

SUMMARY
Patients with germline succinate dehydrogenase subunit D gene (SDHD) pathogenic variants (i.e., paraganglioma 1 syndrome / PGL1) are predominantly affected by head and neck paragangliomas (HNPGLs), which in less than 20% of patients may coexist with PGL arising from other locations (e.g., adrenal medulla, paraaortic, cardiac/thoracic, and pelvic). Given the higher risk of tumor multifocality and bilaterality for pheochromocytomas and paragangliomas (PPGLs) due to SDHD pathogenic variants than for their sporadic and other genotypic counterparts, the management of patients with SDHD PPGLs is clinically complex in terms of imaging, treatment, and management options. Furthermore, locally aggressive disease can be discovered at a young age or late in the disease course, which presents challenges in balancing surgical intervention with various medical and radiotherapeutic approaches. The axiom “first do no harm” should always be considered and an initial period of observation (watchful waiting) is often appropriate to characterize tumor behavior in patients with these pathogenic variants. These patients should be referred to specialized high-volume medical centers. This consensus statement aims to help physicians in clinical decision-making process when caring for patients with SDHD PPGLs.
Keywords: paragangliomas, genetics, guidelines, management, surgery, radiotherapy, SDHD
INTRODUCTION
Hereditary head and neck paragangliomas (HNPGLs) are the most common tumors in patients with germline succinate dehydrogenase subunit D gene (SDHD) pathogenic variants. They are typically slow-growing hypervascular tumors but have the potential to become locally aggressive. HNPGLs can be found in all anatomic sites of distribution of parasympathetic paraganglia such as in the middle ear cleft, in the jugular bulb, along the vagus nerve, and in the carotid body (Figure 1). HNPGLs are rarely functional and are inherited in 40–50% of cases. Patients with SDHD pathogenic variants may also develop thoracic, retroperitoneal, and pelvic PGLs (in less than 20% of cases and rarely in isolation) and, more rarely, gastrointestinal stromal tumors, renal cell carcinoma, and pituitary adenomas.1 These tumors should be screened during the imaging work-up of patients. Multifocality of PGL is observed in approximately 75% of cases; however, the overall risk of metastatic disease is approximately 5%. Disease penetrance is parent-of-origin dependent (genomic imprinting). After a paternal transmission of SDHD pathogenic variants, the proportion of individuals that will develop PPGL throughout their life is 90–100% whereas maternal transmission extremely rarely leads to tumor development. Genetic counseling and testing are recommended for at risk individuals with PPGL screening for those with SDHD pathogenic variants including cases of maternal inheritance.2
Figure 1. Different potential locations of HNPGLs.
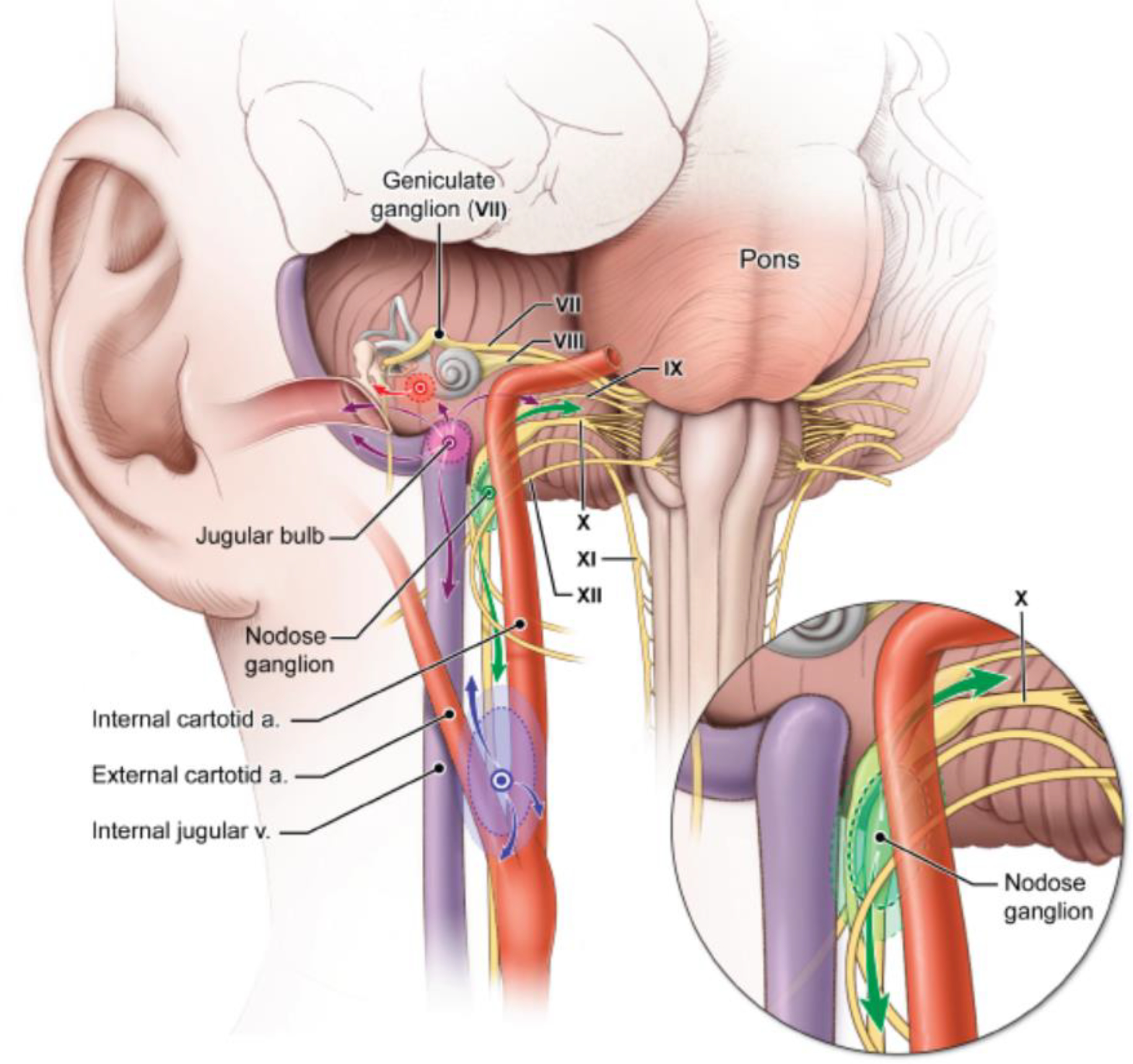
HNPGL may arise from the carotid bifurcation (purple), the nodose ganglion (green), jugular bulb (pink), and middle ear (red) and invade adjacent structures within the head and neck. Arrows indicate the potential patterns of local tumor extension for a given tumor type.
METHODOLOGY
The consensus included three chairpersons (DT, JWML, and KP) and one project manager (LM). The project was initiated in May 2021 and started with the setting-up of the working groups (steering and rating group members). The steering group included 12 members (GBW, MA, CLL, NP, SN, LA, HJLMT, ZGS, ALE, ML, ELP, LV). The rating group members included the remaining coauthors of the consensus. All steering and rating group members participating in the consensus development are experts in PPGL and represent a variety of countries, practice settings and disciplines (Endocrinology, Oncology, Surgery, Radiotherapist, Radiology, Nuclear Medicine, Genetics, Pathology).
A first zoom meeting with the steering group was held on August 18, 2021. During the meeting, the steering group members were requested to conduct their own literature searches using PubMed (US National Library of Medicine) using proposed search strategies (See Search strategy and selection criteria section). The steering group members were requested to perform a review and critical analysis of the available literature, to draft relevant graded recommendations (using GRADE) for each thematic area and supported by a concise paragraph named Evidence with the most relevant references and possible figures and tables that support evidence.
Search strategy and selection criteria.
The steering group members were requested to conduct their own literature searches in PubMed (US National Library of Medicine) using search strategies with controlled vocabulary MeSH terms and keywords for the condition of interest and section topic. Searches were limited to those articles published after 2000. The search strategies combined the term Paraganglioma and the following terms into separate searches for each section topic: “Succinate Dehydrogenase”, SDHD, “positron emission tomography”, PET/CT, “Radiotherapy”[MeSH], “Endocrine Surgical Procedures”[MeSH], “General Surgery”[MeSH], Radiofrequency, Chemoembolization, Cryoablation, “Thermal Ablation”, Surveillance, “Follow-up”, Chemotherapy, Sunitinib, “Temozolomide”, Immunotherapy, “Peptide receptor radionuclide therapy”, PRRT, DOTATATE, DOTATOC, DOTANOC, Somatostatin, MIBG. During screening of the results, articles were excluded if they were animal studies, case reports or case series, and not published in English.
In February 2022, the rating group members received the proposed recommendations with evidence and supplemental Tables but without the ratings of the strength of grading and quality of evidence. Each rating group member voted whether they agreed or disagreed with the narrative forms of the recommendations (strongly agree, agree, neither agree or disagree, disagree, strongly disagree, don’t know/other), and then rated the strength of the proposed grading (1=strong, 2=weak) and quality of the evidence using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) (⊕ to ⊕⊕⊕⊕).3 They could also leave further comments or suggestions about the reason why they agreed/disagreed or why they did do any ratings (e.g. lack of expertise in a specific topic) with the proposal (optional, not required).
The results of the responses from the rating group members were reported in PowerPoint slides and presented to steering and rating group members during two initial zoom meetings (May 2, 2022, May 18, 2022). During these meetings, any discordances between the rating and steering groups were discussed in order to find consensus on the phrasing of the recommendations and grades regarding the strength of the proposal and quality of the evidence. After this initial period, two additional rounds of voting were conducted with the rating group via Google Forms. Rating group members were requested to indicate their agreement or disagreement with the phrasing of the recommendations, strength of recommendation (GRADE) and quality of evidence. A total of two additional virtual meetings (June 22, 2022, September 9, 2022) were conducted with the rating group and steering group members in order to find a consensus. After the last meeting in September 2022, the chairpersons and project manager drafted the final version of the consensus statement and sent the manuscript with supplemental files to all steering and rating group members for final review and approval.
RECOMMENDATIONS
Healthcare environment for patients with SDHD PPGLs (R1)
R1. We recommend that all decisions in patients with SDHD PPGLs be discussed and managed by an expert interdisciplinary tumor team familiar with the disease to ensure favorable outcomes and appropriate follow-up (Grade 1 ⊕○○○).
Evidence
There is no solid evidence from clinical studies showing favorable outcomes if patients are managed by an expert interdisciplinary team with expertise in PPGL. However, PPGL is a complex and heterogeneous disease involving many organs in a variable way. Due to the rarity of these tumors, most physicians will have limited experience in this field, in particular with covering the competencies of the different involved specialties. Therefore, this recommendation is mainly based on the experience and conviction of many international experts that interdisciplinary discussion of management decisions of patients with PPGL is the optimal approach. Such approach facilitates tailoring of clinical management to the individual patient level. This extends beyond diagnosis and treatment, even to offering the most appropriate individualized follow-up and surveillance.4,5 Expert interdisciplinary teams are operative in clinical centers specialized in PPGL. This includes but is not limited to the ENS@T Centers of Excellence and the Clinical Centers of Excellence accredited by the Pheochromocytoma Paraganglioma Alliance. This applies also to any clinical center as long as regular interdisciplinary expert-team discussions are operational.
Initial work-up for patients with SDHD PPGLs (R2-R4)
R2. We recommend that patients with PPGL and germline SDHD pathogenic variants should be evaluated by clinical assessment and biochemical measurements (metanephrines in plasma or urine and plasma 3-methoxytyramine [MTY]) (Grade 1⊕⊕⊕○).
R3. We recommend performing head/neck magnetic resonance imaging (MRI) as the first modality for patients with HNPGL to screen for assessment of multifocality and tumor extension (Grade 1⊕⊕⊕○).
R4. To search for SDHD PPGL in patients on a whole-body scale, we recommend performing whole-body anatomic imaging together with positron emission tomography (PET) (preferably with radiolabeled somatostatin analogs) as the first choice (Grade 1⊕⊕⊕○).
Evidence
In patients with SDHD HNPGL, symptoms and signs are often delayed and related to local mass effects caused by large tumors rather than catecholamine excess.
Patients with HNPGL with plasma normetanephrine levels more than double the upper reference limit are rare (2.3%). Increased normetanephrine levels in SDHD patients are therefore more likely to be related to the presence of PGL outside the head and neck region.6 Furthermore, up to 30% of HNPGLs produce dopamine, as indicated by increases in plasma methoxytyramine (MTY).6–8 Urine MTY is not a useful biomarker of tumoral dopamine production in these tumors since urine dopamine is derived almost exclusively from renal uptake and decarboxylation of circulating 3,4-dihydroxyphenylalanine (DOPA). Biochemical workup should include plasma (usually preferred) or urine metanephrines and plasma MTY.9–11
Imaging plays a vital role in the evaluation of patients with SDHD PPGLs. Imaging should encompass the base of the skull to the pelvis. Magnetic resonance imaging (MRI) with angiography sequences (MRA) are the most sensitive radiological techniques for HNPGL staging.12–15 Various MR acquisition protocols have been described in the literature, including one large study that evaluated MRA in 157 HNPGL patients with germline pathogenic variants (63/157 SDHD). In this study, a combination of contrast-enhanced three-dimensional time-of-flight angiography at arterial phase and axial plane fast spin-echo T1-weighted sequence with fat saturation showed a sensitivity and specificity of 88.7% and 93.7%, respectively.16 For patients with skull base HNPGLs, temporal bone CT provides irreplaceable information on the extent of bone involvement. Cervico-thoraco-abdominalpelvic PPGLs can also be visualized using computed tomography (CT). CT scan with intravenous contrast is less costly and time consuming than MRI and particularly useful for perioperative planning. CT is therefore often preferred over MRI at initial evaluation of patients, with the exception of certain pediatric cases or during pregnancy.
