

Groundbreaking work demonstrated that ectopic expression of four transcription factors, Oct4, Klf4, Sox2, and c-Myc, could reprogram murine somatic cells to induced pluripotent stem cells(iPSCs)(Takahashi and Yamanaka, 2006), and human iPSCswere subsequently generated using similar genetic manipulation (Takahashi et al., 2007; Yu et al., 2007). To address the safety issues arose from harboring integrated exogenous sequences in the target cell genome, several modified genetic methods have been developed and produced iPSCs with potentially reduced risks (for discussion, see Yamanaka, 2009, and references therein). However, all of the methods developed to date still involve the use of genetic materials and thus the potential for unexpected genetic modifications by the exogenous sequences in the target cells. Here we report generation of protein-induced pluripotent stem cells (piPSCs) from murine embryonic fibroblasts using recombinant cell-penetrating reprogramming proteins. We demonstrated that such piPSCs can long-term self-renew and are pluripotent in vitro and in vivo.
One possible way to avoid introducing exogenous genetic modifications to target cells would be to deliver the reprogramming proteins directly into cells, rather than relying on the transcription from delivered genes. Previous studies have demonstrated that various proteins can be delivered into cells in vitro and in vivo by conjugating them with a short peptide that mediates protein transduction, such as HIV tat and poly-arginine (Inoue et al., 2006; Michiue et al., 2005; Wadia and Dowdy, 2002). In addition, various solubilization and refolding techniques for processing inclusion body proteins expressed in E. coli to bioactive proteins have been developed to allow facile and largescale production of therapeutic proteins (Lafevre-Berntetal.,2008).To generate recombinant proteins that can penetrate across the plasma membrane of somatic cells, we designed and fused a poly-arginine(i.e.,11R)protein transduction domain to the C terminus of four reprogramming factors: Oct4, Sox2, Klf4, and c-Myc (see Figure S1A online). These proteins were expressed in E. coli in inclusion bodies, which were then solubilized, refolded, and further purified (Figure S1B). The protein identities were confirmed by mass spectrometry and western blot analysis (Figure S1C). To test the cell permeability and stability of the proteins, we treated mouse embryonic fibroblast (MEF) cells with the recombinant proteins at various concentrations by adding them to the cell culture media for 6–72 hr and examining cell morphology and protein presence by immunocytochemistry. We found that the purified 11R-tagged recombinant transcription factors readily entered cells at concentrations of 0.5–8 mg/ml within 6 hr and could translocate into nucleus (Figure S1D). In addition, we found that the transduced proteins appeared to be stable inside cells for up to 48 hr (Figure S1D).
We then employed this simple protein transduction protocol to reprogram OG2/ Oct4-GFP reporter MEF cells. Because reprogramming through the iPSC mechanism/process typically requires sustained activity of reprogramming proteins for 7–10 days, we devised a strategy that involved treating the cells in four cycles. In each cycle the fibroblasts (initially seeded at the density of 5 3 104 cells/well in a six-well plate) were first treated overnight with the recombinant reprogramming proteins (i.e., Oct4–11R, Sox2–11R, Klf411R, and c-Myc-11R) at 8 mg/ml in the mESC growth media supplemented with or without 1 mM valproic acid (VPA), a HDAC inhibitor that can significantly improve reprogramming efficiency (Huangfu et al., 2008b), followed by changing to the same media without the recombinant reprogramming proteins and VPA, and culturing for additional 36 hr before the next cycle of the treatment. After completing four repeated protein transductions of reprogramming proteins, the treated cells were transferred onto irradiated MEF feeder cells and simply kept in mESC growth media until colonies emerged around day 30–35 (Figure 1A). We obtained three GFP+ colonies per 53104 cells when they were transduced with four proteins and treated with VPA,andoneGFP+colonyper53104cells when they were transduced with only three proteins (i.e., Oct4–11R, Sox2–11R, and Klf4–11R) and treated with VPA. However, we did not obtain stable GFP+ piPSC colonies by transducing the three or four reprogramming proteins only for the same period, although GFP-negative cell colonies were observed. Those GFP-negative cell colonies stained positive with ALP, an early pluripotency marker, suggesting they might be partially reprogrammed cells. The initial GFP+ colonies were subsequently passaged under conventional mESC growth conditions to yield piPSCs and were characterized further.
