

Summary
Heterozygous pathogenic variants in POLR1A, which encodes the largest subunit of RNA Polymerase I, were previously identified as the cause of acrofacial dysostosis, Cincinnati-type. The predominant phenotypes observed in the cohort of 3 individuals were craniofacial anomalies reminiscent of Treacher Collins syndrome. We subsequently identified 17 additional individuals with 12 unique heterozygous variants in POLR1A and observed numerous additional phenotypes including neurodevelopmental abnormalities and structural cardiac defects, in combination with highly prevalent craniofacial anomalies and variable limb defects. To understand the pathogenesis of this pleiotropy, we modeled an allelic series of POLR1A variants in vitro and in vivo. In vitro assessments demonstrate variable effects of individual pathogenic variants on ribosomal RNA synthesis and nucleolar morphology, which supports the possibility of variant-specific phenotypic effects in affected individuals. To further explore variant-specific effects in vivo, we used CRISPR-Cas9 gene editing to recapitulate two human variants in mice. Additionally, spatiotemporal requirements for Polr1a in developmental lineages contributing to congenital anomalies in affected individuals were examined via conditional mutagenesis in neural crest cells (face and heart), the second heart field (cardiac outflow tract and right ventricle), and forebrain precursors in mice. Consistent with its ubiquitous role in the essential function of ribosome biogenesis, we observed that loss of Polr1a in any of these lineages causes cell-autonomous apoptosis resulting in embryonic malformations. Altogether, our work greatly expands the phenotype of human POLR1A-related disorders and demonstrates variant-specific effects that provide insights into the underlying pathogenesis of ribosomopathies.
Keywords: RNA Polymerase I, craniofacial anomalies, limb defects, ribosomal RNA, neural crest cells, acrofacial dysostosis, congenital heart defect, epilepsy, developmental delay, ribosomopathies
Smallwood et al. describe a series of individuals with heterozygous variants in POLR1A in association with variable craniofacial, heart, and brain anomalies. Adverse effects of POLR1A perturbation are demonstrated on mouse development in vivo, as well as on rRNA transcription in vitro.
Introduction
Ribosomopathies are a phenotypically and genetically heterogeneous group of congenital malformation syndromes caused by disruption of ribosome biogenesis. Ribosome biogenesis is a multi-step process that begins with transcription of ribosomal DNA genes by RNA polymerase I (Pol I).1 Pathogenic variants in TCOF1, a transcription factor which binds rDNA promotors and Pol I, are associated with Treacher Collins syndrome (TCS [MIM: 154500]) which is characterized by a specific pattern of anomalous craniofacial development resulting in mandibulofacial dysostosis.2 Pathogenic variants in genes encoding Pol I subunits POLR1A, POLR1B, POLR1C, and POLR1D have been associated with more heterogeneous human phenotypes. Pathogenic variants in POLR1D (MIM: 613715) are associated with dominant3 or recessive4 TCS while pathogenic variants in POLR1C (MIM: 610060) are associated with recessive TCS3 or recessive leukodystrophy (MIM: 616494).5 More recently, pathogenic variants in POLR1B (MIM: 602000) were associated with dominant TCS.6 We previously showed that heterozygous pathogenic variants in POLR1A (MIM: 616404), which encodes the largest Pol I subunit, cause autosomal-dominant acrofacial dysostosis, Cincinnati-type (AFDCIN [MIM: 616462]), which is characterized by craniofacial anomalies reminiscent of TCS, as well as variable limb defects.7
Tcof1 and genes encoding the Pol I subunits A, B, C, and D are broadly expressed during mouse embryogenesis8 and yet their associated human phenotypes range from relatively restricted to the face (TCOF1, POLR1B, POLR1D) to variable craniofacial and limb involvement (POLR1A) and variable craniofacial and neurologic involvement (POLR1C). Of the genes encoding Pol I subunits A–D, POLR1A variants have been described in a relatively small group of individuals without recognized genotype-phenotype correlations and with broader phenotypic heterogeneity compared to individuals harboring variants in POLR1B-D. Since 2015 when 3 individuals with AFDCIN were reported, 4 additional individuals with POLR1A-related disease have been described. Shenoy et al.9 reported a single individual (proband 7) with severe mandibulofacial dysostosis without limb defects and a de novo POLR1A c.1325G>A (p.Arg442His) variant. An additional individual with the same variant as individual 1A2 in Weaver et al.7 was reported by da Rocha et al.10 as part of a series of individuals with limb malformations (individual F12). Finally, a report of neurodegenerative disease in two brothers with homozygous POLR1A variants was published in 2017,11 providing preliminary evidence that POLR1A variants may also lead to neurologic disease without craniofacial or limb involvement.
Here, we describe 17 previously unreported individuals with heterozygous novel (not previously reported in gnomAD) or ultra-rare variants in POLR1A and provide additional clinical details on 1 previously reported individual (F12 in da Rocha et al.).10 While all have some degree of craniofacial dysmorphism, they also present with an increased incidence of cardiac and neurodevelopmental abnormalities, findings which were not appreciated in the small number of previously described individuals. Given that cardiac and neurodevelopmental abnormalities have been associated with other disorders of ribosome biogenesis,2,12,13 we hypothesized that human phenotypes associated with heterozygous POLR1A variants extend beyond the face and limbs, and that there may be genotype- and tissue-specific effects on development. While recent work identified a critical developmental requirement for Polr1a in pre-migratory neural crest cells,8 variant-specific effects and the requirement for Polr1a in other developmental lineages have not been defined. We used a combination of in vitro (cell culture) and in vivo (in mice) modeling to investigate the effects of specific variants, further delineate the role of Polr1a in craniofacial development, and define the requirements for Polr1a in cardiac and neural development.
Material and methods
Identification and characterization of human cohort
The cohort was assembled through collaboration with GeneDx (Gaithersburg, Maryland), GeneMatcher,14 and personal e-mail communication to the corresponding author (K.N.W.). Individuals were included if they had a heterozygous variant in POLR1A interpreted as being of uncertain significance, likely pathogenic, or pathogenic according to American College of Medical Genetics/Association for Molecular Pathology consensus criteria.15 Existing clinical information about each individual was collected from the referring physician or healthcare provider. Individuals or their legal guardians agreed to participate, and consent for publication of photographs (if included) was obtained separately. The Institutional Review Board at Cincinnati Children’s provided oversight of the study (IRB 2021-0535).
In vitro assessment of POLR1A variant effects on rRNA transcription
To generate expression vectors for 8 of the 12 unique POLR1A variants, site-directed mutagenesis was performed to introduce each point mutation into pEF1-FRT-HaloTag-POLR1A (RPA194)16 using primers containing the desired mutation and a PCR protocol that amplifies the entire plasmid template. A genome-edited HCT116 cell line expressing mAID-mClover-POLR1A (RPA194)16 from the endogenous locus was transfected with each pEF1-FRT-HaloTag-POLR1A construct (wild-type and 8 variants). Following transfection, cells were incubated in medium supplemented with doxycycline (1 μg/mL) for 24 h at 37°C under 5% CO2, and then treated with 500 μM indole-3-acetic acid (19119-61, Nacalai), a natural auxin, in the presence of doxycycline for 3 h. As shown previously, this results in rapid knock-down of the cells’ native POLR1A,16 thus reducing the risk of indirect effects and allowing expression of only the transfected (mutant or wild-type) Halo-Tag-POLR1A. After the treatment, 5-ethinyl uridine (EU) incorporation was performed with Click-iT RNA Alexa Fluor 594 imaging kits (C10330, Thermo Fisher Scientific) according to the manufacturer’s instructions. EU incorporation provides a readout of RNA transcription rate, but rRNA transcription specifically can be determined by restricting analysis to the nucleoli. Section images were recorded with a DeltaVision Elite microscope (Applied Precision). For quantitative analysis of rRNA transcription, the nucleolar region of interest was created based on HaloTag-POLR1A signals in Fiji software. To reduce the effect of variation in HaloTag-POLR1A expression level between cells, EU signal intensity was normalized to HaloTag-POLR1A signal intensity in a cell, and the relative ratio of each mutant to wild-type cells was calculated.
Image analysis by the wndcharm machine learning algorithm and fisher linear discriminant analysis (FLDA)
For quantitative image analysis, we first excised nucleolar images by Fiji software17 and subjected them to machine learning software, wndchrm v.1.60.18 Wndchrm (weighted neighbor distances via a compound hierarchy of algorithms representing morphology) is a computational pattern recognition approach that provides a quantitative assessment of the differences between wild-type and mutant morphological phenotypes.18,19 We created 10 image folders, each containing deconvolved nucleolar images from Halo-tagged POLR1A variant-expressing cells. Wild-type and 9 variants were studied: 8 described in the current work (corresponding to POLR1A p.Glu1330del, p.Cys1562Phe, p.Asp59Val, p.Val1631Met, p.Pro1638Leu, p.Met496Ile, p.Val1241Ile, p.Arg393His. and p.Glu593Gln) as well as the previously published POLR1A c.1777G>C (p.Glu593Gln), reported by Weaver et al.7 in an individual with severe craniofacial and limb anomalies. The dataset was tested by 60 rounds of cross validations to measure class similarity which was visualized as a phylogenetic tree by the PHYLIP package.20 The numbers of images used were 93 (wild-type), 113 (p.Glu1330del), 127 (p.Cys1562Phe), 94 (p.Asp59Val), 93 (p.Val1631Met), 72 (p.Pro1638Leu), 79 (p.Met496Ile), 108 (p.Val1241Ile), 67 (p.Arg393His), and 37 (p.Glu593Gln). The options used for image analysis were a large feature set of 2,919 (-l) and multi-processors (-m).
To visualize the relative image similarity individually in 3D scatter plotting, we used a machine learning library called scikit-learn in Python. All the images in each class were applied to wndchrm to compute the Fisher scores of all 2,919 image features implanted in wndchrm, and the top 438 were extracted. The 438 feature values for each image were then used to perform Fisher Linear Discriminant Analysis (FLDA). FLDA finds the vector that maximizes the separation between each variant of the image data and enables dimensionality reduction from 438 to 3 to describe 3D scatter plotting.
Generation of Polr1a mouse alleles
The animal protocol for this work was approved by the Cincinnati Children’s Hospital Medical Center Institutional Animal Care and Use Committee (IACUC #2019-0043) or the Stowers Institute for Medical Research (IACUC #2022-143). Polr1atm1a(EUCOMM)Hmgu/BayMmucd (henceforth Polr1atm1a/+) mice were created as previously described.8,21 The Tm1a allele consists of an FRT sequence followed by the LacZ sequence and a loxP site, a neomycin cassette driven by the human beta-actin promoter, an SV40 polyA, a second FRT site, a second loxP site, and a third loxP site downstream of exon 6 (Figure S5A). These mice were bred with germline Flpase transgenic mice (B6.129S4-Gt (ROSA)26Sor-tm1(FLP1)Dym/RainJ) to generate mice with a conditional allele (Polr1atm1c/+ or Polr1aflox/+). Polr1aflox/+ mice were bred with B6.C-Tg (CMV-cre)1Cgn/J mice to create mice with a germline deletion allele, Polr1atm1d/+. Polr1a allele constructs are shown in Figure S5A. Polr1atm1a and Polr1atm1d are functionally both null alleles (Polr1anull).