Functional imaging complements anatomic imaging for whole-body disease staging and can exclude other potential diagnoses. Patients with SDHD PPGLs typically exhibit strong somatostatin receptor subtype 2 expressions, which is reflected by the high sensitivity of somatostatin-receptor (SSTR)-guided PET/CT using gallium-68 radiolabeled somatostatin analogs.17–21 Its sensitivity approaches 100% for HN parasympathetic primary and metastatic lesions, but it appears to be less sensitive for primary sympathetic tumors.22 However, more extensive imaging data for patients with SDHD pathogenic variants are still lacking. 6-[18F]-L-DOPA (6-[18F]fluoro-l-3,4-dihydroxy-phenylalanine (18F-FDOPA) PET has also shown high sensitivity in the detection of SDHD-related HNPGLs23,24 and is a very good alternative to SSTR PET using 68Ga-radiolabeled somatostatin analogs if these are unavailable.25 18F-FDOPA, however, overlooks some sympathetic (outside head and neck area) PPGLs.26,27 18F-FDG PET has shown excellent results in the evaluation of patients with PPGLs harboring germline pathogenic variants in one of the four genes that encode the SDH complex (collective termed as SDHx)28–31. However, this imaging modality has been surpassed by SSTR PET 19 especially, in the detection of SDHD-related HNPGLs (sensitivity ranging 70–90% for 18F-FDG versus nearly 100% for SSTR PET). Its lower clinical value is partially due to the less favorable tumor-to-background uptake ratio than those of specific radiopharmaceuticals and potential drawbacks due to uptake by brown adipose tissue and reactive lymph nodes. Functional imaging findings should be interpreted by a physician who is experienced in PPGL imaging and should consider possible pitfalls, variants, and caveats (Table 1). Compared with PET/CT imaging, 123I-Metaiodobenzylguanidine (123I-MIBG) and 111In-pentetreotide scintigraphy are suboptimal and should not be employed in a purely diagnostic setting.
Table 1.
| PET radiopharmaceuticals | Pitfalls |
|---|---|
|
| |
| SSTR analogs | Uncinate process Stellate ganglia Splenunculi (accessory spleens), splenosis Pancreatic heterotopia Pancreatic serous cystadenoma Bone hemangioma, enchondroma, fibrous dysplasia Active chronic inflammation (e.g., sarcoidosis, tuberculosis, Hashimoto's thyroiditis) Other tumors (e.g., meningioma, breast cancer, renal cancer, lymphoma, thyroid neoplasms, glioma, neuroblastoma) |
|
| |
| 18F-FDOPA | Solid pseudopapillary tumor of the pancreas Thyroid neoplasm Pituitary adenoma Squamous cell carcinoma Poorly differentiated adenocarcinoma Melanoma Multiple myeloma Hepatocellular carcinoma Schwannoma Chondrosarcoma |
Evaluation of surgical interventions for patients with SDHD PPGLs (R5-R14)
Patients with SDHD-related HNPGLs
Recommendations
R5. We recommend that patients with vagal PGL rarely be considered for resection due to the high risk of resultant vocal cord paralysis (Grade 1⊕⊕○○).
R6. We recommend that newly diagnosed patients with jugular, vagal, and carotid PGL without compelling indications for treatment undergo an initial trial of observation to characterize tumor behavior (Grade 1⊕⊕○○).
Evidence
Due to their slow-growing pattern, the axiom “first do no harm” is particularly relevant to the management of patients with HNPGLs (Figure 2). Newly diagnosed patients with SDHD pathogenic variants, notably those with non-tympanicum HNPGL and those without an urgent indication for resection (severe or progressive symptoms/signs from cranial neuropathy including brainstem compression, severe pain, bleeding, and brain ischemia), are good candidates for an initial observation trial. Patients with tympanic PGL often present with hearing loss and pulsatile tinnitus: resection is safe in experienced hands, often done on an outpatient basis, and has a low incidence of complications.
Figure 2. Current synthesis of the important clinical facts that contribute to the decision-making process.
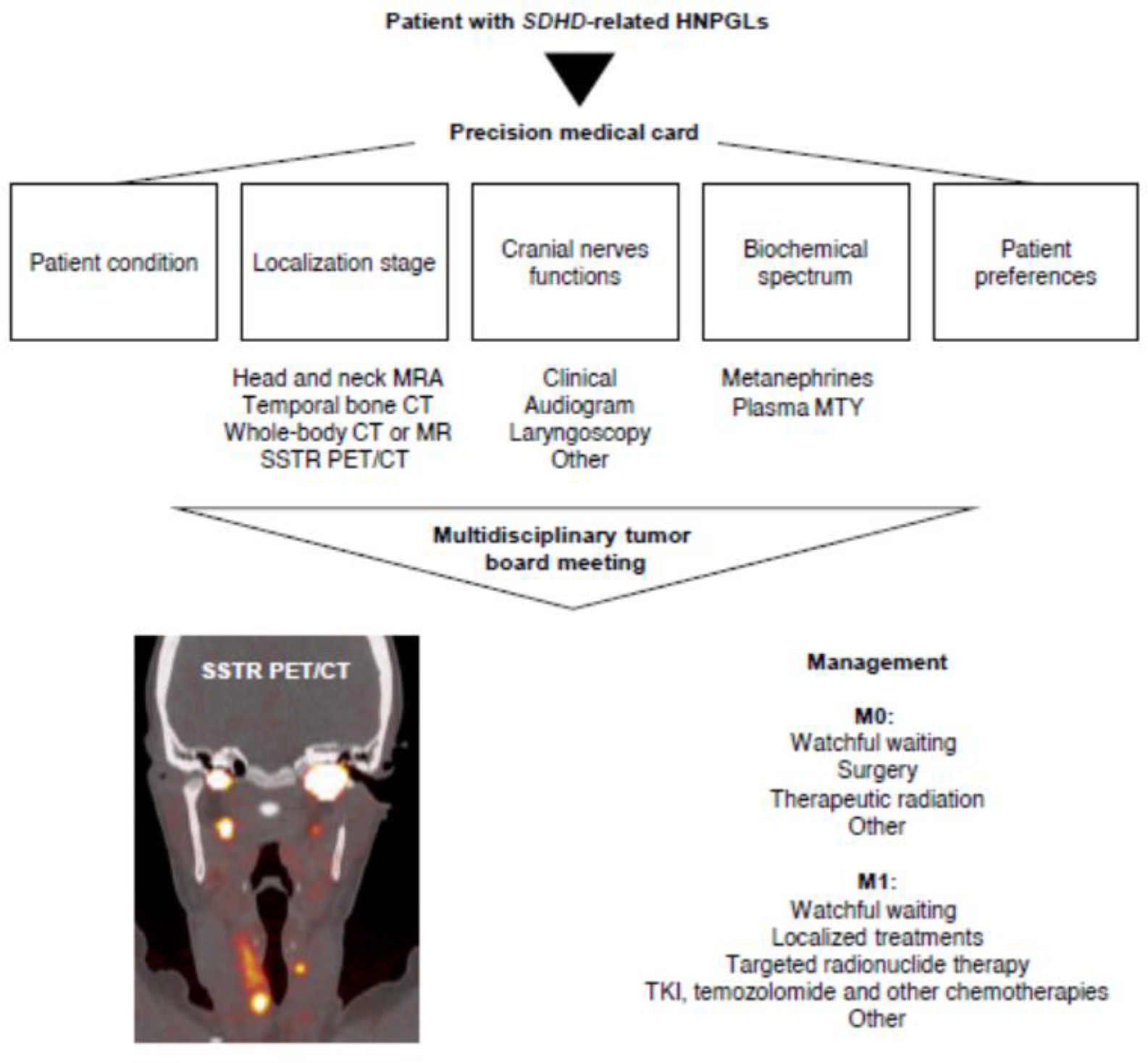
cf means refer to; MRA, magnetic resonance angiography; SSTR PET/CT, somatostatin receptor PET/CT showing tumor multifocality; M0, absence of metastasis; M1, presence of metastasis; MTY, plasma 3-methoxytyramine; TKI, tyrosine kinase inhibitors.
Elective surgical resection of HNPGL should be avoided in elderly and debilitated patients, as well as in those with an inability to tolerate specific cranial neuropathies.32–37 Thus, particular attention must be paid to the patient’s swallowing function and pulmonary reserve as significant dysphagia and aspiration may result from damage to or sacrifice of the lower cranial nerves.33,34,38 As these lesions are typically benign and indolent, a trial of observation in these populations seems to be reasonable and justified.36,39,40 Vagal PGL, in particular, pose a challenge as resection in most patients by default results in vagal nerve sacrifice and resultant vocal cord paralysis.41,42 Surgical intervention on such lesions, should it occur, is generally only performed after the vocal cord is already immobile and is not performed on bilateral lesions as bilateral vocal cord paralysis often leads to the need for tracheostomy.33 Similar caution must be exercised in other scenarios in which bilateral or multifocal lateral skull base disease is present; an extant cranial neuropathy on the unoperated or previously operated side can be devastating for recovery should bilateral paralysis arise after the intervention.32,33,36 Exceptions can be made for patients with bilateral carotid or jugular PGLs in which a staged approach is taken and there is minimal morbidity from resection of the first/initial side (Figures 1 and 2). Primary lesions with distant metastasis and metastases themselves, although rare, should be operated on only in select circumstances; this is generally done with palliative intent, and the proposed intervention should not cause more harm than relief.32 Additional consideration must be given to the patient’s preference. There are no clear size cutoffs for when to refrain from operating on HNPGLs; we recommend that patients be referred to an experienced team.
R7. We recommend intervening (which may include surgical resection) on patients with HNPGL demonstrating sustained growth or compression of vital head and neck structures and in those lesions that progress after radiation (Grade 1⊕⊕○○).
R8. We recommend that for patients with any jugular and large carotid/vagal PGL undergoing surgery, preoperative angiography with embolization be considered. Balloon occlusion testing should be considered if internal carotid sacrifice with reconstruction is contemplated (Grade 1⊕⊕○○).
R9. We recommend an individualized multidisciplinary approach for patients with multifocal HNPGLs, with particular attention paid to avoiding compromising important neurovascular structures. Staging resection is key to minimizing potential morbidity (Grade 1⊕⊕○○○).
Evidence
Indications for surgery include active signs and symptoms, such as compression of head and neck structures, in some cases sustained (especially more rapid) growth, intractable pain, progression after radiation, extent cranial neuropathy, some catecholamine-secreting lesions and/or a low likelihood of postoperative cranial neuropathies, and other sources of morbidity.33,35,42,43 Small tumors in young and otherwise healthy patients are generally ideal, with high local control rates.33 The surgeon must weigh the risk of new cranial neuropathies and make informed decisions with the patient and team. For carotid PGLs, for example, lesions with a higher Shamblin classification (i.e., degree of carotid artery involvement) have a higher risk of cranial neuropathy.42 Additionally, tumors sized >5 cm have a higher cranial neuropathy rate of 67% than lesions sized <5 cm (14%);33,44 when this is compared to the natural history of other HNPGLs, new or progressive deficits are seen in 30–33% of cases.39,40,45 Being able to compare the nerve deficit rates across modalities is an important component of preoperative counseling and decision-making. To standardize reporting and outcomes, jugulotympanic PGLs may be classified according to the Fisch 46 and Glasscock-Jackson staging systems.47
Additionally, anticipating the extent of resection and involving the appropriate surgical services is paramount; collaboration with vascular surgery should occur any time there is a question as to the need for carotid sacrifice,48 and neurosurgery should be available for skull base lesions with an intracranial extent. Carotid stenting or sacrifice with subsequent reconstruction should only be used in select circumstances and those with adequate collateral intracranial circulation.
The classic approach to patients with jugular PGLs requires ear canal overclosure and facial nerve mobilization, resulting in facial paresis and significant conductive hearing loss. To minimize morbidity, subtotal resection, particularly of large jugular PGLs with preservation of the lower cranial nerve, may be considered.35,40
Preoperative angiography with embolization is recommended for all jugular, large (>4 cm), or locally invasive carotid/vagal PGLs. Balloon occlusion testing is recommended for lesions that encase the internal carotid artery and in those where carotid sacrifice and reconstruction are a remote possibility. The primary goal of preoperative embolization of HNPGLs is to help achieve a dry surgical field to visualize key neurovascular structures critical for reducing surgical morbidity and increasing the probability of gross total resection.36,42,49 Angiography and embolization are not without risk, nor are they guaranteed to be reflective of the body’s response to internal carotid artery disruption, and temporary or permanent cranial neuropathy may result even with superselective embolization due to migration of particles to the vasa nervorum of the affected nerves.
There is no simple algorithm that best addresses the therapeutic strategy in patients with multifocal HNPGLs (Figure 3).32 An individualized approach is recommended, as is the use of an experienced multidisciplinary team that includes various surgical teams, endocrinology, radiation oncology, and speech and swallowing therapy. The estimation of when and how to intervene is particularly difficult given that all lesions are not present simultaneously. The possibility of future metachronous lesions further complicates the clinical case.32
Figure 3. Management of patients with SDHD-related HNPGLs with special emphasis on tumor multifocality.
