

Abstract
In recent years, there have been significant advances in understanding the neuronal influence on the biology of solid tumors such as prostate, pancreatic, gastric, and brain cancers. An increasing amount of experimental evidence across multiple tumor types strongly suggests the existence of bidirectional crosstalk between cancer cells and the neural microenvironment. However, unlike cancers affecting many solid organs, brain tumors, namely gliomas, can synaptically integrate into neural circuits and thus can exert a greater potential to induce dynamic remodeling of functional circuits resulting in long-lasting behavioral changes. The first part of the review describes dynamic changes in language, sensory, and motor networks following glioma development and presents evidence focused on how different patterns of glioma-induced cortical reorganization may predict the degree and time course of functional recovery in brain tumor patients. The second part focuses on the network and cellular-level mechanisms underlying glioma-induced cerebral reorganization. Finally, oncological and clinical factors influencing glioma-induced network remodeling in glioma patients are reviewed.
Keywords: cancer related cognitive impairments, central nervous system plasticity, cognition, cortical remodeling, diffuse glioma, neuron network, glioma network
1. Central Nervous System Remodeling
Central nervous system (CNS) remodeling builds on the concept of cortical plasticity which refers to far reaching changes occurring within the CNS throughout the life of an organism in response to internal and external demands. Cerebral plasticity is typically thought to be greatest during early brain development. This process is sometimes referred to as “critical periods” wherein neural circuits change both structurally and functionally according to a new experience and/or learning stimuli.[1,2] This adaptive neural plasticity that occurs throughout the life of an individual is critical to maintain normal neuronal function and support cognitive information processing in the mature CNS.[3] Different types of plasticity mechanisms, namely synaptogenesis, neurogenesis, and myelin remodeling have been demonstrated to underlie the structural and functional-level changes of neural circuits in the adult CNS.[4] Similar to the ability of the brain to adapt to a changing environment during adolescence and adulthood, the neural plasticity processes occurring as a mechanism to compensate for the steady but gradual loss of neurons during aging are significant.[5,6] Although reduced when compared with early developmental stages, evidence of neuronal network remodeling in adult cancer is mainly derived from preclinical and clinical studies of injured CNS where in the surviving neural tissue reacts to brain damage and compensates for the injury occurring in different pathological conditions such as stroke, epilepsy and brain neoplasms. The goal of this article is to review a contemporary understanding of how the nervous system is remodeled by brain cancers and tumor-associated factors. We begin with a review of known mechanisms of cortical and subcortical plasticity in the postacute setting across CNS diseases. We will then focus on the current understanding of brain cancer-induced cortical plasticity.
2. Stroke
Stroke, especially ischemic stroke, is a neurological disorder resulting from a sudden or gradual occlusion of blood flow to the brain. Circulatory disturbances and insufficient brain perfusion further trigger a complex pathophysiological cascade of biochemical and molecular events that are detrimental to neuronal, glial, and endothelial cell function in the ischemic brain.[7] The death of mature and progenitor neurons and glia accompanied by profound changes in neurovascular coupling and neurotransmission subsequently leads to a significant loss of function in the infarcted area as well as in contralateral regions remotely connected to the area of tissue damage.[8,9] Consequently, stroke patients often exhibit a wide range of motor and cognitive impairments, such as aphasia, delayed working memory, and slowed processing speed.[10,11] However, many patients show considerable recovery, with the greatest improvement in cognitive and motor task performance occurring within the first 3 months after the onset of injury.[12] It is believed that this neurological recovery following stroke is attributed to several distinctive, yet overlapping, processes that involve structural changes and functional remodeling of the damaged cortex and surrounding areas.[8] At the structural level, preclinical investigations demonstrate activation of neurogenesis, angiogenesis, axonal sprouting, and the formation of new neuronal connections in the peri-infarct cortical areas following stroke.[13,14] The relevance of these findings in adult human is debatable. Furthermore, it remains to be seen whether these mechanisms offer a direct functional benefit to patients. Advanced CNS imaging techniques, such as positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) have enabled the study of functional constituents of stroke recovery in both preclinical and clinical settings. Among several mechanisms, stroke-related cortical remodeling is reported as the most essential driver of functional recovery in both rodent and nonhuman primate animal models as well as in human stroke patients.[15–18]
3. Epilepsy
Epilepsy is a chronic disorder of electrical excitability characterized by the appearance of spontaneous recurrent seizures generated by an imbalance of cerebral excitatory and inhibitory synaptic transmissions.[19] Similar to stroke, both preclinical and clinical evidence supports dynamic structural and functional changes in cortical activation during and after epileptogenesis. Neuronal injury to mature neurons occurring during seizures may trigger a series of responses, including the sprouting of new axonal collaterals, increasing the number of synaptic connections and neurogenesis in the hippocampal dentate gyrus.[20] The cellular and molecular events associated with epilepsy-induced structural synaptic alterations further contribute to remodeling of neural circuits that could either reestablish normal neuronal functions or aggravate the pathological processes associated with epilepsy.[20,21] Importantly, evidence from animal models of epilepsy, demonstrating the ability of newborn neurons to mature and integrate into nearby neural circuitries implies the strong propensity of changes in the structural connectome to impact functional outcomes and recovery in epilepsy patients.[22] Among several CNS imaging techniques employed, fMRI has emerged as a widely used tool to investigate the long-range neuronal network remodeling occurring in both injured and unaffected hemispheres during seizures and postrecovery. Compared to the general population that are strongly left lateralized, patients with epilepsy are reported to have reduced predominance of language in their left hemisphere and show a more bilateral representation with simultaneous involvement of the contralateral right hemisphere.[23,24] Moreover, compared to acute lesions such as stroke and traumatic brain injury, patients with epilepsy are presented with language sites spanning over a wide area of left lateral cortex, extending well beyond traditional Broca’s and Wernicke’s areas in the dominant left hemisphere.[25] However, regardless of this strong evidence of cerebral language reorganization occurring in the setting of epilepsy, experimental and clinical studies on whether these changes contribute to improved cognitive performance or directly translate to clinical functional recovery remain limited.[26]
4. CNS Remodeling in Adult Glioma
In the past few years, there has been growing interest in the study of cancer-nervous system interactions, which has laid the foundation for the newly emerging scientific discipline of “cancer neuroscience.” Specifically, attention has been paid to the mechanisms by which brain cancers, namely low and high-grade gliomas induce structural and functional remodeling of the central nervous system and how glioma-induced neuronal network remodeling may shape the scope of neurological impairments and functional recovery in patients with brain tumors.[27,28] Unlike the sudden, acute, vascular based lesions caused by acute stroke and traumatic brain injury, brain cancers are characterized by the invasion of neoplastic cells in the brain microenvironment, including non-neoplastic neurons and glia. Brain cancers therefore have a great propensity to cause widespread changes in how short-range neural circuit and long-range neuronal networks operate. Invasive and noninvasive imaging modalities have empowered the study of glioma-induced cortical neuron reorganization using direct electrocortical stimulation (DES), PET, magnetoencephalography (MEG), fMRI, electrocorticography (ECoG), and transcranial magnetic stimulation (TMS) (Figure 1).[27,29,30]
Figure 1.
