

Summary:
Novel agents targeting immune checkpoint molecules or mutated BRAF are active therapeutic options for patients with BRAFV600-mutant melanoma. However, the most effective first-line treatment and the optimal sequencing of these agents have not been well characterized. To explore this, we retrospectively assessed 114 patients from 4 centers with advanced, BRAFV600-mutant melanoma who received anti-programmed cell death-1 (PD-1)/PD-L1 antibodies. We evaluated clinical outcomes, including objective response rate (ORR), overall survival (OS), and progression-free survival (PFS) to initial and subsequent therapies in patients that received anti-PD-1 first (n = 56) versus those that received BRAF ± MEK inhibitors (BRAFi) first (n = 58). Median OS was similar between these groups (27.5 vs. 40.3mo, P = 0.71). Patients who progressed on anti-PD-1 during the study timeframe had worse outcomes after starting subsequent BRAFi than those who had not received prior anti-PD-1 (median PFS 5 vs. 7.4mo, median OS 10.6 vs. 40.3 mo). Similarly, patients who previously progressed on BRAFi had seemingly inferior outcomes after starting anti-PD-1 compared with those without prior BRAFi, including ORR (25% vs. 41%), median PFS (2.8 vs. 10.6 mo) and median OS (8.2 vs. 27.6 mo). Notably, patients who benefited >6 months from BRAFi had superior ORR to subsequent anti-PD-1 compared with those with more rapid progression (<6 mo) on BRAFi (34% vs. 15%, P = 0.04). We conclude that either BRAFi or anti-PD-1 may be effective regardless of treatment sequence in patients with BRAFV600-mutant melanoma, but clinical outcomes to front-line therapy are superior. In addition, we suggest a shared “responder phenotype” between BRAFi and anti-PD-1.
Keywords: BRAF, melanoma, anti-PD-1, nivolumab, pembrolizumab, vemurafenib, dabrafenib, trametinib, sequence, immune
Advances in our understanding of the underlying molecular biology of advanced melanoma have led to the regulatory approval of 9 agents and three combination regimens for this disease since 2011. Immune therapies, including ipilimumab (anti-cytotoxic T-lymphocyte antigen-4) and anti-programmed cell death-1 (PD-1; nivolumab and pembrolizumab) antibodies produce long-lasting responses in a sizable number of patients.1–4 Small molecule inhibitors of mutant BRAF also induce dramatic responses for the 40%–50% of patients whose melanomas harbor the BRAFV600 mutation.5–7 Responses occur in the majority of patients but are limited in duration by the onset of acquired resistance. More recently, combinations of immune therapies (ipilimumab and nivolumab) and targeted therapies (BRAF/MEK inhibitor combinations) have been shown to be more effective than single-agent therapies.8,9
With these remarkable advances that have established 2 divergent treatment strategies, a critical unanswered question is the identification of the optimal front-line therapeutic regimen for patients with BRAFV600 mutations. Importantly, the currently available clinical data suggest that front-line anti-PD-1 and front-line BRAF/MEK inhibitor therapy are associated with approximately similar median overall survivals (OSs), (23–25 mo) when comparing across trials (with all the limitations that entails). Efforts to further dissect these data sets to identify pretreatment characteristics most likely associated with long-term benefit have been performed with the dabrafenib/trametinib phase III trials and have shown that OS approaches 70% for front-line therapy in patients with a normal lactate dehydrogenase (LDH), limited disease sites (≤3), and a preserved performance status [Eastern Cooperative Oncology Group (ECOG 0)].10 Unfortunately, no similar data exist with the anti-PD1 antibodies, nor are there any clinically validated blood or tissue based biomarkers to help with patient stratification and treatment selection between these 2 approaches.