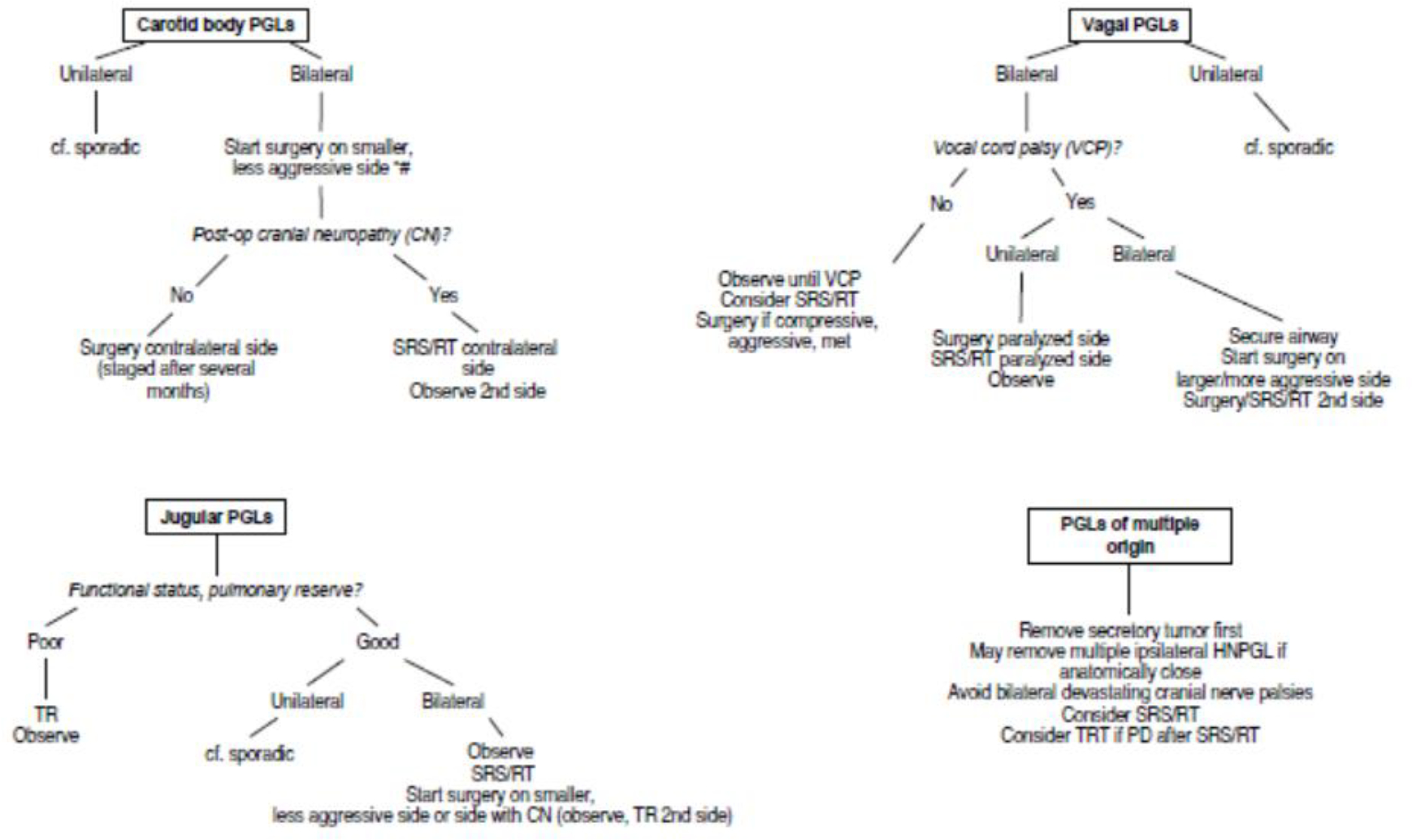
Jugular PGLs are also named jugulotympanic PGLs; SRS, stereotactic radiosurgery (preferred radiotherapeutic option); RT, hypofractionated radiotherapy; CN, cranial neuropathy; PD, progressive disease; MET, metastatic; TRT, targeted radionuclide therapy; Adx, adrenalectomy; HNPGLs, head and neck paragangliomas; PGL, paraganglioma; VCT, vocal cord palsy
*For patients without preoperative neuropathy
#Some authors prefer to start on the side of the larger tumor
Important determinants of treatment include the patient’s life expectancy, tumor behavior, baseline neurological/cranial nerve status, swallowing function, and pulmonary reserve.32,33 The overarching goals of treatment should be to exercise appropriate restraint, minimize the risk of multiple and/or bilateral cranial nerve deficits, and not compromise the major cerebral vasculature. In the case of bilateral tumors, staging should be implemented to minimize bilateral, potentially devastating cranial neuropathies.32,33,42 While there is no wide consensus on special circumstances, some groups advocate resecting multiple head and neck tumors in a single stage if they are ipsilateral and anatomically close.32,42 In the case of bilateral tumors, some authors recommend operating on the side with existing cranial neuropathies and observing or radiating the contralateral side to avoid bilateral nerve palsies. If no neuropathies exist preoperatively, resection of the smaller of the two lesions poses a lower risk to the cranial nerves. If there is no nerve deficit, the contralateral side may be subsequently attempted. If there is a postoperative deficit, the contralateral lesion should be observed or radiated.32,42 Special consideration should be given to avoid baroreflex failure in patients with bilateral carotid PGLs or intracranial hypertension in patients with bilateral jugular PGLs. Resecting one side and staging resection of the contralateral tumor several months later can decrease these complications and allow for compensation.32 If observation is chosen, median growth rates in these lesions may be as low as 1.0 mm/year with a median doubling time of 4.2–5.7 years40, depending on method of comparison (linear measurements versus volumes) and the assumptions of the mathematical models utilized.
R10. We recommend a thorough cranial nerve examination and laryngoscopy before and after surgical intervention or radiotherapy for patients with HNPGL (Grade 1⊕⊕○○).
R11. We recommend that in cases of postoperative facial nerve palsy, corneal protection be prioritized to avoid exposure keratitis or corneal abrasion (Grade 1⊕⊕⊕○).
Evidence
Routine pre- and postoperative screenings for cranial neuropathies are recommended, with a focused evaluation of the nerves at risk from surgical intervention. All patients should be evaluated for palsies of VII-XII, as measured by symmetric facial movement, audiogram, evaluation of swallow/dysphagia, flexible bedside laryngoscopy33, and evaluation of palate rise, shoulder elevation, and tongue mobility. Additional facets of the neurological examination may be incorporated depending on the specific lesion(s).
New cranial neuropathies are rather common after surgical intervention of patients with HNPGLs, with particularly high rates in jugular PGLs with intracranial extension,50–52 large and/or invasive carotid body PGLs,33,44 and virtually all vagal PGLs. However, new or progressive deficits are also observed in 30–33% of patients with observed lesions.39,45 Cranial neuropathies causing dysphagia, aspiration, or facial paralysis after HNPGLs resection may prolong hospitalization and recovery and have a profound effect on the quality of life.
Patients with SDHD-related non-HNPGLs and pheochromocytomas
R12. We recommend that functional PPGLs (which are predominantly retroperitoneal) be resected as an initial priority in patients with multifocal disease including HNPGLs (Grade 1⊕⊕⊕○).
R13. We recommend that patients with non-HNPGL and pheochromocytoma (adrenal, retroperitoneal, pelvic, or thoracic) be offered appropriate surgical consultation with an experienced surgeon with knowledge of this specific disease. Tumor resection should be considered when there are no absolute contraindications, especially when complete tumor removal is possible (Grade 1⊕⊕⊕○).
R14. We recommend a limited role for palliative debulking in patients with locally aggressive, large tumors with high probability of incomplete surgical resection, or metastatic disease, but this can be considered in patients who are not responsive to medical management or have debilitating sequelae such as pain or mass effects that worsen quality of life (Grade 1 ⊕⊕○○).
Evidence
SDHD carriers have a high incidence of multifocality, and a thorough preoperative cross-sectional nuclear medicine evaluation should be performed for complete surgical planning. Biochemically positive PGLs can be present in the context of pathogenic variants and are mostly related to retroperitoneal PPGLs. These tumors should be resected prior to other surgical interventions because of the risk of a perioperative hypertensive crisis (see R21).
The main objective of surgical resection for patients with SDHD-related PPGLs is to improve symptoms by removing the source of excess catecholamine secretion, preventing further tumor growth, and minimizing the risk of metastatic disease. The current estimated rates of metastatic behavior in patients with SDHD-associated PPGLs range between 4.5 and 7.7%.53–55
Pheochromocytoma (PHEO) treatment is typically surgical and often amenable via minimally invasive surgery. The technical approach can be either anterior or posterior (posterior retroperitoneoscopic adrenalectomy) depending on surgical expertise. There are no modalities to ensure complete removal of the medullary tissue unless the entire gland is removed. Because of multifocality and potential for missing small tumors, a cortical-sparing technique may not be always ideal. These factors should be taken into consideration and weighed against the risk of potential adrenal insufficiency if contralateral tumor develops.
An open approach is recommended rather than a laparoscopic approach for most patients with primary SDHD PPGLs when the size is >5–6 cm because of the need to assess the locoregional nodal disease.56,57
Retroperitoneal extra-adrenal SDHD and others PGLs can be locally invasive with major vessel involvement of the inferior vena cava, aorta, renal vein, and superior mesenteric artery/vein. When technically possible, complete resection may require vascular reconstruction. In a retrospective study including 29 patients with PGLs and major blood vessel involvement, the authors report that overall survival was higher in patients who underwent complete tumor resection than in those who underwent only medical management.58 There are no prospective clinical trials directly comparing laparoscopic or robotic versus open adrenalectomy for patients with PGLs. A few case reports have demonstrated the effectiveness and safety of laparoscopic surgery for patients with small tumors and no invasion into any surrounding structure.59 The difficulty lies in lack of preparation for intraoperative identification of tumors that encroach on surrounding structures. These tumors are more likely to be adherent without distinguishable tissue planes and require proximal and distal vascular control, multiple vessel ligation, and potential vascular reconstruction. Safe resection requires manual assessment, palpation, careful retraction, and the ability to cross-clamp large vessels. Common locations of extra-adrenal SDHD PPGLs include the bladder, heart, and the area between or above the aortic bifurcation e.g., the inferior mesenteric artery (the organ of Zuckerkandl). Patients with PGLs that arise in the pelvis can have unique presentations and deserve special attention during surgery, given the proximity of these tumors to the parasympathetic region, especially in males, discussion and consideration of subsequent sexual dysfunction should be considered and appropriately discussed with a patient. Additionally, sexual dysfunction has been reported as a potential complication after other pelvic and aortic surgeries, which must be discussed, especially for patients with a PGL located in the organ of Zuckerkandl.
The management of patients with thoracic SDHD PGLs is complex and technically challenging. Complete anatomical involvement of the tumor should be determined prior to resection. En bloc removal provides the best long-term outcome and freedom from recurrence. Cardiac SDHD PGLs, although rarely malignant, often involve cardiac structures such as the left atrium and ventricle, pulmonary artery, and coronary arteries without a distinct border.60 Imaging often underestimates the actual involvement at the time of operative resection. Multiple cardiac chamber reconstruction may often be required with coronary artery bypass if coronary vessels are involved, and in rare cases, cardiac auto transplantation may be required. En bloc resection of the tumor is required in all cases. For patients with thoracic, para-aortic, and pelvic PGLs, open operations allow for the manual interpretation of two important aspects of the technical procedure: assessment of the extent of vascular wall invasion and presence of lymph node disease. Interpretation of imaging and anticipation of invasion or adherence to vessels are paramount in planning. The reactive formation, particularly in chest structures, seems to be locally more difficult and requires specialized cardiac surgical expertise.
On occasion, SDHD PGLs may occur in a location where surgical resection cannot be safely accomplished, and other therapies are required to control both hormone hypersecretion and tumor growth.
A perioperative hemodynamic management plan should be devised to prevent instability and complications during the perioperative period. Beyond pharmacological preparation, this requires good communication among multiple specialties, including the availability of experienced anesthesiologists. Excellent intraoperative communication with the surgical team and understanding the half-life and effects of pharmacological agents are important factors in the management of intravascular volume, heart rate, and blood pressure.
Palliative debulking rarely grants pharmacological independence; one study showed that aggressive debulking for biochemical management alone may not be effective as only seven of 24 cases had a partial biochemical response, with six of seven cases recurring within 12 months.61 The authors also reported that resection can selectively relieve certain tumor-associated symptoms and signs such as pain.
Therapeutic radiation for patients with SDHD PPGLs (R15-R18)
Patients with SDHD-related HNPGLs
R15. We recommend therapeutic radiation as a treatment for patients with SDHD HNPGLs, more specifically for patients with radiologically progressive or symptomatic SDHD HNPGLs. Older patients with multiple comorbidities, and/or highly complex surgical resectability of tumors with cranial nerve palsies, such as vagus nerve involvement and contralateral lower cranial neuropathies, are strong candidates for primary therapeutic radiation (Grade 1⊕⊕○○).
R16. We recommend therapeutic radiation for patients with post-surgical residual and recurrent SDHD HNPGLs with progressive disease (Grade 1⊕⊕○○).
Evidence
We recommend a multidisciplinary discussion on therapeutic radiation for each patient with SDHD HNPGLs (Figure 2).
