

Abstract
Posttraumatic osteoarthritis (PTOA) results in joint pain, loss of joint function, and impaired quality of daily life in patients with limited treatment options. We previously demonstrated that epidermal growth factor receptor (EGFR) signaling is essential for maintaining chondro-progenitors during articular cartilage development and homeostasis. Here, we used a nonsurgical, loading-induced PTOA mouse model to investigate the protective action of EGFR signaling. A single bout of cyclic tibial loading at a peak force of 6 N injured cartilage at the posterior aspect of lateral femoral condyle. Similar loading at a peak force of 9 N ruptured the anterior cruciate ligament, causing additional cartilage damage at the medial compartment and ectopic cartilage formation in meniscus and synovium. Constitutively overexpression of an EGFR ligand, heparin binding EGF-like growth factor (HBEGF), in chondrocytes significantly reduced cartilage injury length, synovitis, and pain after 6 N loading and mitigated medial side cartilage damage and ectopic cartilage formation after 9 N loading. Mechanistically, overactivation of EGFR signaling protected chondrocytes from loading-induced apoptosis and loss of proliferative ability and lubricant synthesis. Overexpressing HBEGF in adult cartilage starting right before 6 N loading had similar beneficial effects. In contrast, inactivating EGFR in adult cartilage led to accelerated PTOA progression with elevated cartilage Mankin score and synovitis score and increased ectopic cartilage formation. As a therapeutic approach, we constructed a nanoparticle conjugated with the EGFR ligand TGFα. Intra-articular injections of this nanoconstruct once every 3 weeks for 12 weeks partially mitigated PTOA symptoms in cartilage and synovium after 6 N loading. Our findings demonstrate the anabolic actions of EGFR signaling in maintaining articular cartilage during PTOA development and shed light on developing a novel nanomedicine for PTOA.
Keywords: EPIDERMAL GROWTH FACTOR RECEPTOR, MECHANICAL LOADING, NANOPARTICLE, PHOSPHOLIPID MICELLAR, POST-TRAUMATIC OSTEOARTHRITIS
Introduction
Osteoarthritis (OA) is the most common cause of disability and poses the greatest public health threat for over 27 million adults in the United States. Among all OA risk factors, joint trauma is an important one that increases the likelihood of developing OA by 3.86 times.(1) Etiology studies find that approximately 12% of symptomatic OA can be attributed to posttraumatic OA (PTOA) of the hip, knee, or ankle.(2) Specifically, joint injuries in young adults eventually lead to radiographic OA in half of patients within 12–14 years.(3) Most pharmacologic treatments for PTOA are only palliative with adverse side effects. No disease-modifying drugs are available for PTOA. Therefore, there is an unmet demand in understanding the mechanism of PTOA and developing novel therapies to slow down or reverse PTOA progression.
The epidermal growth factor receptor (EGFR) signaling pathway is very important in mammalian cells. Upon ligand binding, EGFR, a 170-kDa kinase receptor, activates a cascade of downstream signaling pathways, including MEK/MAPK, PI3K/Akt, PLCγ, and others, to modulate a variety of cellular functions, such as proliferation, survival, adhesion, migration, and differentiation.(4) EGFR is expressed in most human tissues, including cartilage. Among its ligands, transforming growth factor α (TGFα) and heparin binding EGF-like growth factor (HBEGF) are the two most expressed ones in articular cartilage.(5) Gene profiling found that both of them are regulated in OA cartilage.(6,7) Other studies also linked the TGFA gene locus to human OA,(8–10) although the functional relevance of these findings remains unclear.
Although EGFR is expressed ubiquitously by articular cartilage chondrocytes, EGFR activity, indicated by phosphorylated EGFR (p-EGFR) and phosphorylated ERK (p-ERK), is mainly located at the superficial layer of human and mouse articular cartilage.(11) This activity is drastically reduced during aging and OA initiation, indicating an essential role in OA progression. Previous cell culture studies showed that EGFR had both anabolic and catabolic actions.(12) While promoting proliferation and survival, addition of EGFR ligands to chondrocytes in culture suppresses their expression of cartilage matrix genes, such as Col2a1 and Aggrecan, and stimulates their expression of cartilage degradation proteases, such as Mmp13 and Adamts5. However, knocking down EGFR activity in vivo using a constitutive chondrocyte-specific Cre driver (Col2a1-Cre) greatly reduces the number of superficial chondrocytes in the articular cartilage in adolescent mice and leads to spontaneous knee OA in adult mice.(5) Moreover, these conditional knockout mice are much more susceptible to the destabilization of the medial meniscus (DMM) surgery than wild type (WT) mice.(5) A subsequent study using inducible chondrocyte-specific Aggrecan-CreER demonstrated that EGFR activity is also required for the normal structure and function of articular cartilage and preventing OA initiation after DMM surgery.(11)
In contrast, EGFR overactivation in mice by overexpressing EGFR ligand, HBEGF, in chondrocytes using Col2a1-Cre leads to articular cartilage expansion in young mice and resistance to DMM-induced OA progression in adult mice.(13) Interestingly, HBEGF overexpression stimulates the proliferation and survival of chondrocytes at the top layer of articular cartilage but does not affect the production of cartilage matrix proteins and the synthesis of cartilage degradation proteases.(13) Consistent with these data, depletion of a negative feedback inhibitor of EGFR, mitogen-inducible gene 6 (Mig6),(14) in chondrocytes results in similar articular cartilage thickening in young mice.(15–17) These reports provide strong evidence that EGFR is a potential target for OA therapy.
One limitation of the aforementioned studies is that only one model, DMM, was tested to examine the action of EGFR signaling in PTOA development. Although DMM is commonly used for OA studies, it is an invasive surgical procedure with confounding factors not related to clinically relevant joint degeneration. Compared to DMM, a noninvasive loading model is more accurate in mimicking mechanically induced human PTOA.(18) Therefore, in this study, we subjected the aforementioned EGFR-deficient and overactivating mouse models to a previously established tibial loading protocol(19) and examined whether EGFR signaling protects articular cartilage from one bout of cyclic loading. As a possible therapeutic approach, we further tested whether a newly designed nanoparticle (NP) conjugated with an EGFR ligand, TGFα, attenuates PTOA development in WT mice after loading injury.
Material and Methods
Animals
Animal protocols were approved by the Institutional Animal Care and Use Committees of the University of Pennsylvania. These experiments were performed in the animal facilities of our institution, which implement strict regimens for animal care and use.
To establish a cartilage-specific HBEGF overexpression mouse model, we crossed Col2a1-Cre (Jackson Laboratory, Stock No. 003554)(20) and Aggrecan-CreER (Jackson Laboratory, Stock No. 019148)(21) mice with Rosa-DTR (diphtheria toxin receptor, Jackson Laboratory, Stock No. 007900)(22) mice to generate constitutive (Col2a1-Cre DTR) and inducible (Aggrecan-CreER DTR) DTR overexpression mice, respectively. Since DTR is human full-length HBEGF,(23) these mice are equivalent to HBEGF overexpression (HBEGF OverCol2 and HBEGF OverAcanER). DTR- or Cre-only siblings were used as WT controls. To generate cartilage-specific EGFR-deficient mice, we first crossed Aggrecan-CreER mice with EgfrWa5/+(24) to generate Aggrecan-CreER EgfrWa5/+ mice, which was then crossed with Egfrflox/flox(25) mice to produce Aggrecan-CreER EgfrWa5/flox (Egfr iCKOAcanER) mice and their WT (Aggrecan-CreER Egfrflox/+ and Egfrflox/+) siblings. EgfrWa5/+ and Egfrflox/flox mouse lines were originally acquired from Dr. David Threadgill at the University of North Carolina at Chapel Hill. To induce Cre activity, HBEGF OverAcanER mice, Egfr iCKOAcanER mice, and their respective WT control mice were intraperitoneally injected with 75 mg/kg/day tamoxifen for 5 days prior to loading injury.