Several studies have suggested that patients who progress on single-agent BRAF inhibitors rarely respond to subsequent ipilimumab.11,12 Conversely, patients who fail ipilimumab or other immune therapies may still derive significant benefits from subsequent BRAF inhibition. Whether these data are more generalizable to all sequencing of BRAF-targeted therapy and immunotherapy is unclear, especially given the changes in the standard of care therapy since these retrospective studies were performed. In particular, anti-PD-1 therapy (either alone or in combination with ipilimumab) has supplanted ipilimumab monotherapy as the preferred front-line immune therapy strategy.8,13,14 Similarly, BRAF + MEK inhibitor combination therapy (dabrafenib + trametinib) has proven superior to single-agent BRAF inhibition.15–17 As these novel treatment regimens have demonstrated greater response rates and superior survival compared with their predecessors, it is possible that there will be different results when evaluating the outcomes of various sequences of these therapies. Several recent published and unpublished reports have suggested that anti-PD-1 may be less effective after failure of BRAF-inhibitor therapy than when given before BRAF-inhibitor therapy, though these agents are clearly associated with responses in a substantial minority of patients after BRAF-inhibitor therapy.18–20
To date, however, there is little insight into the most effective treatment sequence for patients with BRAFV600-mutant melanoma. Although randomized clinical trial data are needed to address this question, we sought to particularly address how patients harboring these mutations responded to anti-PD-1 before or following BRAF-directed therapy and to describe the outcomes of BRAF inhibitors after anti-PD-1. To address these questions, we retrospectively assessed patients from 4 large melanoma centers. We investigated the outcomes of 2 cohorts: those who received anti-PD-1 first, and those who received anti-PD-1 after progression of BRAF ± MEK inhibitors.
METHODS
Patients
After IRB approval of all relevant protocols, we retrospectively identified a cohort of 114 patients from 4 large cancer centers: Vanderbilt University Medical Center, Beth Israel Deaconess Medical Center, Dana-Farber Cancer Institute, and Massachusetts General Hospital. All patients were found to have a BRAFV600 mutation by Clinical Laboratory Improvement Amendments certified laboratory assays that varied by institution. All patients received anti-PD-1 or anti-PD-L1, which included nivolumab, pembrolizumab, and atezolizumab (MPDL3280A). Several patients who received ipilimumab + nivolumab were included. Many patients received BRAF-directed therapy, which included vemurafenib, dabrafenib, or encorafenib, either alone or in combination with a MEK inhibitor (cobimetinib, trametinib, or binimetinib). Patients who received other lines of systemic or local therapy either before, in between, or following these treatments were included. Patients received these therapies either as standard of care or as part of clinical trials.
Study Design
We divided patients into 2 cohorts; those who had received anti-PD-1 (± ipilimumab) before BRAF ± MEK inhibition (“anti-PD-1 first”) or those who received BRAF ± MEK inhibition before anti-PD-1 (“BRAFi first”). We compared OS for each cohort, and compared objective response rate (ORR) and progression-free survival (PFS) of each individual therapy. Specifically, we assessed the ORR and PFS of BRAFi before and after anti-PD-1, and assessed these parameters of anti-PD-1 before and after BRAFi. We also assessed whether outcomes were superior following progression to BRAFi monotherapy or after BRAF + MEK inhibition. We did not attempt to assess patients who only received BRAF + MEK inhibitors, given the frequent poor performance status at treatment start, and the unavailability of anti-PD-1 as standard therapy during part of the study timeframe. Finally, we correlated whether duration of benefit on the first-line therapy correlated with benefit from subsequent therapy. ORR was based on Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 criteria and was investigator assessed.21 Patients with unevaluable responses were considered non-responders.
Statistical Analysis
OS was calculated as start of first therapy to time of death for any reason and compared between cohorts using the logrank test. PFS was calculated as time of therapy start to progression by RECIST 1.1 criteria and was compared using the logrank test. ORR and baseline patient characteristics were compared using χ2 testing. Cox proportional hazards models to assess predictors of OS were developed; variables included age, sex, stage, prior therapies (yes or no), and serum LDH levels. All patients were censored for OS and/or PFS at last follow-up. P-values were considered statistically significant at <0.05.