Therapeutic radiation, specifically stereotactic radiosurgery (SRS), should be considered the primary treatment for all patients with SDHD HNPGLs, including in elderly patients, those with significant comorbidities, or those with cranial neuropathies.62–64 Hypofractionated SRS may be preferred in cases of contralateral lower cranial neuropathies or multifocal disease involving the bilateral vagal nerves as it is an effective method to preserve cranial nerves, even in large tumors. 65,66
Therapeutic radiation should be considered as a secondary treatment for progressive lesions after surgical resection33 or planned as an adjuvant treatment 8–12 weeks after subtotal resection.65 Single-fraction SRS is most effective in smaller tumors (maximum diameter <3 cm)66 but has also demonstrated efficacy in residual tumors with volume ≤4 cm3. These series found a median marginal tumor dose of 14 Gy with 80% of cases demonstrating tumor stability and 20% with shrinkage and no clinical progression.67 Gamma knife radiosurgery in patients with post-surgical jugulotympanic PGL patients also showed volumetric tumor control and clinical control in 94.8% and 91.4% of cases, respectively.68
R17. We recommend SRS as the primary or complementary treatment for surgical resection. Hypofractionated radiotherapy may be recommended for patients with larger SDHD HNPGLs (Grade 1⊕⊕⊕○).
Evidence
Radiation therapy is a treatment modality that works with ionizing radiation, generating free radicals causing breaks in DNA and cell death through apoptosis, as well as via mitotic cell death.64,69 Conventional fractionated external beam radiation therapy is typically delivered as intensity-modulated radiation therapy (IMRT). IMRT has been reported to have control rates and toxicities similar to those of SRS.68,70,71 Compared to traditional multi-fraction IMRT occurring over several weeks, SRS is one to five fractions. Each fraction is a single daily fraction, thereby having no more than 5 days of SRS treatment for the same efficacy of radiation therapy compared to IMRT. Furthermore, SRS has several advantages over IMRT. First, SRS has a lower biological effective dose than conventional radiation therapy, thereby reducing the radiation dose to the surrounding normal tissues. Second, SRS also provides submillimeter accuracy of the tumor target with a steep dose gradient, minimizing radiation exposure to nearby critical structures. Single-fraction SRS is considered most effective in smaller tumors (maximum diameter <3 cm) and has also been shown to be equally efficacious, with toxicity rates similar to or lower than those of hypofractionated radiotherapy.66,72
There are multiple equivalents of SRS, with linear accelerator-based types such as linear accelerator (LINAC), Gamma Knife, and CyberKnife being the most common.73 A metaanalysis that examined radiosurgical treatment of patients with jugular PGLs showed that Gamma Knife, LINAC, and CyberKnife technologies all exhibited similarly high rates of tumor control (95%) and clinical control (97%) across all studies.74
A meta-analysis of 15 studies (2018) reviewing the treatment of patients with jugular PGLs with SRS as the primary treatment showed tumor control, symptom control and complications in 92%, 93%, and 8% of patients, respectively. The analysis also showed that smaller tumor volumes predicted symptomatic improvement.75 The North American Gamma Knife Consortium collated the outcomes of eight gamma knife radiosurgical centers that had treated patients with jugular PGLs with a median tumor margin dose of 15 Gy (n=132, 134 procedures) and demonstrated actuarial tumor control of 88%, 5 years after radiosurgery. Improvement in pre-existing cranial nerve deficits was observed in 11% of patients, new or progressive cranial neuropathies were seen in 15%, and no mortality was noted (Supplemental Table 1).76 In other smaller series of patients with jugular PGLs, tumor control was noted to be 94.7–100%.77–79 Additionally, in previous small series reports, LINAC radiosurgery treatment of patients with jugular PGLs showed tumor control rates of 91–100%.80–82 A long-term series of jugular PGLs treated with frameless LINAC-based SRS over nearly two decades demonstrated a 7-year progression-free survival of 97.0% and 7.7% grade 1/2 (Common Terminology Criteria for Adverse Events system) toxicities.83
Because carotid body SDHD PGLs are the most common type of HNPGLs, surgical resection is the most common treatment. To date, only one systematic review has compared the results of surgical resection to those of IMRT (not only including SDHD ones) in 2,302 patients across 80 publications.84 Long-term tumor control was noted in 94.5% of IMRT patients and 93.8% of surgical patients. However, surgically induced cranial neuropathies are four times more common than those induced by IMRT.
In a meta-analysis examining treatment outcomes for patients with vagal PGLs, local tumor control rates were similar between surgery and therapeutic radiation (93.3%), with a mean follow-up of 86.7 months.64
Radiation-induced malignancy rates have been historically difficult to assess because of variability in follow-up, rarity of HNPGLs, and varying methods of radiotherapeutic treatment.85 The Mayo Clinic reviewed the institutional data of all HNPGLs patients who received either external beam radiation therapy or SRS and found no radiation-induced malignancy.86 This is consistent with the historical risk of 0.28%87 and a recent publication indicating that SRS is likely to have a lower risk of radiation-induced malignancy than traditional external beam therapy because of a lower median dose.88
Patients with SDHD-related non-HNPGLs
R18. We suggest considering therapeutic radiation for patients with symptomatic or progressive chest, abdomen, and pelvis SDHD PGLs that cannot be resected (Grade 2⊕○○○).
Evidence
Limited literature supports the role of external beam radiotherapy in the treatment of patients with PGLs below the neck as these tumors are generally managed with surgical resection. Radiation can provide high rates of local control for patients with advanced and unresectable PGLs to both primary and metastatic sites.89–91 While dose and fractionation data can be extrapolated from the HNPGL literature, radiation to the chest and abdomen has unique considerations, including respiratory motion and interfraction deformation of anatomy and organs at risk. Higher radiation doses have been associated with improved local control.89 The use of advanced radiation technologies, including IMRT and stereotactic body radiotherapy with adequate motion management, can allow safe dose escalation in the setting of radiosensitive structures such as the small bowel. Published series on radiation for patients with non-HNPGLs have used standard fractionation (1.8–2 Gy/fraction) or fractionated stereotactic ablative radiotherapy rather than single fraction SRS, as is commonly used in patients with HNPGLs.
Medical management of patients with SDHD PPGLs (R19-R22)
R19. We recommend the use of alpha-adrenoceptor blockers as medical treatment for the management of norepinephrine-associated manifestations (Grade 1 ⊕⊕○○).
R20. We recommend avoiding the use of medications that may elicit a catecholamine crisis in patients with catecholamine-producing SDHD PPGLs who do not receive appropriate adrenoceptor blockade (Grade 1 ⊕○○○).
R21. We recommend the use of alpha-adrenoceptor blockers prior to any surgical and non-surgical treatment interventions in patients with SDHD PPGLs demonstrating norepinephrine production (Grade 1 ⊕⊕○○).
R22. We do not recommend the use of medical treatment prior to interventions for patients with exclusively dopamine-producing SDHD PPGLs (as indicated by isolated elevation of plasma MTY) (Grade 1 ⊕○○○).
Evidence
Preoperative biochemical screening is mandatory to avoid rare but catastrophic perioperative complications in patients treated surgically, regardless of the presence of symptoms and signs. Patients with norepinephrine-producing SDHD PPGLs should be treated with α-adrenoceptor blockade prior to any therapeutic intervention. Norepinephrine production is defined and recognized by an elevation in plasma and/or urine normetanephrine. Some patients also have concurrent elevation of plasma and/or urine norepinephrine. Tumors displaying norepinephrine production (not only secretion as reflected by elevated normetanephrine) require pretreatment. 92
In the case of tachycardia during α-adrenoceptor blockade, a β-adrenoceptor blocker could be added. Metyrosine, which inhibits tyrosine hydroxylase and thereby catecholamine biosynthesis, can be used as an add-on drug where available. Monotherapy with (non-selective) β-adrenoceptor blockers can elicit hypertension and is contraindicated. The exclusive production and subsequent secretion of dopamine by HNPGLs is unlikely to provoke any significant hemodynamic effects.93
Even in rare cases of patients with larger dopamine-only producing SDHD PPGLs, or in isolated metastatic cases of overwhelming dopamine excess patients are typically normotensive or even hypotensive.94–96 Therefore, in patients with dopamine-only SDHD HNPGLs, management with α-adrenoceptor blockers before any type of treatment is not advised.
In contrast, preoperative adrenoceptor blockade can be considered in rare cases of patients with norepinephrine-producing SDHD HNPGLs. It should be ascertained that there are no additional sympathetic PPGLs as a source of norepinephrine for which treatment might be prioritized. In a single-center series of 152 patients with 182 HNGLs97, 7.7% of tumors were deemed clinically significant secretors of catecholamines based on the presence of explicitly documented hyperadrenergic symptoms (sustained or intermittent palpitations, tachycardia, diaphoresis, tremors, or new-onset hypertension in conjunction with at least one of these other symptoms) and/or elevated levels of normetanephrine (i.e., ≥2-fold the upper reference limit). This subgroup was treated with α- or β-adrenoceptor blockade prior to surgery, radiotherapy, or during observation, whereas pretreatment was omitted in patients with clinically insignificant increased normetanephrine levels. A review of anesthesia records showed no instances of hemodynamic instability requiring vasopressors, aggressive fluid resuscitation, or antihypertensives. Although a small number of patients with clinically insignificant elevations of catecholamines did not have any identifiable perioperative complications, whether they should also receive perioperative blockade remains debatable.
To our knowledge, a catecholamine crisis elicited by radiation therapy or systemic therapy for patients with SDHD PPGL is not common. Nevertheless, in the case of norepinephrine production usually reflected by elevated plasma normetanephrine but not necessarily plasma norepinephrine, treatment with appropriate adrenoceptor blockade should be considered before these interventions. Therefore, patients must be carefully monitored before, during, and after any procedure and vigorously treated in cases of hemodynamic instability. Postoperative baroreflex failure is associated with surgery and radiotherapy for patients with bilateral carotid body SDHD PGLs; thus, hemodynamic complications must be carefully considered.98
To control the symptoms and signs of catecholamine excess and to prevent complications of therapeutic interventions, α-adrenoceptor blockers are widely used as the primary treatment. Both α1-selective and competitive adrenoceptor blockers, such as doxazosin, prazosin, or terazosin, and the non-selective and non-competitive α1- and α2-adrenoceptor blocker phenoxybenzamine are used. These drugs are typically started at least 7–14 days preoperatively with gradually increasing dosages until blood pressure targets are achieved.99 The efficacy of phenoxybenzamine and doxazosin has recently been investigated in PRESCRIPT, the first randomized controlled trial on presurgical treatment in patients with PPGLs.100 The primary endpoint was defined as the total time a patient’s blood pressure was outside a predefined range intraoperatively. While there was no difference between the two drugs, there was less intraoperative hemodynamic instability with phenoxybenzamine. Additionally, metyrosine and calcium channel blockers can be used.101 The latter may be used either as an adjunct to α-adrenoceptor blockers to control refractory hypertension or as presurgical monotherapy in cases of normal to mildly elevated blood pressure levels or cases of severe orthostatic hypotension when an α-adrenoceptor blocker is used. Tachycardia is treated with either non-selective β- or β1-selective (preferably) adrenoceptor blocker. To reduce the risk of preoperative orthostatic hypotension and postoperative hypotension, it is common practice to employ a high-sodium diet and administer one to two liters of saline 24 hours prior to surgery, and use compressive stockings.101
Surveillance of patients with non-metastatic SDHD PPGLs (R23-R27)
R23. We suggest that treatment-naïve patients with SDHD PPGLs with no compelling indication for treatment should be imaged at 3 to 6-months and at 1-year following diagnosis to document the disease course and decide on treatment options (Grade 2⊕○○○).
R24. We suggest that patients surgically treated for primary functional PPGLs should undergo measurement of plasma or urine metanephrines and plasma MTY by 8 weeks post-treatment. Imaging could be done at 3–6 months. In patients in whom the PPGL was not functional, imaging should be performed at 3–6 months (Grade 2⊕○○○).
Evidence
After patients with SDHD PPGLs have received a diagnosis, approximately 50% of patients undergo treatment and the remaining undergo surveillance.102
Most patients with functional HNPGLs are recognized by an elevation of plasma and/or urinary normetanephrine and methoxytyramine, even if they do not have catecholamine-related signs and symptoms. Non-functionality is defined as normal plasma and urinary normetanephrine and/or MTY or as plasma metanephrines that are too low according to tumor size. 103
For patients with functional SDHD PPGLs, measurement of metanephrines should be performed 2–8 weeks postoperatively.4,104 In patients with non-functional tumors who have completely normal preoperative biochemistry, imaging should be done at 3–6 months to check whether surgery was complete. Repeat imaging should be done 3–6 months after any therapy.
In patients with non-metastatic (M0) SDHD HNPGL cases, imaging should be repeated at 3–6 months postoperatively since there are no clinically reliable predictors of metastasis. This is particularly important in patients with large SDHD PPGLs. If the disease is stable, annual imaging findings should be considered. The estimated median volume growth rate is 10–12% per year, 105,106 despite with no progression in 60% of SDHD-related PGLs.106
R25. We recommend that patients with SDHD PPGLs (regardless of surgical history) undergo annual blood pressure measurements, clinical assessment, and biochemical measurements to detect new PPGLs or metastases or progression (Grade 1⊕⊕○○).
R26. We recommend that a whole-body MRI be performed at least every 2–3 years to detect new SDHD PPGLs, metastases, or progression. Initially, more frequent imaging follow-up is recommended for patients with unoperated PPGL and/or metastasis (Grade 1⊕⊕○○).
R27. We suggest the use of SSTR PET/CT on an individual basis to screen for disease progression in patients with non-metastatic PPGL (Grade 2⊕○○○).