Figure 1.
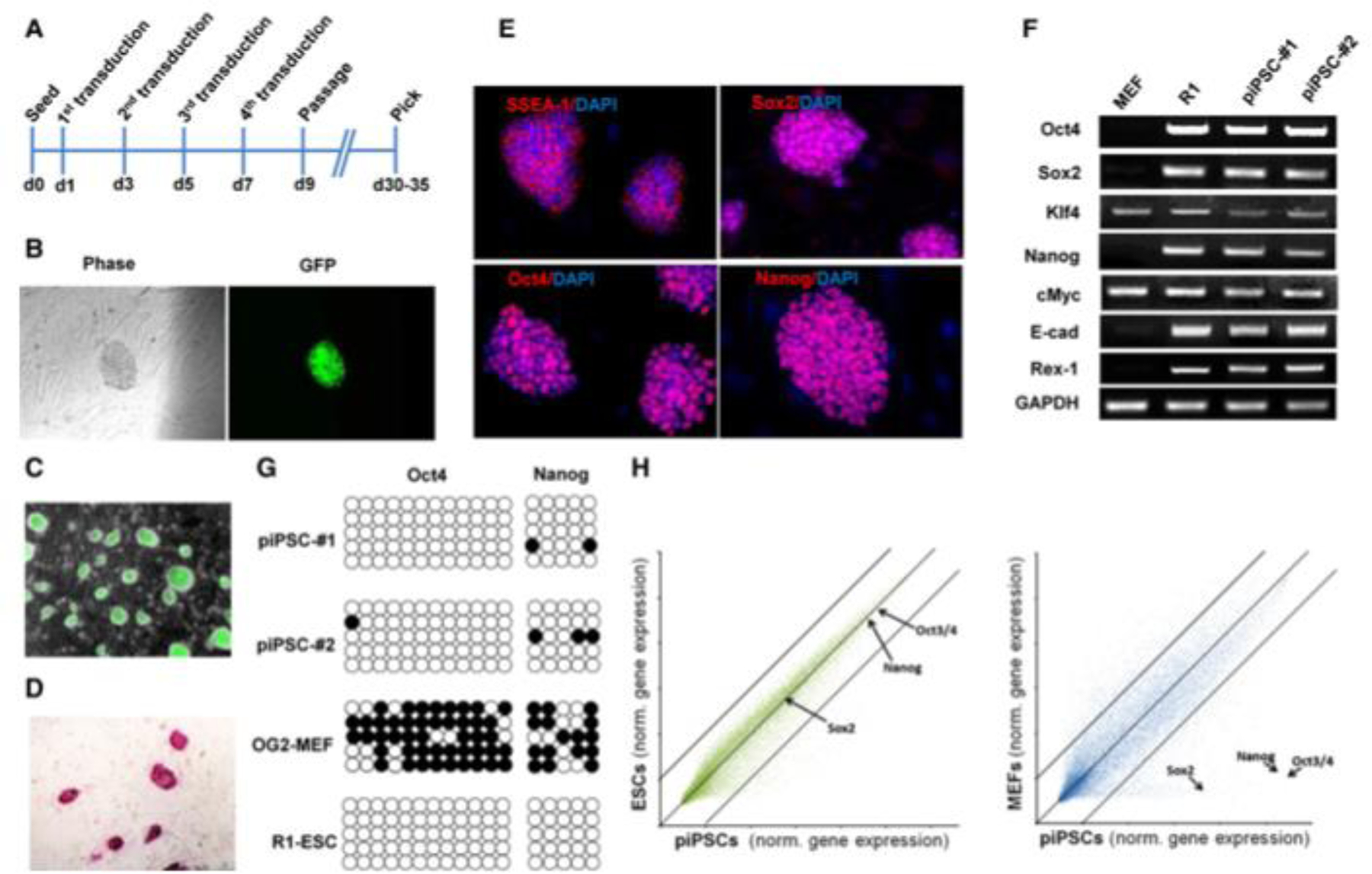
Generation of Protein-Induced Pluripotent Stem Cells by Recombinant Reprogramming Proteins.