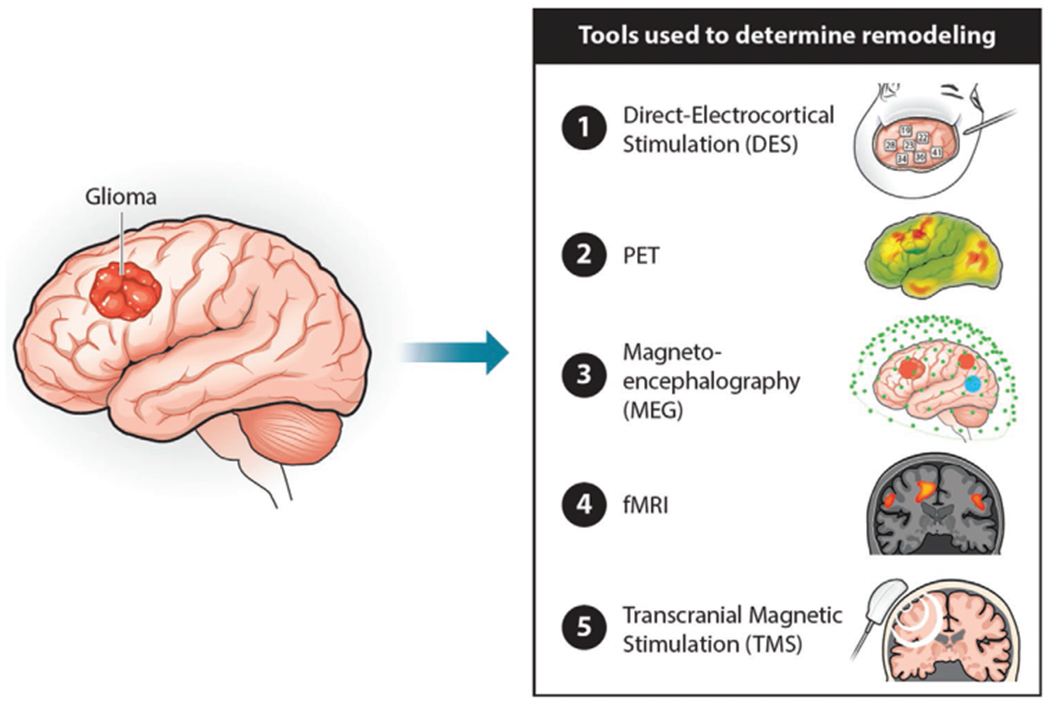
Schematic representation of the different imaging modalities-DES, PET, MEG, fMRI, and TMS to study glioma-induced cortical reorganization in brain tumor patients.
5. Clinical Evidence of Neuroplasticity in Glioma Patients
Unlike other brain pathologies, gliomas represent a unique model system for the study of longitudinal changes in brain reorganization and associated neuroplasticity mechanisms.[27] One of the initial reports of glioma-induced network plasticity was described by Duffau and colleagues in which investigators observed an absence of aphasia in patients with perisylvian dominant hemisphere gliomas despite tumor invasion into presumed functional areas such as the anterior frontal (Broca’s area) or posterior temporal (Wernicke’s area) lobes.[31–33] Equally interestingly, they found that patients exhibited a complete postoperative functional recovery even after the total removal of these presumed critical speech and motor areas that were traditionally considered to be inoperable.[32,33] The authors concluded that glioma invasion of functional areas may activate compensatory mechanisms to preserve normal function and that the location of cortical sites supporting neurological function may not be rigid but rather change over time in the setting of chronic nonstatic brain injury, in this case being diffuse glioma in adults. In agreement with Duffau’s group, several other preoperative brain mapping studies reported either mild or no symptoms of aphasia, suggesting that patients with glioma exhibit a substantial capacity for cerebral reorganization and that the tumor-induced neural compensatory mechanisms could potentially aid in postoperative functional recovery.[34,35]
Further evidence of glioma-induced network remodeling was demonstrated in patients who underwent repeat craniotomy for glioma resection guided by direct cortical stimulation brain mapping. In this study, a neurosurgeon applied a small electric current to the cortex (usually 1–6 Hz depending on the protocol) while patients completed a cognitive or behavioral task, which directly identifies functionally relevant cortical areas based on interpretation of task performance. Out of the 18 patients with WHO 2 and 3 diffuse gliomas undergoing repeat functional mapping craniotomies at the time of tumor recurrence, six demonstrated a change in the functional map between time points. These changes included both motor and language cortical sites identified as critical in the first surgery and were no longer found to be functionally significant during the second surgery.[36] Interestingly, these patients who lost function at the sites that were required for movement or language during the initial surgery exhibited no corresponding motor and speech impairments on neurological examination, indicating that the motor and language functions were relocated and assigned to a different cortical region by the time of the second resection.[36] Similar observations of motor and somatosensory cortical plasticity have been made by two independent groups led by Duffau and Gibb in diffuse low-grade glioma and glioblastoma patients, respectively, who reported a lack of neurological deficit despite the loss of cortical function in the primary motor and somatosensory areas in the second operation that were functionally active during the first surgery.[37,38] Overall, these findings provide the closest direct causal evidence that glioma-induced network reorganization can lead to long-term neuroplastic changes with preservation of function.
6. Neuroplasticity Mechanisms in Glioma
6.1. Network Level
Although the exact structural and functional changes underlying recovery are still under investigation, several imaging studies have been performed that describe different compensatory changes within either the short-range peritumoral region or long-range distant cortical sites (Table 1). Noninvasive imaging such as PET and fMRI have been successfully used to uncover compensatory mechanisms as expressed by an altered activation pattern in the functional networks either at rest or during language or motor task performance.[39] For example, using PET imaging, Thiel et al. investigated language network reorganization by administering a verb generation task in a large cohort of low-grade glioma patients in comparison to healthy controls.[35] Compared to controls in which activations were found predominantly in left Brodmann’s Area (BA)44 and BA45, superior posterior temporal gyrus, and right cerebellum, brain tumor patients showed intrahemispheric compensatory mechanisms by additionally recruiting distant left frontolateral regions, such as BA46, BA47, anterior insula, and left cerebellum during word generation tasks. Similar evidence of cortical redistribution of language function to peritumoral sites in brain tumor patients has been reported by several other studies.[32,34] PET imaging, however, has the known limitation of poor spatial and temporal resolution. Therefore, given that glioma–neuron interactions alter the excitability of local cortical neurons, Aabedi et al. recently demonstrated the extent to which glioma-infiltrated cortex can meaningfully participate in neural computations.[40] Using subdural electrocorticography, they demonstrated that glioma-infiltrated cortex engaged in synchronous activity during task performance in a manner similar to normal-appearing cortex but recruited a diffuse spatial network. However, neuronal activation within glioma-infiltrated language areas suffered from a loss of information storage capacity (entropy). The result in task performance was that glioma-injured cortex may retain the ability to participate in basic cognitive tasks but lose computationally demanding and nuanced aspects of cognition.[40] Taken together, these results illustrate ipsilateral hemispheric glioma-induced network remodeling.
Table 1.
Evidence of glioma-induced cerebral plasticity-Network level studies.