Evidence
Overall, the prognosis of patients with SDHD PPGLs remains excellent, with no substantial increase in mortality observed in a Dutch sample of SDHD variant carriers.107 However, patients with SDHD are at risk of developing recurrence, metastasis, or progression. Therefore, patients should receive lifelong follow-up, and quality of life should be monitored. In one meta-analysis, the pooled risk for metastasis was 4% for patients with SDHD PGLs versus 13% for patients with SDHB PGLs.55 A postoperative analysis of 47 SDHx patients (n=33 SDHD) followed for a median of 2.7 years showed that 11% developed local recurrence.102 Disease penetrance was very high in the presence of SDHD PGLs, but the occurrence of metachronous tumors may be delayed.108–110 A study that included 93 patients with SDHD-associated HNPGLs found that a diagnosis of biochemically positive PPGLs was made in 30% of cases (with five glomus PGLs). The diagnosis was made at the initial screening in 63% of cases, whereas 37% of cases were detected after repeated biochemical screening. In this study, only patients in whom urinary excretion catecholamines and/or metabolites was above the reference limit were subjected to imaging.110 In a follow-up study performed over 22 years, new PGLs were found in 73% of cases, the majority of which were HNPGLs. Eight patients (4%) developed PHEOs, and 12 (5%) developed sympathetic PGLs.111 The diagnosis of a biochemically positive lesion is critical to avoid risks related to catecholamine crises. Therefore, an annual assessment of plasma metanephrines in the follow-up of patients with SDHD should be mandatory.112 To limit radiation exposure to patients, whole-body MRI or multiple standard MRIs should be performed first.112,113 If possible and based on current clinical assessment, the administration of gadolinium-based contrast agents could be avoided because of the risk of deposition in the brain. PET/CT can be performed every three to five years on an individual basis to screen for multifocality and metastasis.114
Surveillance and management of patients with advanced/metastatic SDHD PPGLs (R28-R34)
R28. We recommend the use of SSTR PET/CT to evaluate disease progression in patients with metastatic PPGL (Grade 1⊕⊕○○).
R29. We recommend characterizing disease progression in the setting of an interdisciplinary tumor board using clinical information, biochemistry, and imaging (Grade 1 ⊕⊕○○).
R30. We recommend active surveillance for patients with asymptomatic (or stable symptoms/signs) or stable/very slow-growing metastatic lesions (stable disease >12 months), particularly in patients with low tumor burden (Grade 1 ⊕⊕○○).
R31. We recommend considering local therapies (e.g., surgery, therapeutic radiation, interventional radiology procedures) for patients with symptomatic oligometastatic SDHD PPGL without contraindication who cannot be otherwise controlled or in those with lesions at risk of more severe local complications (Grade 1⊕⊕○○).
R32. We recommend targeted radionuclide therapy as the first-line systemic therapy for SSTR- or MIBG-positive metastatic tumors with moderate-to-high tumor burden and without evidence of rapidly progressive disease (Grade 1 ⊕⊕○○).
R33. We recommend chemotherapy as the first-line therapy in cases of rapid progression or high visceral tumor burden and possibly as second-line therapy if there is rapid progression following other systemic therapies (Grade 1 ⊕⊕○○). In patients in whom CVD is not tolerated, not wished by the patient or if there are contraindications to CVD, tyrosine kinase inhibitors (sunitinib) or temozolomide can be used as alternative agents, carefully evaluating their adverse effects.
R34. We recommend either tyrosine kinase inhibitors (sunitinib) (Grade 1 ⊕⊕⊕○) or temozolomide (Grade 1 ⊕⊕○○) in cases of progressing tumors not eligible for PRRT or MIBG or following progression to radionuclide therapy or CVD.
Evidence
Assessment of disease progression mainly relies on anatomical and functional imaging in selected cases (preferably PET/CT using somatostatin analogs). The Consensus on Molecular Imaging and Theranostics in Neuroendocrine Tumors has recently proposed that detection of new lesion(s) (after exclusion of pitfalls) by functional imaging with the same tracer can be considered sufficient to define progression.115 However, data are too scarce to provide any recommendation in the setting of patients with metastatic SDHx PPGLs.
In a study that included therapy-naïve patients with metastatic PPGLs, 87% of patients who experienced progressive disease at one year had progressive disease at baseline. Therefore, an imaging work-up three months after the diagnosis of metastatic disease may be recommended.116
As SDHD-related PPGL metastases are often associated with slow progression, active surveillance may be reasonable for patients with low tumor burden (Figure 4).
Figure 4.
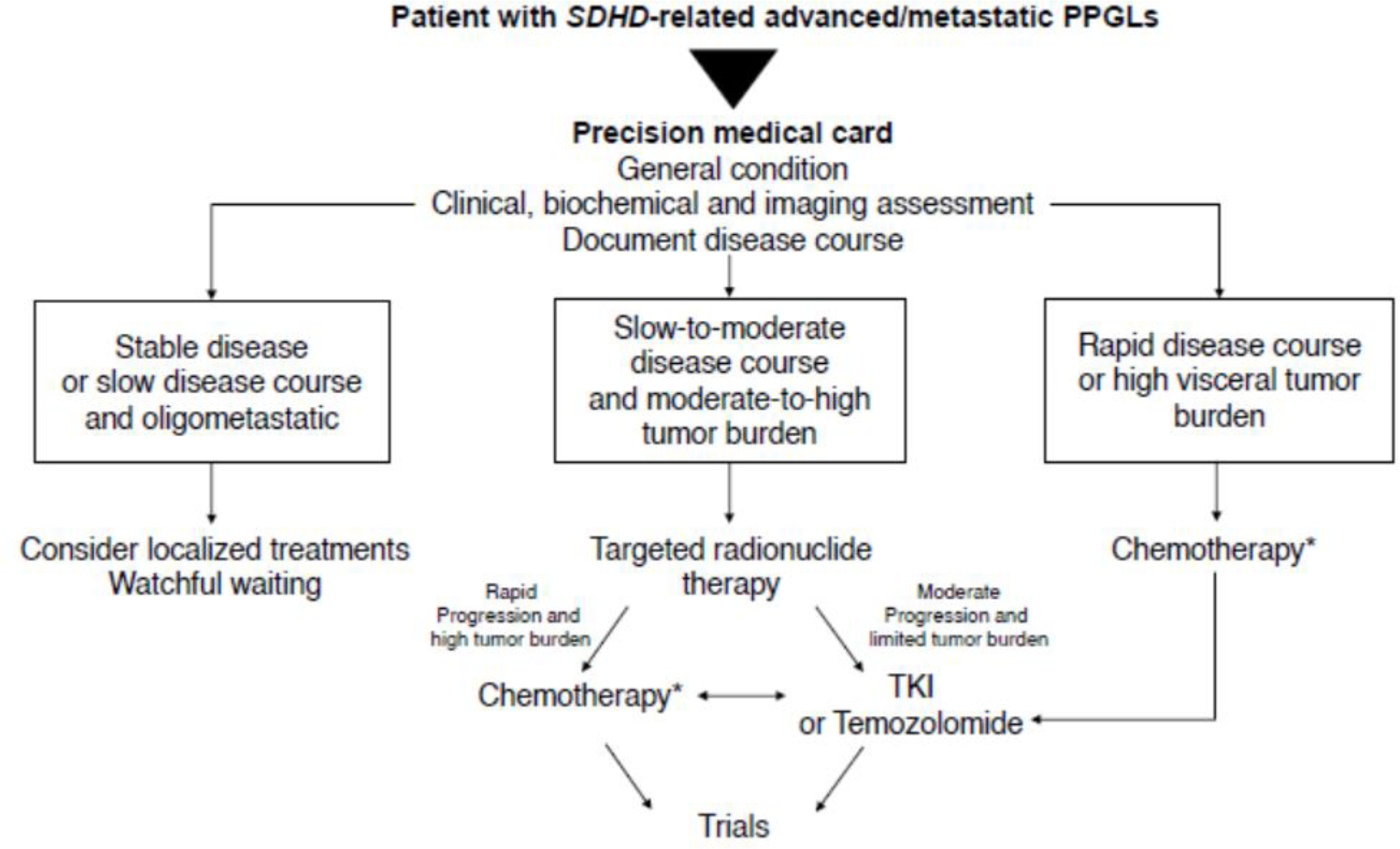
Management of patients with metastatic SDHD-related PPGLs
TKI, tyrosine kinase inhibitors
* In patients in whom CVD is not tolerated, not wished by the patient or if there are contraindications to CVD, tyrosine kinase inhibitors (sunitinib) or temozolomide can be used as alternative agents, carefully evaluating their adverse effects.
Surgery for the primary tumor (see R15) and locoregional treatments should be considered on an individual basis in the setting of a multidisciplinary board. Patient preparation was recommended for all local interventions, similar to that for surgery.
The European Society of Hypertension recommends targeted radionuclide therapy for patients with slow/moderate disease progression with a moderate tumor burden (Figure 4). Targeted radionuclide therapy of metastatic/inoperable PPGLs is a palliative treatment, with rare cases demonstrating complete responses. The goals of therapy are stabilization/regression of progressive, metastatic, or inoperable tumors, amelioration of symptoms, and control of disease-specific cardiovascular effects. The choice between the two systemic radiotherapeutic options (peptide receptor radionuclide therapy-PRRT or 131I-MIBG) is mainly dependent on the imaging phenotypes seen on SSTR PET and 123I-MIBG.117 If a radiopharmaceutical is superior in targeting most or all of a patient’s lesions, that compound is favored. If both radiopharmaceuticals localize similarly, other issues related to toxicity and morbidity must be considered. Additional issues to be considered are reimbursement and inpatient versus outpatient therapies. Peptide Receptor Radionuclide Therapy (PPRT) is favored when the patient has a low marrow reserve or baseline leukopenia/thrombocytopenia due to a lower potential for marrow toxicity (especially compared to 18.5GBq 131I-MIBG). Owing to the less differentiated nature of SDHD PPGLs and their usual parasympathetic origin, PRRT is utilized more than MIBG therapy in this setting. Supplemental Tables 2 and 3 summarize the results from clinical studies investigating PRRT, low- and high-specific-activity MIBG therapy, and potential side effects. The disease control rate (DCR) for patients with PPGLs with PRRT in most retrospective studies was ≥ 80%, and the progression-free survival (PFS) was 17–39 months. In the largest meta-analysis of 234 pooled PPGLs patients treated with PRRT, a high DCR of 90% was reported.118 However, only 71% of patients showed progressive disease at treatment initiation, which complicates drawing conclusions. Another meta-analysis of 201 pooled PPGLs patients treated with PRRT reported an overall response rate of 25% and DCR of 84%.119
For rapidly progressing disease or high visceral tumor burden, chemotherapy with cyclophosphamide, vincristine, and dacarbazine (CVD) is the recommended first-line therapy.4,120–126 CVD should be the second-line therapy following progression to targeted radionuclide therapy in the case of rapid progression or high visceral tumor burden (Figure 4).
Targeted systemic therapies (tyrosine kinase inhibitors [TKIs]) or temozolomide should be considered following progression after targeted radionuclide therapy or subsequent progression after CVD (Figure 4). For sunitinib, the first randomized placebo-controlled clinical trial (FIRST-MAPP) (n=78) showed a DCR of 35.9% over 12 months and a significant improvement in median PFS in the sunitinib group compared to the placebo group (8.9 versus 3.6 months).127 The results of the FIRST-MAPP trial have yet to be published in a peer-reviewed journal. Another prospective clinical trial (n=25) that investigated sunitinib in patients with PPGLs reported a response rate of 13%, stable disease in 70% of patients over 3 months (DCR 83%), and a DCR of 61% over 6 months. All patients with SDHx-related disease showed partial response or stable disease. 128
For temozolomide, two retrospective studies indicate efficacy in patients with metastatic PPGLs including SDHx pathogenic variant cases: one retrospective study in patients with PPGLs (n=14 with 10 SDHB cases) reported an overall DCR of 80% with a partial response rate of 33% (according to RECIST plus PERCIST), with all responders being SDHB pathogenic variants carriers (overall PFS 13.3 months, with a significantly longer PFS of 19.7 versus 2.9 months in SDHB versus non-SDHB pathogenic variants carriers).129 Another retrospective study (n=17 with one SDHA-, one SDHC-, and seven SDHB-pathogenic variant cases, 15 patients evaluable by RECIST, one SDHC, and one SDHB partial response) reported a DCR of 67% (partial response 40%, stable disease 27%, overall median PFS 2.2 years, median PFS 1.3 years for SDH pathogenic variants carriers, and 5.5 years for non-carriers).130 Thus, whether temozolomide has a specific benefit for SDH pathogenic variant carriers remains unclear. Other studies are described in Supplemental Table 2.
It is worth mentioning that TKIs may worsen hypertension; thus, careful follow-up and aggressive antihypertensive dosage adjustment prior to and during TKI therapy are needed. In the case of progression following sunitinib or temozolomide treatment, an alternative treatment can be used. Following the progression of both approaches, inclusion in a clinical trial should be investigated. Similar to other neuroendocrine tumors, anti-resorptive therapies are recommended for patients with SDHD PPGL patients with widespread bone metastases.131
CONCLUSION
All patients with SDHD pathogenic variants should be managed by an expert interdisciplinary team and require clinical, endocrine as well as imaging work-up to screen and diagnose PPGL at a whole-body scale. This can be achieved by combination of morphological imaging and in most patients by SSTR PET/CT. In patients with HNPGL, long-term preservation of cranial nerve function is a main concern when considering treatment. Therapeutic radiation can complement or be an alternative to surgery in certain situations. Life-long surveillance is mandatory to screen for new PPGL, disease progression and/or metastases. Management of metastatic PPGL mainly relies on hormonal secretion, disease extension and pace of growth. This guideline should help standardize high quality care for PPGL patients with SDHD pathogenic variants.
Supplementary Material
Acknowledgements
Thank you to Alicia A. Livinski, National Institutes of Health Library, Bethesda, MD, USA for assistance with this project and manuscript.