Loading procedure
To induce PTOA, 2-month-old male mice were subjected to mechanical loading using a Bose ElectroForce TestBench system (TA Instruments) at the right knees, as previously described.(19) Briefly, under anesthesia, the right tibia was positioned horizontally with the knee in deep flexion between the left loading cup (attached to the actuator) and the right fixed cup (linked to the load cell) (Fig. 1A) and subjected to axial compressive loading at a peak force of 6 or 9 Newtons (N). A 0.5-N preload force was applied to maintain the limb in position between loading cycles. Cyclic loads were applied for 0.34 s with a rise and fall time of 0.17 s each and a baseline hold time of 10 s between cycles for 60 cycles (Fig. 1B). The uninjured left knees were used as controls. We used six mice per time point per genotype for genetically modified mice and five mice per treatment group for NP treatment experiment.
Fig. 1.
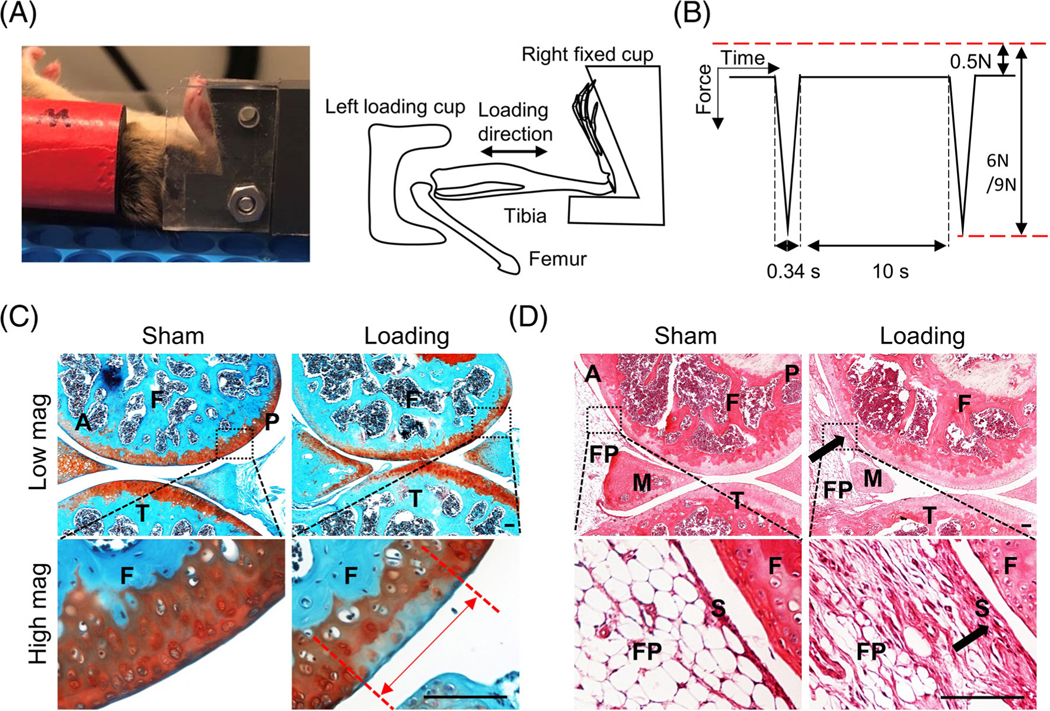
A loading-induced posttraumatic osteoarthritis (PTOA) mouse model. (A) Photograph (left panel) and cartoon (right panel) of a mouse low hindlimb during loading. (B) Waveform of applied hold and peak load magnitudes and rate and time interval of loading. (C) Representative safranin-O/Fast green-stained images of Sham or 6 N-loaded WT mouse joints at 1 month after loading. The red double-headed arrow indicates the cartilage injury length between two red dashed lines. Scale bars, 100 μm. (D) Representative hematoxylin and eosin (H&E)–stained images of Sham or 6 N-loaded mouse joints at 1 month after loading. Arrows point to thickened synovium area for quantification. Scale bars, 100 μm. A, anterior; F, femur; FP, fat pad; M, meniscus; P, posterior; S, synovium; T, tibia. In (C, D), bottom high-magnification images are from the outlined areas in the top low-magnification images.
Histology
After euthanasia, mouse knee joints were harvested and fixed in 4% paraformaldehyde overnight followed by decalcification in either 10% ethylenediaminetetraacetic acid (EDTA, AMRESCO) for 21 days or Formical-2000 decalcifier (StatLab) for 3 days prior to paraffin embedding. A series of 6-μm-thick sagittal sections (about 100) were cut across the entire lateral compartment of the joint. Two sections within every consecutive six sections in the entire section set were stained with safranin-O /Fast green. The section with the longest cartilage lesion was used to quantify cartilage injury length and Mankin score. The cartilage lesion site is normally located at the posterior aspect of lateral femoral condyle (Fig. 1C). Injury length is defined as the longitudinal length of negative safranin-O staining, indicating proteoglycan loss, at the articular cartilage surface.(19) Mankin score is previously defined as a combined score assessing cartilage structure (0–5 points), chondrocyte abnormalities (0–3 points), safranin-O staining (0–5 points), and tidemark integrity (0–1 point).(26) Scoring was performed independently by two blinded examiners.
To calculate the synovial inflammation score, another two sections within every consecutive six sections in the entire section set were stained with hematoxylin and eosin (H&E) and subjected to synovitis scoring based on the following morphological parameters as previously described(27): (i) hyperplasia/enlargement of synovial lining cell layer; (ii) activation of resident cells/synovial stroma. (iii) inflammatory infiltration. All defined histopathological qualities are graded from absent (0), slight (1) and moderate (2) to strong (3), with summaries ranging from 0 to 9.0. Although synovitis occurs throughout the joint, we selected the same anterior location among all mice for quantification (Fig. 1D). Each knee received a single score representing the maximal score of all sections. Scoring was performed independently by two blinded examiners.
To examine anterior cruciate ligament (ACL) rupture-induced cartilage damage, we cut the entire medial compartment of the joints from WT and HBEGF OverCol2 mice at 3 months after loading to determine the Mankin scores. Scoring was performed independently by two blinded examiners.
Immunohistochemistry
Paraffin sections were processed for antigen retrieval, followed by incubation with primary antibodies against p-EGFR (1:100, Abcam, ab40815), Ki67 (1:100, Abcam, ab15580), PRG4 (1:100, Abcam, ab28484), MMP13 (1:100, Abcam, ab219620), or ADAMTS5 (1:100, Abcam, ab41037) at 4°C overnight. After biotinylated secondary antibody (Vector Laboratories, PK-4001) incubation and 3,3′-Diaminobenzidine (DAB) color development (Thermo Fisher Scientific, TA-060-QHDX), images were captured under a light microscope (Eclipse 90i, Nikon).
A terminal deoxynucleotidyl transferase deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) assay was carried out according to the manufacturer’s instructions (ApopTag Fluorescein In Situ Apoptosis Detection Kit, Millipore Sigma, S7110). The number of positive cells was counted in the uncalcified articular cartilage by two blinded observers using ImageJ.
OA pain analysis
Pain after loading injury was evaluated using von Frey filaments as described previously.(13,28) Individual mice were placed on a wire-mesh platform (Excellent Technology Co.) under a 4 × 3 × 7-cm cage to restrict their movements. The mice were trained to become accustomed to this condition every day starting from 7 days before the test. During the test, a set of von Frey fibers (Stoelting Touch Test Sensory Evaluator Kit 2–9, ranging from 0.015 g to 1.3 g force) were applied to the plantar surface of the hind paw until the fibers bowed, and then held for 3 s. The threshold force required to elicit withdrawal of the paw (median 50% withdrawal) was determined five times on each hind paw with sequential measurements separated by at least 5 min.