| Study | Patients [number] | Gliomagrade | Imaging modality | Function/network | Key findings |
|---|---|---|---|---|---|
| Piai et al., 2020 | 14 | LGG, HGG | MEG | Language | Evidence of presurgical plasticity with the right hemisphere performing similar neuronal computations as the left dominant hemisphere |
| Thiel et al., 2001 | 61 | LGG | PET | Language | Evidence of intra- and interhemispheric compensation with recruitment of atypical language areas and frontolateral activation in the nondominant hemisphere |
| Southwell et al., 2016 | 18 | LGG, HGG | DES | Language, sensory, motor | Preservation of neurological function despite cortical function loss in the primary motor and somatosensory areas during repeat surgery |
| Gibb et al., 2020 | 6 | HGG | DES | Somatosensory, motor | Evidence of plasticity in the primary motor and somatosensory cortices of glioblastoma patients |
| Aabedi et al., 2021 | 12 | LGG, HGG | DES | Language | Glioma-infiltrated cortex recruits a diffuse broader network of atypical cortical regions and participates in coordinated neural responses during speech production |
| Rosenberg et al., 2008 | 1 | LGG | fMRI | Language | Compensation exerted by right hemisphere is incomplete; Change in language-related laterality from left to right corresponded to a decline in language function |
| Traut et al., 2019 | 73 | LGG, HGG | MEG | Language | Robust involvement of contralateral homotopic language areas in functional reorganization |
| Bulubas et al., 2020 | 78 | LGG, HGG | MEG | Motor | Both contralateral and ipsilateral motor cortices can compensate for impaired function in the primary motor cortex of brain tumor patients |
| Vassal et al., 2017 | 6 | LGG | fMRI | Sensorimotor | Increased interhemispheric connectivity between the primary sensorimotor cortex ipsilateral to the tumor and the contralateral SMA correlates with rapid motor recovery |
| Acioly et al., 2015 | 1 | LGG | fMRI | Sensorimotor | Functional motor recovery correlated with a shift of SMA activation to the contralateral hemisphere concomitant with an increase in the lateral premotor circuitry |
| Rosenberg et al., 2010 | 26 | LGG, HGG | fMRI | Language, Motor | Greater SMA activation in the lesioned hemisphere correlated with motor and language recovery |
| Duffau et al., 2002 | 3 | LGG | DES | Language, Sensorimotor | Evidence of surgical resection-induced functional reorganization in the peritumoral and infiltrated brain region |
| Bulubas et al., 2016 | 100 | LGG, HGG | nTMS | Motor | Evidence of significant contribution of ipsilateral secondary motor areas such as premotor cortex in patients with motor eloquent brain tumors |
Questions remained, however, regarding whether glioma-induced remodeling included contralateral hemispheric compensation. Thiel et al. demonstrated resting state neuronal activation in the right frontolateral nondominant hemisphere in patients with brain tumors within the left hemisphere, suggesting involvement of the interhemispheric neural compartment to compensate for the increasing demand for speech production in these patients.[35] However, this observed nondominant hemisphere activation was associated with decreased language performance in patients, implying the limited ability of right-sided language area homologues to maintain function and compensate for glioma-induced injury.[35,41] These findings are in agreement with stroke studies demonstrating that the degree of aphasia recovery after brain injury is mainly predicted by the preservation of ipsilesional (left hemisphere) language areas and that the activation of the contralateral connections may be indicative of incomplete language recovery by the affected dominant hemisphere.[25,42,43] Nonetheless, it is worth noting that several other studies have demonstrated robust compensatory recruitment of right-sided language area homologues in mediating functional recovery in patients, and hence the contribution of such reorganization to the preservation of language performance still remains ambiguous.[25,26,44,45]
Similar to language network, several studies have demonstrated evidence of plasticity of motor activation in brain tumor patients with gliomas inducing large-scale alterations of the sensorimotor network at both intra- and interhemispheric levels.[46] The supplementary motor area (SMA) is a segment of the premotor cortex that plays an important role in the planning or initiation of motor activity, including speech function.[47] Patients with tumors invading the dominant hemisphere SMA typically express a disorder called SMA syndrome characterized by immediate postoperative motor and speech dysfunctions that recover almost completely within a few weeks or months of SMA resection.[47] Acioly et al. studied the potential compensatory mechanisms of SMA syndrome and found that postoperative motor recovery occurred in parallel with a shift of SMA activation to the opposite hemisphere (contralateral to the tumor) together with an increased recruitment of the lateral premotor circuitry in the contralesional hemisphere.[48] This finding is in agreement with previous functional MRI studies where the degree of contralateral SMA recruitment was associated with a faster motor recovery time following surgical SMA resection.[49,50] On the contrary, another fMRI study showed that the activation of regions ipsilateral and adjacent to the lesioned SMA is essential for functional recovery from SMA syndrome and that the recruitment of contralateral healthy SMA offers no additional protective role and rather indicates a functional decompensatory effect.[51]
Further evidence of plasticity of the motor cortical network came from patients with primary motor cortex tumors.[52] Using intraoperative stimulation mapping, Duffau and colleagues described functional reshaping of motor areas that occurred in the interval between two consecutive surgeries in a patient with left precentral WHO grade 2 oligodendroglioma.[37] Although the patient exhibited no motor neurological impairments, intraoperative electrical stimulation showed motor function within the tumor, which resulted in incomplete tumor resection during the first surgery. However, at the second operation, motor sites were no longer identified in the tumor parenchyma and primary motor areas of the hand showed a more posterior location than during the first procedure, suggesting that both pre- and postoperative functional plasticity can occur in patients undergoing resection of the brain.[37] Another study by the same group demonstrated acute reorganization in a patient with a left precentral lesion as evidenced by the sudden intrasurgical appearance of additional functional sites (for hand and arm movement) in regions of primary motor cortex that were functionally silent before tumor resection.[53] Functional compensatory mechanisms promoting motor recovery involve the recruitment of a widely distributed motor cortical network such as the ipsilateral nonprimary, or contralateral primary and nonprimary motor cortices.[54,55] The significant contribution of ipsilateral secondary motor areas, including premotor cortex and SMA has been previously reported in well-recovered stroke patients and primates with unilateral motor cortex lesion.[8,56] In line with the findings from these studies, Bulubas et al. observed a broader spatial representation of motor areas using TMS in patients with motor eloquent brain tumors, including the recruitment of superior and middle frontal gyri and premotor areas of the lesioned hemisphere.[54] Another recent study by the same author, which compared changes in the location of cortical motor network of recurrent glioma patients using serial MEG scans acquired before surgery and at tumor relapse, detected a considerable shift of both the ipsi- and contralesional primary motor cortices occurring between first and second MEG scan time points.[55] Furthermore, the authors observed a direct correlation between ipsilesional activation peak shift and the presence of motor impairments and the time period between imaging. Patients with existing motor impairments and/or a longer period between imaging demonstrated a higher degree of cortical reorganization as evidenced by larger primary motor cortex shifts.[55]
Compared to the extensive work demonstrating dynamic reorganization of brain functional areas occurring before, during, and after surgery in patients with infiltrative gliomas, the number of studies which examined language and/or motor cortex plasticity in noninfiltrative brain tumors such as meningiomas or metastases is limited. Avramescu-Murphy et al. studied pre- and postoperative language reorganization in the dominant and nondominant hemispheres of glioma and metastasis patients and found that, unlike in gliomas, the language activation pattern observed before surgery remained unchanged even 2 years after surgery in patients with brain metastasis.[57] They concluded that compared to diffuse gliomas, brain metastases infiltrate white matter structures to a lesser extent and are thus less likely to produce a widespread destructive effect on the language network.[57] In contrast to the absence of post-to-presurgical language reorganization, tumor resection resulted in plasticity of motor cortical areas and facilitated restoration of primary motor area function and recovery of patients with metastatic brain tumors.[58]
Although meningiomas are the most frequently diagnosed primary CNS tumors, little research has been done regarding how meningiomas affect functional networks and influence functional outcomes. Using MEG, van Nieuwenhuizen et al. examined the resting state functional connectivity of meningioma patients including correlations with cognitive performance.[59] They found that compared to other cognitive domains, patients showed working memory impairments, which was in turn associated with lower functional connectivity in the default mode network (DMN).[59] Given that both meningiomas and diffuse low-grade gliomas are slow growing tumors, a more in-depth understanding of the underlying network plasticity mechanisms will be critical to ascertain differences in network reorganization and functional compensation between these two oncologic entities.
6.2. Cellular Mechanisms of Plasticity
Although there is still no consensus regarding whether ipsilateral or contralateral cortical remodeling represents the driving force for functional recovery, collective findings point to a hierarchical model of functional compensation in the setting of diffuse glioma (Figure 2). As demonstrated in stroke induced remodeling, first, an intrinsic reorganization occurs within lesioned language or motor areas—this compensation utilizes eloquent neural networks which remain intact within the tumor and typically correlate to a favorable functional outcome. Next, when reorganization is insufficient, neural networks that are either adjacent to the tumor or even remote to the damaged area in the ipsilesional hemisphere are recruited. Finally, if the above two repair mechanisms are still incapable of yielding sufficient recovery, the functional control is then transferred to the unaffected contralateral hemisphere—this compensation results in maladaptive behavioral responses and demonstrates a poor recovery with neurological impairments.[60] Although the above neuroplastic changes have been demonstrated to support behavioral recovery, a mechanistic understanding of these processes is incompletely understood in humans. Existing evidence on the pathophysiological changes associated with brain lesion-induced cortical plasticity is mostly derived from preclinical animal models.
Figure 2.