RESULTS
Patient Demographics
A total of 114 patients were included; 56 in the anti-PD-1-first cohort and 58 in the BRAFi-first cohort. The PD-1 inhibitor received was pembrolizumab in 61%, nivolumab in 16%, atezolizumab (anti-PD-L1) in 5%, and ipilimumab + nivolumab in 18%. BRAF inhibitors alone were given in 47% and BRAFi + MEKi in 53%. We compared baseline demographics and treatment characteristics between cohorts. Patients in the BRAFi-first cohort tended to have more adverse prognostic features, including elevated LDH (40% vs. 19%, P = 0.054), brain metastases (24% vs. 9%, P = 0.054), and ECOG performance status 1–2 (30% vs. 18%, P = 0.182), with similar average age and sex distribution (Table 1). The number of prior therapies and interval therapies was similar between arms. Many patients in the anti-PD-1 first cohort had not required BRAFi at last follow-up due to continued response to anti-PD-1 (n = 34).
TABLE 1.
Clinical Characteristics of Anti-PD-1 First and BRAFi- First Cohorts
| Variables | n (%) |
|||
|---|---|---|---|---|
| Anti-PD-1 First (N = 56) |
BRAFi First (N = 58) |
P | ||
|
| ||||
| Age (median) (y) | 56.5 | 50 | 0.15 | |
| Sex | ||||
| Male | 37 (66) | 35 (60) | 0.66 | |
| Female | 19 (34) | 23 (40) | ||
| ECOG performance status | ||||
| 0 | 46 (82) | 40 (70) | 0.18 | |
| 1–2 | 10 (18) | 17 (30) | ||
| Brain metastases | ||||
| Yes | 5(9) | 14 (24) | 0.05 | |
| No | 51 (91) | 44 (76) | ||
| Lactate dehydrogenase | ||||
| Normal | 40 (74) | 27 (54) | 0.05 | |
| > ULN | 10 (19) | 20 (40) | ||
| >2xULN | 4(7) | 3(6) | ||
| Anti-PD-1 agent | ||||
| Nivolumab | 10 (18) | 8(14) | < 0.001 | |
| Pembrolizumab | 24 (43) | 45 (78) | ||
| Atezolizumab | 3(5) | 3 (5) | ||
| Ipilimumab + nivolumab | 19 (34) | 2 (3) | ||
| BRAF inhibitor | ||||
| BRAFi monotherapy | 13 (23) | 26 (45) | * | |
| BRAFi + MEKi | 9(16) | 32 (55) | ||
| None | 34 (61) | 0 | ||
| Prior therapy | ||||
| Prior ipilimumab | 12 (21) | 16 (28) | 0.86 | |
| Prior IL-2 | 12 (21) | 12 (21) | ||
| Prior chemotherapy | 3(5) | 4 (7) | ||
Baseline lactate dehydrogenase and ECOG performance status not documented in 10 and 1 patients, respectively.
Denotes no formal statistical testing performed.
ECOG indicates Eastern Cooperative Oncology Group; IL-2, interleukin-2; ULN, upper limit of normal.
Treatment Outcomes
We then compared the OS for patients in each cohort. Patients in the anti-PD-1 first and BRAFi first cohorts had similar OS (median 27.5 vs. 40.3 mo, logrank P = 0.71) and PFS to the front-line treatment regimen (10.6 vs. 7.4, P = 0.1; Fig. 1A). Unsurprisingly, patients who did not progress on anti-PD-1 during the study timeframe had excellent survival (median survival not reached; Fig. 1B). By contrast, patients who failed anti-PD-1 and subsequently received BRAFi ± MEKi had poor outcomes overall, with a median survival of only 14.5 months, compared with 40.3 months for those who received BRAFi ± MEKi followed by anti-PD-1.
FIGURE 1.