A mouse model of POLR1A c.4685G>T (p.Cys1562Phe) was generated by the CCHMC Transgenic Animal and Genome Editing (TG) Core. This variant was chosen because it was identified as a de novo event in two unrelated individuals (15 and 16) with near-identical phenotypes (infantile spasms, hypotonia, and hypertelorism) (Figure 1A). The methods for design of sgRNAs and production of animals were previously described.22 The sgRNA was selected using on- and off-target scores from CRISPOR23 and transcribed in vitro using the MEGAshorscript T7 kit (ThermoFisher). The product was purified with the MEGAclear Kit (ThermoFisher) and stored at −80°C. The injection mix was prepared by incubating sgRNA and Cas9 protein (IDT) at 37°C for 15 min to form ribonucleoproteins and then adding the single-stranded DNA donor oligo (IDT). The final concentrations were 50 ng/μL sgRNA, 100 ng/μL Cas9 protein, and 75 ng/μL donor oligo. The mutant mice were generated by injection of the mix into the cytoplasm of fertilized eggs of C57BL/6 genetic background, using a piezo-driven microinjection technique.24 After injection, eggs were transferred into the oviductal ampulla of pseudopregnant CD-1 females. In addition to the desired c.4676G>T (p.Cys1559Phe) mutation, a silent mutation was also introduced (c.4680_4683delinsTTTA) to enable genotyping with PCR followed by restriction enzyme digest with DraI (New England Biolabs). Three mosaic male mice were born, and all carried the desired Polr1a c.4676G>T (p.Cys1559Phe) variant that is orthologous to POLR1A c.4685G>T (p.Cys1562Phe). One mouse had only the c.4676G>T variant (line known as Cys1559Phe_b), while two mice had the desired variant in trans with deletions on the other allele (lines known as Cys1559Phe_a and Cys1559Phe_c). The offspring of these mosaic founders were sequenced to confirm genotyping and selected for future mating if they had the G>T variant and no additional deletions. There were no observed phenotypic differences between the a/b/c lines, which will subsequently be referred to as Polr1aC1559F. sgRNA and donor sequences used for generation of this allele are shown in Table S1.
Figure 1.

Human cohort phenotypes
(A) Facial photographs of 12 individuals in the cohort.
(B) Diagram of POLR1A with all reported variant locations indicated. Variants of individuals with photos are indicated in green triangles. Variants reported in this work are above and previously published variants are below the protein diagram. Of previously published affected individuals, IA1, IA2, and IA3 were reported by Weaver et al.7 and 7III3 was reported by Shenoy.9 IA3 variant details are c.3895G>T (p.Val1299Phe). Details for all other variants are contained within the manuscript and supplemental documents.
(C) Phenotype summary of all individuals with heterozygous POLR1A variants.
A mouse model of POLR1A c.4913C>T (p.Pro1638Leu) was generated using the same method as above, with the exception that the sgRNA was purchased from Synthego. This variant was chosen because it was identified in the individual who had the most severe congenital heart defect among the entire cohort (individual 18). Two mosaic male mice were produced. One had only the desired c.4904C>T (p.Pro1635Leu) variant, orthologous to POLR1A c.4913C>T (p.Pro1638Leu). The other had the desired variant as well as an additional substitution (c.4895C>T [p.Ala1632Val]) which was confirmed to be in cis with c.4904C>T. The offspring of both founders were sequenced to confirm genotyping before proceeding with any further mating. sgRNA and donor sequences used for generation of these alleles are shown in Table S1. The mice used for experiments are subsequently referred to as Polr1aP1635L.
Mouse maintenance and genotyping
Mutant embryos were generated using breeding pairs as shown in Table S2. Noon on the day after mating (as judged by vaginal plug) was designated as embryonic day (E) 0.5, and somites were counted in young (E9.5 and earlier) embryos to ensure staging was consistent and accurate. Polr1a animals were genotyped using the primers listed in Table S3. The PCR product from Polr1aC1559F was digested by restriction enzyme DraI (New England Biolabs) to produce 173 bp and 140 bp products in mutants. The PCR product from Polr1aP1635L was digested by restriction enzyme MboII (New England Biolabs), producing 146 bp and 127 bp products.
For conditional deletion of Polr1a in specific cell lineages of interest, we used four different Cre recombinase-expressing alleles. Wnt1Cre (B6; Tg (Wnt1-cre)11Rth) was used to delete Polr1a in pre-migratory neural crest cells and was genotyped as previously described.25 Sox10Cre (B6; CBA-Tg(Sox10-cre)1Wdr/J) was used to delete Polr1a in migratory neural crest cells and was genotyped as previously described.26 Mef2c-AHF-Cre (Tg(Mef2c-cre)2Blk/Mmnc) was used to delete Polr1a in the anterior heart field and was genotyped as previously described.27 Foxg1-IRES-Cre (B6.129T(SJL)-Foxg1tm1.1(cre)Ddmo/J) was used to delete Polr1a in the telencephalon and was genotyped as previously described.28 For lineage tracing, we used a ROSAdTom/EGFP (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) allele, which activates GFP in cells that express Cre recombinase and their lineage descendants.29 For all mutant lines created, a chi-square test was used to compare observed and expected Mendelian genotypes for each cross.
X-gal staining
LacZ transgene activity was visualized in whole embryos by fixing for 15 min at room temperature with 4% paraformaldehyde, washing with PBST three times (5 min each) at room temperature, washing in LacZ Wash Buffer (0.36% sodium phosphate monobasic, 1.094% sodium phosphate dibasic, 0.2% 1M MgCl2, 0.0001% deoxycholate, and 0.0002% nonidet-P40 in ddH2O), and staining overnight with X-gal (1 mg/mL). LacZ transgene activity was visualized in sectioned embryos by fixing for 2 h at room temperature with 4% paraformaldehyde, washing with PBST two times (5 min each) at room temperature, then dehydrating overnight at 4°C with 30% sucrose, and embedding in OCT freezing compound. Embedded embryos were sectioned at 12 μm, post-fixed in 0.5% paraformaldehyde on ice for 10 min, and then rinsed with cold PBS. Sections were then washed in LacZ Wash Buffer on ice for 10 min, stained with X-gal at room temperature, washed again with LacZ Wash Buffer two times (5 min each), and rinsed with PBS for 5 min. Stained sections were sealed with Pro-Long Gold (Invitrogen). Whole embryos and sectioned embryos were viewed on a Zeiss Discovery V8 stereomicroscope and imaged on brightfield with an Axiocam 305 camera.
Histology
Histological analysis was conducted on formalin- or Bouins-fixed tissues, paraffin-embedded, sectioned at 8–10 μm, and stained with hematoxylin and eosin. Histology sections were viewed on a Zeiss Discovery V8 stereomicroscope and imaged with an Axiocam 305 camera.
Myocardial thickness quantification
Histology images were viewed and measured using Zeiss ZEN Blue software. The myocardial thickness for each sample was measured at three positions for each ventricle: the dorsal, lateral, and ventral surface. Seven sections of the ventricles were selected at random for the measurements.
Skeletal preparations
Mouse embryo heads were collected at E17, deskinned, and fixed in 95% ethanol for 2 days, then placed in Alcian blue for cartilage staining overnight at room temperature. The skulls were then washed in 95% ethanol for 2 days and cleared in a 1% KOH solution for 24 h at room temperature. Bone was counterstained with Alizarin Red for 24 h at room temperature. The skulls were then cleared with a 20% glycerol/1% KOH solution (at room temperature, changing daily), stored in 50% glycerol/50% ethanol solution, and photographed.
Mouse echocardiography
Mouse echocardiography was performed by the CCHMC Cardiovascular Imaging Core Research Laboratory (CICRL). Mice were anesthetized using inhaled isoflurane, had their chest hair removed with a depilatory agent, and were secured with tape to a warmed imaging platform. Transthoracic echocardiograms were performed using a Vevo 3100 mouse echocardiography system equipped with a 40 MHz high-frequency transducer. Mice were returned to their cages and observed until they had completely recovered (3–5 min). The echocardiography data were downloaded to Vevo LAB analysis software. All measurements were performed in triplicate on three consecutive cardiac cycles and reported as an average value.
Whole-mount immunohistochemistry
Embryos used for whole-mount immunohistochemistry (WMIHC) for Cleaved Caspase-3 (CC3) were fixed in 4% paraformaldehyde in PBS for 30 min, followed by a 1-min wash in PBS. Samples were then incubated in a WMIHC blocking solution (0.1% NGS, 0.01% sodium azide, 0.001% Triton X-100 in PBS) for 24 h, followed by incubation with primary antibody anti-Cleaved Caspase-3 (CC3 1:300, Cell Signaling Technology #9661, in WMIHC blocking solution) for 72 h, then four washes (1 h each) in PBS, and incubation with secondary antibody in the dark for 48 h. Secondary antibodies included Invitrogen Alexa Fluor 488, Invitrogen Alexa Fluor 555, and Invitrogen Alexa Fluor 594 (all goat anti-rabbit, applied at 1:500). The samples were then washed four times (30 min each) in PBS, incubated in DAPI for 24 h, washed in PBS for 30 min, then incubated in RIMS (88% Histodenz, 0.1% Tween 20, 0.01% sodium azide in 0.02M Phosphate Buffer) for 24 h. All washes and incubations were done on a rocker at room temperature. Images were taken on a Nikon C2+ confocal microscope.
Embryos used for WMIHC with PECAM-1 (platelet endothelial cell adhesion molecule; BD Biosciences) were fixed in 4% PFA overnight, dehydrated through a graded methanol/PBS series into 100% methanol, and stored at −20°C until processed. Embryos were treated with Dent’s bleach for 2 h, rehydrated through a graded methanol/PBS series into PBS, immunostained with a PECAM1 antibody (BD Pharmingen #553370 at 1:500), followed by an HRP-coupled secondary antibody (goat anti-rat HRP, 1:500). Color revelation was performed using the DAB substrate Kit (Vector laboratories, SK-4100) according to the manufacturer’s instructions. The embryos were stained until the desired color intensity was obtained, and then rinsed three times with PBST. Bright-field images of embryos were taken on a Leica MZ16 microscope using a Nikon DS-Ri1 camera. Manual z-stacks were taken and assembled using Helicon Focus software.