Funding statement
This work was supported, by the Intramural Research Program of the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Footnotes
Declaration of Interests
DT has received personal honoraria for lectures and consulting from AAA/Novartis and support for meeting attendance from AAA/Novartis.
GBW has nothing to declare.
MA has nothing to declare.
CLL has received personal honoraria for lectures from Ipsen and support for meeting attendance from Ipsen.
NP has nothing to declare.
SV has received research grant to the institution from German Research Foundation.
LA has received personal honoraria for lectures from Servier and Ipsen.
HJLMT has nothing to declare.
ZGS has nothing to declare.
ALE has received fees for consulting from WL Gore and fees for advisory board from Artivion.
ML has received research grants to the institution from Arbor, BMS, Accuray, Biohaven, Urogen, honoraria for research consulting from VBI, InCephalo Therapeutics, Merck, Pyramid Bio, Insightec, Biohaven, Sanianoia, Hemispherian, Novocure, Noxxon, InCando, Century Therapeutics, CraniUs, honoraria for non-research consulting from Stryker. ML is a shareholder of Egret Therapeutics. ML holds various patents. ML is a member of the Data and Safety Monitoring Board of Cellularity.
ELP has received fees for advisory board from Vysioneer.
LV has nothing to declare.
IB has nothing to declare.
RTC has received personal honoraria for lectures from Novartis, support for meeting attendance from Ipsen. RTC serves as a board member for Society for Endocrinology clinical committee UKINETS clinical committee.
FC has nothing to declare.
RCB has nothing to declare.
EPMC has nothing to declare.
RRDK has nothing to declare.
JDR has nothing to declare.
GE has nothing to declare.
HKG has nothing to declare.
APGR has nothing to declare.
AG has nothing to declare.
AI has nothing to declare.
JCJ has nothing to declare.
AJ has nothing to declare.
MK has nothing to declare.
HPMK has nothing to declare.
JKL has received honoraria for lectures from Stryker and is a consultant for Stryker.
ERM has received fees for consulting from MSD, personal honoraria for lecture from MSD.
DM has nothing to declare.
LBMA has nothing to declare.
OM has nothing to declare.
MN has nothing to declare.
NN has received an intramural research grant from the NIH.
NPT has received research grants to the institution from Innervate, Clarity pharma, fees for consulting from Progenics, Lantheus, innervate. MPT is a member of the Data and Safety Monitoring Board of Progenics, Lantheus. MPT serves as a board member for SNMMI.
FS has nothing to declare.
AT has nothing to declare.
JW has nothing to declare.
LM has nothing to declare.
JWML has nothing to declare.
KP has nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Maleeha Ahmad, Department of Neurosurgery, Stanford University School of Medicine, Palo Alto, California, USA..
Prof Laurence Amar, Université Paris Cité, Inserm, PARCC, Equipe Labellisée par la Ligue contre le Cancer, F-75015 Paris, France; Hypertension Unit, Hôpital Européen Georges Pompidou, Assistance Publique - Hôpitaux de Paris, 75015 Paris, France..
Prof Isabelle Bourdeau, Division of Endocrinology, Department of Medicine and Research Center, Centre Hospitalier de l’Université de Montréal, Montréal, Quebec, Canada.
Ruth T. Casey, University of Cambridge Department of Medical Genetics, NIHR Cambridge Biomedical Research Centre, Cancer Research UK Cambridge Centre, Cambridge Biomedical Campus, Cambridge, UK..
Prof Frédéric Castinetti, Department of Endocrinology, Aix-Marseille University, Conception Hospital, Marseille, France..
Prof Roderick Clifton-Bligh, Department of Endocrinology and Cancer Genetics Laboratory, Royal North Shore Hospital Kolling Institute, St Leonards NSW Australia, University of Sydney, Australia..
Eleonora P.M. Corssmit, Center of Endocrine Tumors Leiden, Department of Endocrinology, Leiden University Medical Centre, Leiden, The Netherlands..
Ronald R. de Krijger, Department of Pathology, University Medical Center Utrecht, 3584 CX Utrecht, The Netherlands..
Jaydira Del Rivero, Developmental Therapeutics Branch, Rare Tumor Initiative, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, Maryland, USA..
Prof Graeme Eisenhofer, Institute of Clinical Chemistry and Laboratory Medicine, University Hospital Carl Gustav Carus at the TU, Dresden..
Prof Anthony L. Estrera, Department of Cardiothoracic and Vascular Surgery UTHealth Houston, McGovern Medical School Memorial Hermann Hospital Heart and Vascular Institute..
Hans K. Ghayee, Division of Endocrinology and Metabolism, Department of Medicine, University of Florida and the Malcom Randall VA Medical Center, Gainesville, Florida, USA..
Prof Anne-Paule Gimenez-Roqueplo, Université Paris Cité, Inserm, PARCC, Equipe Labellisée par la Ligue contre le Cancer, F-75015 Paris, France; Département de Médecine Génomique des Tumeurs et des Cancers, Assistance Publique - Hôpitaux de Paris, Hôpital Européen Georges Pompidou, F-75015 Paris, France..
Prof Ashley Grossman, Green Templeton College, University of Oxford, UK and NET Unit, Royal Free Hospital, London, UK..
Prof Alessio Imperiale, Department of Nuclear Medicine and Molecular Imaging – Institut de Cancérologie de Strasbourg Europe, IPHC, UMR 7178, CNRS, University of Strasbourg, France..
Prof Jeroen C. Jansen, Department of Otorhinolaryngology, Leiden University Medical Center, Leiden, The Netherlands..
Abhishek Jha, Section on Medical Neuroendocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland, USA..
Michiel Kerstens, Department of Endocrinology, University Medical Center Groningen, Groningen, The Netherlands..
Prof Henricus P.M. Kunst, Department of Otolaryngology and Head & Neck Surgery - Dutch Academic Alliance Skull Base Pathology, Radboud University Medical Center, Maastricht University Medical Center+, Nijmegen / Maastricht, The Netherlands..
Prof Jacques W.M. Lenders, Department of Internal Medicine, Radboud University Medical Center, Nijmegen, The Netherlands. and Department of Medicine ΙΙI, University Hospital Carl Gustav Carus at the TU Dresden, Germany.
Prof Michael Lim, Department of Neurosurgery, Stanford University School of Medicine, Palo Alto, California, USA..
Prof James K. Liu, Department of Neurosurgical Surgery, Rutgers New Jersey Medical School, Newark, New Jersey, USA..
Prof Charlotte Lussey-Lepoutre, Université Paris Cité, Inserm, PARCC, Equipe Labellisée par la Ligue contre le Cancer, F-75015 Paris, France; Department of Nuclear Medicine, Pitié-Salpêtrière Hospital, Sorbonne University, Paris, France..
Prof Eamonn R. Maher, University of Cambridge Department of Medical Genetics, Cambridge Biomedical Campus, Cambridge, UK..
Prof Daniele Marchioni, Department of Otorhinolaryngology and Head and Neck Surgery, University Hospital of Verona, Piazzale Aristide Stefani 1, 37126, Verona, Italy..
Prof Leilani B. Mercado-Asis, Section of Endocrinology and Metabolism, Department of Medicine, Faculty of Medicine & Surgery, University of Santo Tomas Hospital, University of Santo Tomas, Espana, Manila, Philippines..
Prof Ozgur Mete, Department of Laboratory Medicine and Pathobiology, University of Toronto President, Endocrine Pathology Society, Toronto, Ontario, Canada..
Leah Meuter, Stanford University School of Medicine, Department of Physician Assistant Studies, Stanford, California, USA..
Mitsuhide Naruse, Department of Endocrinology and Metabolism, National Hospital Organization Kyoto Medical Center, Kyoto, Japan..
Prof Naris Nilubol, Surgical Oncology Program, National Cancer Institute, NIH, Bethesda, Maryland, USA..
Prof Svenja Nölting, Department of Endocrinology, Diabetology and Clinical Nutrition, University Hospital Zurich and University of Zurich, Zurich, Switzerland..
Prof Karel Pacak, Section on Medical Neuroendocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, Bethesda, Maryland, USA..
Prof Nancy D. Perrier, Department of Surgical Oncology, MD Anderson Cancer Center, Houston, Texas, USA..
Prof Neeta Pandit-Taskar, Department of Radiology, Molecular Imaging and Therapy Service, Memorial Sloan Kettering Cancer Center, New York. USA..
Erqi Liu Pollom, Department of Radiation Oncology, Stanford University School of Medicine, Palo Alto, California, USA..
Prof Frédéric Sebag, Department of Endocrine Surgery, Aix-Marseille University, Conception University Hospital, Marseille, France..
Zachary G. Schwam, Icahn School of Medicine at Mount Sinai, Department of Otolaryngology-Head and Neck Surgery, New York, USA..
Prof David Taïeb, Department of Nuclear Medicine, Aix-Marseille University, La Timone University Hospital, Marseille, France..
Akiyo Tanabe, Division of Diabetes and Endocrinology, National International Center for Global Health and Medicine, Tokyo, Japan..
Prof Henri J. L. M. Timmers, Department of Internal Medicine, Radboud University Medical Center, Nijmegen, The Netherlands..
Lucas Vitzthum, Department of Radiation Oncology, Stanford University School of Medicine, Palo Alto, California, USA..
Prof George B. Wanna, Icahn School of Medicine at Mount Sinai, Department of Otolaryngology-Head and Neck Surgery, New York, USA..
Prof Jiri Widimsky, Third Department of Medicine, Department of Endocrinology and Metabolism of the First Faculty of Medicine, Charles University and General University Hospital in Prague, Prague, Czech Republic..
References
- 1.Mannelli M, Canu L, Ercolino T, et al. DIAGNOSIS of ENDOCRINE DISEASE: SDHx mutations: beyond pheochromocytomas and paragangliomas. Eur J Endocrinol 2018; 178(1): R11–R7. [DOI] [PubMed] [Google Scholar]
- 2.Burnichon N, Mazzella JM, Drui D, et al. Risk assessment of maternally inherited SDHD paraganglioma and phaeochromocytoma. J Med Genet 2017; 54(2): 125–33. [DOI] [PubMed] [Google Scholar]
- 3.Atkins D, Best D, Briss PA, et al. Grading quality of evidence and strength of recommendations. BMJ 2004; 328(7454): 1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenders JWM, Kerstens MN, Amar L, et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens 2020; 38(8): 1443–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99(6): 1915–42. [DOI] [PubMed] [Google Scholar]
- 6.Richter S, Qiu B, Ghering M, et al. Head/neck paragangliomas: focus on tumor location, mutational status and plasma methoxytyramine. Endocr Relat Cancer 2022; 29(4): 213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darr R, Pamporaki C, Peitzsch M, et al. Biochemical diagnosis of phaeochromocytoma using plasma-free normetanephrine, metanephrine and methoxytyramine: importance of supine sampling under fasting conditions. Clin Endocrinol (Oxf) 2014; 80(4): 478–86. [DOI] [PubMed] [Google Scholar]
- 8.Rao D, Peitzsch M, Prejbisz A, et al. Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol 2017; 177(2): 103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisenhofer G, Prejbisz A, Peitzsch M, et al. Biochemical Diagnosis of Chromaffin Cell Tumors in Patients at High and Low Risk of Disease: Plasma versus Urinary Free or Deconjugated O-Methylated Catecholamine Metabolites. Clin Chem 2018; 64(11): 1646–56. [DOI] [PubMed] [Google Scholar]
- 10.Andrews KA, Ascher DB, Pires DEV, et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gimenez-Roqueplo AP, Caumont-Prim A, Houzard C, et al. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL.EVA Investigators. J Clin Endocrinol Metab 2013; 98(1): E162–73. [DOI] [PubMed] [Google Scholar]
- 12.Gravel G, Niccoli P, Rohmer V, et al. The value of a rapid contrast-enhanced angio-MRI protocol in the detection of head and neck paragangliomas in SDHx mutations carriers: a retrospective study on behalf of the PGL.EVA investigators. Eur Radiol 2015. [DOI] [PubMed] [Google Scholar]
- 13.van den Berg R, Schepers A, de Bruine FT, et al. The value of MR angiography techniques in the detection of head and neck paragangliomas. Eur J Radiol 2004; 52(3): 240–5. [DOI] [PubMed] [Google Scholar]
- 14.Neves F, Huwart L, Jourdan G, et al. Head and neck paragangliomas: value of contrast-enhanced 3D MR angiography. AJNR Am J Neuroradiol 2008; 29(5): 883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gimenez-Roqueplo AP, Caumont-Prim A, Houzard C, et al. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL.EVA Investigators. J Clin Endocrinol Metab 2013; 98(1): E162–73. [DOI] [PubMed] [Google Scholar]
- 16.Gravel G, Niccoli P, Rohmer V, et al. The value of a rapid contrast-enhanced angio-MRI protocol in the detection of head and neck paragangliomas in SDHx mutations carriers: a retrospective study on behalf of the PGL.EVA investigators. Eur Radiol 2016; 26(6): 1696–704. [DOI] [PubMed] [Google Scholar]
