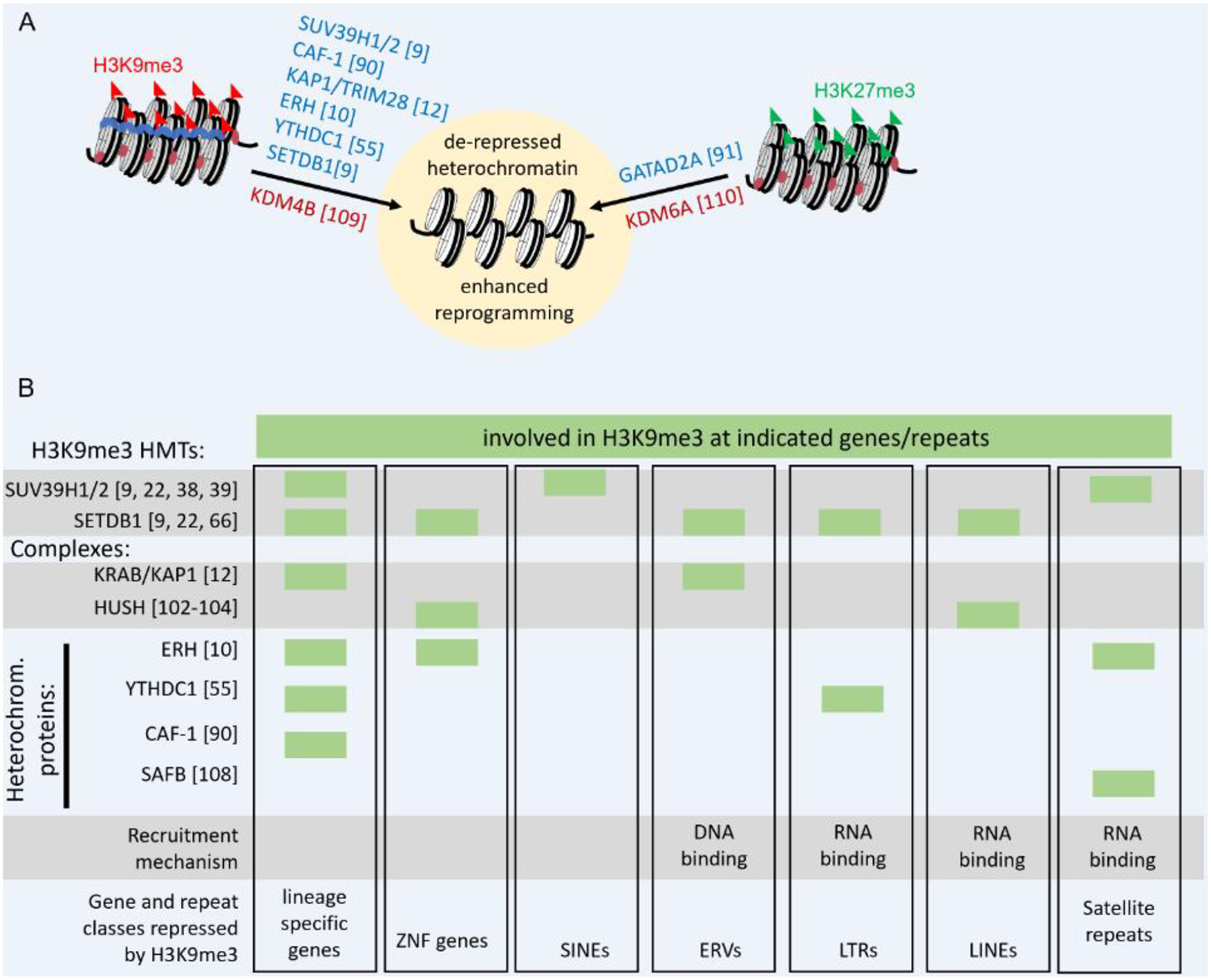
Groups of heterochromatin proteins regulate distinct classes of heterochromatin and can be disrupted to facilitate gene activation. (A) Published results showing knockdowns/knockouts (blue) and overexpression (red) experiments that lead to de-repression of heterochromatin and enhanced reprogramming. (B) Regulation of H3K9me3 at genes and repeat classes by H3K9me3 HMTs, protein complexes and selected heterochromatin proteins. Green boxes indicate the indicated protein or complex has been experimentally demonstrated to regulate H3K9me3 at the designated gene or repeat class.