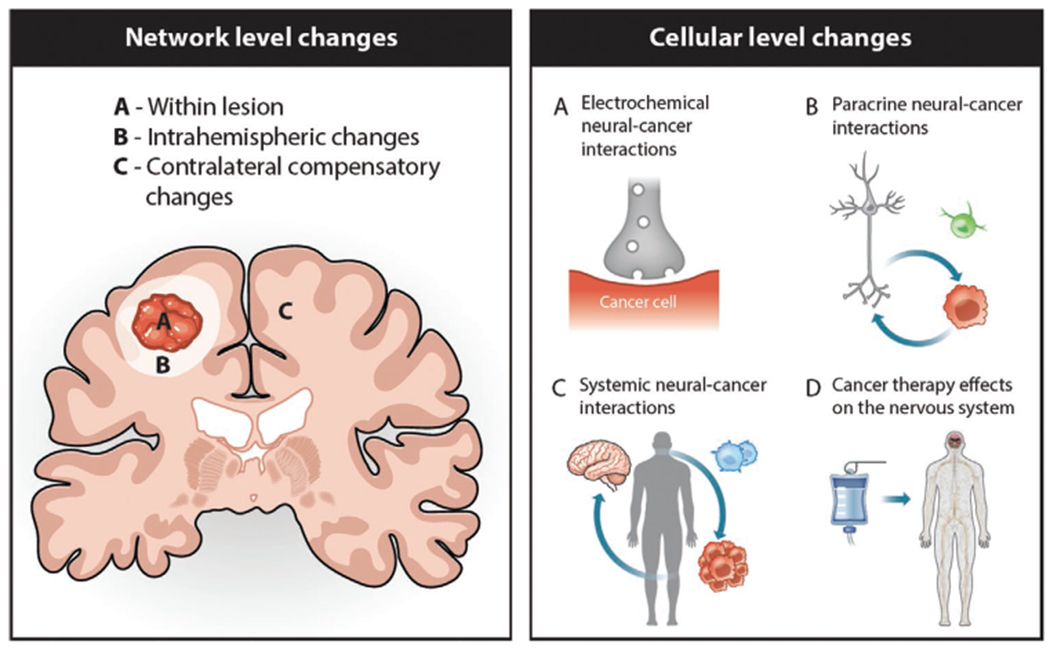
Schematic representation of the network and cellular-level mechanisms underlying glioma-induced cerebral reorganization. Left panel demonstrates a hierarchical model of functional compensation in the setting of diffuse glioma wherein compensation initially utilizes eloquent neural networks which remain intact within the tumor A) or in the peritumoral area B) before the recruitment of the unaffected contralateral hemisphere C). Right panel A, B) illustrates interaction between nervous system (gray) and cancer cells (red) and neuron-glioma communication as a key cellular mechanism underlying glioma-induced cortical neuroplasticity. C) In addition to the direct neuron-cancer interaction, nervous system also influences immune responses and promotes tumor growth and progression via expression of cytokines and tumor-associated immune cells (blue) in the tumor microenvironment. D) Chemotherapy or radiotherapy may also have significant toxic effect on the nervous system and the extent to which therapy-induced neuropathy may modulate nerve-cancer interaction and glioma growth needs to be investigated.
In the healthy brain, redundant functional synapses, which are present throughout the cortex, are normally silenced by GABAergic inhibitory interneurons. However, in the event of an acute or chronic cortical injury such as stroke, this inhibition is lost and the pre-existing latent excitatory connections can be activated to compensate for the injured neurons, thus helping to restore normal function.[61] Decreased intracortical inhibition with consequent unmasking of redundant excitatory cortical circuits is thought to represent one of the primary cellular compensation strategies in glioma-induced neuroplasticity. In support of this concept, application of a GABA antagonist to a small region of the primary motor cortex in adult rats rapidly elicits new representational patterns in the motor cortex area adjacent to the affected region.[62] The authors further demonstrated that a decrease in intracortical inhibition and an associated local increase in cortical excitability can unveil latent redundant connections and that the balance between excitatory and inhibitory circuits is a strong dictator of successful brain reorganization.[62] Similar unmasking processes and surgical resection-induced regional hyperexcitability have been demonstrated using intracortical mapping in glioma patients. For instance, rapid functional reorganization was observed during surgery in the primary motor cortex of glioma patients using intraoperative DES mapping.[53] The authors hypothesized that such acute functional reshaping could be due to the local increase in cortical excitability induced by surgery, which further allows for the unmasking of redundant motor network connections.[53] The same notion of decreased intracortical inhibition, more specifically a suppression of transcallosal inhibition, has been implicated in the recruitment of contralesional healthy hemisphere in brain tumor patients.[60] Prior studies have demonstrated that inhibitory fibers connect the two primary motor cortices and that motor activation in one hemisphere suppresses activation of the corresponding motor cortex in the contralateral hemisphere. However, in gliomas and other brain lesions such as stroke or traumatic injuries that could cause direct damage to the primary motor region, this interhemispheric inhibition is lost, which allows for increased activation of the pre-existing contralateral connections to compensate for the affected motor function.[63]
Another proposed mechanism to explain glioma-induced cortical neuroplasticity, is the recruitment of new neural networks from regions that are either adjacent or connected to the area of lesion.[64] At a cellular level, the growth of new neural functional connections has been demonstrated to be complemented by alterations in synaptic strength via long-term potentiation or depression, neuronal excitability, and structural changes including increased turnover of dendritic spines, and enhanced neuro-and gliogenesis.[65] In line with this theory, increased dendritic loss and associated reduction in neuronal excitability in the peri-infarct cortex and adjacent cortical areas have been attributed to the neurological impairments observed during the early (hyperacute and acute) stages of stroke in humans and rodent models.[18] Interestingly, a genetic or pharmacological blockade of inhibitory GABAergic signaling in the peri-infarct region has been shown to restore neuron excitability and promote functional plasticity and poststroke recovery in rodent models.[18] While several experimental and clinical studies in stroke research have validated the strong link between the formation of new synaptic connections, neurogenesis, and functional recovery, data suggesting similar mechanisms in the brain tumor patient population, especially in the context of language and motor recovery, are still largely lacking. This may be explained by the differences in the kinetics and type of brain injury inflicted by the two pathologies; while brain tumors, especially low-grade gliomas, are slow-growing and progressive in nature, stroke results in the sudden death of neurons and acute loss of function, which could rapidly recruit neurogenic signaling pathways to compensate for injured cortical regions.[66]
Enhanced neuronal excitability and increased functional connectivity between neurons is a key cellular feature underlying poststroke plasticity and thus, several drugs that positively modulate glutamate-induced AMPAR-gated excitatory synaptic currents, such as AMPAkines have been successfully used to improve motor recovery in rodent models.[18] Although gliomas are traditionally considered an ablative process, evidence suggests the presence of non-neoplastic astrocytes and mature neurons within gliomas interacts with neoplastic cells within the tumor. Interestingly, in recent years, an increasing number of studies have been published investigating the interaction of brain tumor cells and active neurons in the tumor microenvironment and how this interplay alters cortical excitability. In contrast to the beneficial role of excitatory neurotransmission in stroke, preclinical evidence indicates a cancer-promoting effect of excitatory neuronal activity in malignant gliomas wherein increased neuronal activity and hyperexcitability stimulate glioma cell proliferation and invasion.[67–69] Nervous system-cancer crosstalk occurs both through direct synaptic communication between neurons and malignant glioma cells and neuronal-activity-dependent secretion of growth factors, such as neuroligin-3 and brain-derived neurotrophic factor (BDNF) and cancer cell-derived release of glutamate and glypican-3 in the tumor microenvironment (Figure 2).[67–69] Among these factors, BDNF deserves special attention because of its established role in promoting myelination, synaptic connectivity, and synaptic strength in the normal healthy brain. In health, BDNF regulates synaptic plasticity via signaling through its membrane receptor tropomyosin receptor kinase B (TrkB) to recruit calcium signaling pathways and promote AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptor trafficking to the postsynaptic neuronal membrane.[70] Interestingly, a recent preclinical study demonstrated that BDNF-TrkB signaling also similarly regulates the number of malignant synapses formed between neurons and glioma cells and that the blockade of neuronal activity-regulated BDNF secretion can significantly decrease glioma cell proliferation and growth both in vitro and in vivo.[71] Nervous system activity also strongly influences adaptive myelination by regulating oligodendrocyte precursor cell (OPC) proliferation and differentiation.[72] Hence, it is possible that in the presence of aberrant neuronal activity associated with gliomas, these normal plasticity mechanisms may be hijacked to facilitate maladaptive myelination, contributing to aggressive tumor growth, network hypersynchrony, and epilepsy progression.[72,73]
Increasing evidence indicates that neurons within the tumor microenvironment can also robustly regulate the growth and progression of cancers that metastasize to the brain. For example, Chen et al. revealed that metastatic cells from breast and lung carcinomas selectively express a cell adhesion molecule called protocadherin-7 to establish gap junctions with normal astrocytes and further benefit from the neural microenvironment for enhanced tumor growth and brain metastasis.[74] More interestingly, another group which studied breast-to brain metastasis showed that, unlike the direct synaptic interactions between presynaptic neurons and postsynaptic glioma cells, metastatic breast cancer cells engage in perisynaptic contact with neurons, forming pseudotripartite synapses.[75] Notably, they found that in the pseudotripartite configuration, metastatic breast cancer cells express high affinity NMDA receptor, which then utilize glutamate secreted by adjacent presynaptic neurons to promote breast cancer cell proliferation and invasive tumor growth.[75]
In addition to investigating glioma–neuron interactions, several other groups have examined neurovascular responses during glioma progression and found profound disruption of neurovascular coupling in tumor-burdened regions and whole-brain functional connectivity.[76,77] Given this strong preclinical evidence, it is thus possible that glioma cells can integrate into the broader neuronal network and influence neural circuit dynamics in brain tumor patients. In a recent study, using subdural electrocorticography, Aabedi et al. sampled both normal-appearing and glioma-infiltrated cortex during speech production to assess the language task-related circuit dynamics of IDH-wild-type glioblastoma patients.[40] Interestingly, although glioma-infiltrated cortex was found to participate in coordinated neural responses during speech production similar to normal-appearing cortex, tumor-infiltrated brain regions showed a diffuse broader network of atypical cortical regions and a decreased ability to encode information when challenged on nuanced aspects of speech processing, such as vocalization of mono- versus polysyllabic words.[40] These findings demonstrate the retained, albeit reduced, ability of glioma-infiltrated cortex to participate in cognitive processing and underscore the functional consequences of glioma-network integration on neural circuit dynamics. Further studies are warranted to better understand the role of glioma-induced neuronal changes in cognitive impairments and recovery in brain cancer and to determine the mechanistic significance of neuron-glioma communication.