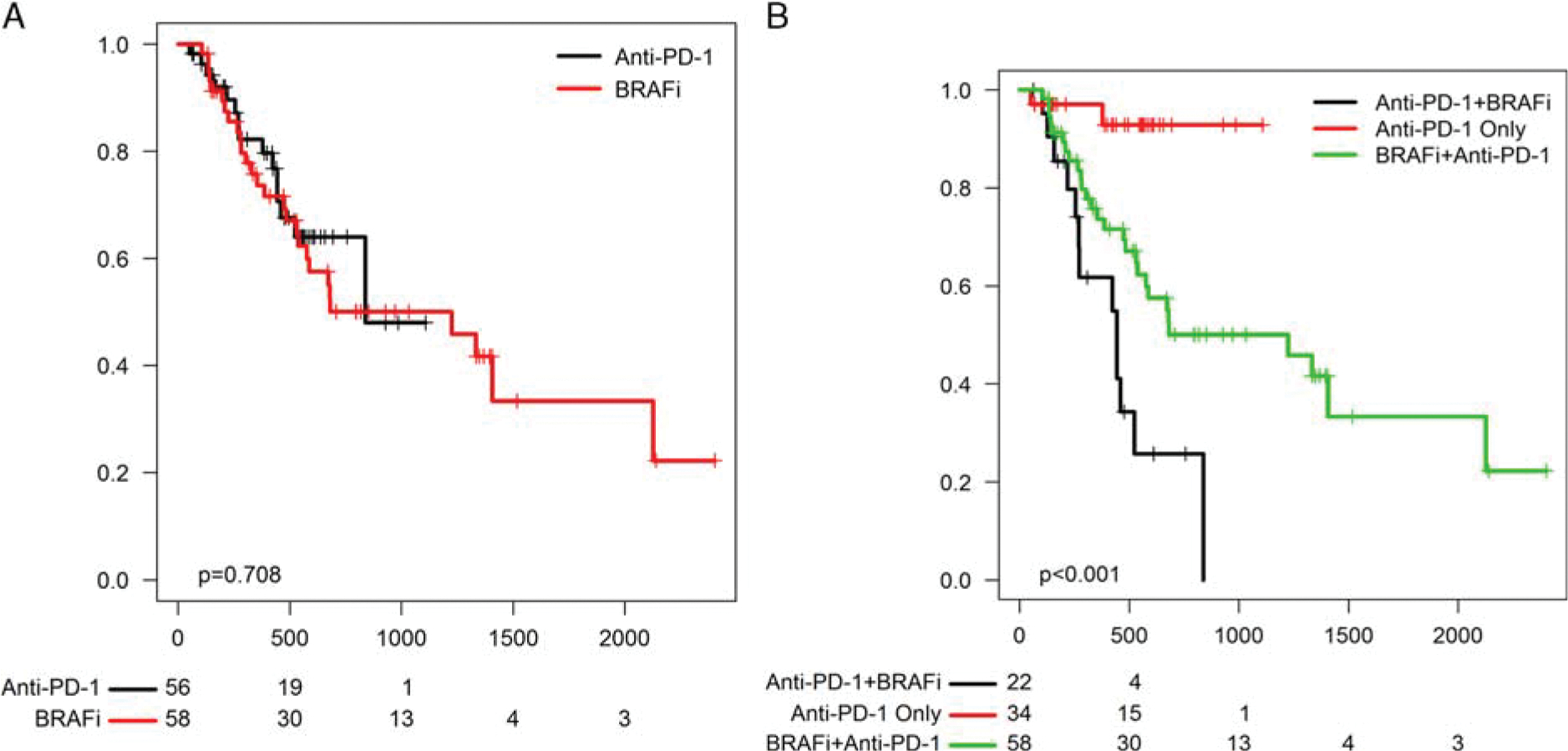
A, Overall survival of anti-programmed cell death-1 (PD-1) first versus BRAFi-first group. B, Survival of patients who received anti-PD-1 only, those who received BRAFi followed by anti-PD-1, and those who received anti-PD-1 followed by BRAFi.
We then assessed clinical outcomes to anti-PD-1 in BRAFi-naive versus BRAFi-pretreated patients. Since by definition these comparisons assessed patients who were more heavily pretreated to those less heavily pretreated, we reported these results descriptively, without P-values. ORR to anti-PD-1 appeared to be slightly higher in the BRAFi naive (anti-PD-1 first) group (41% vs. 25%). ORR to anti-PD-1 was similar in the BRAFi pretreated group, regardless of whether patients had previously received BRAFi alone or BRAFi + MEKi (29% vs. 23%). We then assessed PFS and OS to each individual therapy. PFS to anti-PD-1 appeared superior in the anti-PD-1 first group (10.6 vs. 2.8 mo) (Fig. 2A). OS from the time of anti-PD-1 initiation also seemed superior in the anti-PD-1 first group (median, 27.6 vs. 8.2 mo; Fig. 2B). Similarly, patients treated with BRAF ± MEK inhibitors initially had superior PFS (median, 7.4 vs. 5.0 mo) and OS (median, 40.3 vs. 10.6 mo) after BRAFi initiation compared with those who received them after anti-PD-1 failure (Figs. 2C, D).