Chondrocyte culture and immunoblotting
Chondrocytes were harvested from 3-day-old mouse knee joints. Briefly, the distal femoral and proximal tibial cartilage tissues were carefully collected under the microscope and digested with 0.25% trypsin–EDTA (30 min, Invitrogen) and 600 U/mL type I collagenase (2 h, Worthington Biochemical), and cells were maintained in DMEM medium supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin.
To perform Western blot, cell lysate was solubilized in radioimmunoprecipitation assay buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 1% sodium deoxycholate, 1% Triton-X 100, and 0.1% SDS) with protease inhibitor (P8340–1ML, Sigma-Aldrich). Cell lysate (50 mg) was separated by SDS-PAGE and transferred onto polyvinylidene fluoride membrane. Immunoreactive protein bands were visualized using rabbit anti-ERK (1:1000; CST, 4695), anti-p-ERK (1:1000; CST, 4370), anti-β-actin (1:3000; CST, 4970) antibodies, and secondary antibodies (Vector Laboratories), followed by chemiluminescence (Amersham ECLTM Western Blotting Detection Reagents, GE healthcare).
TGFα-NP synthesis
The human TGFA gene sequence (50 amino acids) was ordered from Integrated DNA Technologies (IDT) and cloned into the Sortase-Tag Expressed Protein Ligation (STEPL) system.(29) In brief, TGFA was fused sequentially with a linker (GGSx2), sortase A (Srt A) substrate (LPETG), another linker (GGSx5), Srt A enzyme, and His12-tag (TGFA-GGSx2-LPETG-GGSx5-SrtA-His12). The sequence-confirmed plasmid construct was heat-shot transformed into Escherichia coli T7 express competent cells (New England BioLabs). On the next day, colonies were cultured in autoinduction medium (Formedium) containing 100 μg/ml ampicillin (Corning) plus 0.6% glycerol and shaken at 150 rpm for 2 days at 25°C. The cultures were pelleted down by centrifugation at 5000 × g for 15 min and lysed with 1% (w/v) octylthioglucoside (OTG, GoldBio) in PBS containing protease inhibitor and end-over-end rotated at 25°C for 30 min. The lysate was centrifuged again at the maximum speed for 20 min to collect supernatant, which was then loaded onto a cobalt resin (Thermo Fisher Scientific) to capture the TGFα fusion protein via His12 tag. After washing by 10 mM imidazole in PBS, the resin was incubated with 100 μM CaCl2 and 2 mM Gly-Gly-Gly (GGG, Santa Cruz Biotechnology) in PBS at 37°C for 1 h or 100 μM CaCl2 and 600 μM Gly-Gly-Gly-DBCO (GGG-DBCO, LifeTein) in PBS at 37°C for 4 h. During the incubation, either GGG or GGG-DBCO was attached to the TGFα protein by sortase A enzyme catalytic reaction. The resultant TGFα (TGFα-GGG) or TGFα-DBCO (TGFα-GGG-DBCO) was collected from the flow through and passed through spin filters (Amicon Ultra-4, 3000 MWCO) to remove excess GGG or GGG-DBCO. Protein concentrations were quantified by bicinchoninic acid (BCA) assay (Thermo Fisher Scientific).
TGFα-NPs were prepared via a click reaction. In brief, a chloroform solution containing 65 M% DSPE-PEG2K (1 mg, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000])/10 M% DOTAP (1,2-dioleoyl-3-trimethylammonium-propane)/25 M% DSPE-PEG5K-N3 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[azido(polyethylene glycol)-5000] (ammonium salt)) was prepared in a roundbottom flask. The solvent was removed using a direct stream of nitrogen followed by vacuum desiccation for a minimum of 4 h. NPs were prepared by adding PBS to the dried film in a 55°C water bath for 5 min, followed by vortex and filter through a 0.22 μm cellulose acetate membrane Nalgene filter (Thermo Fisher Scientific). NPs were then mixed with TGFα-DBCO (1:1 TGFα-DBCO to DSPE-PEG5K-N3 molar ratio) overnight to make TGFα-NPs. To test the stability of TGFα-NPs, TGFα-NPs were stored in PBS for 7 days or bovine synovial fluid for 4 days (Lampire Biological Laboratories, Inc). Dynamic light scattering (DLS) (Zetasizer, Nano-ZS, Malvern) was used to determine the diameter and size distribution of the TGFα-NPs.
NP administration
The right knee joints of 2-month-old male C57Bl/6 mice (Jackson Laboratory) were fixed in a flexed position under anesthesia and subjected to intra-articular injection of 10 μl PBS, TGFα-DBCO (10 μM TGFα content), or TGFα-NPs (10 μM TGFα content) using a 30G Hamilton needle (five mice/treatment). Injections were repeated once every 3 weeks starting right after loading.
Statistics
Results in Fig. 2–6, 8, and Fig. S1, except Fig. 3D, are presented as box plots with median, interquartile range, and maximum and minimum values. The results in Fig. 3D and Fig. 7B–D are presented as mean ± SD. For comparisons between two groups, one-way ANOVA was applied, followed by Tukey–Kramer multiple comparison tests. For comparisons among multiple groups across two fixed-effect factors (e.g., genotype and age), two-way ANOVA was applied, followed by Tukey–Kramer multiple comparison tests using Prism 8 software (GraphPad Software). In all tests, the significance level was set at α= 0.05.
Fig. 2.
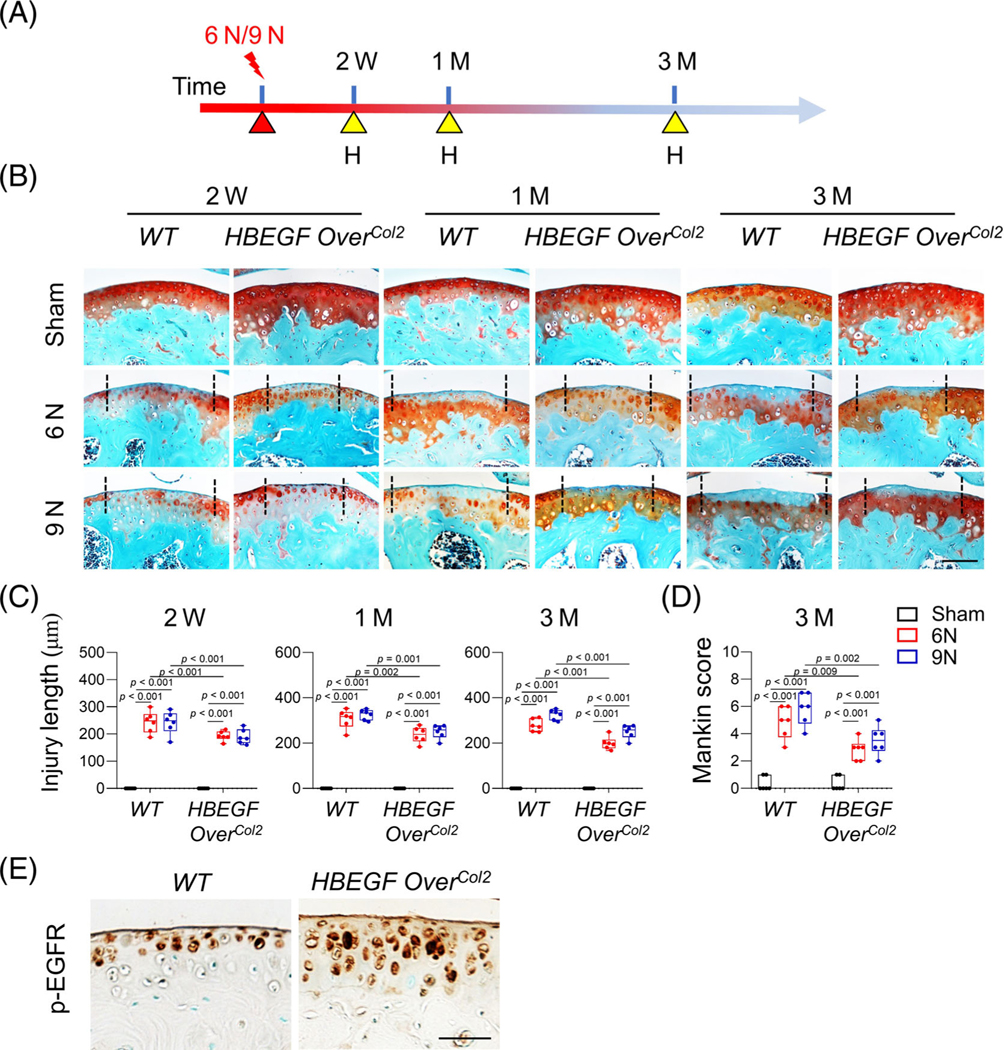
Constitutive epidermal growth factor receptor (EGFR) overactivation attenuates cartilage damage caused by loading. (A) The study protocol shows that 2-month-old WT and HBEGF OverCol2 mice were subjected to 6 or 9 N loading at their right tibiae, followed by joint harvest later. H, harvest. (B) Representative safranin-O/Fast green-stained sections of WT and HBEGF OverCol2 cartilage at 2 weeks, 1 month and 3 months after loading. Two black dashed lines indicate a cartilage lesion site between them. Scale bars, 100 μm. (C) Cartilage injury length was quantified (n = 6/group). (D) OA severity at 3 months after loading was assessed by Mankin score (n = 6/group). (E) Immunostaining of p-EGFR in uninjured articular cartilage from WT and HBEGF OverCol2 mice. Scale bars, 50 μm. The comparisons were conducted by two-way ANOVA with Tukey–Kramer multiple comparison test.