Besides the complex interplay occurring with active neurons, neoplastic glioma cells also interact with infiltrating immune cells in the tumor microenvironment and create a tumor-promoting neuroinflammatory niche that acts in favor of the proliferation, invasion, and survival of tumor cells.[78] Unlike other neuroinflammatory CNS pathologies like multiple sclerosis and encephalitis where the neuro-immune axis is in a hyperactive state, in the context of glioma, neoplastic cells secrete a variety of cytokines, chemokines, and growth factors into the microenvironment, which actively shifts the neuro-immune axis to an immunosuppressive phenotype.[79] However, as the tumor grows and evolves, competent immune cells, namely microglia, natural killer cells, as well as reactive astrocytes, release proinflammatory mediators, such as transforming growth factor and tumor necrosis factor, further exacerbating the inflammatory milieu of the peritumoral microenvironment.[78] Importantly, this cellular inflammation has been identified as a key cellular mechanism contributing to the cortical network excitability progression in the context of tumor-related epilepsy. Specifically, reactive microgliosis accompanied by higher levels of microglial infiltration has been found to correlate positively with cortical hyperexcitability and tumor progression in preclinical animal models.[80] Apart from the tumor-associated inflammatory responses, chemo-and radiation therapy has also been demonstrated to induce neuroinflammation and is attributed as a key mechanism underlying the functional network connectivity disruption and cognitive decline observed in glioma patients.[81–84]
Given this strong evidence of tumor-immune interactions, modulation of the immune system offers a promising approach to regulate cancer growth and progression. As a result, cancer immunotherapies aimed to enhance the endogenous immune response and restore immunity in the tumor microenvironment has become an emerging therapeutic option for gliomas. Extensive research carried out over the past several years has also demonstrated the influence of active neurons on glioma cells and their bidirectional relationship. Therefore, various approaches that could target neuron-glioma signaling and disrupt the integration of brain cancer into functional circuits opens another growing area of research that allows for new developments in the diagnosis and treatment of brain cancers.
7. Oncological and Patient Factors Impacting Glioma-Induced Remodeling and Functional Recovery
7.1. Tumor Grade
Among many tumor-related characteristics, tumor-grade has long been considered as a significant factor influencing glioma-induced remodeling and functional recovery. It is well identified that patients with WHO 2 diffuse low-grade glioma often present with mild language and motor impairments, while patients with high-grade glioma typically exhibit severe impairments.[27,85] In a recent study, Yuan et al. studied resting-state functional connectivity of language networks in low- and high-grade left cerebral glioma patients. They found diminished intra- and interhemispheric language network connections in patients with high-grade glioma compared with patients with low-grade; moreover, the severity of network disruption positively correlated with tumor grade.[86] It is worth noting that the high-grade glioma cohort in this study displayed worse language task performance relative to low-grade glioma patients even after controlling for tumor location. The authors concluded that gliomas, depending on tumor grade, induce different levels of neuroplastic responses and that the behavioral impairments seen in high-grade glioma patients may be suggestive of poor network reorganization and functional compensation. The difference in the extent of functional network disruption and behavioral outcomes caused by low- and high-grade gliomas could be explained by differing tumor growth kinetics. While rapid infiltration and migration of high-grade gliomas into language or motor areas may overwhelm naturally occurring organic remodeling efforts, slow progression of low-grade gliomas may permit sufficient time for functional network reorganization, resulting in normal or only slightly impaired neurocognitive functions.[27]
7.2. Tumor Location
Similar to tumor grade, location of the brain lesion appears to be another important correlate of functional impairment severity and extent of recovery in brain tumor patients. Ghumman et al. observed asymmetric responses between left-and right hemispheric tumors on the functional connectivity of DMN, while patients with right-sided tumors had no effect on DMN activity. Furthermore, individuals with left-sided tumors showed significantly reduced connectivity in the resting state network.[87] In another study, Deverdun et al. compared patients with left temporal and frontal low-grade gliomas and found that only the tumor location in the left temporal lobe overlapping cortical areas of the middle temporal gyrus and underlying white matter was associated with lower task performance.[88] Similar findings of behavioral impairments or lack of functional recovery have been reported in cases when tumors invade “eloquent” brain regions, affecting subcortical pathways. For instance, Smit and colleagues examined diffuse low-grade glioma patients and reported a significant correlation between neurological impairments and tumor localization in eloquent cortico-subcortical regions as opposed to tumors with strictly cortical location.[89] Besides cortical plasticity, subcortical remodeling may also play a critical role in functional recovery in the setting of diffuse glioma. However, compared to cortical regions, plasticity of the white matter is thought to be limited, and therefore, injury to the subcortical white matter tracts could lead to irreversible damage and result in permanent functional deficits. Hence, the extent of tumor infiltration of white matter tracts and preservation of white matter pathways important for motor and language functions during surgery may predict long-term impairments in glioma patients.[90] The concept of glioma-induced myelin plasticity remains largely unexplored to date.