The rationale here is that a transient diminution of heterochromatin proteins can allow the reprogramming factors to activate new genetic networks, and the subsequent restoration of heterochromatin proteins, after transient diminution, can allow a new genetic network to re-establish heterochromatin appropriate for the new cell type.
However, such manipulations can be a dangerous game. Heterochromatin opening during reprogramming can lead to activation of off-target lineages and repeat elements [10]. To lessen this problem, recent findings reveal that groups of heterochromatin proteins co-repress distinct sets of genes located in heterochromatin and each gene set possesses a particular chromatin signature [10]. While H3K9me3 HMTs and complexes such as HUSH target H3K9me3 to broad classes of genes and repeat elements for repression, recent findings have identified heterochromatin proteins necessary for subsets of H3K9me3 targets (Figure 3B). Thus, to more precisely open heterochromatin domains and lessen the undesired consequences, it is necessary to learn more about the mechanisms by which the heterochromatin machinery is targeted in a locus- and gene-specific manner.
Recently it was demonstrated that depletion of Enhancer of Rudimentary Homolog (ERH) in human cells, the S. pombe homolog of which is a known regulator of H3K9 methylation [96,97], leads to global H3K9me3 loss in human cells, activation of heterochromatic protein coding genes during induced hepatocyte reprogramming, and activation of satellite repeats [10] (Figure 3A). In S. pombe, Erh1 interacts with the YTH domain-containing protein Mmi1 and is recruited in an RNA-dependent manner to meiotic genes, to maintain H3K9me3 heterochromatin and silencing [96,97]. Despite a conserved protein sequence with Erh1 [96] and H3K9me3 regulatory function [10], the mechanism of ERH recruitment in humans is unknown, as the direct ortholog of Mmi1 is absent in mammals [98]. Surprisingly human ERH was found to repress genes in heterochromatic and euchromatic H3K9me3 domains, indicating that it may function in the targeting of many or most of H3K9me3 deposition mechanisms [10].
Although heterochromatin is partially defined by its transcriptionally silent nature, recent findings demonstrate a role for RNAs in heterochromatin establishment and maintenance, beyond the canonical role of the XIST RNA in X inactivation [37]. RNA-directed heterochromatin establishment is of particular interest due to the potential for uncovering target specificity, which could allow specific RNAs to be disrupted to unlock specific heterochromatin domains. An example of such sequence specificity can be observed in RNAi-directed post-transcriptional gene silencing by which nuclear Argonaute proteins establish repression in S. pombe, D. melanogaster, A. thaliana and C. elegans [22,41], nuclear Argonaute proteins in mammals, however, may be involved in both activation and repression [99].
Euchromatic H3K9me3 regions are transcriptionally dampened but not fully silenced by the HUSH complex, which recruits SETDB1 to repress evolutionarily young L1 retrotransposons, naïvely integrated lentiviruses, and tissue specific genes including ZNF gene clusters [100]. The HUSH complex is recruited by intronless RNAs, a feature of retroelements, to repress transgenes and mobile elements [101]. In turn, the repression by HUSH also produces shorter non-polyadenylated transcripts, favorable for nuclear exosome targeting (NEXT) degradation [102]. HUSH complex suppression of L1 elements is required for self-renewal of ground-state pluripotent stem cells [103], but depletion of HUSH complex component Periphilin 1 enhanced activation of genes in heterochromatin during reprogramming to hepatocytes [10]. How HUSH is targeted to genes with introns such as the ZNF clusters remains unclear.
In parallel, heterochromatic H3K9me3 domains are highly enriched for HP1 proteins, which can bind RNA through HP1’s hinge domain [62]. Recent in vitro modeling suggests that the affinities of IAP and satellite RNAs for HP1 proteins are five-fold higher than for Mediator complexes, therefore partially explaining the different recruitment mechanisms to repeats versus gene promoters and enhancers [104]. Depletion of HP1 proteins during reprogramming destabilizes H3K9me3 heterochromatin domains that repress pluripotency genes, and therefore enhances reprogramming efficiency [105]. SAFB, a nuclear matrix associated protein, binds major satellite RNA to promote phase separation at the boundaries of H3K9me3 marked heterochromatin domains [106]. Interestingly SAFB has been demonstrated to interact with ERH [107] and may cooperate in miRNA processing [108]. Deletion of YTHDC1, which targets the RNA modification m6A to direct SETDB1 H3K9me3 to retrotransposons and totipotent genes, in mouse ESCs initiated reprogramming to a 2-cell like totipotent state [55] (Figure 3A).