Section immunohistochemistry
Embryos used for section immunohistochemistry (E9.25–E12.25) were fixed overnight in 4% paraformaldehyde at 4°C, dehydrated in 30% sucrose at 4°C, cryo-embedded in OCT compound, and sectioned at a thickness of 10 μm. Sections were collected on slides and placed in boiling citrate buffer (1M sodium citrate, 1M citric acid at pH 6.0) and allowed to cool on bench top for 40 min. Sections were then blocked in 4% Normal Goat Serum in PBST for 30 min and incubated overnight at 4°C with primary antibody in a humid chamber. Primary antibodies included anti-Cleaved Caspase-3 (1:300, Cell Signaling Technology #9661) and anti-phospho-Histone H3 (1:500, Sigma-Aldrich H0412). Slides were washed in PBS, and then secondary antibodies were added for 1 h at room temperature. Secondary antibodies included Invitrogen Alexa Fluor 488, Invitrogen Alexa Fluor 555, and Invitrogen Alexa Fluor 594 (goat anti-rabbit, applied at 1:500). Slides were then washed in PBS, incubated in DAPI for 15 min at room temperature, washed again with PBS, sealed with ProLong Gold (Invitrogen), and imaged on a Nikon C2+ confocal microscope.
Quantification of cells positive for CC3 or pHH3 immunostaining was performed with Nikon Elements software. Cells were counted using bright spot detection on respective channels. Positive cell numbers were compared to total cells counted (DAPI-stained cells) of the specific area. Regions of interest (ROI) were manually drawn when required and are noted in figures.
Intravascular/intracardiac ink injection
E11.5 mutant Polr1aflox/flox; Wnt1-Cre and their littermate controls were dissected free from the uterus with their yolk sacs intact in Tyrode’s buffer solution at room temperature. Embryos were then incubated in pre-warmed DMEM/F12 media at 37°C and 20% O2; 5% CO2; 75% N2 in roller culture bottles (BTC engineering) for 30 min to ensure maintenance of a robust heartbeat. The embryos were then transferred in warm media to a petri dish and the vitellin vein of the yolk sac, which feeds through the umbilical cord into the sinus venosus of the heart, was injected with undiluted highlighter ink from a liquid Sharpie pen30 using a mouth pipette and pulled glass needle. The embryos were then returned to roller culture for a few minutes to ensure the highlighter ink had circulated throughout the entirety of the embryo and yolk sac, before visualizing with a GFP filter on Leica stereoscope, followed by fixation with 4% PFA and overnight staining in DAPI. Embryos were imaged the next day on a Zeiss 700 upright confocal microscope and processed with Zeiss Zen Blue software.
RNA sequencing and analysis
Telencephalons were dissected from E12.5 Polr1aC1559F/flox; Foxg1-Cre and Polr1anull/flox; Foxg1-Cre embryos as well as control littermates. Dissected tissue was flash-frozen using dry ice and stored at −80°C. RNA was extracted from frozen tissue using Trizol and cleaned post-isolation through an ethanol and sodium acetate precipitation. RNA sequencing was performed by Novogene (Novogene, Davis Lab). The RNA-seq analysis was modeled after the DIYtranscriptomics pipeline. Raw reads were checked for quality control through FastQC. Raw reads were pseudoaligned to the reference mouse transcriptome (Ensembl v.39) in the Ubuntu operating system using Kallisto. Kallisto outputs were imported using tximport, and subsequent analysis was carried out in the statistical computing environment R. Various R packages were used throughout the analysis; notably tidyverse, edgeR, limma, and gprofiler2. Genes expressed less than 1 count per million (CPM) in two or more samples were removed, and the remaining genes were normalized using Trimmed Mean of M-values (TMM). To identify differentially expressed genes, precision weights were first applied to each gene using voom, then data were normalized using the TMM method. Linear modeling and Bayesian stats were used to find genes that were up- or down-regulated by 2-fold or more, with a false-discovery rate (FDR) less than or equal to 0.1. Genes were divided into four modules, based on genotype and up- or down-regulation. Gene ontology (GO) analysis was carried out through the gprofiler2 package for each module.
Statistical analysis
Statistical analysis and data plotting were carried out in the statistical computing environment R. Outliers (>1.5∗IQR) were identified and removed. Unless otherwise stated, statistical significance was determined with t tests: Student’s t test when groups had equal variances and Welch’s t test when groups had unequal variances. When the cause of unequal variance is known (e.g., litter effect), it is stated in the figure legend.
Results
Phenotypes of individuals with POLR1A variants
Clinical or research exome or genome sequencing identified 13 heterozygous rare or novel (previously unreported in gnomAD) variants in POLR1A (GenBank: NM_015425.6) in 18 individuals. This included one sibling pair (individuals 1 and 2 are brothers), two mother/child duos (individuals 4 and 5 and individuals 6 and 7), four individuals with two recurrent de novo variants (individuals 13 and 14 share a variant, as do individuals 15 and 16), and eight additional unique variants each found in one individual. Available facial photographs, location of each variant, and a summary of clinical phenotypes are shown in Figures 1A–1C. Additional, detailed clinical information about each individual can be found in the supplemental informationand Table S5. Nine variants were missense and four were nonsense or frameshift. Seven variants were confirmed to be de novo, three were maternally inherited, four were paternally inherited, and four were of undetermined inheritance. The most common phenotypes present in these individuals were craniofacial anomalies (18/18 with anomalies ranging from isolated hypertelorism to clefting and/or cranial bone abnormalities as described in Figure 1C and Table S5) and abnormal neurodevelopment (14/18 with developmental delay, epilepsy, and/or abnormal muscle tone). One individual had cervico-thoracic syringomelia and ventriculomegaly (individual 6, Figure S1A) and one individual had almost complete acalvaria (individual 8, Figures S1B and S1C). Overall, 6/18 (33%) had epilepsy, 11/18 (61%) had developmental delay, 8/18 (44%) had a congenital heart defect, and 6/18 (33%) had a limb defect. The most severe congenital heart defect was identified in individual 18 (bicuspid aortic valve, aortic and pulmonary artery aneurysm requiring surgical repair, partial atrioventricular canal, and cleft mitral valve). She also had significant craniofacial malformation as well as developmental delay.
There were two recurrent variants identified in our cohort. POLR1A c.4685G>T (p.Cys1562Phe) was identified as a de novo variant in two unrelated individuals with hypertelorism, infantile spasms, and hypotonia. Both progressed to multi-drug resistant epilepsy and have severe global developmental delay. POLR1A c.3988_3990delGAG (p.Glu1330del) was identified as a de novo variant in two unrelated individuals with hypertelorism, epilepsy, and developmental delay. POLR1A c.3649delC (p.Gln1217Argfs∗10) is a third recurrent variant previously reported by Weaver et al.7 and also in individual 1010 in this series, paternally inherited in both cases.
In vitro assessment of POLR1A variants
POLR1A variants affect rRNA transcription in vitro
To identify the functional effects of different variants in POLR1A, we assessed rRNA transcription, nucleolar morphology, and nucleolar patterning in vitro in cells over-expressing wild-type or mutant POLR1A. Prior work using a similar system demonstrated that POLR1A derived from POLR1A c.1777G>C (p.Glu593Gln) (described by Weaver et al.7) not only sequesters wild-type POLR1A and causes it to dissociate from rDNA chromatin, but also itself binds to rDNA chromatin and prevents normal POLR1A from binding. Collectively, this leads to a dominant-negative effect with reduction in rRNA transcription and the formation of large nuclear bodies (nucleolar caps) containing POLR1A.16,31 We studied eight additional variants through transient expression of each variant. As shown in Figures 2A and S2A, endogenous mAID-mClover-tagged POLR1A was knocked down with auxin after transfection with HALO-tagged wild-type and mutant POLR1A. In each of the eight transfection experiments, HALO-tagged POLR1A demonstrated correct localization to the nucleolus but had variable effects on rRNA synthesis. The largest effect on rRNA synthesis as determined by the EU incorporation assay was observed in cells expressing POLR1A c.176A>T(p.Asp59Val), c.3988_3990delGAG (p.Glu1330del), and c.4685G>T (p.Cys1562Phe), all of which clearly demonstrated increased transcription compared to wild-type POLR1A (p value < 2.2e−16, Figure 2B). Expression of two additional variants, c.1488G>T (p.Met496Ile) and c.3721G>A (p.Val1241Ile), also solicited a statistically significant (though quantitatively mild) increase in rRNA transcription (p value = 4.6e−4 and 0.038, respectively). In contrast, expression of c.1178G>A (p.Arg393His) led to a decrease in rRNA transcription (p value = 3.9e−9) similar to but less severe than c.1777G>C (p.Glu593Gln) (p value < 2.2e−16). Expression of the remaining two variants, c.4913C>T (p.Pro1638Leu) and c.4891G>A (p.Val1631Met), did not result in a significant difference in rRNA transcription compared to wild type.
Figure 2.

Variant POLR1A effects on rRNA synthesis and nucleolar morphology
(A) Scheme of rRNA synthesis analysis in a genome-edited HCT116 cell line. rRNA synthesis was monitored by incorporating 5-ethynyl-uridine (EU) in nucleoli after transfecting the plasmid containing a POLR1A variant fused with a HaloTag and subsequently adding the plant auxins, indole-3-acetic acid (IAA), for rapid degradation of endogenous POLR1A fused with mini-AID (mAID) and the fluorescent protein mClover (mAID + mClover). The scale bar represents 5 μm.
(B) Boxplot of fluorescence intensity of EU normalized to HaloTag-POLR1A intensity in cells transiently expressing POLR1A corresponding to wild-type, p.Glu1330del, p.Cys1562Phe, p.Asp59Val, p.Val1631Met, p.Pro1638Leu, p.Met496Ile, p.Val1241Ile, p.Arg393His, and p.Glu593Gln POLR1A. Median values are indicated. p values for statistically significant difference between wild type and each variant are indicated above the boxplot of the mutant (Wilcoxon rank-sum test). n = 782 cells (wild-type POLR1A); n = 551 cells (Glu1330del), p < 2.2 × 10−16; n = 409 cells (Cys1562Phe), p < 2.2 × 10−16; n = 267 cells (Asp59Val), n = 381 cells (Val1631Met), p = 0.26; n = 384 cells (Pro1638Leu), p = 0.91; n = 317 cells (Met496Ile), p = 4.6 × 10−4; n = 496 cells (Val1241Ile), p = 0.038; n = 439 cells (Arg393His), p = 3.9 × 10−9; and n = 28 cells (Glu593Gln), p < 2.2 × 10−16.
(C) Phylogeny shows the similarity of the nucleolar morphologies of wild type and nine POLR1A variants. HaloTag-POLR1A image sets were analyzed with the wndchrm program.
(D) Localization of fluorescently labeled HaloTag-POLR1A variants. Each merged image of DAPI (cyan) and HaloTag-POLR1A variants (magenta) is the image with the average feature score out of a set of HaloTag-POLR1A variant images classified by FLDA with Top 438 features out of 2,919 image features computed by the wndchrm program. The scale bar represents 5 μm.