Not all cortical areas share the same potential for remodeling in response to glioma-induced injury. For instance, using connectivity-based cluster analysis, Herbet et al. described the presence of varying neuroplastic potential within the cortical regions of low-grade glioma patients.[91] Specifically, primary areas that lacked alternative functional circuits for processing sensorimotor information, such as the dorsal part of the precentral gyrus (the motor cortex and the underlying corticospinal tract), and the postcentral gyrus (the somatosensory cortex) were found to have a low functional compensation index compared to other cortical regions.91] Accordingly, tumors invading primary sensory and/or motor cortices are more likely to cause permanent behavioral impairments or delayed postoperative recovery.[27] Along this line, another potential factor that could influence glioma-induced cerebral reorganization is the degree of connectivity between the lesioned brain region and its associated functional networks and the type of function affected. Compared to sensorimotor cortex, language processing relies on broad dorsal and ventral networks which are strongly interconnected. Hence tumor-induced disruption of the language network can recruit parallel functional circuits, resulting in more efficient compensation effects and faster recovery in aphasic patients.[60]
7.3. Tumor Volume
The association of tumor volume with neurological impairments and recovery in glioma patients has been explored by multiple groups. Although incomplete, a number of reports have demonstrated minor, or no correlation between tumor volume with the severity of neurological symptoms in low-and high-grade glioma patients.[86,88,89] It is worth noting that the above studies mainly reported neurological outcomes at the time of radiological diagnosis with no further assessment of the influence of preoperative tumor volume on function throughout the disease trajectory. Although fewer in number, reports exist on the significant effect of preoperative tumor volume on the extent of cortical and subcortical remodeling that occurs postsurgically. It was reported that tumor volumes at time of the initial surgery was significantly smaller in patients who exhibited a higher degree of functional reshaping and postoperative plasticity during the second surgery than those with larger tumors.[36]
7.4. Patient-Specific Factors
Besides the above-mentioned tumor-related factors impacting cortical and subcortical remodeling, patient factors exist. Patients’ characteristics not necessarily related to the clinical condition, such as age, sex, and genetics, can influence functional recovery. Although age and gender differences are considered to influence brain plasticity in general, majority of the earlier studies failed to demonstrate significant age and sex effects on neurological impairments and/or functional recovery in brain tumor patients.[55,85,86,92] However, a recent study which investigated a large cohort of patients with left-hemispheric diffuse low-and high- grade gliomas found a significant negative correlation between age and patients’ language task performance even after controlling for tumor grade.[86] Besides influencing plasticity, age is also considered as a significant factor affecting clinical outcomes and survival of the glioma population; elderly patients with high-grade gliomas are traditionally thought to have a poor prognosis than the younger glioma population. Interestingly, one of the earlier studies which compared younger (<65 years) and elderly (>65 years) patients with newly diagnosed glioblastoma failed to demonstrate a significant age-related effect on progression-free survival and overall survival outcomes between the two groups.[93] More importantly, they reported that the above relationship held true only for those elderly and younger patient groups who had extensive tumor resection, and compared with the younger group, the overall survival was significantly worse for elderly patients who underwent tumor biopsy only without any surgical resection.[93] In line with the findings from this study, several other groups identified the role of maximal tumor resection as a major prognostic factor influencing clinical outcomes and reported no significant differences in the functional status and survival times in younger and elderly patients who underwent similar extent of resection.[94–96]
Besides age, prior studies have investigated the prognostic roles of racial and socioeconomic factors in influencing survival outcomes of glioma patients. Liu et al. analyzed a large-scale cancer registry study of glioblastoma patients and found improved overall survival in Asian/Pacific Islanders, Hispanic Whites, and Blacks compared to Non-Hispanic White patients after controlling for tumor, treatment characteristics, and differences in time of symptom onset to hospital presentation.[97] With respect to socioeconomic status, a lower median household income was associated with a poorer outcome in black patients compared to other racial groups. However, this effect was not associated with glioblastoma and was largely driven by comorbid conditions such as cardiovascular and respiratory diseases, and other nonbrain malignant cancers in the black population.[97] Another study which examined the differences in incidence and survival rates of glioma in adults reported similar results with a higher tumor incidence (regardless of glioma subtypes) and lower survival rates in non-Hispanic white patients compared to other racial or ethnic groups.[98] This observed higher risk for glioma and poor survival outcomes in non-minority patients could be attributed to the differences in environmental exposures as well as different molecular and genetic makeup of tumors between races.[99,100] However, further studies are necessary to conclude a causal association between the above factors and glioma risk disparities among races and ethnicities.
With respect to genetics, similar to what has been documented in stroke-induced deficits and poststroke plasticity research, a number of synaptic plasticity-related genes have been suggested to be important to the observed variability in the severity of cognitive decline, as well as the rate of recovery, between glioma patients.[101,102] Specifically, single-nucleotide polymorphisms in the human genes coding for brain-derived neurotrophic factor, dopamine receptor 2, catechol-O-methyltransferase, and apolipoprotein E have been reported to significantly alter the neurocognitive performance of brain tumor patients when assessed several months after surgery without ongoing chemotherapy as well as increase the vulnerability of patients to cognitive and language dysfunction following chemoradiation.[103,104]
8. Conclusion
Here, we have described evidence of brain tumor-induced CNS remodeling focused specifically on cortical remodeling. Although the majority of the reports detailed in this paper reinforce our knowledge of glioma-induced functional reorganization, a number of pressing unanswered questions remain, including how patient outcomes may benefit from effects. Along this line, future work is warranted to uncover whether all forms of CNS plasticity are beneficial to the patient and how each pattern of reorganization differentially affects functional outcomes throughout the disease trajectory.
Acknowledgements
The authors would like to thank Noel Sirivansanti for contributing to the original artwork included in this work. This study was supported by the NIH grants K08NS110919, the Robert Wood Johnson Foundation grant 74259, the UCSF LoGlio Collective, and Resonance Philanthropies to S.H-J.
Biographies

Saritha Krishna, Ph.D., her research interests focus on understanding how glial cells influence brain physiology and, ultimately, human behavior. She completed her Ph.D. at the University of Georgia, where she studied the role of environmental triggers in the modulation of glial-neuron communication in health and disease. In her current role as a postdoctoral fellow at the University of California, San Francisco, she applies the principles of glial-neuron communication to brain tumors and studies the mechanisms by which gliomas interface with the neuronal microenvironment and affect functional cognitive networks.