- 17.Jha A, Ling A, Millo C, et al. Superiority of (68)Ga-DOTATATE over (18)F-FDG and anatomic imaging in the detection of succinate dehydrogenase mutation (SDHx )-related pheochromocytoma and paraganglioma in the pediatric population. Eur J Nucl Med Mol Imaging 2018; 45(5): 787–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janssen I, Blanchet EM, Adams K, et al. Superiority of [68Ga]-DOTATATE PET/CT to Other Functional Imaging Modalities in the Localization of SDHB-Associated Metastatic Pheochromocytoma and Paraganglioma. Clinical cancer research : an official journal of the American Association for Cancer Research 2015; 21(17): 3888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janssen I, Chen CC, Taieb D, et al. 68Ga-DOTATATE PET/CT in the Localization of Head and Neck Paragangliomas Compared with Other Functional Imaging Modalities and CT/MRI. J Nucl Med 2016; 57(2): 186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archier A, Varoquaux A, Garrigue P, et al. Prospective comparison of (68)Ga-DOTATATE and (18)F-FDOPA PET/CT in patients with various pheochromocytomas and paragangliomas with emphasis on sporadic cases. Eur J Nucl Med Mol Imaging 2016; 43(7): 1248–57. [DOI] [PubMed] [Google Scholar]
- 21.Han S, Suh CH, Woo S, Kim YJ, Lee JJ. Performance of (68)Ga-DOTA-Conjugated Somatostatin Receptor-Targeting Peptide PET in Detection of Pheochromocytoma and Paraganglioma: A Systematic Review and Metaanalysis. J Nucl Med 2019; 60(3): 369–76. [DOI] [PubMed] [Google Scholar]
- 22.Gild ML, Naik N, Hoang J, et al. Role of DOTATATE-PET/CT in preoperative assessment of phaeochromocytoma and paragangliomas. Clin Endocrinol (Oxf) 2018; 89(2): 139–47. [DOI] [PubMed] [Google Scholar]
- 23.King KS, Chen CC, Alexopoulos DK, et al. Functional imaging of SDHx-related head and neck paragangliomas: comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I-metaiodobenzylguanidine scintigraphy, and 111In-pentetreotide scintigraphy. J Clin Endocrinol Metab 2011; 96(9): 2779–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amodru V, Romanet P, Scemama U, et al. Tumor multifocality with vagus nerve involvement as a phenotypic marker of SDHD mutation in patients with head and neck paragangliomas: A (18) F-FDOPA PET/CT study. Head Neck 2019; 41(6): 1565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taieb D, Hicks RJ, Hindie E, et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2019; 46(10): 2112–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabriel S, Blanchet EM, Sebag F, et al. Functional characterization of nonmetastatic paraganglioma and pheochromocytoma by (18) F-FDOPA PET: focus on missed lesions. Clinical endocrinology 2013; 79(2): 170–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miederer M, Fottner C, Rossmann H, et al. High incidence of extraadrenal paraganglioma in families with SDHx syndromes detected by functional imaging with [18F]fluorodihydroxyphenylalanine PET. European journal of nuclear medicine and molecular imaging 2013; 40(6): 889–96. [DOI] [PubMed] [Google Scholar]
- 28.Blanchet EM, Gabriel S, Martucci V, et al. (18) F-FDG PET/CT as a predictor of hereditary head and neck paragangliomas. Eur J Clin Invest 2014; 44(3): 325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timmers HJ, Chen CC, Carrasquillo JA, et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J Natl Cancer Inst 2012; 104(9): 700–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timmers HJ, Chen CC, Carrasquillo JA, et al. Comparison of 18F-Fluoro-L-DOPA, 18F-Fluoro-Deoxyglucose, and 18F-Fluorodopamine PET and 123I-MIBG Scintigraphy in the Localization of Pheochromocytoma and Paraganglioma. Journal of Clinical Endocrinology and Metabolism 2009; 94(12): 4757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Timmers HJ, Kozupa A, Chen CC, et al. Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. Journal of Clinical Oncology 2007; 25(16): 2262–9. [DOI] [PubMed] [Google Scholar]
- 32.Alvarez-Morujo RJ, Ruiz MA, Serafini DP, Delgado IL, Friedlander E, Yurrita BS. Management of multicentric paragangliomas: Review of 24 patients with 60 tumors. Head Neck 2016; 38(2): 267–76. [DOI] [PubMed] [Google Scholar]
- 33.Moore MG, Netterville JL, Mendenhall WM, Isaacson B, Nussenbaum B. Head and Neck Paragangliomas: An Update on Evaluation and Management. Otolaryngol Head Neck Surg 2016; 154(4): 597–605. [DOI] [PubMed] [Google Scholar]
- 34.Sniezek JC, Netterville JL, Sabri AN. Vagal paragangliomas. Otolaryngol Clin North Am 2001; 34(5): 925–39, vi. [DOI] [PubMed] [Google Scholar]
- 35.Wanna GB, Sweeney AD, Carlson ML, et al. Subtotal resection for management of large jugular paragangliomas with functional lower cranial nerves. Otolaryngol Head Neck Surg 2014; 151(6): 991–5. [DOI] [PubMed] [Google Scholar]
- 36.Wanna GB, Sweeney AD, Haynes DS, Carlson ML. Contemporary management of jugular paragangliomas. Otolaryngol Clin North Am 2015; 48(2): 331–41. [DOI] [PubMed] [Google Scholar]
- 37.Lloyd S, Obholzer R, Tysome J, Group BC. British Skull Base Society Clinical Consensus Document on Management of Head and Neck Paragangliomas. Otolaryngol Head Neck Surg 2020; 163(3): 400–9. [DOI] [PubMed] [Google Scholar]
- 38.Netterville JL, Jackson CG, Miller FR, Wanamaker JR, Glasscock ME. Vagal paraganglioma: a review of 46 patients treated during a 20-year period. Arch Otolaryngol Head Neck Surg 1998; 124(10): 1133–40. [DOI] [PubMed] [Google Scholar]
- 39.Carlson ML, Sweeney AD, Wanna GB, Netterville JL, Haynes DS. Natural history of glomus jugulare: a review of 16 tumors managed with primary observation. Otolaryngol Head Neck Surg 2015; 152(1): 98–105. [DOI] [PubMed] [Google Scholar]
- 40.Jansen JC, van den Berg R, Kuiper A, van der Mey AG, Zwinderman AH, Cornelisse CJ. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer 2000; 88(12): 2811–6. [PubMed] [Google Scholar]
- 41.Langerman A, Athavale SM, Rangarajan SV, Sinard RJ, Netterville JL. Natural history of cervical paragangliomas: outcomes of observation of 43 patients. Arch Otolaryngol Head Neck Surg 2012; 138(4): 341–5. [DOI] [PubMed] [Google Scholar]
- 42.Taieb D, Kaliski A, Boedeker CC, et al. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev 2014; 35(5): 795–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu K, Persky MS. Multidisciplinary management of paragangliomas of the head and neck, Part 1. Oncology (Williston Park) 2003; 17(7): 983–93. [PubMed] [Google Scholar]
- 44.Papaspyrou K, Mewes T, Rossmann H, et al. Head and neck paragangliomas: Report of 175 patients (1989–2010). Head Neck 2012; 34(5): 632–7. [DOI] [PubMed] [Google Scholar]
- 45.Prasad SC, Mimoune HA, D’Orazio F, et al. The role of wait-and-scan and the efficacy of radiotherapy in the treatment of temporal bone paragangliomas. Otol Neurotol 2014; 35(5): 922–31. [DOI] [PubMed] [Google Scholar]
- 46.Fisch U, Mattox D. Microsurgery of the Skull Base. Stuttgart/New York: Georg Thieme Verlag; 1988: 148–281. [Google Scholar]
- 47.Jackson CG, Glasscock ME 3rd, Harris PF. Glomus Tumors. Diagnosis, classification, and management of large lesions. Arch Otolaryngol 1982; 108(7): 401–10. [DOI] [PubMed] [Google Scholar]
- 48.Kasper GC, Welling RE, Wladis AR, et al. A multidisciplinary approach to carotid paragangliomas. Vasc Endovascular Surg 2006; 40(6): 467–74. [DOI] [PubMed] [Google Scholar]
- 49.Murphy TP, Brackmann DE. Effects of preoperative embolization on glomus jugulare tumors. Laryngoscope 1989; 99(12): 1244–7. [DOI] [PubMed] [Google Scholar]
- 50.Fayad JN, Keles B, Brackmann DE. Jugular foramen tumors: clinical characteristics and treatment outcomes. Otol Neurotol 2010; 31(2): 299–305. [DOI] [PubMed] [Google Scholar]
- 51.Jackson CG, McGrew BM, Forest JA, Netterville JL, Hampf CF, Glasscock ME, 3rd. Lateral skull base surgery for glomus tumors: long-term control. Otol Neurotol 2001; 22(3): 377–82. [DOI] [PubMed] [Google Scholar]
- 52.Moe KS, Li D, Linder TE, Schmid S, Fisch U. An update on the surgical treatment of temporal bone paraganglioma. Skull Base Surg 1999; 9(3): 185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006; 91(3): 827–36. [DOI] [PubMed] [Google Scholar]
- 54.Lee H, Jeong S, Yu Y, et al. Risk of metastatic pheochromocytoma and paraganglioma in SDHx mutation carriers: a systematic review and updated meta-analysis. J Med Genet 2020; 57(4): 217–25. [DOI] [PubMed] [Google Scholar]
- 55.van Hulsteijn LT, Dekkers OM, Hes FJ, Smit JW, Corssmit EP. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: a systematic review and meta-analysis. J Med Genet 2012; 49(12): 768–76. [DOI] [PubMed] [Google Scholar]
- 56.Schovanek J, Martucci V, Wesley R, et al. The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: a retrospective cohort study. BMC Cancer 2014; 14: 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hamidi O, Young WF Jr., Iniguez-Ariza NM, et al. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J Clin Endocrinol Metab 2017; 102(9): 3296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu H, Huang B, Zhao J, Yuan D, Yang Y, Xiong F. En Bloc Resection with Major Blood Vessel Reconstruction for Locally Invasive Retroperitoneal Paragangliomas: A 15-Year Experience with Literature Review. World J Surg 2017; 41(4): 997–1004. [DOI] [PubMed] [Google Scholar]
- 59.Castillo OA, Vitagliano G, Olivares R, Soffia P, Contreras M. Laparoscopic resection of an extra-adrenal pheochromocytoma. Surg Laparosc Endosc Percutan Tech 2007; 17(4): 351–3. [DOI] [PubMed] [Google Scholar]
- 60.Ramlawi B, David EA, Kim MP, et al. Contemporary surgical management of cardiac paragangliomas. Ann Thorac Surg 2012; 93(6): 1972–6. [DOI] [PubMed] [Google Scholar]
- 61.Ellis RJ, Patel D, Prodanov T, et al. Response after surgical resection of metastatic pheochromocytoma and paraganglioma: can postoperative biochemical remission be predicted? J Am Coll Surg 2013; 217(3): 489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lieberson RE, Adler JR, Soltys SG, Choi C, Gibbs IC, Chang SD. Stereotactic radiosurgery as the primary treatment for new and recurrent paragangliomas: is open surgical resection still the treatment of choice? World Neurosurg 2012; 77(5–6): 745–61. [DOI] [PubMed] [Google Scholar]
- 63.Varma A, Nathoo N, Neyman G, et al. Gamma knife radiosurgery for glomus jugulare tumors: volumetric analysis in 17 patients. Neurosurgery 2006; 59(5): 1030–6; discussion 6. [DOI] [PubMed] [Google Scholar]
- 64.Suarez C, Rodrigo JP, Bodeker CC, et al. Jugular and vagal paragangliomas: Systematic study of management with surgery and radiotherapy. Head Neck 2013; 35(8): 1195–204. [DOI] [PubMed] [Google Scholar]
- 65.Maarouf M, Voges J, Landwehr P, et al. Stereotactic linear accelerater-based radiosurgery for the treatment of patients with glomus jugulare tumors. Cancer 2003; 97(4): 1093–8. [DOI] [PubMed] [Google Scholar]
- 66.Hu K, Persky MS. Treatment of Head and Neck Paragangliomas. Cancer Control 2016; 23(3): 228–41. [DOI] [PubMed] [Google Scholar]
- 67.Navarro Martin A, Maitz A, Grills IS, et al. Successful treatment of glomus jugulare tumours with gamma knife radiosurgery: clinical and physical aspects of management and review of the literature. Clin Transl Oncol 2010; 12(1): 55–62. [DOI] [PubMed] [Google Scholar]
- 68.Gandia-Gonzalez ML, Kusak ME, Moreno NM, Sarraga JG, Rey G, Alvarez RM. Jugulotympanic paragangliomas treated with Gamma Knife radiosurgery: a single-center review of 58 cases. J Neurosurg 2014; 121(5): 1158–65. [DOI] [PubMed] [Google Scholar]
- 69.Dupin C, Lang P, Dessard-Diana B, et al. Treatment of head and neck paragangliomas with external beam radiation therapy. Int J Radiat Oncol Biol Phys 2014; 89(2): 353–9. [DOI] [PubMed] [Google Scholar]
- 70.Anderson JL, Khattab MH, Anderson C, et al. Long-term Outcomes for the Treatment of Paragangliomas in the Upfront, Adjuvant, and Salvage Settings With Stereotactic Radiosurgery and Intensity-modulated Radiotherapy. Otol Neurotol 2020; 41(1): 133–40. [DOI] [PubMed] [Google Scholar]
- 71.Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer cell 2005; 8(2): 89–91. [DOI] [PubMed] [Google Scholar]
- 72.Pollock BE. Stereotactic radiosurgery in patients with glomus jugulare tumors. Neurosurg Focus 2004; 17(2): E10. [DOI] [PubMed] [Google Scholar]
- 73.Fatima N, Pollom E, Soltys S, Chang SD, Meola A. Stereotactic radiosurgery for head and neck paragangliomas: a systematic review and meta-analysis. Neurosurg Rev 2021; 44(2): 741–52. [DOI] [PubMed] [Google Scholar]