POLR1A demonstrates variant-specific differences in nucleolar patterning
To test for POLR1A variant-specific effects on localization of the corresponding proteins, we quantified the nucleolar patterning of POLR1A generated from variant and wild-type HALO-tagged POLR1A using the machine-learning algorithm wndchrm. A selection of images used for this analysis is shown in Figure S3. Wndchrm classified the localization patterns of POLR1A foci from wild-type and nine variants (including c.1777G>C [p.Glu593Gln] from Weaver et al.)7 into three groups. These groupings roughly aligned with the rRNA transcription levels quantified by EU incorporation described in the previous section. The first group consists of POLR1A p.Cys1562Phe, p.Glu1330del, and p.Asp59Val, all of which showed increased transcription. The second group includes wild-type, p.Pro1638Leu, and p.Val1631Met (no change in transcription), and p.Val1241Ile and p.Met496Ile (mild increase in transcription). The third group includes p.(Arg393His) and p.Glu593Gln, both of which demonstrated reduced transcription compared to wild type (Figure 2C).
We then further investigated the image features representative of each variant POLR1A’s localization, particularly those in the transcription-upregulated group (p.Cys1562Phe, p.Glu1330del, and p.Asp59Val) and the transcription-downregulated group (p.Arg393His and p.Glu593Gln), using 3D scatter plotting based on Fisher’s linear discriminant analysis (FLDA) (Figure S2B, material and methods). The plot used 438 discriminative image features that were computed and extracted by wndchrm (Figure S2B). Representative images of the average morphology of cells expressing each of the POLR1A variants are found in Figure 2D. The variants that elicited decreased rRNA synthesis (c.1178G>A [p.Arg393His] and c.1777G>C [p.Glu593Gln]) also generated larger and fewer POLR1A foci than wild-type controls, with POLR1A p.Glu593Gln forming the largest and fewest foci. This phenotype is consistent with nucleolar dysfunction (Figure S3). However, these parameters did not yield common features for the transcription-upregulated variants (c.176A>T [p.Asp59Val], c.3988_3990delGAG [p.Glu1330del], and c.4685G>T [p.Cys1562Phe]). Therefore, we selected outlier images from each of these image sets for additional analysis to see whether they might reveal exaggerated morphology. Outlier Halo-tagged POLR1A p.Glu1330del, p.Asp59Val, and p.Cys1562Phe images were chosen based on their distance from others in the 3D scatter plotting (Figures S2B and S4). Analysis of their morphology revealed that foci number is increased compared to wild type and that the foci are extended and linear (indicated as a line-like foci pattern in Figure S4). Taken together, modeling of POLR1A variant in vitro shows differences in nucleolar morphology (based on POLR1A foci) has some correlation with rRNA transcription capacity or status.
In vivo assessment of spatiotemporal requirement for Polr1a
Polr1a expression exhibits spatiotemporal variation throughout embryonic development
To examine whether there are distinct requirements for Polr1a across the different developing tissues that contribute to the diverse phenotypes observed in our human cohort, we first examined the expression of Polr1a in mouse embryos. The murine Polr1atm1a allele consists of the first four exons of Polr1a followed by a splice acceptor, internal ribosomal entry sequence, and LacZ cassette (Figure S5A), allowing for analysis of Polr1a expression via X-gal staining. Embryonic expression of Polr1a is initially ubiquitous at E7.5 and E8.5 but becomes selectively enriched in certain tissues including the branchial arches and frontonasal prominences by E9.5 (Figure S5B), consistent with other recently published data.8 At E10.5, Polr1a remains broadly expressed throughout the embryo with persistent enrichment in the frontonasal prominences, branchial arches, and limb buds as well as the developing heart. This demonstrates the global activity of Polr1a in all tissues during development and the dynamics of elevated expression in the structures affected in the human cohort—face, heart, brain, and limbs.
Polr1anull and Polr1aC1559F homozygosity causes early embryonic lethality
To investigate whether different Polr1a alleles have unique effects on mouse development, we initially chose three distinct alleles for study: a null allele (Polr1atm1a/Polr1atm1d) and two missense variants orthologous to variants in the human cohort, Polr1aC1559F and Polr1aP1635L (orthologous to POLR1A p.Cys1562Phe and p.Pro1638Leu found in individuals 15/16 and 18, respectively). Polr1atm1a is a null allele due to presence of a stop codon after the LacZ cassette and Polr1atm1d contains a multi-exon deletion (Figure S5A). Of note, four individuals in the cohort had variants with predicted premature protein truncation (individuals 3, 9, 10, and 12).
Polr1anull/+ adult mice are healthy and fertile. We intercrossed Polr1anull/+ heterozygotes and identified no Polr1anull/null mutants among 22 E9.5–E10.5 embryos, consistent with a recent study that observed pre-implantation lethality of homozygous null embryos by E3.5 (Table S4).8 Heterozygous animals appeared in expected Mendelian ratios (14/22 heterozygotes, 8/22 WT, chi-square p = 0.7). Polr1anull/+ animals occurred in the expected sex distribution with no apparent abnormal phenotypes (n = 100, chi-square p = 0.258).
Next, we assessed survival and phenotypes of mice with missense variants, Polr1aC1559F and Polr1aP1635L. Adult heterozygotes for both lines appeared at the expected sex distribution and were healthy and fertile. To determine whether animals homozygous for either variant exhibited abnormal phenotypes, we crossed heterozygous mice and collected offspring at E7.5, E8.5, E9.5, P0, and post-weaning. No Polr1aC1559F/C1559F embryos were found (n = 33 embryos, Table S4), consistent with data for homozygous null embryos. In contrast, we found Polr1aP1635L/P1635L mice in expected Mendelian ratios for genotype and sex at weaning (P21) and with normal phenotypes at 6 and 12 months (n = 20, Table S4). We then crossed Polr1aP1635L/+ and Polr1anull/+ mice and found Polr1aP1635L/null adult animals in the expected Mendelian ratios and with normal phenotypes (n = 40). We found similar results with the Polr1aA1632V_P1635L line, with expected Mendelian ratios at weaning for the Polr1aA1632V_P1635L × Polr1aA1632V_P1635L cross (n = 29) as well as homozygous Polr1aA1632V_P1635L/A1632V_P1635L × Polr1anull/+ cross (n = 28). Body morphometrics and echocardiograms of adult Polr1aP1635L/P1635L and Polr1aA1632V_P1635L/A1632V_P1635L animals showed no difference from controls (Figure S6). We therefore concluded that Polr1aC1559F/C1559F causes embryonic lethality (<E7), like the null allele, while Polr1aP1635L/P1635L is well tolerated in mice.
Tissue-specific effects of Polr1a alleles
To circumvent the early lethality of Polr1anull/null embryos and examine tissue-specific effects of loss of Polr1a, we generated Polr1anull/flox; Cre conditional loss-of-function embryos. Further, to circumvent the early lethality of our Polr1aC1559F/C1559F embryos and study tissue-specific effects of Cys1559Phe in vivo, we generated Polr1aC1559F/flox; Cre embryos. Cre drivers were chosen based on the tissues affected in the human cohort. The predominant phenotypes present included craniofacial, cardiac, and neurodevelopmental anomalies. We therefore chose Wnt1-Cre and Sox10-Cre to examine pre-migratory and migratory neural crest cells (NCCs), respectively, as NCCs are critical for craniofacial morphogenesis and also contribute to the cardiac outflow tract.32 Mef2c-AHF-Cre is expressed in the anterior heart field,27 while Foxg1-Cre is expressed in the forebrain.28 Over the course of our experiments, we utilized multiple null alleles of Polr1a (tm1a, tm1d) in combination with the conditional (flox) allele, as well as flox/flox, all of which we confirmed to have identical phenotypes (Figure S5C) and thus for simplicity we will use “null” interchangeably for tm1a, tm1d, and flox in combination with a second flox allele from this point forward. For each cross, we first determined the survival of mutant embryos (Polr1aC1559F/flox; Cre or Polr1anull/flox; Cre) and then proceeded with phenotyping. Survival characteristics for all mutants are summarized in Table S4.
Wnt1-cre-mediated ablation of Polr1a results in craniofacial abnormalities, persistent truncus arteriosus, and mid-gestation lethality
Neural crest cells (NCCs) are a multipotent cell population that are induced at the neural plate border, migrate throughout the embryo, and differentiate into multiple cell types. NCCs contribute to several tissues including the craniofacial cartilage and bone, outflow tract of the heart, vascular smooth muscle cells, and enteric nervous system, among others.33 Wnt1-Cre is expressed in pre-migratory neural crest cells and is used for functional testing and tracing of the NCC lineage.25 Polr1anull/flox; Wnt1-Cre embryos were observed in Mendelian ratios until E12.5 but were not observed at later stages (Table S4). Polr1anull/flox; Wnt1-Cre embryos demonstrate hypoplastic facial prominences evident as early as E9.25, and their appearance becomes more discrepant from wild-type littermates with age progression (Figures 3A and 3B). Lineage tracing using a ROSAdTom/GFP allele revealed a reduction in GFP-marked NCCs in E9.5 Polr1anull/flox; Wnt1-Cre mutants (Figure S7), with almost complete loss of NCCs by E12 (Figure 3A, right). These results are congruent with recently described Polr1aflox/flox; Wnt1-Cre mutants.8 In contrast, Polr1aC1559F/flox; Wnt1-Cre embryos exhibit a less severe phenotype, with some preservation of GFP-labeled NCCs evident at E12.25 (Figure 3A, middle ), suggesting that Polr1a c.4676G>T (p.Cys1559Phe) has a hypomorphic effect compared to Polr1a null. Of note, the abnormal phenotypes of both mutants demonstrated complete penetrance with minimal variability among mutants of same genotype.
Figure 3.

Polr1a craniofacial model
(A) Polr1anull/flox; Wnt1-Cre embryos demonstrate more hypoplastic craniofacial primordia at E12 compared to Polr1aC1559F/flox; Wnt1-Cre. The scale bar represents 1,000 μm.
(B) At E9, cell death (indicated by CC3 accumulation) is prominent in the first arch (dashed white line) of the Polr1aC1559F/flox; Wnt1-Cre mutant. The first arch of the Polr1anull/flox; Wnt1-Cre mutant has less CC3 but overall is smaller than in Polr1aC1559F/flox; Wnt1-Cre at the same stage. The scale bar represents 100 μm.
(C) The outflow tract of Polr1anull/flox; Wnt1-Cre E12 mutants is not septated while the Polr1aC1559F/flox; Wnt1-Cre outflow tract is.
(D) PECAM1 staining shows abnormal vasculature in Polr1anull/flox; Wnt1-Cre embryos at E11.5. The scale bar represents 100 μm.
(E) Ink injections demonstrating Polr1anull/flox; Wnt1-Cre embryos have vascular leakage at E11.5. The scale bar represents 100 μm.
(F) Compact myocardium of Polr1anull/flox; Wnt1-Cre left ventricle is thinner than control at E12 (left). There is no difference in compact myocardium thickness between control and Polr1aC1559F/flox; Wnt1-Cre mutants at E12 (right). Points are individual myocardium measurements and filled diamonds are the average for each embryo, n = 3 for each genotype. Of note, two diamonds are superimposed on the C1559F/flox plot. a, aorta; p, pulmonary trunk. p values calculated via Student’s t test (equal variance, two tailed, two sample). ∗p < 0.05. The scale bar represents 100 μm.