Heterochromatin opening can be facilitated by the active removal of repressive marks and addition of activating marks to histones. Ectopic lysine demethylases, KDM6A and KDM4B, targeting to H3K27me3 and H3K9me3 domains, respectively, improved reprogramming [109,110] (Figure 3A). Similarly, increased histone acetylation, triggered through pathways downstream of MAP2K6 phosphorylation, can lead to improvements in Sox2 and Klf4 binding and reprogramming to pluripotency [111].
These studies reveal that heterochromatin de-repression can be triggered by disrupting maintenance functions or active heterochromatin removal, making target chromatin more permissible and improving reprogramming by transcription factors.
Selectively de-repressing heterochromatin domains
The activation of unintended transcripts, including repeat elements and alternative lineage genes [10], as well as increased genome instability associated with widespread heterochromatin de-repression [17,93,112], remains a major barrier to heterochromatin diminution for cell therapy applications. The goal remains to selectively de-repress specific heterochromatic gene sets or domains while maintaining repression of repeat regions and undesired genes. Further work to understanding how the HUSH complex [100–102], ERH [10,96], or YTHDC1 [55] are recruited or maintained in chromatin will be key. It is important to note that, as best we understand, disrupting H3K9me3 heterochromatin maintenance still requires either dilution through cell division [113] or the action of demethylases, for the H3K9me3 mark to go away [114]. Understanding which H3K9me3 HMTs are targeted and how this targeting can be disrupted is complicated by their ability to function redundantly and compensate for partial losses of the other of the three H3K9me3 HMTs [13,22,93]. Another approach involves the identification of highly specific repressors, such as sequence specific ZNFs [71–74], or the design of synthetic de-repressors, which has been done recently by fusing epigenetic regulators to transcription activator-like effectors [112] and dCas9 [115].
The transcriptional outcome of derepressing H3K9me3 domains may be influenced by other marks that are either coincident with H3K9me3 or that are established in a compensatory manner. For example, in mouse ESCs, dual H3K36me3/H3K9me3 domains, but not H3K9me3-only domains, gained interaction with upregulated genes upon a SETDB1 knockout [116]. H3K9me2 [15] and compensation by H3K27me3 [10,13,93] have been shown to maintain repression at a subset of sites after loss of H3K9me3. Better understanding the complex landscape of heterochromatin will be key to enabling precise and selective de-repression.
Concluding Remarks
Despite the extensive rearrangement of heterochromatin during development, genome stability and repression of repeats are maintained, indicating that different types of heterochromatin can be selectively modulated. Different heterochromatin complexes, directed by RNA and transcription factors, appear at different chromatin domains to accommodate various developmental needs. By discovering the mechanisms by which heterochromatin is selectively targeted during development, we hope to selectively derepress key genes in heterochromatin for reprogramming to diverse cell types, without activating repetitive regions and off-target genes that are seen with global heterochromatin loss [10,93]. Recent advances in human iPSC reprogramming suggests that the route(s) to pluripotency transiently goes through a totipotent state [117], which was recently captured in vitro [118], offering an opportunity to reconstitute early human development in vitro and investigate the heterochromatin remodeling underlying cell fate transitions in greater detail. Future studies are required to dissect upstream signaling pathways, and examine the functional consequences of disrupting different heterochromatin associated proteins and complexes in various developmental and reprogramming contexts to establish a more unifying principle that governs heterochromatin functions (see Outstanding Questions). Finally, understanding how pioneering factors interact with silenced chromatin will also inspire novel designs for synthetic reprogramming factors that combine the chromatin binding capacities of pioneer factors with chromatin effector domains that modulate repressive heterochromatin environments to improve reprogramming.
Outstanding Questions Box.
How are heterochromatin proteins dynamic at particular genomic locations in a cell type specific manner, e.g., during development?
What are the roles of RNA binding proteins and RNAs in regulating heterochromatin at lineage specific genes and how they can be targeted to enhance reprogramming?
How can heterochromatin at genes be de-repressed while maintaining repression of repeats and transposable elements?
How can manipulating heterochromatin be used to improve cellular reprogramming?
Highlights.
Various categories of heterochromatin exist and are regulated by different proteins, RNAs, and mechanisms to restrict access by transcription factors in different ways and degrees.