- 74.Guss ZD, Batra S, Limb CJ, et al. Radiosurgery of glomus jugulare tumors: a metaanalysis. Int J Radiat Oncol Biol Phys 2011; 81(4): e497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shapiro S, Kellermeyer B, Ramadan J, Jones G, Wiseman B, Cassis A. Outcomes of Primary Radiosurgery Treatment of Glomus Jugulare Tumors: Systematic Review With Meta-analysis. Otol Neurotol 2018; 39(9): 1079–87. [DOI] [PubMed] [Google Scholar]
- 76.Sheehan JP, Tanaka S, Link MJ, et al. Gamma Knife surgery for the management of glomus tumors: a multicenter study. J Neurosurg 2012; 117(2): 246–54. [DOI] [PubMed] [Google Scholar]
- 77.Eustacchio S, Trummer M, Unger F, Schrottner O, Sutter B, Pendl G. The role of Gamma Knife radiosurgery in the management of glomus jugular tumours. Acta Neurochir Suppl 2002; 84: 91–7. [DOI] [PubMed] [Google Scholar]
- 78.Sheehan J, Kondziolka D, Flickinger J, Lunsford LD. Gamma knife surgery for glomus jugulare tumors: an intermediate report on efficacy and safety. J Neurosurg 2005; 102 Suppl: 241–6. [DOI] [PubMed] [Google Scholar]
- 79.Sharma MS, Gupta A, Kale SS, Agrawal D, Mahapatra AK, Sharma BS. Gamma knife radiosurgery for glomus jugulare tumors: therapeutic advantages of minimalism in the skull base. Neurol India 2008; 56(1): 57–61. [DOI] [PubMed] [Google Scholar]
- 80.Lior U, Rotem H, Uzi N, Roberto S. LINAC radiosurgery for glomus jugulare tumors: retrospective - cohort study of 23 patients. Acta Neurochir (Wien) 2020; 162(4): 839–44. [DOI] [PubMed] [Google Scholar]
- 81.Wegner RE, Rodriguez KD, Heron DE, Hirsch BE, Ferris RL, Burton SA. Linac-based stereotactic body radiation therapy for treatment of glomus jugulare tumors. Radiother Oncol 2010; 97(3): 395–8. [DOI] [PubMed] [Google Scholar]
- 82.Hurmuz P, Cengiz M, Ozyigit G, et al. Robotic stereotactic radiosurgery in patients with unresectable glomus jugulare tumors. Technol Cancer Res Treat 2013; 12(2): 109–13. [DOI] [PubMed] [Google Scholar]
- 83.Patel AK, Rodriguez-Lopez JL, Hirsch BE, Burton SA, Flickinger JC, Clump DA. Long term outcomes with linear accelerator stereotactic radiosurgery for treatment of jugulotympanic paragangliomas. Head Neck 2021; 43(2): 449–55. [DOI] [PubMed] [Google Scholar]
- 84.Suarez C, Rodrigo JP, Mendenhall WM, et al. Carotid body paragangliomas: a systematic study on management with surgery and radiotherapy. Eur Arch Otorhinolaryngol 2014; 271(1): 23–34. [DOI] [PubMed] [Google Scholar]
- 85.Capatina C, Ntali G, Karavitaki N, Grossman AB. The management of head-and-neck paragangliomas. Endocr Relat Cancer 2013; 20(5): R291–305. [DOI] [PubMed] [Google Scholar]
- 86.Krych AJ, Foote RL, Brown PD, Garces YI, Link MJ. Long-term results of irradiation for paraganglioma. Int J Radiat Oncol Biol Phys 2006; 65(4): 1063–6. [DOI] [PubMed] [Google Scholar]
- 87.Springate SC, Weichselbaum RR. Radiation or surgery for chemodectoma of the temporal bone: a review of local control and complications. Head Neck 1990; 12(4): 303–7. [DOI] [PubMed] [Google Scholar]
- 88.Lekovic GP, Mehta GU, Maxwell AK, Peng KA, Brackmann DE. Radiation-Induced Malignant Peripheral Nerve Sheath Tumor of the Vagus Nerve Following Radiation Treatment of Cervical Paraganglioma. J Neurol Surg Rep 2020; 81(4): e66–e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Breen W, Bancos I, Young WF Jr., et al. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Adv Radiat Oncol 2018; 3(1): 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vogel J, Atanacio AS, Prodanov T, et al. External beam radiation therapy in treatment of malignant pheochromocytoma and paraganglioma. Front Oncol 2014; 4: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fishbein L, Bonner L, Torigian DA, et al. External beam radiation therapy (EBRT) for patients with malignant pheochromocytoma and non-head and -neck paraganglioma: combination with 131I-MIBG. Horm Metab Res 2012; 44(5): 405–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Isaacs M, Lee P. Preoperative alpha-blockade in phaeochromocytoma and paraganglioma: is it always necessary? Clin Endocrinol (Oxf) 2017; 86(3): 309–14. [DOI] [PubMed] [Google Scholar]
- 93.Van Der Horst-Schrivers AN, Osinga TE, Kema IP, Van Der Laan BF, Dullaart RP. Dopamine excess in patients with head and neck paragangliomas. Anticancer Res 2010; 30(12): 5153–8. [PubMed] [Google Scholar]
- 94.Bonnet S, Durand X, Baton O, et al. [Malignant hereditary paraganglioma: problems raised by non-functional forms management]. Ann Chir 2006; 131(10): 626–30. [DOI] [PubMed] [Google Scholar]
- 95.Eisenhofer G, Goldstein DS, Sullivan P, et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab 2005; 90(4): 2068–75. [DOI] [PubMed] [Google Scholar]
- 96.Foo SH, Chan SP, Ananda V, Rajasingam V. Dopamine-secreting phaeochromocytomas and paragangliomas: clinical features and management. Singapore Med J 2010; 51(5): e89–93. [PubMed] [Google Scholar]
- 97.Smith JD, Ellsperman SE, Basura GJ, Else T. Re-evaluating the prevalence and factors characteristic of catecholamine secreting head and neck paragangliomas. Endocrinol Diabetes Metab 2021; 4(3): e00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shah-Becker S, Pennock M, Sinoway L, Goldenberg D, Goyal N. Baroreceptor reflex failure: Review of the literature and the potential impact on patients with head and neck cancer. Head & neck 2017; 39(10): 2135–41. [DOI] [PubMed] [Google Scholar]
- 99.Berends AMA, Kerstens MN, Lenders JWM, Timmers H. Approach to the Patient: Perioperative Management of the Patient with Pheochromocytoma or Sympathetic Paraganglioma. The Journal of clinical endocrinology and metabolism 2020; 105(9). [DOI] [PubMed] [Google Scholar]
- 100.Buitenwerf E, Osinga TE, Timmers H, et al. Efficacy of alpha-Blockers on Hemodynamic Control during Pheochromocytoma Resection: A Randomized Controlled Trial. J Clin Endocrinol Metab 2020; 105(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pacak K Preoperative management of the pheochromocytoma patient. The Journal of clinical endocrinology and metabolism 2007; 92(11): 4069–79. [DOI] [PubMed] [Google Scholar]
- 102.Sen I, Young WF Jr., Kasperbauer JL, et al. Tumor-specific prognosis of mutation-positive patients with head and neck paragangliomas. J Vasc Surg 2020; 71(5): 1602–12 e2. [DOI] [PubMed] [Google Scholar]
- 103.Constantinescu G, Preda C, Constantinescu V, et al. Silent pheochromocytoma and paraganglioma: Systematic review and proposed definitions for standardized terminology. Front Endocrinol (Lausanne) 2022; 13: 1021420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Plouin PF, Amar L, Dekkers OM, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016; 174(5): G1–G10. [DOI] [PubMed] [Google Scholar]
- 105.Michalowska I, Cwikla JB, Michalski W, et al. Growth Rate of Paragangliomas Related to Germline Mutations of the Sdhx Genes. Endocr Pract 2017; 23(3): 342–52. [DOI] [PubMed] [Google Scholar]
- 106.Heesterman BL, de Pont LMH, Verbist BM, et al. Age and Tumor Volume Predict Growth of Carotid and Vagal Body Paragangliomas. J Neurol Surg B Skull Base 2017; 78(6): 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.van Hulsteijn LT, Heesterman B, Jansen JC, et al. No evidence for increased mortality in SDHD variant carriers compared with the general population. Eur J Hum Genet 2015; 23(12): 1713–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van Hulsteijn LT, den Dulk AC, Hes FJ, Bayley JP, Jansen JC, Corssmit EP. No difference in phenotype of the main Dutch SDHD founder mutations. Clin Endocrinol (Oxf) 2013; 79(6): 824–31. [DOI] [PubMed] [Google Scholar]
- 109.Saie C, Buffet A, Abeillon J, et al. Screening of a Large Cohort of Asymptomatic SDHx Mutation Carriers in Routine Practice. J Clin Endocrinol Metab 2021; 106(3): e1301–e15. [DOI] [PubMed] [Google Scholar]
- 110.Havekes B, van der Klaauw AA, Weiss MM, et al. Pheochromocytomas and extraadrenal paragangliomas detected by screening in patients with SDHD-associated head-and-neck paragangliomas. Endocr Relat Cancer 2009; 16(2): 527–36. [DOI] [PubMed] [Google Scholar]
- 111.Heesterman BL, de Pont LMH, van der Mey AG, et al. Clinical progression and metachronous paragangliomas in a large cohort of SDHD germline variant carriers. Eur J Hum Genet 2018; 26(9): 1339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Amar L, Pacak K, Steichen O, et al. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endocrinol 2021; 17(7): 435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tufton N, Sahdev A, Akker SA. Radiological Surveillance Screening in Asymptomatic Succinate Dehydrogenase Mutation Carriers. J Endocr Soc 2017; 1(7): 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tufton N, Sahdev A, Drake WM, Akker SA. Can subunit-specific phenotypes guide surveillance imaging decisions in asymptomatic SDH mutation carriers? Clin Endocrinol (Oxf) 2019; 90(1): 31–46. [DOI] [PubMed] [Google Scholar]
- 115.Ambrosini V, Kunikowska J, Baudin E, et al. Consensus on molecular imaging and theranostics in neuroendocrine neoplasms. Eur J Cancer 2021; 146: 56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hescot S, Leboulleux S, Amar L, et al. One-year progression-free survival of therapy-naive patients with malignant pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2013; 98(10): 4006–12. [DOI] [PubMed] [Google Scholar]
- 117.Jha A, Taieb D, Carrasquillo JA, et al. High-Specific-Activity-(131)I-MIBG versus (177)Lu-DOTATATE Targeted Radionuclide Therapy for Metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res 2021; 27(11): 2989–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Taieb D, Jha A, Treglia G, Pacak K. Molecular imaging and radionuclide therapy of pheochromocytoma and paraganglioma in the era of genomic characterization of disease subgroups. Endocr Relat Cancer 2019; 26(11): R627–R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Satapathy S, Mittal BR, Bhansali A. ‘Peptide receptor radionuclide therapy in the management of advanced pheochromocytoma and paraganglioma: A systematic review and meta-analysis’. Clin Endocrinol (Oxf) 2019; 91(6): 718–27. [DOI] [PubMed] [Google Scholar]
- 120.Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer 2008; 113(8): 2020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med 1988; 109(4): 267–73. [DOI] [PubMed] [Google Scholar]
- 122.Niemeijer ND, Alblas G, van Hulsteijn LT, Dekkers OM, Corssmit EP. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf) 2014; 81(5): 642–51. [DOI] [PubMed] [Google Scholar]
- 123.Asai S, Katabami T, Tsuiki M, Tanaka Y, Naruse M. Controlling Tumor Progression with Cyclophosphamide, Vincristine, and Dacarbazine Treatment Improves Survival in Patients with Metastatic and Unresectable Malignant Pheochromocytomas/Paragangliomas. Horm Cancer 2017; 8(2): 108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Deutschbein T, Fassnacht M, Weismann D, Reincke M, Mann K, Petersenn S. Treatment of malignant phaeochromocytoma with a combination of cyclophosphamide, vincristine and dacarbazine: own experience and overview of the contemporary literature. Clin Endocrinol (Oxf) 2015; 82(1): 84–90. [DOI] [PubMed] [Google Scholar]
- 125.Tanabe A, Naruse M, Nomura K, Tsuiki M, Tsumagari A, Ichihara A. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Horm Cancer 2013; 4(2): 103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jawed I, Velarde M, Darr R, et al. Continued Tumor Reduction of Metastatic Pheochromocytoma/Paraganglioma Harboring Succinate Dehydrogenase Subunit B Mutations with Cyclical Chemotherapy. Cell Mol Neurobiol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Baudin E, GOICHOT B, Berruti A, et al. First International Randomized Study in Malignant Progressive Pheochromocytoma and Paragangliomas (FIRSTMAPPP): An academic double-blind trial investigating sunitinib. Annals of Oncology 2021; 32 (suppl_5): S621–S625. 10.1016/annonc/annonc700. [DOI] [Google Scholar]
- 128.O’Kane GM, Ezzat S, Joshua AM, et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial. Br J Cancer 2019; 120(12): 1113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hadoux J, Favier J, Scoazec JY, et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer 2014; 135(11): 2711–20. [DOI] [PubMed] [Google Scholar]
- 130.Perez K, Jacene H, Hornick JL, et al. SDHx mutations and temozolomide in malignant pheochromocytoma and paraganglioma. Endocr Relat Cancer 2022; 29(9): 533–44. [DOI] [PubMed] [Google Scholar]
- 131.Ayala-Ramirez M, Palmer JL, Hofmann MC, et al. Bone metastases and skeletal-related events in patients with malignant pheochromocytoma and sympathetic paraganglioma. J Clin Endocrinol Metab 2013; 98(4): 1492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.