Fig. 6.

Chondrogenic EGFR deficiency accelerates PTOA progression in adult mice. (A) WT and Egfr iCKOAcanER mice received tamoxifen injections right before 6 N loading at 2 months of age. Their knee joints were harvested 2 weeks and 1 month later. H, harvest; T, tamoxifen injection. (B) Immunostaining of p-EGFR in uninjured articular cartilage from WT and Egfr iCKOAcanER mice at 2 weeks after loading. Scale bars, 50 μm. (C) Safranin-O/Fast green-stained sections of WT and Egfr iCKOAcanER cartilage at indicated time points. Scale bars, 100 μm. (D) Cartilage injury length was quantified (n = 6/group). (E) Mankin score of WT and Egfr iCKOAcanER mouse joints at 1 month after loading was quantified (n = 6/group). (F) H&E-stained sections of WT and Egfr iCKOAcanER synovium at indicated time points. Scale bars, 50 μm. (G) Synovitis score was quantified (n = 6/group). (H) Representative safranin-O/Fast green-stained sections of WT and Egfr iCKOAcanER joints at 1 month after loading. Arrows point to heterotopic ossification area in synovium. Scale bars, 100 μm. F, femur; M, meniscus; S, synovium; T, tibia. The comparisons were conducted by two-way ANOVA with Tukey–Kramer multiple comparison test.
Fig. 8.

Intra-articular injections of TGFα-NPs delays PTOA development. (A) 2-month-old WT mice received 6 N loading, followed by intra-articular injections of PBS, TGFα-DBCO, or TGFα-NPs starting right after loading and then once every 3 weeks for 12 weeks. Knee joints were harvested at 2 weeks and 3 months after loading for further histology analysis. (B) Representative safranin-O/Fast green images of articular cartilage at 2 weeks and 3 months after loading. Scale bars, 100 μm. (C) Cartilage injury length was quantified (n = 5/group). (D) OA severity at 3 months after loading was assessed by Mankin score (n = 5/group). (E) Representative H&E-stained sections of synovium at 2 weeks and 3 months post loading. Scale bars, 50 μm. (F) Synovitis score was quantified (n = 5/group). The comparisons were conducted by ordinary one-way ANOVA with Tukey–Kramer multiple comparison test.
Fig. 3.
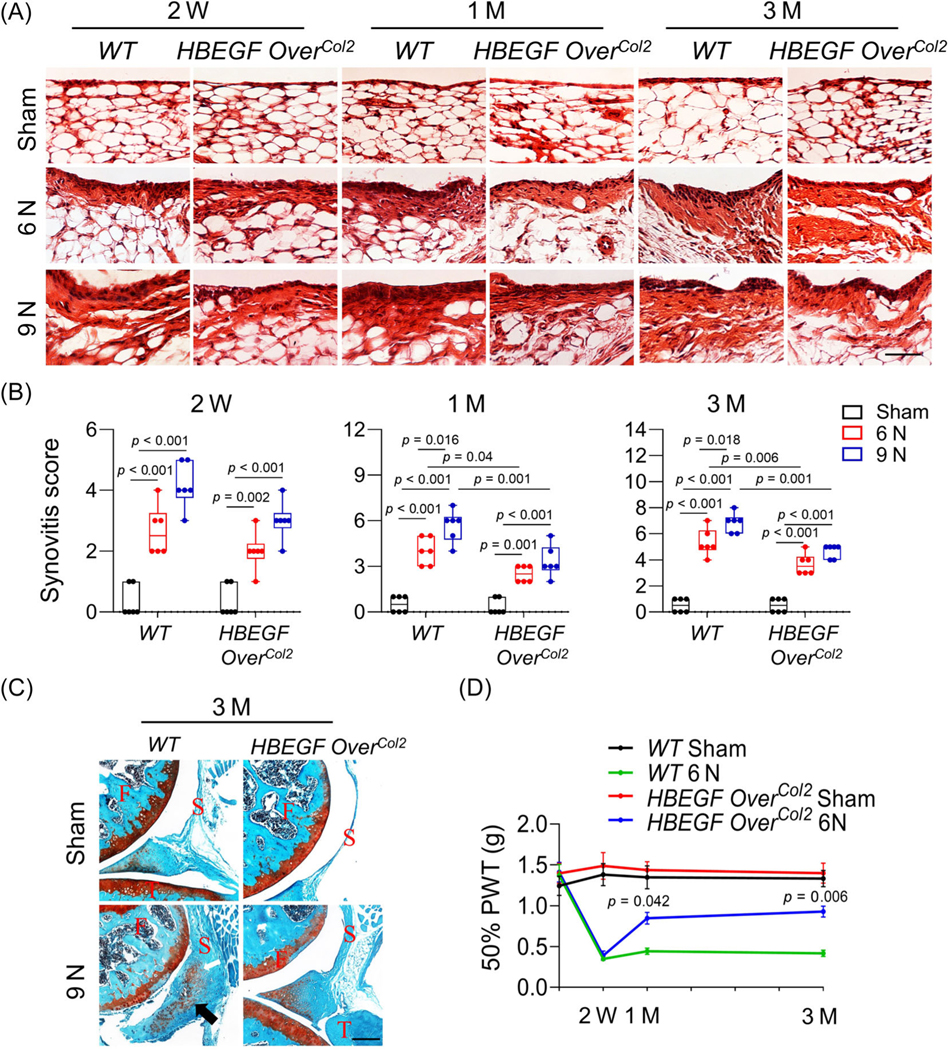
Constitutive epidermal growth factor receptor (EGFR) overactivation mitigates synovitis and ectopic cartilage formation in joint after loading. (A) Representative H&E-stained sections of WT and HBEGF OverCol2 mouse synovium at 2 weeks, 1 month, and 3 months after 6 or 9 N loading. Scale bars, 100 μm. (B) Synovitis score was quantified (n = 6/group). (C) Representative safranin-O/Fast green-stained sections of WT and HBEGF OverCol2 mouse joints at 3 months post 9 N loading. An arrow points to the heterotopic ossification area in synovium tissue. F, femur; S, synovium; T, tibia. Scale bars, 100 μm. (D) At 2 weeks, 1 month, and 3 months after 6 N loading, joint allodynia (50% PWT) was measured by von Frey assay. Value at Day 0 was obtained before loading. PWT, paw withdrawal threshold. (n = 5/group). P values indicate significant difference of 50% PWT between WT 6 N and HBEGF OverCol2 6 N. The comparisons were conducted by two-way ANOVA with Tukey–Kramer multiple comparison test.