Activation of a gene during reprogramming that was in heterochromatin requires both the opening of the heterochromatin and activating factors.
Disrupting mechanisms required for maintenance of heterochromatin makes sites permissive to transcription factor binding and activation, but can cause activation of off-target genes and repeat elements.
Acknowledgements
R.L.M. was supported by an NIH K01 postdoctoral fellowship DK117970-01, J.Z. was supported by an HFSP fellowship LT000761/2019-L to J.Z., and K.S.Z. was supported by NIH R01GM36477.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
No competing interests are declared.
References
- 1.Gurdon JB (1962) The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol 10, 622–640 [PubMed] [Google Scholar]
- 2.Davis RL et al. (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000 [DOI] [PubMed] [Google Scholar]
- 3.Xie H et al. (2004) Stepwise reprogramming of B cells into macrophages. Cell 117, 663–676 [DOI] [PubMed] [Google Scholar]
- 4.Takahashi K and Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- 5.Huang P et al. (2014) Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell 14, 370–384 [DOI] [PubMed] [Google Scholar]
- 6.Ieda M et al. (2010) Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vierbuchen T et al. (2010) Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker JS et al. (2017) Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol. Cell 68, 1023–1037.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soufi A et al. (2012) Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarthy RL et al. (2021) Diverse heterochromatin-associated proteins repress distinct classes of genes and repetitive elements. Nat Cell Biol 23, 905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onder TT et al. (2012) Chromatin-modifying enzymes as modulators of reprogramming. Nature 483, 598–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miles DC et al. (2017) TRIM28 is an Epigenetic Barrier to Induced Pluripotent Stem Cell Reprogramming. Stem Cells 35, 147–157 [DOI] [PubMed] [Google Scholar]
- 13.Nicetto D et al. (2019) H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science 363, 294–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takikita S et al. (2016) A Histone Methyltransferase ESET Is Critical for T Cell Development. J Immunol 197, 2269–2279 [DOI] [PubMed] [Google Scholar]
- 15.Methot SP et al. (2021) H3K9me selectively blocks transcription factor activity and ensures differentiated tissue integrity. Nat Cell Biol 23, 1163–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balmer P et al. (2021) SUV39H2 epigenetic silencing controls fate conversion of epidermal stem and progenitor cells. J Cell Biol 220, e201908178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen A et al. (2018) Heterochromatin: Guardian of the Genome. Annu Rev Cell Dev Biol 34, 265–288 [DOI] [PubMed] [Google Scholar]
- 18.Ou HD et al. (2017) ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai S et al. (2018) The in situ structures of mono-, di-, and trinucleosomes in human heterochromatin. Mol Biol Cell 29, 2450–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haws SA et al. (2022) 3D genome, on repeat: Higher-order folding principles of the heterochromatinized repetitive genome. Cell 185, 2690–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo C et al. (2018) Dynamic DNA methylation: In the right place at the right time. Science 361, 1336–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Padeken J et al. (2022) Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat Rev Mol Cell Biol DOI: 10.1038/s41580-022-00483-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X et al. (2016) Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537, 558–562 [DOI] [PubMed] [Google Scholar]
- 24.Chen Z et al. (2021) Distinct dynamics and functions of H2AK119ub1 and H3K27me3 in mouse preimplantation embryos. Nat Genet 53, 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poleshko A et al. (2017) Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell 171, 573–587.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Steensel B and Belmont AS (2017) Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poleshko A et al. (2019) H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. eLife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C et al. (2018) Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol 20, 620–631 [DOI] [PubMed] [Google Scholar]
- 29.See K et al. (2019) Lineage-specific reorganization of nuclear peripheral heterochromatin and H3K9me2 domains. Development 146, dev174078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wongtawan T et al. (2011) Histone H4K20me3 and HP1alpha are late heterochromatin markers in development, but present in undifferentiated embryonic stem cells. J Cell Sci 124, 1878–90 [DOI] [PubMed] [Google Scholar]