FIGURE 2.
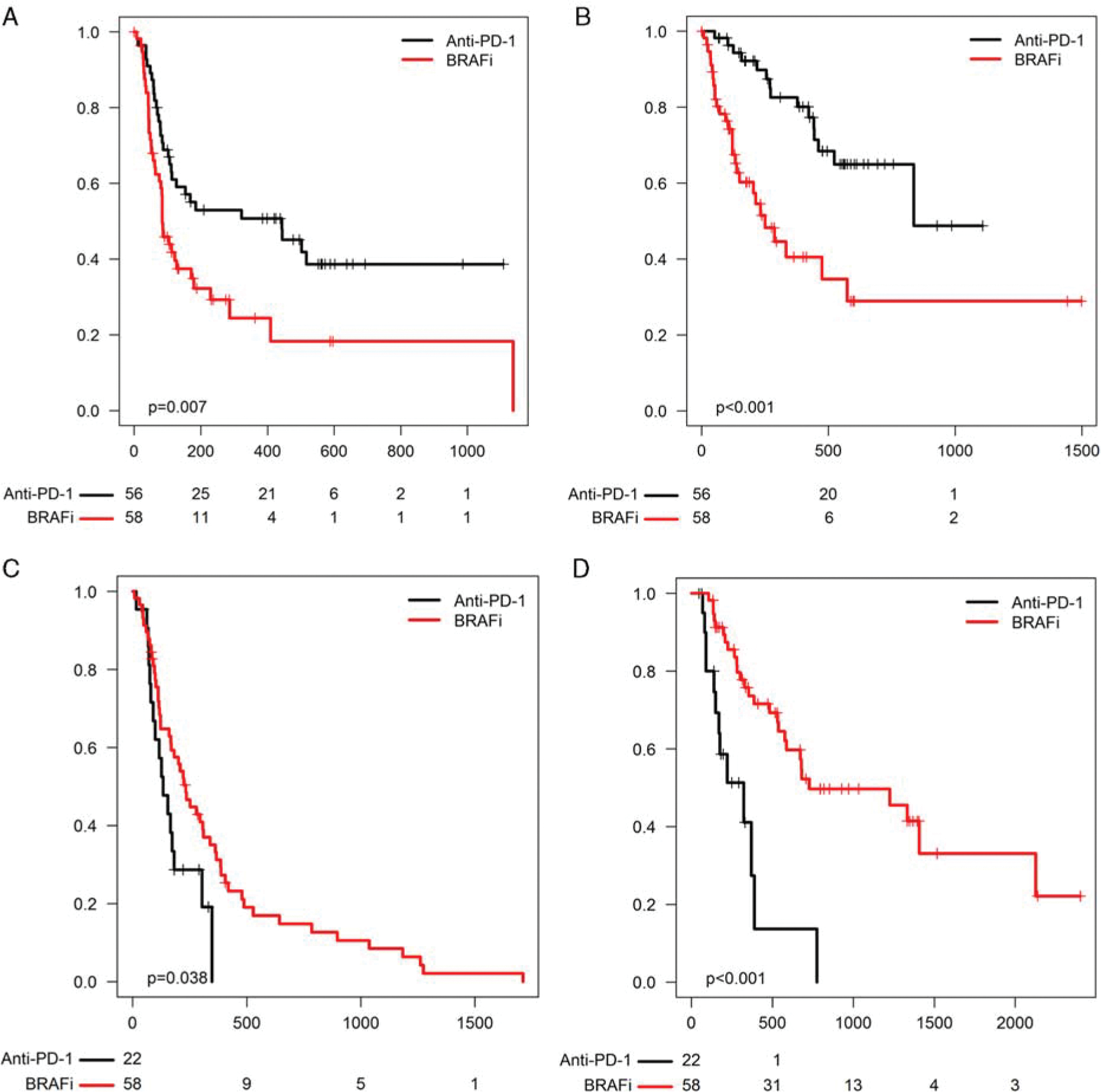
(A) PFS and (B) OS from the start of anti-programmed cell death-1 (PD-1) in the anti-PD-1 first group (labeled “anti-PD-1,” black) versus the BRAFi-first group (labeled “BRAFi,” red). (C) PFS and (D) OS from the start of BRAFi in the anti-PD-1 first group versus the BRAFi-first group.