Fig. 7.

Synthesis and characterization of novel nanoparticle conjugated with TGFα. (A) Schematic diagram of TGFα-NP. (B) Size distribution of TGFα-NP as evidenced by DLS. (C) The diameter of TGFα-NP was measured in PBS for up to 7 days. (D) The diameter of TGFα-NP was measured in bovine synovial fluid for up to 96 h. (E) Western blot of p-ERK, an EGFR downstream target, in primary chondrocytes at 15 min after TGFα-NP (15 ng/ml of TGFα content) treatment. Cells treated by vehicle (PBS), free TGFα (15 ng/ml), and empty-NP (i.e., no TGFα conjugation) were used as controls. The comparisons were conducted by ordinary one-way ANOVA with Tukey–Kramer multiple comparison test.
Results
Constitutive EGFR overactivation mitigates loading-induced joint damage
To upregulate EGFR activity in chondrocytes, we constructed HBEGF OverCol2 mice. At 2 months of age, HBEGF OverCol2 mice and their WT controls were subjected to compressive joint loading at a peak force of 6 or 9 N for 60 cycles at their right tibiae in a single loading period. ACL rupture was observed in all mice within the 9-N group but none within the 6-N group. Knee joints were harvested at 2 weeks, 1 month, and 3 months after loading for histological analyses (Fig. 2A). Loading caused a lesion at the posterior aspect of lateral femoral condyle in WT joints 2 weeks later in both groups (Fig. 2B). This cartilage lesion was characterized by a loss of safranin-O staining, indicating proteoglycan depletion. Over time, this loss became more and more obvious, but the length of the lesion, i.e., injury length, did not significantly increase (Fig. 2C). At 3 months after loading, we observed surface fibrillations in both loading groups. Quantification by Mankin score indicated moderate OA development (Fig. 2D). Compared to the 6-N group, the 9-N group showed a trend of increases in injury length at all three time points and in Mankin score at 3 months after injury.
Compared to the WT controls, HBEGF OverCol2 joints exhibited less cartilage damage after loading. Their injury length was significantly reduced by 20.7%, 22.8%, and 28.5% at 2 weeks, 1 month, and 3 months, respectively, in the 6-N group. These joints also had higher safranin-O staining intensity and smoother cartilage surface, leading to a 41.4% reduction in the Mankin score. Similar protective effects of HBEGF overexpression were also observed in the 9-N group. Immunohistochemistry (IHC) confirmed that HBEGF OverCol2 mice had higher EGFR activity (p-EGFR staining) at the top layer of articular cartilage in the uninjured area than WT siblings (Fig. 2E).
PTOA is associated with synovitis. In both the 6-N and 9-N WT groups, the thickening of synovium was prominent at 2 weeks and further increased at 1 month and 3 months after loading (Fig. 3A, B). The 9-N group had significantly higher synovitis scores than the 6-N group at 1 month and 3 months. Overexpression of HBEGF did not affect the synovium morphology in the Sham group but significantly reduced the thickening of synovium in the loading groups. At 3 months of age, synovitis scores of HBEGF OverCol2 joints were decreased by 31.2% and 31.7% in comparison with WT joints in the 6- and 9-N groups, respectively.
Loading at 9 N, but not at 6 N, promoted ectopic cartilage formation, characterized by neocartilage tissue in the region of meniscus and synovium, in 66.6% of WT mice (four out of six mice) 3 months later (Fig. 3C). Interestingly, no HBEGF OverCol2 mice developed ectopic cartilage after loading. We next performed a von Frey assay to evaluate loading-induced mechanical allodynia, a proxy for PTOA pain (Fig. 3D). After 6 N loading, WT mice experienced a 74.8% decrease in 50% paw withdrawal threshold (PWT) 2 weeks later. This decrease was maintained throughout the experimental period. However, although HBEGF OverCol2 mice showed a similar level of pain initially, their 50% PWT was significantly increased at 1 and 3 months later, indicating less pain than WT after loading. Taken together, the foregoing data demonstrate that joint damage caused by loading is partially attenuated by overactivating EGFR signaling in chondrocytes.
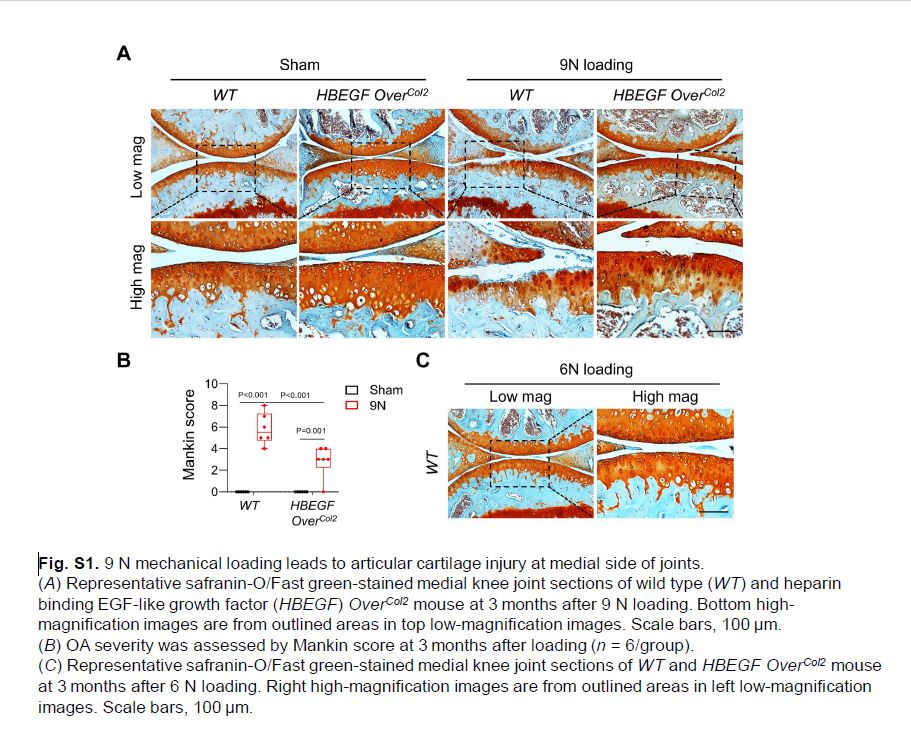
Since ACL transection is an established surgical OA model in mice,The aforementioned mouse model(30) we next examined whether 9 N loading caused additional cartilage damage due to ACL rupture. As expected, we found that articular cartilage degeneration occurred at the medial joint compartment at 3 months after loading, resulting in moderate OA with a Mankin score of 5.8 (Fig. S1A, B). Such damage was not found in 6 N–loaded joints (Fig. S1C), suggesting that only 6 N loading should be considered as a pure loading-induced OA model. Thus, we excluded 9 N in subsequent studies. Nevertheless, it is important to point out that the damage on the medial cartilage due to 9 N loading–induced ACL rupture is attenuated in HBEGF OverCol2 joints (Fig. S1A, B). These data are in line with our previous report that HBEGF OverCol2(5) joints were resistant to DMM-induced OA.