- 31.Ren W et al. (2021) DNMT1 reads heterochromatic H4K20me3 to reinforce LINE-1 DNA methylation. Nat Commun 12, 2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamburri S et al. (2020) Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol Cell 77, 840–856.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martire S and Banaszynski LA (2020) The roles of histone variants in fine-tuning chromatin organization and function. Nat Rev Mol Cell Biol 21, 522–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Healton SE et al. (2020) H1 linker histones silence repetitive elements by promoting both histone H3K9 methylation and chromatin compaction. Proc Natl Acad Sci U S A 117, 14251–14258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burge NL et al. (2022) H1.0 C Terminal Domain Is Integral for Altering Transcription Factor Binding within Nucleosomes. Biochemistry DOI: 10.1021/acs.biochem.2c00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machida S et al. (2018) Structural Basis of Heterochromatin Formation by Human HP1. Mol Cell 69, 385–397.e8 [DOI] [PubMed] [Google Scholar]
- 37.Minajigi A et al. (2015) Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson WL et al. (2017) RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Velazquez Camacho O et al. (2017) Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA-nucleosome association and RNA:DNA hybrid formation. Elife 6, e25293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scarola M et al. (2015) Epigenetic silencing of Oct4 by a complex containing SUV39H1 and Oct4 pseudogene lncRNA. Nat Commun 6, 7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lev I et al. (2019) H3K9me3 is required for inheritance of small RNAs that target a unique subset of newly evolved genes. Elife 8, e40448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Villaseñor R et al. (2020) ChromID identifies the protein interactome at chromatin marks. Nat Biotechnol 38, 728–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burton A and Torres-Padilla ME (2014) Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat Rev Mol Cell Biol 15, 723–34 [DOI] [PubMed] [Google Scholar]
- 44.Puschendorf M et al. (2008) PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat Genet 40, 411–20 [DOI] [PubMed] [Google Scholar]
- 45.Daujat S et al. (2009) H3K64 trimethylation marks heterochromatin and is dynamically remodeled during developmental reprogramming. Nat Struct Mol Biol 16, 777–81 [DOI] [PubMed] [Google Scholar]
- 46.Burton A et al. (2020) Heterochromatin establishment during early mammalian development is regulated by pericentromeric RNA and characterized by non-repressive H3K9me3. Nat Cell Biol 22, 767–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Probst AV et al. (2010) A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev Cell 19, 625–38 [DOI] [PubMed] [Google Scholar]
- 48.Matoba S et al. (2014) Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell 159, 884–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sankar A et al. (2020) KDM4A regulates the maternal-to-zygotic transition by protecting broad H3K4me3 domains from H3K9me3 invasion in oocytes. Nat Cell Biol 22, 380–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Probst AV et al. (2007) Structural differences in centromeric heterochromatin are spatially reconciled on fertilisation in the mouse zygote. Chromosoma 116, 403–15 [DOI] [PubMed] [Google Scholar]
- 51.Izzo A et al. (2017) Dynamic changes in H1 subtype composition during epigenetic reprogramming. The Journal of cell biology DOI: 10.1083/jcb.201611012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burton A et al. (2020) Heterochromatin establishment during early mammalian development is regulated by pericentromeric RNA and characterized by non-repressive H3K9me3. Nat Cell Biol 22, 767–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reik W et al. (2001) Epigenetic Reprogramming in Mammalian Development. Science 293, 1089–1093 [DOI] [PubMed] [Google Scholar]
- 54.Fadloun A et al. (2013) Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat Struct Mol Biol 20, 332–8 [DOI] [PubMed] [Google Scholar]
- 55.Liu J et al. (2021) The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 591, 322–326 [DOI] [PubMed] [Google Scholar]
- 56.Percharde M et al. (2018) A LINE1-Nucleolin Partnership Regulates Early Development and ESC Identity. Cell 174, 391–405 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J et al. (2020) N 6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chelmicki T et al. (2021) m(6)A RNA methylation regulates the fate of endogenous retroviruses. Nature 591, 312–316 [DOI] [PubMed] [Google Scholar]
- 59.A Field Guide to Eukaryotic Transposable Elements | Annual Review of Genetics [Online]. Available: https://www.annualreviews.org/doi/10.1146/annurev-genet-040620-022145. [Accessed: 16-Jan-2023] [DOI] [PMC free article] [PubMed]
- 60.Rodriguez-Terrones D and Torres-Padilla M-E (2018) Nimble and Ready to Mingle: Transposon Outbursts of Early Development. Trends in Genetics 34, 806–820 [DOI] [PubMed] [Google Scholar]
- 61.Rowe HM et al. (2010) KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–40 [DOI] [PubMed] [Google Scholar]