Prior work demonstrated neuroepithelial cell apoptosis in polr1a-null zebrafish7,34 and NCC apoptosis in Polr1aflox/flox; Wnt1-Cre embryos.8 Therefore, we hypothesized that the less severely affected Polr1aC1559F/flox; Wnt1-Cre mutants would exhibit less NCC apoptosis in facial primordia compared to Polr1anull/flox; Wnt1-Cre mutants. Whole-mount immunohistochemistry (WMIHC) with cleaved caspase-3 (CC3) antibody confirmed variably increased apoptosis in the branchial arches and frontonasal prominences of E8.25–9.5 Polr1aC1559F/flox; Wnt1-Cre mutants (n = 3) compared to wild-type embryos. There was variability in CC3 detection with age, with the most robust localization occurring at E9.5 (Figure 3B, middle). Less CC3 was observed at 11-somite and 18-somite stages (Figure S7A). In contrast, Polr1null/flox; Wnt1-Cre embryos have lost almost all NCCs by E10 (n = 4), with markedly smaller branchial arches and frontonasal prominences and fewer CC3-positive cells (Figure 3B, right). However, at E9 (n = 4), there is clearly increased localization of CC3 in the remaining hypoplastic arch, which contains some GFP-marked NCCs (Figure S7B).
The mid-gestation lethality of Polr1anull/flox; Wnt1-Cre mutant embryos is suggestive of cardiac or vascular pathology,35 which we hypothesized was due to perturbed NCC contribution to these organ systems. NCCs form the septum of the outflow tract and contribute to aortic arch arteries. As ablation of NCCs causes outflow tract and looping defects,36 we examined whether Polr1anull/flox; Wnt1-Cre or Polr1aC1559F/flox; Wnt1-Cre mutants displayed structural cardiac anomalies. Cardiac sections from E12 mutants (n = 3) revealed persistent truncus arteriosus in Polr1anull/flox; Wnt1-Cre mutants (Figure 3C, right) while the outflow tract was normally septated in Polr1aC1559F/flox; Wnt1-Cre embryos (Figure 3C, middle). This is consistent with the IHC data, suggesting that Polr1aC1559F causes NCC apoptosis at a slightly later developmental stage than the null allele. To further investigate the cardiovascular status of mutants with conditional deletion of Polr1a in pre-migratory NCCs, we pursued additional investigation of the myocardium in Polr1anull/flox; Wnt1-Cre and Polr1aC1559F/flox; Wnt1-Cre embryos, and vasculature in Polr1aflox/flox; Wnt1Cre embryos.
Immunostaining for PECAM1, a marker of endothelial cells, revealed the presence of hypoplastic branchial arch arteries, and ink injections uncovered diffuse vascular leakage in Polr1aflox/flox; Wnt1Cre embryos at E11.5 (Figure 3D, n = 5; and Figure 3E, n = 3). Given a previous report of ventricular myocardium thinning due to outflow tract anomalies in NCC mutants,37 we quantified myocardial thickness in Polr1anull/flox; Wnt1-Cre and Polr1aC1559F/flox; Wnt1-Cre mutants at E12.5. We observed thinning of the left ventricular wall only in Polr1anull/flox; Wnt1-Cre mutants versus control (n = 3 embryos per genotype, p = 0.022, Figure 3F). Therefore, mid-gestation lethality of Polr1anull/flox; Wnt1-Cre mutants is likely due to abnormal cardiovascular development, which also stems from perturbed NCC development. In contrast, the less severely affected Polr1aC1559F/flox; Wnt1-Cre mutants do not have ventricular thinning. We did not determine the exact timepoint of lethality for the Polr1aC1559F/flox; Wnt1-Cre mutants, but with completely penetrant clefting, the mutants would not survive after birth. Overall, these results indicate that: (1) Polr1a c.4676G>T (p. Cys1559Phe) has a less severe effect than the null allele when expressed in combination with Polr1a conditional allele and Wnt1-Cre; (2) a complete loss of Polr1a is detrimental to pre-migratory NCCs, including those that populate the OFT; and (3) NCC death underlies the hypoplastic craniofacial structures in both mutants.
Sox10-cre-mediated ablation of Polr1a results in less severe phenotypes
Migrating NCCs exhibit a lower rate of rRNA transcription compared to pre-migratory NCCs and thus should be less susceptible to disrupted ribosome biogenesis and NCC death.8 We therefore conditionally deleted Polr1a in migratory NCCs using Sox10-Cre26 and hypothesized that Polr1anull/flox; Sox10-Cre and Polr1aC1559F/flox; Sox10-Cre mutants would have less severe phenotypes than the corresponding Wnt1-Cre mutants.
Polr1aflox/null; Sox10-Cre mutants were recovered in expected Mendelian ratios through E17, demonstrating improved survival relative to Wnt1-Cre mutants. These mutants have completely penetrant yet slightly variable phenotypes. The majority had cleft faces with cleft or absent palate as well as severely under-developed nasal, frontal, and squamosal bones (n = 5 at E17 and n = 3 at E16 mutants, Figures S8A and S8B). Among 19 E14–E17 mutants, 6 had a median mandibular cleft (Figure S8A, bottom). The mandibular length of E17 Polr1anull/flox; Sox10-Cre mutants was significantly shorter than controls (Figure S8B). Concordantly, WMIHC in E9.5 Polr1anull/flox; Sox10-Cre embryos (n = 4 mutants) demonstrated robust CC3 localization in the first branchial arch (Figure S8C). In contrast to Polr1anull/flox; Wnt1-Cre mutants, the outflow tract of the Polr1anull/flox; Sox10-Cre mutants appeared normally septated (Figure S8D).
Given our observation that Polr1aC1559F/flox; Wnt1-Cre embryos are less severely affected than Polr1anull/flox; Wnt1-Cre embryos, we hypothesized the same trend would be observed with Sox10-Cre. Polr1aC1559F/flox; Sox10-Cre embryos had a less severe and more variable phenotype than Polr1anull/flox; Sox10-Cre mutants. Among seven E14–E15 mutants, five had a cleft palate, two of which also had median lip notches and one of which had a cleft face (Figure S8E). The remaining two had normal faces and intact palates (data not shown). This provides further support for Polr1aC1559F having a less severe effect on neural crest cells than Polr1anull.
In summary, NCC death in Sox10-Cre mutants occurs slightly later than in Wnt1-Cre mutants, coincident with later activation of Sox10-Cre in migratory NCCs versus pre-migratory NCCs with Wnt1-Cre. Further, the Polr1a c.4676G>T (p.Cys1559Phe) allele has a less severe effect than the null allele in both migratory and pre-migratory NCCs. This supports the concept of variant-specific effects contributing to phenotypic variability in humans.
Polr1a ablation in the anterior heart field results in loss of right sided structures due to cell death
As described above, Polr1anull/flox; Wnt1-Cre embryos exhibit truncus arteriosus (Figure 3C) consistent with the known role of NCCs in outflow tract septation.36 The heart defects observed in our cohort were primarily septal defects, although individual 18 had more complex congenital heart defects (bicuspid aortic valve, aortic and pulmonary aneurysm, partial AV canal). The second heart field (SHF) is a population of cells adjacent to the primary heart field that migrate into the heart during early organogenesis and are required for formation of the right ventricle, interventricular septum, and smooth muscle of the proximal outflow tract.38 Reciprocal interactions between SHF and NCCs are required for correct patterning of the cardiac valves, chambers, and outflow tract.39 We therefore hypothesized that disruption of the SHF may contribute to congenital heart defects in our cohort. To determine whether Polr1a is required by the SHF, we generated Polr1anull/flox; Mef2c-AHF-Cre mutants. Mef2c is a transcription factor that is expressed specifically by cells in the SHF beginning at approximately E7.5.40 Polr1anull/flox; Mef2c-AHF-Cre mutants survive until E18/P0 (n = 4) but have enlarged hearts (due to blood pooling) with a single ventricle (Figure 4A). Hypoplastic right-sided cardiac structures become evident at mid-gestation, with a shortened outflow tract at E10 (Figure 4C) and primordial right ventricle with a large ventricular septal defect (black arrowhead) at E12 (Figure 4B). Although statistically not significant when quantified, immunohistochemistry with CC3 antibody in E10 cardiac sections does demonstrate an observable focal area of increased apoptosis in the SHF of mutants (Figure 4D) (n = 4 embryos per genotype). Therefore, loss of Polr1a causes cells of the SHF to die, leading to structural cardiac anomalies that reflect the loss of this cell population. While the cardiac phenotype in Polr1anull/flox; Mef2c-AHF-Cre mutants is more severe than in any of the human individuals, the results overall support the association of congenital heart defects with POLR1A variants.
Figure 4.
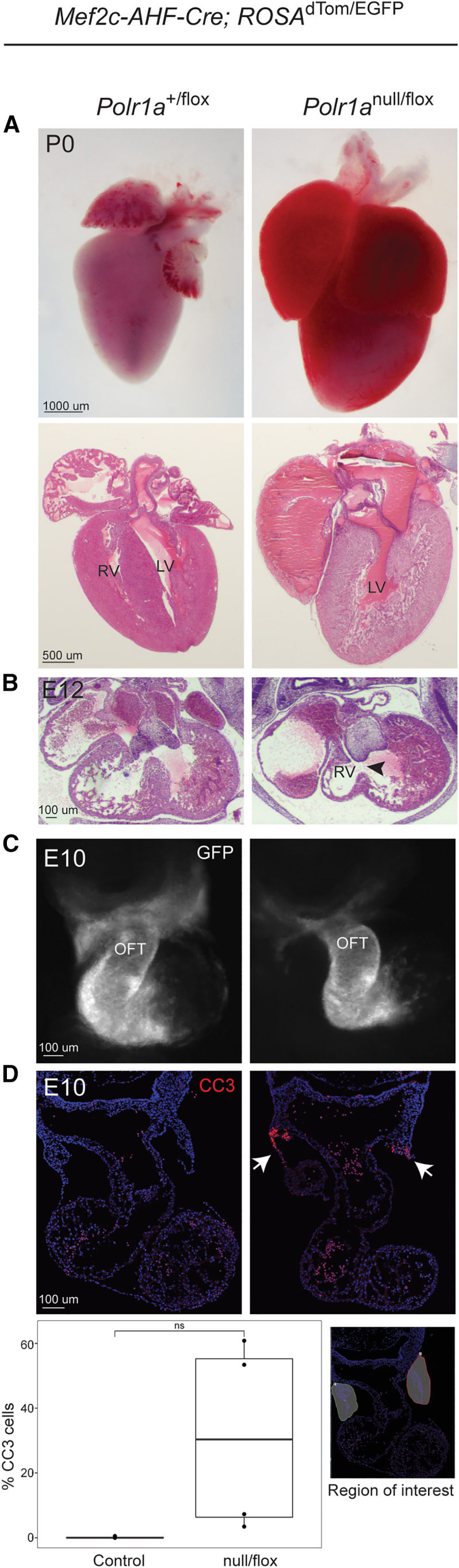
Polr1a cardiovascular model
(A) Hearts from Polr1anull/flox; Mef2c-AHF-Cre neonates (P0) demonstrate gross enlargement compared to control, and histology reveals only a single ventricle is present. The scale bar represents 1,000 μm (top) and 500 μm (bottom).