EGFR signaling protects chondrocytes but does not prevent matrix degradation
To investigate the protective mechanism of EGFR signaling after loading injury, we next performed IHC on mouse articular cartilage at 2 weeks after 6 N loading. In WT mice, loading caused prominent chondrocyte death, as shown by TUNEL staining (Fig. 4A) and reduced chondrocyte proliferation and lubricant production, as shown by Ki67 (Fig. 4B) and PRG4 staining (Fig. 4C), respectively. Note that most affected cells are in the top layer of articular cartilage. Although it did not completely block these adverse loading effects on cells, overexpressing HBEGF partially reversed the damage by decreasing the percentage of apoptotic cells (53.8%) and increasing the percentages of cells with proliferation ability (55.7%) and lubricant production (56.2%). In sham knees, we also observed increases of Ki67+ and PRG4+ chondrocytes in the HBEGF OverCol2 mice. Hence, these data suggest that EGFR signaling protects chondrocytes, especially superficial chondrocytes, from loading-induced damage.
Fig. 4.

EGFR signaling promotes the survival, proliferation, and lubrication of chondrocytes in articular cartilage. (A) TUNEL staining of WT and HBEGF OverCol2 articular cartilage at 4 days after 6 N loading. White dashed lines in the images outline uncalcified zone. The percentage of TUNEL+ cells in uncalcified cartilage was quantified and is shown at the right (n = 5/group). Scale bars, 100 μm. (B–E) Ki67 (B), PRG4 (C), MMP13 (D), and ADAMTS5 (E) staining of WT and HBEGF OverCol2 articular cartilage at 2 weeks after 6 N loading. In each panel, the percentage of positive cells in the articular cartilage is quantified at the right (n = 5/group). Scale bars, 50 μm. The comparisons were conducted by two-way ANOVA with Tukey–Kramer multiple comparison test.
MMP13 and ADAMTS5 are two major proteases responsible for cartilage degradation in OA.(31) We noticed that loading strongly elevated their amounts throughout the entire articular cartilage in WT mice (Fig. 4D, E). However, EGFR overactivation did not alter the percentage of cells staining positive for these proteases, suggesting that EGFR signaling does not affect matrix degradation in loading-induced PTOA.
Inducible EGFR overactivation in adult mice also reduces loading-induced joint damage
The aforementioned mouse model, HBEGF OverCol2 under the control of the Col2a1-Cre driver, overexpresses HBEGF in chondrocytes starting from the embryonic stage. To eliminate confounding developmental effects, we next examined the joint phenotype of an inducible mouse model, HBEGF OverAcanER, after loading (Fig. 5A). At 2 months of age, these mice, along with WT controls, received tamoxifen injections immediately before 6 N loading on the right tibiae. Joints were harvested 2 weeks or 3 months after loading for analysis. At 2 weeks, HBEGF OverCol2 mice displayed much higher EGFR activity, as shown by p-EGFR staining, in the uninjured articular cartilage region than WT siblings (Fig. 5B).
Fig. 5.

Chondrocyte-specific overactivation of EGFR signaling in adult mice hinders PTOA progression. (A) Study design showing WT and HBEGF OverAcanER mice received tamoxifen injections right before 6 N loading. Their knee joints were harvested 2 weeks and 3 months later. H, harvest; T, tamoxifen injection. (B) Immunostaining of p-EGFR in uninjured tibial articular cartilage from WT and HBEGF OverAcanER mice at 2 weeks after loading. Scale bars, 50 μm. (C) safranin-O/Fast green-stained sections of WT and HBEGF OverAcanER cartilage at indicated time points. Scale bars, 100 μm. (D) Cartilage injury length was quantified (n = 6/group). (E) Mankin score of WT and HBEGF OverAcanER mouse joints at 3 months after loading was quantified (n = 6/group). (F) H&E-stained sections of WT and HBEGF OverAcanER synovium at indicated time points. Scale bars, 50 μm. (G) Synovitis score was quantified (n = 6/group). The comparisons were conducted by two-way ANOVA with Tukey–Kramer multiple comparison test.
Similar to HBEGF OverCol2 mice, the HBEGF OverAcanER mice had significantly reduced cartilage injury length compared to WT (30.9% and 27.9% at 2 weeks and 3 months after loading, respectively, Fig. 5C, D). At 3 months, we also observed greater retention of safranin-O staining at the cartilage injury site in HBEGF OverAcanER joints, leading to a 41.4% reduction in the Mankin score (Fig. 5E). Moreover, the synovitis score was decreased by 26.5% in these mice at 3 months (Fig. 5F, G). Thus, inducible EGFR overactivation demonstrated that EGFR signaling in adult cartilage plays a protective role against loading injury.
Inactivating EGFR in chondrocytes accelerates PTOA development
To complement our study, we established an inducible chondrocyte-specific EGFR-deficient mouse model, Egfr iCKOAcanER (Aggrecan-CreER Egfr flox/Wa5 ). Our previous studies indicated that Wa5, a dominant negative allele of Egfr, is required for maximum reduction of EGFR activity in vivo.(5,32,33) We subjected these mice as well as their WT controls at 2 months of age to tamoxifen injections followed by 6 N loading (Fig. 6A). Compared to WT, Egfr iCKOAcanER mice had reduced p-EGFR activity in the uninjured articular cartilage (Fig. 6B).
After loading, the injury length in Egfr iCKOAcanER cartilage was 20.4% and 38.5% longer than WT cartilage at 2 weeks and 1 month, respectively (Fig. 6C, D). Although WT mice maintained a relatively smooth cartilage surface at these early time points, Egfr iCKOAcanER mice had already developed surface fibrillations at 2 weeks and significant cartilage erosion, fissures, and clefts at 1 month (Fig. 6C), leading to a drastic increase in the Mankin score (Egfr iCKOAcanER 6.8 versus WT 3.5, Fig. 6E). Synovitis was also more severe in the Egfr iCKOAcanER joint at both time points (Fig. 6F, G). WT joints normally did not develop ectopic cartilage formation at 1 month after 6 N loading. However, three out of six Egfr iCKOAcanER joints showed neocartilage formation (Fig. 6H), further indicating that joint damage is exaggerated in EGFR-deficient mice.
Synthesis and characterization of TGFα-NPs
The aforementioned mouse genetic studies suggested that manipulating chondrogenic EGFR activity could protect cartilage against PTOA. To develop a potential therapeutic approach, we synthesized a novel NP carrying TGFα, a potent EGFR ligand, as an activator. As a small peptide (5.5 kD), intra-articular injected TGFα suffers from rapid clearance from the joint space. Conjugating the ligand to a nanocarrier could prolong its joint retention and restrict it at the delivery site. TGFα-NPs were prepared by mixing TGFα-DBCO with azide-functionalized phospholipid micellar NPs with lipid composition of 65 M% DSPE-PEG2K/10 M% DOTAP/25 M% DSPE-PEG5K-N3 (Fig. 7A). DLS measurements revealed that TGFα-NPs possessed a mean hydrodynamic diameter of 13.5 nm and a relatively narrow size distribution (Fig. 7B). A stability assay showed no observable change in the hydrodynamic diameter after being present in PBS for 7 days (Fig. 7C) or in bovine joint fluid for 96 h (Fig. 7D). Western blot demonstrated that TGFα-NPs, but not empty NPs, activated the EGFR downstream target ERK in mouse primary chondroprogenitors as effectively as free TGFα (Fig. 7E).