- 62.Meehan RR et al. (2003) HP1 binding to native chromatin in vitro is determined by the hinge region and not by the chromodomain. EMBO J 22, 3164–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allan RS et al. (2012) An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature 487, 249–53 [DOI] [PubMed] [Google Scholar]
- 64.Pace L et al. (2018) The epigenetic control of stemness in CD8(+) T cell fate commitment. Science 359, 177–186 [DOI] [PubMed] [Google Scholar]
- 65.Mochizuki K et al. (2021) Repression of germline genes by PRC1.6 and SETDB1 in the early embryo precedes DNA methylation-mediated silencing. Nat Commun 12, 7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ecco G et al. (2017) KRAB zinc finger proteins. Development 144, 2719–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolf D and Goff SP (2009) Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458, 1201–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pontis J et al. (2019) Hominoid-Specific Transposable Elements and KZFPs Facilitate Human Embryonic Genome Activation and Control Transcription in Naive Human ESCs. Cell Stem Cell 24, 724–735.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jacobs FMJ et al. (2014) An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 516, 242–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jachowicz JW et al. (2017) LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nature genetics 49, 1502–1510 [DOI] [PubMed] [Google Scholar]
- 71.Playfoot CJ et al. (2021) Transposable elements and their KZFP controllers are drivers of transcriptional innovation in the developing human brain. Genome Res. 31, 1531–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johansson PA et al. (2022) A cis-acting structural variation at the ZNF558 locus controls a gene regulatory network in human brain development. Cell Stem Cell 29, 52–69.e8 [DOI] [PubMed] [Google Scholar]
- 73.Venturini L et al. (2016) The stem cell zinc finger 1 (SZF1)/ZNF589 protein has a human-specific evolutionary nucleotide DNA change and acts as a regulator of cell viability in the hematopoietic system. Experimental Hematology 44, 257–268 [DOI] [PubMed] [Google Scholar]
- 74.Franco ED et al. (2021) Primate-specific ZNF808 is essential for pancreatic development in humansmedRxiv, 2021.08.23.21262262
- 75.Ecco G et al. (2016) Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Developmental Cell 36, 611–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Imbeault M et al. (2017) KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554 [DOI] [PubMed] [Google Scholar]
- 77.Cahan P et al. (2014) CellNet: network biology applied to stem cell engineering. Cell 158, 903–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu F et al. (2018) The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soufi A et al. (2015) Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fernandez Garcia M et al. (2019) Structural Features of Transcription Factors Associating with Nucleosome Binding. Mol Cell 75, 921–932.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Balsalobre A and Drouin J (2022) Pioneer factors as master regulators of the epigenome and cell fate. Nat Rev Mol Cell Biol DOI: 10.1038/s41580-022-00464-z [DOI] [PubMed] [Google Scholar]
- 82.Iwafuchi-Doi M et al. (2016) The Pioneer Transcription Factor FoxA Maintains an Accessible Nucleosome Configuration at Enhancers for Tissue-Specific Gene Activation. Mol Cell 62, 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lerner J et al. (2020) Two-Parameter Mobility Assessments Discriminate Diverse Regulatory Factor Behaviors in Chromatin. Mol Cell 79, 677–688.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roberts GA et al. (2021) Dissecting OCT4 defines the role of nucleosome binding in pluripotency. Nat Cell Biol 23, 834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tanaka H et al. (2020) Interaction of the pioneer transcription factor GATA3 with nucleosomes. Nat Commun 11, 4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dodonova SO et al. (2020) Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 580, 669–672 [DOI] [PubMed] [Google Scholar]
- 87.Chen K et al. (2020) Heterochromatin loosening by the Oct4 linker region facilitates Klf4 binding and iPSC reprogramming. EMBO J 39, e99165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen J et al. (2013) H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat Genet 45, 34–42 [DOI] [PubMed] [Google Scholar]
- 89.Mayran A et al. (2018) Pioneer factor Pax7 deploys a stable enhancer repertoire for specification of cell fate. Nat Genet 50, 259–269 [DOI] [PubMed] [Google Scholar]