The generated murine piPSCs have been stably and homogenously expanded for over 30 passages and are morphologically indistinguishable from classic mESCs, forming compact domed small colonies (Figure 1C). They express typical pluripotency markers by immunocytochemistry and staining, including ALP (Figure 1D), Oct4, Nanog, Sox2, and SSEA1 (Figure 1E). RT-PCR analysis confirmed endogenous gene expression of these and additional pluripotency genes (Figure 1F). A single cell survival assay also demonstrated that piPSCs clonally expand efficiently as Oct4-positive colonies in feeder-free and N2/B27-chemically defined conditions (Figure S2A). Bisulfite genomic sequencing analyses of the Oct4 and Nanog promoters revealed that both were demethylated in piPSCs as in mESCs, while in MEFs they were hypermethylated (Figure 1G). This result provides further evidence for reactivation of the pluripotency transcription program in the piPSCs. Global gene expression analysis of piPSCs, OG2-MEFs, and mESCs showed that piPSCs are distinct from OG2-MEFs (Pearson correlation value: 0.895) and most similar to mESCs (Pearson correlation value: 0.969) (Figure 1H), consistent with previous reports.
To examine the developmental potential of piPSCs, standard in vitro differentiation using embryoid bodies (EBs) or monolayer chemically defined stepwise differentiation, and in vivo chimerism assays were performed. piPSCs could efficiently form EBs in suspension and differentiate into cells in the three primary germ layers, including endoderm derivatives (cells expressing AFP, Sox17, GATA4, or FoxA2; pancreatic cells [Pdx1]; and hepatic cells [Albumin]), mesoderm derivatives (cells expressing Brachyury and mature beating cardiomyocytes [CT3 and MHC; Movie S1]), and ectoderm derivatives (neural [Sox1, Pax6] and characteristic mature neuronal [bIII-tubulin, MAP2ab] cells) (Figures 2A, 2B, and S2B). Most importantly, such piPSCs could efficiently incorporate into the inner cell mass of a blastocyst following aggregation with an eight-cell embryo and led to high-level chimerism with apparent germline contribution in vivo after the aggregated embryos were transplanted into mice, as confirmed by GFP genotyping in multiple three germ layer tissues of E13.5 fetuses (Figures 2D and S2C) and observation of Oct4-GFP+ cells in the gonad tissue in 3 out of 17 fetuses (Figure 2C). These in vitro and in vivo characterizations collectively confirm that the purified cell-penetrating recombinant reprogramming proteins are sufficient to reprogram MEFs to become piPSCs, which are molecularly, morphologically, and functionally similar to conventional mESCs.
Figure 2.