TGFα-NPs delays OA progression after loading
Next, we evaluate the in vivo effects of TGFα-NPs on PTOA. Two-month-old WT mice were subjected to 6 N loading on the right tibia and intra-articular injection of PBS, TGFα-DBCO, or TGFα-NPs once every 3 weeks starting right after the injury (Fig. 8A). At 2 weeks after loading, the cartilage injury length was decreased by 25.9% in TGFα-NP-treated joints compared to PBS-treated joints (Fig. 8B, C). At 3 months after loading, TGFα-NP-treated joints retained more safranin-O staining and had less surface fibrillations, leading to a 40.0% reduction in the Mankin score (Fig. 8B, D). Moreover, synovitis was also lessened after TGFα-NP treatment (Fig. 8E, F). TGFα-DBCO alone did not have an obvious effect on cartilage injury. Overall, these data provide proof-of-principle evidence that targeting EGFR signaling using nanotechnology is effective for reducing loading-induced PTOA.
Discussion
In this work, using several genetically modified mouse models with chondrogenic EGFR overactivation and deficiency, we demonstrated the chondroprotective action of EGFR in loading-induced PTOA progression. These data complement our previous reports showing that EGFR signaling attenuates OA development in a surgical DMM model.(5,11,13) A single loading episode of mouse tibia induced lesions, indicated by cell death, in articular cartilage. Mechanistically, we found that activating EGFR signaling promoted the survival and lubrication of chondrocytes at the lesion site, thereby mitigating the loading impact on cartilage. Additionally, we designed a novel NP carrying an EGFR ligand and provided proof-of-principle evidence for its therapeutic action on loading-induced PTOA.
The EGFR signaling pathway is essential for cell growth, survival, proliferation, and differentiation. The general function of EGFR in mammalian organs, such as skin,(34) brain,(35) intestine,(36) and bone,(37) for example, is to maintain tissue-specific stem and progenitors by promoting their proliferation and inhibiting their apoptosis. Studies from our group and others in the past several years have demonstrated that EGFR plays a similar role in articular cartilage by preserving chondroprogenitors in the superficial zone during cartilage homeostasis and OA development.(12) However, those studies mainly utilized spontaneous and DMM-induced OA models. In this work, we validated our previous conclusions by investigating the action of EGFR signaling in a noninvasive mechanical tibial compression model. Unlike DMM injury, a single bout of cyclic tibial loading at a peak force of 6 N does not injure stabilizing tissues in joints. Instead, the mechanical impact rapidly creates a focal lesion at the cartilage surface, which seldom propagates to nearby areas 3 months later. The 9 N peak force is more complicated by additionally rupturing ACL, a well-established cause of PTOA. The specific joint damage seen in our data could be a combination of the immediate impact, overloading, and the joint instability induced by ACL rupture, potentially dominated by the latter. Compared to loadi, DMM induced no morphological changes in articular cartilage within the first few weeks but caused significant cartilage erosion in a much wider region at a later stage.(38) The loading-induced OA model captured the features of traumatic joint injury in human patients and recapitulated some hallmarks of PTOA, including cartilage degeneration, synovitis, ectopic cartilage formation, and pain.(19,39,40) Our data demonstrating the beneficial effects of EGFR signaling in this model provide strong support for developing novel OA therapies that target EGFR pathway.
In healthy adult cartilage, chondrocytes are in a mostly quiescent phase characterized by a fine balance between anabolic (matrix synthesis) and catabolic (matrix degradation) activities. In PTOA cartilage, this balance is tipped toward more catabolic activity and less anabolic activity, leading to cell death and matrix degradation.(41) Our data showed that activating EGFR signaling by HBEGF overexpression stimulates anabolic activity by preserving chondrocytes and their lubrication but does not affect the catabolic activities (MMP13 and ADAMTS5) after loading. These results might explain why intra-articular injection of TGFα-NP partially mitigates, but does not completely reverse, cartilage lesion and synovitis at 3 months after loading. Chronic inflammation at a low level plays a critical role in stimulating catabolic activities during PTOA.(42) We previously designed a phospholipid micellar nanoparticles conjugated with secreted phospholipase A2 inhibitor (sPLA2i).(43) sPLA2 is a heterogeneous group of enzymes that specifically hydrolyzes the sn-2 ester bond of membrane phospholipids to release free fatty acids, such as arachidonic acid and lysophospholipids, which are upstream mediators of inflammation in many chronic inflammatory diseases.(44) We demonstrated that these sPLA2i-NPs are effective in reducing the amounts of p-P65 and p-P100, two downstream reporters of the NF-κB inflammatory pathway, in mouse joints after loading. Research from another group also showed that directly targeting these two NF-κB reporter genes using siRNA strategy partially mitigate PTOA development in a mouse loading model.(45) To further restore articular cartilage to a healthy state and relieve other PTOA-related joint symptoms, we believe that a two-pronged approach targeting both anabolic and catabolic activities of cartilage is more effective for PTOA treatment. For this purpose, in this work, we conjugated TGFα to phospholipid-based micellar NPs, the same nanocarriers we previously used for constructing sPLA2i-NPs. In the future, we will synthesize phospholipid micellar NPs loaded with both TGFα and sPLA2i and test the efficacy of this single reagent in various OA models.
One limitation of our study is that we only study male mice at 2 months of age. In human populations, joint injuries can occur before and after skeletal maturity and in both sexes. Although the use of 2-month-old male mice for PTOA study was common in previous studies,(18,46,47) further investigation should be carried out in female mice as well as skeletally mature mice to identify any possible gender and age effects. Another limitation is that we only study the efficacy of EGFR overactivation immediately after PTOA initiates. Future studies should include a late treatment regimen, for example, injecting tamoxifen in HBEGF OverAcanER mice or TGFα-NPs in WT mice 1 month after loading injury, to match the clinical scenario in which patients often receive treatment long after an injury occurs. Lastly, although we initially intended to adjust PTOA severity using different loading forces, we later discovered that the high loading group (9 N) was a complex PTOA model combining loading injury and ACL rupture that creates cartilage damage sites at both medial and lateral components of joints. However, it is important to point out that HBEGF overexpression is able to attenuate cartilage degeneration at both sites. Taken together, our data strongly support the critical anabolic actions of EGFR signaling in maintaining articular cartilage during PTOA development and shed new light on developing a novel nanomedicine to treat millions of PTOA patients.
Supplementary Material
{kind=link}
Acknowledgments
For their technical assistance we thank Dr. Waixing Tang in the Histology Core and Drs. Wei-Ju Tseng and Yilu Zhou in the Imaging Core of the Penn Center for Musculoskeletal Disorders.
Funding Information
This study was supported by National Institutes of Health Grants R01AG067698, R21AR078650 (to LQ), P30AR069619 (to Penn Center for Musculoskeletal Disorders), and R01AR080820, Health Research Formula Fund from the Pennsylvania Department of Health (to ZC).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4717.
Data Availability Statement
The data that supports this study is available from the authors upon reasonable request.