(B) At E12, Polr1anull/flox; Mef2c-AHF-Cre mutants have an under-developed right ventricle and large ventricular septal defect (black arrowhead). The scale bar represents 100 μm.
(C) At E10 the primordial right ventricle is smaller in width compared to control. The scale bar represents 100 μm.
(D) At E10, section immunohistochemistry demonstrates a trend toward increased abundance of CC3 in the anterior heart field of Polr1anull/flox; Mef2c-AHF-Cre mutants (white arrows). Each genotype had n = 4 and p values were calculated via Welch’s t test (unequal variance, two tailed, two sample). Unequal variance was observed as a litter effect. ns, not significant (p > 0.05). OFT, outflow tract. The scale bar represents 100 μm.
Polr1a ablation in forebrain progenitors results in hypoplastic telencephalon due to cell death
Given that neurodevelopmental abnormalities were observed in 14/18 individuals in our cohort, we next sought to determine the effects of mutant (null and c.4676G>T [p.Cys1559Phe]) Polr1a in neurological lineages. Polr1a c.4676G>T (p.Cys1559Phe) was chosen because the orthologous human variant, c.4685G>T (p.Cys1562Phe), was found as a de novo variant in two unrelated individuals with infantile spasms, hypotonia, and hypertelorism (individuals 15 and 16). Foxg1 is expressed by progenitor cells destined to form the telencephalon,28 and disruptions in FOXG1 are a cause of congenital Rett syndrome.41 Thus, we generated Polr1anull/flox; Foxg1-Cre and Polr1aC1559F/flox; Foxg1-Cre embryos to study forebrain development.
Both mutant genotypes were found in Mendelian ratios at E17. At E17, Polr1anull/flox; Foxg1-Cre embryos demonstrated marked reduction in the size of their telencephalons compared to controls. Polr1aC1559F/flox; Foxg1-Cre embryos demonstrated a less severe but still visually evident and quantifiable reduction (Figure 5A). This same pattern was observed at E14, though less discrepancy in size was present. At E12, there was not a significant difference in forebrain area between the Polr1aC1559F/flox; Foxg1-Cre mutants and controls, though both were significantly larger than Polr1anull/flox; Foxg1-Cre mutants (Figure 5A). The numbers of embryos per genotype used for analysis at each stage are depicted on the corresponding graphs in Figure 5A. Thus, conditional deletion of Polr1a in the telencephalon leads to a marked decrease in telencephalon size, while the presence of Polr1aC1559F ameliorates the phenotype, consistent with the Polr1aC1559F and null alleles having variable effects.
Figure 5.

Polr1a central nervous system model
(A) At E12, cerebral cortex area is reduced in Polr1anull/flox; Foxg1-Cre compared to both Polr1aC1559F/flox; Foxg1-Cre and control. At E14 and E17, the Polr1anull/flox mutant cortex is smaller than Polr1aC1559F/flox, and both are smaller than control. Numbers of embryos per genotype are shown on the graph for each stage. Comparisons were made via Welch’s t test (unequal variance, 2-tailed, 2-sample) for E17 and E14 embryos and Student’s t test (equal variance, 2-tailed, 2-sample) for E12 embryos. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005, and ∗∗∗∗p < 0.00005. The scale bars represent 1,000 μm (top), 500 μm (middle), and 1,000 μm (bottom).
(B) At E12, there is a statistically significant increase in CC3 detected in Polr1aC1559F/flox; Foxg1-Cre cortex compared to control (∗p < 0.05). There is also increased CC3 detected in Polr1anull/flox; Foxg1-Cre cortex compared to control though it does not reach statistical significance (ns, not significant). The Polr1anull/flox; Foxg1-Cre comparison had n = 4 embryos per genotype, and the Polr1aC1559F/flox; Foxg1-Cre comparison had n = 5 embryos per genotype. p values were calculated by Welch’s t test (unequal variance, two tailed, two sample). Bottom row shows close-up images of CC3 (red channel) in the areas indicated by white boxes in the top row images. The scale bars represent 100 μm (top) and 50 μm (bottom).
(C) Bulk RNA-seq of brains from both mutants at E12 demonstrates a subset of differentially regulated (DE) genes between each mutant and controls.
(D) Gene ontology analysis of DE genes shows increased expression of genes involved in apoptosis in Polr1aC1559F/flox; Foxg1-Cre mutants and reduced expression of genes involved in forebrain neuron differentiation and transcription regulation in Polr1anull/flox; Foxg1-Cre mutants.
To understand the mechanisms underlying the reduced size of the telencephalon at E17, we examined cell death using IHC for CC3 at E12. We observed a statistically significant increase in CC3 localized to Polr1aC1559F/flox; Foxg1-Cre E12 telencephalons compared to controls, with the pattern consistent with Foxg1 expression (n = 4 mutants, Figure 5B).21 In contrast, although there was a quantifiable increase in the percentage of CC3-positive cells in Polr1anull/flox; Foxg1-Cre mutants compared to controls, the difference was not statistically significant (n = 4 mutants, Figure 5B). Overall, the data demonstrate hypoplastic telencephalons in both mutants compared to controls with a more severe effect in embryos with a null allele compared to those with Polr1aC1559F.
To complement our IHC experiments, we performed bulk RNA sequencing on E12.5 telencephalon RNA to study gene expression differences between wild-type, Polr1anull/flox; Foxg1-Cre, and Polr1aC1559F/flox; Foxg1-Cre embryos (Figures 5C and 5D). Polr1anull/flox; Foxg1-Cre telencephalons show downregulated forebrain neuronal differentiation and transcription, while Polr1aC1559F/flox; Foxg1-Cre telencephalons exhibit upregulation of apoptosis and downregulation of rRNA processing compared to controls. Interestingly, GO analysis did not identify apoptosis as a major pathway in Polr1anull/flox; Foxg1-Cre telencephalon. This is consistent with our IHC results that identified a significant increase in CC3 in Polr1aC1559F/flox; Foxg1-Cre but not Polr1anull/flox; Foxg1-Cre telencephalons at the same timepoint. This may also align with our results in the Wnt1-Cre and Sox10-Cre mutants, wherein mutants with Polr1aC1559F exhibited NCC apoptosis later than mutants with Polr1anull. These experiments demonstrate a deleterious effect of both null and Polr1aC1559F alleles on forebrain development and further support Polr1aC1559F having a distinct effect from the null allele.
Discussion
To date, heterozygous pathogenic variants in POLR1A have been reported in five individuals with mandibulofacial dysostosis and variable limb defects, collectively referred to as acrofacial dysostosis, Cincinnati-type.7,9,10 Here we report a detailed phenotypic analysis of 17 previously unreported individuals and additional information on one previously reported individual, significantly expanding the human phenotypes associated with heterozygous variants in POLR1A. Consistent with prior reports, we note that craniofacial differences are the most prevalent phenotype and limb defects are variably present. However, we also note a significant rate of neurologic abnormalities ranging from mild delays to refractory epilepsy. While none of the individuals in our cohort were diagnosed with leukodystrophy, bi-allelic pathogenic variants in POLR1A have been described in two brothers with leukodystrophy providing some prior evidence of POLR1A involvement in the central nervous system.11 Interestingly, bi-allelic pathogenic variants encoding another subunit of Pol I, POLR1C, are associated with both TCS and leukodystrophy. Most often the craniofacial and leukodystrophy phenotypes have been mutually exclusive, although there was recently a single case of an individual with bi-allelic variants in POLR1C who had both craniofacial and leukodystrophy phenotypes.42 This provides further evidence for the role of Pol I-mediated rRNA transcription and ribosome biogenesis in the combined pathogenesis of abnormal craniofacial and neurologic development.
In addition to neurologic abnormalities, we also note an increased incidence of congenital heart defects in our expanded cohort. This primarily consist of septal defects although individual 18 had more significant disease: aortic and pulmonary artery aneurysms requiring surgical repair, and polyvalvular abnormalities. Compared to the general population, the incidence of congenital heart defects is also higher among individuals with TCS2 and with Diamond-Blackfan anemia (DBA), which are caused by pathogenic variants in genes encoding Pol I subunits and associated factors or ribosomal proteins, respectively.13,43,44,45,46
Compromised cellular function of individual POLR1A variants was determined by expression of mutant POLR1A in cell culture. Ribosomal RNA transcription, which is performed by Pol I in the nucleolus, is a rate-limiting step for ribosome biogenesis.47 POLR1A is the largest subunit of Pol I and together with POLR1B forms the catalytic core of the polymerase. Our in vitro assessment of eight POLR1A variants revealed variable effects on the rate of rRNA synthesis and configuration of POLR1A in the nucleoli in six of the eight variants tested. Increased rRNA synthesis was clearly observed with expression of variants encoding POLR1A p.Asp59Val, p.Cys1562Phe, and p.Glu1330del. Further, the nucleoli of cells expressing these variants show more foci with a more linear configuration than wild type. While the significance of the linear configuration is unknown, machine learning analysis confirmed these nucleolar morphologies to be different from wild type and other variants tested that did not have increased rRNA synthesis. Two of these “transcription-up” variants, which correspond to POLR1A p.Cys1562Phe and p.Glu1330del, each occurred independently in two individuals with striking, primarily neurologic phenotypes (individuals 13, 14, 15, and 16). The third, p.Asp59Val, occurred in two brothers with primarily craniofacial phenotypes (individuals 1 and 2). Individuals 13, 14, 15, and 16 all have craniofacial dysmorphisms but did not have structural craniofacial defects, and all four have epilepsy, described as intractable in three individuals. Thus, their phenotype can be described as primarily neurologic in comparison to individuals 1 and 2 who have clefting and craniosynostosis without neurologic abnormalities. The cells expressing POLR1A variants encoding POLR1A p.Met496Ile and p.Val1241Ile demonstrated only a slight increase in rRNA transcription compared to wild type and had minimal effect on nucleolar morphology. POLR1A p.Met496Ile was found in an individual with severe craniofacial and neurologic anomalies (individual 8 with acalvaria, infantile spasms, and developmental delay) while the p.Val1241Ile variant was found in an individual with a primarily neurologic phenotype (individual 11 with spastic dystonia). Cells expressing the remaining variant, corresponding to POLR1A p.Arg393His, had reduced rRNA transcription and formed fewer and larger nucleolar foci than wild-type protein, consistent with nucleolar dysfunction. Interestingly, this resembles (but is less severe than) the in vitro effect of POLR1A p.Glu593Gln, which was previously investigated in vitro by Ide et al.16 This variant occurred as a de novo event in one of the first individuals described with AFD-CIN who had a severe craniofacial phenotype. The craniofacial phenotypes of individuals 4 and 5 (mother and son) with POLR1A p.Arg393His are much less severe than the individual with p.Glu593Gln (1A1 in Weaver et al.),7,16 which may be consistent with a less severe reduction in rRNA synthesis. Putting this all together, we propose that variants causing increased rRNA transcription may be more likely to cause neurologic phenotypes while those causing decreased rRNA transcription are more associated with craniofacial phenotypes. However, combinations of phenotypes in individuals with transcription-up or transcription-down variants indicate that there are other mechanisms of pathogenicity. This is further supported by an apparent lack of effect on rRNA transcription or nucleolar morphology for POLR1A p.Pro1638Leu and p.Val1631Met, which were both found in individuals with craniofacial, cardiac, skeletal, and neurologic phenotypes. An alternative and/or complementary possibility is that perturbation of ribosome biogenesis in either direction (increase or decrease) is damaging in early embryogenesis, given evidence that ribosome biogenesis is a dynamic and spatiotemporally regulated process.8,48
Our in vivo mouse studies of an allelic series of Polr1a variants further support the important role of this gene in normal development across multiple tissues including neural crest, second heart field, and neuronal progenitors. Conditional deletion of Polr1a was detrimental to cell survival in all lineages tested, suggesting that loss of function of Polr1a is a plausible mechanism of pathogenicity for the structural birth defects observed in our human cohort. Interestingly, Polr1aC1559F led to different developmental consequences when expressed in combination with conditional knock-out of Polr1a in the same lineages, consistent with the results of in vitro studies and phenotypes of two unrelated individuals with the orthologous variant, POLR1A p.Cys1562Phe. Finally, Polr1aP1635L, orthologous to POLR1A p.Pro1638Leu, is well tolerated by mice, which is distinct from Polr1anull and Polr1aC1559F, both of which are embryonic lethal in homozygosity. In vitro studies similarly did not reveal an effect on rRNA transcription or nucleolar morphology with the corresponding p.Pro1638Leu variant. While this raises the possibility that this variant is not causal for the individual’s phenotype, there is currently no alternative or better explanation identified on her exome. Further, analysis using different inheritance models (i.e., autosomal de novo, homozygous recessive, compound heterozygous, X-linked de novo, X-linked recessive) did not identify other reportable variants.