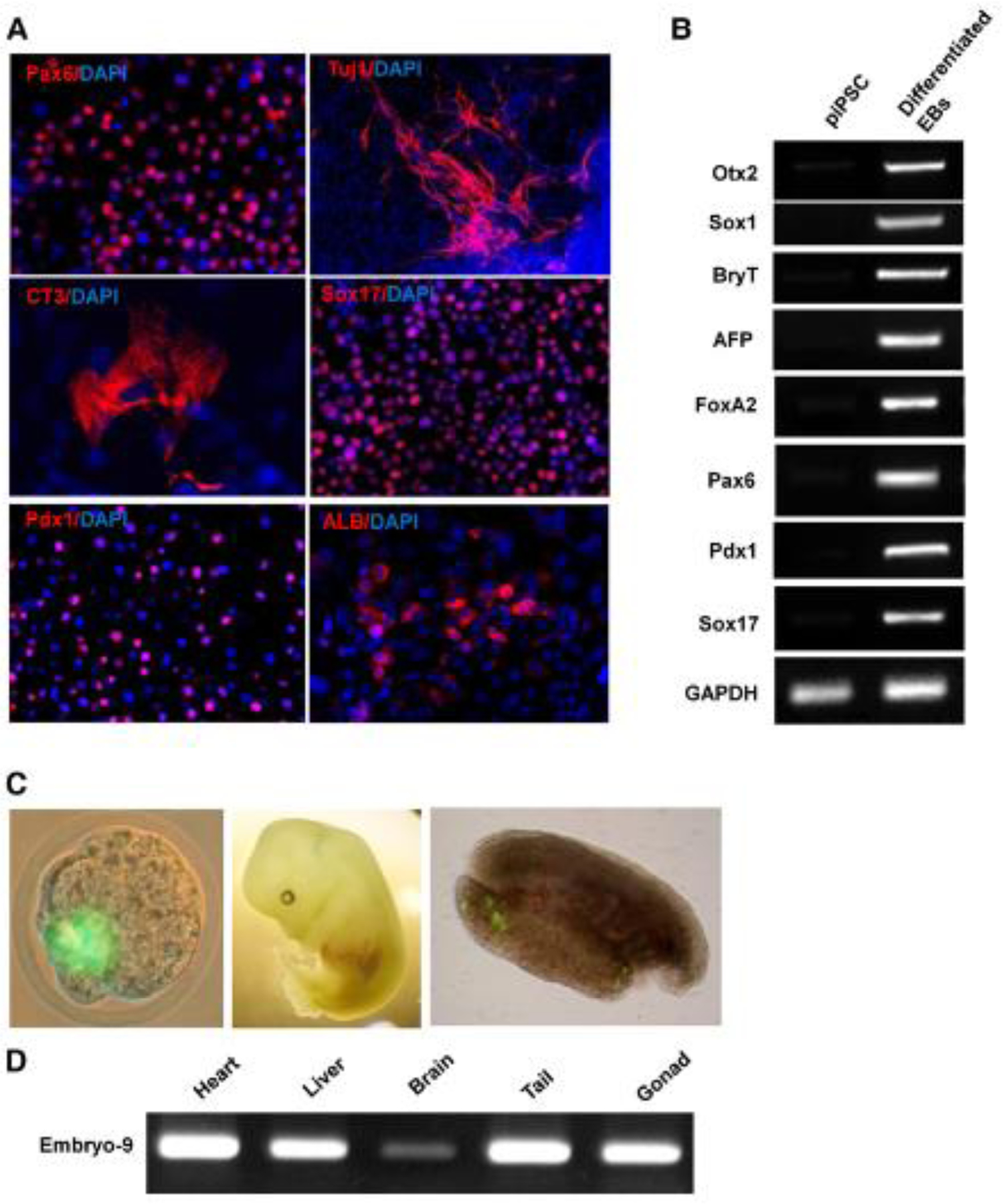
In Vitro and In Vivo Pluripotency of piPSCs.