- 90.Caretti G et al. (2004) The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 18, 2627–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Petruk S et al. (2017) Delayed Accumulation of H3K27me3 on Nascent DNA Is Essential for Recruitment of Transcription Factors at Early Stages of Stem Cell Differentiation. Mol Cell 66, 247–257.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Donaghey J et al. (2018) Genetic determinants and epigenetic effects of pioneer-factor occupancy. Nature genetics 50, 250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Montavon T et al. (2021) Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nat Commun 12, 4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cheloufi S et al. (2015) The histone chaperone CAF-1 safeguards somatic cell identity. Nature 528, 218–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mor N et al. (2018) Neutralizing Gatad2a-Chd4-Mbd3/NuRD Complex Facilitates Deterministic Induction of Naive Pluripotency. Cell Stem Cell 23, 412–425.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xie G et al. (2019) A conserved dimer interface connects ERH and YTH family proteins to promote gene silencing. Nat Commun 10, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sugiyama T et al. (2016) Enhancer of Rudimentary Cooperates with Conserved RNA-Processing Factors to Promote Meiotic mRNA Decay and Facultative Heterochromatin Assembly. Mol. Cell 61, 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hazra D et al. (2019) m6A mRNA Destiny: Chained to the rhYTHm by the YTH-Containing Proteins. Genes (Basel) 10, E49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nazer E et al. (2022) Seeking the truth behind the myth: Argonaute tales from “nuclearland.” Mol Cell 82, 503–513 [DOI] [PubMed] [Google Scholar]
- 100.Robbez-Masson L et al. (2018) The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 28, 836–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seczynska M et al. (2022) Genome surveillance by HUSH-mediated silencing of intronless mobile elements. Nature 601, 440–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Garland W et al. (2022) Chromatin modifier HUSH co-operates with RNA decay factor NEXT to restrict transposable element expression. Molecular Cell 82, 1691–1707.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Müller I et al. (2021) MPP8 is essential for sustaining self-renewal of ground-state pluripotent stem cells. Nat Commun 12, 3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Asimi V et al. (2022) Hijacking of transcriptional condensates by endogenous retroviruses. Nat Genet 54, 1238–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sridharan R et al. (2013) Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1gamma in reprogramming to pluripotency. Nature cell biology 15, 872–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huo X et al. (2020) The Nuclear Matrix Protein SAFB Cooperates with Major Satellite RNAs to Stabilize Heterochromatin Architecture Partially through Phase Separation. Mol Cell 77, 368–383.e7 [DOI] [PubMed] [Google Scholar]
- 107.Drakouli S et al. (2017) Enhancer of rudimentary homologue interacts with scaffold attachment factor B at the nuclear matrix to regulate SR protein phosphorylation. FEBS J 284, 2482–2500 [DOI] [PubMed] [Google Scholar]
- 108.Fang W and Bartel DP (2020) MicroRNA Clustering Assists Processing of Suboptimal MicroRNA Hairpins through the Action of the ERH Protein. Mol Cell 78, 289–302.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wei J et al. (2017) KDM4B-mediated reduction of H3K9me3 and H3K36me3 levels improves somatic cell reprogramming into pluripotency. Sci Rep 7, 7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou C et al. (2019) H3K27me3 is an epigenetic barrier while KDM6A overexpression improves nuclear reprogramming efficiency. FASEB J 33, 4638–4652 [DOI] [PubMed] [Google Scholar]
- 111.Xing G et al. (2021) MAP2K6 remodels chromatin and facilitates reprogramming by activating Gatad2b-phosphorylation dependent heterochromatin loosening. Cell Death Differ DOI: 10.1038/s41418-021-00902-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Decombe S et al. (2021) Epigenetic rewriting at centromeric DNA repeats leads to increased chromatin accessibility and chromosomal instability. Epigenetics Chromatin 14, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Escobar TM et al. (2019) Active and Repressed Chromatin Domains Exhibit Distinct Nucleosome Segregation during DNA Replication. Cell 179, 953–963 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ragunathan K et al. (2015) Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348, 1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yu H et al. (2022) Dynamic reprogramming of H3K9me3 at hominoid-specific retrotransposons during human preimplantation development. Cell Stem Cell 29, 1031–1050.e12 [DOI] [PubMed] [Google Scholar]
- 116.Barral A et al. (2022) SETDB1/NSD-dependent H3K9me3/H3K36me3 dual heterochromatin maintains gene expression profiles by bookmarking poised enhancers. Mol Cell 82, 816–832 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang Y et al. (2018) Unique molecular events during reprogramming of human somatic cells to induced pluripotent stem cells (iPSCs) at naïve state. eLife 7, e29518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rolling back human pluripotent stem cells to an eight-cell embryo-like stage | Nature [Online]. Available: https://www.nature.com/articles/s41586-022-04625-0. [Accessed: 26-Sep-2022] [DOI] [PubMed]