Our large cohort allows us to detect evidence of reduced penetrance or extreme variability in the phenotypes of individuals with heterozygous POLR1A variants. Most notably, five individuals (1, 2, 9, 10, and 12) inherited their variant from a reportedly unaffected parent. Given the still-small numbers of reported affected individuals, it remains to be determined whether some parents heterozygous for a POLR1A variant found in their child truly are non-penetrant, versus having minor craniofacial features that may be hard to appreciate clinically. Extremely mild phenotypes in dominant genetic syndromes are well established for multiple genes, including TCOF1 and POLR1D.2,3 Consistent with these ideas, the results of our in vitro and in vivo studies demonstrate that specific genetic variants cannot fully explain phenotypic expression, and as with other dominant genetic conditions, genetic and environmental modifiers likely impact disease expression in humans. The identification of a second de novo likely pathogenic variant in a gene associated with variable neurologic phenotypes (ATP1A1) in individual 13 is an exemplary case, as we suspect that there may be some contribution to his phenotype from both (POLR1A and ATP1A1) variants.
It is of interest that the loss-of-function POLR1A variants seem to be associated with more extreme phenotypic variability, in that among the five individuals who inherited four variants from four asymptomatic parents (individuals 1 and 2 are siblings), three of four variants are predicted to cause loss of function (premature termination). However, these truncating variants all affected the distal (C-terminal) portion of the protein, whereas the other individual in the cohort with a truncating variant (individual 3) died in infancy from medical complications but had a de novo early-truncating variant.
Our study has a number of limitations. Individual phenotype information was collected retrospectively over a number of years at different institutions and thus individuals were not systematically evaluated by the same medical provider(s), an inherent problem when collating information about individuals with rare disease. Given the early embryonic lethality of homozygous null and homozygous missense mice, we used a combined in vivo (conditional deletion) and in vitro approach to evaluate the effects of Polr1a alleles on development, rRNA transcription, and nucleolar morphology. This contrasts with the individuals in this cohort, who are all heterozygous for their respective POLR1A variants. POLR1A demonstrates significant constraint against loss-of-function variants in the general population; therefore, it is likely that bi-allelic null variants in POLR1A would be incompatible with survival in humans as well.49 In further support of this idea, few reports of individuals with bi-allelic variants in POLR1A exist. There is one report of two brothers who are homozygous for missense POLR1A variants. Finally, the majority of the variants were studied in vitro which by definition removes developmental spatiotemporal variables. However, it is not economically feasible to generate knock-in mouse models for such a large number of variants. Our prioritized approach thus likely represents the best way to evaluate developmental and functional effects of an allelic series of POLR1A variants.
In conclusion, we have described a large cohort of individuals with heterozygous variants in POLR1A and used a combination of in vivo and in vitro models to demonstrate that reduced function of POLR1A leads to craniofacial, neurologic, and cardiovascular abnormalities. Our work represents a comprehensive assessment of the role of Polr1a in mammalian development and provides evidence that there are at least some variant-specific effects contributing to phenotypic variability among individuals with heterozygous variants in POLR1A. The phenotype originally described as acrofacial dysostosis, Cincinnati-type, constitutes only a subset of presentations now associated with heterozygous variants in POLR1A. Further work in our Polr1aC1559F mouse model could lead to targeted therapies, particularly for the drug-resistant epilepsy found in some individuals.
Acknowledgments
We thank the individuals and families for their willingness to be included in this manuscript, the CCHMC TG Core for generation of knock-in Polr1a lines, and the CICRL for performing echocardiography. We thank Katherine Yutzey, PhD, for her critical review of the manuscript and insightful discussions concerning data and conclusions. We thank Gang Wu, PhD, for assistance with RNA-seq analysis. We thank Hiroko Ochi and Hiroshi Tamano for sample preparation for the in vitro assessment of rRNA transcription and assistance with wndchrm’s initial setup, respectively.
This work was funded by the National Institutes of Health National Heart Lung and Blood Institute K08 HL143177-01A1 (K.N.W.), the National Institute of Child Health and Human Development K12 HD028827 (K.N.W.), Cincinnati Children’s Research Foundation Center for Pediatric Genomics Pilot Proposal (K.N.W.), the Cleft Palate Foundation’s Paul W. Black Grant for Emerging Researchers (K.N.W.), the National Institute of Dental and Craniofacial Research K99 (K.E.N.W.), and the Stowers Institute for Medical Research 1008 (P.A.T.). Operational Program Integrated Infrastructure, funded by the ERDF (ITMS2014+:313011V455) was provided to M.S., and M.Z. receives research support from the German Research Foundation (DFG 458949627; ZE 1213/2-1). The Acute Care Genomics study is funded by grants from the Australian Government’s Medical Research Futures Fund (GHFM76747) and the Royal Children's Hospital Foundation (2020-1259). In vitro work was funded by the Japan Society for the Promotion of Science (JSPS) grants (22H05606, 21H02535 to S.I.; 19H05273, 20H05936 to K. Maeshima), JST CREST (JPMJCR15G2 to K. Maeshima), the Takeda Science Foundation (K. Maeshima), and the Uehara Memorial Foundation (K. Maeshima).
Declaration of interests
A.B., M.J.G.S., K. McWalters, R.P., and R.S. are employees of GeneDx.
Published: April 18, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.03.014.
Web resources
DIY.transcriptomics, https://doi.org/10.1128/mBio.01214-21
Ensembl, https://doi.org/10.1093/nar/gkab1049
FastQC, v.0.11.9, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
gprofiler2, https://doi.org/10.12688/f1000research.24956.2
Image J, https://imagej.net/software/fiji/
Kallisto, v.46.2, https://doi.org/10.1038/nbt.3519
knitr, https://yihui.org/knitr/
Nikon Elements Software, https://www.microscope.healthcare.nikon.com/products/software/nis-elements
Python, https://www.python.org/
R, v.4.2.0, https://www.r-project.org/
rmarkdown, https://rmarkdown.rstudio.com
scikit-learn, https://scikit-learn.org/stable/
tidyverse, https://doi.org/10.21105/joss.01686
TMM method, https://doi.org/10.1186/gb-2010-11-3-r25
Vevo LAB, v.5.6.1 (Build 2257), 2021, https://www.visualsonics.com/product/software/vevo-lab
Zeiss Zen Blue Software, https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html
Supplemental information
Data and code availability
The accession number for the raw RNA-seq reads and metadata reported in this paper is located in the Gene Expression Omnibus (GEO: GSE206197). All Linux and R scripts, and their outputs, have been archived to the Zenodo repository (Zenodo: 10.5281/zenodo.7022932).
Original data underlying this manuscript that was generated at the Stowers Institute can be accessed from the Stowers Original Data Repository (Stowers: LIBPB-2376).