iPSCs (and especially patient-specific ones), which are similar to ESCs but are much easier to create and, in the case of human cells, less controversial, present unprecedented opportunities for biomedical research and clinical applications. Realization of the promise of iPSCs will require improved methods of directed differentiation for generating homogenous populations of lineage-specific cell types as well as elimination of the risks and drawbacks associated with the current iPSC protocol, including genetic manipulation, and the low-efficiency/slow kinetics of induction. Recent advances in using various genetic approaches have addressed some of those iPSC challenges, including using nonintegrating adenoviruses to deliver reprogramming genes (Stadtfeld et al., 2008), transient transfection of reprogramming plasmids (Okita et al., 2008), a piggyBac transposition system (Woltjen et al., 2009; Kaji et al., 2009), Cre-excisable viruses (Soldner et al., 2009), and oriP/EBNA1-based episomal expression system (Yu et al., 2009). In addition, strategies of exploiting endogenous gene expression in certain cell types also allowed easier reprogramming and/or fewer required exogenous genes (Shi et al., 2008b; Aasen et al., 2008; Kim et al., 2008). Moreover, small molecules have been identified that enhance reprogramming efficiency and replace certain reprogramming factors (Shi et al., 2008a, 2008b; Li et al., 2009; Huangfu et al., 2008a, 2008b). However, all of those methods have yet to produce iPSCs without the use of any genetic material. Our present study is the first to demonstrate that somatic cells (i.e., murine fibroblasts) can be fully reprogrammed into pluripotent stem cells by direct delivery of recombinant reprogramming proteins. This protein transduction method represents a significant advance in generating iPSCs and has several major advantages over previous iPSC methods. First, it effectively eliminates any risk of modifying the target cell genome by exogenous genetic sequences, which are associated with all previous iPSC methods, and consequently offers a method for generating safer iPSCs. Second, the protein transduction method provides a substantially simpler and faster approach than the currently most advanced genetic method, which requires time-consuming sequential selection of potentially integration-free iPSCs. And finally, given the robustness and wide availability of large-scale recombinant protein production, our demonstrated completely chemically defined reprogramming regimen could potentially enable broader and more economical application of reprogramming methodology.
Supplementary Material
References
- Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, et al. (2008). Nat. Biotechnol 26, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, and Melton DA (2008a). Nat. Biotechnol 26, 795–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, and Melton DA (2008b). Nat. Biotechnol 26, 1269–1275. [DOI] [PubMed] [Google Scholar]
- Inoue M, Tomizawa K, Matsushita M, Lu YF, Yokoyama T, Yanai H, Takashima A, Kumon H, and Matsui H (2006). Eur. Urol 49, 161–168. [DOI] [PubMed] [Google Scholar]
- Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, and Woltjen K (2009). Nature 458, 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Zaehres H, Wu G, Gentile L, Ko K, Sebastiano V, Arauzo-Bravo MJ, Ruau D, Han DW, Zenke M, et al. (2008). Nature 454, 646–650. [DOI] [PubMed] [Google Scholar]
- Lafevre-Bernt M, Wu S, and Lin X (2008). Mol. Cancer Ther 7, 1420–1429. [DOI] [PubMed] [Google Scholar]
- Li W,Wei W,Zhu S,Zhu J,Shi Y,Lin T,Hao E, Hayek A, Deng H, and Ding S (2009). Cell Stem Cell 4, 16–19. [DOI] [PubMed] [Google Scholar]
- Michiue H,Tomizawa K,Wei FY,Matsushita M, Lu YF, Ichikawa T, Tamiya T, Date I, and Matsui H (2005). J. Biol. Chem 280, 8285–8289. [DOI] [PubMed] [Google Scholar]
- Okita K, Nakagawa M, Hyenjong H, Ichisaka T, and Yamanaka S (2008). Science 322, 949–953. [DOI] [PubMed] [Google Scholar]
- Shi Y, Desponts C, Do JT, Hahm HS, Scholer HR, and Ding S (2008a). Cell Stem Cell 3, 568–574. [DOI] [PubMed] [Google Scholar]
- Shi Y, Do JT, Desponts C, Hahm HS, Scholer HR, and Ding S (2008b). Cell Stem Cell 2, 525–528. [DOI] [PubMed] [Google Scholar]
- Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, et al. (2009). Cell 136, 964–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Nagaya M, Utikal J, Weir G, and Hochedlinger K (2008). Science 322, 945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Wadia JS, and Dowdy SF (2002). Curr. Opin. Biotechnol 13, 52–56. [DOI] [PubMed] [Google Scholar]
- Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hamalainen R, Cowling R, Wang W, Liu P, Gertsenstein M, et al. (2009). Nature 458, 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S (2009). Cell 137, 13–17. [DOI] [PubMed] [Google Scholar]
- Yu J,Vodyanik MA,Smuga-Otto K,AntosiewiczBourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. (2007). Science 318, 1917–1920. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, and Thomson JA (2009). Science Published online March 26, 2009. 10.1126/science.1172482. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.