Shawn L. Hervey-Jumper, MD, is an associate professor at the University of California San Francisco. He is a neurosurgeon researcher with a clinical practice focused on the surgical management of patients with brain cancer within functional areas using physiological mapping. His research laboratory seeks to understand how brain tumor cells influence functional circuits and cognition. His ultimate goal is to better understand the mechanisms of cognitive impairments, thereby leading to novel treatment options focused on improving network recovery and, therefore, cognitive outcomes for patients.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Saritha Krishna, Department of Neurological Surgery, University of California, San Francisco, CA 94143, USA.
Shawn L. Hervey-Jumper, Department of Neurological Surgery, University of California, San Francisco, CA 94143, USA; Weill Neurosciences Institute, University of California, San Francisco, CA 94143, USA; Helen Diller Comprehensive Cancer Center, University of California, San Francisco, CA 94143, USA
References
- [1].Berardi N, Sale A, Maffei L, Dev. Med. Child Neurol 2015, 57, 4. [DOI] [PubMed] [Google Scholar]
- [2].Hubener M, Bonhoeffer T, Cell 2014, 159, 727. [DOI] [PubMed] [Google Scholar]
- [3].Tau GZ, Peterson BS, Neuropsychopharmacology 2010, 35, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].O’Rourke M, Gasperini R, Young KM, Neural Regener. Res 2014, 9, 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bayona NA, Bitensky J, Teasell R, Top Stroke Rehabil. 2005, 12, 1. [DOI] [PubMed] [Google Scholar]
- [6].Berlucchi G, Buchtel HA, Exp. Brain Res 2009, 192, 307. [DOI] [PubMed] [Google Scholar]
- [7].Xing C, Arai K, Lo EH, Hommel M, Int. J. Stroke 2012, 7, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pekna M, Pekny M, Nilsson M, Stroke 2012, 43, 2819. [DOI] [PubMed] [Google Scholar]
- [9].Presa JL, Saravia F, Bagi Z, Filosa JA, Front. Physiol 2020, 11, 584135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Al-Qazzaz NK, Ali SH, Ahmad SA, Islam S, Mohamad K, Neuropsychiatr. Dis. Treat 2014, 10, 1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hatem SM, Saussez G, Della Faille M, Prist V, Zhang X, Dispa D, Bleyenheuft Y, Front. Hum. Neurosci 2016, 10, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Desmond DW, Moroney JT, Sano M, Stern Y, Stroke 1996, 27, 1798. [DOI] [PubMed] [Google Scholar]
- [13].Carmichael ST, Kathirvelu B, Schweppe CA, Nie EH, Exp. Neurol 2017, 287, 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liang H, Zhao H, Gleichman A, Machnicki M, Telang S, Tang S, Rshtouni M, Ruddell J, Carmichael ST, Proc. Natl. Acad. Sci. USA 2019, 116, 13621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gerstenecker A, Lazar RM, Clin. Neuropsychol 2019, 33, 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Grefkes C, Fink GR, Neurol. Res. Pract 2020, 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jones TA, Adkins DL, Physiology 2015, 30, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Joy MT, Carmichael ST, Nat. Rev. Neurosci 2021, 22, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Scharfman HE, Curr. Neurol. Neurosci. Rep 2007, 7, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jarero-Basulto JJ, Gasca-Martinez Y, Rivera-Cervantes MC, Urena-Guerrero ME, Feria-Velasco AI, Beas-Zarate C, Pharmaceuticals 2018, 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yue ZW, Wang YL, Xiao B, Feng L, Neurochem. Res 2018, 43, 878. [DOI] [PubMed] [Google Scholar]
- [22].Scharfman HE, Neuroscientist 2002, 8, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cousin E, Baciu M, Pichat C, Kahane P, Le Bas JF, Neuropsychiatr. Dis. Treat 2008, 4, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rosenberger LR, Zeck J, Berl MM, Moore EN, Ritzl EK, Shamim S, Weinstein SL, Conry JA, Pearl PL, Sato S, Vezina LG, Theodore WH, Gaillard WD, Neurology 2009, 72, 1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hamberger MJ, Cole J, Neuropsychol. Rev 2011, 21, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chamoun RB, Mikati MA, Comair YG, Epilepsy Behav. 2007, 11, 384. [DOI] [PubMed] [Google Scholar]
- [27].Kong NW, Gibb WR, Tate MC, Neural. Plast 2016, 2016, 2365063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Monje M, Borniger JC, D’Silva NJ, Deneen B, Dirks PB, Fattahi F, Frenette PS, Garzia L, Gutmann DH, Hanahan D, Hervey-Jumper SL, Hondermarck H, Hurov JB, Kepecs A, Knox SM, Lloyd AC, Magnon C, Saloman JL, Segal RA, Sloan EK, Sun X, Taylor MD, Tracey KJ, Trotman LC, Tuveson DA, Wang TC, White RA, Winkler F, Cell 2020, 181, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ghinda CD, Duffau H, Front. Surg 2017, 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Piai V, De Witte E, Sierpowska J, Zheng X, Hinkley LB, Mizuiri D, Knight RT, Berger MS, Nagarajan SS, J. Cogn. Neurosci 2020, 32, 1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Duffau H, Acta Neurochir. 2012, 154, 575; discussion 84. [DOI] [PubMed] [Google Scholar]
- [32].Duffau H, Bauchet L, Lehericy S, Capelle L, Neuroreport 2001, 12, 2159. [DOI] [PubMed] [Google Scholar]
- [33].Sarubbo S, Le Bars E, Moritz-Gasser S, Duffau H, Neurosurg. Rev 2012, 35, 287; discussion 92. [DOI] [PubMed] [Google Scholar]
- [34].Meyer PT, Sturz L, Schreckenberger M, Spetzger U, Meyer GF, Setani KS, Sabri O, Buell U, Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 951. [DOI] [PubMed] [Google Scholar]
- [35].Thiel A, Herholz K, Koyuncu A, Ghaemi M, Kracht LW, Habedank B, Heiss W-D, Ann. Neurol 2001, 50, 620. [DOI] [PubMed] [Google Scholar]
- [36].Southwell DG, Hervey-Jumper SL, Perry DW, Berger MS, J. Neurosurg 2016, 124, 1460. [DOI] [PubMed] [Google Scholar]
- [37].Duffau H, Denvil D, Capelle L, J. Neurol. Neurosurg. Psychiatry 2002, 72, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gibb WR, Kong NW, Tate MC, Neural. Plast 2020, 2020, 8893708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Heiss WD, Thiel A, Kessler J, Herholz K, Neuroimage 2003, 20, S42. [DOI] [PubMed] [Google Scholar]
- [40].Aabedi AA, Lipkin B, Kaur J, Kakaizada S, Valdivia C, Reihl S, Young JS, Lee AT, Krishna S, Berger MS, Chang EF, Brang D, Hervey-Jumper SL, Proc. Natl. Acad. Sci. USA 2021, 118, e2108959118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rosenberg K, Liebling R, Avidan G, Perry D, Siman-Tov T, Andelman F, Ram Z, Fried I, Hendler T, Neurocase 2008, 14, 465. [DOI] [PubMed] [Google Scholar]
- [42].Ko SB, Yoon BW, Front. Neurol. Neurosci 2013, 32, 1. [DOI] [PubMed] [Google Scholar]
- [43].Saur D, Lange R, Baumgaertner A, Schraknepper V, Willmes K, Rijntjes M, Weiller C, Brain 2006, 129, 1371. [DOI] [PubMed] [Google Scholar]
- [44].Fisicaro RA, Jost E, Shaw K, Brennan NP, Peck KK, Holodny AI, Top Magn. Reson. Imaging 2016, 25, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Traut T, Sardesh N, Bulubas L, Findlay A, Honma SM, Mizuiri D, Berger MS, Hinkley LB, Nagarajan SS, Tarapore PE, Hum. Brain Mapp 2019, 40, 1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Vassal M, Charroud C, Deverdun J, Le Bars E, Molino F, Bonnetblanc F, Boyer A, Dutta A, Herbet G, Moritz-Gasser S, Bonafé A, Duffau H, De Champfleur NM, J. Neurosurg 2017, 126, 1181. [DOI] [PubMed] [Google Scholar]
- [47].Rostomily RC, Berger MS, Ojemann GA, Lettich E, J. Neurosurg 1991, 75, 62. [DOI] [PubMed] [Google Scholar]
- [48].Acioly MA, Cunha AM, Parise M, Rodrigues E, Tovar-Moll F, J. Neurol. Surg. A Cent. Eur. Neurosurg 2015, 76, 508. [DOI] [PubMed] [Google Scholar]
- [49].Krainik A, Duffau H, Capelle L, Cornu P, Boch AL, Mangin JF, Le Bihan D, Marsault C, Chiras J, Lehéricy S, Neurology 2004, 62, 1323. [DOI] [PubMed] [Google Scholar]
- [50].Sailor J, Meyerand ME, Moritz CH, Fine J, Nelson L, Badie B, Haughton VM, AJNR Am. J. Neuroradiol 2003, 24, 1837. [PMC free article] [PubMed] [Google Scholar]
- [51].Rosenberg K, Nossek E, Liebling R, Fried I, Shapira-Lichter I, Hendler T, Ram Z, J. Neurosurg 2010, 113, 1152. [DOI] [PubMed] [Google Scholar]