We then assessed whether a shared “responder” phenotype existed. We found that patients who benefited from Anti-PD-1 BRAF-directed therapy for ≥6 months had a 34% ORR to subsequent anti-PD-1 (11 of 32). By contrast, patients who benefited for <6 months subsequently had a 15% ORR to anti-PD-1 (4 of 26; P = 0.04). We did not perform this analysis in the anti-PD-1 cohort since nearly all patients who responded to anti-PD-1 continued to respond during the study timeframe and had not yet required salvage BRAFi therapy.
In view of the widely divergent outcomes within the anti-PD-1 first and the BRAFi first cohorts, we hypothesized that particular clinical characteristics may help identify whether patients would have better outcomes with initial anti-PD-1 versus initial BRAFi. We performed logistic regression to assess whether distinct clinical features correlate with OS in the anti-PD-1 first versus the BRAFi first group. In this study, LDH, stage, prior therapies did not correlate with OS to either therapy in a statistically significant fashion.
DISCUSSION
The proliferation of effective therapies for advanced melanoma has greatly improved the outcomes of patients, but poses challenges in selecting front-line treatment in the absence of clinical data to guide these decisions. In this study, we showed that patients with BRAFV600-mutated melanoma can benefit from anti-PD-1 either before receiving, or after progression on BRAF±MEK inhibitors. Outcomes to anti-PD-1 appeared superior when patients received therapy in the untreated setting, similar to previous published and unpublished data that has consistently reflected this finding.18–20 It is interesting to note that the converse was true as well for patients who received BRAF ± MEK inhibitors. Median PFS and OS to BRAF inhibition in the salvage setting were quite poor (5 and 10.6 mo, respectively) compared with those who received these treatments in the anti-PD-1 naive setting (7.6 and 40.3 mo). This finding seems to contradict the general feeling that BRAF ± MEK inhibitors have equivalent efficacy following immune therapy failure as in the first line. To our knowledge, this is the first study describing the activity of BRAF ± MEK inhibitors following anti-PD-1. We also observed a group of patients that appeared to benefit more from both classes of therapies. Specifically, patients who benefited from BRAF inhibition for >6 months had dramatically higher response rates to subsequent anti-PD-1 (34% vs. 15%).
These findings corroborate several recent molecular studies. Notably, tumors with innate resistance to anti-PD-1 therapy share many common features to the BRAFi-resistant state, including T-cell depletion, upregulated wound healing signatures, and exuberant angiogenesis (Innate anti-PD-1 resistance gene signatures; innate anti-PD-1 resistance).22,23
Conversely, these studies also suggest that BRAFi resistance induces an immune suppressed tumor microenvironment, potentially contributing to a lack of response to subsequent anti-PD-1. Dissecting molecular features of these resistant states may inform strategies to combat both immune and targeted therapy resistance. A number of molecular studies are ongoing to unravel these features and identify markers of response to immune and targeted therapies, including large-scale next-generation sequencing studies to evaluate total mutational load and RNA-expression signatures.