References
- 1.Blagojevic M, Jinks C, Jeffery A, Jordan KP. Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthr Cartil. 2010;18(1):24–33. [DOI] [PubMed] [Google Scholar]
- 2.Brown TD, Johnston RC, Saltzman CL, Marsh JL, Buckwalter JA. Posttraumatic osteoarthritis: a first estimate of incidence, prevalence, and burden of disease. J Orthop Trauma. 2006;20(10):739–744. [DOI] [PubMed] [Google Scholar]
- 3.Roos EM. Joint injury causes knee osteoarthritis in young adults. Curr Opin Rheumatol. 2005;17(2):195–200. [DOI] [PubMed] [Google Scholar]
- 4.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. NatRev Mol Cell Biol. 2006;7(7):505–516. [DOI] [PubMed] [Google Scholar]
- 5.Jia H, Ma X, Tong W, et al. EGFR signaling is critical for maintaining the superficial layer of articular cartilage and preventing osteoarthritis initiation. Proc Natl Acad Sci U S A. 2016;113(50):14360–14365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Appleton CT, Usmani SE, Bernier SM, Aigner T, Beier F. Transforminggrowth factor alpha suppression of articular chondrocyte phenotype and Sox9 expression in a rat model of osteoarthritis. Arthritis Rheum. 2007;56(11):3693–3705. [DOI] [PubMed] [Google Scholar]
- 7.Long DL, Ulici V, Chubinskaya S, Loeser RF. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is increased in osteoarthritis and regulates chondrocyte catabolic and anabolic activities. Osteoarthr Cartil. 2015;23(9):1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castano-Betancourt MC, Evans DS, Ramos YF, et al. Novel genetic variants for cartilage thickness and hip osteoarthritis. PLoS Genet. 2016; 12(10):e1006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui G, Wei R, Liu D, et al. Association of Common Variants in TGFA with increased risk of knee osteoarthritis susceptibility. Genet Test Mol Biomarkers. 2017;21(10):586–591. [DOI] [PubMed] [Google Scholar]
- 10.Zengini E, Hatzikotoulas K, Tachmazidou I, et al. Genome-wide analyses using UK biobank data provide insights into the genetic architecture of osteoarthritis. Nat Genet. 2018;50(4):549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei Y, Ma X, Sun H, et al. EGFR signaling is required for maintain adult cartilage homeostasis and attenuating osteoarthritis progression. J Bone Miner Res. 2022;37(5):1012–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin L, Beier F. EGFR signaling: friend or foe for cartilage? JBMR Plus. 2019;3(2):e10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei Y, Luo L, Gui T, et al. Targeting cartilage EGFR pathway for osteo-arthritis treatment. Sci Transl Med. 2021;13(576):eabb3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450(7170):741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pest MA, Russell BA, Zhang YW, Jeong JW, Beier F. Disturbed cartilage and joint homeostasis resulting from a loss of mitogen-inducible gene 6 in a mouse model of joint dysfunction. Arthritis Rheumatol. 2014;66(10):2816–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shepard JB, Jeong JW, Maihle NJ, O’Brien S, Dealy CN. Transient anabolic effects accompany epidermal growth factor receptor signal activation in articular cartilage in vivo. Art Ther. 2013;15(3):R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staal B, Williams BO, Beier F, Vande Woude GF, Zhang YW. Cartilage-specific deletion of Mig-6 results in osteoarthritis-like disorder with excessive articular chondrocyte proliferation. Proc Natl Acad Sci U S A. 2014;111(7):2590–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christiansen BA, Guilak F, Lockwood KA, et al. Non-invasive mouse models of post-traumatic osteoarthritis. Osteoarthr Cartil. 2015; 23(10):1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu P, Holguin N, Silva MJ, Fu M, Liao W, Sandell LJ. Early response ofmouse joint tissue to noninvasive knee injury suggests treatment targets. Arthritis Rheumatol. 2014;66(5):1256–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26(2):145–146. [PubMed] [Google Scholar]
- 21.Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis. 2009;47(12):805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buch T, Heppner FL, Tertilt C, et al. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods. 2005;2(6):419–426. [DOI] [PubMed] [Google Scholar]
- 23.Iwamoto R, Higashiyama S, Mitamura T, Taniguchi N, Klagsbrun M, Mekada E. Heparin-binding EGF-like growth factor, which acts as the diphtheria toxin receptor, forms a complex with membrane protein DRAP27/CD9, which up-regulates functional receptors and diphtheria toxin sensitivity. EMBO J. 1994;13(10):2322–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee D, Cross SH, Strunk KE, et al. Wa5 is a novel ENU-induced antimorphic allele of the epidermal growth factor receptor. Mamm Genome. 2004;15(7):525–536. [DOI] [PubMed] [Google Scholar]
- 25.Lee TC, Threadgill DW. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis. 2009;47(2):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aigner T, Cook JL, Gerwin N, et al. Histopathology atlas of animalmodel systems-overview of guiding principles. Osteoarthr Cartil. 2010;18(Suppl 3):S2–S6. [DOI] [PubMed] [Google Scholar]
- 27.Krenn V, Morawietz L, Haupl T, Neidel J, Petersen I, Konig A. Gradingof chronic synovitis--a histopathological grading system for molecular and diagnostic pathology. Pathol Res Pract. 2002;198(5):317–325. [DOI] [PubMed] [Google Scholar]
- 28.Piel MJ, Kroin JS, Im HJ. Assessment of knee joint pain in experimental rodent models of osteoarthritis. Methods Mol Biol. 2015;1226: 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warden-Rothman R, Caturegli I, Popik V, Tsourkas A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Anal Chem. 2013;85(22): 11090–11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang H, Beier F. Mouse models of osteoarthritis: modelling risk factors and assessing outcomes. Nat Rev Rheumatol. 2014;10(7): 413–421. [DOI] [PubMed] [Google Scholar]
- 31.Martel-Pelletier J, Welsch DJ, Pelletier JP. Metalloproteases and inhibitors in arthritic diseases. Best Pract Res Clin Rheumatol. 2001;15(5): 805–829. [DOI] [PubMed] [Google Scholar]
- 32.Jia H, Ma X, Wei Y, et al. Reduction in Sclerostin as a mechanism of subchondral bone plate sclerosis in mouse knee joints during late-stage osteoarthritis. Arthritis Rheumatol. 2018;70(2):230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Tamasi J, Lu X, et al. Epidermal growth factor receptor plays an anabolic role in bone metabolism in vivo. J Bone Miner Res. 2011; 26(5):1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nanba D, Toki F, Barrandon Y, Higashiyama S. Recent advances in the epidermal growth factor receptor/ligand system biology on skin homeostasis and keratinocyte stem cell regulation. J Dermatol Sci. 2013;72(2):81–86. [DOI] [PubMed] [Google Scholar]
- 35.Galvez-Contreras AY, Alfredo Q-H, Gonzalez-Perez O. The role of EGFR and ErbB family related proteins in the oligodendrocyte specification in germinal niches of the adult mammalian brain. Front Cell Neurosci. 2013;7:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stenson WF, Ciorba MA. Nonmicrobial activation of TLRs controls intestinal growth, wound repair, and radioprotection. Front Immunol. 2021;11:617510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandra A, Lan S, Zhu J, Siclari VA, Qin L. Epidermal growth factor receptor (EGFR) signaling promotes proliferation and survival in osteoprogenitors by increasing early growth response 2 (EGR2) expression. J Biol Chem. 2013;288(28):20488–20498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doyran B, Tong W, Li Q, et al. Nanoindentation modulus of murine cartilage: a sensitive indicator of the initiation and progression of post-traumatic osteoarthritis. Osteoarthr Cartil. 2017;25(1):108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ter Heegde F, Luiz AP, Santana-Varela S, et al. Noninvasive mechanical joint loading as an alternative model for osteoarthritic pain. Arthritis Rheumatol. 2019;71(7):1078–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rai MF, Duan X, Quirk JD, et al. Post-traumatic osteoarthritis in micefollowing mechanical injury to the synovial joint. Sci Rep. 2017;7: 45223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldring MB, Marcu KB. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res Ther. 2009;11(3):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. 2013;5(2):77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei Y, Yan L, Luo L, et al. Phospholipase A(2) inhibitor-loaded micellar nanoparticles attenuate inflammation and mitigate osteoarthritis progression. Sci Adv. 2021;7(15):eabe6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011; 111(10):6130–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan H, Duan X, Pan H, et al. Suppression of NF-kB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc Natl Acad Sci U S A. 2016;113(41):E6199–E6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Christiansen BA, Anderson MJ, Lee CA, Williams JC, Yik JH, Haudenschild DR. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthr Cartil. 2012;20(7):773–782. [DOI] [PubMed] [Google Scholar]
- 47.Ko FC, Dragomir C, Plumb DA, et al. In vivo cyclic compression causes cartilage degeneration and subchondral bone changes in mouse tibiae. Arthritis Rheum. 2013;65(6):1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports this study is available from the authors upon reasonable request.