- [52].Nakajima R, Kinoshita M, Nakada M, Front. Hum. Neurosci 2020, 14, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Duffau H, J. Neurol. Neurosurg. Psychiatry 2001, 70, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bulubas L, Sabih J, Wohlschlaeger A, Sollmann N, Hauck T, Ille S, Ringel F, Meyer B, Krieg SM, J. Neurosurg 2016, 125, 1431. [DOI] [PubMed] [Google Scholar]
- [55].Bulubas L, Sardesh N, Traut T, Findlay A, Mizuiri D, Honma SM, Krieg SM, Berger MS, Nagarajan SS, Tarapore PE, Front. Hum. Neurosci 2020, 14, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liu Y, Rouiller EM, Exp. Brain Res 1999, 128, 149. [DOI] [PubMed] [Google Scholar]
- [57].Avramescu-Murphy M, Hattingen E, Forster MT, Oszvald A, Anti S, Frisch S, Russ MO, Jurcoane A, Clin. Neuroradiol 2017, 27, 299. [DOI] [PubMed] [Google Scholar]
- [58].Shinoura N, Suzuki Y, Yamada R, Kodama T, Takahashi M, Yagi K, AJNR Am. J. Neuroradiol 2006, 27, 1275. [PMC free article] [PubMed] [Google Scholar]
- [59].van Nieuwenhuizen D, Douw L, Klein M, Peerdeman SM, Heimans JJ, Reijneveld JC, Stam CJ, Hillebrand A, J. Neurooncol 2018, 140, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cargnelutti E, Ius T, Skrap M, Tomasino B, Neuroimage Clin. 2020, 28, 102435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Desmurget M, Bonnetblanc F, Duffau H, Brain 2007, 130, 898. [DOI] [PubMed] [Google Scholar]
- [62].Jacobs KM, Donoghue JP, Science 1991, 251, 944. [DOI] [PubMed] [Google Scholar]
- [63].Kong NW, Gibb WR, Badhe S, Liu BP, Tate MC, Neural. Plast 2020, 2020, 3648517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sharma N, Classen J, Cohen LG, Handb. Clin. Neurol 2013, 110, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Liu Y, Hu G, Yu Y, Jiang Z, Yang K, Hu X, Li Z, Liu D, Zou Y, Liu H, Chen J, Front. Oncol 2020, 10, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ohab JJ, Fleming S, Blesch A, Carmichael ST, J. Neurosci 2006, 26, 13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, Körber C, Kardorff M, Ratliff M, Xie R, Horstmann H, Messer M, Paik SP, Knabbe J, Sahm F, Kurz FT, Acikgöz AA, Herrmannsdörfer F, Agarwal A, Bergles DE, Chalmers A, Miletic H, Turcan S, Mawrin C, Hänggi D, Liu H-K, Wick W, Winkler F, Kuner T, Nature 2019, 573, 532. [DOI] [PubMed] [Google Scholar]
- [68].Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, Tam LT, Espenel C, Ponnuswami A, Ni L, Woo PJ, Taylor KR, Agarwal A, Regev A, Brang D, Vogel H, Hervey-Jumper S, Bergles DE, Suvà ML, Malenka RC, Monje M, Nature 2019, 573, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yu K, Lin CJ, Hatcher A, Lozzi B, Kong K, Huang-Hobbs E, Cheng Y-T, Beechar VB, Zhu W, Zhang Y, Chen F, Mills GB, Mohila CA, Creighton CJ, Noebels JL, Scott KL, Deneen B, Nature 2020, 578, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jin W, J. Clin. Med 2020, 9, 257.31963575 [Google Scholar]
- [71].Taylor KR, Barron T, Zhang H-C, Hui A, Hartmann GG, Ni L, Venkatesh HS, Du P, Mancusi R, Yalçin B, Chau I, Ponnuswami A, Aziz-Bose R, Monje M, bioRxiv 2021. [Google Scholar]
- [72].Gibson EM, Geraghty AC, Monje M, Dev. Neurobiol 2018, 78, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Knowles JK, Xu H, Soane C, Batra A, Saucedo T, Frost E, Tam LT, Fraga D, Ni L, Villar K, Talmi S, Huguenard JR, Monje M, Nat. Neurosci 2022, 25, 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob LS, Patwa R, Shah H, Xu K, Cross JR, Massagué J, Nature 2016, 533, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, Mccabe BD, Galvan JA, Robinson HPC, Zlobec I, Ciriello G, Hanahan D, Nature 2019, 573, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hahn A, Bode J, Kruwel T, Solecki G, Heiland S, Bendszus M, Tews B, Winkler F, Breckwoldt MO, Kurz FT, Sci. Rep 2019, 9, 11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Montgomery MK, Kim SH, Dovas A, Zhao HT, Goldberg AR, Xu W, Yagielski AJ, Cambareri MK, Patel KB, Mela A, Humala N, Thibodeaux DN, Shaik MA, Ma Y, Grinband J, Chow DS, Schevon C, Canoll P, Hillman EMC, Cell Rep. 2020, 31, 107500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Al-Kharboosh R, ReFaey K, Lara-Velazquez M, Grewal SS, Imitola J, Quinones-Hinojosa A, Mayo Clin. Proc. Innov. Qual Outcomes 2020, 4, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mitchell D, Shireman J, Sierra Potchanant EA, Lara-Velazquez M, Dey M, Front. Cell Neurosci 2021, 15, 716947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hatcher A, Yu K, Meyer J, Aiba I, Deneen B, Noebels JL, J. Clin. Invest 2020, 130, 2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Balentova S, Adamkov M, Int. J. Mol. Sci 2015, 16, 27796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Berg TJ, Pietras A, Semin. Cancer Biol 2022, 10.1016/j.semcancer.2022.02.011. [DOI] [PubMed] [Google Scholar]
- [83].Mitchell TJ, Seitzman BA, Ballard N, Petersen SE, Shimony JS, Leuthardt EC, Brain Connect. 2020, 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Tannock IF, Ahles TA, Ganz PA, Van Dam FS, J. Clin. Oncol 2004, 22, 2233. [DOI] [PubMed] [Google Scholar]
- [85].Zhang N, Xia M, Qiu T, Wang X, Lin CP, Guo Q, Lu J, Wu Q, Zhuang D, Yu Z, Gong F, Farrukh Hameed NU, He Y, Wu J, Zhou L, Hum. Brain Mapp 2018, 39, 4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yuan B, Zhang N, Yan J, Cheng J, Lu J, Wu J, Cortex 2020, 129, 141. [DOI] [PubMed] [Google Scholar]
- [87].Ghumman S, Fortin D, Noel-Lamy M, Cunnane SC, Whittingstall K, J. Neurooncol 2016, 128, 437. [DOI] [PubMed] [Google Scholar]
- [88].Deverdun J, van Dokkum LEH, Le Bars E, Herbet G, Mura T, D’Agata B, Picot M-C, Menjot N, Molino F, Duffau H, Moritz Gasser S, Brain Imaging Behav. 2020, 14, 1779. [DOI] [PubMed] [Google Scholar]
- [89].Smits A, Zetterling M, Lundin M, Melin B, Fahlstrom M, Grabowska A, Larsson E-M, Berntsson SG, Front. Neurol 2015, 6, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ortiz KJ, Hawayek MI, Middlebrooks EH, Sabsevitz DS, Garcia DP, Quinones-Hinojosa A, Chaichana KL, World Neurosurg. 2021, 146, 64. [DOI] [PubMed] [Google Scholar]
- [91].Herbet G, Maheu M, Costi E, Lafargue G, Duffau H, Brain 2016, 139, 829. [DOI] [PubMed] [Google Scholar]
- [92].Liu D, Chen J, Hu X, Hu G, Liu Y, Yang K, Xiao C, Zou Y, Liu H, J. Neurosurg 2020, 10, 1. [DOI] [PubMed] [Google Scholar]
- [93].Oszvald A, Guresir E, Setzer M, Vatter H, Senft C, Seifert V, Franz K, J. Neurosurg 2012, 116, 357. [DOI] [PubMed] [Google Scholar]
- [94].Barbagallo GMV, Altieri R, Garozzo M, Maione M, Di Gregorio S, Visocchi M, Peschillo S, Dolce P, Certo F, Front. Oncol 2020, 10, 631255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Molinaro AM, Hervey-Jumper S, Morshed RA, Young J, Han SJ, Chunduru P, Zhang Y, Phillips JJ, Shai A, Lafontaine M, Crane J, Chandra A, Flanigan P, Jahangiri A, Cioffi G, Ostrom Q, Anderson JE, Badve C, Barnholtz-Sloan J, Sloan AE, Erickson BJ, Decker PA, Kosel ML, Lachance D, Eckel-Passow J, Jenkins R, Villanueva-Meyer J, Rice T, Wrensch M, Wiencke JK, et al. , Jama Oncol. 2020, 6, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Renovanz M, Hickmann AK, Nadji-Ohl M, Keric N, Weimann E, Wirtz CR, Singer S, Ringel F, Coburger J, Support Care Cancer 2020, 28, 5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liu EK, Yu S, Sulman EP, Kurz SC, J. Neurooncol 2020, 149, 55. [DOI] [PubMed] [Google Scholar]
- [98].Ostrom QT, Cote DJ, Ascha M, Kruchko C, Barnholtz-Sloan JS, JAMA Oncol. 2018, 4, 1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chen P, Aldape K, Wiencke JK, Kelsey KT, Miike R, Davis RL, Liu J, Kesler-Diaz A, Takahashi M, Wrensch M, Cancer Res. 2001, 61, 3949. [PubMed] [Google Scholar]
- [100].Persaud-Sharma D, Burns J, Trangle J, Castro G, Barengo N, Moulik S, Lozano JM, Medicina 2019, 55, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Krishna S, Kakaizada S, Almeida N, Brang D, Hervey-Jumper S, Neurosurgery 2021, 89, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Pearson-Fuhrhop KM, Cramer SC, PM R 2010, 2, S227. [DOI] [PubMed] [Google Scholar]
- [103].Altshuler DB, Wang L, Zhao L, Miklja Z, Linzey J, Brezzell A, Kakaizada S, Krishna S, Orringer DA, Briceño EM, Gabel N, Hervey-Jumper SL, Neurooncol. Pract 2019, 6, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Correa DD, Satagopan J, Baser RE, Cheung K, Richards E, Lin M, Karimi S, Lyo J, Deangelis LM, Orlow I, Neurology 2014, 83, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]