References
- 1.Misiaszek A.D., Girbig M., Grötsch H., Baudin F., Murciano B., Lafita A., Müller C.W. Cryo-EM structures of human RNA polymerase I. Nat. Struct. Mol. Biol. 2021;28:997–1008. doi: 10.1038/s41594-021-00693-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vincent M., Geneviève D., Ostertag A., Marlin S., Lacombe D., Martin-Coignard D., Coubes C., David A., Lyonnet S., Vilain C., et al. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet. Med. 2016;18:49–56. doi: 10.1038/gim.2015.29. [DOI] [PubMed] [Google Scholar]
- 3.Dauwerse J.G., Dixon J., Seland S., Ruivenkamp C.A.L., van Haeringen A., Hoefsloot L.H., Peters D.J.M., Boers A.C.d., Daumer-Haas C., Maiwald R., et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 2011;43:20–22. doi: 10.1038/ng.724. [DOI] [PubMed] [Google Scholar]
- 4.Schaefer E., Collet C., Genevieve D., Vincent M., Lohmann D.R., Sanchez E., Bolender C., Eliot M.M., Nürnberg G., Passos-Bueno M.R., et al. Autosomal recessive POLR1D mutation with decrease of TCOF1 mRNA is responsible for Treacher Collins syndrome. Genet. Med. 2014;16:720–724. doi: 10.1038/gim.2014.12. [DOI] [PubMed] [Google Scholar]
- 5.Thiffault I., Wolf N.I., Forget D., Guerrero K., Tran L.T., Choquet K., Lavallée-Adam M., Poitras C., Brais B., Yoon G., et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat. Commun. 2015;6:7623. doi: 10.1038/ncomms8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez E., Laplace-Builhé B., Mau-Them F.T., Richard E., Goldenberg A., Toler T.L., Guignard T., Gatinois V., Vincent M., Blanchet C., et al. POLR1B and neural crest cell anomalies in Treacher Collins syndrome type 4. Genet. Med. 2020;22:547–556. doi: 10.1038/s41436-019-0669-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weaver K.N., Watt K.E.N., Hufnagel R.B., Navajas Acedo J., Linscott L.L., Sund K.L., Bender P.L., König R., Lourenco C.M., Hehr U., et al. Acrofacial Dysostosis, Cincinnati Type, a Mandibulofacial Dysostosis Syndrome with Limb Anomalies, Is Caused by POLR1A Dysfunction. Am. J. Hum. Genet. 2015;96:765–774. doi: 10.1016/j.ajhg.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falcon K.T., Watt K.E.N., Dash S., Zhao R., Sakai D., Moore E.L., Fitriasari S., Childers M., Sardiu M.E., Swanson S., et al. Dynamic regulation and requirement for ribosomal RNA transcription during mammalian development. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2116974119. e2116974119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shenoy R.D., Shetty V., Dheedene A., Menten B., Pandyanda Nanjappa D., Chakraborty G., Sips P., de Paepe A., Callewaert B., Chakraborty A. Phenotypic and Molecular Heterogeneity in Mandibulofacial Dysostoses: A Case Series From India. Cleft Palate Craniofac J. 2021;59:1346–1351. doi: 10.1177/10556656211050006. [DOI] [PubMed] [Google Scholar]
- 10.da Rocha L.A., Pires L.V.L., Yamamoto G.L., Magliocco Ceroni J.R., Honjo R.S., de Novaes França Bisneto E., Oliveira L.A.N., Rosenberg C., Krepischi A.C.V., Passos-Bueno M.R., et al. Congenital limb deficiency: Genetic investigation of 44 individuals presenting mainly longitudinal defects in isolated or syndromic forms. Clin. Genet. 2021;100:615–623. doi: 10.1111/cge.14041. [DOI] [PubMed] [Google Scholar]
- 11.Kara B., Köroğlu Ç., Peltonen K., Steinberg R.C., Maraş Genç H., Hölttä-Vuori M., Güven A., Kanerva K., Kotil T., Solakoğlu S., et al. Severe neurodegenerative disease in brothers with homozygous mutation in POLR1A. Eur. J. Hum. Genet. 2017;25:315–323. doi: 10.1038/ejhg.2016.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edvardson S., Nicolae C.M., Agrawal P.B., Mignot C., Payne K., Prasad A.N., Prasad C., Sadler L., Nava C., Mullen T.E., et al. Heterozygous De Novo UBTF Gain-of-Function Variant Is Associated with Neurodegeneration in Childhood. Am. J. Hum. Genet. 2017;101:267–273. doi: 10.1016/j.ajhg.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gazda H.T., Sheen M.R., Vlachos A., Choesmel V., O'Donohue M.F., Schneider H., Darras N., Hasman C., Sieff C.A., Newburger P.E., et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet. 2008;83:769–780. doi: 10.1016/j.ajhg.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ide S., Imai R., Ochi H., Maeshima K. Transcriptional suppression of ribosomal DNA with phase separation. Sci. Adv. 2020;6:eabb5953. doi: 10.1126/sciadv.abb5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shamir L., Orlov N., Eckley D.M., Macura T., Johnston J., Goldberg I.G. Wndchrm - an open source utility for biological image analysis. Source Code Biol. Med. 2008;3:13. doi: 10.1186/1751-0473-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsumori H., Watanabe K., Tachiwana H., Fujita T., Ito Y., Tokunaga M., Sakata-Sogawa K., Osakada H., Haraguchi T., Awazu A., et al. Ribosomal protein L5 facilitates rDNA-bundled condensate and nucleolar assembly. Life Sci. Alliance. 2022;5:e202101045. doi: 10.26508/lsa.202101045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felsenstein J. PHYLIP: Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 21.Skarnes W.C., Rosen B., West A.P., Koutsourakis M., Bushell W., Iyer V., Mujica A.O., Thomas M., Harrow J., Cox T., et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan C.L., Hu Y.C. A Transgenic Core Facility's Experience in Genome Editing Revolution. Adv. Exp. Med. Biol. 2017;1016:75–90. doi: 10.1007/978-3-319-63904-8_4. [DOI] [PubMed] [Google Scholar]
- 23.Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott M.A., Hu Y.C. Generation of CRISPR-Edited Rodents Using a Piezo-Driven Zygote Injection Technique. Methods Mol. Biol. 2019;1874:169–178. doi: 10.1007/978-1-4939-8831-0_9. [DOI] [PubMed] [Google Scholar]
- 25.Danielian P.S., Muccino D., Rowitch D.H., Michael S.K., McMahon A.P. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 26.Matsuoka T., Ahlberg P.E., Kessaris N., Iannarelli P., Dennehy U., Richardson W.D., McMahon A.P., Koentges G. Neural crest origins of the neck and shoulder. Nature. 2005;436:347–355. doi: 10.1038/nature03837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verzi M.P., McCulley D.J., De Val S., Dodou E., Black B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005;287:134–145. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 28.Kawaguchi D., Sahara S., Zembrzycki A., O'Leary D.D.M. Generation and analysis of an improved Foxg1-IRES-Cre driver mouse line. Dev. Biol. 2016;412:139–147. doi: 10.1016/j.ydbio.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muzumdar M.D., Tasic B., Miyamichi K., Li L., Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 30.Takase Y., Tadokoro R., Takahashi Y. Low cost labeling with highlighter ink efficiently visualizes developing blood vessels in avian and mouse embryos. Dev. Growth Differ. 2013;55:792–801. doi: 10.1111/dgd.12106. [DOI] [PubMed] [Google Scholar]
- 31.Ide S., Tamura S., Maeshima K. Chromatin behavior in living cells: Lessons from single-nucleosome imaging and tracking. Bioessays. 2022;44:e2200043. doi: 10.1002/bies.202200043. [DOI] [PubMed] [Google Scholar]
- 32.Trainor K., Watt K. In: Neural Crest Cells: Evolution, Development and Disease. Trainor P.A., editor. Elsevier; 2014. Neurocristopathies; pp. 361–394. [Google Scholar]
- 33.Gandhi S., Bronner M.E. Insights into neural crest development from studies of avian embryos. Int. J. Dev. Biol. 2018;62:183–194. doi: 10.1387/ijdb.180038sg. [DOI] [PubMed] [Google Scholar]
- 34.Watt K.E.N., Neben C.L., Hall S., Merrill A.E., Trainor P.A. tp53-dependent and independent signaling underlies the pathogenesis and possible prevention of Acrofacial Dysostosis-Cincinnati type. Hum. Mol. Genet. 2018;27:2628–2643. doi: 10.1093/hmg/ddy172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conway S.J., Kruzynska-Frejtag A., Kneer P.L., Machnicki M., Koushik S.V. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35:1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- 36.Yelbuz T.M., Waldo K.L., Kumiski D.H., Stadt H.A., Wolfe R.R., Leatherbury L., Kirby M.L. Shortened outflow tract leads to altered cardiac looping after neural crest ablation. Circulation. 2002;106:504–510. doi: 10.1161/01.cir.0000023044.44974.8a. [DOI] [PubMed] [Google Scholar]
- 37.Stottmann R.W., Choi M., Mishina Y., Meyers E.N., Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–2218. doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waldo K.L., Hutson M.R., Ward C.C., Zdanowicz M., Stadt H.A., Kumiski D., Abu-Issa R., Kirby M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005;281:78–90. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 39.Kimberly E., Inman M.E., Bronner-Fraser B., Trainor P.A. In: Heart Development and Regeneration. Rosenthal N., Harvey R., editors. Elsevier; 2010. Role of Cardiac Neural Crest Cells in Morphogenesis of the Heart and Great Vessels; pp. 417–439. [Google Scholar]
- 40.Edmondson D.G., Lyons G.E., Martin J.F., Olson E.N. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994;120:1251–1263. doi: 10.1242/dev.120.5.1251. [DOI] [PubMed] [Google Scholar]
- 41.Ariani F., Hayek G., Rondinella D., Artuso R., Mencarelli M.A., Spanhol-Rosseto A., Pollazzon M., Buoni S., Spiga O., Ricciardi S., et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 2008;83:89–93. doi: 10.1016/j.ajhg.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gauquelin L., Cayami F.K., Sztriha L., Yoon G., Tran L.T., Guerrero K., Hocke F., van Spaendonk R.M.L., Fung E.L., D'Arrigo S., et al. Clinical spectrum of POLR3-related leukodystrophy caused by biallelic POLR1C pathogenic variants. Neurol. Genet. 2019;5:e369. doi: 10.1212/NXG.0000000000000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gripp K.W., Curry C., Olney A.H., Sandoval C., Fisher J., Chong J.X.L., UW Center for Mendelian Genomics. Pilchman L., Sahraoui R., Stabley D.L., Sol-Church K. Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am. J. Med. Genet. 2014;164A:2240–2249. doi: 10.1002/ajmg.a.36633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landowski M., O'Donohue M.F., Buros C., Ghazvinian R., Montel-Lehry N., Vlachos A., Sieff C.A., Newburger P.E., Niewiadomska E., Matysiak M., et al. Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond-Blackfan anemia. Hum. Genet. 2013;132:1265–1274. doi: 10.1007/s00439-013-1326-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pradat P., Francannet C., Harris J.A., Robert E. The epidemiology of cardiovascular defects, part I: a study based on data from three large registries of congenital malformations. Pediatr. Cardiol. 2003;24:195–221. doi: 10.1007/s00246-002-9401-6. [DOI] [PubMed] [Google Scholar]
- 46.Willig T.N., Niemeyer C.M., Leblanc T., Tiemann C., Robert A., Budde J., Lambiliotte A., Kohne E., Souillet G., Eber S., et al. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Societe d'Hematologie et d'Immunologie Pediatrique (SHIP), Gesellshaft fur Padiatrische Onkologie und Hamatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI) Pediatr. Res. 1999;46:553–561. doi: 10.1203/00006450-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 47.Laferté A., Favry E., Sentenac A., Riva M., Carles C., Chédin S. The transcriptional activity of RNA polymerase I is a key determinant for the level of all ribosome components. Genes Dev. 2006;20:2030–2040. doi: 10.1101/gad.386106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chau K.F., Shannon M.L., Fame R.M., Fonseca E., Mullan H., Johnson M.B., Sendamarai A.K., Springel M.W., Laurent B., Lehtinen M.K. Downregulation of ribosome biogenesis during early forebrain development. Elife. 2018;7:e36998. doi: 10.7554/eLife.36998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the raw RNA-seq reads and metadata reported in this paper is located in the Gene Expression Omnibus (GEO: GSE206197). All Linux and R scripts, and their outputs, have been archived to the Zenodo repository (Zenodo: 10.5281/zenodo.7022932).
Original data underlying this manuscript that was generated at the Stowers Institute can be accessed from the Stowers Original Data Repository (Stowers: LIBPB-2376).