Ultimately, the clinical decision between BRAF-targeted and immune therapy for patients with advanced, BRAF mutated melanoma will require a higher level of evidence, generated through a randomized clinical trial, and ideally clinically validated biomarkers that identify who is most likely to benefit from one therapy but not the other. In pursuit of higher level evidence, an intergroup (ECOG-American College of Radiology Imaging Network; ECOG ACRIN)-sponsored trial, EA6134 is ongoing, comparing ipilimumab + nivolumab with dabrafenib + trametinib with crossover to the opposite group at time of progression. Pending these results, the clinical decision is currently made based on physician preference, and most experts favor a trial of immune therapy with a goal of achieving a durable response. Our data suggest that BRAFi as a salvage strategy may not be highly active in the subgroup that fails anti-PD-1 and front-line BRAF/MEK inhibitor therapy should be considered. Although these results are unlikely to strongly influence therapy selection at this time, they do address a key prognostic question for clinicians and patients.
This study has several limitations. Patients were treated with various anti-PD-1 agents and BRAF-directed therapies at large academic institutions. Follow-up was relatively short in many cases, limiting long-term survival conclusions. Some imbalances were present between the BRAFi and anti-PD-1 first cohorts (eg, more negative prognostic markers in the BRAFi group) reflecting clinical practice (patients with more aggressive disease may receive BRAFi more commonly). Also, more patients in the anti-PD-1 first group received ipilimumab + nivolumab combination therapy. Otherwise, the patient population, treatment options, and clinical decisions mirror standard practice.
In conclusion, either BRAF-targeted or anti-PD-1-directed therapy can result in clinical activity for patients with BRAFV600-mutated melanoma, either in first-line, or in previously treated patients. A subgroup of patients appears to have aggressive disease and suboptimal responses to both therapies. Understanding drivers of resistance and optimal sequences of therapy remain critical objectives.
Acknowledgments
Supported by K12 CA 0906525 (D.B.J.).
Footnotes
CONFLICTS OF INTEREST/FINANCIAL DISCLOSURES
D.B.J. serves on advisory boards for Genoptix and BMS. The other authors declare no conflicts of interest.
REFERENCES
- 1.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (Anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. [DOI] [PubMed] [Google Scholar]
- 4.Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. [DOI] [PubMed] [Google Scholar]
- 8.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–451. [DOI] [PubMed] [Google Scholar]
- 10.Long GV, Weber JS, Infante JR, et al. Overall survival and durable responses in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib combined with trametinib. J Clin Oncol. 2016;34:871–878. [DOI] [PubMed] [Google Scholar]
- 11.Ackerman A, McDermott DF, Lawrence DP, et al. Outcomes of patients with malignant melanoma treated with immunotherapy prior to or after vemurafenib. J Clin Oncol. 2012;30:8569. [Google Scholar]
- 12.Ascierto PA, Simeone E, Giannarelli D, et al. Sequencing of BRAF inhibitors and ipilimumab in patients with metastatic melanoma: a possible algorithm for clinical use. J Transl Med. 2012;10:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532. [DOI] [PubMed] [Google Scholar]
- 14.Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK Inhibition versus BRAF inhibition alone in melanoma. Lancet. 2015;386:444–451. [DOI] [PubMed] [Google Scholar]
- 16.Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. [DOI] [PubMed] [Google Scholar]
- 17.Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–1876. [DOI] [PubMed] [Google Scholar]
- 18.Larkin J, Lao CD, Urba WJ, et al. Efficacy and safety of nivolumab in patients with BRAF V600 mutant and braf wild-type advanced melanoma: a pooled analysis of 4 clinical trials. JAMA Oncol. 2015;1:433–440. [DOI] [PubMed] [Google Scholar]
- 19.Ramanujam S, Lo S, Kong B, et al. Anti-PD1 responses in BRAF mutated advanced melanoma patients with prior BRAF inhibitor (BRAFi) or combined BRAF and MEK inhibitor (Combi) therapy. Pigment Cell Melanoma Res. 2015. [Abstract]. [Google Scholar]
- 20.Puzanov I, Ribas A, Daud A, et al. Pembrolizumab for advanced melanoma: effect of BRAFV600 mutation status and prior BRAF inhibitor therapy. Pigment Cell Melanoma Res. 2015. [Abstract]. [Google Scholar]
- 21.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 22.Hugo W, Zaretsky JM, Sun L, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hugo W, Shi H, Sun L, et al. Non-genomic and immune-evolution of melanoma acquiring MAPKi resistance. Cell. 2015;162:1271–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]